 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A lithium–air battery and gas handling system demonstrator†
Jack W.
Jordan
 ab,
Ganesh
Vailaya
a,
Conrad
Holc
a,
Max
Jenkins
bc,
Rory C.
McNulty
ab,
Constantin
Puscalau
d,
Begum
Tokay
d,
Andrea
Laybourn
d,
Xiangwen
Gao
c,
Darren A.
Walsh
ab,
Graham N.
Newton
ab,
Peter G.
Bruce
bc and
Lee R.
Johnson
*ab
ab,
Ganesh
Vailaya
a,
Conrad
Holc
a,
Max
Jenkins
bc,
Rory C.
McNulty
ab,
Constantin
Puscalau
d,
Begum
Tokay
d,
Andrea
Laybourn
d,
Xiangwen
Gao
c,
Darren A.
Walsh
ab,
Graham N.
Newton
ab,
Peter G.
Bruce
bc and
Lee R.
Johnson
*ab
aNottingham Applied Materials and Interfaces Group, School of Chemistry, University of Nottingham, Nottingham, NG7 2TU, UK. E-mail: lee.johnson@nottingham.ac.uk
bThe Faraday Institution, Harwell Campus, Didcot, OX11 0RA, UK
cDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
dFaculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK
First published on 18th July 2023
Abstract
The lithium–air (Li–air) battery offers one of the highest practical specific energy densities of any battery system at >400 W h kgsystem−1. The practical cell is expected to operate in air, which is flowed into the positive porous electrode where it forms Li2O2 on discharge and is released as O2 on charge. The presence of CO2 and H2O in the gas stream leads to the formation of oxidatively robust side products, Li2CO3 and LiOH, respectively. Thus, a gas handling system is needed to control the flow and remove CO2 and H2O from the gas supply. Here we present the first example of an integrated Li–air battery with in-line gas handling, that allows control over the flow and composition of the gas supplied to a Li–air cell and simultaneous evaluation of the cell and scrubber performance. Our findings reveal that O2 flow can drastically impact the capacity of cells and confirm the need for redox mediators. However, we show that current air–electrode designs translated from fuel cell technology are not suitable for Li–air cells as they result in the need for higher gas flow rates than required theoretically. This puts the scrubber under a high load and increases the requirements for solvent saturation and recapture. Our results clarify the challenges that must be addressed to realise a practical Li–air system and will provide vital insight for future modelling and cell development.
Introduction
The lithium–air (Li–air) battery has a theoretical material-level specific energy of 3500 W h kg−1, making it a leading next-generation electrochemical energy storage technology for high-energy applications.1–4 The Li–air battery exploits the two-electron reduction of O2 at a porous, carbon positive electrode, forming Li2O2, with the concomitant oxidation of Li metal at the negative electrode.5 During charging, the reactions are reversed, reforming O2 and Li. However, the battery faces significant challenges,3,6,7 such as degradation of the electrolyte during operation,8–12 slow electrochemistry due to the insulating nature of Li2O2,13–15 resulting in the need for homogeneous redox mediators to oxidise Li2O2,16–22 instability of Li metal23 and the need for anode-protection layers.24 Significant progress has been made in these areas, however, most studies of the Li–air battery involve the use of pure O2 gas as the feedstock for the positive electrode and often only low-capacity systems (<1 mA h cm−2) are explored. Some examples of more practical cell configurations, albeit without gas-handling systems, have been reported.25 Kubo and co-workers described a multilayer pouch cell that could store 150 W h kgcell−1 at 0.5 mA h cm−2,26 while Zhao and co-workers reported a double-layer pouch cell with a capacity of >750 W h kgcell−1.27 More recently, Lee and co-workers demonstrated a 1200 W h kgcell−1 folded pouch cell configuration that greatly exceeds the specific energy density possible by most battery technologies.28,29 Practical, “real-world” Li–air batteries will operate in air, exposing the electrolyte to H2O and CO2, which can react with Li2O2 to yield LiOH and Li2CO3 respectively.30 LiOH can cause electrolyte degradation, and both salts have high oxidation potentials, which would significantly limit the coulombic efficiency of the cell.31,32 Due to the challenges associated with atmospheric gases on the operation of Li–air cells, “real-world” open devices will incorporate gas-handling systems to “scrub” the air33 of H2O and CO2 and it is assumed that concentrations of <10 ppm are needed for both.34Gallagher et al. proposed theoretical system models for a practical air-breathing (open) battery comprising a gas-feed stream and an air scrubber system. The gas-handling system significantly impacted the system-level performance of the Li–air pack according to the BatPac model.34 No experimental data on the requirements of a practical gas purification system have been reported. Some work has probed the effects of parameters such as gas composition,35 O2 partial pressure36 and gas flow37,38 on the capacity of Li–air cells, but usually in cells operating at unrealistically low current densities (<100 μA cm−2) and large flow rates. Progress towards the development of a practical Li–air battery requires a holistic view of the challenges involved, including not only the chemical and electrochemical processes occurring within the cell, but also of the effects of the gas-handling parameters on device performance. Delivery of air and removal of CO2 and H2O from the input stream are inherently linked; efficient delivery of air to the cell will require higher flow rates, which will increase the workload of the scrubber system. Such effects must be considered in tandem during device testing to provide realistic estimates of cell performance.
