
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtic ionic liquids for sustainable uses
Josh
Bailey
 ,
Emily L.
Byrne
,
Peter
Goodrich
,
Paul
Kavanagh
and
Małgorzata
Swadźba-Kwaśny
*
,
Emily L.
Byrne
,
Peter
Goodrich
,
Paul
Kavanagh
and
Małgorzata
Swadźba-Kwaśny
*
The QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK. E-mail: m.swadzba-kwasny@qub.ac.uk
First published on 8th December 2023
Abstract
This review provides an overview of the current state-of-the-art and major trends in the application of protic ionic liquids (PILs) to sustainable chemistry. Following a brief description of the distinguishing properties of PILs, there are four application areas reviewed: acid catalysis, biomass transformations, energy storage and conversion, and electrocatalysis. The aim of this contribution is to showcase applications in which the properties of PILs are the key enabling factor for a particular sustainable chemistry challenge. In addition, the challenges and future directions in sustainable applications of PILs are discussed, highlighting challenges as well as areas for future development.
Introduction
Protic ionic liquids (PILs) are typically prepared by the neutralisation of a Lewis base with a Brønsted acid, which leads to the formation of a cation (protonated Lewis base) and an anion (conjugate base of the Brønsted acid), as shown in eqn (1). PILs are often described in contrast to “conventional” aprotic ionic liquids (AILs), in which the cation is generated by alkylation, rather than by proton transfer.| HA + B ⇄ [HB]+ + [A]− | (1) |
An excellent, in-depth introduction to the structure, properties, and applications of PILs can be found in comprehensive reviews published in 2008 and 2015 by Greaves and Drummond.1,2 They are complemented by a historical perspective comparing the development of PILs and AILs, published in 2007 by an early pioneer of PIL studies, C. Austen Angell, and his co-workers.3 Further to these, a number of more recent reviews discuss specific aspects of PILs, from fundamental insights into dielectric properties4 and NMR spectroscopic studies,5 to applications in proton exchange membranes,6,7 and fuel cell electrolytes.8,9 These and other review papers are signposted in appropriate sections discussing the applications of PILs.
In this contribution, we have set out to review growing areas of applications of PILs within the past decade (2013–2023), in the context of Green Chemistry, that is – PILs for sustainable uses. This calls for an introductory discussion of two aspects: the definition of “sustainable uses”, and a brief overview of the key properties that distinguish PILs as a group.
The question of sustainable use
The ionic liquids (ILs) community has long dispensed with the notion that ILs are intrinsically green and/or sustainable.10 In the case of PILs, even the argument of negligible volatility, often presented as the key advantage of ILs over volatile organic solvents, does not hold true, as discussed further in this review. However, as pointed out by Greer, Jacquemin, and Hardacre in their review on the industrial applications of ILs,11 when implemented correctly, ILs have demonstrated the potential to improve the green metrics of processes in terms of both environmental and economical sustainability.In this review, we have focussed on applications of PILs that meet three key criteria. Firstly, they must enable an aspect of chemistry that directly delivers on one of the Sustainable Development Goals (SDGs).12 Secondly, the nature of PILs must be the key enabling factor; either the same chemistry cannot be achieved in water or common organic solvents, or the use of a PIL offers a well-demonstrated advantage in terms of sustainable chemistry. Finally, true sustainability must be realised across green metrics,13 considering people (societal impact), planet (environmental impact) and profit (the cost of the process in question, which does not equate to high/low cost of the PIL itself).11 It would have been ideal to refer to specific green metrics, such as E-factor, process mass intensity or life cycle analysis, but unfortunately, these metrics are not routinely included in publications on PILs.
With these considerations in mind, we have identified four areas of applications: (1) catalytic transformations, with the focus on esterifications and CO2 activation; (2) biomass treatment, centred on lignocellulosic biomass, but touching upon marine sources; (3) energy storage and conversion, which is the most extensive part of this review, spanning various types of batteries, supercapacitors and fuel cells; and finally (4) electrocatalysis, relatively brief but expected to grow rapidly in the upcoming years. In all these, the nature of PILs is the key enabling factor, enhancing an aspect of sustainable chemistry. Within each section, we have provided critique of the current state-of-the-art, along with predictions of future developments and areas that currently appear to be underexplored.
Definition of protic ionic liquids
PILs are formed by proton transfer, upon neutralisation of a Lewis base with a Brønsted acid (eqn (1)). This has two consequences: (1) most PILs are easy and inexpensive to synthesise; (2) their physico-chemical properties are dictated by the presence of labile protons, a dense network of hydrogen bonds, and often incomplete proton transfer.Typical PILs are synthesised by the addition of a Brønsted acid to a Lewis base, which generates heat. When using weak acids, such as carboxylic acids, PILs are often synthesised at ambient temperature under solventless conditions.14 Solventless synthesis of PILs based on strong acids may be achieved using a specialised set-up, such as that developed by Martinelli and co-workers,15 and/or ice-bath cooling,15,16 to prevent evaporation or thermal decomposition of the base due to the exothermic reaction; Angell, Davidowski and co-workers suggest the use of an ice bath for acids weaker than sulfuric acid and an acetone/dry ice bath for those that are stronger.17 In many instances, however, at least one component tends to be dissolved in water, or an organic solvent, to manage the exothermic effect.18 The reaction can be stoichiometric, or with base in excess; in the latter case, base is removed in the drying step.19,20 The removal of solvent, residual water, and/or base is usually carried out under reduced pressure, but some authors advise freeze-drying to avoid unintentional removal of the basic component (and leaving an excess of acid).18,21
This simple synthetic procedure yields ILs that are potentially very cheap, as exemplified by ILs based on sulfuric acid. Their price can be on par with common organic solvents, and their properties are vastly tuneable by altering amines and stoichiometry.22–25 These advantages have inspired a rapidly growing body of research on acid catalysis and biomass processing using these PILs. These applications, critically compared to extant technologies, are discussed in the respective sections of this review.
In electrochemical applications, minute imbalances in stoichiometry have a major impact on electrolyte performance, which invites greater care in their synthesis. Walsh and co-workers, who developed an electroanalytical method to study molecular impurities in PILs based on trifluoromethanesulfonic acid (triflic acid, HOTf), have stressed that whereas excess base can be easily removed under reduced pressure, it is impossible to remove excess acid from a PIL.19 Furthermore, they emphasised that a minute excess of acid has a major influence on the performance of a PIL. This may necessitate more careful preparatory techniques and more involved analysis to ensure sufficient purity of each batch. Furthermore, electrochemical applications tend to require more costly acids, such as triflic acid or bis(trifluoromethane)sulfonimide (bistriflimide acid, HTFSI), and sometimes more expensive bases, such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). All these factors will inevitably increase the price tag.
Another category to be considered in this review, distinct in terms of their syntheses, are ILs that are essentially aprotic (they are synthesised through quaternisation, not proton transfer) but act as “latent PILs”; that is, under certain conditions they display “protic behaviour” through the equilibrium shown in eqn (1).26
A borderline case between protic and aprotic ILs are alkylsulfonic-acid-functionalised ILs. Although Greaves and Drummond were hesitant to include them within their reviews,2 they in fact fit within the definition of PILs in that they are synthesised through acid–base reaction, here involving a zwitterion acting as a base combined with a Brønsted acid (Fig. 1b). However, this step is preceded by alkylation with a sulfone (Fig. 1a), an expensive alkylating agent, which leads to the relatively high cost of these PILs. Furthermore, it must be noted that the acid–base reaction (Fig. 1b) does not generate a cationic centre, in contrast to eqn (1). Considering the immense popularity of alkylsulfonic-acid-functionalised ILs in Brønsted acid catalysis, the authors have included them in this review, with appropriate commentary.
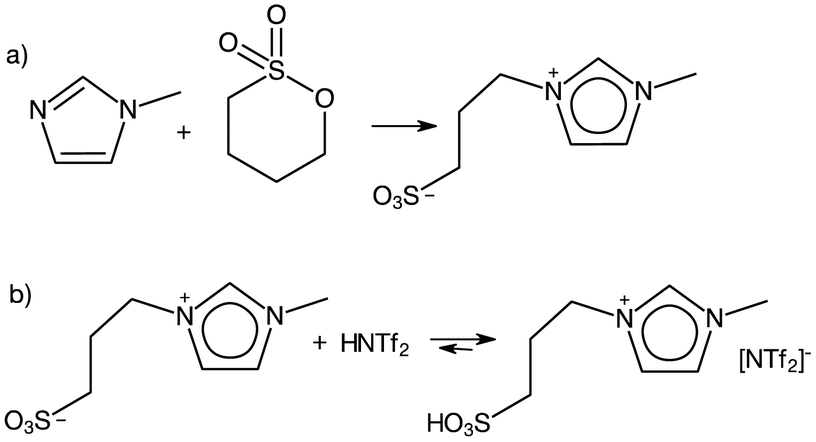 | ||
| Fig. 1 Synthesis of ionic liquids with alkylsulfonic-acid-functionalised cations. | ||
An even more “controversial” case are aprotic ILs synthesised via the standard procedure for AILs: quaternisation followed by anion exchange, that under certain conditions act as PILs, displaying acid–base equilibrium (eqn (1)).26 This is exemplified by 1-ethyl-3-methylimidazolium acetate, [C1C2im][OAc] (Fig. 2a) and trihexyl(tetradecyl)phosphonium bistriflimide, [P666,14][OAc] (Fig. 2b).
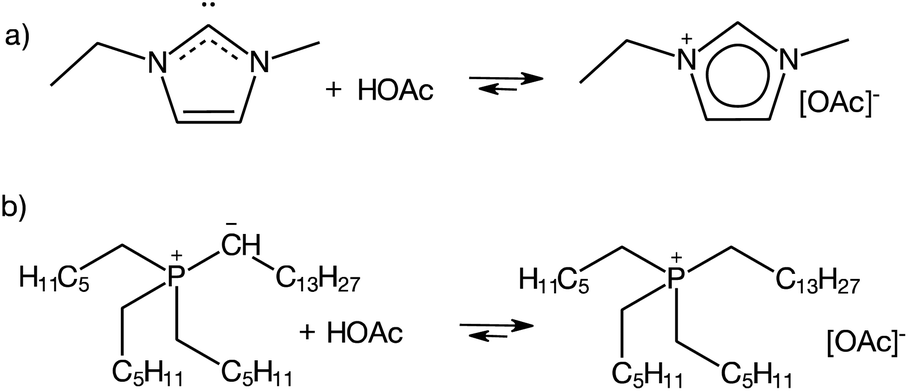 | ||
| Fig. 2 Examples of protic behaviour in aprotic ILs.26–28 | ||
Imidazolium-based ILs combined with basic anions, such as acetate, are a well-known source of N-heterocyclic carbenes (Fig. 2a).27,28 Although their detection remains elusive due to strongly right-shifted equilibrium constants,29 carbenes generated in situ have been used in transition metal catalysis,30 organocatalysis,31 as well as synthesis and stabilisation of nanoparticles.32 In phosphonium ILs, basic anions have been reported to “induce protic behaviour”,26 leading to ylide formation, which has been studied in-depth by Brennecke and co-workers in the context of CO2 capture.33–36 Under certain conditions, discussed elsewhere in this review, even AILs with a very weakly basic [eFAP]− anion have been found to exhibit protic-like behaviour, through ylide formation (Fig. 2b), an important step in the electrocatalytic activation of nitrogen.37
In this review, we have been guided by the evidence for a labile proton (eqn (1)) and relevance to recent developments in sustainable uses, rather than limited by the traditional view of PILs and stringent definitions, although the focus remains on conventional PILs.
Key fundamental aspects protic ionic liquids
Physical properties of PILs, such as viscosity, density and conductivity, as well as structure–property relationships found in PILs are discussed in great depth and detail by Greaves and Drummond.1,2 Here, we provide a brief overview of the key physico-chemical aspects of PILs that directly impact their performance in sustainable applications. These are: degree of proton transfer; acid-to-base ratio; and the nature of PILs doped with small molecules (water or organic solvents).In a recent paper, Mariani, Passerini, and co-workers43 point out a distinction between ionicity and proton transfer. Proton transfer is concerned with the number of ions in the system, as per the definition by MacFarlane and Seddon.38 Ionicity, on the other hand, is concerned with the number of free charge carriers, and will be decreased by – for example – close-contact ion pairing. Several studies have suggested that the relationship between ΔpKa is arbitrary and other parameters, such as proton affinity44 and Laity resistance coefficients,45 may be better descriptors of ionicity.
A very high degree of proton transfer results in PILs most similar to aprotic ILs (viz. negligible vapour pressure). However, the highest degree of proton transfer is not synonymous with the best performance in each application. For example, in a study of ILs based on 1-methylimidazole and one of four acids: HTFSI, HOTf, H2SO4 and H3PO4, it was found that the frequency of cation–anion proton exchange was inversely proportional to degree of proton transfer (Fig. 3).46 The high frequency proton exchange lowered the H+ concentration gradient, thereby reducing potential loss at the electrode, making PILs derived from medium-strength acids, such as H3PO4, the optimum choice for electrolytes in fuel cells and electrolysers.
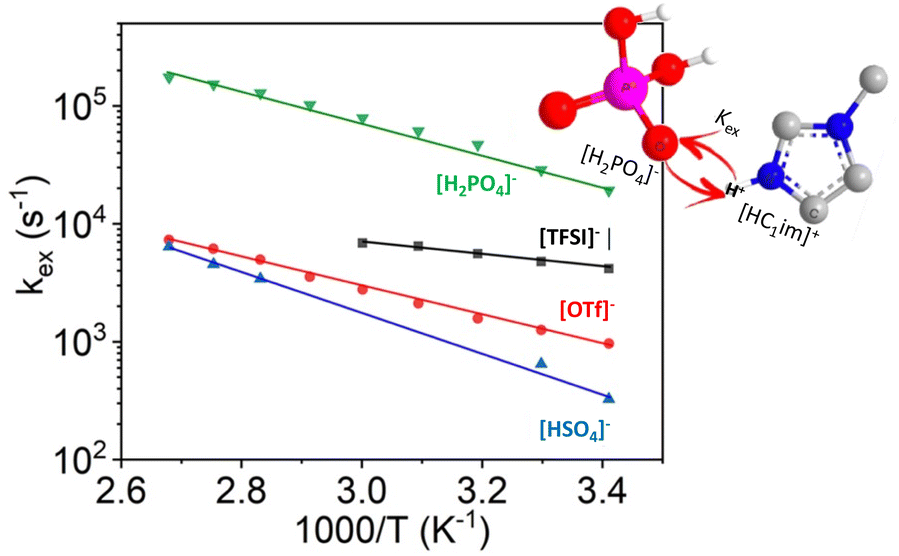 | ||
| Fig. 3 Arrhenius plot of the proton exchange rate in PILs based on the methylimidazolium cation and a range of anions, adapted from ref. 46 with permission from American Chemical Society, copyright 2023. | ||
Mixtures of amines and carboxylic acids are often characterised by very limited proton transfer; Umebayashi and co-workers described such system (a mixture of 1-methylimidazole and acetic acid) as a “conductive liquid with no ions” and named it a “pseudo-protic ionic liquid”.47 Such PILs became an object of many curiosity-driven experimental48–50 and computational51–54 studies on the mechanism of conductivity (Fig. 4).
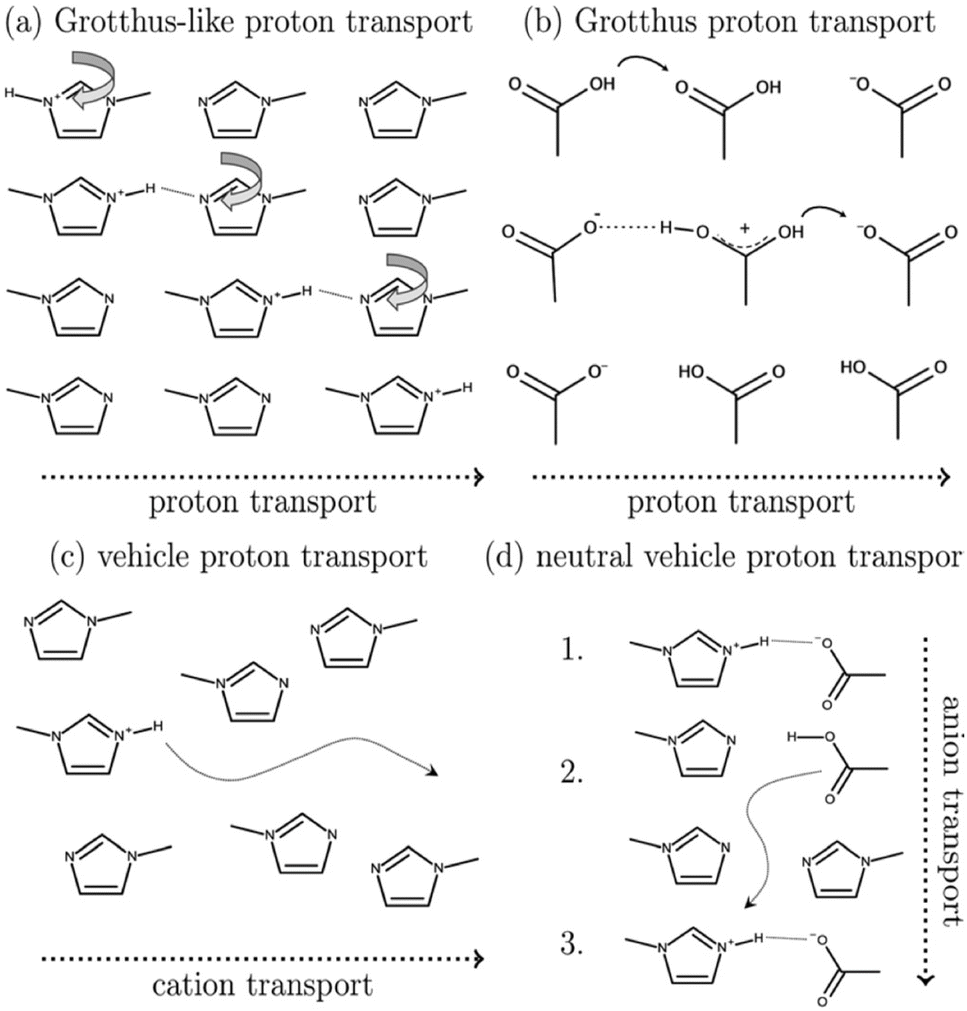 | ||
| Fig. 4 Schematic showing four potential charge-transport mechanisms in PILs. Reproduced from ref. 51 with permission from Royal Society of Chemistry, copyright 2023. | ||
Carboxylic-acid-based PILs have relatively low thermal stabilities, but this drawback is counterbalanced by very low cost, enhanced biodegradability, and safety, which are appealing factors when economic sustainability is considered.
Even low-ionicity PILs, with mostly un-ionised components, have properties distinctly different from those estimated by linear addition. Joerg and Schröder used polarisable molecular dynamics (MD) simulation to study “1-methylimidazolium acetate”, which contained ca. 30% ions.53 The dielectric spectrum of this PIL could not be reproduced by linear combination of spectra for isolated ionic and neutral species, indicating “strong correlations between all species, altering their individual dynamic properties”. Reid et al. measured Kamlet–Taft solvatochromic parameters for acetic acid and N,N-dimethylethanolamine, across compositional range.55 The studied mixtures had β-hydrogen bond basicities varying across a wide range, both above and below the values measured for the starting materials, demonstrating the value of exploring such PILs as tuneable solvents.
Although this recently growing interest stems from PILs, the study of such systems is a very old and well-researched subject, vastly predating the term “ionic liquids”. For example, an extensive publication on the physico-chemical studies of acetic acid-pyridine mixtures, across their full compositional range, was presented by Venkatesan and Suryanarayana in 1956.59 The authors reported significant departure from ideal mixing behaviour, incomplete proton transfer, and maxima in conductivity and volume contractions (indicators of maximal ionicity) at molar ratios of the acid of about χHOAc = 0.83 (Fig. 5). In the same year, Barrow used IR spectroscopy to study protonation of pyridine by carboxylic acidss (in chloroform).60
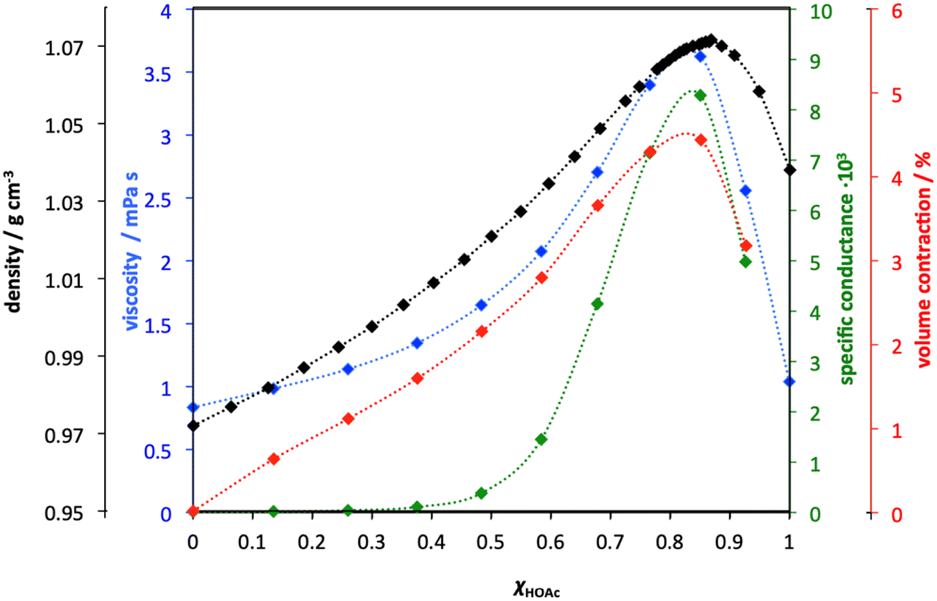 | ||
| Fig. 5 A graphical representation of all physicochemical properties of pyridine-acetic acid mixtures as a function of molar ratio of the acid, χHOAc, known in 1956. Adapted from ref. 61. | ||
These results, although decades old, have lost nothing of their validity and are convergent with trends reported in recent papers on PILs.50,56,57,62 It is therefore very valuable to screen older literature on acid–base mixtures.
