
The promotion of fatty acid β-oxidation by hesperidin via activating SIRT1/PGC1α to improve NAFLD induced by a high-fat diet†
Tong
Nie
 a,
Xin
Wang
a,
Aqun
Li
a,
Anshan
Shan
b and
Jun
Ma
*ac
a,
Xin
Wang
a,
Aqun
Li
a,
Anshan
Shan
b and
Jun
Ma
*ac
aCollege of Veterinary Medicine, Northeast Agricultural University, Harbin, 150030, P. R. China. E-mail: majun@neau.edu.cn
bCollege of Animal Science and Technology, Northeast Agricultural University, Harbin, 150030, P. R. China
cHeilongjiang Provincial Key Laboratory of Pathogenic Mechanism for Animal Disease and Comparative Medicine, Harbin, 150030, P.R. China
First published on 1st December 2023
Abstract
Reducing fat deposits in hepatocytes is a direct treatment for nonalcoholic fatty liver disease (NAFLD) and the fatty acid metabolic processes mediated by fatty acid β-oxidation are important for the prevention of NAFLD. In this study, we established high-fat-diet models in vitro and in vivo to investigate the mechanism by which hesperidin (HDN) prevents NAFLD by modulating fatty acid β oxidation. Based on LC-MS screening of differential metabolites, many metabolites involved in phospholipid and lipid metabolism were found to be significantly altered and closely associated with fatty acid β-oxidation. The results from COIP experiments indicated that HDN increased the deacetylation of PGC1α by SIRT1. In addition, the results of CETSA and molecular docking experiments suggest that HDN targeting of SIRT1 plays an important role in their stable binding. Meanwhile, it was found that HDN reduced fatty acid uptake and synthesis and promoted the expression of SIRT1/PGC1α and fatty acid β-oxidation, and the latter process was inhibited after transfection to knockdown SIRT1. The results suggest that HDN improves NAFLD by promoting fatty acid β-oxidation through activating SIRT1/PGC1α. Thus, the findings indicate that HDN may be a potential drug for the treatment of NAFLD.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a pathological syndrome characterized by the excessive deposition of fat in hepatocytes1 and is not caused by alcohol or other definite factors, but it is closely associated with metabolic syndromes, such as obesity,2 dyslipidemia,3 hypertension, and cardiovascular circulatory diseases.4 NAFLD first presents simple hepatic steatosis, then gradually develops into nonalcoholic steatohepatitis and cirrhosis and eventually into hepatocellular carcinoma,5 thus posing a great threat to human health. Many kinds of research have shown that NAFLD influences the fatty acid transport, synthesis,6,7 and β-oxidation process8,9 in hepatocytes, making it impossible to completely break down and metabolize excessive fat; therefore, a large amount of fat accumulates in hepatocytes, which causes liver damage.10 Therefore, reducing hepatic fat deposition is an effective treatment for NAFLD,6,11,12 and it is important to investigate the mechanism.Peroxisome proliferator-activated receptor gamma activator-1α (PGC1α), a member of the peroxisome proliferator-activated receptor gamma cofactor-1 family, plays an important role in regulating mitochondrial function,13 lipid metabolism,14 and fatty acid β-oxidation15 by interacting with peroxisome proliferator-activated receptor α (PPARα). Silent mating-type information regulation 2 homolog 1 (SIRT1) is a nicotinamide adenine dinucleotide-dependent deacetylase that is involved in regulating various physiological functions by deacetylating a number of transcriptional regulators.16,17 Many reports have shown that PGC1α is deacetylated by SIRT1,18,19 affecting various PGC1α-mediated biological processes. Therefore, we suggest that liver injury caused by fat deposition in NAFLD hepatocytes may be associated with changes in SIRT1/PGC1α expression, investigating which may provide a potential method for pharmacological intervention to improve NAFLD treatment.
Hesperidin (C28H34O15, HDN) is a flavonoid that is widely found in nature and is an effective component of the traditional Chinese medicine Chen Pi, which is widely available and can be isolated in large quantities from the peels of some citrus fruits.20 HDN positively impacts various sites, such as the nerves,21 cardiovascular system,22 heart,23 and liver. Many studies have reported that flavonoids, such as quercetin,24 galangin,25 and naringenin,26 can treat liver diseases through the SIRT1 pathway. It is speculated that HDN, also a flavonoid, may improve NAFLD by activating SIRT1. Excessive fatty acid transport to the liver increases the burden of fatty acid beta-oxidation, while excessive fat deposition in the liver leads to liver damage. SIRT1/PGC1α has been reported to improve NAFLD by regulating autophagy,27 fibrosis,28 endoplasmic reticulum stress,29 oxidative stress,30 and apoptotic31 pathways. Therefore, we speculated that HDN improves NAFLD by activating SIRT1/PGC1α by promoting fatty acid β oxidation. In this study, we established the NAFLD model by feeding a high-fat diet (HFD) to mice and palmitic (PA) and oleic acid (OA) complex (PO) to induce AML12 lipid accumulation, and then measured the aspartate transaminase (AST), alanine transaminase (ALT), total cholesterol (TC), triglyceride (TG), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) levels to clarify the mechanism of the effect of HDN in reducing liver and cellular lipid deposition to improve NAFLD, with an aim to provide new clinical ideas to improve NAFLD.
2. Materials and methods
2.1 Animals and diet treatments
Male C57BL/6J mice (8 weeks old) were purchased from Changsheng Biological Technology (Changchun, China). All the animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Northeast Agricultural University and approved by the Animal Ethics Committee of Northeast Agricultural University. After acclimation for 1 week, the mice were divided into 5 groups (n = 8) at random, including the control group (CON), HDN group (300 mg kg−1 d−1 HDN), high-fat-diet group (HFD), HDN low-dose-treatment groups (HFD + 150 mg kg−1 d−1 HDN, DHL), and HDN high-dose-treatment groups (HFD + 300 mg kg−1 d−1 HDN, DHH). The doses administered to the mice were selected with reference to Makan Cheraghpour's research32 and were converted according to the body surface area method.The C57BL/6 mice were fed a normal chow diet (CON; 10% kcal fat, 70% carbohydrates, 20% protein) or a high-fat diet (HFD; 60% kcal fat, 20% carbohydrates, 20% protein) for 16 weeks. Meanwhile, the HFD group, DHL group, and DHH group were orally administered with HDN (150, 300 mg kg−1 d−1) or vehicle once per day for 16 weeks. HDN was dissolved in 0.5% CMC-Na (Sigma-Aldrich, Germany), and the 0.5% CMC-Na was used as a vehicle for the CON group.
2.2 Intraperitoneal glucose tolerance and insulin tolerance test
The intraperitoneal glucose tolerance test (IPGTT) was performed at week 15. The mice were fasted for 12 h and then injected intraperitoneally with glucose (1 g kg−1). Blood samples were collected from the tail vein of the mice, and blood glucose was measured using a glucometer (Yuwell, China) at 0 min before glucose injection, and at 15, 30, 60, 90, and 120 min after glucose injection in each group of mice. The intraperitoneal insulin tolerance test (IPITT) was performed three days after the IPGTT test and blood was taken before insulin injection to measure the plasma insulin levels according to the ELISA kit (AD3184Mo, Chenglin Biological, China) instructions. The mice were fasted for 6 h and then injected intraperitoneally with insulin (1 U kg−1). Blood samples were collected from the tail vein of the mice, and blood glucose was measured. The area under the blood glucose curve (AUC) for IPGTT and IPITT was calculated.2.3 Liquid chromatography tandem mass spectrometry (LM-MS)
Tissues (100 mg) were individually ground with liquid nitrogen and the homogenate was resuspended with prechilled 80% methanol by good vortexing. The samples were incubated on ice for 5 min and were then centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and 4 °C for 20 min. Some of supernatant was diluted to a final concentration containing 53% methanol by LC-MS grade water. The samples were subsequently transferred to a fresh Eppendorf tube and were then centrifuged at 15000g and 4 °C for 20 min. Finally, the supernatant was injected into the LC-MS/MS system for analysis.
000g and 4 °C for 20 min. Some of supernatant was diluted to a final concentration containing 53% methanol by LC-MS grade water. The samples were subsequently transferred to a fresh Eppendorf tube and were then centrifuged at 15000g and 4 °C for 20 min. Finally, the supernatant was injected into the LC-MS/MS system for analysis.
We used univariate analysis (t-test) to calculate the statistical significance (P-value), and metabolites with VIP > 1 and P-value < 0.05, fold change ≥ 2, or FC ≤ 0.5 were considered differential metabolites. Partial least-squares discriminant analysis (PLSDA) was performed using R version 4.0.3. For the clustering heat maps, the data were normalized using Z-scores of the intensity areas of the differential metabolites and were plotted using the pheatmap package in R language.
2.4 Immunohistochemical staining (IHC)
Paraffin sections of liver tissue were deparaffinized and dehydrated with gradient alcohol. After antigen repair, endogenous catalase was blocked with 3% H2O2 and then incubated overnight at 4 °C with a primary antibody after sealing with serum. The next day, the secondary antibody was incubated at room temperature for 1 h. After incubation, the color was developed with DAB (Beyotime, China) and the nuclei were stained with hematoxylin (Biosharp, China). Sections were dehydrated and visualized under a light microscope. Positive areas were counted using Image J Fiji.2.5 AML12 cell viability analysis and in vitro intervention assay
AML12 cells were cultured in 10% FBS, 1% penicillin–streptomycin, and 1% ITS (Beyotime, China) containing DMEM/F12 (Gibco, USA) medium in a 5% CO2 humidified incubator at 37 °C. The cultural medium was changed every other day.AML12 cells were seeded at 2 × 103 cells per well in 96-well dishes. The cells were stimulated as described in the figure legend (Fig. 6A). AML12 cells were incubated with DMEM/F12 and treated with different HDN (>98%, Yuanye, China) concentrations for 24 h. Then, the cell viability was measured by the CCK-8 assay (Biosharp, China), then the liquid in the dishes was changed to DMEM/F12 with 1% penicillin–streptomycin solution and 1% ITS without HDN, and CCK-8 was added to each well, and then incubated for 3 h at 37 °C. Finally, the cell viability was analyzed at a wavelength of 450 nm.
The optimal dosing concentrations of HDN were selected for the subsequent experiments. The experiment was divided into five groups: control group (CON), HDN group (200 μM HDN), model group (PO), HDN low-dose-treatment groups (PO + 100 μM HDN, PHL), and HDN high-dose-treatment groups (PO + 200 μM HDN, PHH).
First, 0.5 mM PO (PA:OA = 1:2) was prepared to induce lipid accumulation in AML12 cells. Also, the AML12 cells were incubated with PO and treated with different concentrations of HDN for 24 h.
2.6 Biochemical analyses
Serum biochemical indexes of TC, TG, HDL, LDL, AST, and ALT were measured using an automatic biochemical analyzer (Mindray, Shenzhen, China). TG (A110-1-1), TC (A111-1-1), HDL (A112-1-1), and LDL (A113-1-1) in the liver tissue homogenates and TG, TC in the AML12 cell lysates were measured using commercial kits (Nanjing Jiancheng, China).2.7 Hematoxylin–eosin and Oil Red O staining
Liver tissues were fixed using 4% paraformaldehyde and then embedded in paraffin to make wax blocks. The wax blocks were cut to a thickness of 5 μm for the hematoxylin and eosin (H&E) staining (Solarbio, China), and frozen liver tissues were embedded in OCT to make frozen sections, which were then sliced at 10 μm, followed by 0.5% Oil Red O (Solarbio, China) staining. AML12 cells were fixed in 4% paraformaldehyde for 30 min, rinsed with PBS, stained with Oil Red O for 30 min, and then isopropyl alcohol was used to wash away the floating color. The sections and cells were observed under a microscope (Nikon, Tokyo, Japan).2.8 Immunofluorescence staining (IF)
Paraffin sections of liver tissue were dewaxed and then dehydrated with gradient alcohol. After antigen repair, the sections were blocked with secondary antibody homologous serum (Beyotime, China) for 30 min and then the sections were incubated with primary antibody overnight. The next day, the sections were washed with PBS and incubated with the secondary antibodies Alexa Fluor 488 (green) and Alexa Fluor 594 (red) (Bioss, China) for 1 h and DAPI (blue) (Biosharp, China) for 5 min.AML12 cells were fixed with 4% paraformaldehyde (Biosharp, China) at 4 °C for 3–6 h. The cells were then permeabilized with 0.25% Triton X-100 for 20 min and blocked with secondary antibody homologous serum for 30 min. After blocking, the cells were incubated with primary antibody overnight at 4 °C and the next day incubated with secondary antibody for 1 h and stained with DAPI for 5 min.