Here we describe the first example of an integrated Li–air battery demonstrator with in-line gas handling system, consisting of the cell, atmospheric control and gas scrubber chamber. The cell is based on a fuel cell design and the gas handling system controls the flow and pressure of the gas to the cell. The cell incorporates a flow field to distribute the gas flow over the positive electrode. The impact of gas flow rate and composition is explored and we show that this has a drastic impact on cell performance. Our analysis highlights some deficiencies in the design of current flow field plates, gas diffusion electrodes, and gas scrubber materials when used in Li–air cells, thus highlighting where further innovation is needed if we are to achieve a practical air-breathing Li–air system.
Results and discussion
To evaluate the impact of gas composition, flow rates and scrubber material on the performance of the Li–air cell and gas handling system, we developed a demonstrator battery system (Fig. 1) in which the composition (including the humidity), pressure, and flow rate of the gas delivered to the cell can be controlled. The gas handling system was constructed from stainless steel Swagelok tubing and the cell was based on a fuel cell stack, which contains a technologically mature gas delivery design, albeit optimised for aqueous systems.39 The three inlet gas compositions used were 100% O2, 20% O2 (balanced with N2) and compressed air. The gas could either be routed through the scrubber or passed directly to the cell. The scrubber consisted of a steel tube (5 cm diameter, 15 cm length), which could be heated and placed under vacuum to regenerate the scrubber media. The scrubber volume far exceeded that of the cell, an unrealistic and best-case scenario for the practical system, and thus the limiting factor should be the performance of the materials used to remove H2O and CO2. Two in-line mass flow controllers were used to control flow rates to the cell; one was positioned before the scrubber and one before the cell. A back-pressure regulator was positioned after the scrubber to compensate for the pressure drop as gas was routed through the scrubber. Pressure gauges placed at three locations allowed continuous monitoring of the system pressure. For all experiments, the gas inlet section was pressurised to about 1400 mbar, with a pressure of about 1000 mbar before the cell. A sensor measured the CO2 content and humidity of the gas entering the cell.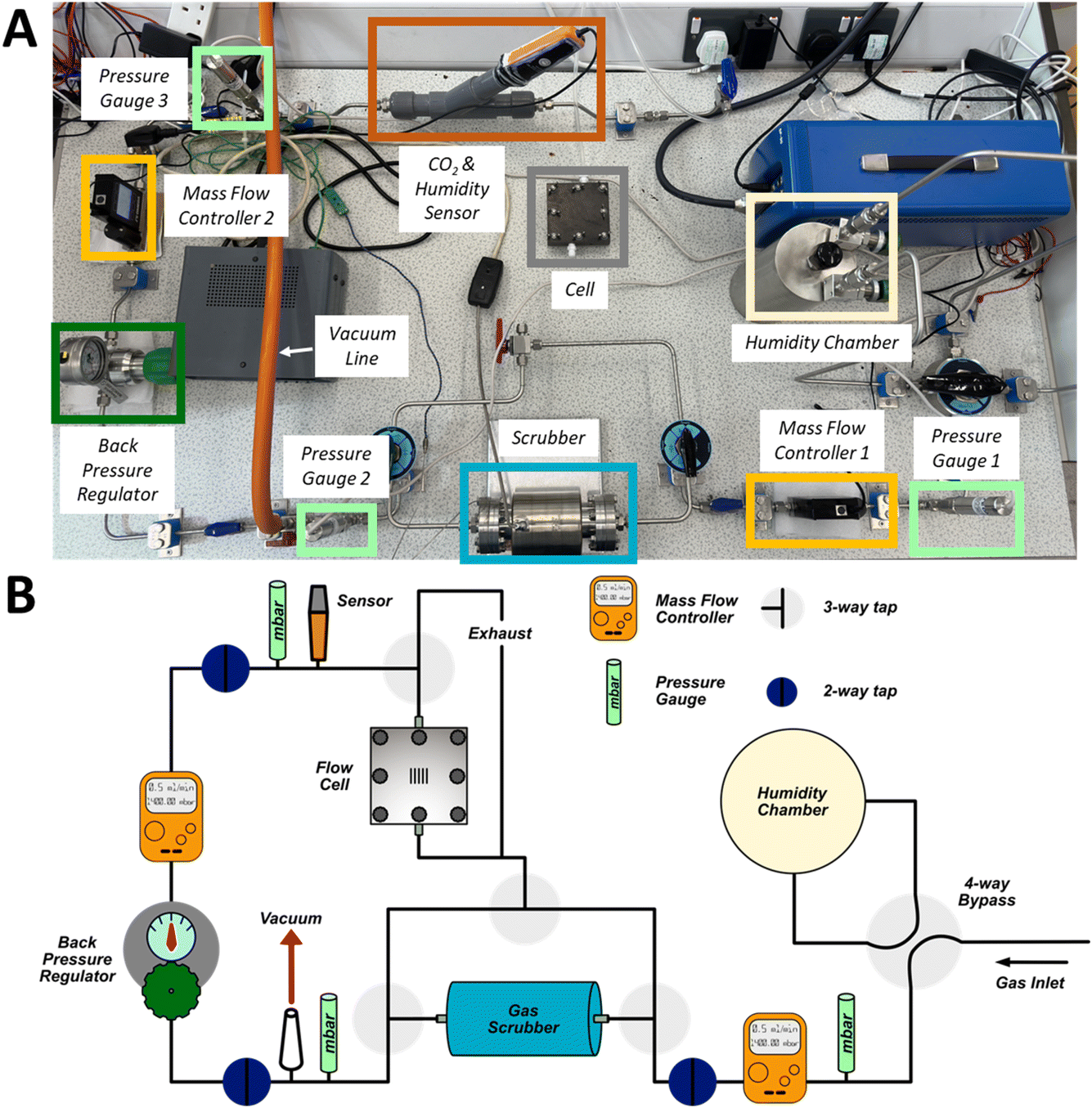 | ||
| Fig. 1 (A) Photograph of the Li–air demonstrator system with labelled components. (B) A schematic diagram of the Li–air demonstrator system. A description of the design can be found in the text. | ||
The open cell (Fig. 2A and B) was composed of a graphite plate with serpentine-type flow field for gas delivery to the positive electrode, a steel plate current collector for the negative electrode, and a stainless-steel mesh current collector for the positive electrode. A polytetrafluoroethylene (PTFE) gasket was used to separate the two plates. As is typical in the field, freestanding pre-charged LiFePO4 (LFP) was used as the negative electrode (350 μm thickness, 22 mm × 22 mm) rather than Li, as the latter requires the development of a protected Li anode to avoid reactions with the electrolyte. Free standing 20 × 20 mm Super P cathodes (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 wt% Super P:PTFE) were used as the positive electrode. A glass fibre separator was placed between the electrodes. The electrolyte for all experiments was 150 μL cm−2 of 1.0 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) dissolved in tetraethylene glycol dimethyl ether (tetraglyme) unless otherwise stated.
:20 wt% Super P:PTFE) were used as the positive electrode. A glass fibre separator was placed between the electrodes. The electrolyte for all experiments was 150 μL cm−2 of 1.0 M lithium bis(trifluoromethane)sulfonimide (LiTFSI) dissolved in tetraethylene glycol dimethyl ether (tetraglyme) unless otherwise stated.