The nature of the acid or base dissolved in stoichiometric PILs, leading to non-stoichiometric compositions, has been a matter of some debate. A full description of the liquid structure of all stoichiometric and non-stoichiometric PILs is far beyond the scope of this review. Here, we focus on acid-rich systems with no long alkyl chains, as featuring most prominently in sustainable applications. In some studies, the formation of discrete anionic or cationic clusters, such as [Hx−1(OAc)x]−,62 or [(HSO4)·xH2SO4]−,16 (Fig. 6) has been suggested, while others have proposed a three-dimensional, hydrogen-bonded network with no discrete clustering.63,64
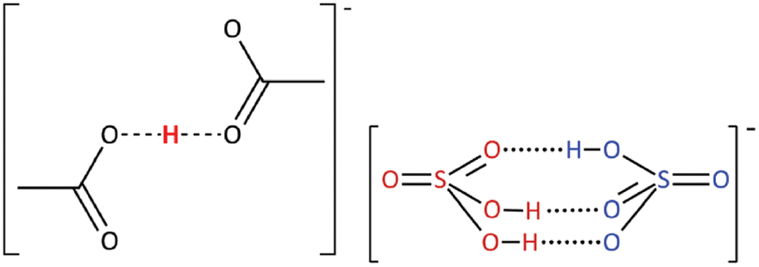 | ||
| Fig. 6 The postulated structure of anionic clusters suggested to be present in PILs with an excess of acid. Reproduced from ref. 16 with permission from Royal Society of Chemistry, copyright 2023. | ||
These clusters have been proposed as a visual representation of hydrogen-bonding motifs observed through vibrational spectroscopy,16 and to justify increased protonation of bases at higher acid concentrations: [(OAc)·xHOAc]− is a weaker conjugate base than [OAc]−, therefore it is easier to protonate a base at four-fold excess of acid – even though the pKa of the acid remains the same (Fig. 5).65
In contrast, neutron scattering studies of non-stoichiometric PILs, which allow for the probing of long-range liquid structure, demonstrate that long-lived discrete clusters are not observed, but 3D networks of rich hydrogen bonds, found in the parent acids, were retained in PILs.63,64 In pyridine – sulfuric acid PIL (χH2SO4 = 0.67), there was a 3D network of [HSO4]− anions and H2SO4 acid molecules, with pyridinium cations also hydrogen-bonded to these networks, but residing in “holes” (Fig. 7).64 In the pseudo-protic pyridine–acetic acid system, the equimolar composition (χHOAc = 0.50) contained exclusively a network of hydrogen-bonded acid and pyridine sitting in the “holes”; only χHOAc = 0.75 showed a small degree of protonation, with acetate anions and charge-neutral acid molecules in a strongly hydrogen-bonded network.63
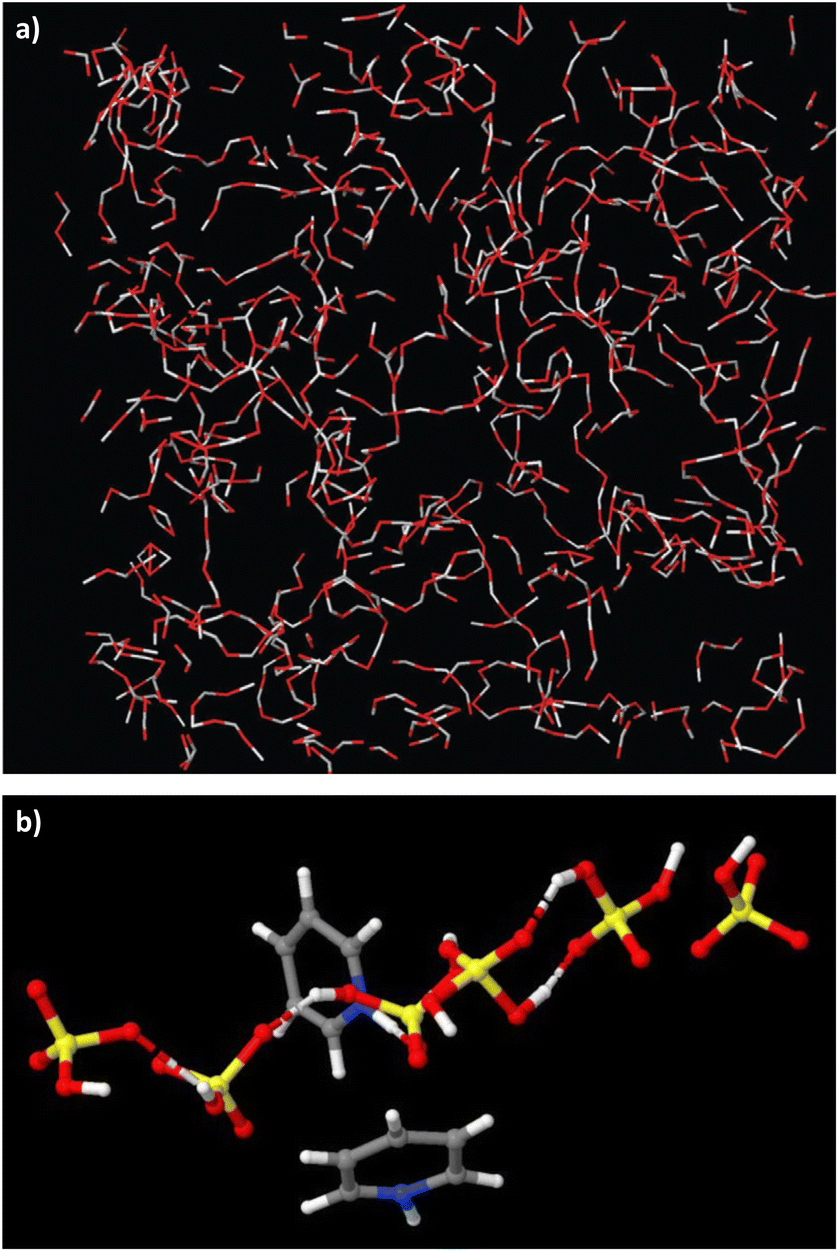 | ||
| Fig. 7 Hydrogen-bonded chains of acids and their conjugate bases in two PILs: (a) pyridine–acetic acid (χHOAc = 0.67), reproduced from ref. 63 with permission from Royal Society of Chemistry, copyright 2023. And pyridine–sulfuric acid (χH2SO4 = 0.67) – unpublished image from our neutron scattering studies of this system.64 | ||
We suggest that these two descriptions: clusters vs. 3D networks are not contradictory, but complementary. Clusters shown in Fig. 6 are transient, fragmentary representations of dominant hydrogen-bonding motifs (Fig. 8) that together constitute the long-range 3D liquid structure. What is important from the applications viewpoint is that each acid–base composition is a distinctive PIL, with a unique set of properties, which can be fine-tuned to suit an application, adding an additional degree of freedom in the design of ILs.55
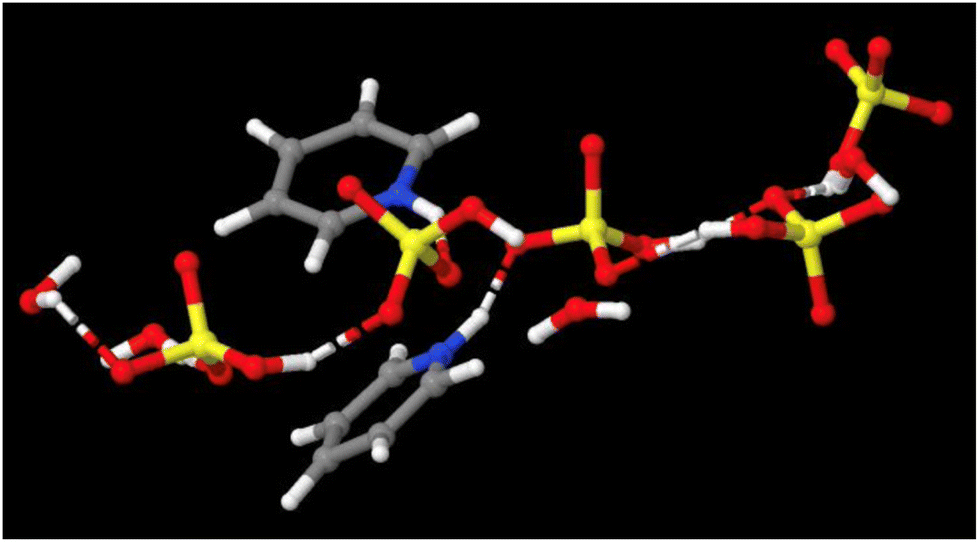 | ||
| Fig. 8 Hydrogen-bonded chains of [HSO4]− and H2O in pyridine – sulfuric acid (χH2SO4 = 0.50), doped with 17 wt% of water – unpublished image from our neutron scattering studies of this system.64 | ||
In carboxylic acid PILs, the preference for hydrogen-bonded, acid-rich networks may be so strong that excess base is phase-separated from the mixture, particularly in the case of ternary amines, devoid of hydrogen-bond-donating {N–H} motifs. In-depth studies show that the two phases are: an acid-rich PIL on the bottom, and a phase containing mainly the amine (or very base-rich PIL) on top.21,43,66 Although well-reported, this has caused some confusion in the field. Some publications report, somewhat inaccurately, that certain carboxylic acids and ternary bases “do not mix”.14 Others report on the existence of homogeneous PILs formulated from equimolar amounts of triethylamine and acetic acid; as suggested by Mariani, Passerini and co-workers, this homogeneous PIL is probably the acid-rich phase, while the upper phase was removed under reduced pressure.43 This is in line with the earlier discussion that it is easy to remove excess base from a PIL, but nearly impossible to remove an excess acid.19 Finally, some authors have called the entire system a “biphasic ionic liquid”,66 which is likely an overly liberal use of the term “ionic liquid”. In conclusion, despite the very easy synthetic procedure, it is crucial to adhere to full spectroscopic and thermal characterisation of prepared PILs, to ensure that their composition is correctly understood.
Water is the most common impurity in ILs, and PILs are difficult to dry due to strong hydrogen-bonding and the potential of accidentally removing one of the components. On the other hand, intentional doping of PILs with water is increasingly practiced, to improve their physico-chemical properties.68 Doping PILs with water dramatically decreases viscosity, decreases density, increases conductivity, and enhances transport properties (viz. the example of 1-butylpyrrolidiniun bistriflimide doped with 1–3.8% of water).69 Interactions of H2O molecules with components of PILs depend very much on the nature of PILs, as shown through three examples below.
Kirchner and co-workers modelled methylammonium nitrate, containing 1.6 wt% of water, which revealed strong interactions of H2O molecules with cations (hydrogen-bond donors bearing three {N–H} motifs) and somewhat weaker interactions with anions as hydrogen-bond acceptors, leading to a tetrahedral hydrogen-bonding motif. Properties of both PIL and water molecules were altered; H2O molecules had lower dipole moments, and the structure of the PIL (mainly the anion–anion orientation) was altered.70 A similar conclusion was reached by Atkin and co-workers from their neutron scattering study of ethylammonium nitrate doped with a six-fold excess of water molecules.71 Water did not act as a diluent, but interacted with all charged moieties of the PIL, forming a new liquid with a distinct nanostructure.
In pyridinium hydrogensulfate, doped with 17 wt% of water, the H2O molecules were incorporated into hydrogen-bonded chains of [HSO4]−, forming a water-in-salt solvent system.64 This can be attributed to the hydrogensulfate anion being quite basic, with both hydrogen-bond donor {H–S–O} and hydrogen-bond acceptor {S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O} motifs.
O} motifs.
Lastly, 1-butylpyrrolidinium bistriflimide doped with 1–3.8 wt% of water was modelled by classical MD simulation, with division of the entire system into: polar, nonpolar, fluorous, and aqueous domains69 In this PIL, neither cation nor anion was a particularly strong hydrogen-bond donor, and water was found to form clusters: about three H2O molecules for 1–2 wt% water concentration, and much larger clusters of 15–16 H2O molecules at 3.8% loading (Fig. 9).
 | ||
| Fig. 9 Snapshots from MD simulations of 1-butylpyrrolidinium bistriflimide with three different concentrations of water, showing clustering of water (blue). Reproduced from ref. 69 with permission from Wiley-VCH, copyright 2023. | ||
In conclusion, varying the acid, the base, the stoichiometry, and/or dopants produces infinite possibilities for solvent modification. This offers enormous opportunities, but also experimental challenges in finding the optimum solvent by traditional experimental methods. The last innovation highlighted in this introduction is the use of high-throughput screening, and artificial intelligence (e.g., neural networks) for the efficient study of vast matrices of PILs. For example, Greaves and co-workers screened 208 solvent compositions based on just eight PILs, by changing the ratio of acid, base, and water.72 The surface tension, apparent pH, and liquid nanostructure were all measured and powerful relationships between the nature of each component and its influence on each property were revealed, as exemplified in Fig. 10.
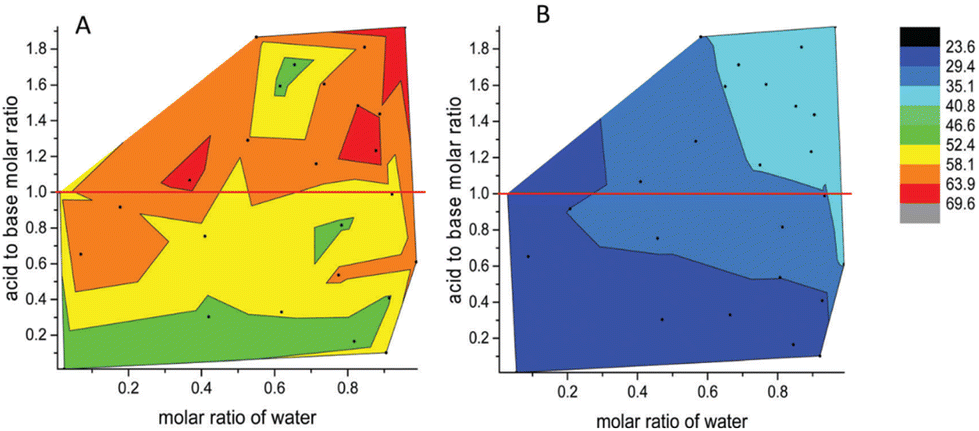 | ||
| Fig. 10 Contour view of surface tension values of PILs based on (A) ethanolammonium nitrate and (B) pentylammonium nitrate. Black dots are experimental values. Reproduced from ref. 72 with permission from Royal Society of Chemistry, copyright 2023. | ||
Catalytic transformations
In the early years of research on air- and water-stable ionic liquids, there has been much interest in their use in a diverse range of homogeneous and heterogeneous, liquid–liquid, and solid–liquid catalysis. Therein, ILs were used to stabilise transition metal complexes and nanoparticles, stabilise enzymes, act as solvents and catalysts, as well as modify solid–liquid interfaces for catalytic applications. Chemistries explored spanned mainly C–C bond forming reactions, oxidations, and reduction reactions, predominantly using aprotic ionic liquids, and have been subject to several excellent reviews.73–76 This area was almost automatically perceived as an intrinsic part of green chemistry, given the importance of catalysis in sustainable chemical transformations, and the safety gains associated with ionic liquids (low vapour pressure, low flammability), when compared to volatile organic solvents.Over the past decade, however, there has been a significant decline in research on ILs for catalytic reactions – especially in comparison with the growing interest in other fields, such as electrochemical applications in Li-ion batteries, supercapacitors, as electrolytes, as well as for natural product dissolution and gas storage.11 Aside from biomass conversion and electrocatalysis, discussed in subsequent sections of this review, there has been relatively little interest in ionic liquids as catalyst/solvent systems for conventional catalytic transformations.
Protic ionic liquids were mainly used as Brønsted acid and/or Lewis base catalysts, in some cases also acting as solvents. They have demonstrated the potential to replace conventional catalysts, operate in solvent-free conditions, and accelerate many organic transformations, such as Knoevenagel77 and Michael additions,78 Beckmann rearrangement to generate ε-caprolatam,79,80 as well as the upgrading of waste materials through hydrolysis of polyethylene terephthalate (PET).81
An interesting fundamental study, although not truly catalytic, is the use of the multi-functionality of PILs as a regenerable solvent, Brønsted acid, and source of a nucleophile. This has been demonstrated by Andreev et al.82 in the case of 1-methylimidazolium thiocyanate [HC1im][NCS] which was successfully used as a safe source of thiocyanic acid, HNCS, and facilitated (4 + 2)-annulation, epoxide ring openings, and other organic transformations. Yields using this PIL were significantly higher than those using conventional routes that involve NaSCN/DMF, although loss of the [NCS]− anion means that a large excess of PIL was required. This is a very valuable contribution to the field, offering mechanistic insight into the influence of the basis and the acidic components of PIL on the reaction pathway.
The two decidedly prominent areas of research focus were: Fischer-type esterification reactions and the chemical transformation of carbon dioxide.
Esterification reactions
The use of PILs as homogeneous acidic catalysts for esterification and transesterification reactions has gathered some momentum in the past decade.There has been significant interest in the use of PILs in the synthesis of biodiesel from refined virgin oils, waste cooking oils, and fatty acids. Such processes feed into SDG7 – affordable and clean energy, but also support SDG10 – reduced inequalities, by generating fuels from resources of much more even global distribution than oil.12 Advantages of PILs are highlighted in two reviews by Zhang et al.83 (2016) and by Ullah et al.84 (2018). Compared to conventional inorganic acids, Brønsted acidic PILs have the advantage of easier separation of hydrophobic products, enabling better recycling and reducing waste disposal treatment. The focus in terms of PILs design has been the control of acidity through judicious choice of acid–base pairing, manipulating their stoichiometry, or through the addition of acidic functional groups (chiefly alkanesulfonic acid, R–SO3H).
Chiappe et al. demonstrated that simple PILs based on pyrrolidinium, imidazolium, or piperidinium cations (Fig. 11), associated with mildly Brønsted acidic anions, e.g., hydrogensulfate, produced higher yields in acetic acid esterification, when compared to aprotic anions such as triflate, chloride, and nitrate.85 There was a correlation between Hammet acidity of the ILs and their catalytic performance, which explains this observation. However, the authors pointed out that hydrophilicity of the ionic liquid medium also affected the process.
 | ||
| Fig. 11 Protic cations and anions used for the esterification of acetic acid. Reproduced from ref. 86 with permission from Royal Society of Chemistry, copyright 2023. | ||
It was reported that PIL acidity, and therefore activity, could be increased by changing the cationic centre from imidazolium to ammonium. For example, triethylammonium hydrogensulfate, [HN222][HSO4], was used to produce biodiesel from crude palm oil in 97% yield at 170 °C, albeit in a two-step esterification-transesterification process.87 Waste edible oils were also successfully converted into ethyl esters (98% yield) in supercritical ethanol using methylimidazolium hydrogensulfate, [HC1im][HSO4]. Interestingly, no conversion was observed in the absence of PILs, even at 250 °C and 9.62 MPa.88
Amide-type PILs, based on N-methyl-2-pyrrolidone, have shown excellent performance in the transesterification of β-ketoesters (80 °C, 3 h).89 Compared to aprotic ionic liquids, [HNMP][CH3SO3] and [HNMP][HSO4] were easy to synthesise, from reportedly inexpensive starting materials and gave higher conversions. However, not all cyclic amide motifs gave such promising results; for example, PILs based on caprolactam showed little difference in comparison to mineral acids, unless appended with sulfonic acid groups.90
Although PIL acidity can be modified by switching cationic centres, recent efforts to manipulate acidity have largely focused on synthesising Brønsted ILs containing –R–SO3H appendages on the cation. These stronger acids allowed lower reaction temperatures while maintaining high conversion. For example, Das et al. reported the synthesis of biodiesel at 70 °C in 94% yield from Jatropha oil, using 1-methyl-3-(4-sulfobutyl)imidazolium hydrogensulfate, although a reaction time of 6 h was required.91 Further studies investigated increasing the acidity of sulfonic-acid-type PILs with modification of the cation. The more acidic 1-methyl-3-(4-sulfobutyl)benzimidazolium triflate outperformed the lass acidic imidazolium counterpart in the conversion of castor oil into methyl ester at 40 °C, giving 96% yield after 14 h.92 Unfortunately, genuine contribution of such studies to furthering the sustainable agenda remains controversial. As shown in Fig. 1, sulfonic acid-functionalised ILs are prepared from sultones, which are expensive starting materials. Combined with the relatively expensive, fluorinated triflate anion, their cost is orders of magnitude higher than that of mineral acids, and it is extremely unlikely that such systems would ever be implemented in practice – especially given the extremely long reaction times that are required.
Another strategy to improve PIL catalytic performance has involved the tethering of more than one sulfonic acid group to the cationic centre, increasing the effective concentration of acidic protons. Linear alkylammonium and cyclic alkylammonium dicationic PILs containing two sulfonic acid functional groups per cation, coupled with either [HSO4]− or [TfO]− anions, were investigated for the conversion of oleic acid to methyloleate, under mechanical stirring and ultrasonic conditions.93 Ultrasonic radiation resulted in improved mixing with higher yields; however, this did require post-reaction centrifugation due to emulsification. In contrast, mechanical stirring gave significantly lower yields, but after the reaction, a triphasic mixture was formed, very easy to separate. The dicationic PILs were catalytically more efficient compared to the corresponding monocationic PILs. Using Hammett acidity functions, the authors determined that the cyclic alkylammonium dicationic ILs based on 1,4-diazabicyclo[2.2.2]octane (DABCO) were the most acidic, and therefore the most active in producing methyl oleate (achieving 90% yield vs. 78% yield achieved using HOTf).
Although much effort has focused on the development of cationic centres with sulfonic acid motifs, simple acid–base mixtures have also shown excellent catalytic activity in esterification reactions, perhaps negating the need for convoluted synthetic procedures. In 2009, Zhang et al. studied the catalytic impact of various PILs for the conversion of fatty acids into methyl esters.94 The impact of the cationic centre: imidazolium, pyridinium, pyrrolidinium or alkylammonium, the counterion: [BF4]−, [H2PO4]−, and [CH3SO3]−, and the presence or absence of sulfonic acid functionalities, have been investigated (Fig. 12). The yields of the methanesulfonate-based ILs at 70 °C were higher (76–94%) than those observed using either sulfuric or sulfonic acid, which both showed ca. 67% yield. Non-imidazolium-based PILs also produced slightly higher yields than their imidazolium counterparts. The highest yield however was observed with N-methyl-2-pyrrolidonium methylsulfonate, [HNMP][CH3SO3], which gave 96% conversion, albeit over 8 h. Once again, this was related to higher PIL acidity through electronic interactions. Remarkably, these ILs were also active at room temperature with the alkylammonium PIL showing similar conversions to that observed with the inorganic acids at 70 °C. This implies that, in terms of energy consumption, PILs could provide advantages over conventional acid catalysts for industrial operations.
 | ||
| Fig. 12 Structures of cations and anions used for esterification reactions. Reproduced from ref. 95 with permission from Elsevier, copyright 2023. | ||
The role of increasing the Brønsted acidity of PILs by choice of cation–anion interactions has been discussed by Martini and co-workers.96 By combining cyclic voltammetry with catalytic reactions, they concluded that sulfonic-acid-functionalised cations with [HSO4]− anions were simply acting as a source of free sulfuric acid delivered by reverse auto-pyrolysis, and this was catalysing the esterification. Although it is yet to be confirmed, the use of amide-based PILs could also be acting as potential reservoirs for neat acids and this warrants further investigation.
Finally, acidity of PILs can be enhanced by a move away from conventional acid–base ‘neutral’ PILs to acid-rich media. Utilising sulfuric acid in a χH2SO4> 0.50 versus the corresponding amine base resulted in a PIL with an extended anionic hydrogen-bonded network.64 At χH2SO4 = 0.67, these PILs showed acidity levels similar to those of analogous PILs based on HOTf. Chrobok and co-workers reported that these PILs outperformed neat sulfuric acid and the product could be easily separated by decantation. In comparison to sulfuric acid, reaction equilibrium has been shifted towards the products, driven by the inclusion of water within hydrogen-bonded PIL, and enhanced expulsion of the organic ester.16,97 Other structural motifs have included the use of dicationic and tricationic amine-based hydrogensulfate (χH2SO4> 0.50) PILs. They catalysed the esterification of oleic acid at temperatures up to 60 °C, over 1 h, with yields higher than that of sulfuric acid.98 Interestingly, these polycationic systems are also less expensive than those based on monoamines. In contrast to sulfonic acid functionalisation, this approach is extremely low cost, and presents no synthetic challenge, which makes it much more amenable to real-world application.
While most publications have focused on the use of small molecule PILs, a few papers have explored polymeric and solid-supported PIL systems. Polymeric ionic liquids, poly(ILs), synthesised through quaternisation of polyethylenimine (PEI) dendrimer with sulfonic acid, were explored as catalysts for esterification of cinnamic acids, showing excellent catalytic activity (50 °C, 3 h).99 The large hydrodynamic radii due to the branched architecture of PEI dendrimers facilitated the separation process promoting these PILs as easily recoverable and reusable catalysts.
Protic pyridinium-based ionic liquid hybrid mesoporous materials (PMO-py-IL) were successfully prepared by the introduction of a 3-propyl triethoxysilane side chain to the pyridine ring and formation of a pyridine polysiloxane network, followed by the addition of trifluoroacetic acid and subsequent IL organosilica formation. The resultant solid PMO-py-IL catalysed esterification reactions between a broad range of free fatty acids and short chain alcohols.100 A polymer-supported sulfonic acid-functionalised benzimidazolium poly(IL) has been used as a heterogeneous catalyst in the esterification of levulinic acid with ethanol.101 Again, a high conversion to ethyl levulinate (99%) was obtained (9 h at 70 °C).
Supported PILs bring an additional level of complexity (and cost), but they do offer unparalleled process advantages in terms of catalyst separations. It would be extremely beneficial if such reports were followed by engineering papers, analysing techno-economical aspects of supported vs. liquid PILs in different processes, thereby informing future directions of study.
