 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing assembly/disassembly of ordered molecular hydrogels†
Susana M.
Ramalhete
a,
Karol P.
Nartowski‡
 ab,
Hayley
Green
c,
Jesús
Angulo
d,
Dinu
Iuga
e,
László
Fábián
a,
Gareth O.
Lloyd
*f and
Yaroslav Z.
Khimyak
*ab
ab,
Hayley
Green
c,
Jesús
Angulo
d,
Dinu
Iuga
e,
László
Fábián
a,
Gareth O.
Lloyd
*f and
Yaroslav Z.
Khimyak
*ab
aSchool of Pharmacy, University of East Anglia, Norwich Research Park, Norwich, NR2 1TS, UK. E-mail: Y.Khimyak@uea.ac.uk
bDepartment of Drug Form Technology, Wroclaw Medical University, Borowska 211A, 50-556 Wroclaw, Poland
cInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, UK
dInstituto de Investigaciones Químicas (CSIC-US), Avda. Américo Vespucio, 49, Sevilla 41092, Spain
eDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK
fSchool of Chemistry, University of Lincoln, Lincoln, LN6 7DL, UK. E-mail: GLloyd@lincoln.ac.uk
First published on 23rd May 2024
Abstract
Supramolecular hydrogels have a wide range of applications in the biomedical field, acting as scaffolds for cell culture, matrices for tissue engineering and vehicles for drug delivery. L-Phenylalanine (Phe) is a natural amino acid that plays a significant role in several physiological and pathophysiological processes (phenylketonuria and assembly of fibrils linked to tissue damage). Since Myerson et al. [Chem. Eng. Commun., 2002, 189(8), 1079–1090] reported that Phe forms a fibrous network in vitro, Phe's self-assembly processes in water have been thoroughly investigated. We have reported structural control over gelation by introduction of a halogen atom in the aromatic ring of Phe, driving changes in the packing motifs, and therefore, dictating gelation functionality. The additional level of control gained over supramolecular gelation via the preparation of multi-component gel systems offers significant advantages in tuning functional properties of such materials. Gaining molecular-level information on the distribution of gelators between the inherent structural and dynamic heterogeneities of these materials remains a considerable challenge. Using multicomponent gels based on Phe and amino-L-phenylalanine (NH2-Phe), we will explore the patterns of ordered/disordered domains in the gel fibres and will attempt to come up with general trends of interactions in the gel fibres and at the fibre/solution interfaces. Phe and NH2-Phe were found to self-assemble in water into crystalline hydrogels. The determined faster dynamics of exchange between the gel and solution states of NH2-Phe in comparison with Phe were correlated with weaker intermolecular interactions, highlighting the role of head groups in dictating the strength of intermolecular interactions. In the mixed Phe/NH2-Phe systems, at a low concentration of NH2-Phe, disruption of the network was promoted by interference of the aliphatics of NH2-Phe with the electrostatic interactions between Phe molecules. At high concentrations of NH2-Phe, multiple-gelator hydrogels were formed with crystal habits different from those of the pure gel fibres. NMR crystallography approaches combining the strengths of solid- and solution-state NMR proved particularly suitable to obtain structural and dynamic insights into the “ordered” fibres, solution phase and fibre/solution interfaces in these gels. These findings are supported by a plethora of experimental (diffraction, rheology, microscopy and thermal analysis) and computational methods.
Introduction
Supramolecular gels are colloidal dispersions formed by a rigid three-dimensional structure.1 They possess solid-like rheological properties, despite their high contents of solvent.1 The building blocks of supramolecular gels that have a molecular weight of less than 1000 Da are referred to as low-molecular-weight (LMW) gelators.2 Their self-assembly occurs through unidirectional non-covalent forces, leading to formation of entangled fibrillar networks that arrest the solvent via surface tension and capillary forces.3,4 The resulting gels are stimuli responsive due to the reversible nature of the interactions, so external stimuli can prompt gel-to-solution transitions, a change of shape or release of entrapped molecules.5 This feature is behind their broad range of biomedical applications, with them being developed as scaffolds for tissue engineering,6 matrices for cellular growth7 or vehicles for advanced drug delivery.6–8 The design and discovery of novel structures capable of gelling in a variety of solvents has been of great interest due to their unparalleled properties as soft materials.Multi-component gel systems are composed of two or more molecules.9,10 They can form gels only when combined, gel independently (Fig. 1a), or have their properties modified in the presence of non-gelling additives (Fig. 1b).9 When both molecules form gel networks on their own, the resulting mixed system may be formed by interpenetrated structures of the pure gelators, termed self-sorting, or might give rise to new mixed architectures, a process designated as co-assembly (Fig. 1a).9 Understanding, at the molecular level, the structure of multi-component gels provides opportunities to design customised soft materials.
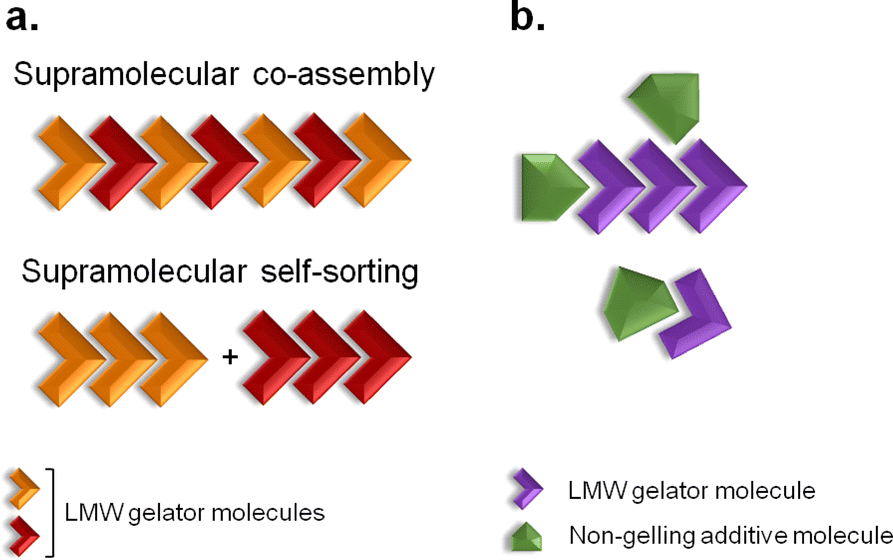 | ||
| Fig. 1 Two-component self-association. (a) Supramolecular co-assembly and self-sorting processes can occur when both molecules are LMW gelators. (b) When the additive is a non-gelling molecule, the physical properties of the resulting gel material might be modified. Adapted from ref. 9. | ||
Phenylketonuria is an autosomal recessive disease that originates from mutations in the gene coding for phenylalanine hydroxylase.11,12 The absence of this enzyme leads to large accumulation of L-phenylalanine (Phe) in the plasma, brain tissue and cerebral fluids.11 The resultant accumulation of Phe leads to formation of stable toxic aggregates, which have been detected at micromolar concentrations in vivo.11–13 It has also been determined previously that Phe self-assembles into long fibres that give rise to a supramolecular crystalline hydrogel at millimolar concentrations in vitro.11,13–16