 | ||
| Fig. 2 (A) Photographs of the cell housing used in the study, with a magnified image of the flow field plate shown. (B) A schematic of the open Li–air cell design showing the stack composition and flow field position. (C) Discharge profiles of cells discharged under flowing 100% O2 (0.50 mL min−1) with and without the addition of 50 mM DBBQ. (D) SEM image of the positive electrode after discharge with DBBQ. The inset shows the pristine positive electrode. The scale bar is 20 μm long. The cell was discharged at a rate of 0.1 mA cm−2. | ||
Cells discharged at 0.5 mA cm−2 under a flowing excess of 100% O2 (0.50 mL min−1) yielded a relatively low capacity of 0.7 mA h cm−2 to a cut-off of 2.3 V vs. Li/Li+ (Fig. 2C). Low capacities have been observed previously during analysis of ether-based Li–air cells, and have been improved by the use of redox mediators.40 The discharge was repeated with the addition 50 mM di-tert-butyl dibenzoquinone (DBBQ) resulting in an areal capacity of 6.6 mA h cm−2, 9.4 times greater than in the absence of the redox mediator, and among the highest areal capacities recorded for a Li–air cell.40,41 A cell containing the same electrode components in a Swagelok cell filled with a static headspace of 100% O2 gave an areal capacity of 3.5 mA h cm−2, demonstrating the improvement possible by the use of flowing gas (Fig. S1†). The result also supports the need for dissolved redox mediators in the cell to reach significant capacities at high current densities, even under a high flow of pure O2. As such, 50 mM DBBQ was added to all subsequent cells unless otherwise stated. Fig. 2D shows scanning electron microscopy (SEM) images of the discharge product from a DBBQ-containing cell. The appearance of toroidal structures indicates that solution-mediated Li2O2 formation occurred on discharge.40,42 The Li2O2 yield for the DBBQ-containing cell was determined to be 82% using the method developed by Hartmann et al.43 (other yield measurements can be found in Table S1†). These data confirm that the Li–air demonstrator discharge performance was consistent with coin and Swagelok cells, but that the use of flowing gas improved capacity significantly.
To evaluate the performance of the open cell architecture under various operating conditions, cells were discharged under 100% O2, 20% O2 and air at a range of flow rates (Fig. 3). Pure O2 represents optimal performance conditions of the open cell and matches the conditions in most studies in this field, which use a static headspace/flow of pure O2. However, 20% O2 better reflects the atmospheric concentration of O2 and operation under optimum scrubber conditions (H2O and CO2 are completely removed). Operation under air reflects the performance in the absence of a scrubber. When using 100% O2, the capacity of the cell was almost directly proportional to the gas flow rate (Fig. 3A). At the highest rate of 0.5 mL min−1, a maximum capacity of 6.6 mA h cm−2 was achieved, compared to 0.8 mA h cm−2 at the lowest rate of 0.05 mL min−1. The cell voltage (determined from the mid-point of the discharge plateau, Table S2†) was also dependent on the flow rate, indicating that O2 depletion occurred at lower flow rates, lowering the discharge potential. When using 20% O2 and a current density of 0.5 mA cm−2, the capacity initially increased with increasing flow rate (Fig. 3B). However, beyond 0.75 mL min−1 the capacity did not increase, and some cells displayed a lower capacity. As expected, the discharge plateaus were lower than those observed under flows of pure O2. Despite the use of flow rates an order of magnitude higher than those used with pure O2, the maximum capacity that could be achieved was approximately 2.6 mA h cm−2. Discharging using air gave a similar trend to that obtained using 20% O2 (Fig. 3C and D).