Transformations of CO2
The capture and conversion of CO2 to value-added products are key technologies required to address the climate emergency (SDG13).12 However, transformations of CO2 are technically difficult, owing to its thermodynamic stability and the resulting need for high energies or potent catalysts to activate it. They are also economically very challenging, largely due low efficiency of reactions and limited use of CO2 adducts in the chemical industry. CO2 conversion into cyclic carbonate or carbamates can be achieved thermally, electrochemically, and photochemically, as outlined in a recent review of strategies for the capture and utilisation of CO2 in ILs.102Early studies focused on the use of ILs with a metal co-catalyst, usually a transition metal halide, to achieve good selectivity for the conversion of CO2 and epoxides into cyclic carbonates. Mechanistic studies revealed that a proton source could behave similarly to a Lewis acidic metal centre to activate the epoxide. As a result, there has been a move away from metal salt co-catalyst, and towards the use of hydroxyl-functionalised ILs3 and carboxyl-functionalised ILs,4 with halide counterions to achieve good transformations. Since then, several PILs with halide anions have been investigated.
Hydroxyl-functionalised PILs based on guanidinium103 and thioguanadinium104 cations have been investigated for carbonate formation. The synergistic effect of epoxide activation by a proton and nucleophilicity of bromide anions suggested that both types of PIL could be used for solvent-free synthesis of cyclic carbonates with high selectivity (130–140 °C, CO2 atmosphere, 2.0–2.5 MPa). In addition, it was postulated that the tertiary nitrogen atom of guanidinium cation could coordinate with CO2 to further enhance the catalysis. In a similar manner, a series of protic carboxyl-functionalised imidazolium ILs were used to catalyse a solvent-free reaction of CO2 and propylene oxide.105 Lower temperatures and pressures (120 °C, 1.5 MPa CO2) reduced the yield to 92%. Again, the deployment of a bromide anion resulted in the highest catalytic activity versus chloride analogues.
Wang et al. reported on a DBU-based PIL for the addition of CO2 to propargyl alcohols (Fig. 13).106 Whereas 3-phenyl propargylic alcohol (R = Ph in Fig. 13) and other phenyl-substituted alcohols yielded the expected, corresponding cyclic carbonates, the reaction of 1,1-dimethyl-2-propyn-1-ol (R = H in Fig. 13) led to the formation of an open-chain acyclic carbonate. Mechanistic studies conducted by 1H and 13C NMR spectroscopy and density functional theory (DFT) calculations confirmed that both the cation and the anion of the IL were crucial for driving the reaction via cooperative hydrogen-bond donor/acceptor activation. Again, this exemplifies a valuable study whereby the study of an underpinning reaction mechanism leads to better understanding and, in consequence, opens up the potential for informed design of new catalytic systems.
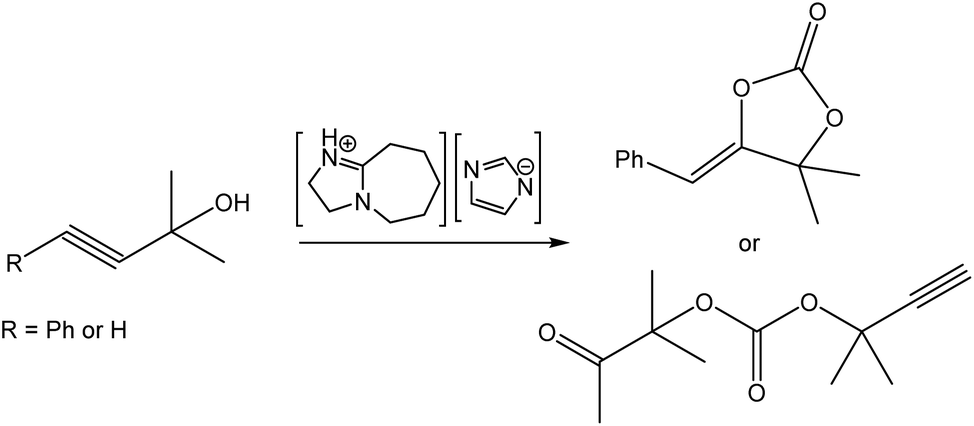 | ||
| Fig. 13 Conversion of phenyl propargyl alcohol into the cyclic carbonate. Adapted from ref. 106 with permission from Wiley-VCH, copyright 2023. | ||
In another study, a DBU-based PIL was used to directly synthesise aliphatic and aromatic carbamates. Using 10 mol% catalyst under a CO2 pressure of 5 MPa in acetonitrile at 150 °C, carbamate was isolated in up to 96% yield. The DBU-based PILs outperformed their analogues based on DABCO, imidazolium, and pyridinium. Other functional groups within the substrates remained passive, affording exclusive chemoselectivity for amine activation. In addition, a chemical shift corresponding to a mixture of aniline and [HDBU][OAc] was observed in the 1H NMR spectrum, related to the formation of hydrogen bonds between aniline and [OAc]−. In conclusion, a well-performing PIL required a protonated cation and a basic anion.
The use of superbase PILs has been taken one step further through the formation of a switchable DBU-based PIL for the synthesis of industrially relevant esters, such as methyl propionate (MP) and methyl methacrylate (MMA).107 Khokarale and Mikkola reported on a highly selective room-temperature synthesis through a reversible CO2 capture approach, involving DBU and methanol. When these components were mixed, [HDBU][MeCO3] was formed, and subsequently reacted with the corresponding substrate anhydride to form MP or MMA, respectively (Fig. 14).
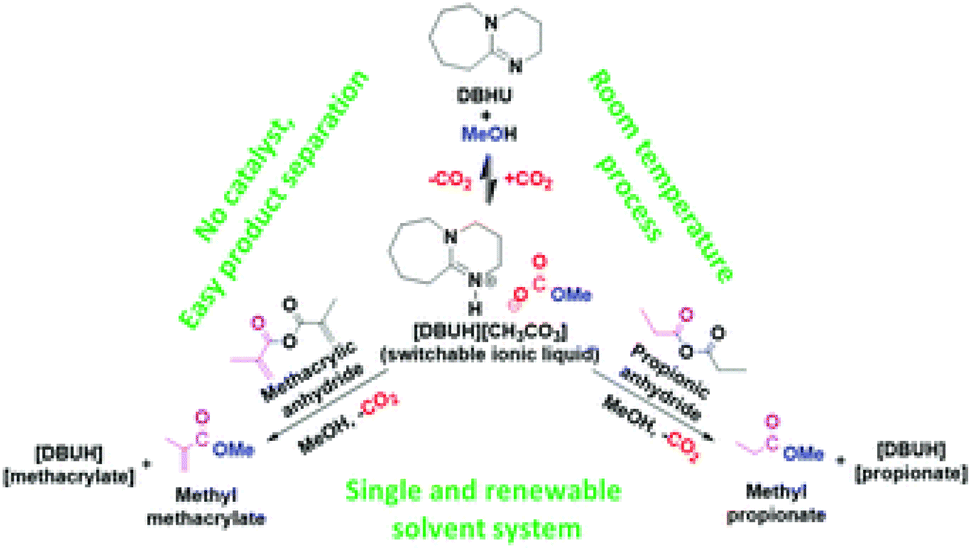 | ||
| Fig. 14 Use of the switchable PIL [HDBU][MeCO3] for the conversion of CO2 into MP and MMA. Reproduced from ref. 107 with permission from Royal Society of Chemistry, copyright 2023. | ||
These reactions could be carried out in various solvents, but in methanol, the product monomer esters phase-separated from the in situ formed DBU derivatives, such as [HDBU][propionate] or [HDBU][methacrylate]. This resulted in yields of 85% and 92% for MP and MMA, respectively. The DBU could be recovered by washing with NaCl solution, before being recycled. The recovered MMA with methanol was also polymerised to poly-MMA using a benzoyl-peroxide-induced free radical polymerisation process.
Other examples of catalysis for sustainable uses
Although less prominent, the uses of PILs for catalytic depolymerisation (key in chemical recycling of polymers), biomass transformations and processing other than CO2 gaseous contaminants are exemplified in this section.DBU-based PILs have been used for the efficient breakdown of waste polylactic acid to lactate esters,108 and [Hpy][HSO4] – for the breakdown of xylose and cellulose into furfural.109 The higher acidity of [Hpy][HSO4] versus its alkylated analogue resulted in higher reaction yields of 95%. The catalytic upgrading of waste PET via glycolysis has also been investigated. The use of the protic salt based on TBD and methanesulfonic acid was shown to effectively promote depolymerisation at 180 °C.110 Interestingly the same PIL could also promote the repolymerisation reaction at 250–270 °C with the resultant PET having similar characteristics to that of a virgin polymer produced using a conventional catalyst. A DBN-phenoxide PIL has also been investigated for PET depolymerisation. Up to 85% yield of BHET could be obtained from PET at 190 °C in 30 minutes.111 These are relatively recent reports, but given the dynamically changing situation in the polymer market, with rapid, policy-driven emphasis on recyclability (for example, according to European Green Deal, all packaging in the EU will have to be reusable or recyclable by 2030), the authors see this strand of research as a likely area of rapid growth.
Kumar et al. conducted a hydrothermal conversion of glucose into levulinic acid using a range of imidazolium-based PILs.112 Levulinic acid formation was improved with the addition of NiSO4 as a co-catalyst with a SO3H-tethered PIL resulting in 56% yield. Levulinic acid was also synthesised from cellulose (41–55 mol%) in the presence of 1,1,3,3-tetramethylguanidinium hydrogensualfate–FeCl2 mixtures; however, yields were lower than the 69 mol% observed for titanium (oxy)sulfate.113 This strand of research is possibly less promising, given the very expensive catalyst to generate low-value platform chemicals in the former case, and in the latter – low yields when compared to the benchmark.
Upgrading of hydrogen sulfide into value-added products has also been studied in PILs. Liu et al.114 studied a series of super-base PILs, with 2,4,6-tris(dimethylaminomethylphenol) coupled with DBN being the most promising. The PIL was able to dissolve high amounts of hydrogen sulfide and transform it into a range of thiols in high yield under mild conditions at 30 °C. Zhang et al. have also utilised a PIL derived from bis(2-dimethylaminoethyl) ether and HTFSI for the capture and conversion of hydrogen sulfide into mercaptan acids.115
Summary and outlook
While there has been an overall decline in the breadth of research on PILs for catalysis, growth has been seen in select fields associated with esterifications/transesterifications and CO2 transformations. PILs for catalysis have been developed from conventional imidazolium or alkylamine precursors, and from organic superbases: 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and DBU. A sub-set of PILs was functionalised with –SO3H appendages (Fig. 12) to achieve increased acidity. Anions spanned from highly fluorinated: [TFSI]−, [OTf]−, [BF4]− and [PF6]−, through those derived from inexpensive mineral acids, chiefly [HSO4]− and [H2PO4]−, to conjugated bases of carboxylic acids, mainly [OAc]−. A sub-set of these are non-stoichiometric systems, with excess of acid (most often sulfuric acid) for increased acidity.A positive development in PIL catalysis is the pronounced focus on several fields of interest: esterifications/transesterifications and CO2 transformations, along with potentially very promising research on the recycling of polymers. Arguably, this demonstrates increasing maturity of research on ionic liquids in catalysis: from very broad scoping in the past decades, to a focus on fields of research where there are clear advantages to using certain types of ionic liquids, and the potential for further growth.
There have been several excellent fundamental studies, that go beyond comparative reporting of catalytic activity, and draw on a wide range of techniques to understand the mechanistic considerations when using PILs, such as: Andreev et al.82 studying the multimodal influence of [HC1im][NCS] on organic transformations, the neutron scattering study on sulfuric acid-based PILs, leading to understanding enhanced phase separation in esterifications,64 or the combined NMR spectroscopy and DFT investigation of DBU-based PIL in the reaction of CO2 and propargyl alcohols, proving the cooperative role of both the anion and the cation.106 These insights have real potential to enable rational design of improved catalytic systems.
Finally, there have been encouraging developments of systems that have at least the potential to compete with the industrial benchmarks. Non-stoichiometric PILs with acid excess may offer inexpensive and highly acidic systems, with phase behaviour different from the parent acid, while supported PILs promise “the best of both worlds” in terms of liquid-like mass transfer, combined with solid state-like ease of phase separation and catalyst recycling.
Despite the progress, there is much to be addressed in this area of research. Many papers still propose the use of fluorinated anions: [TFSI]−, which is very expensive, or [BF4]− and [PF6]−, which are hydrolytically unstable and pose the hazard of releasing HF. While fundamental studies should be free to explore any materials irrespective of cost or potential of implementation, researchers working towards sustainable applications must take these into account.
Though many PILs can be recycled, a significant number of publications report a decline in activity over multiple cycles. Issues such as leaching, long-term stability, and regeneration remain unresolved. Other factors such as potential corrosivity, waste generation, and biodegradability also need to be further investigated. As is often highlighted, the role of water appears prominent, but is not fully understood and needs further study.
Overall, although there are a great many initial, discovery-oriented studies on PILs in catalysis, the area lacks follow-up research, moving from chemistry towards chemical engineering, that would truly demonstrate and quantify the sustainability claims. Most papers lack comparisons to industrial benchmarks, even if relevant industrial processes exist (viz. esterifications). There is little reference to SDGs, let alone more involved calculations of green metrics, or robust techno-economic analysis. Such studies would inform further development and potentially promote greater industrial uptake.
Looking forward, the landscape around the adoption of PILs for catalysis reactions should be considered. In addition to esterifications, there are other acid-catalysed processes that may benefit from non-stoichiometric PILs used as a drop-in solution to replace liquid mineral acids. Otherwise, supported PILs may be used in niche applications, such as flow chemistry in the synthesis of fine chemicals. The CO2 transformations are studied at a very fundamental level, and more research is needed to understand their potential. An interesting and seemingly very promising approach is coupling such studies with electrocatalysis, which is discussed further on in this review. Finally, upgrading and valorisation of waste materials, for example PET, is expected to grow in prominence, driven by the industrial need to recycle polymers, which in turn is mandated by national and international policies.
Biomass treatment
Biomass sources, current issues, and prospects
There are three main types of biorefineries focused on making biofuels: first generation (1G), using human-edible feedstocks, second generation (2G), using lignocellulosic feedstocks which are non-edible for humans and third generation (3G), using algal biomass (Fig. 15).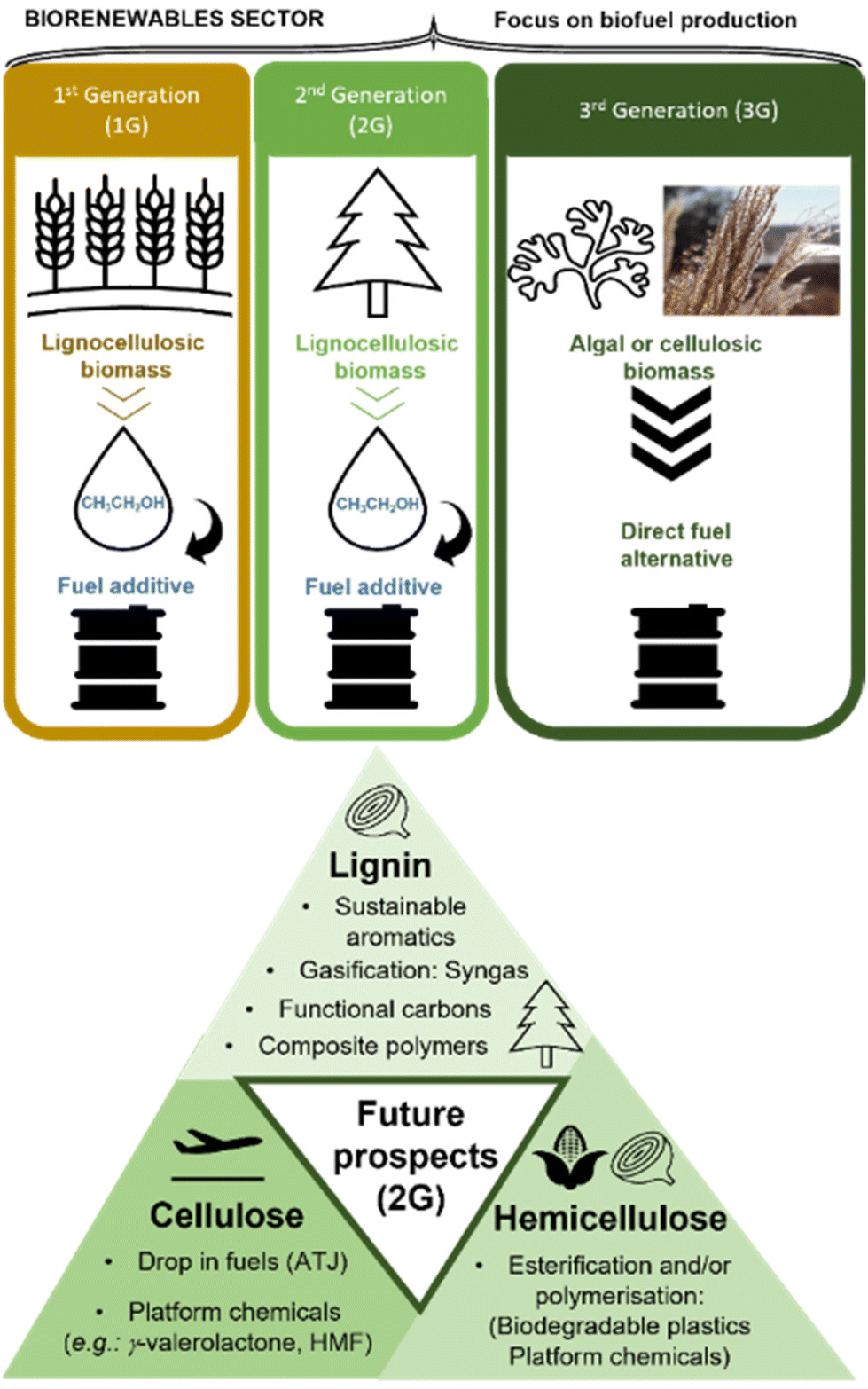 | ||
| Fig. 15 Three main types of biorefineries using renewable feedstocks and their primary products (e.g. bio-alcohols or direct fuel alternatives). The triangle outlines the major future prospects of 2G biorefineries which would utilise all three major bio-polymer constituents of lignocellulosic biomass (cellulose, lignin and hemicellulose). | ||
The oldest 1G biorefineries posed ethical issues, as competing with food supply chains and therefore in potential conflict with the delivery of SDG2 (zero hunger) and the societal aspect of sustainable chemistry. They were superseded by 2G biorefineries, which benefit from low-cost, high abundance feedstock, of more uniform global distribution compared to G1. The 2G feedstocks may include waste and contaminated products of agricultural or forestry origin, contributing to more sustainable use of forest resources (SDG7 – affordable and clean energy, SDG12 – responsible consumption and production; SDG15 – life on land).111,112 The major drawback of both 1G and 2G biorefineries is the end product – ethanol. For example, and average petroleum-fuelled engine in a UK car can tolerate up to 10% ethanol by volume (E10), thereby requiring 90% of petroleum in the blend – a problem known as the ‘blendwall’.113 Furthermore, other fuel types, such as jet fuels, are incompatible with ethanol. These problems could be addressed by 3G biorefineries based on algae, which may generate drop-in fuels, but this feedstock remains very expensive to grow.
Currently, the 3P sustainability triad (benefitting people and planet, while generating profit), is most likely to be achieved by comprehensive processing of all parts of lignocellulosic feedstocks (Fig. 15), identified as the most appropriate for the sustainable production of biofuels and biochemicals.116,117 The cellulosic fraction can be further processed to drop-in fuels, chemically identical to crude-oil-derived jet fuel, diesel, petroleum etc. An example of such process is the alcohols-to-jet (ATJ) method,118 with or without the conversion of ethanol to butanol using the Guerbet reaction.120,121 Although both the academia and the industry have explored the development of the ATJ method using 1G cellulosic feedstocks,118 the more complicated 2G lignocellulosic feedstock has not been used in this context, to the best of our knowledge.
Lignin (15–30 wt% of lignocellulosic biomass),122 must be valorised, preferably to high-added value materials or chemicals (in contrast to currently dominating low-value products), while hemicellulose should be upgraded, for example by conversion to biodegradable plastics, as recently shown using waste corn cobs,123 containing 30% hemicellulose.124 This bears the promise of economically-viable upgrading of other agricultural wastes such as onion or garlic skins (26–39% lignin, 16–26% hemicellulose).125
Processing of lignocellulosic biomass
There is a number of currently operated biomass processing methods, including Organosolv,126 steam,127 ammonia,128 and acid,129 some of which have been discussed in previous reviews.145,146 Pre-treatment processes based on protic ionic liquids are typically benchmarked against the most common: the Kraft pulping process, used globally to isolate cellulose from lignin and hemicellulose in lignocellulosic biomass. In the Kraft process, biomass is treated with a solution of sodium hydroxide and sodium sulfide (pH 13–14) at 170 °C.119,130 Cellulose (the product) is isolated as a solid pulp, while lignin (the by-product/waste) is collected as a solid residue downstream. In the highly caustic environment of the Kraft process, lignin undergoes depolymerisation (required) and condensation reactions (unwanted).119 The condensation reactions result in increased number of chemically resistant C–C bonds in Kraft lignin, hindering its further processing. Combined with the fact that Kraft lignin is often also contaminated with carbohydrates (1–2 wt%)131 and/or functionalised with thiols (1–3 wt%),132 this lignin is typically burned to release heat and generate steam for turbines. This means that 15–30 wt% of the biomass cannot be used to its full potential as a higher value-added product.The “standard” IonoSolv process (Fig. 16) uses PILs, such as [HN222][HSO4], doped with water.23,133,134,138 The addition of ca. 20 wt% water acts as an antisolvent, allowing to fractionate lignin from cellulose and hemicellulose.112,128,133,134 This approach follows the “4th evolution of ILs” strategy, whereby properties of ionic liquids are modified, easily and cheaply, by doping with small molecules, rather than by tedious synthetic modification to the structures of cation or anion.67 It has been demonstrated that loading of ca. 20% of water results in its incorporation into the hydrogen-bonded anionic network, generating a new, distinct liquid structure,64 which is strongly hydrophilic and lipophobic.16
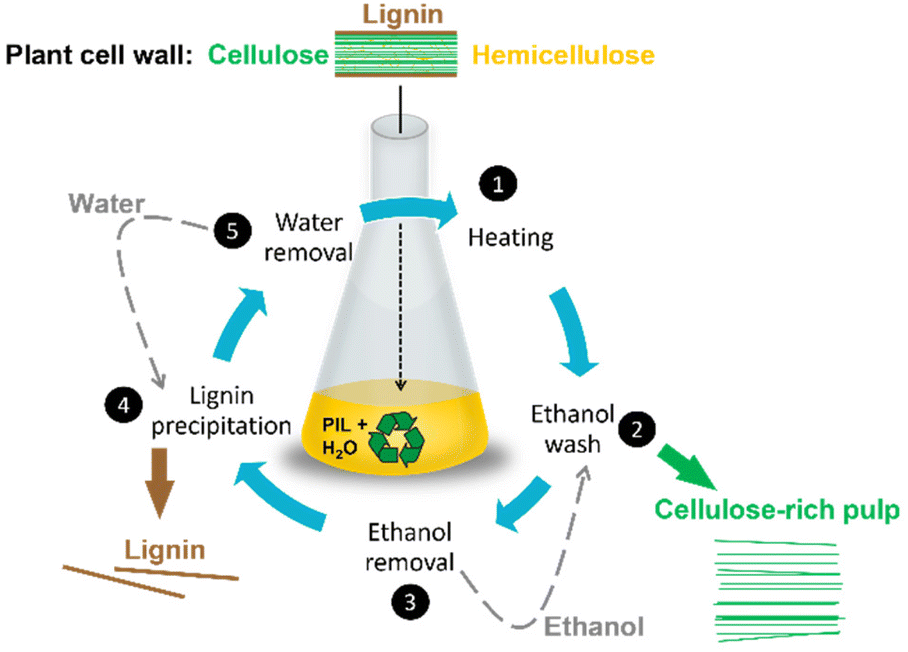 | ||
| Fig. 16 IonoSolv process flow diagram adapted from ref. 23. Typical water content: 10–40 wt%. Typical pre-treatment conditions: 120 °C, 9 h. | ||
This effective fractionation underpinned three major advantages of the IonoSolv process.133,138,140,141 Firstly, highly crystalline cellulose fraction could be obtained, which has the potential to be used as bulking agents in the food or pharmaceutical industry (in contrast to paper-grade Kraft cellulose).133,135 Secondly, enzymatic hydrolysis of cellulose to hexoses (saccharification) could be carried out achieving high yields with lower enzyme loadings, due to the absence of hemicellulose and lignin, that act as cellulase inhibitors.135,142,143 Finally, a high-quality lignin fraction was isolated, as a potential sustainable source of chemical building blocks for renewable aromatics or functional carbon materials. The value added from increased cellulose accessibility and lignin isolation could be vital to ensuring the economic feasibility of lignocellulosic biorefineries.128,137,139–142,144 This is particularly true for softwoods, hardwoods, or certain agricultural residues, with higher average lignin content than lignocellulosic grasses (Fig. 17).