The sections and cells were observed under fluorescence microscopy (Nikon, Tokyo, Japan).
2.9 Cellular thermal shift assay (CETSA)
The CON blank control group and HDN-H (200 μM) drug control group were set up, and the cells were treated with HDN-H for 6 h. The cell lysate was collected, and the protein concentration was measured using the BCA kit. Next, it was heated to at 37–67 °C for 3 min, then loading buffer was added followed by heating at 100 °C for 10 min, and finally storage at −80 °C for subsequent western blot experiments.2.10 siRNA transfection
AML12 cells were seeded at 1 × 104 cells per well in 24-well dishes to achieve a cell density of 30%–50% at the time of transfection. Two hours before transfection, the medium was changed (to without 1% penicillin–streptomycin). The AML12 cells were transfected with 50 nM of SIRT1 siRNA and negative control siRNA using riboFECTTM CP reagent (Ribo, China). The cells were incubated with riboFECTTM CP reagent-siRNA complex for 6 h and then replaced with fresh experimental medium after incubating the cells for 24 h for subsequent experiments.2.11 RNA preparation and qRT-PCR of the cells
We used the qRT-PCR method to detect the mRNA level of SIRT1 after transfection. Total RNA was isolated from cells using Trizol. The RNA concentration reached the test requirement when the A260/A280 of the total RNA was between 1.9 and 2.1. We used the master hiSensi cDNA first-strand synthesis kit (Bioer, China) and qRT-PCR kit (Bioer, China) for reverse transcription and fluorescence quantification of the total RNA from the AML12 cells. The primers for quantification were designed by Shenggong (Shanghai, China). The sequences were Forward 5′-CGCTGTGGCAGATTGTTATTAA-3′ and reverse 5′-TTGATCTGAAGTCAGGAATCCC-3′ for mouse SIRT1; forward 5′-CTACCTCATGAAGATCCTGACC-3′ and Reverse 5′-CACAGCTTCTCTTTGATGTCAC-3′ for β-actin, which was used as a standardized endogenous control.2.12 Co-immunoprecipitation (COIP)
The interaction between endogenous SIRT1 and PGC1α protein and the degree of acetylation of PGC1α were detected by COIP. We extracted the total cellular protein lysate and quantified the protein concentration to 1 μg μL−1 using the BCA kit, and then aspirated some of the lysate as the input group. The remaining lysate was combined with anti-SIRT1 antibody (1:200, Proteintech, China), anti-PGC1α antibody (1:200, Proteintech, China), or control lgG antibody (1:100, Abclonal, China) by rotating at 4 °C overnight. The next day, the lysate was supplemented with protein A/G-Agarose (Abmart, China), followed by rotating at 4 °C for 2 h, and then subjected to western blot analysis.
2.13 Western blot
Following our previous protocol,33 the expressions of the proteins in the livers and AML12 cells were measured, and the primary antibodies were SIRT1 (1:1000, Proteintech, China), PGC1α (1:5000, Proteintech, China), Acetylated-Lysine Mab (1:500, Abmart, China), PPARα (1:500, Wanlei, China), cluster of differentiation 36 (CD36) (1:500, Wanlei, China), carnitine palmitoyltransferase 1A (CPT1) (1:1000, Abclonal, China), acyl coenzyme A Oxidase 1 (ACOX1) (1:1000, Abclonal, China), enoyl coenzyme A (EHHADH) (1:1000, Abclonal, China), stomatal cytokinesis-defective 1 (SCD1) (1:1000, Abclonal, China), and fatty acid binding protein 1 (FABP1) (1:1000, Abclonal, China), while the secondary antibodies were goat anti-rabbit IgG (1:10000, Bioss, China), and goat anti-mouse IgG (1:10000, Bioss, China). Finally, the ECL kit (Meilunbio, China) was used to visualize the bands on the membrane by a Tanon 5200 system, and Image J Fiji was used to quantify the protein bands.
2.14 Protein–protein interaction networks
The String database (https://cn.string-db.org/) is a database of both known and predicted protein–protein interactions. We uploaded the proteins involved in our experiments to the String database to predict their interactions.2.15 Molecular docking
We used ZDOCK software to dock SIRT1 and PGC1α to analyze the possible amino acid binding sites. The protein structure files of SIRT1 (PDB ID: 5BTR) and PGC1α (PDB ID: 1HTR) were downloaded from the PDB database (https://www.rcsb.org/). After importing the protein structure into PyMol 2.3.0 to remove crystal water and small molecules, the protein structure file was uploaded to ZDOCK software for the calculations. The generated results were sorted according to the ZDOCK scores, with higher scores yielding better results. Finally, PyMol 2.3.0 was applied to observe the interaction pattern of the top-ranked docking model.We downloaded the molecular structure of HDN from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Molecular docking of HDN and SIRT1 was performed using AutoDockVina-1.1.2. Based on the position of the protein ligand, the coordinates of the protein active site were determined, and the conformation with the lowest docking binding energy was selected for molecular docking binding mode analysis to obtain the free energy of the molecular docking binding, and the results were then graphed using PyMol.
2.16 Statistical analysis
In the present study, we performed one-way ANOVA tests followed by Tukey's multiple range tests in GraphPad Prism 8.0 software to confirm whether there were differences between each group, where ANOVA p-values less than 0.05 were considered statistically significant. Data were expressed as the mean ± standard error. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.3. Results