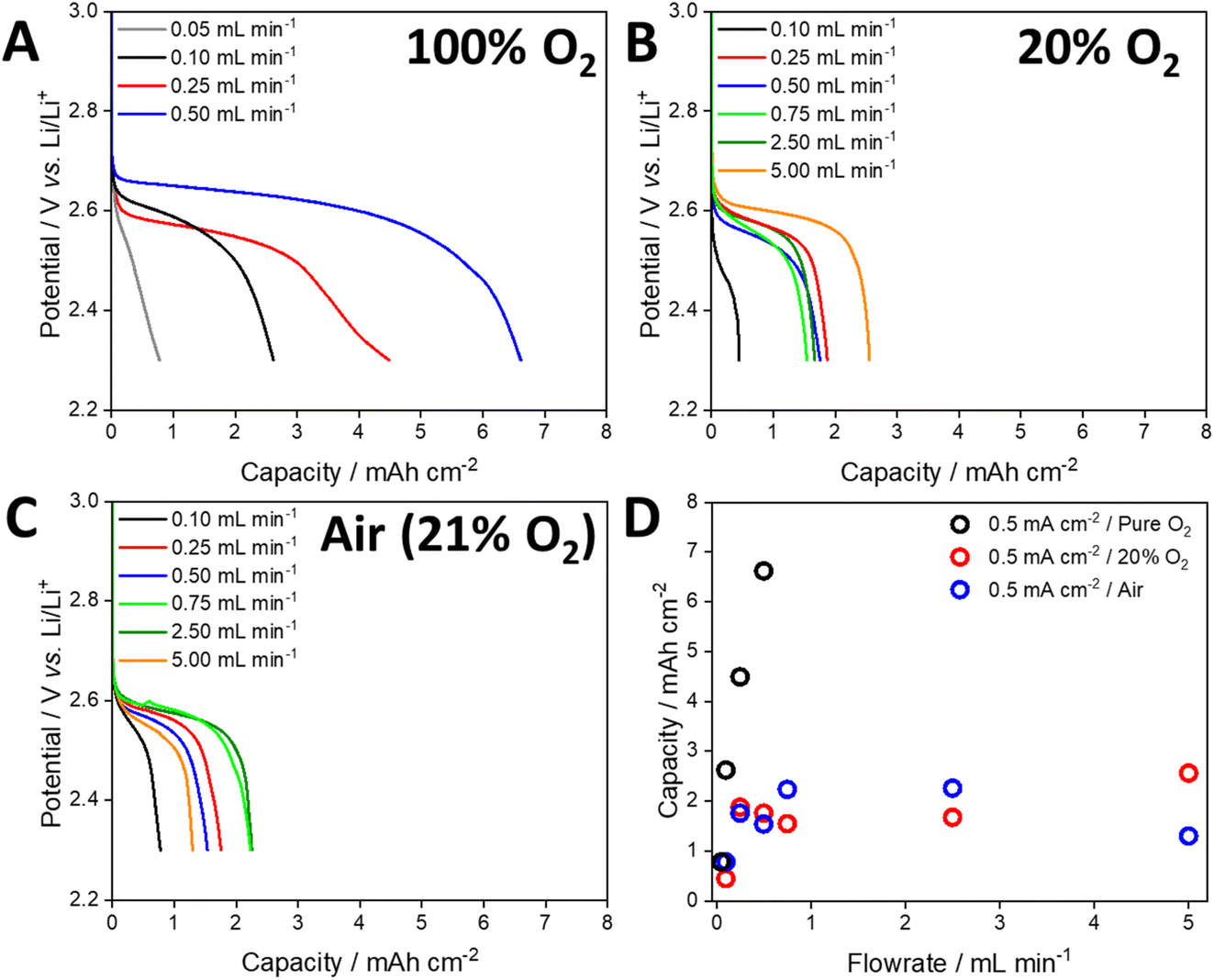 | ||
| Fig. 3 Discharge profiles of cells discharged at a current density of 0.5 mA cm−2 under a flow of (A) 100% O2, (B) 20% O2 with N2 balance and (C) air. (D) Shows the areal capacity of these cells. | ||
Based on a 2e− reduction of O2 and the applied current density, the rate of O2 consumption was calculated and used to develop plots of normalised flow rate versus capacity (Fig. 4A, see the ESI and Table S3† for details of the calculations). When using pure O2 and a current density of 0.5 mA cm−2, the maximum capacity (6.6 mA h cm−2) was achieved with a flow rate that was 33 times higher than the theoretical rate of O2 consumption. In contrast, applying a flow rate factor of 3.3 gave an areal capacity of 0.8 mA h cm−2 (Fig. 4A). Despite the use of 100% O2, this demonstrates that significant excess gas flow may be required at the positive electrode. For a flow rate factor of 33 using 20% O2, the capacity of the cell decreased to 1.8 mA h cm−2 at a current density of 0.5 mA cm−2 (Fig. 4A). This capacity could not be significantly increased (2.6 mA h cm−2) by increasing the flow rate factor up to 66. By reducing the applied current density to 0.1 mA cm−2 the cell using 20% O2 was able to achieve a capacity similar to that using 100% O2 (5.7 mA h) at the same flow rate factor (Fig. 4B), while noting that the absolute flow rates were different due to different gas compositions and applied current densities. These data suggest that the rate of O2 dissolution and transport within the electrolyte solution limits the capacity of the cell under open conditions, which is particularly significant for atmospheric O2 concentrations. While increasing the flow rate increased the capacity, very high flow rates had the opposite effect. Post-cycling analysis of the cell suggested that this was due to loss of electrolyte solution from the air electrode, which has been observed previously,37,38 reconfirming the need for a solvent-management system.34
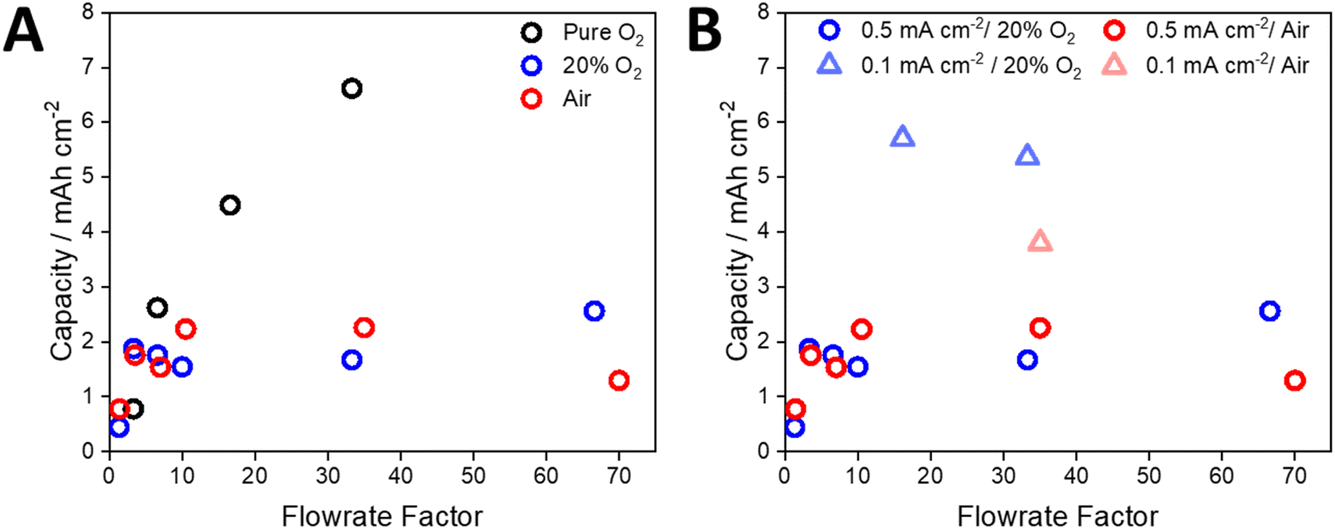 | ||
