 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNafion coated nanopore electrode for improving electrochemical aptamer-based biosensing†
Grayson F. Huldinab,
Junming Huanga,
Julius Reitemeier a and
Kaiyu X. Fu*ac
a and
Kaiyu X. Fu*ac
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: kfu@nd.edu
bMaterials Science and Engineering Program, University of Notre Dame, Notre Dame, Indiana 46556, USA
cBerthiaume Institute for Precision Health, University of Notre Dame, Notre Dame, Indiana 46556, USA
First published on 14th August 2024
Abstract
The transition to a personalized point-of-care model in medicine will fundamentally change the way medicine is practiced, leading to better patient care. Electrochemical biosensors based on structure-switching aptamers can contribute to this medical revolution due to the feasibility and convenience of selecting aptamers for specific targets. Recent studies have reported that nanostructured electrodes can enhance the signals of aptamer-based biosensors. However, miniaturized systems and body fluid environments pose challenges such as signal-to-noise ratio reduction and biofouling. To address these issues, researchers have proposed various electrode coating materials, including zwitterionic materials, biocompatible polymers and hybrid membranes. Nafion, a commonly used ion exchange membrane, is known for its excellent permselectivity and anti-biofouling properties, making it a suitable choice for biosensor systems. However, the performance and mechanism of Nafion-coated aptamer-based biosensor systems have not been thoroughly studied. In this work, we present a Nafion-coated gold nanoporous electrode, which excludes Nafion from the nanoporous structures and allows the aptamers immobilized inside the nanopores to freely detect chosen targets. The nanopore electrode is formed by a sputtering and dealloying process, resulting in a pore size in tens of nanometers. The biosensor is optimized by adjusting the electrochemical measurement parameters, aptamer density, Nafion thickness and nanopore size. Furthermore, we propose an explanation for the unusual signaling behavior of the aptamers confined within the nanoporous structures. This work provides a generalizable platform to investigate membrane-coated aptamer-based biosensors.
Introduction
A long-standing goal in the field of bioanalytical chemistry is determining if a treatment is effective over time in a biological environment. Most treatments can be confirmed using a standard endpoint measurement,1 while treatments for chronic diseases, such as cancer, where drug dosing and metabolism vary from person to person, would ideally be constantly monitored to determine if a treatment is effective.2,3 So continuous in vivo biosensors for long-term measurements are needed to monitor the target concentration profile and kinetics.4,5 Electrochemical aptamer based (EAB) biosensors have been developed over the past two decades; these sensors offer multiple advantages over traditional electrochemical biosensors that rely on direct redox reactions of analytes. Aptamers are short segments of single-stranded RNA or DNA selected with high affinity for a specific target and they undergo a conformation change in the presence of the target which produces a measurable readout signal.6–8 Unlike other affinity-based biosensors, such as electrochemical ELISA, EAB biosensors are tunable, allowing researchers to optimize the sensitivity, selectivity and to detect the selected target in a reversible manner for continuous and long-term biosensing.To monitor treatments continuously it is important for in vivo electrochemical biosensors to detect targets inside living organisms with high fidelity while maintaining a small device footprint.9–11 Advances in nanoelectrochemistry over the past decade have enabled electrochemical biosensors to achieve lower detection limits and higher signal-to-noise ratios by scaling down the electrode dimension to the nanometer scale.12–21 Early pioneering works used nanostructured electrodes to enhance biosensors’ signals through increased surface area and these biosensors have shown orders of magnitude improved detection performance for DNA and RNA targets by controlling the electrode morphologies and compositions.22 Recently, we designed nanoporous gold electrodes in conjunction with aptamer-based biosensors, demonstrated superior detection performance and enhanced stability of doxorubicin (DOX) measurement in the tumor environment compared to planar gold electrodes.23 In addition to signal enhancement, using a combination of experimental and computational approaches, we observed faster electron transfer, which contributes to a better detection limit due to the extension of the Debye volume within the confined nanoporous environment.24
Another hurdle to implementing in vivo biosensing using electrochemical biosensors is the sensor's stability in a complex biological environment where device implantation often triggers the foreign body reactions leading to chronic biofouling issues.25–28 Biocompatible membrane coatings have been widely used to passively protect implanted biomaterials and extend the lifetime of the materials.29,30 In addition, the membrane coated onto various biosensors must be deliberately designed to maintain or even enhance the biosensing functionality without compromising the antifouling performance.31 Nafion, a commercially available cation exchange membrane with a polyfluorinated carbon backbone and negatively charged sulfonate group, exhibits chemical inertness, anti-biofouling capabilities, cation permeability and has attracted particular interest for in vivo biosensing over the years. Early works showed that a Nafion coated planar electrode can enhance the electrochemical signal of ruthenium bipyridine (Ru(bpy)3).32,33 Also Nafion coated polyelectrolyte multilayers have been applied to enhance the cation selectivity for electrodialysis application.34 We furthered those studies by using Nafion as an ionic gate on a nanopore electrode array, resulting in the exclusive rejection of negatively charged ferricyanide while greatly enhancing the transport of the positively charged ruthenium hexaamine.35 Nafion has also been utilized in blood glucose sensors to block interfering molecules that could also interact with glucose oxidase, leading to stable glucose sensing over time.36,37 Thus, it is natural to anticipate that membrane coatings will play an essential role in protecting EAB biosensors and with further study to endow them with additional transport selectivity and increased sensitivity.
In this work, we expand on our previous studies on membrane-coated nanoporous electrodes and investigate the interaction of the Nafion membrane and nanoporous electrode that ultimately affect the biosensing performance. Specifically, we designed a Nafion-coated EAB biosensor for the detection of cationic DOX, a chemotherapeutic drug. Initially, we investigated the Nafion coating on both planar gold (pAu) electrodes and nanoporous gold (npAu) electrodes and evaluated the Nafion's impact on DOX sensing performance. Nafion-coated planar gold (n-pAu) electrodes were incapable of detecting the target due to membrane blockage of aptamer active sites. On the contrary, Nafion-coated nanoporous gold (n-npAu) electrodes exhibit void volumes that excluded Nafion from the pore interior, enabling the biosensor to detect DOX, as shown in Scheme 1. Furthermore, we fabricated a series of npAu EAB biosensors by varying pore size and electrode thickness and compared their target responses, allowing us to determine the degree of Nafion penetration into the pores under various conditions. The npAu height and pore size were optimized to ensure a sufficient active sensing area uncovered by Nafion while maintaining membrane protection on the top side. We then carefully examined the impact of membrane thickness and the method of membrane preparation on DOX transport across the membrane to evaluate the Nafion exclusion outside the tiny nanoporous structures. We further studied the morphological effects on the npAu electrodes, resulting in optimized coating conditions where the Nafion membrane has minimal impact on preventing the sensor from functioning. Finally, the confinement provided by the Nafion encapsulated nanopore electrode has shown additional benefits to study mass-limited analytes in a biologically relevant concentration. We evaluated the ionic strength, DOX diffusion time and aptamer surface density on biosensing performance. In conclusion, these experiments collectively support that Nafion-coated nanoporous electrodes can prevent membrane from interfering with inner nanopores that could serve as a robust biosensing platform to detect small molecules with good sensitivity, specificity and stability, where it will lead to potential applications in in vivo biosensing over a longer period.