In this report, we discuss our findings on amino-L-phenylalanine (NH2-Phe) and its solution- and solid-state interactions with Phe (Fig. 2). The main purpose of this research project was to study single and multi-component hydrogels of Phe and NH2-Phe, and to pinpoint the interactions responsible for the disruption of Phe hydrogels upon the addition of NH2-Phe. Hence, the present study describes the mechanism of disruption of Phe hydrogels upon adding low concentrations of NH2-Phe, and how this information provides an insight into the structure and dynamics of single- and multiple-gelator hydrogels (which are formed at high concentrations of NH2-Phe). The resulting materials were characterised using microscopy, rheology, diffraction and advanced nuclear magnetic resonance (NMR) spectroscopy, methodologies that are able to probe different regimes of mobility, levels of self-organisation and intermolecular connectivities.
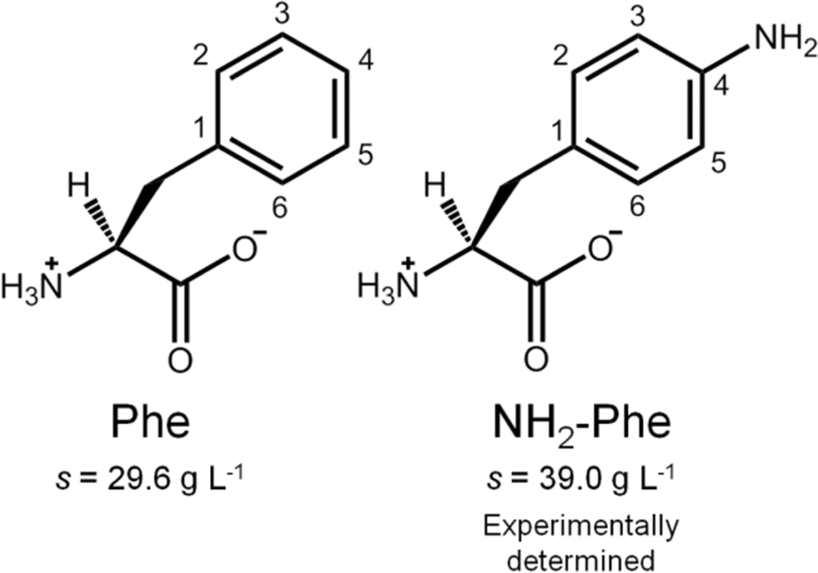 | ||
| Fig. 2 Molecular structures of zwitterionic Phe and NH2-Phe and their water solubilities at 298 K. | ||
Results and discussion
Macroscopic and morphological characterisation
When Phe and NH2-Phe were mixed in water, different products were obtained depending on the concentration and molar ratio of the gelator molecules (Fig. 3 and 4). A detailed description of the concentration and molar ratio of hydrogels under study can be found in the ESI (Table S1†). For comparison purposes, the concentration of Phe was maintained at 303 mM, which corresponds to the concentration at which the pure monohydrate form of Phe is obtained.16 | ||
Fig. 3 Images of hydrogels of (a) Phe (303 mM), (b) Phe/NH2-Phe (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05), (e) Phe/NH2-Phe (1:2) and (f) NH2-Phe (606 mM), (c) the suspension of Phe/NH2-Phe (1:0.4) and (d) the solution of Phe/NH2-Phe (1:1). :0.05), (e) Phe/NH2-Phe (1:2) and (f) NH2-Phe (606 mM), (c) the suspension of Phe/NH2-Phe (1:0.4) and (d) the solution of Phe/NH2-Phe (1:1). | ||
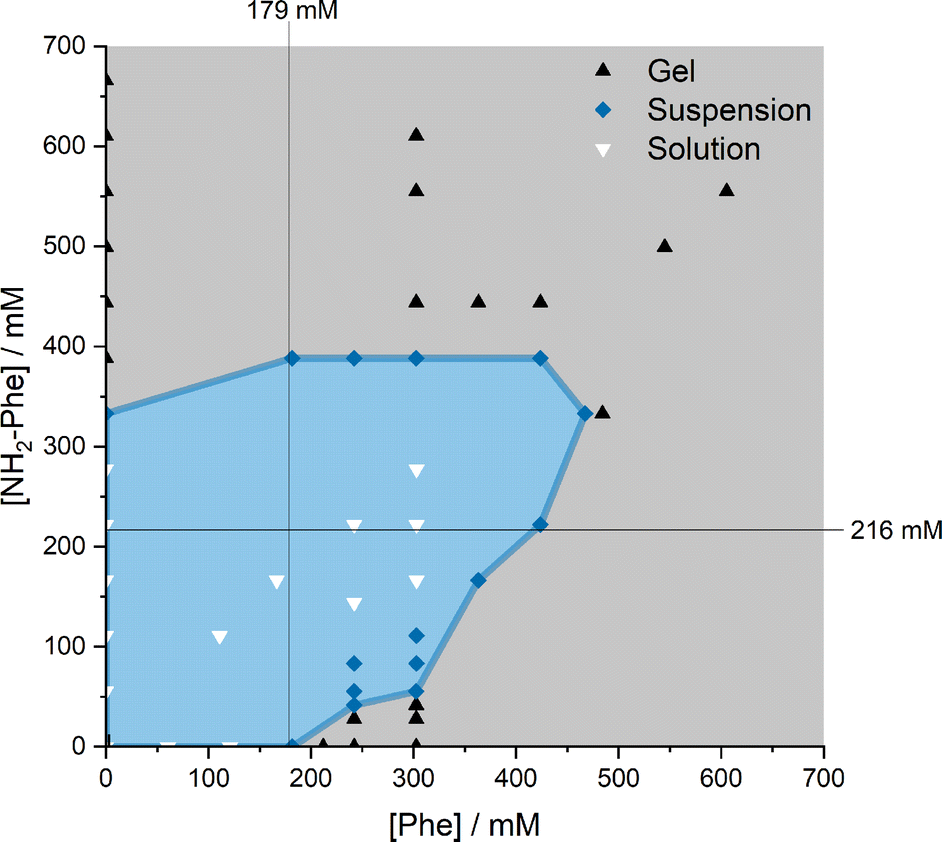 | ||
| Fig. 4 Phase diagram of the products obtained depending on the concentration and molar ratio of Phe and NH2-Phe in water. The water solubilities of Phe (179 mM) and NH2-Phe (216 mM) are highlighted with black lines. | ||
Phe gives rise to white opaque hydrogels (Fig. 3a), composed of long hair-like fibres (Fig. 5a), at concentrations higher than 212 mM.16 When small concentrations of NH2-Phe were added (up to 1:0.2), a brownish colouration appeared, but the self-sustaining properties of the material were kept (Fig. 3b). Between the ratios of 1:0.2 and 1:0.4, a heterogeneous sample was obtained, containing white bulky clouds in suspension. These samples exhibited flow when inverted. When the concentration of NH2-Phe was increased further, thin white particles were observed in suspension (Fig. 3c). Above a 1:1 ratio, these particles were fully solubilised and a clear brown solution was obtained (Fig. 3d). When both molecules were mixed above their individual critical gelation concentrations (CGC) (CGCPhe = 212 mM and CGCNH2-Phe = 388 mM), brown hydrogels containing white crystalline structures were formed (Fig. 3e and 5b). The white elements were attributed to the long hair-like fibres belonging to Phe, interpenetrated with shorter and thicker needle-like crystals belonging to NH2-Phe (Fig. 5b). The presence of distinguishable morphologies is commonly associated with self-sorted materials, composed of intertwined fibres of the pure hydrogels.9
 | ||
| Fig. 5 SEM images of (a) Phe (303 mM), (b) Phe/NH2-Phe (1:2) and (c) NH2-Phe (606 mM) dried hydrogels. The hair-like fibres of Phe had an average width of 1.1 μm (ranging between 0.4 and 1.7 μm), whereas the average width of the needle-like fibres of NH2-Phe was 25.3 μm (ranging between 2.6 and 47.9 μm). | ||
Throughout these studies, we discovered that NH2-Phe is also able to self-assemble in water into organised structures, forming brown opaque crystalline hydrogels (Fig. 3f) composed of wide needle-like fibres (Fig. 5c) at concentrations above 388 mM. The gelation process of NH2-Phe was found to be very dependent on quenching and agitation rates. Gel materials were successfully obtained only when the hot solutions were immediately cooled down in an ice bath with constant agitation, as slow cooling rates and lack of agitation favoured precipitation of crystalline needle-like components over gel formation. The three-dimensional habit of these needles corresponded to the crystal structure of the NH2-Phe gel fibres.
Mechanical properties of hydrogels
With the goal of understanding the consequences of combining Phe and NH2-Phe, we investigated initially how the mechanical properties of these crystalline gels were modulated. The strength of the hydrogel fibres was assessed by determining the materials' viscoelastic parameters during frequency-sweep studies. The phase angle (δ) formed between the phases of stress and strain was below 10° for pure hydrogels, reflecting the solid-like nature of these materials.17 The storage moduli (G′) for Phe and NH2-Phe hydrogels were on the order of 105 Pa (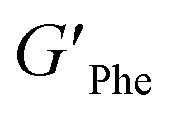 = 4.7 × 105 Pa and
= 4.7 × 105 Pa and 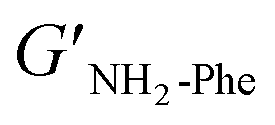 = 5.1 × 105 Pa) and these values were one order of magnitude greater than the loss moduli (G′′) (
= 5.1 × 105 Pa) and these values were one order of magnitude greater than the loss moduli (G′′) (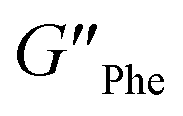 = 3.2 × 104 Pa and
= 3.2 × 104 Pa and 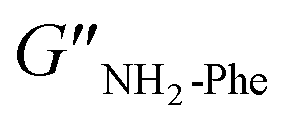 = 3.1 × 104 Pa) (Fig. 6), characteristic values of robust gels.18 These results confirmed the viscoelastic nature of the materials.
= 3.1 × 104 Pa) (Fig. 6), characteristic values of robust gels.18 These results confirmed the viscoelastic nature of the materials.
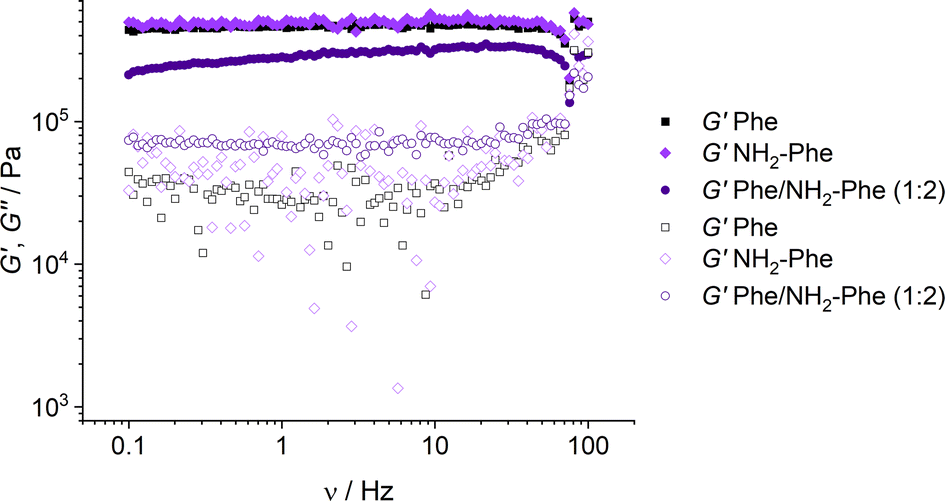 | ||
| Fig. 6 Storage (G′) and loss (G′′) moduli for Phe (303 mM), Phe/NH2-Phe (1:2) and NH2-Phe (606 mM) hydrogels in frequency-sweep experiments. | ||
Interestingly, weaker gel fibres with lower resistance to deformation were found for the mixed hydrogel Phe/NH2-Phe (1:2). The higher values of the phase angle (δ > 10) in combination with the lower elastic response (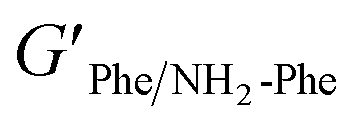 = 2.7 × 105) and the higher inelastic response (
= 2.7 × 105) and the higher inelastic response (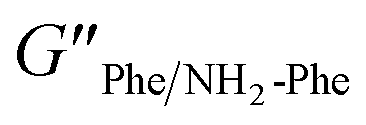 = 7.1 × 104 Pa) for these hydrogels compared to the pure materials pointed towards a system showing less elastic behaviour. These variations in the bulk properties of the pure and mixed gels reflected differences in their molecular and supramolecular level arrangements.
= 7.1 × 104 Pa) for these hydrogels compared to the pure materials pointed towards a system showing less elastic behaviour. These variations in the bulk properties of the pure and mixed gels reflected differences in their molecular and supramolecular level arrangements.