| Fig. 4 (A) The capacity of the cells discharged with a current density of 0.5 mA cm−2 as a function of the flow rate factor, where a flow rate factor of 1 is equivalent to the rate of oxygen consumption within the cell based on the applied current density. (B) The effect of current density on the capacity of the cell when discharged under a flow of 20% O2 and air, as a function of flow rate factor (note that data points for the 0.1 mA cm−2 data set correspond to absolute flow rates of 0.25 and 0.50 mL min−1 respectively). | ||
To explore the challenge of removing CO2 and H2O from the gas stream of an open-architecture cell, we tested the scrubber filled with either activated charcoal or molecular sieves using a flow rate factor of 35.7. Both were activated within the device by holding them at 10−4 mbar at 120 °C for 72 hours, approximating the conditions expected in a real Li–air gas handling system. A stream of air with relative humidity of 35% at 20 °C was used for all tests. When passing this gas composition through the scrubbing media at 20 °C, the relative humidity was almost unchanged, dropping by ca. 2% and 4% for activated charcoal and molecular sieves, respectively. In contrast, CO2 levels dropped by ca. 32% and 41% for activated charcoal and molecular sieves, respectively (Fig. 5A). It is important to note that the volume of the scrubber far exceeded that of the headspace of the cell, indicating the need for significant innovation in scrubbing media/architecture, but the removal of significant amounts of CO2 is promising nevertheless. Cells were discharged at 0.1 mA cm−2 (Fig. 5B) with and without the molecular-sieve scrubber (selected due to its ability to remove more CO2) and the discharged cathodes were analysed by Fourier transform infra-red (FTIR) spectroscopy. In both cases, notable carbonate peaks were observed, and the intensities were similar, regardless of the lower incoming CO2 concentration, but were much greater than that seen when using a cell discharged with 100% O2 (Fig. 5C). The similar carbonate peak intensities, despite the drop in the gas stream CO2 concentration, indicates a non-linear relationship between CO2 and Li2CO3 formation, suggesting that near-absolute removal of CO2 will be required for open Li–air devices.