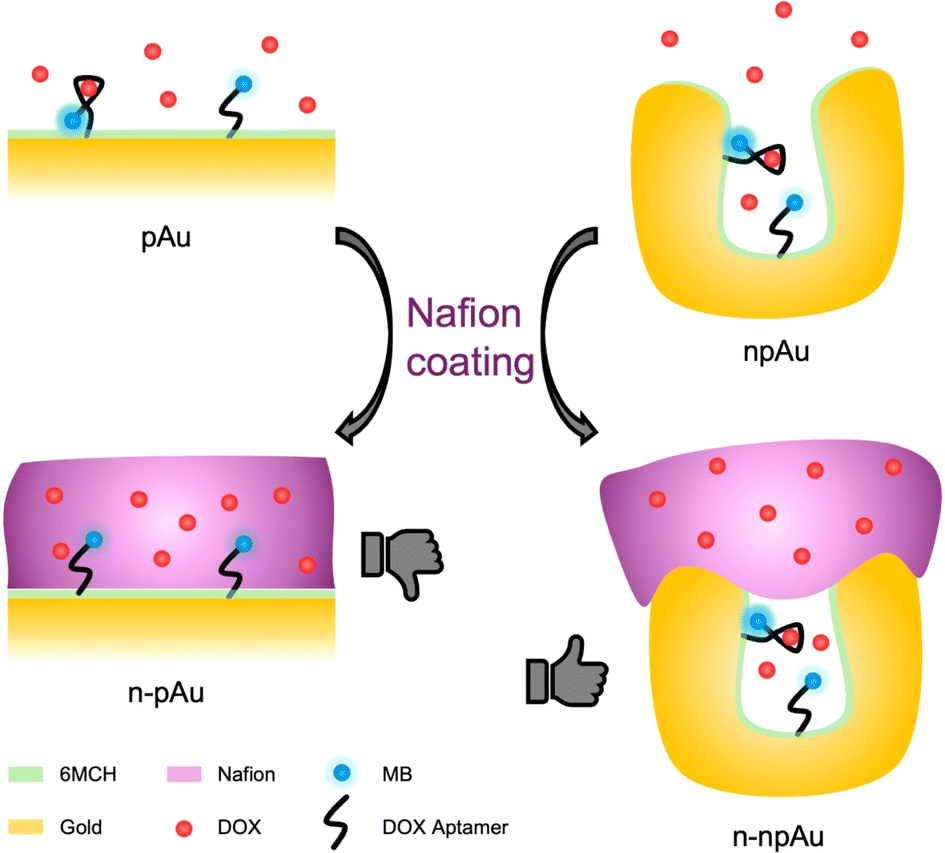 | ||
| Scheme 1 Schematic illustrations of Nafion-coated planar gold (n-pAu) electrode and Nafion-coated nanoporous gold (n-npAu) electrode for electrochemical aptamer-based biosensing. Nafion blocks the aptamer from changing conformation in the case of n-pAu, while the aptamer retains its redox signal change upon target binding in the case of n-npAu. | ||
Results and discussion
Nafion-coated planar electrode
We first investigated the EAB biosensors fabricated on a Nafion-coated planar gold (n-pAu) electrode, as shown in Scheme 1. Doxorubicin (DOX) is a small molecule chemotherapeutic drug with a positive charge under physiological conditions (pKa of 8.1–8.3). DOX aptamer (DOX apt) has been extensively studied in the literature, showing good sensitivity and high specificity for DOX detection.2,4,23 Methylene blue (MB) was chosen as the redox reporter due to its high reversibility in continuous redox readout and its suitability for attachment to the aptamer via EDC–NHS coupling reaction. DOX apt is a 28-base single-stranded oligonucleotide that has a hydrodynamic radius over a few nanometers. After MB conjugation to the distal end of DOX apt, the DOX apt–MB was immobilized on the pAu electrode. Subsequently, Nafion was coated onto the aforementioned electrode surface by varying spin speeds, resulting in different membrane thicknesses, as shown in Fig. 1A. The Nafion membrane used in this study was formed in situ by spinning a 5 wt% Nafion in a mixture of lower aliphatic alcohols onto the electrode surface. Upon solvent evaporation, Nafion assembles into hydrophilic and narrow (<2 nm) porous structures that allow analyte diffusion through the membrane.35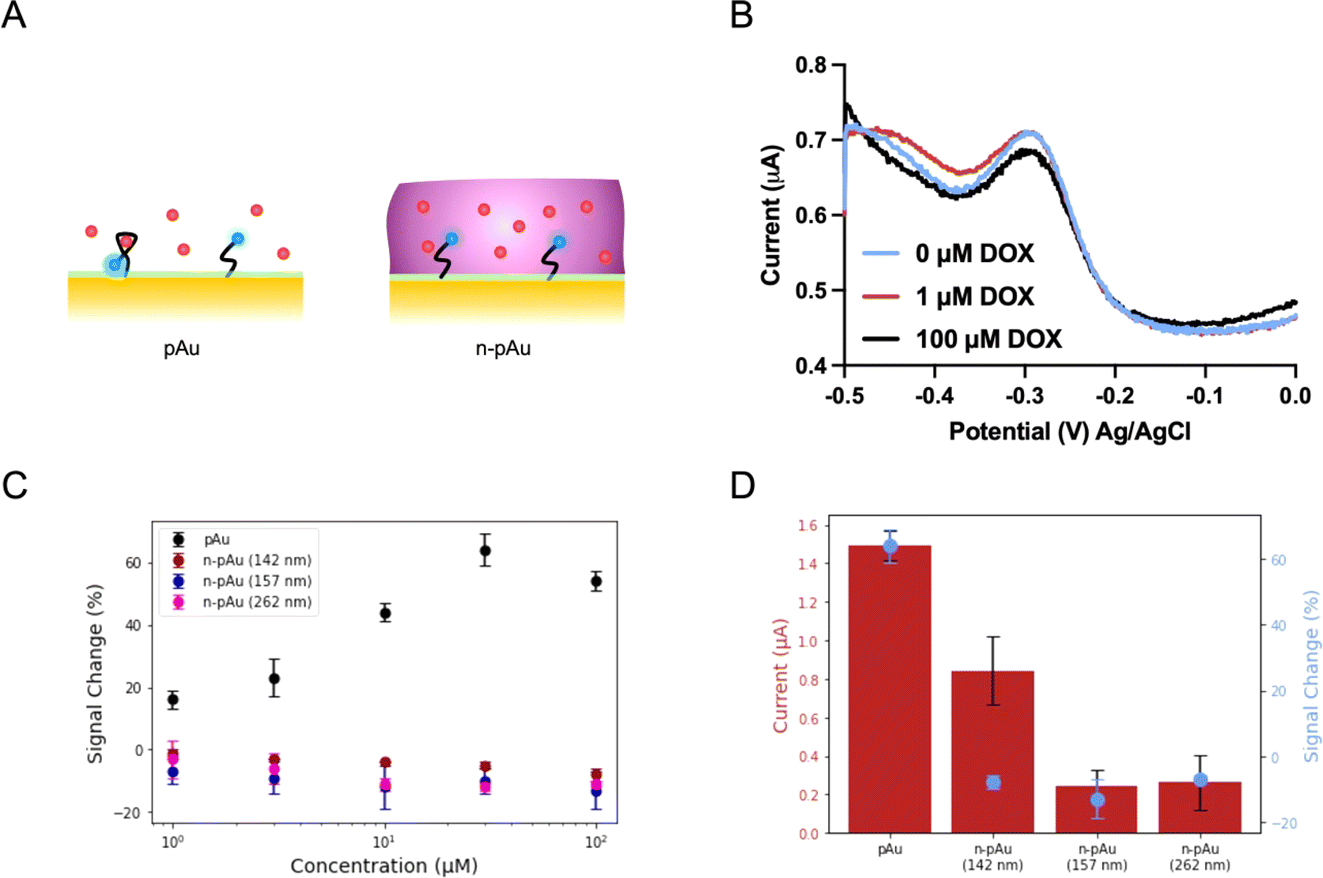 | ||
| Fig. 1 Electrochemical biosensing performance of Nafion-coated planar gold electrode. (A) illustrates both the pAu and the n-pAu interacting with the DOX targets. (B) shows SWV plots of n-pAu with different DOX concentrations. (C) displays the signal changes of pAu, n-pAu (6000 rpm, 142 nm), n-pAu (4000 rpm, 157 nm) and n-pAu (1500 rpm, 262 nm) after DOX addition (n = 3). (D) lists the current levels on the left y-axis and the maximum signal changes on the right y-axis of pAu, n-pAu (6000 rpm, 142 nm), n-pAu (4000 rpm, 157 nm) and n-pAu (1500 rpm, 262 nm) upon the addition of DOX (n = 3). | ||
Following previously reported protocols, square wave voltammetry (SWV) was used as the electrochemical detection method to measure MB redox current signals before and after DOX target binding to DOX apt–MB.23,24 Maximum peak values are derived from SWV curves, where the MB redox reporter has a redox potential near −0.25 volt (V) to −0.30 V versus the Ag/AgCl reference electrode. The signal change (in percent) was then calculated by comparing the peak current before and after the addition of the DOX target to the test solution. The aptamer is referred to as a signal ON aptamer if the folding of the aptamer upon target binding brings the redox reporter closer to the electrode surface, thereby increasing the redox current signal.38 Since SWV measurements are voltage pulse dependent, we optimized the pulse frequency (Fig. S1†) from low to high frequency range, such that the well-balanced frequency was selected to achieve the maximum signal change with minimum current variation and high signal-to-noise ratio. For example, a signal OFF response was observed at 60 Hz because the target binding pushed the aptamer closer to the electrode, resulting in electron transfer occurring before the current measurement for the target-bound aptamer, than the unbound aptamer at low frequency. While 300 Hz and 500 Hz both produced signal ON and 300 Hz was selected as the frequency for subsequent tests due to both strong signal ON behavior and lower standard deviation than 500 Hz.