Characterisation of the structure of the gel fibres
The identification of diffraction peaks in the powder X-ray diffraction (PXRD) patterns of the hydrogels (Fig. 8) confirmed their crystalline nature. Phe hydrogel fibres are composed of the Phe monohydrate, as identified in our previous studies using single X-ray diffraction and corroborated with solid-state NMR and DFT calculations.16 Using single-crystal and powder X-ray diffraction experiments (Fig. S12‡), the crystal structure of the NH2-Phe gel fibres was determined successfully (Fig. 7 shows the packing of the structure as determined by single-crystal diffraction). It is clear that the Phe monohydrate and the (NH2-Phe)2(H2O)3 structures are not isostructural and therefore should not easily form solid solutions, according to the Kitaigorodsky studies and design rules for solid solutions.19–25 This is important to note here and will be discussed further in the manuscript. What follows is a description of the NH2-Phe crystal form and the structural similarities to the Phe monohydrate structure.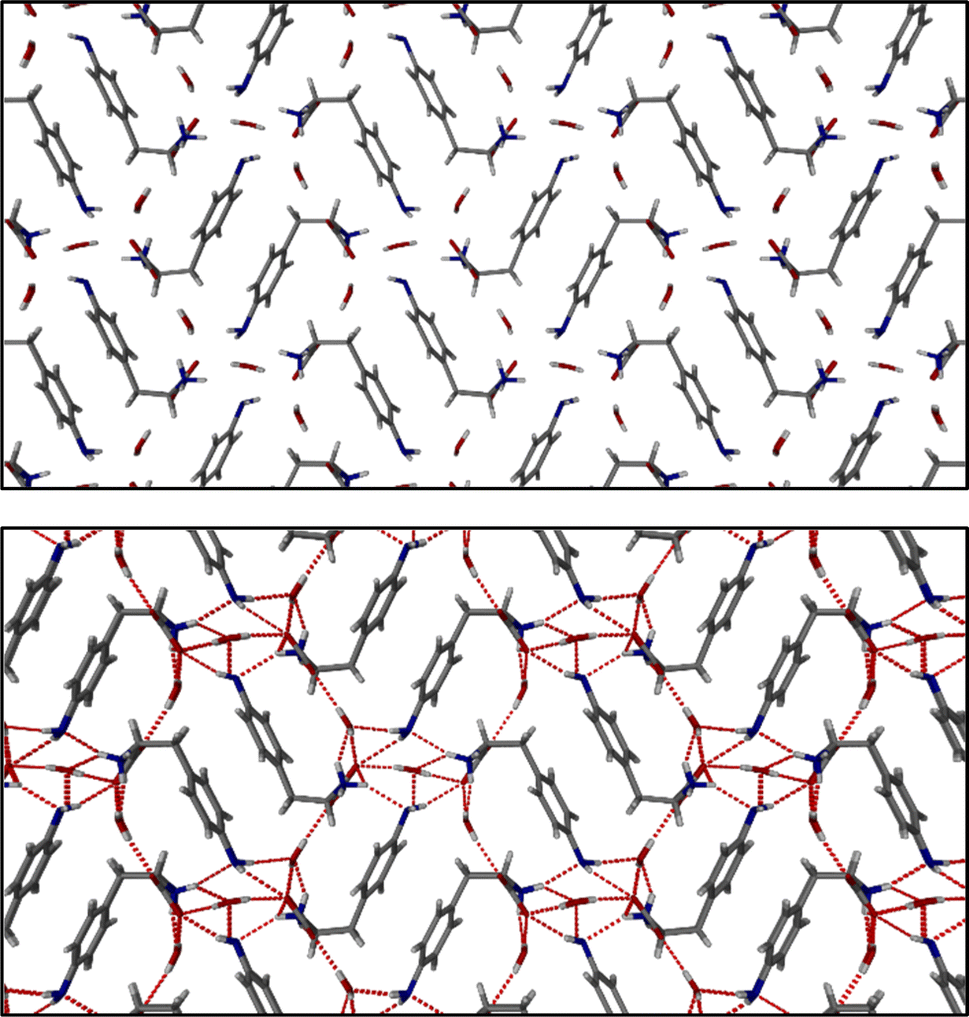 | ||
| Fig. 7 Molecular packing of NH2-Phe shown along the a axis. | ||
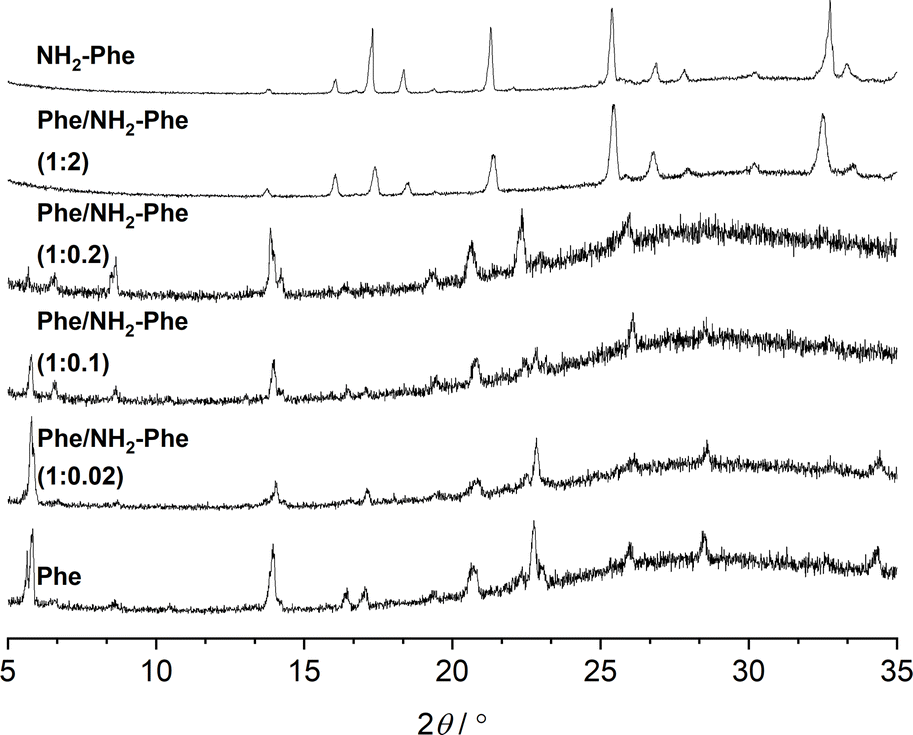 | ||
| Fig. 8 PXRD patterns of Phe (303 mM), Phe/NH2-Phe (1:0.02), Phe/NH2-Phe (1:0.1), Phe/NH2-Phe (1:0.2), Phe/NH2-Phe (1:2) and NH2-Phe (606 mM) hydrogels. | ||
The structure determined from samples of NH2-Phe in water was ascertained to be a NH2-Phe hydrate [the stoichiometry of the asymmetric unit is (NH2-Phe)2(H2O)3]. The structure of the gel fibres of the Phe gels has a crystallographic stoichiometry of (Phe)2(H2O)2, from the asymmetric unit. The “extra hydration” of the NH2-Phe hydrate is not unexpected, as the amino group provides both hydrogen-bond donation and acceptor character, but these components are not stoichiometric. The amino groups of both molecules in the asymmetric unit only interact with water as a donor hydrogen-bond group (the amino groups also donate hydrogen bonding to the carboxylate groups), with the acceptor hydrogen-bonding property of the amino groups interacting with the ammonium group only, and not water. To better understand the interactions of NH2-Phe within the crystal structure, we turned to the determination of not only the hydrogen bonding, but estimates of the interaction energies.26–29
Interaction maps of Phe and NH2-Phe were calculated to determine if there was a preferential direction associated with intermolecular interactions. This preferred-direction hypothesis has been utilised by a number of researchers to understand the relationship between crystal structures and gelation/fibre formation.3,30–33 Both single crystal structures' hydrogen positions were normalised using Mercury (CCDC). The Crystallographic Information Files (CIFs) were then analysed utilising the Crystal Explorer software and Tonto. The Phe structure reveals a strong unidirectional preference for the dimer of Phe molecules. The dimers interact through a total interaction energy of 168.1 kJ mol−1, but this dimer interaction does not form a periodic pattern. However, the two molecules in the asymmetric unit strongly interact with each other through directional periodic interactions (along the b axis) of 125.3 and 125.5 kJ mol−1. These interaction strengths are a combination of zwitterion-based ionic interactions and charge-assisted hydrogen bonds, as we and others have determined previously.16 Although the preference of the interactions is not as large in the (NH2-Phe)2(H2O)3 structure, there is still some degree of directional preference. The stacking of the NH2-Phe zwitterions is still clearly visible and dominant, with calculated interaction energies of 115.6 kJ mol−1 and 110.1 kJ mol−1 for the two molecules in the asymmetric unit. These interactions are directed along the a axis and were found to be the strongest interaction between neighbouring NH2-Phe molecules. The next-strongest interactions are more than 20 kJ mol−1 weaker, are associated with the hydrogen bonding between the amine groups and the zwitterion end group, and are found perpendicular to the stronger interactions. The next-strongest interactions (ca. 50 kJ mol−1) are associated with water hydrogen bonding. What these calculations highlight is that the one-dimensional hypothesis often utilised in connecting gelation with crystal forms can be applied here as well, even though the hydrogen bonding and π–π interactions do not clearly show a preferential direction (the point of the energy framework analysis). Coupling these interactions with the Bravais–Friedel–Donnay–Harker (BFDH) morphology prediction (performed in the CSD Mercury program), we can clearly see how the fibrous material is generated through the intermolecular interactions of NH2-Phe, in association with water.
The addition of low amounts of NH2-Phe to the Phe hydrogels did not affect the three-dimensional molecular arrangement of the Phe gel fibres, as the PXRD patterns (Fig. 8) and 1H–13C CP/MAS NMR spectra (Fig. 9) were identical to those of pure Phe. The crystalline structures of particles in suspension – Phe/NH2-Phe (1:0.4) – also matched the monohydrate form of Phe. However, a further increase in the concentration of NH2-Phe led to different results. The PXRD pattern of the mixed hydrogel was very similar to the one of pure NH2-Phe (Fig. 8), suggesting an analogous supramolecular arrangement. This is not surprising, as NH2-Phe is present at a higher concentration, imposing its crystalline organisation.
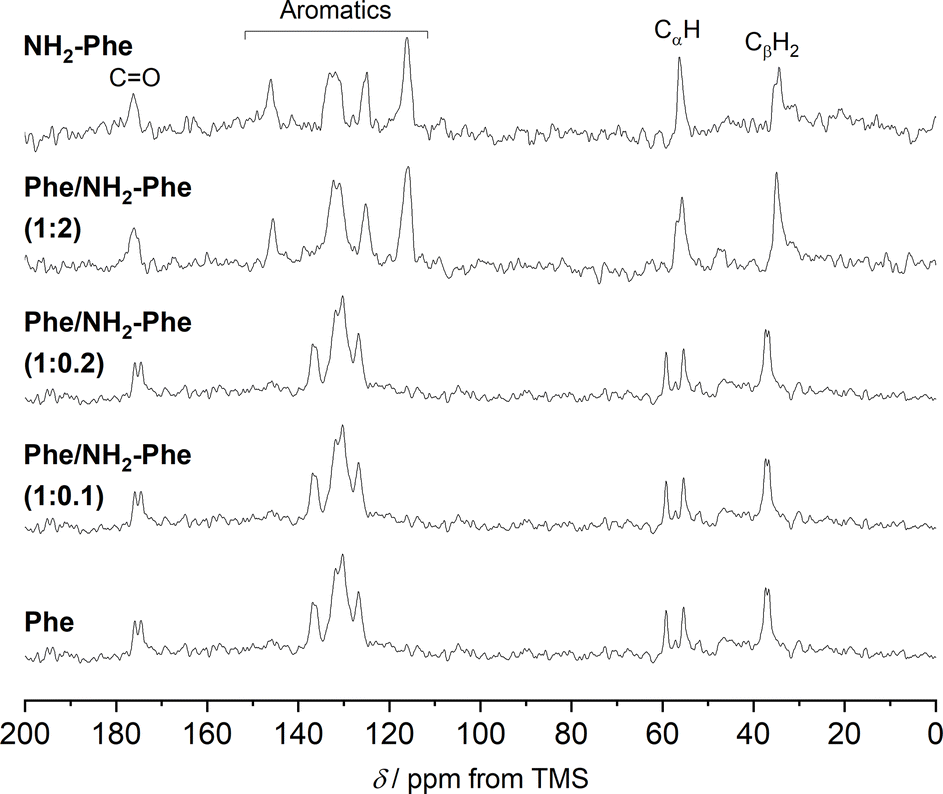 | ||
| Fig. 9
1H–13C CP/MAS NMR spectra of Phe (303 mM), Phe/NH2-Phe (1:0.1), Phe/NH2-Phe (1:0.2), Phe/NH2-Phe (1:2) and NH2-Phe (606 mM) hydrogels acquired with an MAS rate of 8.5 kHz using a 400 MHz solid-state NMR spectrometer. | ||
To help interpret the observed differences, the structure of the rigid components of the fibres was assessed using 1H–13C CP/MAS solid-state NMR spectroscopy. This method relies on the efficient transfer of polarisation from 1H to strongly dipolar-coupled 13C nuclei,34 and only the rigid gel fibres satisfy this condition. 1H–13C CP/MAS NMR spectra of Phe hydrogels showed the characteristic peak splitting of the monohydrate form, with two peaks per carbon site (Fig. 9 and 10), corresponding to two non-equivalent magnetic environments. Regarding the NH2-Phe hydrogel, the very good agreement between the 13C chemical shift values determined experimentally and those predicted using CASTEP for NH2-Phe hydrogels (Fig. 11) allowed us to confidently confirm the molecular packing motif within the gel fibres.