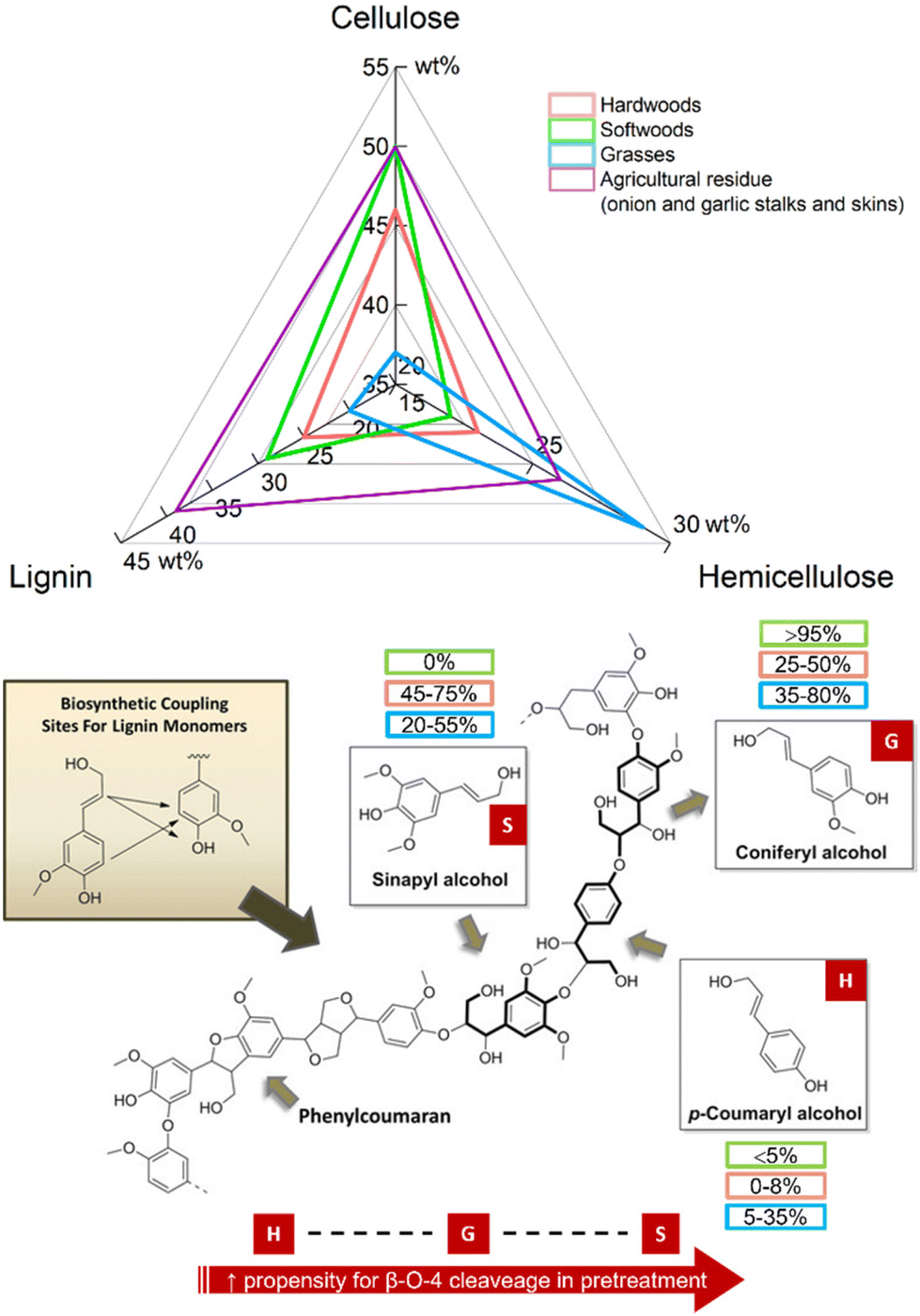 | ||
| Fig. 17 Top: Composition of the three main constituents of hardwoods (red), grasses (blue), and softwood (green) lignocellulosic biomass as well as lignocellulosic biomass from agricultural residues such as garlic and onion stalks and skins. Bottom: Monolignol components present in lignin polymers and their compositions in softwoods (green), hardwoods (red), and grasses (blue). Figure adapted from ref. 151. Values (top and bottom) taken from upper compositional limits in ref. 125, 152 and 153. | ||
Numerous scoping studies demonstrated broad applicability of IonoSolv. Gschwend et al.147 showed that asymmetric N,N-dimethyl-N-butylammonium hydrogensulfate, [HN114][HSO4], was more effective at softwood (pine – Pinus Sylvestris) fractionation than the symmetric [HN222][HSO4]. However, it should be noted that a significant degree of lignin polycondensation was still observed. The IonoSolv process has also been used to successfully fractionate lignocellulosic components of organic-148 and metal-149 contaminated waste wood. Further, successful fractionation of lignocellulosic material from UK invasive species150 has also been addressed. However, the lignin isolated in this case was simply burned for energy instead of further upgrading.
A significant challenge for the PILs pre-treatment technology remains the valorisation of hemicellulose. Typically, the hemicellulose fraction is entrained in the PIL,147,154 but it can be isolated via column chromatography as pentoses, humins, or furfurals.155 Such processes are expensive and poorly scalable; therefore an in situ valorisation approach would be preferred. For example, in 2020, Bukowski et al.156 used an IonoSolv pre-treatment step in combination with polyoxometallates and molecular oxygen (OxFA process), to in situ oxidise hemicellulose to formic acid.
A second challenge is greater control over the degree of condensation reactions which occur in the lignin, to generate high-quality products from a variety of non-food crop feedstock. Softwoods from forestry residues are a cheap and sustainable lignocellulosic source, with the potential to supply 1.3 million dry tonnes per annum in the UK alone.157 Lignins in softwoods contain guaiacyl (G) units, and in hardwoods – syringyl (S) units; both G and S units are beneficial for lignin fractionation, increasing the propensity for β-aryl ether (β-O-4) cleaveage. However, condensation products related to G–G units are more stable than those formed from a combination of G–S units,158,159 therefore recalcitrant C–C bonds formed via condensation reactions in softwood biomass tend to generate low quality of lignin.
Combined IonoSolv-organosolv process
Organosolv pre-treatment with high ethanol concentrations was reported to dramatically reduce lignin condensation and other degradation reactions, at least in biomass from the easily fractionated grass, Miscanthus giganteus.160 Ovejero-Pérez et al.161 have reviewed and compared IonoSolv and organosolv methods, recommending the combination of the two technologies for synergistic benefits.Implementation of this suggestion, that is biomass pre-treatment using 4th generation PILs with mixed dopants (water and/or alcohols), is still in its infancy. In 2020, Chen et al.162 found that, using [HN114][HSO4] with 40 wt% ethanol or butanol, provided multi-faceted advantages when compared to traditional IonoSolv. The energy consumption in the PIL regeneration process was lower when using alcohol dopants. In the presence of alcohols, β-O-4 ether linkages were converted to α-alkoxylated ether linkages, which enhanced delignification and hindered lignin condensation. Finally, a highly enzyme-accessible cellulose fraction was produced. Thus, this method has been identified as a potential route towards a higher value-added lignin product and a more economically viable lignocellulosic biorefinery.
PIL pre-treatment of Kraft lignin to produce high value chemicals
PILs have been used as pre-treatment solvents to upgrade low-quality Kraft lignin produced from the Kraft process.163 Here, [HN122][HSO4] was used as a pre-treatment solvent (160 °C, 1 h, mass ratio lignin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PIL = 1:9) to depolymerise and deoxygenate Kraft lignin prior to a hydrothermal liquefaction (280 °C, 4 h). This produced bio-oils which contained up to 59% vanillin. Importantly, including the PIL pre-treatment stage resulted in a 27% higher bio-oil yield when compared to direct hydrothermal liquefaction.
:PIL = 1:9) to depolymerise and deoxygenate Kraft lignin prior to a hydrothermal liquefaction (280 °C, 4 h). This produced bio-oils which contained up to 59% vanillin. Importantly, including the PIL pre-treatment stage resulted in a 27% higher bio-oil yield when compared to direct hydrothermal liquefaction.
Marine biomass processing methods
Moving away from lignocellulosic, land-grown biomass, chitosan and chitin are typically found in marine biomass, as a part of cephalopods’ endoskeletons (e.g. octopus, squid) and crustacean shells (e.g. crabs, lobsters).164,165 Although these are amongst the most abundant biopolymers after cellulose, there have been relatively few attempts to fractionate and valorise these components using PILs. This is likely due to the recalcitrant nature of these biopolymers, which are linked together by strong β-1–4 glycosidic bonds.166In 2015,167 methylimidazolium hydrogensulfate, [HC1im][HSO4], was shown to promote the conversion of chitosan and chitin to HMF in reasonable yields. Aqueous solution of [HC1im][HSO4] (4 wt%) was reacted with chitosan or chitin (180 °C, 5 h) to yield 30 or 19 mol% of HMF, respectively. A 5% decline in yield was observed from run 1 to run 5. Pure HMF was isolated through a simple procedure of washing with ethyl acetate and subsequent vacuum distillation. In 2017, the same PIL was used at a higher mass loading (10 wt%) in a 3:2 wt:wt ratio of H2O:DMSO.168 Reaction with chitosan and chitin (180 °C, 6 h) gave HMF at yields of 35 and 26 mol%, respectively. Although these conditions increased HMF yield by ca. 5–7 mol%, the HMF purification step was more intensive and required column chromatography making the water-based process far more appealing.
In conclusion, there is much room for improvement in this research area, and there may be an opportunity for PILs to advance the field in the same way as they have in the treatment of lignocellulosic biomass.
Summary and outlook
The current ‘state of the art’ for lignocellulosic biomass processing is the Kraft process used in the paper pulping industry. The Kraft process requires highly caustic processing conditions, and yields low-quality, recalcitrant, and contaminated lignin. Between 15 and 30 wt% of the biomass is of such low quality that it is simply burned as a heat source.Research into PIL-based biomass treatment is an excellent example of a sustainable technology that has been developed from fundamental discovery of enhanced biomass fractionation using hydrated PILs, through robust scoping studies and scale-up, to techno-economic analysis and finally to the market. It relies on very low-cost PILs, based on sulfuric acid and inexpensive amines. PILs are the key enabling factor required to achieve circularity in this realm, and their use is aligned with 12 Principles of Green Chemistry, delivering less hazardous chemical synthesis and safer solvents/auxiliaries, in addition to extant benefit of using renewable feedstock.169 A combined IonoSolv-organosolv approach is particularly promising, as there is the potential to use ethanol from the saccharification of the isolated cellulose fraction as an IL cosolvent, in place of water. This reduces greatly the cost of PIL regeneration and improves circularity of the process. Furthermore, alkoxylating the β-O-4 ether linkages in lignin enhances delignification and inhibits condensation, generating a high-value, pure lignin fraction.
The main challenge in the technology remains valorisation of the hemicellulose entrained in the PIL phase, essential for full circularity. Most reported approaches rely on chromatographic separation, which is not cost effective or well-scalable. Only very recently, an in situ catalytic oxidation approach has been reported, but this strand of research is in its naissance, and requires further intensive study to assess its viability.
Another challenge – or possibly an opportunity – is the valorisation of other biomass sources, in particular marine biomass. Chitosan and chitin have been reported to dissolve in aprotic ionic liquids,170 but there is paucity of research on their dissolution and processing in protic ILs. This is particularly puzzling in the case of chitosan, which is known to dissolve in solutions of mild organic acids. Existing research in the area is focused on the catalytic conversion of these biopolymers to HMF; again, the papers are few and far between, and use aqueous solutions of weakly acidic PILs, such as [HC1im][HSO4].
Looking into the future, it is predicted that the combined IonoSolv-organosolv process has the potential for large-scale industrial implementation in plant biomass processing. Furthermore, it could potentially be used in marine biomass processing, as alkoxylation of the glycosidic ether bonds in chitin and chitosan may be the key required to unlock many potential uses of these biopolymers. Analysing gaps in the knowledge, broader research into the processing of marine biopolymers with inexpensive and recyclable protic ionic liquids is an exciting area, waiting to be explored.
Energy storage and conversion
Concerns about climate change, pollution, and energy security motivate extensive research into energy storage and conversion devices, aiming for net-zero global emissions by 2050. These activities are strongly aligned with several SDGs: SDG7 – affordable and clean energy, SDG11 – sustainable cities and communities and SDG13 – climate action. Energy storage is particularly important for balancing electrical grids, which are increasingly supplied with electricity from renewable resources, which are intermittent and non-dispatchable. Although most energy storage is currently pumped hydroelectricity (ca. 96% of global capacity),171 electrochemical energy storage (ESS) is being increasingly deployed around the world (∼300 GW h by 2030 according to the US Department of Energy)172 and the International Energy Agency predicts almost a seven-fold increase from 2020 to 2026.173 In fact, to meet net-zero emissions targets, it is estimated that 600 GW are required by 2030.Broadly, ESS comprises various types of batteries and supercapacitors, whilst energy conversion devices constitute fuel cells and electrolysers. Both categories are explored here in the context of using PILs to make functional improvements to current state-of-the-art technologies.
Alkali-ion batteries
Batteries based on alkali-ions, predominantly lithium-ions, are now ubiquitous in everyday life: in portable electronics, electric vehicles, and stationary energy storage.174 Despite widespread commercialisation of lithium-ion battery (LIB) technology since the 1990s, safety concerns remain due to the inherent volatility and flammability of commercially used carbonate-based organic electrolytes.175 Several high-profile cases involving thermal runaway events, as well as limited operating temperatures, have driven research towards alternative electrolyte systems, whilst accelerating research into other alkali-ions, such as sodium-, potassium-, magnesium- and calcium-ion batteries.176 Indeed, these less-studied technologies have received increasing attention with a view to increasing energy density, whilst also seeking cheaper, more sustainable routes to mass production.With regards to alternative electrolyte systems, ILs have long promised safety advantages due to their intrinsically low volatility and high thermal stability,177 but most research to date has been on aprotic ionic liquids,178 as reviewed by Balducci.179 The presence of “free” protons in PILs discouraged their use as electrolytes in alkali-ion batteries, but recently it has been recognised that the availability of these protons could be controlled (and reduced) through the difference in pKa of the Brønsted acid and Lewis base constituting the PIL.180,181 For the last decade, research has highlighted the advantages and limitations of using PILs in alkali-ion electrolytes.182
Early proof-of-concept work by Balducci and co-workers demonstrated both the use of PILs in mixtures with traditional LIB electrolytes, as in the case of 0.5 M LiNO3 in [HHpyrr][NO3]/propylene carbonate (PC),180 as well as the implementation of a PIL as the sole electrolyte, with [HN222][TFSI] containing 1.0 M LiTFSI.183 Although the water content was high for LIBs (<80 ppm) in the former case, the charge–discharge process for a LiFePO4 (LFP) cathode was demonstrated to occur at a potential in which the mixed electrolyte was stable.180 Despite displaying reasonable discharge capacities at low C-rates, a constant capacity fade was observed at room temperature, and sufficient stability was only found at reduced temperatures. The latter case demonstrated that PILs could be employed as sole electrolytes (with low water content of <10 ppm), and by use of Li4Ti5O12 as an anode, due to the low cathodic stability of LiTFSI in [HN222][TFSI]. A low discharge capacity of 60 mA h g−1 was observed, representing only 40% of the capacity seen in the analogous propylene carbonate (PC) system, but this capacity was shown to be stable for a total of 300 cycles.183 A subsequent Raman and FT-IR spectroscopic study184 showed that coordination of added Li-ions by anions of the IL is greater in aprotic than in protic systems, due to greater inter-ionic interactions in the latter, potentially leading to improved mobility of Li+ and greater ionic conductivity. Early MD calculations of [HN2HH][NO3] strengthened this view, suggesting that the structure of PILs is only marginally disrupted by the addition of lithium salts.185 Moreover, mixtures of PILs and PC were further shown to suppress anodic dissolution of aluminium current collectors.186
Most subsequent research has been focused on LIBs and has highlighted both the lower cost and easier synthesis of PILs with respect to similar AILs.187–189 These investigations have centred on their use as sole electrolytes, combined with an appropriate alkali-ion salt, investigating ionic conductivity, viscosity, electrochemical stability window (ESW), and charge–discharge behaviour in coin-cell batteries. Given the high viscosity of ILs with respect to carbonate-based electrolytes, Li-ion transport was thought to be much reduced, particularly affecting LIB performance at high current densities. However, in 2015, Menne et al. showed that the unique Li+ environment in PILs based on [FSI]−, such as [HC4pyrr][FSI], gave access to higher performance than analogous AILs (Fig. 18).190 The viscosity of these PILs was shown to be lower than those containing [TFSI]− anions and, importantly, ca. 40% lower than that of the equivalent AIL. Most importantly, the discharge capacities of LFP-graphite LIBs were shown to be only slightly reduced versus the incumbent technology and indeed capacity at high C-rates was greater than that observed in the AIL system.
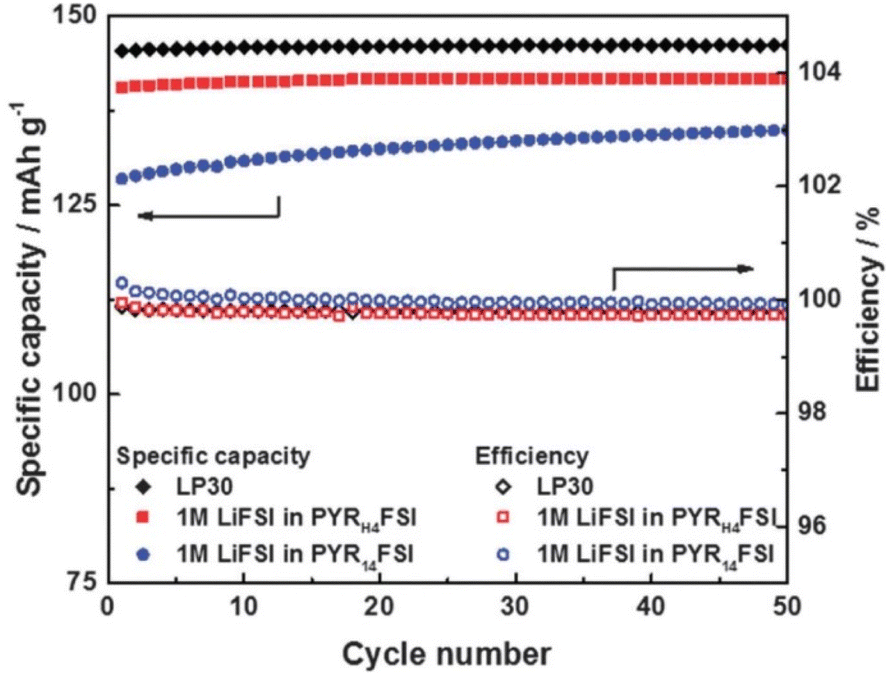 | ||
| Fig. 18 Plot showing specific capacity as a function of cycle number for LIBs with PIL and AIL electrolytes containing [FSI]− anions, compared with traditional LP30 electrolyte, reproduced from ref. 182 with permission from Elsevier, copyright 2023. | ||
Further work from the same group investigated PILs formed from [TFSI]− anions and various cations, concluding that PILs containing imidazolium and pyrrolidinium cations generally have higher conductivities than those containing piperidinium and di-pyrrolidinium cations.191 Although increasing alkyl chain length was shown to decrease ionic conductivity, cation ring size had a greater influence. The PILs explored in this study demonstrated higher ionic conductivity than analogous AILs, with evidence suggesting that coordination numbers of [TFSI]− around Li+ was as low as 0.4–0.5, as compared to ca. 2 for AILs (Fig. 19). The different trends in viscosity and ionic conductivity across the different cations highlighted the importance of taking both viscosity and lithium coordination number into account to optimise battery performance. However, it is important to note that the performance of PIL electrolytes was not compared to traditional carbonate-based electrolytes such that its competitiveness with the incumbent technology could not be directly assessed.