3.1 HDN reduced the body weight and blood lipid levels in high-fat-diet mice
It is known that HDN has a positive effect on hyperlipidemia and alleviates hepatocyte fat deposition, and so to investigate the effect of HDN on NAFLD, we established the NAFLD model with HFD mice for 16 weeks (Fig. 1A and B). The results showed that compared with the CON group, the body weight, liver weight, and liver index of the HFD group were significantly increased (P < 0.001). Interestingly, HDN supplementation in the HFD mice significantly reduced the body weight, liver weight, and liver index (P < 0.01) (Fig. 1C–E). The serum TG, TC, and LDL in the HFD group were significantly higher (P < 0.01) while HDL was significantly lower than in the CON group (P < 0.01). However, HDN intervention significantly improved the lipid levels in the HFD mice (P < 0.05), but there was no significant difference in TC (P > 0.05) (Fig. 1F–I). These results suggest that HDN intervention reduces body weight and lipid levels in high-fat-diet mice.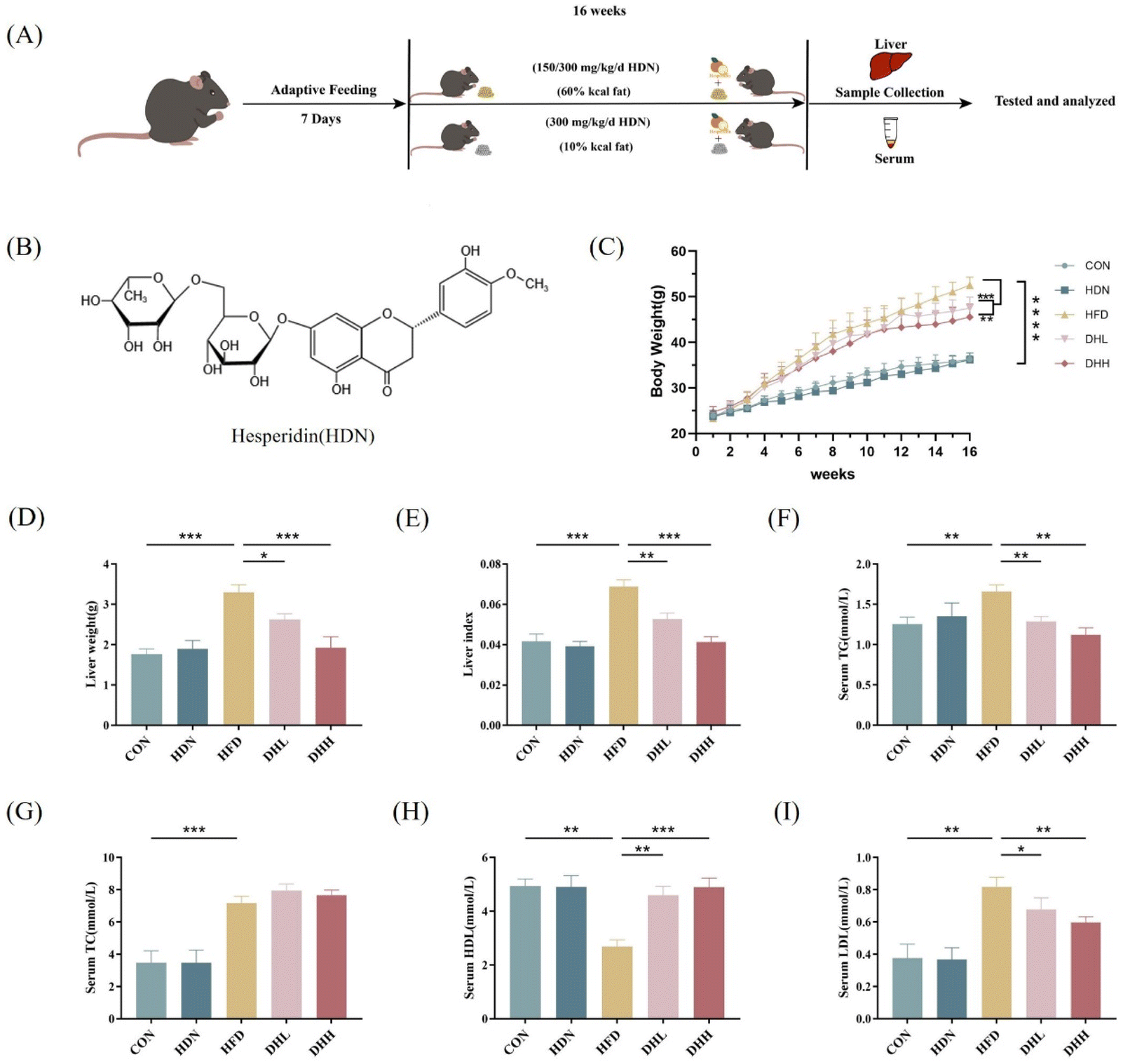 | ||
| Fig. 1 The effect of HDN on body weight, liver weight, liver index, and lipid levels in high-fat-diet mice. (A) Flow chart of the animal model establishment. (B) Chemical structure of HDN. (C) Whole-body weight gain curves after 16 weeks of HDN administration in high-fat-diet mice. (D) Liver weight of the mice. (E) Liver index (mice's liver weight/body weight at week 16). (F) The level of TG in the mice serum. (G) The level of TC in the mice serum. (H) The level of HDL in the mice serum. (I) The level of LDL in the mice serum. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
3.2 HDN improved hepatic lipid accumulation and liver injury induced by the high-fat diet
To further investigate the effects of HDN on the liver of HFD mice, we examined the liver function, and liver lipid levels and observed the structural changes in the liver. By testing the liver function and liver lipid levels we discovered that compared with the CON group, the serum AST and ALT were significantly increased in the HFD group (P < 0.001) (Fig. 2A and B); and TG, TC, and LDL were significantly increased in the liver (P < 0.0001) (Fig. 2C, D and F), while HDL was significantly decreased (P < 0.0001) (Fig. 2E). HDN intervention significantly reversed the changes in these indices. We observed from the H&E results that compared with the CON group, the hepatocytes in the HFD group were in steatosis as shown by the swelling of the hepatocytes (black arrows) and vacuoles in the cytoplasm (red arrows). At the time of HDN intervention, hepatocellular steatosis was alleviated in the DHL group and even approached the level of the CON group in the DHH group (Fig. 2G). The above results suggest that HDN could significantly improve liver structure and function and reduced liver lipid levels in the HFD mice.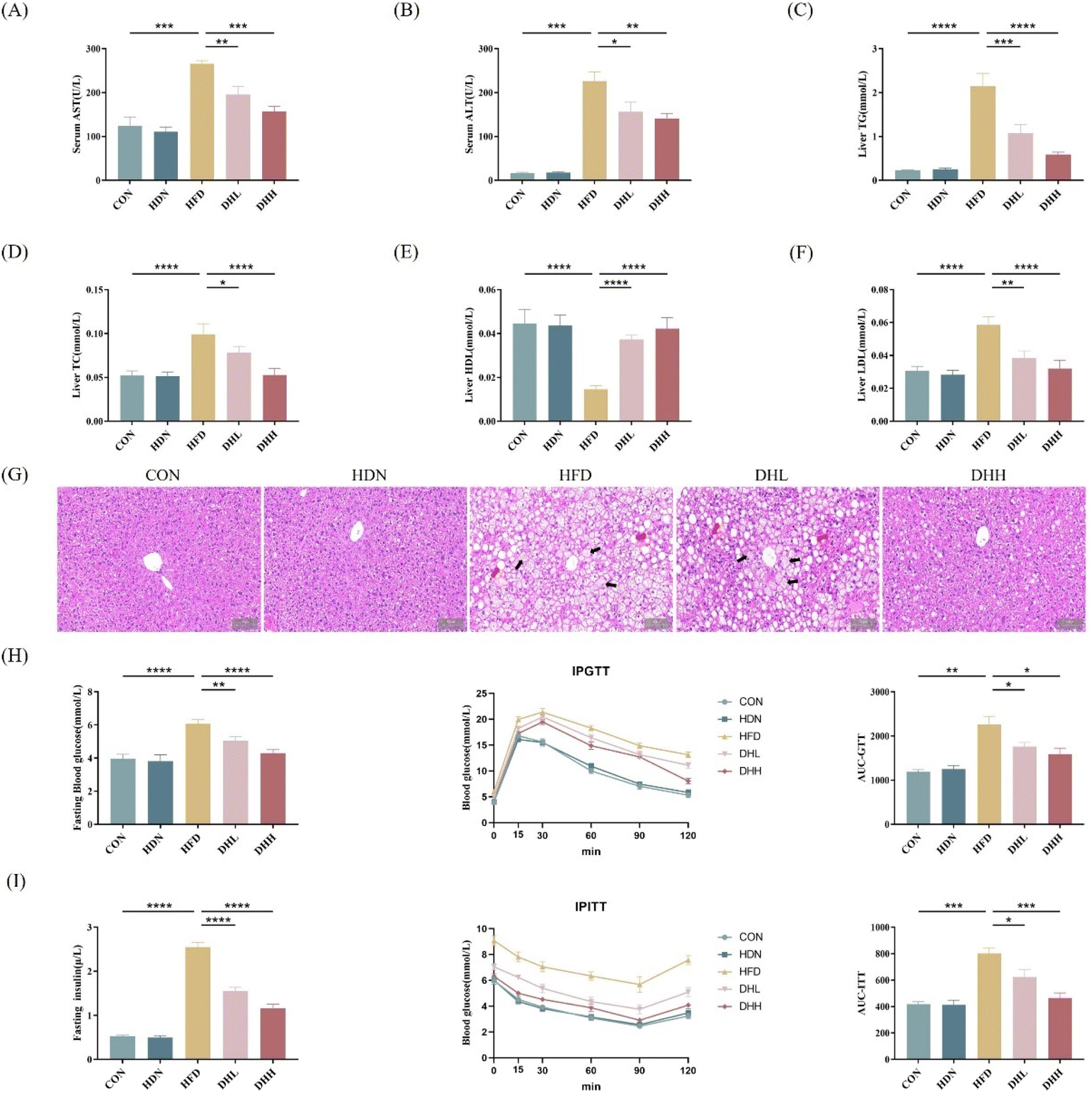 | ||
| Fig. 2 The effects of HDN on liver function, hepatic lipid levels, and structural changes in the liver of high-fat-diet mice. (A) The levels of AST in the mice serum. (B) The level of ALT in the mice serum. (C) The levels of TG in the mice liver. (D) The levels of TC in the mice liver. (E) The levels of HDL in the mice liver. (F) The levels of LDL in mice liver. (G) Representative histological images of H&E staining in the liver. (H) Blood glucose, IPGTT, and the calculation of the corresponding AUC. (I) Plasma insulin in mice after fasting for 6 h, IPITT, and the calculation of the corresponding AUC. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
3.3 HDN improved insulin resistance caused by the high-fat diet
Insulin resistance has been recognized as a key risk factor for NAFLD. To investigate whether HDN plays a role in altering insulin resistance, we performed IPGTT and IPITT on each group of mice to assess their glucose homeostasis and insulin sensitivity. Compared with the CON group, the HFD group had significantly increased fasting glucose and insulin levels at baseline (P < 0.0001) and significantly decreased glucose and insulin levels after the HDN intervention (P < 0.01). After the intraperitoneal administration of glucose to the mice, the reduced response to glucose loading in HDN-treated HFD group mice was reversed, which suggested that HDN had an effect on improving glucose tolerance (Fig. 2H). Similarly, the HDN-treated HFD group mice had a significantly faster glucose response to insulin loading (Fig. 2I). Therefore, HDN intervention significantly improved insulin sensitivity and glucose tolerance in the HFD mice.3.4 HDN regulated the expression of lipid-related metabolites