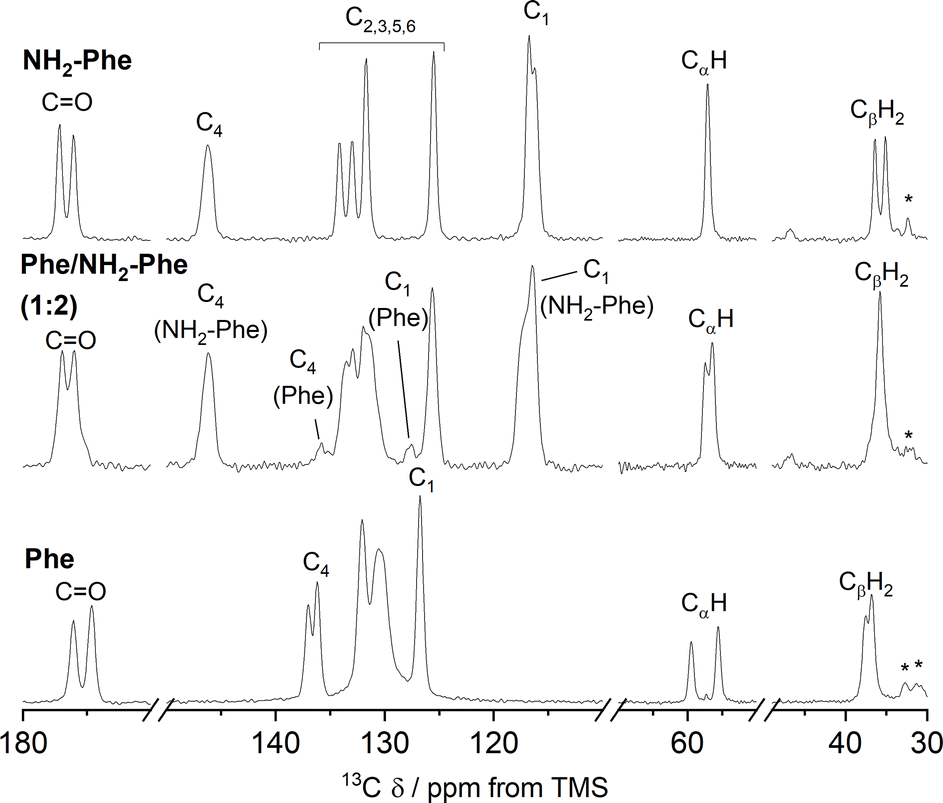 | ||
| Fig. 10 Amplification of 1H–13C CP/MAS NMR spectra of Phe (303 mM), Phe/NH2-Phe (1:2) and NH2-Phe (606 mM) dry hydrogels acquired with an MAS rate of 10 kHz using a 400 MHz solid-state NMR spectrometer. | ||
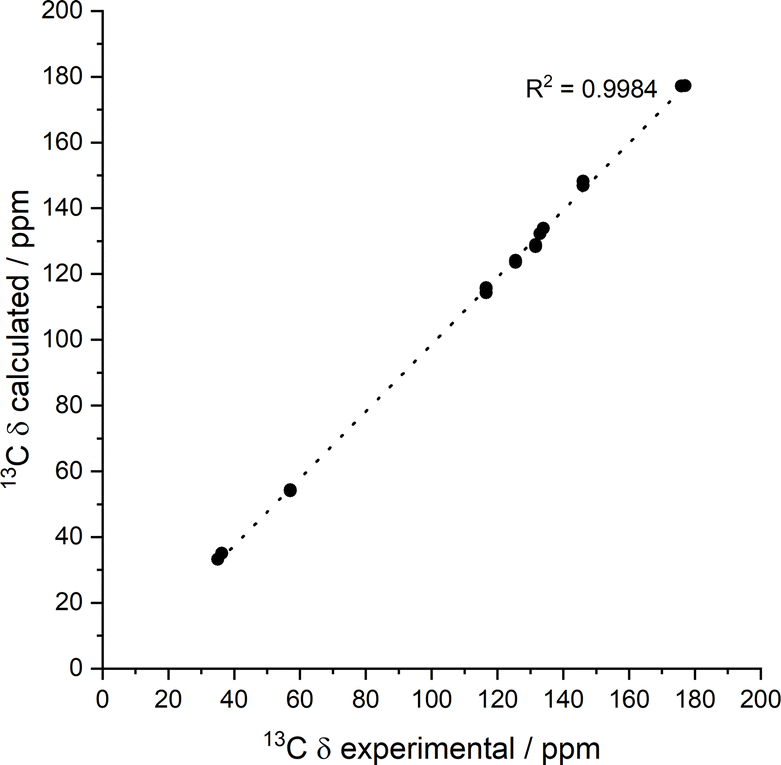 | ||
| Fig. 11 Experimental 13C chemical shift values for the NH2-Phe (606 mM) dry hydrogel acquired with an MAS rate of 10 kHz vs. calculated values for the predicted structure. Calculated isotropic chemical shieldings were converted to chemical shifts by matching the calculated and observed chemical shift of the peak at 168.25 ppm. | ||
Even though the PXRD pattern of the Phe/NH2-Phe (1:2) hydrogel presented a great resemblance to that of pure NH2-Phe, differences were identified in the CP/MAS studies (Fig. 10). The 1H–13C CP/MAS NMR spectrum was not a simple superposition of both spectra of the pure hydrogels (Fig. S14†). Instead, chemical shift variation was observed for the carbonyl, aromatic and CβH2 carbons (Table S6†). Moreover, the aromatic carbons of the mixtures were significantly broadened with different line shapes in comparison with the spectra of the single-gelator hydrogels. These studies showed that NH2-Phe is not imposing its supramolecular organisational preference, as suggested by PXRD data, but both molecules are intricately modifying each other's packing motifs.
15N MAS NMR experiments further supported the previous findings. 15N is an NMR-active nucleus sensitive to changes in the local environments of N-bearing groups and geometry of hydrogen bonds; therefore, it contains specific structural information.35 Due to its poor NMR sensitivity and negative gyromagnetic ratio,3515N-labelled Phe was used when monitoring the local environment of 15NH3+-motifs in single- and multiple-gelator hydrogels. The high-field 1H–15N CP/MAS spectrum of the Phe hydrogel showed two sharp peaks corresponding to the two molecules per asymmetric unit of the monohydrate form (Fig. 12). Interestingly, 15N-NMR peaks in the spectrum of the mixed gel system were significantly broadened with an additional 15N peak. The differences in peak intensities reflected that these 15N sites were structurally different. Furthermore, the presence of a variety of magnetically non-equivalent environments was consistent with the line broadening observed in the corresponding 1H–13C CP/MAS NMR spectrum, and showed that new 15NH3+-Phe environments were formed within the rigid fibres of multiple-gelator hydrogels. Altogether, these data show that molecules within the supramolecular structures of mixed gel systems have different local environments in comparison with those in pure hydrogels.
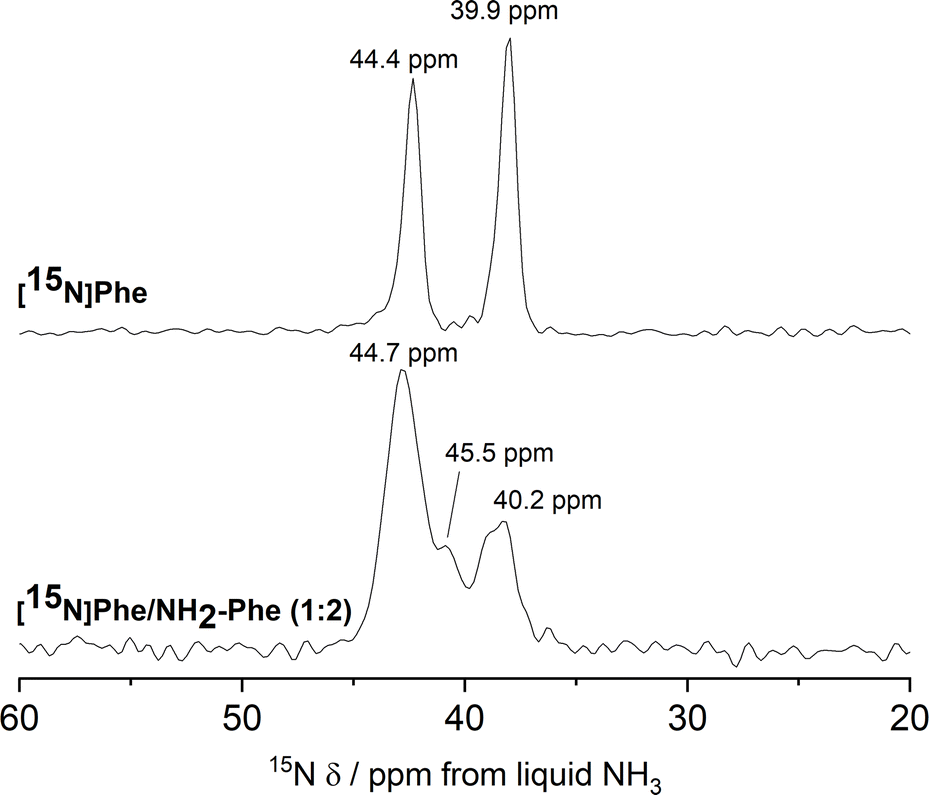 | ||
| Fig. 12
1H–15N CP/MAS NMR spectra of 15N-labelled [15N]Phe (303 mM) and [15N]Phe/NH2-Phe (1:2) dried hydrogel samples, acquired with MAS rates of 10 kHz, using an 850 MHz solid-state NMR spectrometer. | ||
Investigation of interactions responsible for aggregation
Understanding the dynamics of disruption and identifying the structure of the products may shed light on the composition of the solid-state components that pre-empt multiple-gelator hydrogelation. The supramolecular arrangement of the particles suspended in the Phe/NH2-Phe (1:0.4) mixed system corresponded to the monohydrate form with Phe and NH2-Phe present in the aggregates in a 1:1 ratio (equimolar). Interestingly, when studying spatial correlations between both molecules in the suspension and solution regimes, no cross-peaks were found between Phe and NH2-Phe in 2D 1H–1H NOESY spectra (Fig. S22†). However, these through-space interactions were detected in the hydrogel samples (Fig. 14), which are characterised by different cross-relaxation and relaxation rates. Hence, it was proposed that the lifetime of these interactions in solution and suspension is too short on the time scale of the experiment, i.e., faster than the cross-relaxation rate.
Identification of intermolecular interactions responsible for network disruption
In solution, gelation and crystallisation have the common starting point of nucleation followed by fibre or crystal growth.36 These processes affect the local environments of nuclear spins. NMR is sensitive to molecular environments and conformational changes, frequently translated by changes in chemical shifts. Experiments monitoring the chemical shift values of Phe with gradual addition of NH2-Phe were unsuccessful. Working under gel-forming conditions does not provide a clear trend in the modification of local 1H environments, since the peaks are significantly broadened and correspond to an average of multiple species in both solution and gel states.37,38 The mechanism of disruption was therefore investigated via dilution studies of the Phe/NH2-Phe (1:0.15) hydrogel. Several 1H-NMR spectra were acquired at variable concentration to identify which proton sites were most affected by aggregation processes. The most significant chemical shift variation was observed for the CαH and CβH2 protons of both Phe and NH2-Phe (Fig. 13), meaning the aliphatic region of both molecules was the most involved in the formation of intermolecular interactions. These findings also pointed towards participation of NH2-Phe in pre-gelation aggregation processes.