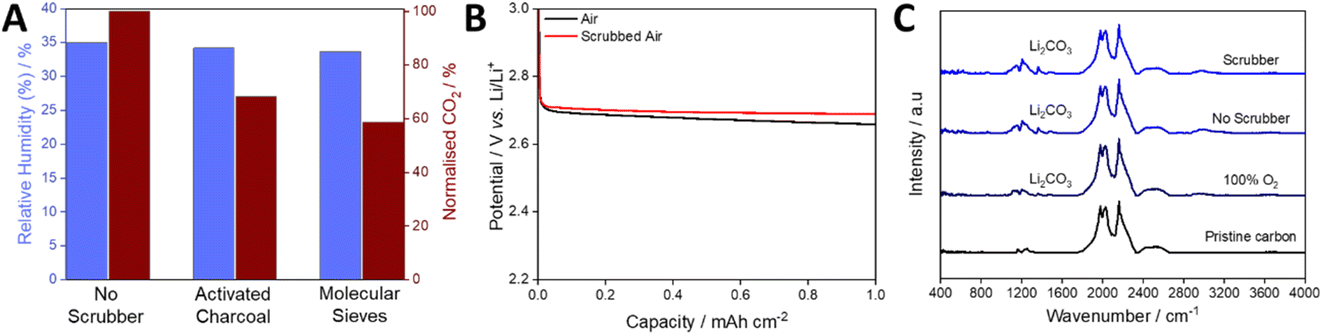 | ||
| Fig. 5 (A) Relative humidity and normalised CO2 levels of compressed air at 35% RH passed through the scrubber in the demonstrator system. (B) Discharge profiles of cells discharged in air and air that has been scrubbed using molecular sieves at 0.5 mL min−1 (flow rate factor 35.7) and at 0.1 mA cm−2. (C) FTIR spectra of the cathodes extracted from cells discharged in 100% O2, air and scrubbed air. Cells were discharged at a current density of 0.1 mA cm−2 to 5 mA h at a gas flow of 0.5 mL min−1. | ||
Implications for the Li–air system
These data demonstrate that moving from a static head space to an open system with gas flow could offer significant increases in capacity and rate. However, when using flow fields designed for aqueous systems, the quantity of gas (O2 or air) to be compressed, scrubbed and supplied may be significantly higher than the volume theoretically required by the cell,44 and the higher flow rates required can lead to cell drying by evaporating the solvent. This will increase the need for saturation and capture of solvent from the gas stream.37,38,45 For Li–air to become a commercially viable battery technology, we anticipate the need for a system-specific energy of >400 W h kg−1.3,33 Our calculations suggest that if a positive electrode capable of storing 40% vol. Li2O2 (approximately 30 mA h cm−2) can be achieved, then a system-level energy density of an “open” architecture Li–air cell could reach >450 W h kg−1 (see Table S4†). Achieving this target will require the development of better gas diffusion electrodes and flow fields for electrodes, as well as organic solvents that can sustain O2 delivery to the cell at low flow rates.Considering the gas scrubber, neither molecular sieves nor activated carbon was able to scrub H2O and CO2 from the gas stream, despite the scrubber being significantly larger than the cell. This highlights the need for further innovation in the development of scrubber materials or membranes to selectively allow O2 transport. We note that improvements in the efficiency of gas delivery to the electrode could lessen the burden on the scrubber and that the removal of CO2 was better than that of H2O, potentially simplifying its removal. An alternative approach is to redesign the cell chemistry to tolerate both H2O and CO2.46,47 It is known that H2O can be tolerated at greater levels than originally thought, and can even be beneficial.42,48 Some progress has also been made in understanding the impact of LiOH and Li2CO3 within metal–air cells,35,49–51 but further optimisation is required for operation in air.
Conclusion
Here we have described an integrated Li–air battery with in-line gas handling system that achieves areal capacities of ca. 7 mA h cm−2 at 0.5 mA cm−2 when using redox mediators. The capacity of the cell is directly proportional to the gas flow when using pure O2, but the volume of gas required by the cell during discharge far outweighs the theoretical gas consumption at the positive electrode. This phenomenon is exacerbated in air due to its lower O2 concentration and may be due to the use of flow-field plates designed for aqueous systems. No scrubber material tested in this study successfully removed H2O and CO2 from the gas stream, but a marked decrease in CO2 concentration was observed. These data highlight the challenges that must be overcome if we are to achieve a practical air-breathing Li–air battery, including the need for better flow field plates, gas diffusion electrodes that can support 30 mA h cm−2 at lower flow rates, and new gas-scrubbing materials to more efficiently remove H2O and CO2. Critically, advances in one area will lower the requirements elsewhere. For example, improved gas delivery to the cell will reduce the volume of gas required and put less pressure on the performance of the scrubber. Alternatively, improving the cell's tolerance to H2O and CO2 will similarly lessen, or even remove, reliance on the gas scrubber entirely.Author contributions
All authors contributed to the conception and design of the study. JWJ, GV, CH, MJ, RCM, and CP performed the experiments. All authors contributed to manuscript writing. BT, AL, XG, DAW, GNN, PGB and LRJ supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support of this research by the Faraday Institution's Seed, Degradation and LiSTAR projects (EP/S003053/1 FITG001, FIRG014, FIRG024, FIRG051 and EP/S514901/1), an EPSRC Fellowship (EP/S001611/1), and the University of Nottingham's Propulsion Futures Beacon of Excellence. The authors would like to thank the University of Nottingham School of Chemistry workshop team, particularly Mr Paul Gaetto for assistance with the development of the demonstrator.References
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS.