The SWV plots shown in Fig. 1B represent three different concentrations (0 μM, 1 μM and 100 μM) of DOX solutions placed onto the n-pAu electrode. No discernible signal difference was observed between the three plots, indicating that DOX apt–MB did not respond to the target even at high DOX concentration (100 μM). We collected the electrochemical responses from the pAu electrode at the same target concentrations as a comparison, which shows a reasonable signal change (∼18%) at the lowest concentration (1 μM) of DOX and a substantial signal increase (∼58%) at 100 μM of DOX (Fig. 1C). We hypothesized that a free-standing aptamer on the electrode surface would be constrained by the surrounding Nafion membrane, preventing the aptamer from folding and interacting with DOX. A Nafion coated electrode reported in the literature for direct redox reaction was less affected than our aptamer based biosensors, which needs to exclude Nafion away from the electrode surface for aptamer structural switching upon target binding. To verify the interaction between Nafion and DOX apt–MB, n-pAu with three different Nafion membrane thicknesses (262 nm, 157 nm and 142 nm) were prepared by varying the spin speed (Fig. S2†). As shown in Fig. 1C, the DOX apt–MB on all n-pAu electrodes showed almost no response to a variety of DOX concentrations. These results imply that the Nafion, regardless of its thickness, restricts the mobility of the aptamer causing no change in current upon DOX addition. More importantly, Fig. 1D shows that the target-free currents from different n-pAu electrodes were significantly lower than that the current from the pAu electrode. This evidence further suggests that the aptamer was more likely to adopt a stretched conformation forcing the conjugated redox reporter further away from the electrode surface.39
Nafion-coated nanoporous electrode
By designing nanoporous gold electrodes with pores in the range of 10–30 nanometers, we aimed to obtain a randomly distributed nanoporous structure that excludes Nafion from entering the nanopores, allowing the target to pass through the membrane and interact with the aptamers immobilized on the inner pores, as shown in Fig. 2A. The nanoporous gold electrodes were fabricated according to our previous protocol in which gold and silver were co-sputtered in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, resulting in a three-dimensional network interconnected by randomly distributed nanopores.23 Both the npAu electrodes and the pAu electrodes used in this study have the same device footprint, while the intrinsic nanoporous structures of npAu electrodes enlarged the surface area, forming an area 41 times larger than that of the pAu electrodes. As a result, the current from the DOX apt–MB immobilized npAu electrode was two orders of magnitude greater than the current from the same footprint aptamer-immobilized pAu electrode. As shown in Fig. 1B, the n-pAu electrodes show similar peak currents at all DOX concentrations. On the contrary, n-npAu electrodes show different current levels at different DOX concentrations (Fig. 2B), suggesting that DOX diffused through the Nafion membrane and interacted with the aptamer. It also implies that the aptamer is capable of free movement because of the exclusion of the Nafion due to the nanoporous structure and the aptamers within the nanopore undergo a conformational change upon target binding.40
:2 ratio, resulting in a three-dimensional network interconnected by randomly distributed nanopores.23 Both the npAu electrodes and the pAu electrodes used in this study have the same device footprint, while the intrinsic nanoporous structures of npAu electrodes enlarged the surface area, forming an area 41 times larger than that of the pAu electrodes. As a result, the current from the DOX apt–MB immobilized npAu electrode was two orders of magnitude greater than the current from the same footprint aptamer-immobilized pAu electrode. As shown in Fig. 1B, the n-pAu electrodes show similar peak currents at all DOX concentrations. On the contrary, n-npAu electrodes show different current levels at different DOX concentrations (Fig. 2B), suggesting that DOX diffused through the Nafion membrane and interacted with the aptamer. It also implies that the aptamer is capable of free movement because of the exclusion of the Nafion due to the nanoporous structure and the aptamers within the nanopore undergo a conformational change upon target binding.40
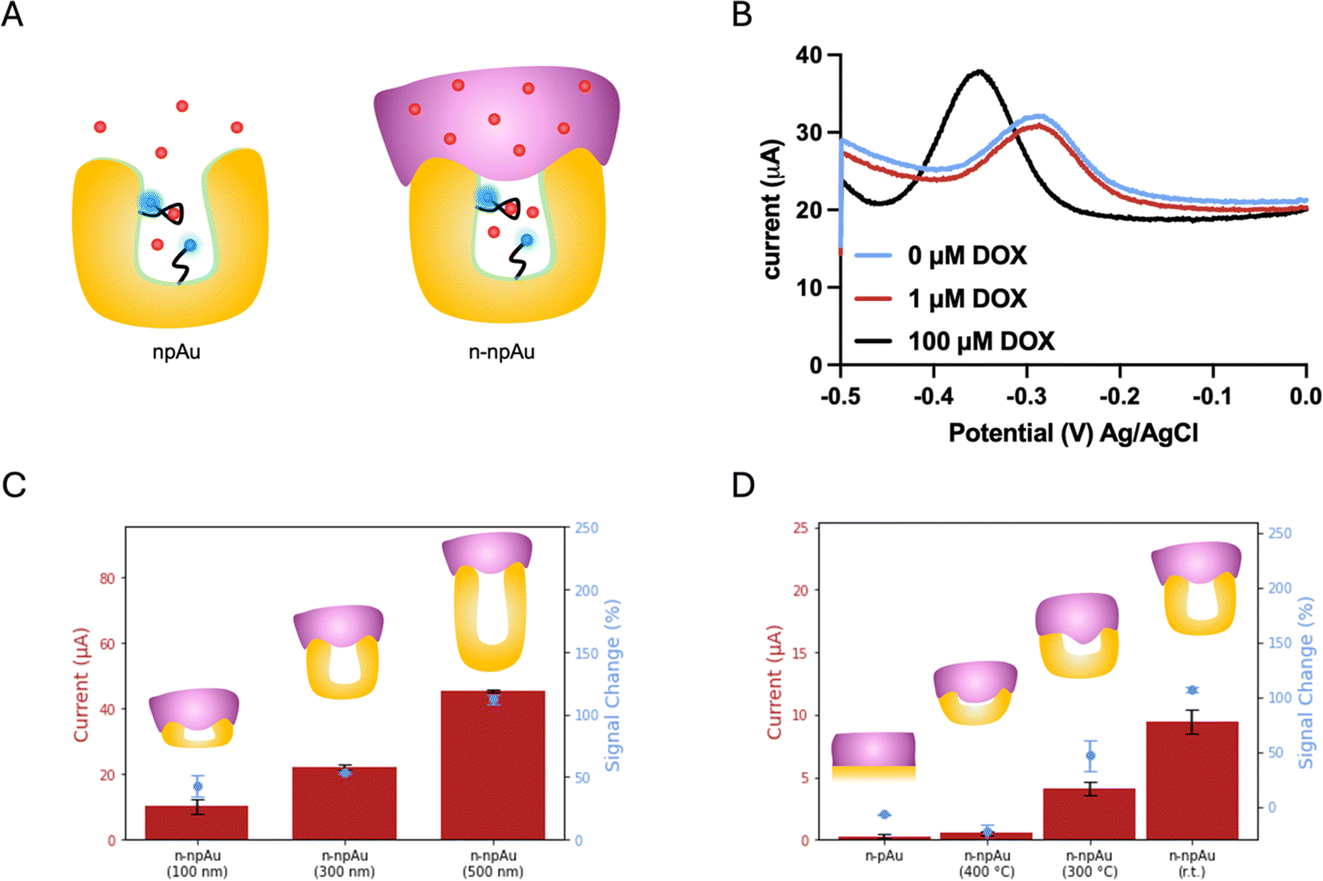 | ||