To further investigate the effect of HDN on the HFD mice, we analyzed the metabolites by untargeted metabolomics. We found that many metabolites associated with the lipid, amino acid, and energy metabolic pathways showed significant changes in the HFD mice, and HDN intervention reversed these changes (Fig. 3A and B). At the same time, we screened the common differential metabolites in HFD vs. CON, HFD-H vs. HFD (Fig. 3C) for analysis, which showed few intra-group and significant inter-group differences (Fig. 3D), and then we chose the metabolites that had significant differences and were of interest to us for analysis (Fig. 3E). Compared with the CON group, taurochenodeoxycholic acid (sodium salt), glycoursodeoxycholic acid, and hypotaurine were significantly reduced in the HFD group, while 9-KODE and acetoacetate were significantly increased, and HDN intervention reversed the expression of these metabolites, all of which were related to lipid formation and lipid lowering, suggesting that HDN intervention affected the synthesis and metabolism of the lipids. Compared with the CON group, metabolites of phosphatidylcholine, such as LPC 16:1 and cytidine 5′-diphosphocholine, were significantly decreased in the HFD group. The high-fat diet affected phospholipid metabolism in the mice. Meanwhile, compared with the CON group, the HFD group had significantly decreased levels of 13-OxoODE and nicotinamide, while 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5-monophosphate was significantly increased, and HDN intervention reversed the expression of 13-OxoODE, and nicotinamide, and the changes in these metabolites related to fatty acid β-oxidation suggested that fatty acid β-oxidation was inhibited in the HFD mice, and that fatty acid β-oxidation was mitigated after HDN intervention. Therefore, HDN may alleviate lipid metabolism abnormalities induced by a high-fat diet through a fatty acid β-oxidation process.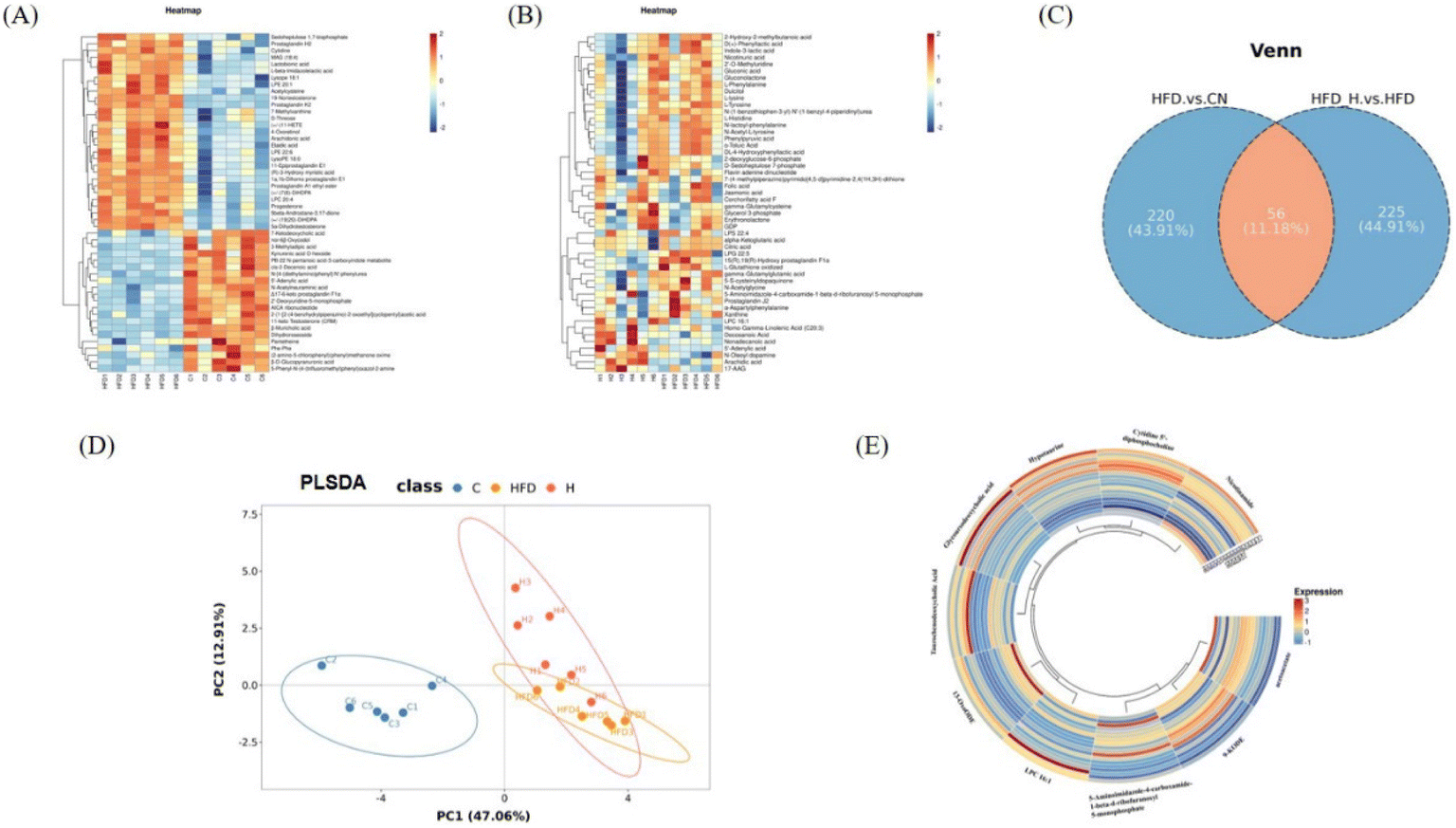 | ||
| Fig. 3 HDN regulated the expression of lipid-related metabolites. (A) Top 50 differential metabolites in HFD vs. CON. (B) Top 50 differential metabolites in HFD-H vs. HFD. (C) Venn diagram representing the number of differential metabolites common to HFD vs. CON and HFD-H vs. HFD. (D) PLSDA plot of the differential metabolites common to HFD vs. CON and HFD-H vs. HFD. (E) Heatmap of selected common differential metabolites in HFD vs. CON and HFD-H vs. HFD. | ||
3.5 HDN improved hepatic lipid accumulation by activating SIRT1/PGC1α
According to the above results, we found that HDN could improve HFD-induced NAFLD in mice, so we further investigated the mechanism by which HDN improved NAFLD. The obtained results showed that compared with the CON group, the expression levels of SIRT1 and PGC1α proteins were significantly decreased in the HFD group (Fig. 4A), and the IF double-staining and IHC results of SIRT1 and PGC1α showed the same changes (P < 0.05) (Fig. 5A and B). Also, the expression levels of proteins related to fatty acid transport synthesis of CD36, SCD1, and FABP1 were significantly increased (P < 0.05) (Fig. 4B); while the expression levels of proteins related to fatty acid β-oxidation PPARα, CPT1, ACOX1, and EHHADH were significantly reduced (P < 0.05) (Fig. 4C). HDN intervention significantly reversed the expression of these proteins. The PPI protein interaction network results showed that the above proteins were closely related and regulated by each other (Fig. 4D). The results of the Oil Red O staining showed that liver lipid droplets were significantly increased in the HFD group compared with in the CON group (P < 0.001) and significantly decreased after HDN intervention (P < 0.05) (Fig. 4E). These results suggest that HDN promoted fatty acid β-oxidation through the activation of SIRT1/PGC1α, thereby ameliorating hepatic lipid accumulation in NAFLD.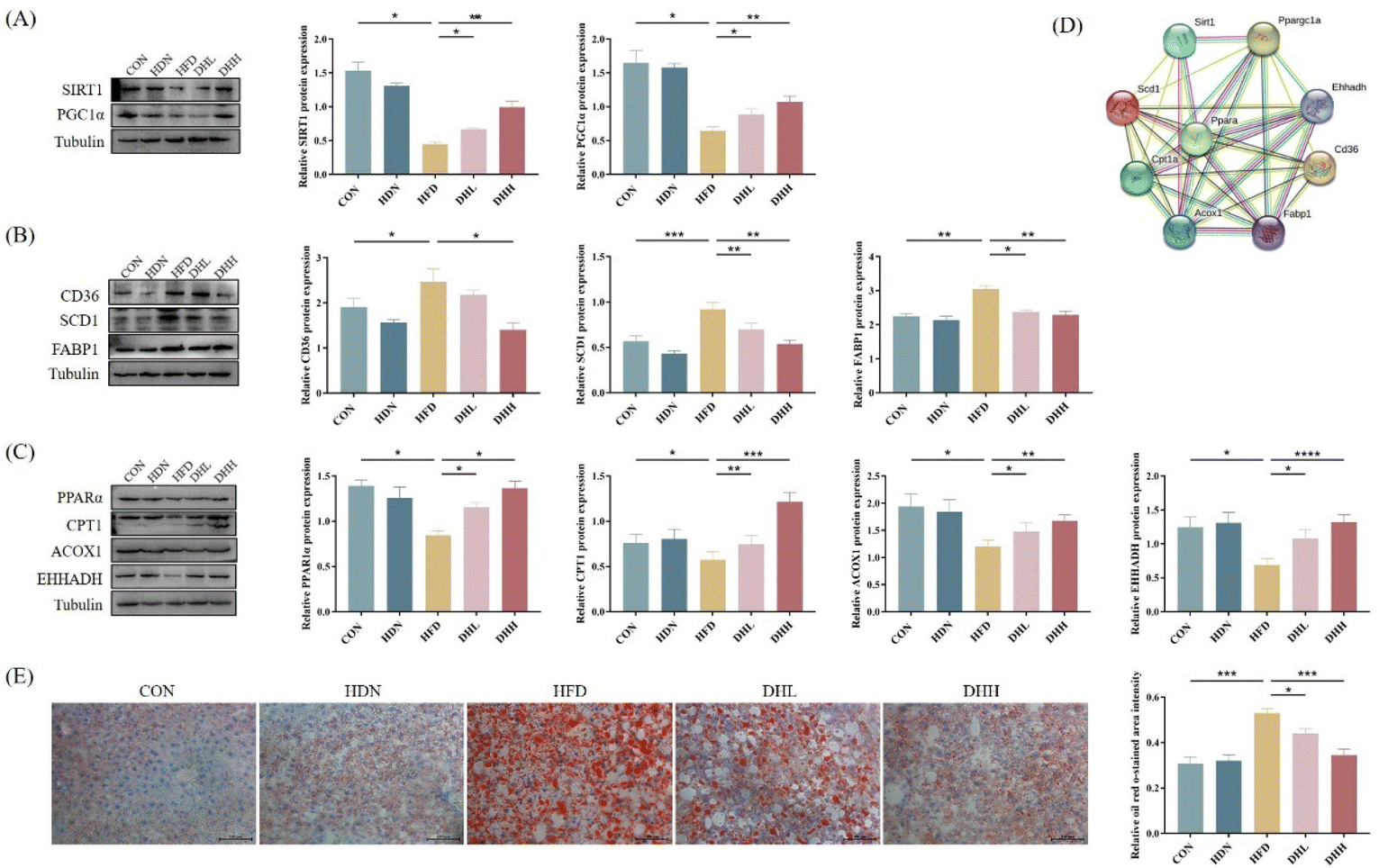 | ||
| Fig. 4 Effect of HDN on SIRT1/PGC1α. (A) Protein bands and quantitative results of SIRT1 and PGC1α. (B) Protein bands and quantitative results of CD36, SCD1, and FABP1. (C) Protein bands and quantitative results of PPARα, CPT1, ACOX1, and EHHADH. (D) PPI protein network interactions map. (E) Oil Red O staining of the liver and the quantitative results. *P < 0.05, **P< 0.01, ***P < 0.001, and ****P < 0.0001. | ||
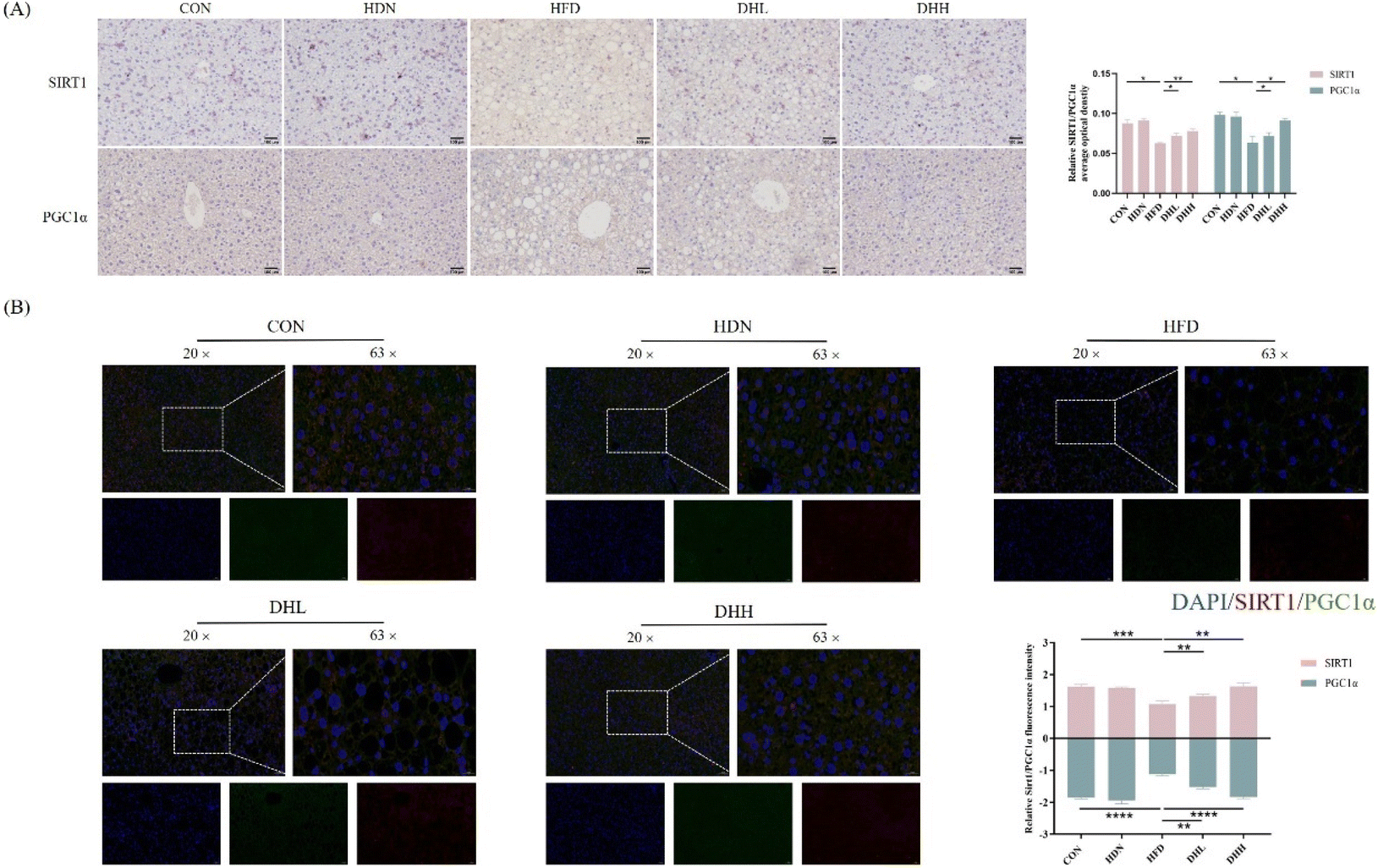 | ||
| Fig. 5 Effect of HDN on SIRT1/PGC1α. (A) Results of IHC staining and the quantification of SIRT1 and PGC1α in mice liver. (B) Results of IF double-staining and quantitative results of the mice liver. SIRT1 (red), PGC1α (green), and DAPI (blue). *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.6 HDN improved PO-induced lipid accumulation in AML12 cells
To further investigate the effect of HDN on high lipid-induced lipid accumulation, we induced a high lipid model in AML12 cells by PO in vitro. First, we conducted the CCK8 assay to select the optimal drug concentration for HDN application to AML12 cells. Based on the results of the CCK8 assay, we observed a significant decrease in cell viability at a HDN concentration of 400 μM compared to the CON group (P < 0.001) (Fig. 6A), so we selected two drug concentrations of 100 μM and 200 μM HDN for the subsequent experiments. After that, we induced AML12 lipid accumulation with PO and performed HDN intervention. The obtained results showed that compared with the CON group, TC and TG were significantly increased in the PO group (P < 0.01) and significantly decreased in the HDN intervention groups (P < 0.05) (Fig. 6B and C). The results of Oil Red O staining showed that compared with the CON group, the lipid droplets significantly increased in the PO group (P < 0.001) and significantly decreased in the HDN intervention groups (P < 0.05) (Fig. 6D and E), which was consistent with the results obtained for TC and TG. The above results suggest that HDN attenuated PO-induced AML12 lipid accumulation in vitro.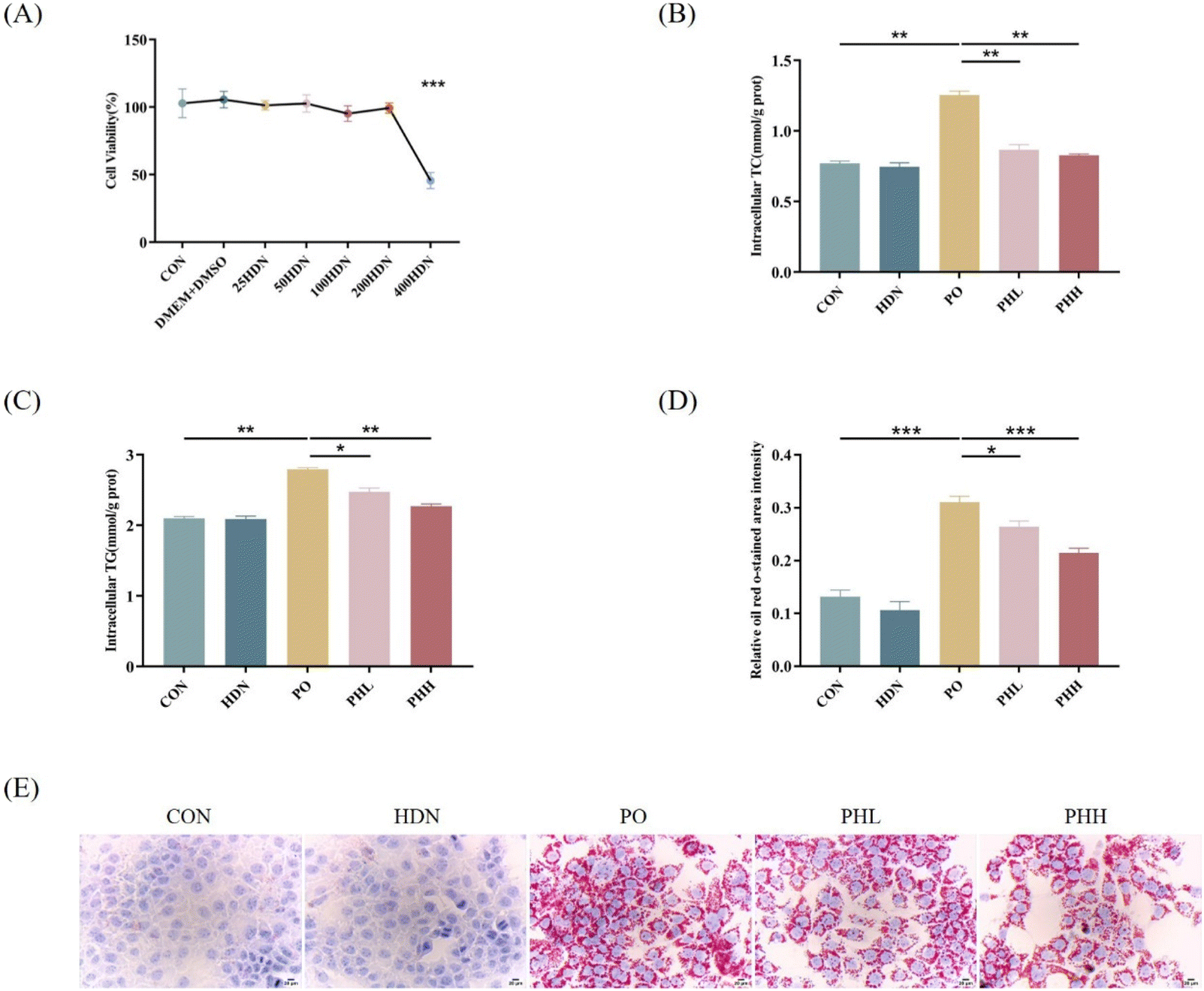 | ||