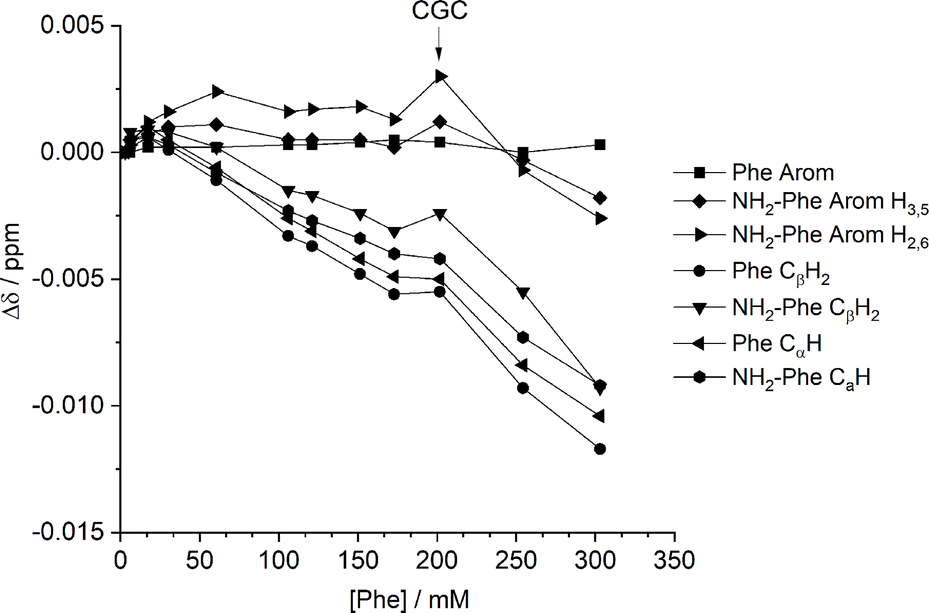 | ||
| Fig. 13 Chemical shift variation (Δ) in 1H-NMR spectra in dilution studies of the Phe/NH2-Phe (1:0.15) hydrogel, measured at 298 K. | ||
The dynamic character of supramolecular hydrogels is an advantageous feature, as molecules on the surface of fibres carry information from the network when returning to solution. The phenomenon of solution-state NMR spectra containing information from the hydrogel fibres due to fast molecular exchange between solution and gel states has been described previously.39,40 Therefore, the network can be investigated indirectly. 2D 1H–1H NOESY NMR experiments were conducted on the Phe/NH2-Phe (1:0.15) hydrogel to determine the interactions responsible for disruption of the network and probable solubilisation of Phe molecules in water. Negative cross-peaks, characteristic of medium-to-large molecules,41 were observed between all protons (Fig. 14). The resulting map of through-space connectivities allowed calculation of interproton distances (Table 1). In combination with the evolution of nOe enhancements with mixing time (Fig. S22†), interproton distances enabled the conclusion that the most significant interaction was between NH2-Phe aliphatic protons and Phe CαH. NH2-Phe and Phe probably interact in solution via their electrostatic moieties, suggesting that the mechanism of disruption of Phe dimers16 occurs via mixed H-bonding of the zwitterionic parts.
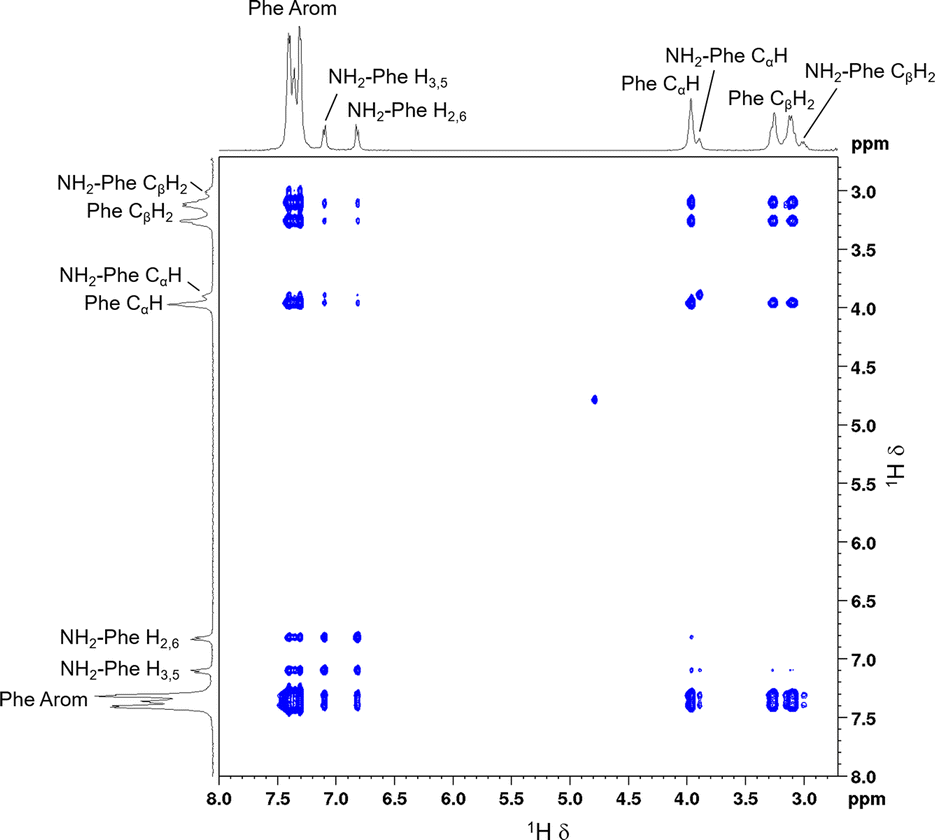 | ||
| Fig. 14 2D 1H–1H NOESY NMR spectrum of the Phe/NH2-Phe (1:0.15) hydrogel with a mixing time of 0.5 s, measured at 298 K. | ||
:0.15) hydrogel with a mixing time 0.01 s, measured at 298 K, using the H2,6–H3,5 distance from NH2-Phe as a reference. Average errors of 7% were assumed as for fast-tumbling molecules in viscous solvents27
| Correlation | r (Å) | Error (Å) |
|---|---|---|
| a Distance used as reference. | ||
| NH2-Phe Arom H2,6:NH2-Phe Arom H3,5 |
2.28a | — |
| NH2-Phe CβH2:Phe CβH2 |
2.48 | 0.20 |
| NH2-Phe Arom H3,5:Phe Arom H3,5 |
2.68 | 0.21 |
| NH2Phe CαH:Phe CβH2 |
2.70 | 0.22 |
| NH2-Phe Arom H3,5:Phe Arom H2,6 |
2.80 | 0.22 |
| NH2-Phe Arom H3,5:Phe CβH2 |
3.23 | 0.26 |
| NH2-Phe Arom H2,6:Phe Arom H3,5 |
3.24 | 0.26 |
| NH2-Phe Arom H2,6:Phe CβH2 |
3.37 | 0.27 |
| NH2-Phe Arom H2,6:Phe Arom H2,6 |
3.50 | 0.28 |
| NH2-Phe Arom H3,5:Phe CαH |
3.60 | 0.29 |
| NH2-Phe Arom H2,6:Phe CαH |
3.68 | 0.29 |
Characterisation of dynamics of disruption and gel formation
Since 1H-NMR is a quantitative analytical method, it allows the determination of the ratio between molecules dissolved in water and molecules forming a rigid gel network. The latter's short transverse relaxation times, strong dipolar couplings and chemical shift anisotropy are responsible for these components being NMR ‘silent’.39,42 Measurements of 1H-NMR peak intensity can give an indication of the concentration of NMR ‘silent’ vs. NMR ‘visible’ gelator molecules in solution-state NMR spectra. This technique was used to monitor the self-assembly processes of Phe in the presence of NH2-Phe. The 1H-NMR peaks of Phe and NH2-Phe became broader and less intense throughout the gelation process of a hot solution of Phe/NH2-Phe (1:0.1) (Fig. 15), consistent with aggregation and formation of solution-state NMR ‘silent’ components.42 Interestingly, NH2-Phe protons showed reduced peak intensities, a strong indication that ca. 35% of NH2-Phe molecules were also entrapped in the rigid gel fibres (Table S7†).
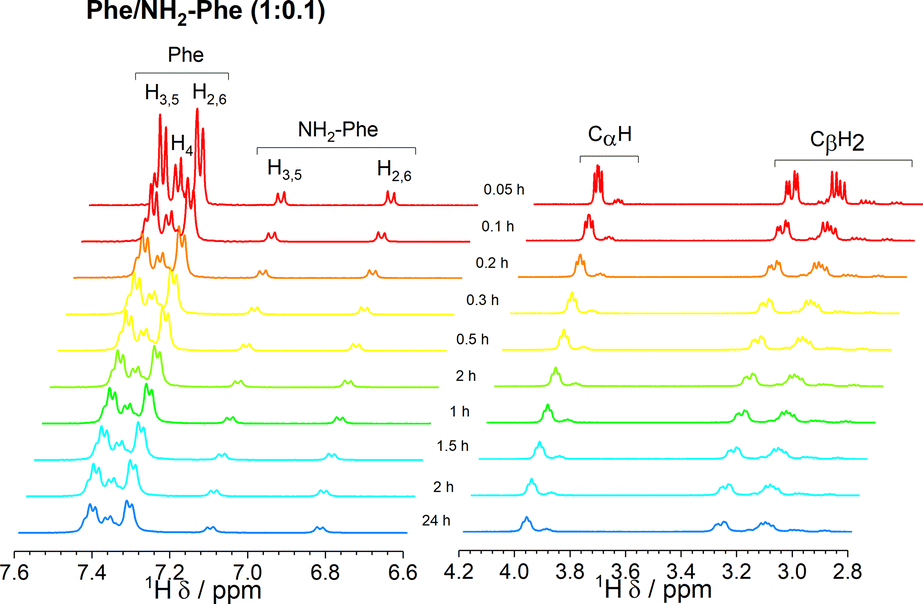 | ||
| Fig. 15 Kinetics of gelation monitored via the acquisition of 1H-NMR spectra over time, immediately after cooling down a hot solution of Phe/NH2-Phe (1:0.1), with gradual formation of a hydrogel. Colour scheme represents the temperature of the solution, as all spectra were acquired at 298 K. | ||
As the concentration of NH2-Phe in the Phe hydrogels was continuously raised, an increased peak intensity for Phe protons was recorded, reflecting the higher concentration of Phe dissolved in solution (Fig. 16 and Table S7†). Sharp and intense 1H peaks revealed the molecular variations associated with the formation of a suspension at Phe/NH2-Phe (1:0.3), exhibiting spectral features characteristic of isotropic solutions. It was concluded that the co-existence of Phe and NH2-Phe (at these molar ratios and concentrations) increased their water solubilities and led to disruption of the supramolecular network.