| Fig. 2 Electrochemical biosensing performance of Nafion-coated nanoporous gold electrode. (A) illustrates n-npAu before and after target binding. (B) shows SWV plots of n-npAu with different DOX concentrations. (C) displays the current levels on the left y-axis and the maximum signal changes on the right y-axis of n-npAu (100 nm), n-npAu (300 nm) and n-npAu (500 nm), upon the addition of DOX (n = 3). (D) lists the current levels on the left y-axis and the maximum signal changes on the right y-axis of n-pAu, n-npAu (400 °C), n-npAu (300 °C) and n-npAu (r.t.) upon the addition of DOX (n = 3). | ||
Having validated that a n-npAu could serve as a viable EAB biosensor, we proceeded to optimize the device by varying the npAu dimensions, including electrode thickness and nanopore size to create an EAB sensor that can maximize the Nafion exclusion outside the nanopores. Fig. 2C shows the current levels and signal changes of n-npAu with three different electrode thicknesses (100 nm, 300 nm and 500 nm). As the electrode thickness changes from 100 nm to 500 nm, the blank current level increases 4.5-fold, thus proving that DOX apt–MB can be immobilized deeply into the nanoporous network rather than only at the surface of the electrodes. Also, npAu electrodes that are less than 100 nm tall result in small current due to the decreased surface area, as such we focus on the effect of npAu electrode thickness on the signal change between 100 nm and 500 nm. Interestingly, the signal change obtained from three different electrode thicknesses indicates that the interaction of DOX target and DOX apt–MB is less affected by the upper Nafion membrane because n-npAu has a thicker nanoporous layer. The signal change data (Fig. 2C) indicates that the 100 nm n-npAu electrode thickness exhibits a signal change that is one third of the signal change obtained using the 500 nm n-npAu electrode, potentially due to the blockage of the aptamer near the nanopore opening by the Nafion membrane. The DOX apt–MB located adjacent to the Nafion membrane would have a similar electrochemical signaling behavior to that of the n-pAu electrode. n-npAu with electrode thicknesses of 300 and 500 nm produce a distinct signal change, with the latter responding to the DOX when the thickest Nafion membrane in this study (262 nm by 1500 rpm spin speed) is coated on the top of the npAu electrode. While the influence of the Nafion at the top of the pores is still evident in the lower blank current. Sufficient DOX targets pass through the membrane to produce a signal change that is comparable to that of the npAu without any Nafion coating.
Another critical factor affecting the signaling behavior is the pore size of the n-npAu electrode. To determine the threshold size of nanoporous structures to exclude Nafion, we proceeded to a pore size dependency test using thermal annealing at elevated temperatures to vary the pore diameter. For this study, we chose thermal annealing because of its ease of use for a device with multiple individually addressable electrodes. Thermal annealing enlarges the nanopores by slightly melting partial gold materials causing them to bunch together and form larger nanopores during the cooling process. The annealing time and temperature window determine the degree to which the nanoporous network is enlarged. We fixed the annealing time at ten minutes while varying the annealing temperature from room temperature to 400 °C. We hypothesized the Nafion would fill a larger porous volume as the pore radius increased, until the npAu electrode reached a point where the nanopores were large enough to be completely filled and behaved similarly to the pAu electrode. As shown in Fig. 2D, we compared n-pAu, n-npAu (r.t.) and n-npAu (400 °C) and n-npAu (300 °C). This temperature gradient produced nanopore diameters of 15 nm for npAu (r.t.), 56 nm for npAu (300 °C) and 251 nm for npAu (400 °C), as shown in Fig. S3.† The thermally annealed devices were then immobilized with DOX apt–MB and coated with Nafion in the same manner as the non-thermally annealed n-pAu and n-npAu devices. At 400 °C, the signal and current of n-npAu (400 °C) were suppressed to the point that the electrode behaved almost identically to the n-pAu electrode, indicating that the Nafion filled most of the nanoporous structures. At 300 °C, it was evident that both the current level and the signal change of n-npAu (300 °C) had been suppressed by about two thirds of that of n-npAu (r.t.), indicating that the Nafion could penetrate a greater extent of the nanoporous structure when the pores were above 50 nm compared to the untreated nanoporous structures. These experiments strongly support that a larger pore size does not exclude Nafion outside the nanopores, ultimately resulting in a deterioration of biosensing performance.