| Fig. 6 Effect of HDN on lipid accumulation in vitro. (A) Effect of different concentrations of HDN on the viability of AML12 cells. (B) Levels of TC in AML12 cells. (C) Levels of TG in AML12 cells. (D) Quantitative results of Oil Red O staining in AML12 cells. (E) Results of Oil Red O staining of AML12 cells. *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
3.7 HDN promoted the deacetylation of PGC1α by SIRT1
To confirm the specific interaction between SIRT1 and PGC1α and the effect of HDN on this interaction, we detected the binding of SIRT1 to PGC1α and the acetylation level of PGC1α using COIP and IF assays. To initially confirm whether there was a relationship between SIRT1 and PGC1α, we first analyzed the relationship between the interaction of SIRT1 and PGC1α by ZDOCK. According to the ZDOCK results, the Z-scores = 1429.480, and ARG-179, ARG-181, LYS-233, HIS-457, and PRO-456 of SIRT1, respectively, formed hydrogen bonds with TYR-292, GLU-241, TYR-244, TYR-283, and ASN-281 of PGC1α (Fig. 7D). The above results showed that SIRT1 was able to bind tightly to PGC1α and formed stable protein–protein complexes. The results of IF showed that the SIRT1 and PGC1α protein expression levels were significantly reduced after PO treatment, while HDN promoted SIRT1 and PGC1α expression. SIRT1 and PGC1α were strongly correlated (Fig. S1†), and HDN increased the co-localization of SIRT1 and PGC1α. (Fig. 7C). According to the COIP results, SIRT1 interacted directly with PGC1α (Fig. 7A); PO induced a significantly increased acetylation of PGC1α (P < 0.001), while HDN inhibited the increase in the acetylation of PGC1α induced by PO (P < 0.001) (Fig. 7B). In summary, according to the above results, we discovered that HDN promoted the interaction of SIRT1 with PGC1α and increased the deacetylation of PGC1α by SIRT1.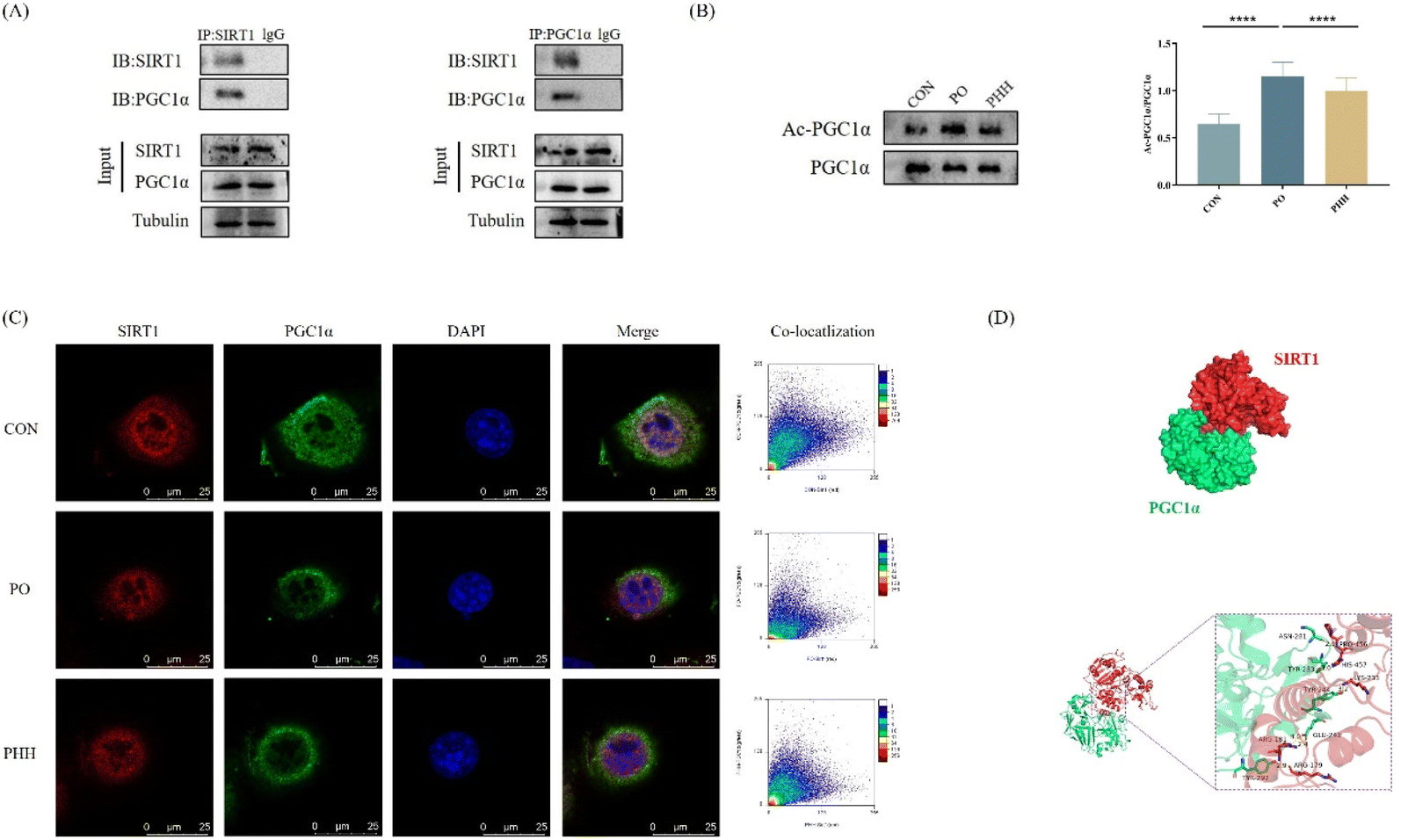 | ||
| Fig. 7 Effect of HDN on the interaction between SIRT1 and PGC1α. (A) COIP detected the interaction of SIRT1 with PGC1α. (B) COIP detected the degree of PGC1α acetylation and the ratio of acetylated PGC1α to total PGC1α. (C) Results of IF double-staining and the co-localization quantification of AML12 cells. SIRT1 (red), PGC1α (green), and DAPI (blue). (D) Protein–protein interaction analysis. ****P < 0.0001. | ||
3.8 HDN improved PO-induced lipid accumulation in AML12 cells via activating SIRT1
After successfully establishing the cellular high-fat model, we further explored the mechanism of HDN to improve lipid accumulation in vitro. According to the results, compared with the control group, the protein expression levels of SIRT1 and PGC1α were significantly decreased in the HFD group (P < 0.01) (Fig. 8A), and the IF results of SIRT1 showed the same changes (P < 0.001) (Fig. 8D); while the protein expression levels of CD36, SCD1, and FABP1 related to fatty acid transport synthesis were significantly increased (P < 0.001) (Fig. 8B); and the protein expression levels of PPARα, CPT1, ACOX1, and EHHADH related to fatty acid β-oxidation were significantly decreased (P < 0.05) (Fig. 8C). HDN intervention significantly reversed the expression of these proteins (P < 0.05).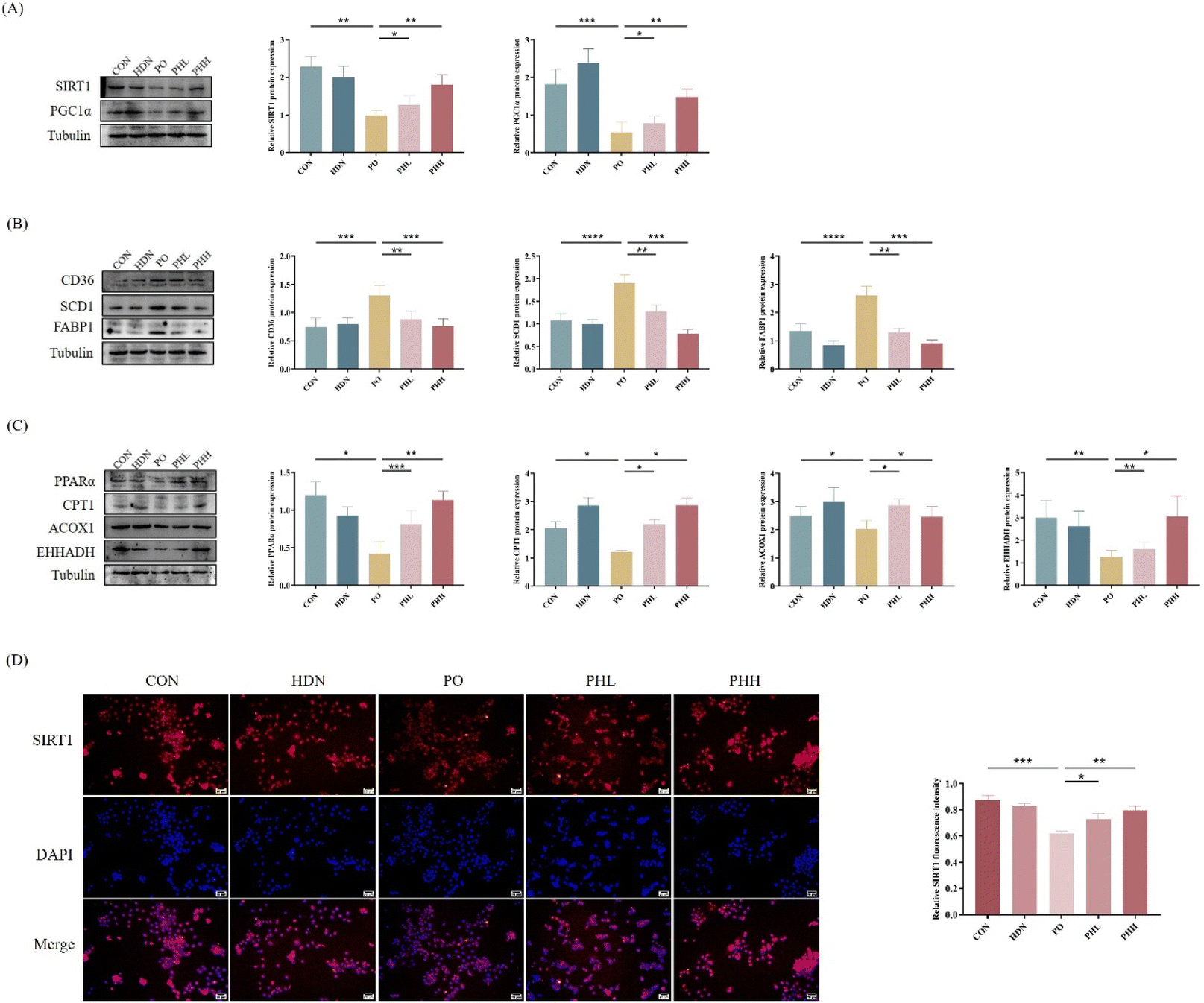 | ||
| Fig. 8 Effect of HDN on SIRT1/PGC1α in vitro. (A) Protein bands and the quantitative results of SIRT1 and PGC1α. (B) Protein bands and the quantitative results of CD36, SCD1, and FABP1. (C) Protein bands and the quantitative results of PPARα, CPT1, ACOX1, and EHHADH. (D) Immunofluorescence staining and the quantitative results of SIRT1. SIRT1 (red), DAPI (blue). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
In order to determine the targeting relationship between HDN and SIRT1, we performed CETSA and molecular docking. The CETSA results showed that compared with CON, when HDN intervened, the SIRT1 expression increased with increasing temperature, while molecular docking with the free energy of binding of −9.1 kcal mol−1 showed the binding energy was lower, indicating a good binding ability. These results suggest a targeting relationship between HDN and SIRT1 (Fig. 9A). To further determine that HDN regulated lipid accumulation in AML12 cells via activating SIRT1, we performed transfection experiments in vitro to knock down SIRT1 expression with a good transfection efficiency (Fig. S2†). After transfection, qRT-PCR and western blot were performed and the results showed that the mRNA and protein expression of SIRT1 were significantly decreased in the siSIRT1 group compared to the CON group (P < 0.05), and there was no significant difference between the siNC and the CON group (P > 0.05) (Fig. 9C and D). Subsequent experiments were performed after determining the successful knockdown of SIRT1. The obtained results showed that SIRT1 protein expression was significantly decreased in AML12 cells after PO and siSIRT1 treatment, and SIRT1 expression was further decreased when siSIRT1 and PO were treated together, and the SIRT1 IF results also showed the same changes (Fig. 8B); meanwhile, its downstream proteins PGC1α, PPARα, CPT1, ACOX1, and EHHADH expressions were significantly decreased (Fig. 9C and H), while CD36, SCD1, and FABP1 were significantly increased (Fig. 9G). SIRT1 expression was significantly increased and reversed the expression of its downstream proteins when HDN intervened. The above results suggest that HDN promoted fatty acid β-oxidation and improved lipid accumulation in AML12 cells in vitro by activating SIRT1.