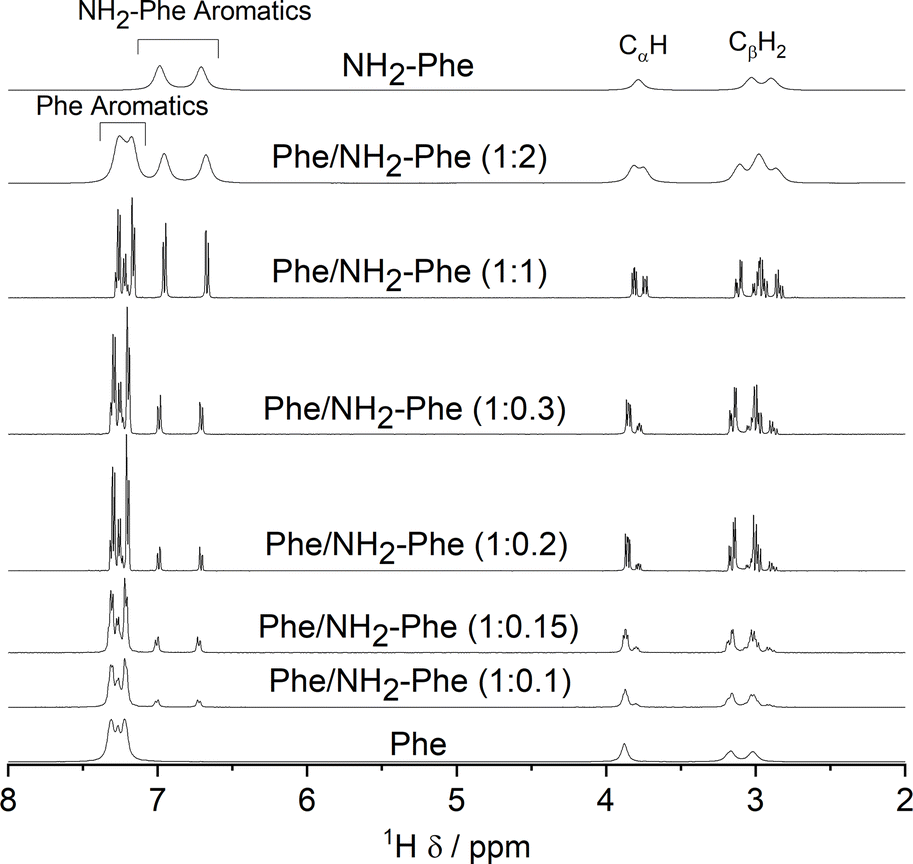 | ||
| Fig. 16
1H solution-state NMR spectra of the Phe (303 mM), Phe/NH2-Phe (1:0.1), Phe/NH2-Phe (1:0.15), Phe/NH2-Phe (1:0.2), Phe/NH2-Phe (1:2) and NH2-Phe (606 mM) hydrogels, Phe/NH2-Phe (1:0.3) suspension and Phe/NH2-Phe (1:1) solution, acquired at 298 K. | ||
Since relaxation processes are affected by 1H mobilities, these could be used to probe molecular motions in supramolecular gels.381H longitudinal relaxation times (T1) were monitored throughout the gel-to-solution transitions of these thermoreversible materials. The 1H T1 times were similar for different 1H species in the Phe hydrogel (Fig. 17), a behaviour associated with fast exchange processes occurring between gel and solution states.37 The resulting 1H T1 values are an average of molecules in both environments.37,38
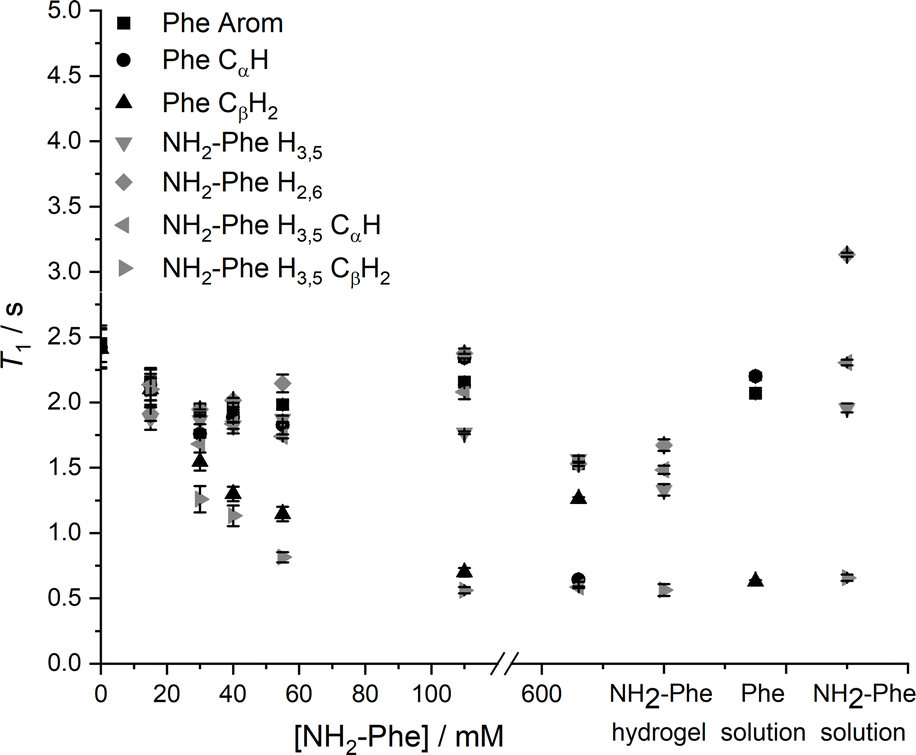 | ||
| Fig. 17 1H solution-state longitudinal relaxation times (T1) of Phe (303 mM) hydrogels with variable concentration of NH2-Phe, NH2-Phe (606 mM) hydrogel, and Phe (100 mM) and NH2-Phe (100 mM) solutions. | ||
When low concentrations of NH2-Phe were added to the Phe hydrogel, the 1H T1 times were similar for different 1H sites of NH2-Phe (Fig. 17). The similarity of the 1H T1 values proved that Phe and NH2-Phe were exchanging between gel and solution states, a phenomenon reported previously by our group.37 After the addition of 40 mM of NH2-Phe (molar ratio of 1:0.15), the similarity between T1 values for different groups was lost. A full distribution of T1 times, typical of solutions, was observed when the concentration of NH2-Phe was over 55 mM (molar ratio of 1:0.2). Above this concentration, the system was dominantly composed of fast-tumbling molecules of Phe and NH2-Phe dissolved in isotropic pools of solvent, a consequence of the destruction of the supramolecular network, in agreement with the sharp and intense peaks detected in the corresponding 1H spectrum (Fig. 16).
Similarly, the 1H T1 times in pure NH2-Phe and mixed Phe/NH2-Phe hydrogels presented a dispersion of values in the gel state similar to solutions of NH2-Phe, contrasting with the pure Phe hydrogel (Fig. 17). The linear dependence of 1H T1 times on temperature (Fig. S17†) pointed towards Phe and NH2-Phe being mainly dissolved throughout the range of temperatures. These results reflected different dynamics of exchange of molecules between gel and solution environments for the pure NH2-Phe gel and mixed gels when compared with the pure Phe gel material.
Saturation transfer difference (STD) NMR experiments were carried out to assess the dynamics of exchange at the gel/solution interfaces.37,42,43 A mono-exponential evolution of build-up of saturation in solution was observed for the Phe hydrogel (Fig. 18), indicative of fast exchange phenomena between gel and solution states on the NMR relaxation time scale.37
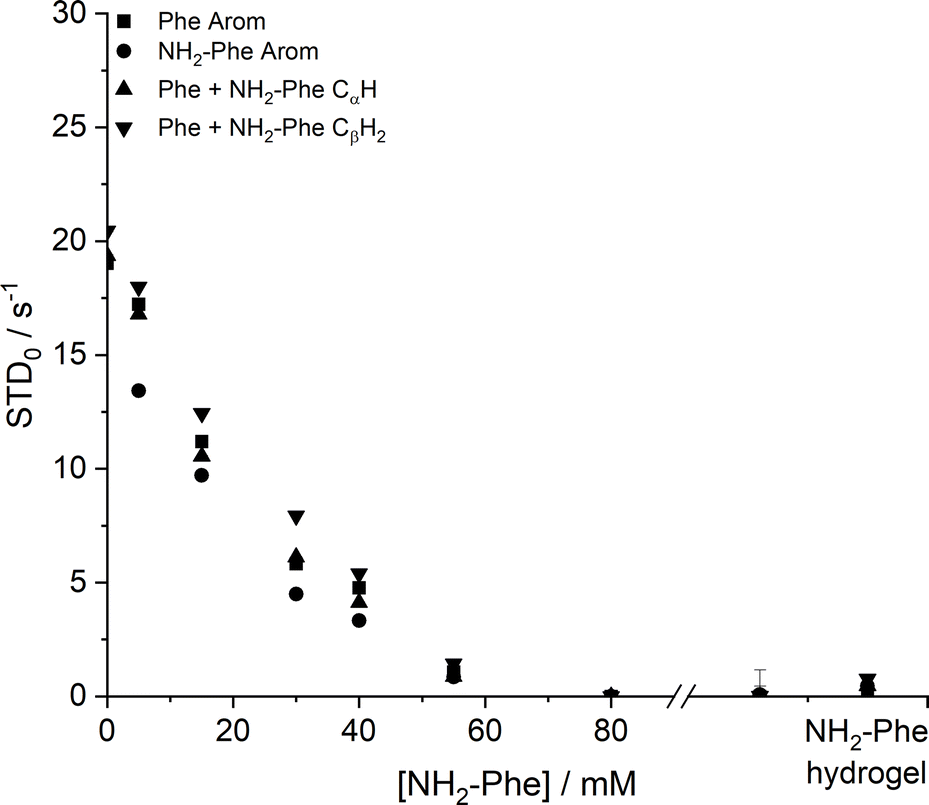 | ||
| Fig. 18 Initial slope of build-up curves (STD0) of Phe hydrogels with increasing concentrations of NH2-Phe, measured at 298 K. | ||
The initial slope for the fractional STD response, STD0, decreased gradually as higher concentrations of NH2-Phe were introduced (Fig. 18). More importantly, no build-up of saturation was detected at concentrations of NH2-Phe higher than 55 mM (molar ratio of 1:0.2). Since STD NMR experiments rely on the transfer of saturation from a large supramolecular network, which acts as a reservoir of magnetisation, to protons in close proximity, these studies proved further that the supramolecular network lost its structural integrity at molar ratios higher than Phe/NH2-Phe (1:0.2). This was in agreement with 1H longitudinal relaxation findings and was consistent with the loss of self-sustaining properties macroscopically observed. These experiments allowed determination of the ‘breaking point’ of the network at the molecular level.
Low initial build-up values (STD0) were observed in the pure NH2-Phe and mixed Phe/NH2-Phe hydrogels (Fig. 18). Accumulation of saturation in solution for Phe was therefore less efficient in the multiple-gelator hydrogel. When variable-temperature NMR measurements were carried out to investigate exchange phenomena, the levels of accumulation of saturation in solution decreased with temperature (Fig. S8 and S9†), showing that the rate of exchange between bound and free states was increased. This allowed the conclusion that, at room temperature, Phe and NH2-Phe exchange at the gel/solution interfaces faster than the NMR relaxation time scale, as a consequence of weak intermolecular interactions.
Summary
Phe self-assembles into organised gels in water.16 However, hydrogelation of Phe can be prevented when this gelator molecule is mixed with NH2-Phe at molar ratios between 1:0.2 and 1:2 (Phe/NH2-Phe), with either a suspension or solution being formed. Macroscopic observations showed that the hydrogels lost their structural integrity when the ratios of Phe/NH2-Phe were above 1:0.2, but solution-state NMR studies showed this was a continuous process. 1H-NMR and longitudinal relaxation studies indicated there was gradual solubilisation of Phe and NH2-Phe in water as NH2-Phe was added, with sharp and intense 1H-NMR peaks being accompanied by a distribution of 1H T1 times resembling those of solutions, which became more marked above a ratio of 1:0.2 (55 mM of NH2-Phe). Consequently, the considerable dissolution of the network promoted by NH2-Phe led to the disappearance of the STD NMR response at this concentration, due to the absence of a supramolecular structure capable of accumulating and transferring saturation. When monitoring both disruption and gelation phenomena, it was found that NH2-Phe was equally involved in pre-gelation aggregation, possibly manifesting its disruption effects in early nucleation processes. This was attributed to the interference of NH2-Phe with the electrostatic interactions between Phe dimers, as CαH and CβH2 protons from both molecules exhibited the most significant chemical shift variation and the strongest intermolecular nOe enhancement in 2D 1H–1H NOESY studies. Such intermolecular interactions between Phe and NH2-Phe increased their water solubilities, explaining the formation of a clear brown solution at a ratio of 1:1. This probably results from solution complexation processes, with the formation of a stable and soluble Phe/NH2-Phe complex in solution.44
When working at higher concentrations of NH2-Phe, a different scenario was encountered. NH2-Phe was mixed with Phe at gel-forming concentrations ([Phe] > 212 mM and [NH2-Phe] > 388 mM), and multiple-gelator hydrogels of Phe/NH2-Phe were obtained. Morphology studies showed the formation of individual fibres of Phe or NH2-Phe, pointing towards a system that maintains the structure of the pure gelators, a phenomenon named self-sorting. Their supramolecular arrangements seemed to be dominated by the NH2-Phe crystal structure, as diffraction experiments showed great similarity between the crystalline components of NH2-Phe and Phe/NH2-Phe hydrogels. However, investigation of local molecular environments via solid-state NMR spectroscopy showed a certain degree of supramolecular disorder. The line broadening observed in 1H–13C CP/MAS NMR spectra and the presence of additional 15N environments for Phe provided evidence that both gelator molecules were differently surrounded within the rigid mixed fibres. Moreover, the dispersion of 1H T1 values and the low STD NMR response reflected modification of their dynamics of exchange at the gel/solution interfaces in comparison with pure hydrogels. These data provided evidence of interaction between both gelator molecules in solution and on the surface of the mixed hydrogel fibres. It is the affinity of Phe for NH2-Phe, and vice versa, that is behind the presence of both molecules in the solid fibres. We hypothesised that multiple-gelator hydrogels were composed of purely self-sorted fibres in a delicate balance with co-assembled structures formed of both Phe and NH2-Phe molecules.