Fig. 3A shows the SEM top view and cross-sectional view of the npAu electrode. Prior to measurement, the entire electrode surface was sputter coated with iridium to enhance surface conductivity for imaging. Cross-sectional images were obtained by focused ion beam (FIB) milling with a protective platinum layer deposited on the top of the npAu electrode for surface protection. The nanopores have an average diameter of 15 nm and formed an interconnected 3D nanoscale network in the vertical direction, allowing small molecules to access while rejecting large molecules. Fig. 3B shows the top view and cross-sectional view of the n-npAu electrode. These images confirm that the Nafion only penetrates the large cracks, but is unable to enter the tiny nanopores, which compose most of the electrode area. This further confirms the current levels observed in Fig. 2D, where the npAu electrode is larger than all of the n-npAu electrodes. As the pores became larger, the Nafion is able to fill the pores more easily, resulting in a reduced number of aptamers that are able to undergo conformational change upon target binding, thereby suppressing the current level and associated signal change.41,42
 | ||
| Fig. 3 Nafion coating optimization and morphological characterization. (A) and (B) are the top view and cross-sectional view of SEM images for npAu and n-npAu, respectively. The Nafion membrane covers the top surface of npAu and the tiny nanoporous structures are essential to exclude the Nafion outside the nanopores. (C) shows the current levels on the left y-axis and the maximum signal changes on the right y-axis of npAu, n-npAu (6000 rpm, 142 nm), n-npAu (4000 rpm, 157 nm) and n-npAu (1500 rpm, 262 nm) upon the addition of DOX (n = 3). (D) shows the current levels on the left y-axis and the signal changes on the right y-axis of n-npAu (dip coating) and n-npAu (spin coating) upon the addition of DOX (n = 3). | ||
Effect of Nafion membrane thickness and preparation methods
As the pore size and electrode thickness of n-npAu were well tuned for Nafion exclusion, we then investigated the Nafion membrane, including its thickness and formation methods, to gain a better understanding of the membrane–electrode interaction on biosensing performance. Unlike commercially available Nafion films with micron thicknesses, we formed the Nafion membrane in situ on both pAu and npAu electrodes by spin coating and dip coating, respectively. Specifically, 5 wt% Nafion solution was spun on the electrode surface at three different speeds (1500 rpm, 4000 rpm and 6000 rpm), resulting in membrane thicknesses of 262 nm, 157 nm and 142 nm, respectively (Fig. S2†). A direct comparison was then conducted to optimize the membrane thickness and investigate the effect of Nafion thickness on current and signal change. As shown in Fig. 3C, the n-npAu (1500 rpm, 262 nm) device has the largest signal change while maintaining the lowest current level. And n-npAu (4000 rpm, 157 nm) and n-npAu (6000 rpm, 142 nm) devices behave slightly different from each other in both signal change and current level. Unlike the n-pAu, where Nafion blocks most of the signal change caused by DOX interacting with the aptamer, the n-npAu (1500 rpm) shows comparable signal change to the npAu.Even though the npAu and the pAu electrodes were fabricated on a flat glass substrate, we wondered about the signaling behavior of the Nafion-coated electrode on an uneven surface. Compared with spin coating, dip coating is a simple and low-cost approach for coating the membrane on a variety of substrate types regardless of the smoothness of the electrode. Another advantage of dip coating on the npAu electrode is determining whether Nafion would fill a greater volume of the nanopore interior.43 To verify this, we dropped Nafion solution onto the npAu electrode surface and allowed the solution to sit for ten minutes before spinning off the excess Nafion solution. As shown in the SEM image in Fig. S4,† Nafion did not penetrate into the nanoporous network, even though the membrane is much thicker than the spin-coated membranes. The Nafion incubation causes an increase in the current level compared to the spin-coated n-npAu (Fig. 3D). However, the signal change diminishes, probably due to the increased length of the Nafion, which prevents the DOX from passing through the membrane within the measurement time frame.
Nanopore confinement effect on biosensing performance
After the optimization of the npAu electrode and Nafion, we investigated the biosensing performance arising from volume confinement inside nanopores.44–48 Previously, we demonstrated that ionic strength affects the signal change in a npAu electrode.23 Unlike the flat and thin Debye layer for pAu electrodes at physiological ionic strength, npAu electrodes have a concave structure, the Debye screening is curved in three dimensions as Debye volume, where the screening of the redox species inside is greatly reduced.49 Considering the 15 nm average diameter of the nanopore, the aptamer immobilized on the inner wall of the nanopore will benefit from the reduced screening and accelerate the electron transfer rate, leading to an increased current when compared to the aptamer immobilized on the pAu electrode. Therefore, we investigated the dependence of ionic strength, DOX diffusion time and aptamer surface density on the biosensing performance.First, we evaluated n-npAu at three different ionic strength conditions (0.1× PBS, 1× PBS and 10× PBS). The data in Fig. 4A shows that the 0.1× PBS has the lowest current level and negligible signal change. The low current is consistent with previous findings, but the low signal change of n-npAu is unexpected.23 In our previous study, the npAu (0.1× PBS) produces the highest signal change using the npAu electrode. One possible explanation is that the reduced ionic strength impairs the ability of the DOX to diffuse across the Nafion membrane. At 10× PBS, the higher ion concentration within the nanopores limits the extent of the electric field, resulting in a relatively modest signal change at this high ionic strength. At 1× PBS, n-npAu provides a reasonably high enough ionic strength for DOX to diffuse across the Nafion membrane without compromising the Debye screening effect that ultimately resulted in the highest signal change reported.
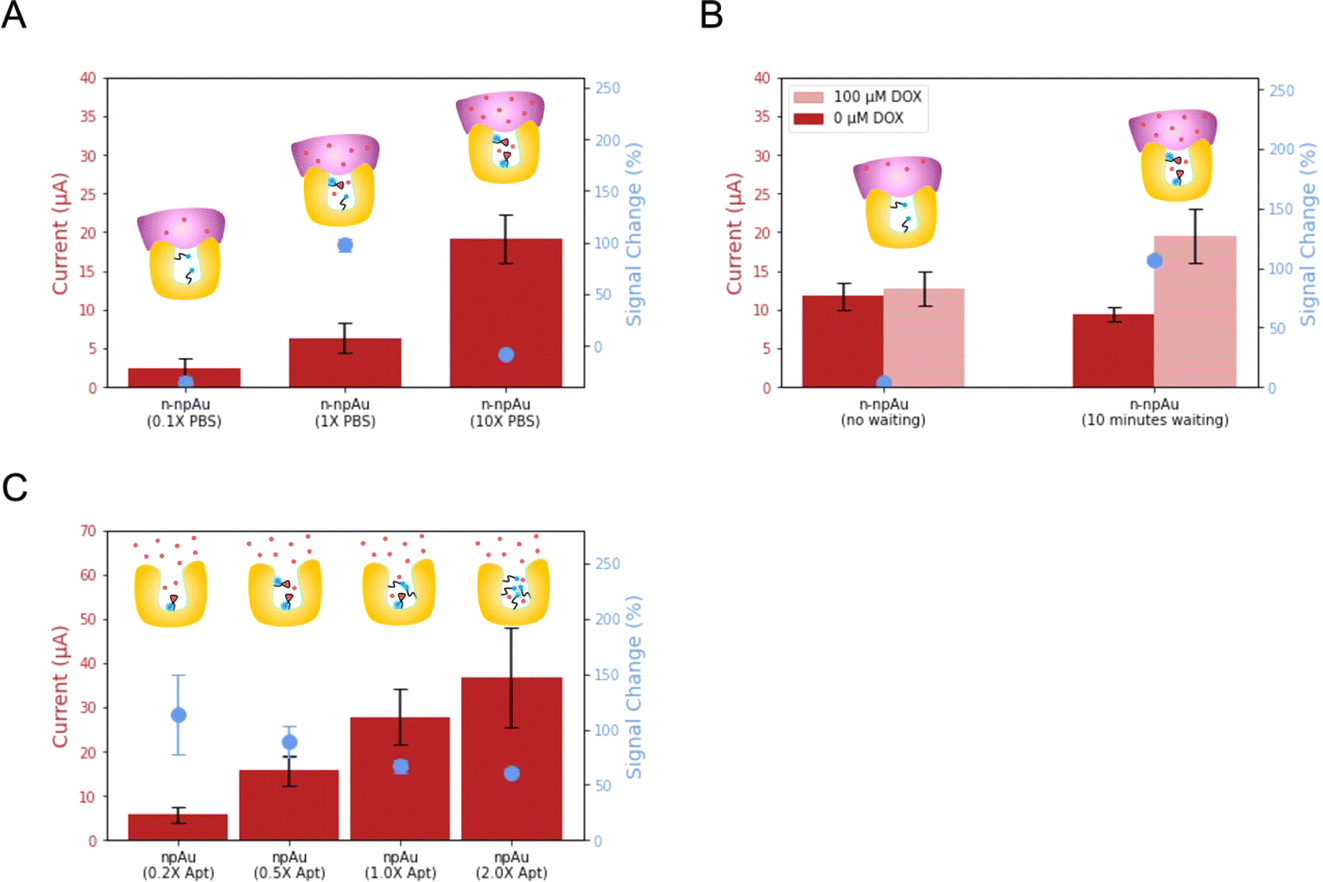 | ||
| Fig. 4 Effect of ionic strength, DOX diffusion time and aptamer surface density on electrochemical biosensing. (A) shows the current levels on the left y-axis and the maximum signal changes on the right y-axis of n-npAu (0.1× PBS), n-npAu (1× PBS) and n-npAu (10× PBS) upon the addition of DOX (n = 3). (B) shows the current levels on the left y-axis with the dark red as the currents of 0 μM DOX and the light red as the currents of 100 μM DOX. The right y-axis shows the signal changes with the addition of 100 μM DOX without (n-npAu (no waiting)) and with ten minute waiting period (n-npAu (10 minutes waiting)) (n = 3). (C) shows the current levels on the left y-axis and the signal changes on the right y-axis of npAu (0.2× DOX apt–MB), npAu (0.5× DOX apt–MB), npAu (1× DOX apt–MB) and npAu (2× DOX apt–MB) upon the addition of DOX (n = 3). | ||
The unusual signal behavior of 0.1× PBS indirectly proves that the presence of a membrane on the electrode surface results in a different mass transport behavior between npAu electrodes and n-npAu electrodes. To investigate this effect, the diffusion time is tuned to reflect the changes in current levels and signal changes for DOX detection, as illustrated in Fig. 4B and S5.† For the npAu electrodes, the signal change is nearly instantaneous over time, indicating that a steady state is reached almost immediately. While in the n-npAu, the signal change shows a tiny signal change after DOX addition. After a waiting period of ten minutes, the signal change increases to a level approaching the signal change of npAu, indicating the complete diffusion of DOX across the membrane.