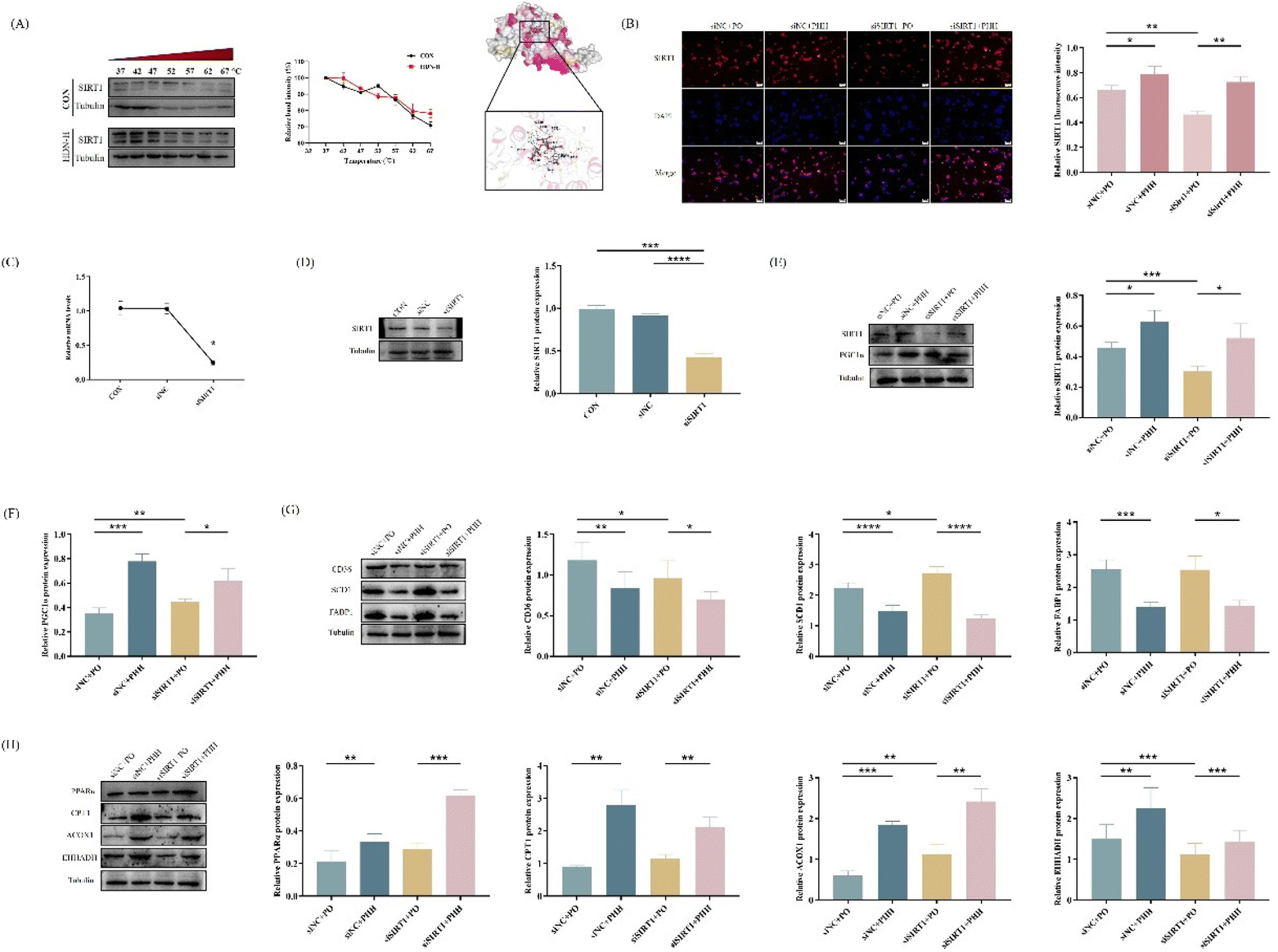 | ||
| Fig. 9 Effect of HDN on SIRT1/PGC1α after the knockdown of SIRT1. (A) HDN and SIRT1 molecular docking (binding free energy of −9.1 kcal mol−1) and CETSA experiments. (B) IF staining and the quantitative results of SIRT1 (C) QRT-PCR analysis of the mRNA expression of SIRT1. (D) Protein bands and the quantitative results of SIRT1 after transfection. (E and F) Protein bands and the quantitative results of SIRT1 and PGC1α. (G) Protein bands and the quantitative results of CD36, SCD1, and FABP1. (H) Protein bands and the quantitative results of PPARα, CPT1, ACOX1, and EHHADH. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. | ||
4. Discussion
NAFLD is a complex disease that begins with an abnormal accumulation of TG in the liver developing into liver injury, which leads to liver damage through multiple pathways, such as autophagy,11,34 mitochondrial stress,35 oxidative stress,36 and apoptosis.37 The pathogenesis of NAFLD is a complex and multifactorial process. NAFLD affects not only physiological changes in the liver but also the structural functions of other body systems, such as the heart,38 kidney,39 and cardiovascular system.40 NAFLD is a serious threat to life and health. There is no definite treatment for NAFLD. Therefore, it is important to find a safe and effective treatment drug. HDN is a natural phenolic compound with a wide range of biological effects, including anti-inflammatory,41 antioxidant,42 and anti-cancer43 properties. In this study, we investigated the mechanism by which HDN ameliorated lipid accumulation in nonalcoholic fatty liver by establishing a high-fat model in vitro and in vivo. The results showed that HDN ameliorated the HFD-induced reduction in SIRT1 expression, inhibited the acetylation of PGC1α, and promoted fatty acid β-oxidation.The inhibition of lipid accumulation in the liver is the key to improving NAFLD. In the present study, we found hepatocyte steatosis, hepatocellular enlargement, and cytoplasmic vacuolization in the high-fat-diet NAFLD mice, which is consistent with previous studies.44–46 In the in vitro experiments, AML12 cells were induced by PO with increased TC and TG contents and lipid droplet quantities. Our current data suggested that HDN improved the increase in blood lipids, liver lipids (TC, TG, HDL, LDL), and liver injury (AST, ALT) caused by a high-fat diet and reduced lipid accumulation in vivo and in vitro, as well as improved insulin resistance. These results show that HDN could effectively improve NAFLD induced by a high-fat diet.
As a result of analytical screening based on the results of LC-MS analysis, we found that taurochenodeoxycholic acid (sodium salt), glycoursodeoxycholic acid, and hypotaurine were significantly decreased and 9-KODE and acetoacetate were significantly increased in the high-fat-diet mice. It has been shown that taurochenodeoxycholic acid (sodium salt) is negatively correlated with PPARγ,47 which is known to be a major regulator of adipogenesis and insulin resistance,48 and that changes in the taurochenodeoxycholic acid (sodium salt) content are closely related to PPARγ, affecting adipogenesis and insulin resistance., and our results also showed that HFD affected glucose tolerance and insulin resistance, and could significantly increase the expression of PPARγ (Fig. S3A†); while glycoursodeoxycholic acid could improve HFD-induced insulin resistance and hepatic steatosis,49 and hypotaurine, a precursor of taurine, promoted fat metabolism and reduces fat accumulation.50 Further, 9-KODE is a derivative of linoleic acid; while acetoacetate stimulates triacylglycerol synthesis, and these changes in metabolites responding to lipid synthesis and metabolism are consistent with the embodiment of our present findings. When phospholipid metabolism is abnormal, it usually affects fat metabolism as well, and the two interact to play important roles. Metabolites related to phospholipid metabolism, such as LPC 16:1 and cytidine 5′-diphosphocholine, were significantly reduced and phospholipid metabolism was aberrant under the high-fat diet. HDN intervention reversed the expression of the above metabolites. In addition, compared with CON, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranosyl 5-monophosphate was increased in the high-fat-diet mice, and AICARiboside (an AMPK activator) was its precursor,51 and interestingly we found a significant decrease in AICARiboside from the results of the HFD vs. CON metabolite analysis, and HDN intervention slowed down AICAR and its metabolism. To investigate the effect of HDN on AMPK and the relationship between AMPK and SIRT1, we performed the in vivo detection of AMPK and SIRT1 expression by western blot, and both were found to be significantly reduced in the HFD group compared with CON, which was reversed after HDN intervention. We used AICAR and Compound C (AMPK inhibitor) in vitro and found that when AMPK was activated or inhibited, SIRT1 underwent the same changes (Fig. S4A, B and C†), suggesting that AMPK is the upstream protein of SIRT1 and is positively correlated with it. It has been shown that 13-OxoODE is an activator of PPARα52 and can affect PPARα expression; while nicotinamide affects lipid metabolism by increasing fatty acid β-oxidation activity.53 Here, 13-OxoODE and nicotinamide were significantly decreased in the HFD group, and both were significantly increased after HDN intervention. From the analysis of the LC-MS results, we concluded that HDN affects the fatty acid β-oxidation process and thus improves lipid metabolism.
Recently, SIRT1 has been identified as an important regulator of mitochondrial biogenesis54 and fatty acid oxidation,55 which regulates the mitochondrial state by targeting PGC1α interactions.56 It has been reported that changes in SIRT1/PGC1α expression enhance hepatic antioxidant capacity, and regulate oxidative stress and lipid peroxidation and thus improve NAFLD induced by a high-fat diet.57 Fatty acid β-oxidation is one of the important processes that occur in mitochondria involved in lipid metabolism.58 SIRT1 increases the transcriptional activity of PGC1α by deacetylating PGC1α.59 On the other hand, PGC1α binds and co-activates PPARα,60 which induces many genes that are important for fatty acid metabolism, including CPT1, ACOX1, EHHADH, CD36, SCD1, and FABP1. The results of this study showed that HDN promoted the deacetylation of PGC1α by SIRT1 and increased the expressions of PGC1α and PPARα. The interaction of SIRT1 with PGC1α was verified in AML12 cells by performing forward and reverse pull by COIP. When SIRT1 was knocked down, the SIRT1/PGC1α expression and fatty acid β-oxidation process were inhibited, showing that HDN improved NAFLD mainly through SIRT1/PGC1α. The above results suggest that HDN activates SIRT1 to increase the deacetylation of PGC1α as a key to regulating fatty acid β-oxidation.
The liver is one of the most active tissues for fatty acid oxidation,61 which includes β-oxidation, propionate oxidation, α-oxidation, ω-oxidation, and unsaturated fatty acid oxidation,62 with the most important being β-oxidation. Fatty acid β-oxidation is one of the main pathways of TG metabolism.63 PPARα is highly expressed in the liver and is a target protein that regulates the process of fatty acid β-oxidation,64 while hepatocyte-specific PPARα knockdown impairs fatty acid metabolic catabolism and leads to hepatic lipid accumulation, and PPARα is essential for systemic fatty acid homeostasis.65 CPT1 is the rate-limiting enzyme for fatty acids to enter the mitochondria for β-oxidation, which transports fatty acids to the mitochondria to participate in fatty acid β-oxidation,66 and then catalyzes the desaturation of acyl CoAs to 2-trans-enoyl-CoAs in the first step of β-oxidation under the action of ACOX1,67 while EHHADH mediates the dehydrogenation of hydroxy acyl CoAs to ketoacyl CoAs in the third step of β-oxidation.68 In addition, the change of PPARα expression level affects the expression level of the downstream protein CPT1,69 which in turn affects fatty acid transport into the mitochondria. Consequently, fatty acids that have already entered the cell cannot enter the β-oxidation process, causing a large number of fatty acids to be deposited in the cytoplasm. In this study in vitro and in vivo, we observed that the high-fat diet induced significantly decreased expression levels of PPARα, CPT1, ACOX1, and EHHADH, while the fatty acid uptake synthesis-related proteins CD36, SCD1, and FABP1 were significantly increased and also differentially regulated by PPARα. This phenomenon was reversed after HDN intervention. These results suggested that HDN promotes fatty acid β-oxidation processes, reduces fatty acid uptake and synthesis, and attenuates intracellular lipid accumulation.