- A. Manthiram and L. Li, Adv. Energy Mater., 2015, 5, 1401302 CrossRef.
- D. Aurbach, B. D. McCloskey, L. F. Nazar and P. G. Bruce, Nat. Energy, 2016, 1, 16128 CrossRef CAS.
- J.-H. Kang, J. Lee, J.-W. Jung, J. Park, T. Jang, H.-S. Kim, J.-S. Nam, H. Lim, K. R. Yoon, W.-H. Ryu, I.-D. Kim and H. R. Byon, ACS Nano, 2020, 14, 14549–14578 CrossRef CAS.
- L. Ma, T. Yu, E. Tzoganakis, K. Amine, T. Wu, Z. Chen and J. Lu, Adv. Energy Mater., 2018, 8, 1800348 CrossRef.
- D. Kundu, R. Black, B. Adams and L. F. Nazar, ACS Cent. Sci., 2015, 1, 510–515 CrossRef CAS.
- F. Li, T. Zhang and H. Zhou, Energy Environ. Sci., 2013, 6, 1125–1141 RSC.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Griffin, L. J. Hardwick, F. Barde, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040–8047 CrossRef CAS PubMed.
- D. Sharon, M. Afri, M. Noked, A. Garsuch, A. A. Frimer and D. Aurbach, J. Phys. Chem. Lett., 2013, 4, 3115–3119 CrossRef CAS.
- B. D. McCloskey, A. Valery, A. C. Luntz, S. R. Gowda, G. M. Wallraff, J. M. Garcia, T. Mori and L. E. Krupp, J. Phys. Chem. Lett., 2013, 4, 2989–2993 CrossRef CAS.
- R. C. McNulty, K. Jones, C. Holc, J. W. Jordan, P. G. Bruce, D. A. Walsh, G. N. Newton, H. W. Lam and L. R. Johnson, Adv. Energy Mater., 2023, 13, 2300579 CrossRef CAS.
- N. Mozhzhukhina, F. Marchini, W. R. Torres, A. Y. Tesio, L. P. Mendez De Leo, F. J. Williams and E. J. Calvo, Electrochem. Commun., 2017, 80, 16–19 CrossRef CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J. M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS.
- J. Højberg, B. D. McCloskey, J. Hjelm, T. Vegge, K. Johansen, P. Norby and A. C. Luntz, ACS Appl. Mater. Interfaces, 2015, 7, 4039–4047 CrossRef.
- V. Viswanathan, K. S. Thygesen, J. S. Hummelshoj, J. K. Norskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 214704 CrossRef CAS.
- S. Ahn, C. Zor, S. Yang, M. Lagnoni, D. Dewar, T. Nimmo, C. Chau, M. Jenkins, A. J. Kibler, A. Pateman, G. J. Rees, X. Gao, P. Adamson, N. Grobert, A. Bertei, L. R. Johnson and P. G. Bruce, Nat. Chem., 2023, 15, 1022–1029 CrossRef CAS PubMed.
- Y. Chen, S. A. Freunberger, Z. Peng, O. Fontaine and P. G. Bruce, Nat. Chem., 2013, 5, 489–494 CrossRef CAS PubMed.
- B. J. Bergner, A. Schurmann, K. Peppler, A. Garsuch and J. Janek, J. Am. Chem. Soc., 2014, 136, 15054–15064 CrossRef CAS.
- N. Feng, X. Mu, X. Zhang, P. He and H. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 3733–3739 CrossRef CAS.
- Y. Chen, X. Gao, L. R. Johnson and P. G. Bruce, Nat. Commun., 2018, 9, 767 CrossRef.
- H.-D. Lim, B. Lee, Y. Zheng, J. Hong, J. Kim, H. Gwon, Y. Ko, M. Lee, K. Cho and K. Kang, Nat. Energy, 2016, 1, 16066 CrossRef CAS.
- V. Pande and V. Viswanathan, ACS Energy Lett., 2017, 2, 60–63 CrossRef CAS.
- Q. C. Liu, J. J. Xu, S. Yuan, Z. W. Chang, D. Xu, Y. B. Yin, L. Li, H. X. Zhong, Y. S. Jiang, J. M. Yan and X. B. Zhang, Adv. Mater., 2015, 27, 5241–5247 CrossRef CAS PubMed.