The final aspect of the local environment that significantly affects the biosensing is the aptamer surface density within the nanopore.50 In such a confined nanopore volume, aptamers tend to interact with each other more than on the pAu.51 In this measurement, we tested four different aptamer surface densities by immobilizing aptamer solution with a concentration of 0.2 μM, 0.5 μM, 1 μM and 2 μM, respectively. As shown in Fig. 4C, the current level increases with increasing the aptamer surface density. This is expected as the addition of DOX apt–MB will result in more redox reporters leading to higher redox currents. While the signal change decreases, the 0.20 μM aptamer shows the largest signal change, indicating the least steric hindrance of the aptamer conformational change upon target binding. However, its redox current peak is more difficult to distinguish from the baseline than other aptamer surface densities. Balancing the current level and signal change, we adopted the 0.50 μM aptamer as the optimized aptamer density as it exhibits a noticeable signal change with much less variation while maintaining a large peak current from the baseline for target detection.
Conclusion
In this work, we demonstrated a facile and low-cost approach to fabricate Nafion-coated nanopore electrodes and their ability as EABs to detect small biomolecules with good sensitivity and high specificity. We designed a hierarchical nanostructure that seamlessly coated a cation exchange membrane with nanochannels (<2 nm) on the top of a nanoporous (>15 nm) gold electrode without compromising the biosensing performance for cationic targets. Initially, we characterized the Nafion-coated planar electrode (n-pAu), which showed no response to the DOX target because the Nafion completely passivated the DOX apt–MB immobilized on the planar electrode, preventing the aptamer from undergoing a conformational change upon target binding. However, in the case of our n-npAu electrode, the Nafion was unable to penetrate the 3D random and tiny nanoporous network, resulting in a target response that is comparable to that of the uncovered npAu electrode. To validate our hypothesis, we investigated the Nafion filling behavior in the nanoporous regime by tuning the pore size by thermal annealing and the pore depth by varying the metal co-sputtering protocols. We confirmed that the tiny nanopores of npAu electrodes provided a unique biosensing environment to allow the aptamer to respond freely to the target without the constraints of the Nafion coating on top. On the other hand, sealing the entrance of the npAu electrode altered the target transport and provided a nanoscale confinement for molecular detection. We investigated the local ionic strength, target diffusion time and aptamer surface density and optimized our electrochemical biosensor. The concept of membrane-coated nanoporous electrode for EAB biosensors introduced in this work can serve as a framework for future studies on membranes with different stimulus-response and a variety of biomolecular targets. More importantly, the integration of chemical functionality and anti-biofouling properties illustrated here will pave the way for future in vivo biosensor research for long-term study.Experimental section
Chemicals and materials
Concentrated nitric acid (67–70%), concentrated sulfuric acid (95–98%), concentrated hydrochloric acid (34–37%), hydrogen peroxide (30%), 10× phosphate buffered saline (PBS), sodium chloride, sodium bicarbonate, reagent grade ethanol and nuclease free water were purchased from Avantor and used as received. Polydimethylsiloxane was purchased in a kit from Avantor and prepared according to package instructions. Ultrapure water was purified and collected using the Alto Ultrapure Lab Water Polisher (18.2 MΩ cm). Grafix Frisket film was purchased from Amazon for use as electrode masking tape for laser cutting. Doxorubicin hydrochloride (DOX), tris(hydroxymethyl)aminomethane hydrochloride (TCEP) were purchased from Sigma-Aldrich and used as received. The doxorubicin aptamer, described in our previous study, with a sequence of 5′HS(CH2)6ACCATCTGTGTAAGGGGTAAGGGGTGGT/3AmMC6T, was purchased from Integrated DNA Technologies and diluted to 100 μM using nuclease free water. The DOX aptamer was then modified using an EDC–NHS coupling reaction to attach Atto MB2 NHS ester (a methylene blue (MB) derivative, >90% HPLC, purchased from Sigma-Aldrich) to the distal end as DOX apt–MB. D521 Nafion dispersion (alcohol-based 1100 EW at 5 wt%) was purchased from Ion Power.Electrode fabrication
Planar gold (pAu) and nanoporous gold (npAu) electrodes were fabricated using laser patterned masks and metal sputtering by modification of a previously reported procedure.23 Glass slides were cleaned in Piranha solution (a 3:1 mixture of sulfuric acid (95–98%) and hydrogen peroxide (30%) – caution strong oxidizer, handle and use with extreme care) for 5 minutes and then rinsed with ultrapure water. Electrode mask patterns were prepared by laser cutting the Frisket Film and then were placed on the pre-cleaned glass slides to define the electrode area. The masked glass slides were sputtered with metals using the Oerlikon 450B sputtering system to fabricate the pAu and npAu electrodes. For the pAu electrode, a 10 nm titanium adhesion layer and then a 100 nm gold layer were sputtered onto the unmasked electrode area of the slides. The prepared pAu electrodes were cleaned with Piranha solution for five minutes and immersed in ultrapure water before use. For the npAu electrode, a 10 nm titanium adhesion layer, a 50 nm gold layer and a 1:2 gold/silver bilayer with three different thicknesses (100 nm, 300 nm, 500 nm) were co-sputtered onto the slides. The silver layer was dissolved in concentrated nitric acid (70%) for ninety minutes and then the prepared npAu electrodes were immersed in ultrapure water overnight before use.