We also found out that NH2-Phe is able to independently self-assemble in water to give rise to strong brown gels above 388 mM. The three-dimensional ordering of the needle-like crystalline fibres was determined to be an (NH2-Phe)2(H2O)3 structure using diffraction methods, giving a very good agreement with solid-state NMR experiments. These fibres incorporated ca. 80% of the gelator molecules, with the rest coexisting dissolved in pools of water or partially trapped at the fibre interfaces, and exchanging between both environments. The dynamics of this exchange were faster than in the pure Phe hydrogel, highlighting the importance of the Phe head group in dictating the strength of intermolecular interactions.45
Conclusions
Phe and NH2-Phe were found to self-assemble in water into crystalline hydrogels, and we managed to determine the crystal structure of the NH2-Phe gels. The determined faster dynamics of exchange between gel and solution states of NH2-Phe in comparison with Phe were correlated with weaker intermolecular interactions, highlighting the role of head groups in dictating the strength of intermolecular interactions.When mixed in water, different products were obtained depending on the concentration and molar ratio of the gelator molecules. At low concentrations of NH2-Phe, disruption of the network was promoted by interference of the aliphatics of NH2-Phe with the electrostatic interactions between Phe molecules, which are the anisotropic forces of self-assembly of Phe. The affinity between both molecules in solution, forming a solution complex, most likely is responsible for network disruption and provided clues on their interaction in the solid state. At high concentrations of NH2-Phe, multiple-gelator hydrogels were formed with crystal habits different from those of the pure gel fibres, as new environments were detected using solid-state NMR. Consequently, in these mixed materials, the interactions between Phe and NH2-Phe were different from those present in pure hydrogels, and the phenomenon of exchange at the gel/solution interfaces was modulated. Despite Phe and NH2-Phe forming different crystal structures, their molecular similarity and similar potential to participate in non-covalent bonds allows them to intimately interact in solution during self-assembly processes, which is manifested at larger scales by the modulation of the resulting fibres at gel-forming concentrations. These findings may provide an insight into the mechanisms of prevention of accumulation of Phe, as well as aggregation processes of peptides and proteins in pathological processes.
Experimental section
Materials
Reagent grade (>98%) L-phenylalanine and hexamethylbenzene (HMB) were purchased from Sigma-Aldrich and 4-amino-L-phenylalanine from Fluorochem. L-[13C9,15N]-Phenylalanine and L-[15N]-glycine were purchased from Cortecnet. Deuterium oxide and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) were purchased from Goss Scientific. We note that, in some cases, care should be taken with commercial samples of 4-amino-L-phenylalanine as these are not always sufficiently pure to ensure that gelation is reproducible. Milli-Q water was obtained with a Thermo Scientific Barnstead Nanopure purification system coupled to a Barnstead hollow fibre filter.Methods
Crystal data for (NH2-Phe)2(H2O)3: C18 H30 N4 O7, M = 414.46, clear colourless needle, 0.40 × 0.05 × 0.01 mm3, monoclinic, space group P21 (No. 4), a = 5.9813(9) Å, b = 11.3702(15) Å, c = 14.985(2) Å, β = 93.681(8)°, V = 1017.0(2) Å3, Z = 2, Dc = 1.353 g cm−3, F000 = 444, Bruker APEX-II CCD, MoKα radiation, λ = 0.71073 Å, T = 100.15 K, 2θmax = 51.6°, 2591 reflections collected, 2591 unique (Rint (merged) = 0.0726). Final GooF = 1.086, R1 = 0.0573, wR2 = 0.1576, R indices based on 2295 reflections with I > 3s(I) (refinement on F2), 277 parameters, 15 restraints. Lp and absorption corrections applied, μ = 0.105 mm−1.
Solid-state NMR spectroscopy. Solid-state NMR experiments were performed using a Bruker Avance III spectrometer at a 1H frequency of 400.23 MHz, 13C frequency of 100.65 MHz and 15N frequency of 40.56 MHz, equipped with a 4 mm triple resonance wide-bore probe. 40 μL hot solutions were transferred into Kel-F inserts and allowed to cool down to room temperature, after which gels were obtained. 1H–13C CP/MAS NMR experiments were conducted using a recycle delay of 20 s and a contact time of 2 ms. 1H–13C CP/MAS NMR spectra of reference solid powders, dried gel samples and hydrogels were acquired using 128, 2048 or 8192 scans, respectively. A magic-angle spinning (MAS) rate of 10 kHz was used for dried powder and gel samples and a rate of 8.5 kHz was used for hydrogels. 1H–15N CP/MAS experiments were conducted using 4096 scans, a recycle delay of 10 s and a contact time of 2 ms. An MAS rate of 10 kHz was used for dried gel samples and a rate of 8.5 kHz was used for hydrogels. High-field solid-state NMR experiments were carried out using a Bruker Avance III NMR spectrometer operating at a 1H frequency of 850.22 MHz, 13C frequency of 231.81 MHz and 15N frequency of 86.15 MHz, equipped with a 3.2 mm triple resonance probe. Dried samples were packed directly into 3.2 mm zirconia rotors. 1H–15N CP/MAS spectra were acquired using 1024 scans, a recycle delay of 10 s, a contact time of 2 ms and an MAS rate of 10 kHz. 1H and 13C spectra were referenced to tetramethylsilane (TMS). 15N spectra were referenced to liquid NH3. Hartmann-Hahn conditions were matched using hexamethylbenzene (HMB) for 1H–13C experiments and L-[15N]-glycine for 1H–15N experiments. All experiments were conducted at 298 K.
Solution-state NMR spectroscopy. Solution-state NMR experiments were performed using a Bruker Avance I spectrometer at a 1H frequency of 499.69 MHz equipped with a 5 mm probe. 600 μL hot solutions were transferred into NMR tubes and allowed to cool down to room temperature, after which gels were obtained. Variable temperature (VT) experiments were carried out from 278 to 353 K, allowing thermal stabilisation of the sample for 15 min. 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) was used as an internal NMR standard inside a coaxial insert.
1H-NMR spectra were acquired with excitation sculpting for water suppression (zgespg), a recycle delay of 10 s and 16 scans. 1H longitudinal relaxation times (T1) were measured using a standard inversion recovery pulse sequence with a recycle delay of 10 s and 8 scans. 16 points were recorded at variable time delays ranging from 0.1 to 20 s. The evolution of intensities was fitted mathematically to the mono-exponential function 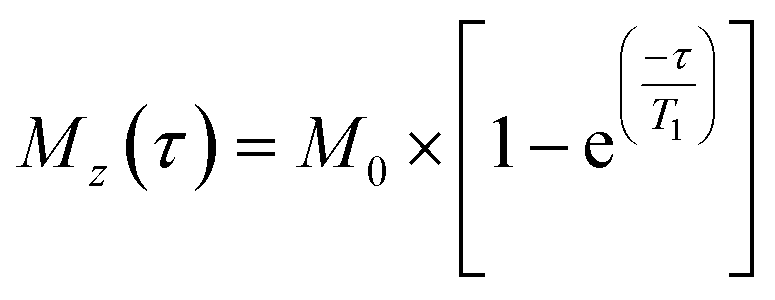 , where Mz is the z-component of magnetisation, M0 is the equilibrium magnetisation and τ is the time delay.46
, where Mz is the z-component of magnetisation, M0 is the equilibrium magnetisation and τ is the time delay.46
2D 1H–1H Nuclear Overhauser effect spectroscopy (NOESY) experiments were recorded using a phase-sensitive 2D NOESY pulse sequence with WATERGATE for water suppression (noesygpph19). 16 points were recorded using variable mixing times (τm = 0.01, 0.1, 0.25 and 0.5), a recycle delay of 2 s and 32 scans. Internuclear distances were calculated according to the Initial Rate Approximation, which establishes that the initial build-up of NOE enhancements with mixing time is approximately linear. The cross-relaxation rate could therefore be determined from the initial slope of the build-up curve (IIS as a function of tm), where the NOE enhancement (IIS) was defined as the ratio between the intensity of the cross-peak and the intensity of the sum of the diagonal peaks, an approach that has been applied to organogels by Canet et al. (2012).39 In turn, the cross-relaxation rate (sIS) was proportional to the inverse sixth power of the internuclear distance,
| sIS = ζrIS−6. | (1) |
This relationship between intensity and distance allowed the observed nOe intensities to be calibrated relative to a known internuclear distance (NH2-Phe H2,6 – NH2-Phe H3,5) within the supramolecular system.39,47,48
Saturation transfer difference NMR experiments were performed with selective saturation of a determined 1H frequency using a train of 40 Gaussian pulses with a duration of 50 ms (stddiffgp19.2), acquired with a recycle delay of 6 s and 16 scans. STD spectra were created by the subtraction of an on-resonance spectrum (STDon), in which a spectral region was selectively saturated, to an off-resonance spectrum (STDoff), acquired with no selective saturation. Interleaved acquisition of STDon and STDoff spectra was performed as pseudo-2D experiments to minimise artefacts caused by variations throughout the experiment. STDon spectra were acquired at a saturation frequency of 1 ppm (where only resonances of the network could be encountered), whereas the STDoff saturation frequency was set to 40 ppm. Each pair of experiments was carried out at variable saturation times ranging from 0.25 to 6 s. The signal intensity in the STD spectrum relative to the STDoff spectrum was used to determine the fractional STD response, ηSTD:
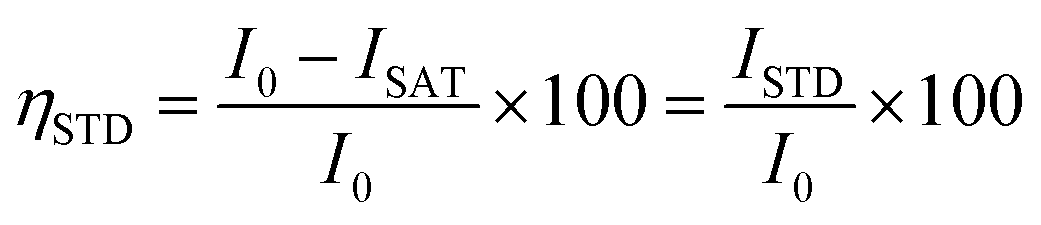 | (2) |
Author contributions
Susana M. Ramalhete: conceptualization, formal analysis, investigation, writing – original draft, review and editing, visualization, funding acquisition; Karol P. Nartowski: conceptualization, methodology, investigation, writing – original draft, review and editing; Hayley Green: analysis, investigation, writing – review and editing; Jesús Angulo: analysis, conceptualization, methodology, investigation, writing – review & editing, supervision; Dinu Iuga: analysis, investigation, writing – review and editing; László Fábián: analysis, conceptualization, methodology, investigation, writing – review and editing, supervision; Gareth O. Lloyd: analysis, conceptualization, methodology, investigation, writing – review and editing, supervision, funding acquisition; Yaroslav Z. Khimyak: analysis, conceptualization, methodology, investigation, writing – original draft, review and editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The UK 850 MHz solid-state NMR Facility used in this research was funded by the EPSRC and BBSRC (contract reference PR140003), as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). SMR also thanks Mr B. Lézé for conduction of SEM and PXRD measurements. SMR thanks the University of East Anglia for the postgraduate studentship. GOL thanks the Heriot-Watt University and the Royal Society of Edinburgh/Scottish Government Personal Research Fellowship scheme for funding. GOL and YZK are grateful to EPSRC Directed Assembly Network for financial support. We acknowledge the use of the 850 MHz solid-state NMR facility at the University of Warwick for the high-field and very fast MAS NMR experiments.References
- J. W. Steed, Supramolecular gel chemistry: developments over the last decade, Chem. Commun., 2011, 47(5), 1379–1383 RSC.