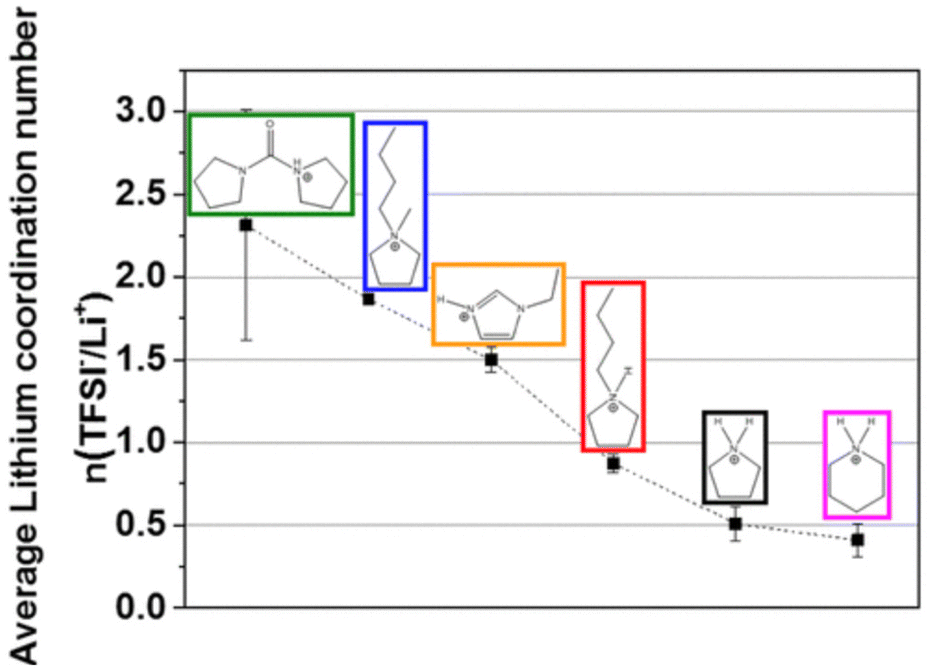 | ||
| Fig. 19 Plot showing the average lithium coordination number with respect to the [TFSI]− anion for various PIL cations, reproduced from ref. 191 with permission from American Chemiscal Society, copyright 2023. | ||
Further work in 2017 that compared [HHpyrr][TFSI] with [HC4pyrr][TFSI] showed that the charge transfer resistance associated with lithium insertion-extraction was significantly lower in PILs compared to similar AILs.192 The study went further by investigating a mixture of PILs, demonstrating stability over 50 cycles, even at low temperatures (0 and −20 °C). However, discharge capacities at these reduced temperatures were relatively low, when compared with other studies, again reflecting fundamental insights gained but without demonstrating a viable, competing system (Table 1). Expanding the pool of cations for use in PILs as LIB electrolytes, Stettner etal. investigated two imidazolium cations, combined with either [FSI]− or [TFSI]−, and showed increased ionic conductivity versus the analogous pyrrolidinium systems.193 This work corroborated the finding that [FSI]−-based PILs had lower viscosities than their [TFSI]− counterparts,190 although the best battery performance was found to be from 1,2dimethylimidazolium bis(trifluoromethanesulfon)imide, [HC1C1im][TFSI], that showed over 50% capacity retention at a high current density of 5C. Again, however, illustrating a prevalent limitation of studies in this area, neither the absolute capacity or its retention with C-rate increase or cycle number were tested with typical carbonate-based electrolytes in the same cell set-up. Without this comparison, it is challenging to contextualise the performance or fairly assess the durability of the [HC1C1im][TFSI] system.
| Year | PIL | AIL | Salt | σ (mS cm−1) | Visco. (mPa s) | ESW (V) | Cathode | Anode | Theoretical discharge capacity (mA h g−1) | Max. discharge capacity/mA h g−1 | Stability cycles | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <1C | 1C | 2C | 5C | 10C | 20C | >20C | ||||||||||||
| ESW: electrochemical stability window; AC: activated carbon; G: graphite; LTO: lithium titanium oxide; HC: hard carbon. LFP: lithium iron phosphate; LVP: lithium vanadium phosphate; NFM: NaNixFeyMnzO2; NVP: sodium vanadium phosphate. | ||||||||||||||||||
| 2013 | [HHpyrr][NO3]/PC | — | 0.5 M LiNO3 | ∼16 | — | 2.6 | LFP | AC | 174 | 134 (C/2) | 126 | 116 | 90 | 68 | — | 39 (25C) | 100 | 180 |
| 2013 | [HN222][NO3] | — | 1.0 M LiTFSI | ∼3 | — | 4.0 | LFP | LTO | 174 | 115 (C/10) | 60 | — | — | 27 | — | — | 300 | 183 |
| 2014 | [HHpyrr][TFSI] | [C1C4pyrr][TFSI] | 0.5 M LiTFSI | ∼2 | 130 | — | LVP | AC | 133 | — | 120 | 116 | 109 | 90 | 54 | — | 100 | 162 |
| [HC4pyrr][TFSI] | 96 | AC | — | 125 | 121 | 112 | 91 | 50 | — | |||||||||
| 2014 | [HC4pyrr][TFSI]/PC | — | LiTFSI | 2–7.5 | 5–95 | 3.5–4.0 | LFP | AC | 174 | — | — | — | — | — | — | — | — | 163 |
| 2014 | [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.5 MLiTFSI | 2 | 100 | 3.8 | LFP | G | 372 | 240 (C/10) | 65 | — | 20 | — | — | — | 20 | 189 |
| 2015 | [HC4pyrr][FSI] | [C1C4pyrr][FSI] | 1.0 M LiFSI | 7.4 | 25 | 3.0 | LFP | AC | 174 | — | 142 | 132 | 115 | 96 | — | — | 50 | 190 |
| 2015 | [HHpyrr][TFSI] | — | 1.0 M LiPF6/EC:DMC | 1–10 | — | — | LVP | Li | 133 (4.3 V) | — | 131 | — | — | 125 | — | 100 (100C) | 100 | 201 |
| 197 (4.8 V) | — | 158 | — | — | 140 | — | 126 (50C) | — | ||||||||||
| 2016 | [Cation][TFSI] | [C1C4pyrr][TFSI] | 0.5 M LiTFSI | 1–4.3 | 45–178 | 4.1 | LFP | AC | 170 | — | 153 | 148 | 135 | 110 | 72 | — | — | 191 |
| 2016 | [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.3 M NaTFSI | 2.5 | 69 | 3.5 | NVP | AC | 118 | — | 93 | 74 | 40 | 20 | 12 | — | 100 | 199 |
| 2017 | [HHpyrr][TFSI] | — | 0.5 M LiTFSI | 2.1 | <69 | — | LFP | AC | 170 | — | 140 | 130 | 88 | 57 | 20 | — | 100 | 192 |
| [HC4pyrr][TFSI] | ||||||||||||||||||
| 2018 | [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.5 M LiTFSI | 2 | 99.7 | 4.0 | LFP | HC | — | 185 (C/5) | 122 | — | 58 | — | — | — | ∼30 | 195 |
| [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.3 M NaTFSI | 2.8 | 70 | 3.5 | NFM | HC | — | — | — | — | — | — | — | — | — | ||
| 2019 | [HC1C1im][TFSI] | — | 0.5 M LiTFSI | — | 27.2 | 2.8 | LFP | AC | 174 | 158 (C/2) | 131 | — | 79 | — | — | — | 100 | 193 |
| [HC1C1im][FSI] | 6 | 16 | 2.6 | 167 (C/2) | 155 | — | 70 | — | — | — | 25 | |||||||
| [HC1im][TFSI] | 3 | 22.3 | 2.8 | 113 (C/2) | 97 | — | 40 | — | — | — | — | |||||||
| [HC1im][FSI] | 9 | 15.6 (60 °C) | 2.8 | 143 (C/2) | 110 | — | 36 | — | — | — | — | |||||||
| 2019 | [HN222][TFSI] | [N1222][TFSI] | 0.3 M LiTFSI | 2.5 | 68 | — | — | — | — | — | — | — | — | — | — | — | — | 181 |
| 2019 | [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.5 M KTFSI | 2.5 | 78 | 4.0 | HC | AC | — | — | — | — | — | — | — | — | — | 202 |
| 2020 | [HC4pyrr][TFSI] | [C1C4pyrr][TFSI] | 0.1 M Ca(TFSI)2 | 4 | 39 | 3.2 | TiS2 | AC | 1340 | 383 (C/50) | — | — | — | — | — | — | 1 | 194 |
Another consideration is the ESW. For instance, the use of [FSI]− in place of [TFSI]− decreased the anodic stability of the IL.193 Comparing the use of analogous PILs and AILs, the protonated cations have inherently reduced cathodic stability, resulting in a narrower ESW190 due to deprotonation and reduction of the cationic proton.191 This decrease in the ESW has been seen in other experimental studies,194 and is slightly lesser for imidazolium than pyrrolidinium cations.193 PILs have also been employed in alkali-ion batteries using hard carbons in place of graphite as the anode, in order to increase accessible capacities.195 Previous experiments had shown that AILs could enable safer use of hard carbons, but suffered from poor performance at high Crates.196 However, the 0.5 M Li[TFSI] solution in [HC4pyrr][TFSI] was shown to outperform the 0.5 M Li[TFSI] solution in [C1C4pyrr][TFSI], at current densities from 1C up to 5C (although the trend is reversed at low C-rates).195 In this study, however, a film-forming agent, vinyl ethylene carbonate (VEC), was needed, due to the limited cathodic stability of [HC4pyrr]+.
Complementary to the experimental investigations into the use of PIL electrolytes in LIB, MD197 and DFT188 simulations have been used to investigate their structure, inter-ionic interactions, Li+ solvation, and ionic mobility. In 2017, Ray et al. showed that inter-ionic interactions in pyrrolidinium-based AILs are weaker than their PIL counterparts and that the PIL structures were resilient to the addition of Li salts.197 Their work concluded that Li[TFSI] coordination took place by gradual destruction of the H-bonding network in PILs, matching the 2014 report that the Li+ coordination number is lower for PILs than AILs. Li-ions were shown to be trapped in cages formed by [TFSI]− anions and the authors proposed tuning of the interactions between the IL anion and cation, as well as the Li+ salt concentration, to optimise electrolyte performance. It is worth noting that although their classical and ab initio MD simulations displayed self-consistency, no experimental validation of these models was presented. Follow-up work illustrated that [TFSI]− anions approach Li-ions more closely in AILs than PILs, giving rise to greater conductivities in the latter.187 Moreover, the simulations highlighted the need to minimise the formation of Li-ion aggregates, and that the ease of solvation of Li+ ions is inversely proportional to the strength of inter-ionic interactions. In this case, the Li+–Li+ networks described by the MD simulations in this work could be used to rationalise experimentally determined coordination numbers, providing some validation for the approach used. Nasrabadi et al. supported the proportionality between Li+ mobility and interionic interactions, but extended simulations to include the triethylammonium cation, [HN222]+.181 Again, cation–anion distances in the PIL barely changed upon the addition of Li+ salt, but this study recorded the differences between the trans (monodentate) and cis (bidentate) configurations of [TFSI]− around Li+, with the monodentate binding more highly favoured in the PIL electrolyte, due to hydrogen-bonding of protonated cations to [TFSI]− (Fig. 20). It should be noted however, that at the chosen charge-scaling factor, significant deviation between their simulated values and experimentally determined values were observed for the cation self-diffusion coefficient, viscosity, and ionic conductivity. Moreover, neutron scattering experiments could be used to provide greater validation the simulated pair radial distribution functions presented in this work.
 | ||
| Fig. 20 Diagram showing the monodentate and bidentate configurations of the [TFSI]− anion wherein lithium, oxygen, nitrogen, sulfur, carbon, and fluorine are shown in green, red, blue, yellow, cyan, and pink, respectively. Reproduced from ref. 181 with permission from American Chemical Society, copyright 2023. | ||
This work nonetheless gave further theoretical backing to the higher Li+ conduction observed in the PIL versus the equivalent AIL, shown to be maximised at an xLi = 0.2. Recent DFT calculations investigated the graphite surface in the presence of a range of PILs, comparing these electrode–electrolyte interactions with those of similar AILs.188 It was shown that the shortest hydrogen-bonds formed between the cationic protons and the O, N, and F atoms of the anions, supporting stronger inter-ionic interactions in PILs versus analogous AILs. In PILs, anions tend to be located close to the cationic proton, but this additional characteristic appears not to affect the cation's adsorption on graphite significantly. The anions differ in their distribution, with anions in the protic pyrrolidinium systems located above cations, not interacting with the electrode surface, whereas there is some anion–graphite interaction in AILs. However, large differences were seen when changing between pyrrolidinium, ammonium, and imidazolium cations, highlighting the intricate relationship between PILs and graphite surfaces. This suggests further theoretical and experimental work should be carried out to understand electrode–electrolyte interfaces, particularly with other electrode materials.
Alongside investigations into alternative electrolyte systems for LIBs, work has been undertaken to examine the potentially advantageous use of PILs in other alkali-ion technologies. General research into sodium-ion batteries (SIBs) gained traction in 2000 with the discovery of a highly reversible system using hard carbon,198 but it was not until 2016 that the first study of PILs in SIBs compared their performance to similar AILs for two traditional sodium electrode materials, sodium vanadium phosphate (NVP) and sodium manganese magnesium oxide (NaMM).199 The authors found that, despite the lower cathodic stability of the PIL, a higher discharge capacity could be attained for NVP electrodes cycled at the same C-rates. Nevertheless, this PIL still performed worse than the incumbent carbonate–electrolyte system, for which the authors should be commended for their inclusion, as this enables a more holistic assessment of the potential for PILs in SIBs (Fig. 21). Interestingly, despite higher initial capacity using traditional electrolytes compared to PILs, greater capacity fade was observed in the former case. Both PILs and AILs demonstrated a greater coulombic efficiency than the carbonate, illustrating that these systems may present reduced performance but greater durability, indicating a need for longer-term cycling studies.
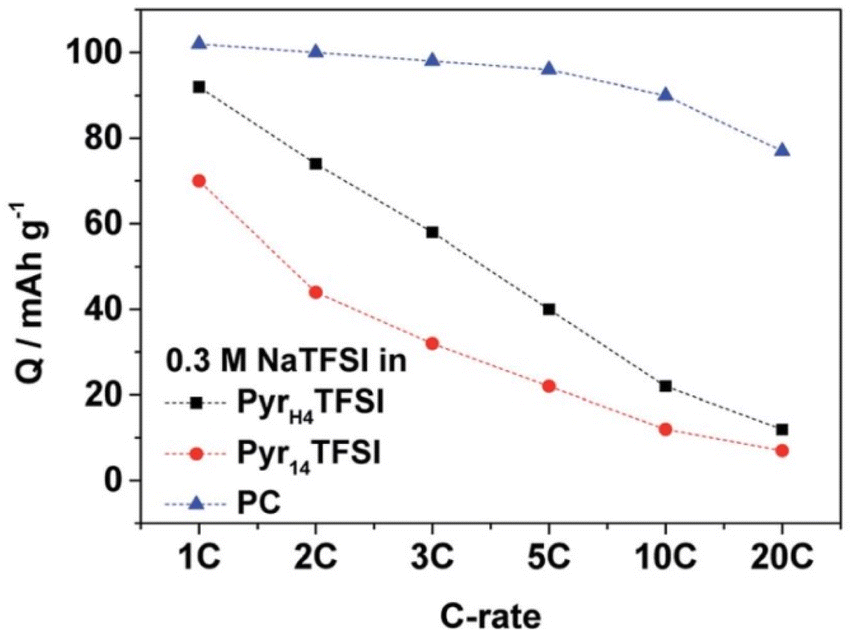 | ||
| Fig. 21 Plot of sodium-ion battery discharge capacities in PIL, AIL and traditional electrolytes, reproduced from ref. 199 with permission from Royal Society of Chemistry, copyright 2023. | ||
The discharge capacities at high C-rates were greater for the PIL than for the AIL, but unlike the LIB case, the performance at high current densities comparable to traditional electrolytes was not observed in the SIB system. Unfortunately, rapid capacity fade was seen in NaMM when using the PILs in this study, again highlighting the importance of pairing the appropriate electrode material with the PIL electrolyte in question.
Further work on SIBs was carried out in combination with hard carbons, but the PIL investigated, [HC4pyrr][TFSI], did not give a working battery. Even with a film-forming additive, the electrolyte was unstable below ca. 1.2 V.195
Given the low potential of K/K+, weak solvation energy of K-ions, and its high transport number, potassium-ion batteries are seen as appealing alternatives to LIBs, given environmental and societal challenges in sourcing both lithium and cobalt.200 Recently, Arnaiz et al. investigated the use of both AILs and PILs in combination with hard carbon and K[TFSI]. Although [C1C4pyrr][TFSI] demonstrated reversible K+ insertion/extraction without the use of VEC, the [HC4pyrr][TFSI] electrolyte did not work even in the presence of VEC, failing to form a stable solid-electrolyte interphase (SEI). This is, however, the only study of PILs in potassium-ion batteries. Similarly, a study on calcium-ion devices, combining 20 mol% PC with [HC4pyrr][TFSI] or [C1C4pyrr][TFSI], demonstrated that only the aprotic electrolyte sustained stable cycling.194
Overall, the application of PILs in alkali-ion batteries is only a decade old, but promising signs relative to their AIL counterparts have been observed, simulated, and rationalised in the Li-ion case. Studies have successfully demonstrated improved performance at higher C-rates and lesser capacity fade in some cases using PILs. Early indicators from sparse work on K-ion and Ca-ion batteries are less encouraging, but the potential improvements associated with the substitution of an AIL with a PIL in SIBs using NVP cathodes and activated carbon anodes are promising, and more research should be directed to uncovering the full potential of PILs in this area, particularly by taking a holistic approach to long-term stability, cost and safety credentials.
Despite improved Li-ion transport in most PILs when compared to AILs, the performance of such devices rarely matches the incumbent technology using traditional carbonate electrolyte systems. Nonetheless, when the improved safety credentials of these non-flammable electrolytes are considered, slight decreases in performance, seen for some PIL systems, could be a compelling compromise. Indeed, more robust and detailed investigations into safety gains and techno-economic assessment of these systems is needed to quantify green metrics for these applications and understand the potential for PILs to be used in future LIBs and SIBs.
To conclude this section, the authors would like to highlight unconventional uses of PILs in alkali-ion batteries, quite apart from their application as alternative electrolytes. A unique example of the application of PILs in alkali-ion batteries was demonstrated in 2015, wherein Zhang et al. used PIL precursors to produce carbon-coated cathode nanoparticles of LVP for use in high-power LIBs.201 LVP suffers from intrinsically low electronic conductivity and capacity fade, arising from side reactions with electrolytes at high potential. Here, a nanoparticulate cathode active material was synthesised within a micron-sized carbon matrix, restricting particle growth, and increasing electronic conductivity. Only a small amount of PIL was required, keeping costs low and high C-rates of 50C were shown to yield high capacities which were maintained with less than 30% loss over 10000 cycles. Certainly, this avenue of study could be expanded to cover other high-capacity electrodes that suffer from low electronic conductivity.
Metal-based batteries
In a bid to access higher energy and power densities, metal-based battery technologies are being pursued.176 This includes Zn–air secondary batteries,203 safer solid-state Li metal batteries,204 and improving the durability of novel chemistries such as Li–S205 and Li–O2.206An early example of the use of PILs in metal-based batteries can be found in the work of Liu et al., who investigated their use in Zn–air batteries.207 Barriers to this technology include short circuits caused by dendritic growth of zinc, passivating ZnO films growing on Zn electrodes, and both evaporation and carbonate precipitation in alkaline aqueous electrolytes. Although there were previous examples of AILs used to demonstrate dendrite-free Zn–air batteries, this work first presented the use of a PIL, [HC1im][TfO], to dissolve ZnO and produce both pure PIL and PIL/H2O mixtures as electrolytes. The study stopped short of investigating full charge–discharge cycling behaviour, but the authors were able to show that quasi-reversible Zn plating/dissolution was possible when using 70:30 PIL:H2O mixtures. This again demonstrates ionic liquid–solvent mixtures as a separate group of formulated, 4th generation ionic liquids.67 Follow-up work that implements this and similar PILs should be carried out in full cell zinc–air batteries to examine electrolyte-electrode interactions in an operating cell.
A 2019 study on the plating behaviour of Na metal, by Jankowski et al.,208 employed a proton on the PIL anion, in a so-called anion amphiprotic ionic liquid, originally designed for use as proton-conducting materials in fuel cells. 1-Ethyl-3-methylimidazolium trifluoromethylsulfonylamide, [C1C2im][TFSA], was shown to have superior ionic conductivity versus [C1C2im][HSO4], and comparable to that of its analogous AIL, [C1C2im][TFSI]. Addition of Na[TFSI] decreased the overall ionic conductivity, but a reduction in overpotentials at high concentrations was observed in Na plating/stripping tests. This improvement was attributed to enhanced Na+ conductivity in highly aggregated clusters. The authors also speculated that the proton on the anion increased Na+ transport due to H+/Na+ exchange, although further studies are required to confirm this.
Recently, the focus has shifted to the potential of testing PILs in full cells containing alkali-metals,209,210 such as Li and Na, for newer technologies beyond LIBs. AILs have shown some promising performance in polymer-based batteries, as well as Li–air and Li–sulfur batteries,211 but due to the narrower ESW of PILs, electrolyte degradation and the hydrogen evolution reaction (HER) can occur, if not mitigated, at the metal anode. Attempts to limit the contact between PILs and Li metal surfaces have included both the envelopment of PILs inside cross-linked polymers (Fig. 22)209 and the use of vinylene carbonate (VC).209,210 VC was shown to form an effective protective layer that minimised contact between [HC4pyrr][TFSI] and Li metal, and although only at a low C-rate of C/20, specific discharge capacities of >150 mA h g−1 were achieved.209 In a follow-up study, an increase in stability due to the formation of poly(VC) on the anode surface was demonstrated, whereby the PIL-based electrolyte was shown to be stable at the potential at which Li plating takes place.210 Cells with Li-metal anodes and LFP cathodes were shown to retain 75% capacity after 50 cycles, and when using the [FSI]− anion, high-voltage nickel–manganese–cobalt cathodes could be used. Nonetheless, further investigations to optimise the VC content are required to reach significant cycle stability, as the long-term durability of these systems is yet to be proven.
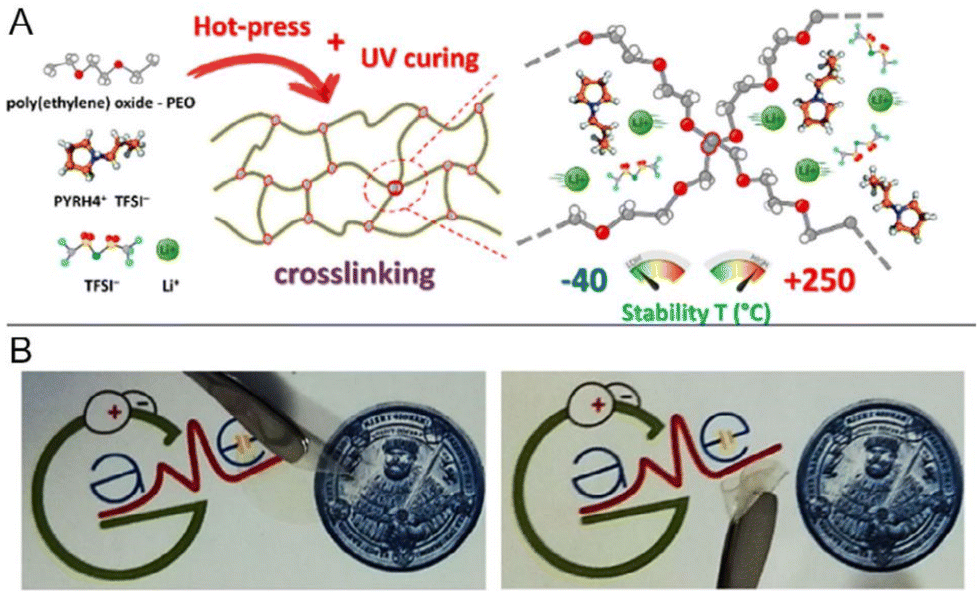 | ||
| Fig. 22 A – Schematic showing the formation of cross-linked polymer with PIL and Li salt, B – Photographs of polymer electrolyte membrane displaying robustness and elasticity. Reproduced from ref. 209 with permission from Wiley-VCH, copyright 2023. | ||
Another application of PILs in the context of metal-based batteries is as additives to Pb-acid batteries (which are increasingly used in electric vehicles for start-stop and energy recovery systems), to widen the ESW and to potentially inhibit corrosion of current collectors.212 It was found that PILs with shorter chains showed higher thermal stability but that the longer the side chain, the greater the reduction in the potential at which the HER occurred. The oxygen evolution reaction was relatively unchanged, such that [HN11,16][HSO4] could be added to the positive electrode and increase the ESW, which resulted in improved performance. This resulted in reduced corrosion currents, particularly with longer side-chain PILs, but slightly higher self-discharge.212 This work is an example of the application of PILs to an older battery technology. Nonetheless, given the continuous prevalence of Pb-acid batteries in traditional applications (uninterruptible power supplies) and their adoption in many electric vehicles for specific functionality, the use of cheap PILs to increase cycling capacity should be subject to techno-economic analysis to establish industrial viability.
The development and implementation of PILs in electrolyte systems that come into contact with metal electrodes is still at an early stage, but recent promising results suggest that despite their inherent reactivity, there may be sufficient means to engineer passivating layers that enable gains versus their harder-to-prepare AIL analogues. The potential for PILs in non-alkali metal-based batteries (additives in Pb-acid systems, solvents in Zn–air batteries) is yet to be explored comprehensively. Notably, there are no examples of PILs used in Li–sulfur or Li–air battery systems. If this was enabled via the use of appropriate passivating agents, access may be granted to much higher density devices.
Supercapacitors
Supercapacitors (also known as electrochemical capacitors), have a higher energy density than dielectric capacitors, and are increasingly sought after for applications where high power density is required. This includes portable and flexible electronics,213 hybrid electric vehicles,214 and uninterrupted power supplies.215 They can be classified either as electrical double-layer capacitors (EDLC), which store charge purely via non-faradaic processes, or as pseudocapacitors that are dominated by reversible redox reactions (faradaic processes) occurring at electrode–electrolyte interfaces,216 or as hybrids. However, the energy density of most supercapacitors remains low, such that significant research is focused on both increasing their specific capacitance and their accessible voltage window, whilst exploring more sustainable routes to producing electrode and electrolyte materials.ILs were first used in EDLCs as salts dissolved in traditional organic solvents, limited by the ESW of the solvent.217 Subsequent studies focused on pure ILs as electrolyte systems, enabling greater energy densities by accessing higher voltages, but facing issues of low conductivity and high viscosity. Nonetheless, a wide range of AILs have been employed as electrolytes and tested in supercapacitors with various electrode materials, which has been comprehensively reviewed elsewhere.218,219 Lately, PILs as electrolytes have also been the subject of research attention, particularly with regards to pseudocapacitive contributions, though their narrower ESW limits accessible energy densities.220 The use of a range of PILs as liquid221 and solid gel222 electrolytes has been recently reviewed. A separate application of PILs associated with supercapacitors, however, is their use as carbon precursors to produce porous carbons for use as electrode materials.223 This was last reviewed in 2017.224 Here, we have decided to refer the reader to the recent reviews on the use of PILs as electrolytes in supercapacitors, and focus on more recent developments in the synthesis of carbon materials from PIL precursors.