5. Conclusions
In summary, our study found that the protective effect of HDN on NAFLD was related to fatty acid β-oxidation. HDN was concluded to improve hepatic fat accumulation and liver injury by activating SIRT1/PGC1α. Also, in the SIRT1/PGC1α process, SIRT1 interacts directly with PGC1α and promotes the deacetylation of PGC1α, which in turn promotes fatty acid β-oxidation. In addition, these results further revealed the mechanism of HDN in the prevention of NAFLD and provide new ideas for the prevention and treatment of NAFLD.Abbreviations
| NAFLD | Nonalcoholic fatty liver disease |
| HDN | Hesperidin |
| PA | Palmitic |
| OA | Oleic acid |
| PO | Palmitic and oleic acid complex |
| TC | Total cholesterol |
| TG | Triglycerides |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| AST | Aspartate transaminase |
| ALT | Alanine transaminase |
| PGC1α | Peroxisome proliferator-activated receptor gamma activator-1α |
| PPARα | Peroxisome proliferator-activated receptor α |
| SIRT1 | Silent mating-type information regulatory 2 homolog 1 |
| CD36 | Cluster of differentiation 36 |
| CPT1 | Carnitine palmitoyl transferase 1A |
| ACOX1 | Acyl coenzyme A Oxidase 1 |
| EHHADH | Enoyl-coenzyme A |
| SCD1 | Stomatal cytokinesis-defective 1 |
| FABP1 | Fatty acid binding protein 1 |
| COIP | Co-immunoprecipitation |
| IF | Immunofluorescence staining |
| IHC | Immunohistochemical staining |
| IPGTT | Intraperitoneal glucose tolerance |
| IPITT | Intraperitoneal insulin tolerance test |
| CETSA | Cellular thermal shift assay |
Author contributions
Tong Nie: data curation, formal analysis, writing; Xin Wang: data curation; Aqun Li: data curation; Anshan Shan: review; Jun Ma: review, visualization, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the Heilongjiang Postdoctoral Project under grant number LBH-Z21101. The authors sincerely appreciate the members of Veterinary Traditional Chinese Veterinary Laboratory, Key Laboratory of Pathogenic Mechanism for Animal Disease and Comparative Medicine, Northeast Agricultural University.References
- E. Cobbina and F. Akhlaghi, Non-alcoholic fatty liver disease (NAFLD) - pathogenesis, classification, and effect on drug metabolizing enzymes and transporters, Drug Metab. Rev., 2017, 49, 197–211 CrossRef CAS PubMed.
- L. H. S. Lau and S. H. Wong, Microbiota, Obesity and NAFLD, Adv. Exp. Med. Biol., 2018, 1061, 111–125 CrossRef CAS.
- D. Jahn, S. Kircher, H. M. Hermanns and A. Geier, Animal models of NAFLD from a hepatologist's point of view, Biochim. Biophys. Acta, Mol. Basis Dis., 2019, 1865, 943–953 CrossRef CAS PubMed.
- G. Targher, C. D. Byrne and H. Tilg, NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications, Gut, 2020, 69, 1691–1705 CrossRef CAS PubMed.
- S. Raza, S. Rajak, A. Upadhyay, A. Tewari and R. Anthony Sinha, Current treatment paradigms and emerging therapies for NAFLD/NASH, Front. Biosci., 2021, 26, 206–237 CrossRef CAS PubMed.
- J. Mun, S. Kim, H. G. Yoon, Y. You, O. K. Kim, K. C. Choi, Y. H. Lee, J. Lee, J. Park and W. Jun, Water Extract of Curcuma longa L. Ameliorates Non-Alcoholic Fatty Liver Disease, Nutrients, 2019, 11, 2536 CrossRef CAS PubMed.
- T. Yan, Y. Luo, N. Yan, K. Hamada, N. Zhao, Y. Xia, P. Wang, C. Zhao, D. Qi, S. Yang, L. Sun, J. Cai, Q. Wang, C. Jiang, O. Gavrilova, K. W. Krausz, D. P. Patel, X. Yu, X. Wu, H. Hao, W. Liu, A. Qu and F. J. Gonzalez, Intestinal peroxisome proliferator-activated receptor α-fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis, Hepatology, 2023, 77, 239–255 CrossRef PubMed.
- L. Barbier-Torres, K. A. Fortner, P. Iruzubieta, T. C. Delgado, E. Giddings, Y. Chen, D. Champagne, D. Fernandez-Ramos, D. Mestre, B. Gomez-Santos, M. Varela-Rey, V. G. de Juan, P. Fernandez-Tussy, I. Zubiete-Franco, C. Garcia-Monzon, A. Gonzalez-Rodriguez, D. Oza, F. Valenca-Pereira, Q. Fang, J. Crespo, P. Aspichueta, F. Tremblay, B. C. Christensen, J. Anguita, M. L. Martinez-Chantar and M. Rincon, Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation, Nat. Commun., 2020, 11, 3360 CrossRef CAS PubMed.
- J. M. Mato, C. Alonso, M. Noureddin and S. C. Lu, Biomarkers and subtypes of deranged lipid metabolism in non-alcoholic fatty liver disease, World J. Gastroenterol., 2019, 25, 3009–3020 CrossRef CAS PubMed.
- W. Zhu, M. Zhang, L. Chang, W. Zhu, C. Li, F. Xie, H. Zhang, T. Zhao and J. Jiang, Characterizing the composition, metabolism and physiological functions of the fatty liver in Rana omeimontis tadpoles, Front. Zool., 2019, 16, 42 CrossRef.
- S. Tanaka, H. Hikita, T. Tatsumi, R. Sakamori, Y. Nozaki, S. Sakane, Y. Shiode, T. Nakabori, Y. Saito, N. Hiramatsu, K. Tabata, T. Kawabata, M. Hamasaki, H. Eguchi, H. Nagano, T. Yoshimori and T. Takehara, Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice, Hepatology, 2016, 64, 1994–2014 CrossRef CAS PubMed.
- E. S. Lee, M. H. Kwon, H. M. Kim, H. B. Woo, C. M. Ahn and C. H. Chung, Curcumin analog CUR5-8 ameliorates nonalcoholic fatty liver disease in mice with high-fat diet-induced obesity, Metabolism, 2020, 103, 154015 CrossRef CAS PubMed.
- M. R. Lynch, M. T. Tran and S. M. Parikh, PGC1α in the kidney, Am. J. Physiol.: Renal Physiol., 2018, 314, F1–F8 CrossRef PubMed.
- L. Zhang, X. Wang, W. Yu, J. Ying, P. Fang, Q. Zheng, X. Feng, J. Hu, F. Xiao, S. Chen, G. Wei, Y. Lin, X. Liu, D. Yang, Y. Fang, G. Xu and F. Hua, CB2R Activation Regulates TFEB-Mediated Autophagy and Affects Lipid Metabolism and Inflammation of Astrocytes in POCD, Front. Immunol., 2022, 13, 836494 CrossRef CAS PubMed.
- C. Zhang, J. Deng, D. Liu, X. Tuo, L. Xiao, B. Lai, Q. Yao, J. Liu, H. Yang and N. Wang, Nuciferine ameliorates hepatic steatosis in high-fat diet/streptozocin-induced diabetic mice through a PPARα/PPARγ coactivator-1α pathway, Br. J. Pharmacol., 2018, 175, 4218–4228 CrossRef CAS PubMed.
- B. L. Tang, Sirt1 and the Mitochondria, Mol. Cells, 2016, 39, 87–95 CrossRef CAS PubMed.
- Y. Yang, Y. Liu, Y. Wang, Y. Chao, J. Zhang, Y. Jia, J. Tie and D. Hu, Regulation of SIRT1 and Its Roles in Inflammation, Front. Immunol., 2022, 13, 831168 CrossRef CAS PubMed.
- W. Dong, W. Quo, F. Wang, C. Li, Y. Xie, X. Zheng and H. Shi, Electroacupuncture Upregulates SIRT1-Dependent PGC-1α Expression in SAMP8 Mice, Med. Sci. Monit., 2015, 21, 3356–3362 CrossRef CAS PubMed.
- Z. H. Wang, X. G. Bao, J. J. Hu, S. B. Shen, G. H. Xu and Y. L. Wu, Nicotinamide Riboside Enhances Endothelial Precursor Cell Function to Promote Refractory Wound Healing Through Mediating the Sirt1/AMPK Pathway, Front. Pharmacol., 2021, 12, 671563 CrossRef CAS PubMed.
- C. Li and H. Schluesener, Health-promoting effects of the citrus flavanone hesperidin, Crit. Rev. Food Sci. Nutr., 2017, 57, 613–631 CrossRef CAS.
- M. Hajialyani, M. Hosein Farzaei, J. Echeverría, S. M. Nabavi, E. Uriarte and E. Sobarzo-Sánchez, Hesperidin as a Neuroprotective Agent: A Review of Animal and Clinical Evidence, Molecules, 2019, 24, 648 CrossRef CAS PubMed.
- A. Mas-Capdevila, J. Teichenne, C. Domenech-Coca, A. Caimari, J. M. Del Bas, X. Escote and A. Crescenti, Effect of Hesperidin on Cardiovascular Disease Risk Factors: The Role of Intestinal Microbiota on Hesperidin Bioavailability, Nutrients, 2020, 12, 1488 CrossRef CAS PubMed.
- R. Rezaee, A. Sheidary, S. Jangjoo, S. Ekhtiary, S. Bagheri, Z. Kohkan, M. Dadres, A. Oana Docea, K. Tsarouhas, D. A. Sarigiannis, S. Karakitsios, A. Tsatsakis, L. Kovatsi and M. Hashemzaei, Cardioprotective effects of hesperidin on carbon monoxide poisoned in rats, Drug Chem. Toxicol., 2021, 44, 668–673 CrossRef CAS PubMed.
- Z. Cui, X. Zhao, F. K. Amevor, X. Du, Y. Wang, D. Li, G. Shu, Y. Tian and X. Zhao, Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism, Front. Immunol., 2022, 13, 943321 CrossRef CAS PubMed.
- J. J. Lee, S. C. Ng, J. Y. Hsu, H. Liu, C. J. Chen, C. Y. Huang and W. W. Kuo, Galangin Reverses H(2)O(2)-Induced Dermal Fibroblast Senescence via SIRT1-PGC-1α/Nrf2 Signaling, Int. J. Mol. Sci., 2022, 23, 1387 CrossRef CAS PubMed.
- Y. Q. Hua, Y. Zeng, J. Xu and X. L. Xu, Naringenin alleviates nonalcoholic steatohepatitis in middle-aged Apoe(-/-)mice: role of SIRT1, Phytomedicine, 2021, 81, 153412 CrossRef CAS PubMed.
- Y. Jiang, D. Chen, Q. Gong, Q. Xu, D. Pan, F. Lu and Q. Tang, Elucidation of SIRT-1/PGC-1α-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease, Lipids Health Dis., 2021, 20, 40 CrossRef CAS PubMed.
- M. Raffaele, L. Bellner, S. P. Singh, G. Favero, R. Rezzani, L. F. Rodella, J. R. Falck, N. G. Abraham and L. Vanella, Epoxyeicosatrienoic intervention improves NAFLD in leptin receptor deficient mice by an increase in PGC1α-HO-1-PGC1α-mitochondrial signaling, Exp. Cell Res., 2019, 380, 180–187 CrossRef CAS PubMed.
- M. T. Swe, L. Thongnak, K. Jaikumkao, A. Pongchaidecha, V. Chatsudthipong and A. Lungkaphin, Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats, Clin. Sci., 2019, 133, 2415–2430 CrossRef CAS PubMed.
- T. Ren, L. Zhu, Y. Shen, Q. Mou, T. Lin and H. Feng, Protection of hepatocyte mitochondrial function by blueberry juice and probiotics via SIRT1 regulation in non-alcoholic fatty liver disease, Food Funct., 2019, 10, 1540–1551 RSC.
- Z. Derdak, K. A. Villegas, R. Harb, A. M. Wu, A. Sousa and J. R. Wands, Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease, J. Hepatol., 2013, 58, 785–791 CrossRef CAS PubMed.