MB conjugation and aptamer immobilization
The DOX aptamer was first diluted to 100 μM with nuclease-free water and stored in a −20 °C freezer. The DOX aptamer was then modified by attaching a methylene blue (MB) redox reporter to the 3′ end of the DNA through an EDC–NHS coupling reaction. Specifically, 15 μL of the MB NHS ester, 50 μL of the 100 μM aptamer and 6 μL of 75 g L−1 sodium bicarbonate were mixed and allowed to react overnight at 4 °C. Then, 10 μL of 3 M NaCl and 200 μL of reagent grade ethanol were added to the above solution and stored at −20 °C for 30 minutes. The solution was centrifuged at 4 °C and 20000g for 30 minutes by an Eppendorf Centrifuge 5425 and the supernatant was removed to obtain a crude DOX apt–MB pellet at the bottom. This solid was resuspended with reagent grade ethanol and spun down until the supernatant was colorless (MB free). Finally, the DOX apt–MB aptamer was dried in a desiccator under vacuum and dissolved in nuclease-free water before use. The yield of MB conjugation was monitored by measuring the UV-vis absorption ratio of MB and DOX aptamer using a Thermo Fisher NanoDrop.
Aptamer immobilization on the electrode was achieved by incubating 100 μL DOX apt–MB solution with the pAu electrode and npAu electrode, respectively. Prior to incubation, the as-prepared DOX apt–MB stock solution (100 μM) was reduced with excess TCEP to convert the disulfide bond at the 5′ end of the DNA into a thiol group. The DOX apt–MB solution was then diluted to the desired final concentration (0.2, 0.5, 1, 2 μM) with PBS buffer (0.1×, 1×, 10×) for specific measurements. After incubation of the DOX apt–MB solution for one hour, the electrodes were washed and then immersed in a 6-mercapto-1-hexanol (6-MCH) thiol solution (73.1 mM) for three hours to form a self-assembled monolayer on gold and passivate the remaining electrode area. Finally, the electrodes were washed with ultrapure water to remove any excess 6-MCH and stored in 1× PBS buffer before use.
Nafion coating and morphological characterization
The n-pAu electrode and n-npAu electrode were prepared by spinning 5 wt% Nafion solution at different speeds (from 1000 rpm to 8000 rpm) with a Suss MicroTec LabSpin 8 and then dried overnight at room temperature with a cover before use. Nafion thicknesses were measured using a Flimmetrics 40 system. The morphological images of the n-npAu electrode were taken using a FIB SEM (Helios G4 Ux Dual Beam).Electrochemical measurements
All electrochemical measurements were carried out using a four-channel potentiostat MultiEmStat4 (PalmSens). Pt counter electrode and Ag/AgCl reference electrode were purchased from CH instruments. Square wave voltammetry (SWV) measurements were performed at different amplitudes and different frequencies to obtain the optimized electrochemical parameters. The voltage sweep ranged was from −0.5 V to 0 V (vs. Ag/AgCl at 298 K) and SWV data were recorded for analysis. During the measurement, a PDMS well (8 mm in diameter) was placed on top of the electrode array as a solution holder to hold PBS buffer solution and subsequent addition of DOX solution to a desired concentration. All SWV data were collected and analyzed using MultiTrace (Palmsens) and plotted by Python Matplotlib or Graphpad.Data availability
The data supporting this article have been included as part of the ESI.† Additional data are available upon request from the authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by internal funds from the University of the Notre Dame. Fabrication and structural characterization of the devices studied here were performed at the Notre Dame Nanofabrication Facility and the Notre Dame Integrated Imaging Facility, whose generous support is gratefully acknowledged. We thank Victoria Iannelli for graphic design suggestions, Jarek Metro for Nafion discussions and Andrew Trowbridge for writing comments. GFH also acknowledges Jack Huldin for supporting this and many other projects.References
- R. Tavallaie, et al., Nucleic acid hybridization on an electrically reconfigurable network of gold-coated magnetic nanoparticles enables microRNA detection in blood, Nat. Nanotechnol., 2018, 13(11), 1066–1071 CrossRef PubMed.
- B. S. Ferguson, et al., Real-Time, Aptamer-Based Tracking of Circulating Therapeutic Agents in Living Animals, Sci. Transl. Med., 2013, 5(213), 213ra165 Search PubMed.
- P. L. Mage, et al., Closed-loop control of circulating drug levels in live animals, Nat. Biomed. Eng., 2017, 1(5), 0070 CrossRef.
- J.-W. Seo, et al., Real-time monitoring of drug pharmacokinetics within tumor tissue in live animals, Sci. Adv., 2022, 8(1), eabk2901 CrossRef PubMed.
- N. Arroyo-Currás, et al., Real-time measurement of small molecules directly in awake, ambulatory animals, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(4), 645–650 CrossRef PubMed.
- O. Alkhamis, et al., Nuclease-assisted selection of slow-off rate aptamers, Sci. Adv., 2024, 10(24), eadl3426 CrossRef PubMed.
- O. Alkhamis, et al., High-Affinity Aptamers for In Vitro and In Vivo Cocaine Sensing, J. Am. Chem. Soc., 2024, 146(5), 3230–3240 CrossRef PubMed.
- H. Yu, et al., Advances and Challenges in Small-Molecule DNA Aptamer Isolation, Characterization, and Sensor Development, Angew. Chem., Int. Ed., 2021, 60(31), 16800–16823 CrossRef CAS PubMed.
- N. Arroyo-Currás, et al., High Surface Area Electrodes Generated via Electrochemical Roughening Improve the Signaling of Electrochemical Aptamer-Based Biosensors, Anal. Chem., 2017, 89(22), 12185–12191 CrossRef PubMed.
- J. Liu, et al., Achieving Reproducible Performance of Electrochemical, Folding Aptamer-Based Sensors on Microelectrodes: Challenges and Prospects, Anal. Chem., 2014, 86(22), 11417–11424 CrossRef CAS PubMed.
- A. M. Downs, et al., Nanoporous Gold for the Miniaturization of In Vivo Electrochemical Aptamer-Based Sensors, ACS Sens., 2021, 6(6), 2299–2306 CrossRef CAS.
- S. E. Salamifar and R. Y. Lai, Fabrication of Electrochemical DNA Sensors on Gold-Modified Recessed Platinum Nanoelectrodes, Anal. Chem., 2014, 86(6), 2849–2852 CrossRef CAS.
- S. Li, et al., A wrinkled structure of gold film greatly improves the signaling of electrochemical aptamer-based biosensors, RSC Adv., 2021, 11(2), 671–677 RSC.
- L. A. Baker, Perspective and Prospectus on Single-Entity Electrochemistry, J. Am. Chem. Soc., 2018, 140(46), 15549–15559 CrossRef CAS PubMed.
- J. E. Dick, C. Renault and A. J. Bard, Observation of Single-Protein and DNA Macromolecule Collisions on Ultramicroelectrodes, J. Am. Chem. Soc., 2015, 137(26), 8376–8379 CrossRef CAS PubMed.
- K. McKelvey, et al., Nanopipettes as a tool for single nanoparticle electrochemistry, Curr. Opin. Electrochem., 2017, 6(1), 4–9 CrossRef CAS.
- K. Fu, et al., Single Entity Electrochemistry in Nanopore Electrode Arrays: Ion Transport Meets Electron Transfer in Confined Geometries, Acc. Chem. Res., 2020, 53(4), 719–728 CrossRef CAS PubMed.
- K. Liu, et al., Spatial Analysis of Reactive Oxygen Species in a 3D Cell Model Using a Sensitive Nanocavity Electrode, Anal. Chem., 2022, 94(38), 13287–13292 CrossRef CAS.
- S. Ganguli, et al., Nano-Impact Single-Entity Electrochemistry Enables Plasmon-Enhanced Electrocatalysis, Angew. Chem., Int. Ed., 2023, 62(25), e202302394 CrossRef CAS PubMed.