Much effort has focused on the production of carbon electrode materials with controlled structures, using a range of hard and soft templating methods, to attain high surface areas, large pore volumes, and heteroatom doping. It has been known for some time that mesopores (2–50 nm) facilitate mass transport, micropores increase active sites, and heteroatoms could improve wettability and oxidative stability.225 Given the limited availability of suitable precursors for N-doped carbon materials, as most organics evaporate or completely decompose at carbonisation temperatures,225 ILs as such were naturally of interest as potential non-volatile precursors.226 High costs and multi-stage synthetic procedures for AILs led Zhang et al. to explore the use of PILs as low-molecular weight organic precursors for the direct synthesis of porous carbons.227 They demonstrated that easy-to-prepare PILs containing nitrogen atoms could be carbonised to yield N-doped porous carbons and highlighted the tunability of the approach, given the simple acid and base starting materials. Evidence showed that nitrogen doping gave rise to improved conductivity, basicity, and oxidative stability due to interaction of nitrogen's free electron pair with the carbon π-system.225 Although this work surveyed an impressive array of PILs, investigating the influence of precursor structure on the resultant doped carbon material, the study stopped short of applying these electrode materials in supercapacitor devices.
Fechler et al. had highlighted a possible synergistic interaction between sulfur and nitrogen atoms in co-doped carbon materials derived from AILs, suggesting increased surface density of active sites and improved catalytic reactivity.228 Many research groups thus explored the use of PILs containing both N and S atoms to produce co-doped porous carbons for use as electrodes in supercapacitors (Fig. 23).216,229–232
 | ||
| Fig. 23 Schematic illustrating the use of PILs and templating agents to produce hierarchically porous structures for use as supercapacitor electrodes, reproduced from ref. 231 with permission from American Chemical Society, copyright 2023. | ||
Throughout these studies, a templating agent, such as colloidal silica,225 F-127,229,231,232 or sodium dodecyl sulfate,229,232 has been used to create mesopores, whilst micropores were shown to result from the release of SO2 and NH3 during high-temperature carbonisation. There has been a focus on producing hierarchical pore structures that combine the benefits of different pore size regimes: micropores for charge storage, mesopores providing fast ion-transport, and macropores providing large ion reservoirs.229 Thus, the smallest pores are more associated with electrochemical capacitance, whereas mesopores and macropores give rise to high-rate capacitive performance. In addition to the use of soft templates, activation using KOH or other chemical activators233,234 have been used to generate ordered porous carbons with high surface areas (Fig. 24). The implications of a high-temperature template removal step or a harsh chemical activation on the green credentials of these synthetic routes remains unquantified.
 | ||
| Fig. 24 SEM micrographs of hierarchically porous carbons made from carbonisation of PILs based on lysine (a) without activation; (b) activated by NH4Cl; (c) activated by KOH; (d) activated by K2CO3 and (f)–(h) showing EDX mapping of porous carbons derived from lysine with K2CO3 activation, showing uniform heteroatom doping. Reproduced from ref. 233 with permission from American Chemical Society, copyright 2023. | ||
As shown in Table 2, early N/S co-doping work from 2015 to 2018 predominantly used PILs containing the [HSO4]− anion, combining it with a nitrogen-containing cation such as 1,10-phenanthrolinium, [Phen]+,225 phenothiazinium, [Phne]+,230,231 or N-methylglucamine, [Megl]+.229 Supercapacitor testing was carried out in 6 M KOH or in more environmentally benign 1 M Na2SO4. The porous carbon electrodes derived from [Phne][HSO4] had the highest specific capacitance in alkaline conditions, reaching 330 F g−1 at a current density of 1 A g−1,230,231 likely due to high sulfur content deriving from both anion and cation of the PIL precursor. When activated at 700 °C with KOH, this porous carbon material had a significantly higher surface area of 2010 m2 g−1 (cf. 575 m2 g−1 for its unactivated analogue) and displayed better high-rate capability. This approach highlights the potential for a combination of soft templating and chemical activation to achieve high-performance supercapacitor electrodes. On the other hand, in neutral conditions, electrodes derived from [Phen][HSO4] demonstrated higher specific capacitances,225 suggesting that the specific interactions between electrodes and electrolytes requires further study in order to understand how to optimise performance when using a particular electrolyte.
| Year | PIL | Surface area (m2 g−1) | V tot (cm3 g−1) | Heteroatom content (atom wt%) | Electrolyte | Specific capacitance/F g−1 | Stability cycles | Max ED (W h kg−1) | PD at max. ED (W kg−1) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <1 A g−1 | 1 A g−1 | 2 A g−1 | 5 A g−1 | 10 A g−1 | 20 A g−1 | 50 A g−1 | ||||||||||
| V tot = total pore volume; ED = energy density; PD = power density. | ||||||||||||||||
| 2015 | [Phen][HSO4]2 | 1161 | 2.49 | 8.23 (N) | 1 M H2SO4 | — | 156 | 158 | 155 | 153 | 140 | — | 10000 |
— | — | 225 |
| 6 M KOH | — | 145 | 133 | 128 | 120 | 107 | — | 10000 |
9.6 | 493 | ||||||
| 2017 | [Phne][HSO4] | 2010 | 0.76 | 2.43 (N), 6.06 (S) | 6 M KOH | 390 (0.5 A g−1) | 330 | 265 | 248 | 210 | — | — | 5000 | — | — | 230 |
| 2017 | [Phne][HSO4] | 575 | 0.55 | 3.41 (N), 6.65 (S) | 6 M KOH | — | 320 | 260 | 210 | 175 | — | — | 5000 | — | — | 231 |
| 1 M Na2SO4 | — | 54 | 48 | 42 | — | — | — | 3500 | — | — | ||||||
| 2018 | [Megl][HSO4] | 1210 | 0.51 | 5.32 (N), 3.02 (S) | 6 M KOH | — | 251 | — | — | — | 174 | — | 5000 | — | — | 229 |
| 1 M Na2SO4 | 157 (0.5 A g−1) | 150 | 145 | 140 | 133 | 125 | — | 5000 | 15.8 | 212 | ||||||
| 2018 | [pPD][CH3PhSO3]2 | 986 | 0.59 | 2.98 (N), 0.73 (S) | 6 M KOH | — | 280 | 230 | 185 | 176 | 160 | 134 | 10000 |
7.4 | 487 | 216 |
| 2019 | [Chit][HSO4] | 927 | 0.47 | 5.13 (N), 2.51 (S) | 6 M KOH | 302 (0.5 A g−1) | 228 | 200 | 190 | 180 | 173 | — | 5000 | 9.7 | 250 | 232 |
| 1 M Na2SO4 | 130 (0.5 A g−1) | 127 | 120 | 115 | 105 | 99 | — | 5000 | 17.6 | 250 | ||||||
| 2019 | [Lys][HSO4]2 | 900 | 0.38 | 3.82 (N), 1.12 (S) | 6 M KOH | — | 155 | 144 | 135 | 120 | 100 | — | 5000 | 6.9 | 248 | 233 |
| 2 M Na2SO4 | — | 108 | 85 | 38 | 10 | — | — | — | — | |||||||
| 1 M H2SO4 | — | 115 | 106 | 87 | 67 | 28 | — | — | — | |||||||
| [Lys][HSO4]2 | 2959 | 2.00 | 1.07 (N), 0.99 (S) | 6 M KOH | — | 350 | 321 | 300 | 290 | 270 | — | — | — | |||
| K2CO3 activation | 2 M Na2SO4 | — | 213 | 152 | 109 | 86 | 45 | — | — | — | ||||||
| 1 M H2SO4 | — | 310 | 267 | 208 | 175 | 133 | — | — | — | |||||||
| 2021 | [Pyr][HSO4] | — | — | 7.43 (N), 2.26 (S) | 1 M H2SO4 | 290 (0.7 A g−1) | 283 | 263 | 225 | 173 | — | — | 1000 | 6.3 | 280 | 237 |
| 2021 | [Arg][H2PO4]2 | 413 | 0.32 | 2.28 (N), 2.13 (P) | 6 M KOH | — | 150 | 133 | 105 | 61 | 41 | — | — | — | — | 235 |
| [Arg][H2PO4]2 | 788 | 1.06 | 2.37 (N), 2.90 (P) | 6 M KOH | — | 260 | 235 | 205 | 186 | 157 | — | — | 12.5 | 445 | ||
| (NH4)2(HPO4) activation | 1 M Na2SO4 | — | 138 | 130 | 87 | 56 | 12 | — | — | — | — | |||||
| 1 M H2SO4 | — | 168 | 155 | 125 | 90 | 38 | — | 10000 |
— | — | ||||||
| 2021 | [Thr][H3BO3]/CTAB | 946 | 0.40 | 1.6 (N), 0.90 (S) 2.3 (B) | 6 M KOH | — | 242 | 224 | 205 | 180 | 140 | — | 10000 |
5.1 | 250 | 234 |
| 2022 | [Ser][H2PO4] | 845 | 1.41 | 4.19 (N), 0.81 (P) | 6 M KOH | 223 (0.5 A g−1) | 214 | 202 | 187 | 172 | 167 | 165 | 10000 |
13.0 | 163 | 236 |
| F127 + Co(NO3)2·6H2O | ||||||||||||||||
Interestingly, from 2019 onwards, there has been a tendency towards carbonisation of bio-derived PILs, including from chitosan232 and from amino acids by the combination of lysine,233 arginine,235 threonine,234 or serine cations,236 with either [HSO4]− or [H2PO4]2− anions. A comprehensive study of 21 amino-acid PILs was carried out by Zhou et al. that showed that the greatest carbon yields were derived from aromatic amino acids, and of the aliphatic amino acids, lysine-based PILs gave the highest yield.233 This study investigated the use of NH4Cl, K2CO3 and KOH activation, showing that K2CO3-activation of [Lys][HSO4]2-derived electrodes led to highly ordered porous carbons (Fig. 24) with the highest specific capacitance to date, 350 F g−1 (at 1 A g−1). This is particularly encouraging given the more benign nature of K2CO3vs. KOH. On the contrary, due to the lower voltage window (0–1 V) associated with this material, its maximum energy density (6.9 W h kg−1 at 248 W kg−1) is far below the maximum energy density measured in supercapacitors derived from arginine-based PILs (12.5 W h kg−1 at 445 W kg−1)235 and serine-based PILs (13.0 W h kg−1 at 163 W kg−1),236 given their wider voltage windows.
These latter studies demonstrated the inclusion of P atoms by use of phosphate in the place of sulfate anions.235,236 The introduction of phosphorus was shown to increase wettability and decrease charge transfer resistance, while widening the potential window by protecting unstable active sites, enhancing both oxidative and reductive stability.236 In the work by Zhou et al., poly(vinyl alcohol) was used to make a gel electrolyte of the amino-acid PIL and activation by (NH4)2(HPO4) in a 1:2 precursor/activator ratio produced a high-performing device in 6 M KOH, with a P content of ca. 3 at%. A lesser amount of phosphorous was introduced by carbonisation of a serine-based PIL (<1 at%),235 but nonetheless a high maximum energy density was observed due to the wider voltage window (0–1.3 V).235 The potential to introduce multiple heteroatoms that increase specific capacitance and operating voltage windows, whilst simultaneously forming hierarchical porous carbon structures from simple amino-acid PILs, is promising, and more research should focus on morphology optimisation while retaining high heteroatom content from these sustainable precursor systems.
Wang et al. recently demonstrated a means to introduce boron into porous carbons, as B atoms are thought to impart greater oxidative resistance whilst also favouring graphitisation.234 In this work, [Thr][HSO4] was self-assembled into a 3D pore network using a cheap cationic surfactant (cetrimonium bromide) and boric acid. Upon carbonisation, boric acid acted both to introduce boron into the electrode surface but also as a pore-forming agent, yielding an electrode that demonstrated a high specific capacitance of 242 F g−1 (at 1 A g−1) when using 10 wt% CTAB and a 1:1 ratio of threonine and boric acid.234 Nevertheless, a suitable benchmark using state-of-the-art electrode materials fabricated by traditional means was not included in this study, limiting the understanding of the extent to which these boron-doped carbon electrodes might be competitive.
There are several examples of PILs being used effectively to give rise to hierarchical porous carbons for use as supercapacitor electrodes, with significant heteroatom doping, and a recent trend towards amino-acid precursors and the co-doping of a mixture of N and either S, P or B atoms (Table 2). Recent work by Wu et al. stands out, however, for its use of a metal system to grow crystals that are subsequently removed by the addition of HCl (Fig. 25),235 transforming an otherwise suboptimal microstructure into a highly graphitic structure with a more suitable pore size distribution. Although it may be argued that metal-free synthesis is more appealing following green chemistry principles, the use of this removable catalyst may unlock the potential of other amino-acid PILs, with high carbon yields but otherwise unsuitable microstructures. A recent study by Al-Zohbi et al. is also notable for using [HHpyrr][HSO4] rather than an amino-acid-based PIL.237 By combining it with polyaniline and nanomaterials, the authors produced electrodes that performed well in acidic conditions (cf. [Lys][HSO4]2 + K2CO3 activation233), but without requiring an activation step.237 Cycle stability was investigated to only 1000 cycles with capacitance retention at 73% for their best system, but this study invites greater exploration of PILs in combination with conductive polymers, particularly as the PIL was shown to give rise to favourable nanofiber-type morphology with high surface areas that are ideal for supercapacitor applications.
 | ||
| Fig. 25 Schematic showing the fabrication of hierarchical porous carbons by use of PILs and metal catalyst (Co(NO3)2·6H2O), reproduced from ref. 236 with permission from Elsevier, copyright 2023. | ||
Overall, it is apparent that studies in this area rarely benchmark the system under investigation against a more classical porous carbon fabrication process, such that assessing the techno-economic balance between traditional and new PIL-based procedures is challenging. A greater understanding of the dynamics at electrode–electrolyte interfaces in these novel systems is only starting to be probed,214,238 and the exploration of their robustness to use in flexible applications is fledgling.235,236 Finally, a recent report that incorporates PILs into cross-linked polymer electrolytes provides inspiration for further exploration in the fabrication of solid-state EDLCs.209
Redox flow batteries
Redox flow batteries (RFB) are secondary batteries that charge and discharge by the redox processes taking place in two liquids, normally on opposite sides of a membrane, that are circulating between tanks and electrochemical cells. Energy is stored in redox active species rather than electrodes, such that RFB are expected to be more durable than metal and metal-ion batteries. Combining this with independent scaling of energy and power, and potentially lower costs for scale-up, the technology promises to provide long-duration, large-scale energy storage.239 The incumbent chemistry is the all-vanadium RFB which has been commercialised, but is limited by inherently low energy density due to its solubility limit in aqueous solutions (2.0–2.5 M depending on supporting electrolyte)240 and the narrow ESW of water (<2 V).For over a decade, researchers have looked at alternative electrolyte systems, including the use of ILs, mainly of the aprotic type. Recent reviews of their use as supporting electrolytes, solvents, active species, and in the formation of membranes can be found here.241,242 PILs then gained attention because of their cheap and single-stage preparation procedures. Recently, three main avenues have been pursued, that of using PILs as solvents to access higher concentrations of active species,243,244 using PILs as part of the fabrication of novel membranes,245 or indeed using PILs as redox-active species themselves.246
In 2020, Zhou et al. showed that [HN122][TfO] could be used effectively to produce asymmetric porous membranes for use in vanadium-based RFBs.245 The PIL was dissolved in a sulfonated poly(ether sulfone) (SPES) solution and via phase inversion and PIL removal, a membrane with a dense SPES layer and a highly porous layer was fabricated. The greater the PIL concentration, the greater the ionic conductivity of the membrane produced. Importantly, a much lower vanadium-ion permeability was observed versus typical Nafion® membranes. As a result, a higher coulombic efficiency was attained, though a lower voltage efficiency meant a comparable energy efficiency was achieved. Nonetheless, this membrane costs less than one-tenth of the price of Nafion®, so research aimed at minimising its Ohmic resistance and thus maximising voltage efficiencies at high current densites should be pursued, which may be achieved by employing an even thinner membrane.
Nikiforidis et al. showed for the first time that [HHpyrr][MeSO3] can be used as a solvent for vanadium active species, increasing the maximum concentration from 2.5 M in purely aqueous solution to 6 M in a mixed PIL–water solution.243 Using this mixed system also overcomes the limitation of upper operating temperature; V2O5 normally precipitates from aqueous H2SO4 at ca. 45 °C, whereas the authors showed their electrolyte could be stored for over 60 days from −20 to 80 °C. The viscosity of the 2 M V4+ PIL–water solution, was only a little higher than a 2 M V4+ aqueous solution (3.86 mPa s versus 0.9 mPa s), though this increased dramatically upon increasing concentration to 6 M. Upon addition of VOSO4 to the PIL, pH dropped to similar values seen in the incumbent system and it was shown that the species likely exists in a wide variety of complexes whereby deprotonated pyrrolidine may act as a ligand. coulombic efficiencies of ca. 85% were observed, but energy efficiencies did not exceed 67% (Fig. 26), which is appreciably lower than that obtained in the standard case (ca. 80–85%). Nevertheless, an energy density of 77 W h L−1 was demonstrated, representing 70% of the notably high theoretical energy density of 112 W h L−1. The authors recommend further work to identify the most appropriate membrane and investigate longer-term cycling stability as only 30 cycles were presented. A follow-up study from the same authors ran a single cell at 40 °C with 3 M VOSO4, demonstrating a coulombic efficiency of ∼90%, but still a low energy efficiency of ca. 64%.244
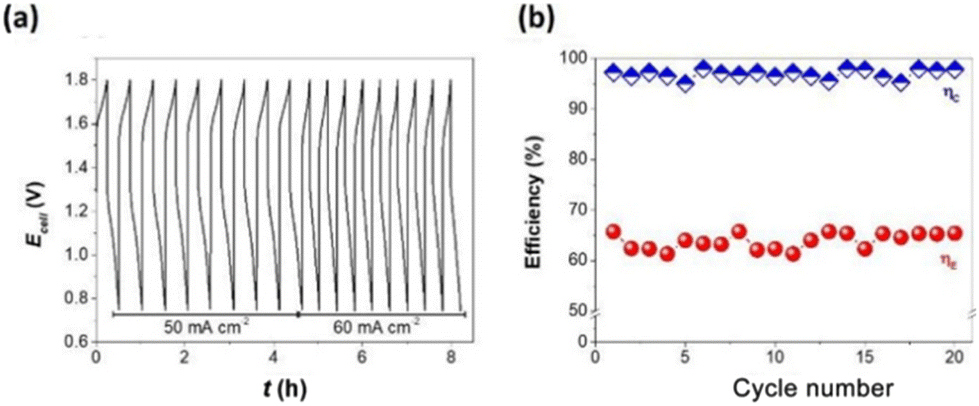 | ||
| Fig. 26 Plots showing (a) the cycling performance of the aqueous PIL at 4 M vanadium concentration; (b) the associated coulombic (blue) and energy (red) efficiency at 60 mA cm−2 and 25 °C, reproduced from ref. 243 with permission from Elsevier, copyright 2023. | ||
A recent investigation of a range PILs as redox-active species for use in RFBs is particularly encouraging in this area.246 The authors state that it is straightforward to synthesise redox-active PILs by combining redox-active organic or organometallic acids with liquid amines and present voltammetric measurements for a range of PILs. Conclusions drawn from this study were that PILs may be able to supply protons to electrochemical reactions even in a basic environment, that it is wise to avoid precursor molecules with acidic functional groups attached directly to aromatic rings, and that is it important to select redox-active species with large free energy differences between both their oxidised and deprotonated forms, and their reduced and protonate forms. The most promising PIL investigated was propylammonium 4-nitrophenylacetate, [H3N3][(NO)2(C6H4)(CO2CH3)], which displayed fully reversible redox behaviour, but full cell testing against a suitable complementary half-cell remains to be explored. Indeed, further work in this area should not only account for electrolyte properties (electrode potential, ionic conductivity, and viscosity etc.), but it is also vital to account for electrolyte–membrane interactions that give rise to cross-over, fouling and therefore battery cycle stability.
Clearly the design and implementation of PILs either as electrolytes or in the fabrication of membranes is at a very early stage. It is promising that mixtures of PILs and water can solubilise typical active species (i.e., vanadium), highlighting the need to explore this strategy for maximising the energy density in non-vanadium systems. Moreover, the recent work highlighting the potential of PILs to act as redox-active species themselves should be inspiration for further analysis of the wide pool of candidate PILs, considering the cautionary conclusions from this work. Lastly, the potential for use of PILs in membraneless designs, as have recently been investigated for their AIL counterparts, should be explored.247
Proton batteries
Given the somewhat labile proton present in PILs, there has also been interest from the proton battery community to utilise them either as components in the battery separator248,249 or as proton-conducting electrolytes.250,251 An early example of the former application was published in 2014 by Mishra et al. in which [HC1C4im][HSO4] was immobilised in a blend of poly(vinylidenefluoride) and poly(vinylpyrrolidone) to afford a gel polymer electrolyte (GPE).245 This proof-of-concept work was the first example of a well-retained PIL within a polymer blend, giving rise to ionic conductivity similar to the liquid state and adequate thermal stability (Td = 130 °C). Proton batteries comprising archetypal Zn/ZnSO4·7H2O anodes and PbO2/V2O5/Graphite/GPE cathodes, sandwiching this GPE as a separator, performed best at low currents, demonstrating a stable cell voltage at 1.54 V with an energy density of 35 W h kg−1, far greater than previous studies (0.1–8 W h kg−1). Nevertheless, at only 50 μA, significant capacity fade was observed over just 10 cycles, which precludes practical use of such a device.An all-organic proton battery was subsequently assembled using a slurry containing substituted pyridine triflates and their corresponding pyridine base, sandwiched between conducting redox polymers with pendant active species (Fig. 27).252 An appreciable discharge capacity of 103 mA h g−1 was observed at 70 mA g−1 (corresponding to a C-rate of C/2) and still had measurable discharge capacity (25 mA h g−1) at a C-rate of 160C. Although the cell potential was limited to ∼0.5 V, this work successful showed that PILs can contribute to novel battery technologies in the absence of metals. Further work on these systems indicated that lower acid-doping level would likely result in improvements in conductivity, processability, and electrochemical stability, thus bolstering future proton battery performance.253
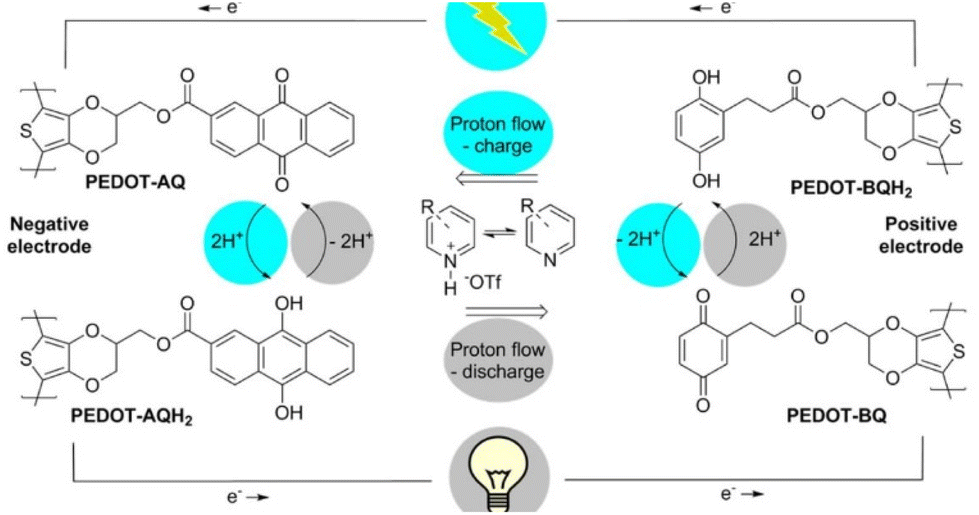 | ||
| Fig. 27 Schematic that shows the chemical structures of the electrodes and electrolyte, and the proton and electron transfer associated with charge (cyan) and discharge (grey) in a proton battery, reproduced from ref. 252 with permission from American Chemical Society, copyright 2023. | ||
A more recent approach has been to polymerise PILs, giving rise to poly(ionic liquids) with high fluidity at RT.248 This study highlighted the importance of non-stoichiometric PILs for effective use in electrochemical devices.252 The authors stated that a low glass transition temperature is needed to ensure high charge carrier mobility and appreciable ionic conductivities in polymer electrolytes. In their work, protic poly(ionic liquids) were made from poly(dimethylsiloxane) backbones with azole groups attached by alkyl thioether linkers, utilising HTFSI as the acid dopant. The resulting electrolytes had ESWs greater than 2 V and good thermal stability (200 °C < Td < 350 °C) but as these electrolytes were not tested in proton battery devices, it is difficult to assess their interactions with the other cell materials, let alone their performance in real-life operating conditions.