- M. Cheraghpour, H. Imani, S. Ommi, S. M. Alavian, E. Karimi-Shahrbabak, M. Hedayati, Z. Yari and A. Hekmatdoost, Hesperidin improves hepatic steatosis, hepatic enzymes, and metabolic and inflammatory parameters in patients with nonalcoholic fatty liver disease: A randomized, placebo-controlled, double-blind clinical trial, Phytother. Res., 2019, 33, 2118–2125 CrossRef CAS PubMed.
- H. Chen, T. Nie, P. L. Zhang, J. Ma and A. S. Shan, Hesperidin attenuates hepatic lipid accumulation in mice fed high-fat diet and oleic acid induced HepG2 via AMPK activation, Life Sci., 2022, 296, 120428 CrossRef CAS PubMed.
- J. Chhimwal, A. Goel, M. Sukapaka, V. Patial and Y. Padwad, Phloretin mitigates oxidative injury, inflammation, and fibrogenic responses via restoration of autophagic flux in in vitro and preclinical models of NAFLD, J. Nutr. Biochem., 2022, 107, 109062 CrossRef CAS PubMed.
- T. Zhou, L. Chang, Y. Luo, Y. Zhou and J. Zhang, Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy, Redox Biol., 2019, 21, 101120 CrossRef CAS PubMed.
- A. Amirinejad, A. S. Totmaj, F. Mardali, A. Hekmatdoost, H. Emamat, M. Safa and F. Shidfar, Administration of hydro-alcoholic extract of spinach improves oxidative stress and inflammation in high-fat diet-induced NAFLD rats, BMC Complementary Med. Ther., 2021, 21, 221 CrossRef CAS PubMed.
- N. Nasiri-Ansari, C. Nikolopoulou, K. Papoutsi, I. Kyrou, C. S. Mantzoros, G. Kyriakopoulos, A. Chatzigeorgiou, V. Kalotychou, M. S. Randeva, K. Chatha, K. Kontzoglou, G. Kaltsas, A. G. Papavassiliou, H. S. Randeva and E. Kassi, Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE((-/-)) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis, Int. J. Mol. Sci., 2021, 22, 818 CrossRef CAS PubMed.
- E. P. Stahl, D. S. Dhindsa, S. K. Lee, P. B. Sandesara, N. P. Chalasani and L. S. Sperling, Nonalcoholic Fatty Liver Disease and the Heart: JACC State-of-the-Art Review, J. Am. Coll. Cardiol., 2019, 73, 948–963 CrossRef PubMed.
- C. D. Byrne and G. Targher, NAFLD as a driver of chronic kidney disease, J. Hepatol., 2020, 72, 785–801 CrossRef PubMed.
- P. Kasper, A. Martin, S. Lang, F. Kutting, T. Goeser, M. Demir and H. M. Steffen, NAFLD and cardiovascular diseases: a clinical review, Clin. Res. Cardiol., 2021, 110, 921–937 CrossRef PubMed.
- S. Tejada, S. Pinya, M. Martorell, X. Capo, J. A. Tur, A. Pons and A. Sureda, Potential Anti-inflammatory Effects of Hesperidin from the Genus Citrus, Curr. Med. Chem., 2018, 25, 4929–4945 CrossRef CAS PubMed.
- S. Ahmad, S. Mittal, R. Gulia, K. Alam, T. K. Saha, Z. Arif, K. A. Nafees, K. Al-Shaghdali and S. Ahmad, Therapeutic role of hesperidin in collagen-induced rheumatoid arthritis through antiglycation and antioxidant activities, Cell Biochem. Funct., 2022, 40, 473–480 CrossRef CAS PubMed.
- V. Aggarwal, H. S. Tuli, F. Thakral, P. Singhal, D. Aggarwal, S. Srivastava, A. Pandey, K. Sak, M. Varol, M. A. Khan and G. Sethi, Molecular mechanisms of action of hesperidin in cancer: Recent trends and advancements, Exp. Biol. Med., 2020, 245, 486–497 CrossRef CAS PubMed.
- L. Li, J. Fu, D. Liu, J. Sun, Y. Hou, C. Chen, J. Shao, L. Wang, X. Wang, R. Zhao, H. Wang, M. E. Andersen, Q. Zhang, Y. Xu and J. Pi, Hepatocyte-specific Nrf2 deficiency mitigates high-fat diet-induced hepatic steatosis: Involvement of reduced PPARγ expression, Redox Biol., 2020, 30, 101412 CrossRef CAS PubMed.
- G. H. Lee, C. Peng, S. A. Park, T. H. Hoang, H. Y. Lee, J. Kim, S. I. Kang, C. H. Lee, J. S. Lee and H. J. Chae, Citrus Peel Extract Ameliorates High-Fat Diet-Induced NAFLD via Activation of AMPK Signaling, Nutrients, 2020, 12, 673 CrossRef CAS PubMed.
- G. Zandani, S. Anavi-Cohen, T. Yudelevich, A. Nyska, N. Dudai, Z. Madar and J. Gorelick, Chiliadenus iphionoides Reduces Body Weight and Improves Parameters Related to Hepatic Lipid and Glucose Metabolism in a High-Fat-Diet-Induced Mice Model of NAFLD, Nutrients, 2022, 14, 4552 CrossRef CAS PubMed.
- M. B. Jimenez-Castro, M. Elias-Miro, M. Mendes-Braz, A. Lemoine, A. Rimola, J. Rodes, A. Casillas-Ramirez and C. Peralta, Tauroursodeoxycholic acid affects PPARgamma and TLR4 in Steatotic liver transplantation, Am. J. Transplant., 2012, 12, 3257–3271 CrossRef CAS PubMed.
- T. Garin-Shkolnik, A. Rudich, G. S. Hotamisligil and M. Rubinstein, FABP4 attenuates PPARγ and adipogenesis and is inversely correlated with PPARγ in adipose tissues, Diabetes, 2014, 63, 900–911 CrossRef CAS PubMed.
- L. Cheng, T. Chen, M. Guo, P. Liu, X. Qiao, Y. Wei, J. She, B. Li, W. Xi, J. Zhou, Z. Yuan, Y. Wu and J. Liu, Glycoursodeoxycholic acid ameliorates diet-induced metabolic disorders with inhibiting endoplasmic reticulum stress, Clin. Sci., 2021, 135, 1689–1706 CrossRef CAS PubMed.
- M. Chen, F. Bai, T. Song, X. Niu, X. Wang, K. Wang and J. Ye, Hepatic Transcriptome Analysis Provides New Insight into the Lipid-Reducing Effect of Dietary Taurine in High-Fat Fed Groupers (Epinephelus coioides), Metabolites, 2022, 12, 670 CrossRef CAS PubMed.
- S. Pirkmajer, S. S. Kulkarni, R. Z. Tom, F. A. Ross, S. A. Hawley, D. G. Hardie, J. R. Zierath and A. V. Chibalin, Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation, Diabetes, 2015, 64, 360–369 CrossRef CAS PubMed.
- M. M. Heintz, J. A. Eccles, E. M. Olack, K. M. Maner-Smith, E. A. Ortlund and W. S. Baldwin, Human CYP2B6 produces oxylipins from polyunsaturated fatty acids and reduces diet-induced obesity, PLoS One, 2022, 17, e0277053 CrossRef CAS PubMed.
- M. Jung, K. M. Lee, Y. Im, S. H. Seok, H. Chung, D. Y. Kim, D. Han, C. H. Lee, E. H. Hwang, S. Y. Park, J. Koh, B. Kim, I. P. Nikas, H. Lee, D. Hwang and H. S. Ryu, Nicotinamide (niacin) supplement increases lipid metabolism and ROS-induced energy disruption in triple-negative breast cancer: potential for drug repositioning as an anti-tumor agent, Mol. Oncol., 2022, 16, 1795–1815 CrossRef CAS PubMed.
- Q. Zhao, Z. Tian, G. Zhou, Q. Niu, J. Chen, P. Li, L. Dong, T. Xia, S. Zhang and A. Wang, SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride, Theranostics, 2020, 10, 4822–4838 CrossRef CAS PubMed.
- A. M. Fritzen, A. M. Lundsgaard and B. Kiens, Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise, Nat. Rev. Endocrinol., 2020, 16, 683–696 CrossRef CAS PubMed.
- R. B. Ding, J. Bao and C. X. Deng, Emerging roles of SIRT1 in fatty liver diseases, Int. J. Biol. Sci., 2017, 13, 852–867 CrossRef CAS PubMed.
- Z. Y. Chen, J. S. Li, J. P. Jiang, M. X. Yan and B. H. He, [Effect of pure total flavonoids from citrus on hepatic SIRT1/PGC-1alpha pathway in mice with NASH], Zhongguo Zhongyao Zazhi, 2014, 39, 100–105 CAS.
- P. Rinaldo, D. Matern and M. J. Bennett, Fatty acid oxidation disorders, Annu. Rev. Physiol., 2002, 64, 477–502 CrossRef CAS PubMed.
- W. Hang, H. Shu, Z. Wen, J. Liu, Z. Jin, Z. Shi, C. Chen and D. W. Wang, N-Acetyl Cysteine Ameliorates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease and Intracellular Triglyceride Accumulation by Preserving Mitochondrial Function, Front. Pharmacol., 2021, 12, 636204 CrossRef CAS PubMed.
- Z. Chen, J. Luo, S. Sun, D. Cao, H. Shi and J. J. Loor, miR-148a and miR-17-5p synergistically regulate milk TAG synthesis via PPARGC1A and PPARA in goat mammary epithelial cells, RNA Biol., 2017, 14, 326–338 CrossRef PubMed.
- P. Nguyen, V. Leray, M. Diez, S. Serisier, J. Le Bloc'h, B. Siliart and H. Dumon, Liver lipid metabolism, J. Anim. Physiol. Anim. Nutr., 2008, 92, 272–283 CrossRef CAS PubMed.
- R. J. Wanders, J. Komen and S. Kemp, Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans, FEBS J., 2011, 278, 182–194 CrossRef CAS PubMed.
- S. Wang, N. Moustaid-Moussa, L. Chen, H. Mo, A. Shastri, R. Su, P. Bapat, I. Kwun and C. L. Shen, Novel insights of dietary polyphenols and obesity, J. Nutr. Biochem., 2014, 25, 1–18 CrossRef CAS PubMed.
- R. G. R. Mooli, J. Rodriguez, S. Takahashi, S. Solanki, F. J. Gonzalez, S. K. Ramakrishnan and Y. M. Shah, Hypoxia via ERK Signaling Inhibits Hepatic PPARα to Promote Fatty Liver, Cell. Mol. Gastroenterol. Hepatol., 2021, 12, 585–597 CrossRef CAS PubMed.
- A. Montagner, A. Polizzi, E. Fouche, S. Ducheix, Y. Lippi, F. Lasserre, V. Barquissau, M. Regnier, C. Lukowicz, F. Benhamed, A. Iroz, J. Bertrand-Michel, T. Al Saati, P. Cano, L. Mselli-Lakhal, G. Mithieux, F. Rajas, S. Lagarrigue, T. Pineau, N. Loiseau, C. Postic, D. Langin, W. Wahli and H. Guillou, Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD, Gut, 2016, 65, 1202–1214 CrossRef CAS PubMed.
- D. Sebastian, M. Guitart, C. Garcia-Martinez, C. Mauvezin, J. M. Orellana-Gavalda, D. Serra, A. M. Gomez-Foix, F. G. Hegardt and G. Asins, Novel role of FATP1 in mitochondrial fatty acid oxidation in skeletal muscle cells, J. Lipid Res., 2009, 50, 1789–1799 CrossRef CAS PubMed.
- A. He, X. Chen, M. Tan, Y. Chen, D. Lu, X. Zhang, J. M. Dean, B. Razani and I. J. Lodhi, Acetyl-CoA Derived from Hepatic Peroxisomal β-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation, Mol. Cell, 2020, 79, 30–42 CrossRef CAS PubMed.
- S. M. Houten, S. Denis, C. A. Argmann, Y. Jia, S. Ferdinandusse, J. K. Reddy and R. J. Wanders, Peroxisomal L-bifunctional enzyme (Ehhadh) is essential for the production of medium-chain dicarboxylic acids, J. Lipid Res., 2012, 53, 1296–1303 CrossRef CAS PubMed.
- Y. F. Zhang, Z. Q. Yuan, D. G. Song, X. H. Zhou and Y. Z. Wang, Effects of cannabinoid receptor 1 (brain) on lipid accumulation by transcriptional control of CPT1A and CPT1B, Anim. Genet., 2014, 45, 38–47 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fo04348g |
| This journal is © The Royal Society of Chemistry 2024 |