- D. J. Adams, Dipeptide and Tripeptide Conjugates as Low-Molecular-Weight Hydrogelators, Macromol. Biosci., 2011, 11(2), 160–173 CrossRef CAS.
- D. K. Kumar and J. W. Steed, Supramolecular gel phase crystallization: orthogonal self-assembly under non-equilibrium conditions, Chem. Soc. Rev., 2014, 43(7), 2080–2088 RSC.
- N. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Hydrogels in pharmaceutical formulations, Eur. J. Pharm. Biopharm., 2000, 50(1), 27–46 CrossRef CAS PubMed.
- R. Fairman and K. S. Åkerfeldt, Peptides as novel smart materials, Curr. Opin. Struct. Biol., 2005, 15(4), 453–463 CrossRef CAS.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Insights into low molecular mass organic gelators: a focus on drug delivery and tissue engineering applications, Soft Matter, 2014, 10(2), 237–256 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani and J. E. Gough, et al., Nanostructured hydrogels for three-dimensional cell culture through self-assembly of fluorenylmethoxycarbonyl–dipeptides, Adv. Mater., 2006, 18(5), 611–614 CrossRef CAS.
- H. Wang, L. Lv, G. Xu, C. Yang, J. Sun and Z. Yang, Molecular hydrogelators consist of Taxol and short peptides/amino acids, J. Mater. Chem., 2012, 22(33), 16933–16938 RSC.
- L. E. Buerkle and S. J. Rowan, Supramolecular gels formed from multi-component low molecular weight species, Chem. Soc. Rev., 2012, 41(18), 6089–6102 RSC.
- A. R. Hirst and D. K. Smith, Two-Component Gel-Phase Materials—Highly Tunable Self-Assembling Systems, Chem.–Eur. J., 2005, 11(19), 5496–5508 CrossRef CAS PubMed.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno and A. Caflisch, et al., Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria, Nat. Chem. Biol., 2012, 8(8), 701–706 CrossRef CAS.
- T. D. Do, W. M. Kincannon and M. T. Bowers, Phenylalanine oligomers and fibrils: the mechanism of assembly and the importance of tetramers and counterions, J. Am. Chem. Soc., 2015, 137(32), 10080–10083 CrossRef CAS PubMed.
- S. Shaham-Niv, A. Ezra, D. Zaguri, S. R. Shotan, E. Haimov and H. Engel, et al., Targeting phenylalanine assemblies as a prospective disease-modifying therapy for phenylketonuria, Biophys. Chem., 2024, 308, 107215 CrossRef CAS PubMed.
- W.-P. Hsu, K.-K. Koo and A. S. Myerson, The gel-crystallization of 1-phenylalanine and aspartame from aqueous solutions, Chem. Eng. Commun., 2002, 189(8), 1079–1090 CrossRef CAS.
- V. Singh, R. K. Rai, A. Arora, N. Sinha and A. K. Thakur, Therapeutic implication of L-phenylalanine aggregation mechanism and its modulation by D-phenylalanine in phenylketonuria, Sci. Rep., 2014, 4, 3875 CrossRef PubMed.
- K. P. Nartowski, S. M. Ramalhete, P. C. Martin, J. S. Foster, M. Heinrich and M. D. Eddleston, et al., The Plot Thickens, Gelation by Phenylalanine in Water and Dimethyl Sulfoxide, Cryst. Growth Des., 2017, 17, 4100–4109 CrossRef CAS.
- H. M. Wyss, Rheology of soft materials, Fluids, Colloids, and Soft Materials: An Introduction to Soft Matter Physics, 2016, vol. 7, p. 149 Search PubMed.
- F. M. Menger and K. L. Caran, Anatomy of a gel. Amino acid derivatives that rigidify water at submillimolar concentrations, J. Am. Chem. Soc., 2000, 122(47), 11679–11691 CrossRef CAS.
- M. Lusi, Solid solutions mixed crystals and eutectics, in Comprehensive Supramolecular Chemistry II, 2017, 109–125 Search PubMed.
- E. Schur, E. Nauha, M. Lusi and J. Bernstein, Kitaigorodsky revisited: polymorphism and mixed crystals of acridine/phenazine, Chem.–Eur. J., 2015, 21(4), 1735–1742 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, M. Lestari and M. Lusi, Cocrystals Help Break the “Rules” of Isostructurality: Solid Solutions and Polymorphism in the Malic/Tartaric Acid System, Cryst. Growth Des., 2018, 18(2), 855–863 CrossRef CAS.
- Y. Yang, H. Zhang, S. Du, M. Chen, S. Xu and L. Jia, et al., Ternary phase diagram and the formation mechanism of two distinct solid solutions of amino acid systems: L-Valine/L-norvaline and L-valine/L-alanine, J. Chem. Thermodyn., 2018, 119, 34–43 CrossRef CAS.
- E. Nauha, P. Naumov and M. Lusi, Fine-tuning of a thermosalient phase transition by solid solutions, CrystEngComm, 2016, 18(25), 4699–4703 RSC.
- O. Shemchuk, D. Braga and F. Grepioni, Alloying barbituric and thiobarbituric acids: from solid solutions to a highly stable keto co-crystal form, Chem. Commun., 2016, 52(79), 11815–11818 RSC.
- M. Lusi, I. J. Vitorica-Yrezabal and M. J. Zaworotko, Expanding the scope of molecular mixed crystals enabled by three component solid solutions, Cryst. Growth Des., 2015, 15(8), 4098–4103 CrossRef CAS.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems, IUCrJ, 2017, 4(5), 575–587 CrossRef CAS PubMed.
- D. E. Braun and U. J. Griesser, Supramolecular organization of nonstoichiometric drug hydrates: dapsone, Front. Chem., 2018, 6, 31 CrossRef.
- C. Wang and C. C. Sun, Identifying slip planes in organic polymorphs by combined energy framework calculations and topology analysis, Cryst. Growth Des., 2018, 18(3), 1909–1916 CrossRef CAS.
- D. Dey, S. Bhandary, S. P. Thomas, M. A. Spackman and D. Chopra, Energy frameworks and a topological analysis of the supramolecular features in in situ cryocrystallized liquids: tuning the weak interaction landscape via fluorination, Phys. Chem. Chem. Phys., 2016, 18(46), 31811–31820 RSC.
- R. Roy, J. Deb, S. S. Jana and P. Dastidar, Exploiting Supramolecular Synthons in Designing Gelators Derived from Multiple Drugs, Chem.–Eur. J., 2014, 20(47), 15320–15324 CrossRef CAS PubMed.
- P. Sahoo, N. Adarsh, G. E. Chacko, S. R. Raghavan, V. G. Puranik and P. Dastidar, Combinatorial Library of Primaryalkylammonium Dicarboxylate Gelators: A Supramolecular Synthon Approach, Langmuir, 2009, 25(15), 8742–8750 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Water gelation by small organic molecules, Chem. Rev., 2004, 104(3), 1201–1218 CrossRef CAS.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Halogen-bonding-triggered supramolecular gel formation, Nat. Chem., 2013, 5(1), 42–47 CrossRef CAS PubMed.
- W. Kolodziejski and J. Klinowski, Kinetics of cross-polarization in solid-state NMR: a guide for chemists, Chem. Rev., 2002, 102(3), 613–628 CrossRef CAS PubMed.
- G. J. Martin, M. L. Martin and J.-P. Gouesnard, 15N-NMR Spectroscopy, Springer Science & Business Media, 2012 Search PubMed.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, The delicate balance between gelation and crystallisation: structural and computational investigations, Soft Matter, 2010, 6(17), 4144–4156 RSC.
- S. M. Ramalhete, K. P. Nartwoski, N. Sarathchandra, J. S. Foster, A. N. Round and J. Angulo, et al., Supramolecular amino acid based hydrogels: probing the contribution of additive molecules using NMR spectroscopy, Chem.–Eur. J., 2017, 23, 8014–8024 CrossRef CAS.
- C. L. Cooper, T. Cosgrove, J. S. van Duijneveldt, M. Murray and S. W. Prescott, The use of solvent relaxation NMR to study colloidal suspensions, Soft Matter, 2013, 9(30), 7211–7228 RSC.
- S. Bouguet-Bonnet, M. Yemloul and D. Canet, New Application of Proton Nuclear Spin Relaxation Unraveling the Intermolecular Structural Features of Low-Molecular-Weight Organogel Fibers, J. Am. Chem. Soc., 2012, 134(25), 10621–10627 CrossRef CAS.
- F. Piana, D. H. Case, S. M. Ramalhete, G. Pileio, M. Facciotti and G. M. Day, et al., Substituent interference on supramolecular assembly in urea gelators: synthesis, structure prediction and NMR, Soft Matter, 2016, 12(17), 4034–4043 RSC.
- G. Clore and A. Gronenborn, Theory and applications of the transferred nuclear Overhauser effect to the study of the conformations of small ligands bound to proteins, J. Magn. Reson., 1982, 48(3), 402–417 CAS.
- B. Escuder, M. LLusar and J. F. Miravet, Insight on the NMR study of supramolecular gels and its application to monitor molecular recognition on self-assembled fibers, J. Org. Chem., 2006, 71(20), 7747–7752 CrossRef CAS PubMed.
- M. D. Segarra-Maset, B. Escuder and J. F. Miravet, Selective Interaction of Dopamine with the Self-Assembled Fibrillar Network of a Molecular Hydrogel Revealed by STD-NMR, Chem.–Eur. J., 2015, 21(40), 13925–13929 CrossRef CAS PubMed.
- A. Jawor-Baczynska, B. D. Moore, H. S. Lee, A. V. McCormick and J. Sefcik, Population and size distribution of solute-rich mesospecies within mesostructured aqueous amino acid solutions, Faraday Discuss., 2013, 167, 425–440 RSC.
- S. Ramalhete, J. S. Foster, H. R. Green, K. P. Nartowski, M. Heinrich and P. Martin, et al., FDHALO17: Halogen effects on the solid-state packing of phenylalanine derivatives and the resultant gelation properties, Faraday Discuss., 2017, 203, 423–439 RSC.
- R. R. Ernst, G. Bodenhausen and A. Wokaun, Principles of Nuclear Magnetic Resonance in One and Two Dimensions, Clarendon Press Oxford, 1987 Search PubMed.
- C. P. Butts, C. R. Jones, E. C. Towers, J. L. Flynn, L. Appleby and N. J. Barron, Interproton distance determinations by NOE–surprising accuracy and precision in a rigid organic molecule, Org. Biomol. Chem., 2011, 9(1), 177–184 RSC.
- D. Neuhaus and M. P. Williamson, The Nuclear Overhauser Effect in Structural and Conformational Analysis, VCH, New York, 1989 Search PubMed.
- A. Viegas, J. Manso, F. L. Nobrega and E. J. Cabrita, Saturation-transfer difference (STD) NMR: a simple and fast method for ligand screening and characterization of protein binding, J. Chem. Educ., 2011, 88(7), 990–994 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental data, structural characterisation of hydrogels including single and powder X-ray diffraction data, NMR spectra recorded in solution and solid state, and data of thermal analysis. CCDC 2350191. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4fd00081a |
| ‡ Deceased author. |
| This journal is © The Royal Society of Chemistry 2025 |