- X. Bi, K. Amine and J. Lu, J. Mater. Chem. A, 2020, 8, 3563–3573 RSC.
- S. Matsuda, M. Ono, S. Yamaguchi and K. Uosaki, Mater. Horiz., 2022, 9, 856–863 RSC.
- Y. Kubo and K. Ito, ECS Trans., 2014, 62, 129–135 CrossRef CAS.
- S. Zhao, L. Zhang, G. Zhang, H. Sun, J. Yang and S. Lu, J. Energy Chem., 2020, 45, 74–82 CrossRef.
- H. C. Lee, J. O. Park, M. Kim, H. J. Kwon, J.-H. Kim, K. H. Choi, K. Kim and D. Im, Joule, 2019, 3, 542–556 CrossRef CAS.
- J. O. Park, M. Kim, J.-H. Kim, K. H. Choi, H. C. Lee, W. Choi, S. B. Ma and D. Im, J. Power Sources, 2019, 419, 112–118 CrossRef CAS.
- T. Zhang and H. Zhou, Nat. Commun., 2013, 4, 1817 CrossRef PubMed.
- D. Cao, C. Tan and Y. Chen, Nat. Commun., 2022, 13, 4908 CrossRef CAS PubMed.
- Z. Zhao, J. Huang and Z. Peng, Angew. Chem., Int. Ed., 2018, 57, 3874–3886 CrossRef CAS.
- A. C. Luntz and B. D. McCloskey, Chem. Rev., 2014, 114, 11721–11750 CrossRef CAS.
- K. G. Gallagher, S. Goebel, T. Greszler, M. Mathias, W. Oelerich, D. Eroglu and V. Srinivasan, Energy Environ. Sci., 2014, 7, 1555–1563 RSC.
- M. Asadi, B. Sayahpour, P. Abbasi, A. T. Ngo, K. Karis, J. R. Jokisaari, C. Liu, B. Narayanan, M. Gerard, P. Yasaei, X. Hu, A. Mukherjee, K. C. Lau, R. S. Assary, F. Khalili-Araghi, R. F. Klie, L. A. Curtiss and A. Salehi-Khojin, Nature, 2018, 555, 502–506 CrossRef CAS PubMed.
- H. J. Kwon, H. C. Lee, J. Ko, I. S. Jung, H. C. Lee, H. Lee, M. Kim, D. J. Lee, H. Kim, T. Y. Kim and D. Im, J. Power Sources, 2017, 364, 280–287 CrossRef CAS.
- J. P. O. Júlio, B. A. B. Francisco, B. P. de Sousa, J. F. Leal Silva, C. G. Anchieta, T. C. M. Nepel, C. B. Rodella, R. Maciel Filho and G. Doubek, Chem. Eng. J. Adv., 2022, 10, 100271 CrossRef.
- B. A. B. Francisco, J. P. O. Júlio, C. G. Anchieta, T. C. M. Nepel, R. M. Filho and G. Doubek, ACS Appl. Energy Mater., 2023, 6, 5167–5176 CrossRef CAS.
- M. Sauermoser, N. Kizilova, B. G. Pollet and S. Kjelstrup, Front. Energy Res., 2020, 8, 1–20 CrossRef.
- X. Gao, Y. Chen, L. Johnson and P. G. Bruce, Nat. Mater., 2016, 15, 882–888 CrossRef CAS PubMed.
- X. Gao, Y. Chen, L. R. Johnson, Z. P. Jovanov and P. G. Bruce, Nat. Energy, 2017, 2, 17118 CrossRef CAS.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS PubMed.
- P. Hartmann, C. L. Bender, J. Sann, A. K. Dürr, M. Jansen, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 11661–11672 RSC.
- L. Grande, E. Paillard, J. Hassoun, J. B. Park, Y. J. Lee, Y. K. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784–800 CrossRef CAS PubMed.
- F. Mohazabrad, F. Wang and X. Li, ACS Appl. Mater. Interfaces, 2017, 9, 15459–15469 CrossRef CAS.
- H. A. Gasteiger, M. Piana, T. Restle, M. Metzger and K. U. Schwenke, J. Electrochem. Soc., 2015, 162, A573–A584 CrossRef.
- D. G. Kwabi, T. P. Batcho, S. Feng, L. Giordano, C. V. Thompson and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2016, 18, 24944–24953 RSC.
- M. C. Policano, C. G. Anchieta, T. C. M. Nepel, R. M. Filho and G. Doubek, ACS Appl. Energy Mater., 2022, 5, 9228–9240 CrossRef CAS.
- S. Ko, Y. Yoo, J. Choi, H.-D. Lim, C. B. Park and M. Lee, J. Mater. Chem. A, 2022, 10, 20464–20472 RSC.
- Z. Gao, I. Temprano, J. Lei, L. Tang, J. Li, C. P. Grey and T. Liu, Adv. Mater., 2023, 35, 2201384 CrossRef CAS PubMed.
- H. Wakita, R. Awata, Y. Maita and T. Takeguchi, J. Phys. Chem. C, 2023, 127, 10012–10024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00137g |
| This journal is © The Royal Society of Chemistry 2024 |