One of the first examples of PILs being employed as electrolytes in proton batteries was by Karlsson et al. in which they used non-stoichiometric triazolium PILs.254 This work underlined the importance of carefully controlling the proton activity of the electrolyte and achieved high conductivities (>20 mS cm−1) at room temperature. Similar electrodes were employed as in the first all-organic battery,252 giving similar electrochemical performance, but with the added advantages of lower toxicity and lower melting point than pyridine-based electrolytes. Further work improved the preparation route of the conducting redox polymers used as electrodes in proton batteries, increasing the operable potential difference to 0.8 V, achieving a discharge capacity of 62 mA h g−1, and a capacity retention of 80% after 500 high C-rate (4.5C) charge–discharge cycles.250 Despite low specific capacity versus alkali-ion battery systems, this early-stage development of a new battery system is broadly promising, given the increase in voltage and decreased capacity fade versus previous systems.
Research into the use of PILs in all-organic proton batteries, as with the use of PILs in redox flow batteries, is in its early stages. However, rapid progress in widening the ESW, improving capacity retention, and widening operating temperature ranges supports greater research intensity, particularly with a view to producing the higher-power density, more sustainable, and more recyclable batteries of the future. Metal-free, durable batteries with improved green and safety credentials are indeed appealing and warrant more research and development.
Hydrogen fuel cells
Hydrogen fuel cells, which convert the chemical energy contained in hydrogen into electrical energy, have long been lauded as a green alternative to traditional fossil-fuel-burning technologies, e.g., the internal combustion engine.255 Their operation relies on the exergonic oxidation of hydrogen at the anode, i.e., the hydrogen oxidation reaction (HOR), coupled to the reduction of oxygen at the cathode, i.e., the oxygen reduction reaction (ORR), yielding water as the sole product. Despite their obvious environmental benefits, their use is limited by economic viability, performance, and durability. Currently the most effective hydrogen fuel cell type is the polymer electrolyte membrane fuel cell (PEMFC). It uses a solid perfluorinated polymer membrane, known as Nafion®, whose dual role is to physically separate the half-cell reactions (necessary to prevent gas mixing) and to facilitate proton diffusion from the anode to the cathode. Although Nafion® performs effectively under ambient conditions, its conductivity diminishes dramatically at elevated temperatures (>80 °C) primarily due to dehydration of the membrane.256 As a result, careful control of heat and humidity levels is necessary when operating PEMFCs at elevated temperatures. PEMFCs also rely on the use of expensive noble metal catalysts, most notably Pt, which are expensive and prone to CO poisoning, thus only high-quality hydrogen (>99.999% purity) can normally be used.255 Moreover, the sluggish electrochemical kinetics of the ORR at the cathode remains a persistent bottleneck which limits the operational voltage of the PEMFC at current densities of practical interest (ca. 1 A cm−2).256Significant recent progress in material design has improved the durability and activity of Pt and Pt-alloy catalysts for the ORR. However, there is a thermodynamic limit to the improvements that can be attained through electrocatalytic material design.257 Beyond that, manipulating the chemical environment at the electrode–electrolyte interface where ORR occurs has been shown to enable further improvements.258 For example, incorporation of a thin, hydrophobic layer of a PIL, [7-methyl-1,5,7-triazabicyclo(4.4.0)dec-5-ene][bis(pentafluoroethyl-sulfonyl)imide], [HMTBD][beti], into Pt–Ni catalyst pores markedly improved their performance and durability toward ORR in acidic media.259–262 The effect was attributed to increased O2 concentration, exclusion of interfacial water, and reduced coverage of adsorbates at active sites. Similar enhancements were reported for Pt,261,263 Fe–N and N-doped carbonaceous264 ORR catalysts. The influence of local proton activity at Pt and Au catalytic active sites on ORR rates was recently investigated by Shao-Horn and coworkers.265 PILs containing protic cations of varying pKa formed thin interfacial layers at the electrode–electrolyte catalytic interface (Fig. 28). The results showed that ORR rates were highly dependent on the strength of hydrogen bonds formed between the PIL and the reaction intermediates at the electrode surface. Optimum rates were observed when the pKa of the PIL cation and that of the reaction intermediates were closely matched, resulting in favourable proton tunnelling dynamics. This study elegantly demonstrates the ability of PILs to selectively tune interfacial hydrogen bonding at catalytic active sites, leading to faster proton coupled electron transfer (PCET) reaction rates. This approach has the potential to be extended to other environmentally significant PCET reactions, such as the electrocatalytic reduction of carbon dioxide to produce fuels.266
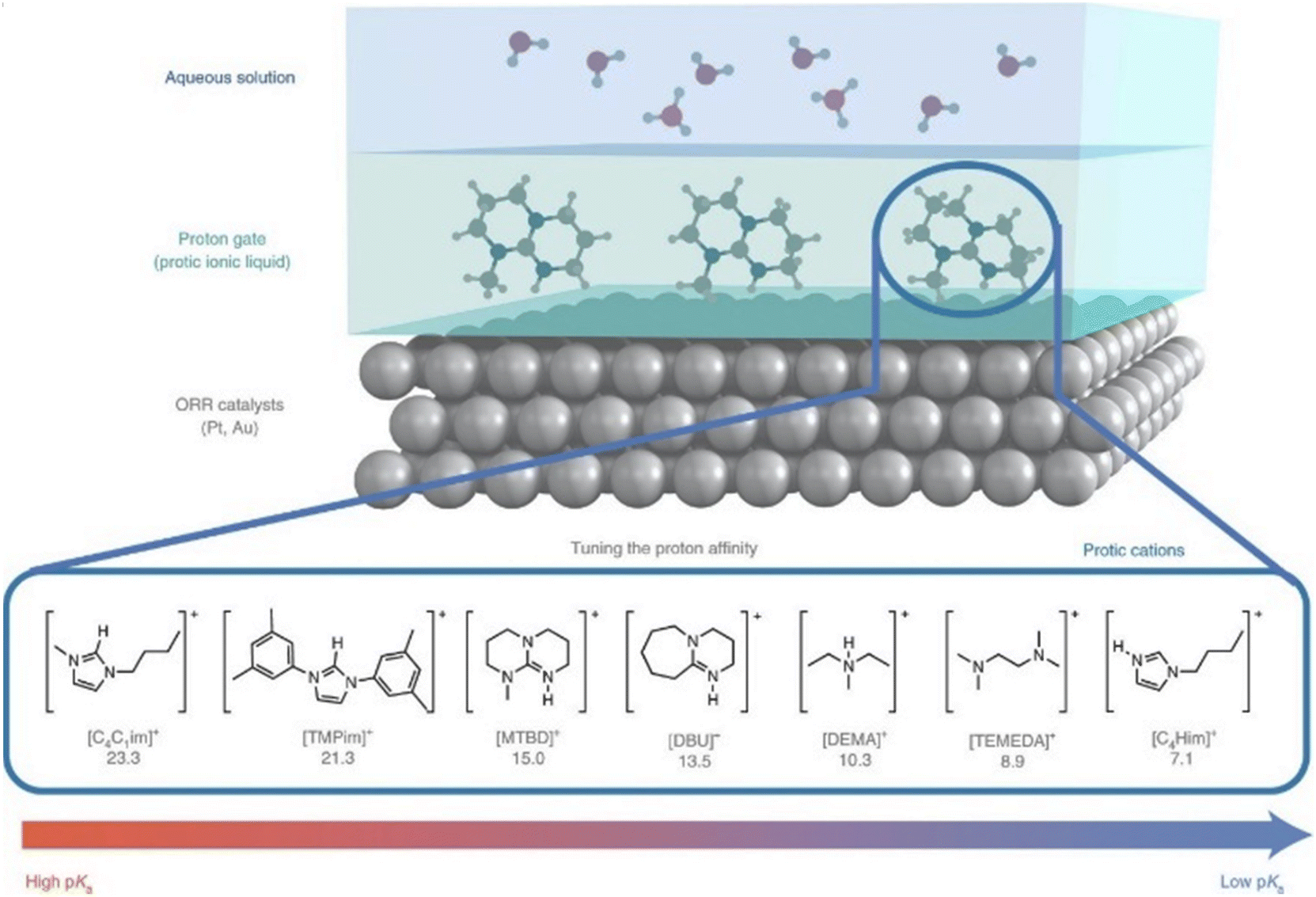 | ||
| Fig. 28 Tuning the local proton activity using PILs at electrocatalytic surfaces for oxygen reduction reaction, reproduced from ref. 265 with permission from Nature Portfolio, copyright 2023. | ||
A separate strategy designed to improve the robustness and activity of the Pt-based catalysts involves raising the operational temperature of the fuel cell beyond 100 °C.7 Under such conditions, the use of anhydrous, thermally stable, proton conducting electrolytes is desirable. In this context, the properties of many PILs, such as low vapour pressure, thermal stability, and availability of active protons, have inspired their use as proton conducting electrolytes, or electrolyte additives, in the development of intermediate temperature (100–300 °C) fuel cells. Progress in this area has been extensively reviewed in the past two years,6,7,267–269 thus our discussion will be limited to a concise overview of both areas, focusing on the current state-of-the-art and future challenges.
Pioneering work by Watanabe and coworkers270–273 demonstrated the proton conducting abilities of neat PIL electrolytes, several exhibiting conductivities >10 mS cm−1 at 120–130 °C. Of these, [HN122][TfO] has emerged as an archetypical PIL electrolyte due to having favourable properties of high ionic conductivity (53 mS cm−1 at 160 °C), low melting point (m. p. = −13.1 °C), high thermal stability (Td = 360 °C) and electroactivity towards HOR and ORR at low overpotentials.272 A fuel cell, using a [HN122][TfO]-Nafion® composite membrane, was shown to produce a power density of 63 mW cm−2 and an open-circuit voltage (OCV) of 0.75 V at 120 °C under non-humidified conditions,274 demonstrating the use of PILs as proton carriers in a practical fuel cell setup. This pioneering work in 2010 laid the foundation for many of the more recent studies shown in Table 3. The introduction of hydrophobic PILs, such as [HN122][TfO] and [HN122][NfO] for example, into the catalyst layer demonstrated a much improved maximum power density of >750 mW cm−2, and decreased Pt/C degradation upon cell reversal conditions.275 On the other hand, only 50 cell reversal cycles were explored and no long-term durability studies were presented. Similar work with another hydrophobic PIL, [MTDB][TFSI], demonstrated slightly lower maximum power density (ca. 600 mW cm−2) but highlighted the importance of selecting the appropriate PIL concentration to avoid blocking catalyst later pores. Indeed, more work is highly recommended in the area of catalyst design and incorporation of hydrophobic PILs in membrane electrode assemblies (MEAs).
| Year | PIL(s) | Catalyst (wt% Pt/C) | PIL content | Catalyst thermal stability (°C) | ECSA (m2 g−1 Pt) | ΔE1/2 (mV) | MA (A mg−1 Pt) | Durability | Pt loading/mg cm−2 | Maximum power density (mW cm−2) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE1/2 change (mV)/MA change (A mg−1 Pt) | Anode | Cathode | ||||||||||
| 2015 | [HMTBD][TFSI] | 20 | 50 vol% | 300 | 81 | 18 | 0.56 (0.90 V) | 9 mV loss in 2000 cycles | — | — | — | 263 |
| 2017 | [HN122][TfO] | 20.5 | 56 wt% | 300 | 74 | 40 | — | 20 mV loss in 50 cycles | 0.15 | 0.25 | 767 | 275 |
| [HN122][NfO] | 74 | 0 | 831 | |||||||||
| 2017 | [HMTBD][beti] | 46.4 | 256 wt% | — | 76 | — | 0.25 (0.90 V) | — | — | — | — | 300 |
| [HMTBD][TFSI] | 73 | — | 0.20 (0.90 V) | — | ||||||||
| [HMTBD][C2F5OC2F4SO3] | 73 | — | 0.22 (0.90 V) | — | ||||||||
| [HMTBD][C6F13SO3] | 76 | — | 0.21 (0.90 V) | — | ||||||||
| [HMTBD][C4F9SO3] | 63 | 20 | 0.24 (0.90 V) | 15% MA loss in 5000 cycles | ||||||||
| 2018 | [HN122][HSO4] + [HDPA][HSO4] | 23.6 | — | — | 42 | −6 | 0.44 (0.85 V) | 14% MA gain in 15000 cycles |
— | — | — | 301 |
| 2018 | [HN122][HSO4] + [HDPA][HSO4] | 23 | — | — | 42 | −6 | 0.44 (0.85 V) | 14% MA gain in 15000 cycles |
— | — | — | 302 |
| [HN122][HSO4] + [PhNH3][HSO4] | 20.4 | — | — | 40 | −17 | 0.34 (0.85 V) | 35% MA gain in 15000 cycles |
— | — | — | ||
| [HN122][HSO4] + 3MT | 21.1 | — | — | 42 | −8 | 0.20 (0.85 V) | 26% MA gain in 15000 cycles |
— | — | — | ||
| 2019 | [HC2im][TFSI] | Fe–NC (non-Pt) | 100 vol% | — | 902 m2 g−1 C | 47 | — | — | — | — | — | 264 |
| 2019 | [HMTBD][TFSI] | 20 | 2 wt% | — | — | — | 0.14 (0.90 V) | — | 0.40 | 0.40 | 605 | 303 |
| 2019 | Poly(DMVBAn-co-Stm) | 20 | 3 wt% | 350 | 82 | 24 | 0.052 (0.90 V) | 16 mV loss in 3000 cycles | — | — | — | 304 |
| 2021 | [HN122][TfO] + EDOT | 10 | — | — | 68 | — | 0.33 (0.90 V) | 31% MA loss in 20000 cycles |
— | — | — | 305 |
| 2021 | [HN122][TfO] + CNT | 24–29 | — | — | 100 | 38.00 | 0.35 (0.90 V) | 6% MA gain in 20000 cycles |
— | — | — | 306 |
| 2021 | [HNRRR][TfO] (PBI) | 60 | — | 540 | 48 | 19 | 0.02 (0.90 V) | 18 mV gain in 2000 cycles | 0.40 | 0.40 | 72 | 307 |
Significant efforts have focused on relating the PIL chemical structure to their thermal and electrochemical properties, as outlined in the introduction. It is increasingly obvious that the physicochemical properties of PILs are not readily predictable based on current models and that a wide range of often overlooked parameters, such as PIL non-stoichiometry (Brønsted acid excess),276 and ion size and symmetry277 are highly influential on their conductive and electroactive properties. Further studies examining the thermal and electrochemical properties of PIL liquid electrolytes, and their relation to PIL composition, are necessary to provide a more complete picture of structure–activity relationships which will aid their design for future fuel cell applications.
For practical application in fuel cells, PILs must be integrated with solid polymeric materials to form thin (ca. 20–50 μm) proton exchange membranes (PEMs) that, ideally, are functional at intermediate temperatures (100–300 °C) under non-humidified conditions. To date, PILs have been integrated with a wide range of conventional polymers, including Nafion®,274,278–280 polybenzimidazole (PBI),279–282 poly(vinylidene fluoride-co-hexafluoropropene) (PVdF-HFP),260 sulfonated polyimide (SPI),283 sulfonated poly(ether ether ketone) (SPEEK)284–286 and, more recently, poly(ionic liquids).287–290 These yield PEMs that, generally, display conductivities in the range 0.1–40 mS cm−1 at intermediate temperatures under non-humidified conditions. Common challenges include PIL leakage from the polymer matrix during operation291 and mechanical weakness (relative to non-PIL functionalised polymers), attributed to plasticising effects of PILs.292,293 Membrane additives, such as silica280,293 and zeolites,281 have been shown to aid PIL retention, whilst mechanical integrity can be improved with judicious choice of PIL, polymer and additive blends for membrane preparation.294–296 Whilst many studies have focused solely on probing the conductive properties of PIL–polymer composite membranes under non-humid conditions, and – in some cases – intermediate temperatures, only a few have examined their performance as MEA components in practical, intermediate-temperature fuel cells.7 In such studies, fuels cells incorporating MEAs based on PIL-modified Nafion,295,297 PBI,282,298 and SPEEK/PBI299 produced impressive current densities (0.3–0.9 A cm−2) and power densities (0.2–0.4 W cm−2) when operated between 100 and 200 °C under anhydrous conditions, as review elsewhere.7 Despite successful fabrication of poly(ionic liquid)-PIL iongel membranes with high conductivity,286–289 these promising materials have yet to be demonstrated in fuel cell devices.
Whilst the performance metrics in the tested systems are encouraging, factors such as long-term stability, gas permeability, response to fluctuating operating temperature, pressure and load variations, catalyst compatibility, and performance using non-purified fuel and oxidant steams (for example <99.99% purity H2 and air) must be thoroughly investigated to establish the feasibility of PIL-integrated PEMs for practical use in next-generation, intermediate temperature fuel cells.
Summary and outlook
Within the area of energy storage and conversion, PILs have been most investigated for their applications in supercapacitors and fuel cells, with an increase in the study of their implementation in lithium-ion batteries over the last decade. Although research into PILs as electrolytes in supercapacitors and as proton conductors in polymer electrolyte membranes in fuel cells continues, interest is growing in both the production of co-doped carbon materials as supercapacitor electrodes and improving electrocatalysis in fuel cells by incorporating PILs at the electrode–electrolyte interface.Despite the ‘labile’ proton in PILs initially discouraging their use in alkali metal-ion batteries, the advantages of lower viscosity, higher conductivity, and potential for cheap and simple synthesis versus analogous AILs have encouraged increasing number of studies. Currently, the main drawback is that their performance does not yet match incumbent technology, even though it surpasses analogous AILs in certain cases. However, advantageous externalities such as safety, cost, and ease of manufacture should be factored in when industrial scale-up is considered.
Pyrrolidinium-based PILs have been most studied, but in many cases, their stable cycling behaviour is only achieved under certain niche conditions, such as at low temperatures,180 with film-forming agents,196 or with specific anode materials.183 Imidazolium systems, although less studied, show more promise and warrant greater investigation.193 Fundamental experimental and theoretical work highlight the lower coordination of Li+ species in PIL versus AIL systems and have shown that the [FSI]− anion is particularly favourable in terms of mass transport, given reduced viscosities. The dominant use of [FSI]− and [TFSI]− anions is, of course, associated with relatively high cost of PILs reported in this section; however, they are used as small scales in contained devices, therefore this high cost is not generally considered to hamstring the implementation of the potential energy storage technologies. At the same time, these electrolytes may be released to the environment, upon device damage or inappropriate disposal, which poses potential threats. This, as well as end-of-life recycling of devices based on highly fluorinated electrolytes, requires more attention from the research community.
This area is undergoing multi-directional, intensive development, with increasing number of applications being explored, and exciting discoveries to be made. Looking into the future – notwithstanding some potential durability improvements during cycling NIBs with NVP electrodes,199 other alkali-ion systems (i.e., K+, Ca2+) have not yet shown promising results, often due to lower cathodic stability, demonstrating greater capacity fade than in traditional electrolytes. Attention should be directed towards the NVP system,199 which indeed presents slightly improved capacity versus a similar AIL system and demonstrates improved cycling stability versus a typical organic electrolyte, with further work should directed at fundamental transport mechanisms. Indeed, increasing durability accelerates the path to commercialisation by meeting application specifications and reducing costs.
Although some examples of PILs having an advantageous effect on metal-plating,207 research into their inclusion in metal-based batteries is fledgling. The use of protective layers to prevent metal-PIL contact is a promising avenue,209 but in-depth compositional and distribution studies involving film-forming agents is required to extend cycling stability to reasonable limits. Indeed, if such micro-engineering were to prove effective for a reasonable number of cycles, the successful implementation of PILs in beyond-Li technologies such as Li–air and Li–S may be unlocked, reducing the need for cobalt in typical high-energy density nickel–cobalt–manganese cathodes.
Research into the use of PIL precursors to produce co-doped carbonaceous electrodes for supercapacitors has grown over the last decade, with a recent focus on bio-based solutions.232,235 Although hydrogensulfate anions are typically used to provide sulfur dopant atoms, achieving maximised specific capacitances was shown to depend on cation selection and activation method, which warrants further study including direct comparison with incumbent activated carbon materials. Of the bio-based approaches, the best-in-class uses a [Lys][HSO4]2 precursor, activated by both potassium carbonate and potassium hydroxide. Although this bio-based precursor direction of study may appear more environmentally sustainable, it is evident that full lifecycle analysis to assess the true benefits of these systems, accounting for their harsh activation steps, is lacking.
Two areas of recent focus have been the use of PILs as flow battery electrolytes and as proton shuttles in proton batteries. In the former case, the lower viscosity and higher conductivity of PILs versus AILs is compelling, although their inherently more viscous nature versus aqueous and common organic electrolytes is a challenge. Recent work highlighting the need to electronically decouple the redox-active centre from any ionizable functional group in PIL systems promises greater investigation in this vein.246 Increasing the accessible energy density and widening the operating temperature window of the vanadium system by selection of the appropriate PIL may also deliver tangible improvements to the incumbent technology if long-term cyclability is established.243
Proton battery technology is nascent but PILs appear to be ideal candidates due to their inherently high proton conductivity. The demonstration of an all-organic proton battery in 2017252 paved the way for further improvements in 2021250 giving rise to a 0.8 V battery with good capacity retention at high C-rate. Nonetheless, longer-term cycle stability studies are still needed to explore its industrial viability.
Although research continues to explore PIL-based membranes for PEMFCs, a promising area of research now focuses on the performance improvements attainable from the inclusion of certain PILs in the catalyst layers of fuel cells. Hydrophobic PILs such as [MTDB][beti] have demonstrated greater longevity and ORR performance when combined with various catalysts in half-cell experiments,259–262,264,265 attributed to increase O2 solubility and greater access to catalytically active sites. Combined with recent studies that highlight the importance of matching the pKa of the PIL with reaction intermediates,265 there is great promise for the implementation of bespoke PILs in real devices, such as operating fuel cells and electrolysers.308,309 The authors recommend further research into PILs in fuel cell catalyst layers, particularly in full-cell systems that more closely mimic real applications.
Overall, there has been increasing research intensity over the past decade in the application of PILs to various electrochemical devices, many of which can be speculated to achieve performance/cost/sustainability balance amenable to deliver a mature, sustainable technology in the future. An important challenge in asserting this lies in prevalent lack of comparative examples of incumbent technologies for a fair assessment of the benefits of these novel approaches (poor benchmarking). Indeed, technoeconomic and life cycle analysis is called for, given the likelihood that some performance and durability compromises may need to be made, albeit with potential sustainability advantages over traditional approaches. It is also incumbent on the field to investigate further the electrode–electrolyte interfaces in these new systems to better understand performance-related micro- and nano-structural changes when these devices are operated in real-world conditions. It is particularly important to investigate the underlying factors that affect the thermal stability and electrochemical performance of PILs to elucidate a more complete picture of structure–activity relationships in these important energy conversion devices.