- X. Xu, et al., The New Era of High-Throughput Nanoelectrochemistry, Anal. Chem., 2023, 95(1), 319–356 CrossRef CAS PubMed.
- Z. Xie, et al., Specific Small-Molecule Detection Using Designed Nucleic Acid Nanostructure Carriers and Nanopores, Anal. Chem., 2024, 96(21), 8528–8533 CrossRef CAS.
- L. Soleymani, et al., Programming the detection limits of biosensors through controlled nanostructuring, Nat. Nanotechnol., 2009, 4(12), 844–848 CrossRef.
- K. Fu, et al., Accelerated Electron Transfer in Nanostructured Electrodes Improves the Sensitivity of Electrochemical Biosensors, Advanced Science, 2021, 8(23), 2102495 CrossRef PubMed.
- V. Kesler, et al., Tailoring Electrode Surface Charge to Achieve Discrimination and Quantification of Chemically Similar Small Molecules with Electrochemical Aptamers, Adv. Funct. Mater., 2023, 33(1), 2208534 CrossRef PubMed.
- Z. Watkins, et al., Week-Long Operation of Electrochemical Aptamer Sensors: New Insights into Self-Assembled Monolayer Degradation Mechanisms and Solutions for Stability in Serum at Body Temperature, ACS Sens., 2023, 8(3), 1119–1131 CrossRef PubMed.
- P. Daggumati, et al., Biofouling-Resilient Nanoporous Gold Electrodes for DNA Sensing, Anal. Chem., 2015, 87(17), 8618–8622 CrossRef.
- Z. Zhang, et al., Incorporating Hydrophobic Moieties into Self-Assembled Monolayers to Enable Electrochemical Aptamer-Based Sensors Deployed Directly in a Complex Matrix, ACS Sens., 2022, 7(9), 2615–2624 CrossRef PubMed.
- A. Piper, D. K. Corrigan and A. R. Mount, An electrochemical comparison of thiolated self-assembled monolayer (SAM) formation and stability in solution on macro- and nanoelectrodes, Electrochem. Sci. Adv., 2022, 2(6), e2100077 CrossRef.
- S. Li, et al., Implantable Hydrogel-Protective DNA Aptamer-Based Sensor Supports Accurate, Continuous Electrochemical Analysis of Drugs at Multiple Sites in Living Rats, ACS Nano, 2023, 17(18), 18525–18538 CrossRef.
- Y. Utagawa, et al., Vasculature-on-a-Chip with a Porous Membrane Electrode for In Situ Electrochemical Detection of Nitric Oxide Released from Endothelial Cells, Anal. Chem., 2023, 95(49), 18158–18165 CrossRef CAS.
- A. J. Trowbridge, et al., Bridging the gap: Polymer materials with controlled architecture and precise functionalization to overcome biofouling challenges for biosensing, drug delivery, and bioseparation, Polymer, 2024, 307, 127230 CrossRef CAS.
- C. R. Martin, I. Rubinstein and A. J. Bard, Polymer films on electrodes. 9. Electron and mass transfer in Nafion films containing tris(2,2'-bipyridine)ruthenium(2+), J. Am. Chem. Soc., 1982, 104(18), 4817–4824 CrossRef CAS.
- G. A. Gerhardt, et al., Nafion-coated electrodes with high selectivity for CNS electrochemistry, Brain Res., 1984, 290(2), 390–395 CrossRef CAS PubMed.
- N. White, et al., Coating of Nafion Membranes with Polyelectrolyte Multilayers to Achieve High Monovalent/Divalent Cation Electrodialysis Selectivities, ACS Appl. Mater. Interfaces, 2015, 7(12), 6620–6628 CrossRef CAS.
- K. Fu, et al., Asymmetric Nafion-Coated Nanopore Electrode Arrays as Redox-Cycling-Based Electrochemical Diodes, ACS Nano, 2018, 12(9), 9177–9185 CrossRef CAS PubMed.
- S. H. Lim, et al., A glucose biosensor based on electrodeposition of palladium nanoparticles and glucose oxidase onto Nafion-solubilized carbon nanotube electrode, Biosens. Bioelectron., 2005, 20(11), 2341–2346 CrossRef CAS.
- D. Chen, et al., PVDF-Nafion nanomembranes coated microneedles for in vivo transcutaneous implantable glucose sensing, Biosens. Bioelectron., 2015, 74, 1047–1052 CrossRef CAS PubMed.
- P. Dauphin-Ducharme and K. W. Plaxco, Maximizing the Signal Gain of Electrochemical-DNA Sensors, Anal. Chem., 2016, 88(23), 11654–11662 CrossRef CAS PubMed.
- J. Lu and J. Li, Monitoring DNA conformation and charge regulations by plasmonic-based electrochemical impedance platform, Electrochem. Commun., 2014, 45, 5–8 CrossRef CAS.
- Y.-L. Ying, et al., Monitoring of an ATP-Binding Aptamer and its Conformational Changes Using an α-Hemolysin Nanopore, Small, 2011, 7(1), 87–94 CrossRef CAS PubMed.
- C. Chau, et al., Probing RNA Conformations Using a Polymer–Electrolyte Solid-State Nanopore, ACS Nano, 2022, 16(12), 20075–20085 CrossRef CAS PubMed.
- J. Wu, et al., Single-Molecule Identification of the Conformations of Human C-Reactive Protein and Its Aptamer Complex with Solid-State Nanopores, ACS Appl. Mater. Interfaces, 2022, 14(10), 12077–12088 CrossRef CAS PubMed.
- C. Chen, et al., Full wetting of plasmonic nanopores through two-component droplets, Chem. Sci., 2015, 6(11), 6564–6571 RSC.
- M. Shen, et al., Quantitative Imaging of Ion Transport through Single Nanopores by High-Resolution Scanning Electrochemical Microscopy, J. Am. Chem. Soc., 2012, 134(24), 9856–9859 CrossRef CAS PubMed.
- Y. Nashimoto, et al., Evaluation of mRNA Localization Using Double Barrel Scanning Ion Conductance Microscopy, ACS Nano, 2016, 10(7), 6915–6922 CrossRef CAS.
- H. Ren, et al., Single-Molecule Titration in a Protein Nanoreactor Reveals the Protonation/Deprotonation Mechanism of a C:C Mismatch in DNA, J. Am. Chem. Soc., 2018, 140(15), 5153–5160 CrossRef CAS PubMed.
- X. Li, et al., Nanopore Opening at Flat and Nanotip Conical Electrodes during Vesicle Impact Electrochemical Cytometry, ACS Nano, 2018, 12(3), 3010–3019 CrossRef CAS.
- T. Xiong, et al., Neuromorphic functions with a polyelectrolyte-confined fluidic memristor, Science, 2023, 379(6628), 156–161 CrossRef CAS PubMed.
- V. Kesler, B. Murmann and H. T. Soh, Going beyond the Debye Length: Overcoming Charge Screening Limitations in Next-Generation Bioelectronic Sensors, ACS Nano, 2020, 14(12), 16194–16201 CrossRef CAS PubMed.
- C. C. Chau, et al., Macromolecular Crowding Enhances the Detection of DNA and Proteins by a Solid-State Nanopore, Nano Lett., 2020, 20(7), 5553–5561 CrossRef CAS.
- X. Chen, et al., Impact of the Coverage of Aptamers on a Nanoparticle on the Binding Equilibrium and Kinetics between Aptamer and Protein, ACS Sens., 2021, 6(2), 538–545 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00144c |
| This journal is © The Royal Society of Chemistry 2024 |