Electrocatalysis
There has been a growing interest in the use of PILs as electrolytes or electrolyte additives in electrocatalysis, extending beyond reactions relevant to hydrogen fuel cells. Preliminary investigations have demonstrated the reactivity of PIL protons in HOR and OER at Pt electrode surfaces,270,272 indicating their active involvement in PCET reactions. This discovery holds considerable significance as it opens up new possibilities for harnessing this property in various environmentally important PCET reactions, including water electrolysis, lignin oxidation, and electrochemical carbon dioxide and nitrogen fixation. Scaling up such electrochemical reactions in a sustainable and cost-effective manner would be transformative, paving the way for replacing traditional, polluting, chemical production processes like the Haber–Bosch process for NH3 production which is estimated to account for 1.2% of the global anthropogenic CO2 emissions.310 Such ambitions rely on delivering not only superior electrocatalytic materials but highly conductive protic electrolytes with tuneable reactivity and high solvating power, especially for gases. Within this framework, PILs might present significant benefits over the conventional aqueous electrolytes currently in use, particularly in terms of precise proton activity control, which is especially crucial in electroreduction reactions where minimising competitive HER is necessary. Voltammetric studies have demonstrated that the electrochemical reactivity of PIL electrolytes can be effectively modulated by carefully selecting specific anion and cation combinations.311,312 Despite their controllable reactivity, the ability to stabilise reactive intermediates through ionic or hydrogen bond interactions, and capacity for proton transfer, it is surprising that only a limited number of studies have explored these characteristics for PCET electrocatalytic reactions to date. In this summary, we will provide an overview of recent research on the use of PILs in such reactions and offer recommendations for future studies.Water electrolysis
The production of hydrogen through water electrolysis is an attractive pathway for sustainable fuel generation. Currently, only about 4% of worldwide hydrogen production comes from water electrolysis, with the remainder predominantly sourced from steam reforming, a fossil fuel dependent process that is energy-demanding and emits CO as a byproduct.313 Ideally, the electricity powering the electrolyser should come from renewable resources, like solar or wind, ensuring the production of truly emission-free ‘green hydrogen’. Currently, advanced electrolysers function under highly acidic or alkaline conditions, which can have adverse effects on the structural integrity of the electrolyser components, such as electrode corrosion and membrane degradation. Researchers have explored the use of ILs as electrolyte additives to facilitate efficient and stable water electrolysis without the use of strong acids or bases.314 De Souza and colleagues were the first to investigate the use of AILs in the form of alkylimidazolium salts as electrolyte additives for water electrolysis.315 Promisingly, mixtures of these AILs and water, with varying proportions, underwent successful electrolysis to produce hydrogen. The most effective combination, in a 70:30 volumetric ratio, exhibited efficiencies exceeding 95%. However, high overpotentials were observed at low current densities in the region of 15 mA cm−2. Subsequent research conducted by the same group demonstrated that substituting the imidazolium-based AIL with an alkylsulfonic acid-functionalized PIL, specifically 3-triethylammonium-propanesulfonic acid tetrafluoroborate, [N222(CH2)3SO3H][BF4], resulted in an enhanced current density of >1 A cm−2 whilst retaining a similar efficiency (93%) for hydrogen production. Remarkably, these results surpassed those achieved with a 0.7 M KOH electrolyte, indicating the superior performance of the [N222(CH2)3SO3H][BF4] electrolyte when present at an equivalent concentration.316 However, a question remains about the stability of this system and the nature of acidic species within. The [BF4]− anion is known to hydrolyse in both acidic and basic aqueous environments,317 releasing HF in quantities sufficient to etch laboratory glassware, which renders any aqueous applications of [BF4]− systems practically impossible.318
Li et al. provided the first report of a PIL, prepared through a Brønsted acid and Lewis base neutralisation reaction, for water electrolysis.319 Using diethylammonium formate, [H2N22][HCOO], as an electrolyte additive, the authors observed hydrogen evolution at a lower onset potential (−0.002 V vs. RHE) and at an enhanced rate (ca. 0.21 A cm−2) compared to aqueous electrolytes that contained imidazolium-based AILs or inorganic salts at equivalent concentrations. Thimmappa et al. examined the mechanism of hydrogen evolution at a Pt cathode in mixtures composed of [HN122][TfO] and water.320 Voltammetric measurements revealed the occurrence of two distinct HER processes. The first process was ascribed to the reduction of hydronium ions at an onset potential of −0.225 V vs. SHE, while the second process was hypothesised to arise from the reduction of [HN122]+ cations, occurring at an onset potential of −0.475 V vs. SHE. Despite the limited number of studies conducted, the existing research highlights the co-catalytic characteristics of PILs when used as electrolyte additives for hydrogen generation at Pt electrodes. While these studies offer valuable insights, the precise mechanism underlying the observed improvements remains uncertain. It is possible that the enhancements may be linked to the formation of favourable hydrogen bonding networks at the electrode–electrolyte interface, which may lower the energy barrier for hydrogen formation, but this remains a speculation. It would be an important next step to examine whether the durability of water electrolysis cell components, and the catalytic activity of electrodes, are preserved over extended periods in the presence of electrolytes containing PIL additives.
Biomass oxidation
The combination of the capability to dissolve biomass and high conductivity renders ionic liquids as suitable media for the electrochemical conversion of lignocellulosic biomass into value added chemicals. Hempelmann and co-workers first suggested the use of PILs as electrolytes for lignin oxidation, highlighting the core benefits of PILs: labile proton and low cost.321 The authors identified that triethylammonium methanesulfonate, [HN222][CH3SO3], exhibits excellent performance as an electrolyte for lignin solubilisation. When used in conjunction with electrodes coated with ruthenium–vanadium–titanium mixed oxide, [HN222][CH3SO3] facilitates the oxidative cleavage of lignin, resulting in a diverse range of fragmentation products including guaiacol, syringol, and vanillin. A subsequent study showed that addition of water to this electrolyte led to enhanced degradation efficiencies and yielded a higher mass of low molecular weight compounds.322 Ma et al. examined the electrochemical oxidation of phenolic lignin model compounds in electrolytes composed of sulfonic acid-functionalised imidazolium-based IL and water. This study revealed that PIL-containing electrolytes demonstrated vastly superior faradaic efficiencies in the oxidation of these model compounds compared to electrolytes containing ILs without the sulfonic acid group. This superiority was partly attributed to labile protons enabling higher conductivity. The authors also emphasised the role of reactive oxygen species generated during the ORR in inducing an indirect oxidation effect on substrates, which was more pronounced in the case of PILs.323This area is relatively new, and while promising, there are several hurdles to be expected in its applications. Firstly, moving on from model systems to actual biomass will introduce variability of feedstock and impurities that are certain to influence the electrocatalytic oxidation, which necessitates robust feedstock scoping studies. Further on, a common challenge in processing of biomass with ionic liquids is the separation of small, polar molecules from the ionic liquid matrix; judicious choice of PIL systems and target products will be required to assess which processes are practical in terms of product separation and PIL recycling.
Nitrogen reduction
Electrochemical reduction reactions that involve proton transfer from the electrolyte to a substrate, such as the conversion of nitrogen to ammonia, are often hampered by non-selective proton reduction, producing H2 at the cathode which dramatically lowers faradaic efficiency. PILs present an advantageous characteristic of allowing the fine-tuning of proton activity. This capability proves valuable in mitigating undesired parasitic side reactions, such as HER, which are commonly encountered in water-based electrolytes.324 Ideally the proton on the PIL cation should possess sufficient stability to avoid direct reduction at the electrode surface, while also exhibiting the necessary activity to participate in the selective PCET reaction. A notable example demonstrating this principle was shown by MacFarlane and co-workers37 utilising an phosphonium-based IL, trihexyl(tetradecyl)phosphonium tris(pentafluoroethyl)-trifluorophosphate [P666,14][eFAP], known for its high nitrogen solubility,325,326 which served the dual role of electrolyte and co-catalyst for the electroreduction of nitrogen to ammonia in the presence of hydrogen gas (Fig. 29). Whilst this IL would typically be characterised as aprotic, the C–H bond adjacent to the phosphorus cationic centre is activated in the presence of organolithium. The activated protons are combined with electrons and lithium-activated N2 to form NH3. The ylide formed in the process is subsequently reduced back to the phosphonium cation by protons liberated by hydrogen oxidation at the anode, thereby completing the catalytic cycle. The weakly acidic character of the IL suppresses hydrogen evolution at the cathode, enabling ammonia production at a rate of 53 nM s−1 cm−2 with a remarkable faradaic efficiency of 69%. This represents a significant advancement compared to previous studies, where reported values typically fall within the range of 1 nM s−1 cm−2 at faradaic efficiencies of about 30%.37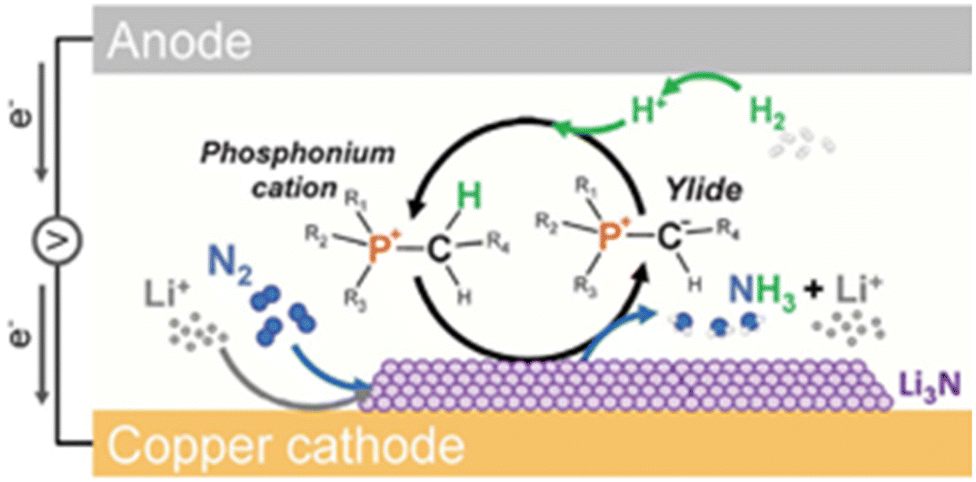 | ||
| Fig. 29 Schematic illustration of the use of a ‘proton shuttling’ phosphonium ionic liquid electrolyte to facilitate the electrochemical reduction of nitrogen to ammonia. Reproduced from ref. 37 with permission from American Association for the Advancement of Science, copyright 2023. | ||
Low-temperature nitrogen activation is one of the Holly Grails of sustainable catalysis, since the current technology to generate ammonia (Haber–Bosch process) consumes 1–2% of global human energy consumption and is associated with enormous CO2 emissions. Recent pioneering papers from the MacFarlane group have attracted enormous interest, as they offer a pathway that is – at least potentially – scalable. From the ionic liquids perspective, the main challenge is posed by the [eFAP]− anion, which is extremely costly and, even if it can be afforded, its salts are difficult to procure in appreciable quantities. It is interesting to see whether the demand for this new application will drive increased availability of this anion, and drive the price somewhat lower (although the level of fluorination poses a barrier to significant cost reduction). While recent reports demonstrate that common alternative anions failed to offer the same performance, it is anticipated that new alternatives will be investigated by the community.
Carbon dioxide reduction
Although the electroreduction of carbon dioxide in IL electrolytes has been the subject of extensive research,327,328 to our knowledge, only Rosenthal and co-workers329,330 explore PIL-containing electrolytes for this purpose. The authors describe the electroreduction of carbon dioxide to formate at a bismuth electrode in an electrolyte composed of acetonitrile, [HDBU][PF6], and tetrabutylammonium perchlorate. During controlled potential electrolysis experiments, formate was generated with an impressive selectivity of ca. 80% at current densities ranging from 20 to 45 mA cm−2.329 Significantly, by substituting the [HDBU]+ cation (pKa = 24) with a less acidic [C4C1im]+ (pKa = 32), the reaction pathway was altered, leading to the dominant production of carbon monoxide, thus highlighting the influence of proton activity on the composition of the generated products (Fig. 30). As always with this family of anions, the question remains whether [PF6]− remains stable under the reaction conditions, or are small quantities of HF being released, which would certainly affect the reaction.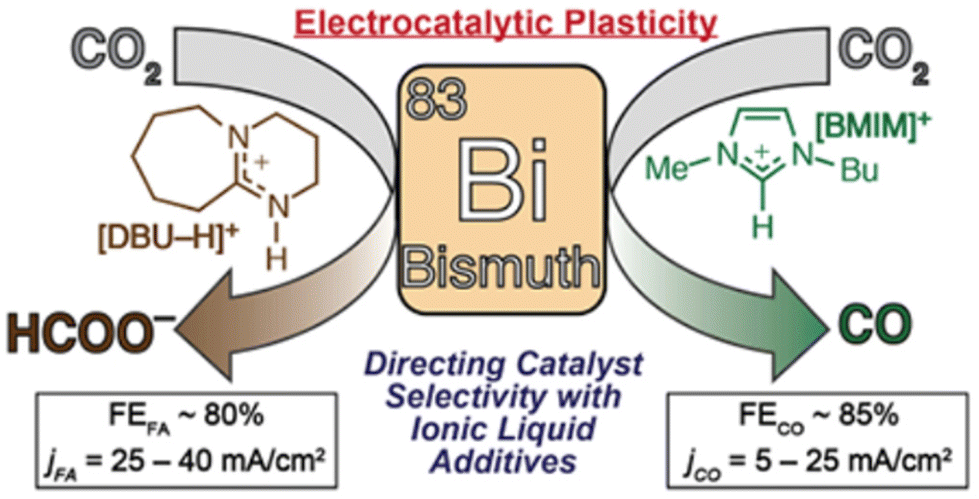 | ||
| Fig. 30 Cation-dependent selectivity in electrocatalytic reduction of CO2. Reproduced from ref. 329 with permission from American Chemical Society, copyright 2023. | ||
In a subsequent study, the group examined the ability to fine-tune product selectivity, using this electrolyte, by adjusting the Ag-to-Sn ratio in silver–tin composite films immobilised on a glassy carbon electrode. The results revealed that films with a higher proportion of Ag favoured the production of carbon monoxide, while films with a higher proportion of Sn resulted in a nearly equimolar mixture of carbon monoxide and formate.330 This again supports the importance of simultaneous development of both the electrolyte and the electrode composition.
Homogeneous molecular electrocatalysis
The previously described studies focus on achieving chemical transformations in PIL-containing electrolytes at catalytically active metallic electrodes. A less explored area of research involves the coupling of PILs with homogeneous (molecular) catalysts to effect chemical transformations at carbon-based electrodes. Frequently, molecular catalysts imitate the role of natural enzymatic active sites, which typically comprise Earth-abundant metal centres and amino acid residues, enabling them to catalyse PCET reactions. In enzymes like hydrogenases, amine groups present in the active site are postulated to act as proton relays and/or serve as localised proton sources, thus reducing the energy barrier required for protonation of the catalytic metal centre.331 It is conceivable that protic alkylammonium cations could fulfil a similar function when present in the coordination sphere of a molecular catalyst. Along this vein, Pool et al.332 conducted a study where they combined N,N-dibutylformamidinium bis(trifluoromethanesulfon)imide, [HDBF][TFSI], with a nickel-based molecular catalyst333 that mimics the function of a hydrogenase for HER. The PIL, whose pKa value closely matched that of the catalyst, served the triple role of solvent (Ni catalyst is water insoluble), electrolyte and proton donor. Significantly, whilst low turnover frequencies were reported for proton reduction in both neat PIL and in PIL–acetonitrile mixtures, rates increased by four orders of magnitude in PIL–water solvent systems. The optimum turnover frequency of 53000 s−1 was achieved when the mole fraction of water was 0.72. While the exact mechanism of catalyst protonation in the PIL–water mixture remains uncertain, it is hypothesised that protons might be transferred to the catalyst either directly from hydronium ions or through a hydrogen bonding network, where water acts as a mediator to relay protons from the PIL to the catalyst.333 This study elegantly demonstrates that the rates of PCET reactions can be dramatically improved through simple solvent engineering using PILs.
Summary and outlook
The utilisation of PILs in electrocatalytic reactions is in its early stages; pioneering papers in the areas such as nitrogen activation and biomass oxidation have been published only recently. The authors are certain that these areas will be developing rapidly, and that further areas of PIL-assisted electrocatalysis will be developed. Currently, it is difficult to point out obvious advantages and disadvantages of PILs in electrocatalysis, since much of the work is very exploratory in its nature, and the full picture is yet to emerge. All the areas reviewed in this section have a significant potential to develop into sustainable technologies, especially if coupled with sustainable sources of electrical energy.Looking into the near future of the field, it is apparent that the attributes of PILs extend well beyond their role as mere proton donors and/or a source of conductive ions in electrolytes. Elucidating the underlying mechanisms that give rise to the apparent co-catalytic properties of PILs in PCET reactions could lead to a more informed search for target applications, thereby unlocking new opportunities. In particular, the growing recognition of the impact of surface activity of PILs on the efficiency of electrochemical PCET reactions334,335 is the likely area of future growth, and a potential source of valuable fundamental understanding. Furthermore, their potential to function as corrosion inhibitors336 and heterogeneous electrocatalyst stabilisers,259,260 renders them as attractive electrolyte components to boost the durability of electrocatalytic reactions, which often face stability challenges.
Conclusions
This review covers four areas of applications: (1) catalytic transformations; (2) biomass treatment; (3) energy storage and conversion; and (4) electrocatalysis, preceded by an extensive introduction, covering the definition and nature of protic ionic liquids. Each of the four application is concluded with a detailed, field-specific “Summary and outlook” part, outlining challenges and opportunities in each field. Rather than repeating these, this section serves as a summary of overall trends in sustainable uses of protic ionic liquids, points out the main pitfalls and indicates future directions of predicted growth.There appear to be two main drivers for the use of PILs in sustainable applications: (1) economic: protic ionic liquids are seen as cheaper and easily synthesised alternatives to their aprotic counterparts, and (2) chemical: the presence of labile proton, which required for example in Brønsted acid catalysis, H+-conducting electrolytes or some electrocatalytic processes.
Each of the four reviewed areas: catalytic transformations, biomass treatment, energy storage and conversion, and electrocatalysis, is at a different stage of development, and relies different families of PILs.
In catalysis, the use as solvent and co-catalyst in transition metal catalysis (traditional angle of research in 2000s) is minimal, with the focus on CO2 capture and activation, as well as Fisher esterification and some other Brønsted acid-catalysed processes, which is a potential growth area. The focus is on the use of inexpensive acids, combined in stoichiometric or non-stochiometric ratios with amines or nitrogen superbases. There are still many studies involving sulfonic acid-functionalised ILs as catalysts, but if any Brønsted acid catalysis is to compete with the industrial benchmark (mineral acids, acidic zeolites), it is hard to envisage an economically-viable process using those, with a possible exception of supported/polymerised systems for niche applications.
Biomass processing a relatively small, but a rapidly growing area, with demonstrated early potential for commercialisation. Virtually all studies use water-doped or alcohol-doped PILs, based on sulfuric acid and cheap amines, which appears to be a very good direction, considering the scale of biomass processing industry.
Energy storage and generation is by far the most prolific research area, with numerous research groups pursuing interesting research in PIL electrolytes for alkali-ion batteries, metal batteries, supercapacitors, redox flow stationary batteries, all-organic proton batteries and hydrogen fuel cells. PILs are also explored in the context of materials for energy storage, such growing research on composite membranes. Since PILs in energy storage devices are to be used in enclosed systems for a long time, there is much less constrain on the price, but more on the electrochemical stability and maximising performance: there is a reliance on highly fluorinated anions, chiefly [OTf]−, [TFSI]−, and [FSI]−, combined with simple amines such as triethylamine, superbases such as DBU, or more exotic bases, such as 7-methyl-1,5,7-triazabicyclo(4.4.0)dec-5-ene (MTBD).
Finally, PILs uses in electrocatalysis are rapidly growing, much like the entire field of electrocatalysis. Although relatively young, the field has already seen some successes in the electrolysis of water and very promising results in the reduction of CO2 and N2. An emergent and very exciting area of interest is homogenous electrocatalysis, where coordination complexes of Earth-abundant metals catalyse biomimetic reactions.
There are some pitfalls common to all areas of PILs research. An important one is developing systems that perform well at a first glance, in that they indeed catalyse a reaction or work well as an electrolyte in the particular experiment, but their design contains an inherent flaw that is bound to prevent their practical application. Examples include the use of expensive alkylsulfonic acid-functionalised ILs in liquid-phase catalysis (which can be conceptualised as a drop-in replacement of sulfuric acid) or the use of [BF4]− ionic liquids in aqueous conditions, although it is known that in the long term, they will hydrolyse to release HF. These examples illustrate chemistries that, for economic or technological reason, can never be implemented and therefore cannot be considered truly sustainable.
Another shortfall is the lack of appropriate benchmarking; although it is not always possible, when developing a sustainable alternative to existing process, it is very important to compare the newly reported chemistry to incumbent solvent, catalyst, electrolyte or material, such as organic electrolytes in Li-ion batteries, or mineral acids in Brønsted acid catalysis. It should be recognised and accepted, that new solutions do not have to outperform the state-of-the art; it is perfectly acceptable to report on sustainable innovations that do not match the incumbent technology, but offer an important advantage, and to propose the strategy for future improvements.
Lastly, both a current shortcoming and a future opportunity is the tendency to report on early-stage discoveries, even very promising, without following up with further studies that would demonstrate robustness of proposed solutions. Only the area of biomass processing has produced appreciable number of studies around process economy and life cycle analysis. In all other cases, there are a few or no articles on green metrics or techno-economic aspects of research. Some areas, such as PILs in electrocatalysis, are still nascent, which explains paucity of such studies, but this does not apply to esterifications or Li-ion batteries research. It would be very beneficial if promising electrolytes were studied in devices and compared to extant technologies; if catalytic studies were scaled-up, or studied in flow, and complemented with techno-economical and lifecycle analysis. It is possible that lack of interdisciplinary collaboration that would lead to such studies is one of the key barriers in translating these new discoveries to industrial applications.
While recognising the need of collaborative research to take the new discoveries towards higher technology readiness levels (TRLs), there is an equally important need for fundamental studies. Science is enabled by insights into mechanisms of reactions, liquid structure and speciation, as well as surface interactions of PILs, in order to understand their chemistry. To this end, collaboration with spectroscopists, computational chemists and other specialisms is most beneficial. In reviewing existing literature, we note that a greater alignment of fundamental studies and applications would be very beneficial. For example, there is a plethora of excellent fundamental studies which focus on nitrate PILs, that have a very limited scope of applications, while less attention has been given to PILs extensively studied in applied chemistry – ionic liquids with [HSO4]−, [OTf]−, [TFSI]− and [FSI]− anions, especially the systems doped with solvents or excess of acid. To summarise, the most exciting future research lies at the cross-section of fields and specialisms, enabling synergistic discoveries.
Author contributions
JB – writing (energy storage and conversion), visualisation; ELB – writing (biomass treatment), visualisation, PG – writing (catalytic transformations), visualisation, PK – writing (hydrogen fuel cells and electrocatalysis), visualisation, MSK – conceptualisation, writing (introduction, conclusions), visualisation.Conflicts of interest
There are no conflicts to declare.References
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed
.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2015, 115, 11379–11448 CrossRef CAS PubMed
- C. A. Angell, N. Byrne and J. P. Belieres, Acc. Chem. Res., 2007, 40, 1228–1236 CrossRef CAS PubMed
- Z. Wojnarowska and M. Paluch, J. Phys.: Condens. Matter, 2015, 27, 20 CrossRef PubMed
- V. Overbeck and R. Ludwig, Annu. Rep. NMR Spectrosc., 2018, 95, 147–190 CrossRef CAS
- C. Y. Wong, W. Y. Wong, K. S. Loh and K. L. Lim, React. Funct. Polym., 2022, 171, 105160 CrossRef CAS
- H. A. Elwan, M. Mamlouk and K. Scott, J. Power Sources, 2021, 484, 229197 CrossRef CAS
- U. A. Rana, M. Forsyth, D. R. Macfarlane and J. M. Pringle, Electrochim. Acta, 2012, 84, 213–222 CrossRef CAS
- D. R. Macfarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC
- T. Welton, Green Chem., 2011, 13, 225 RSC
- A. J. Greer, J. Jacquemin and C. Hardacre, Molecules, 2020, 25, 5207 CrossRef CAS PubMed
- United Nations 17 Sustainability Goals. https://sdgs.un.org/goals.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS
- J. Stoimenovski, E. I. Izgorodina and D. R. Macfarlane, Phys. Chem. Chem. Phys., 2010, 12, 10341–10347 RSC
- E. M. Morais, I. Abdurrokhman and A. Martinelli, J. Mol. Liq., 2022, 360, 119358 CrossRef CAS
- K. Matuszek, A. Chrobok, F. Coleman, K. R. Seddon and M. Swadźba-Kwaśny, Green Chem., 2014, 16, 3463–3471 RSC
- M. Hasani, S. A. Amin, J. L. Yarger, S. K. Davidowski and C. A. Angell, J. Phys. Chem. B, 2019, 123, 33 CrossRef PubMed
- I. Vázquez-Fernández, K. Drużbicki, F. Fernandez-Alonso, S. Mukhopadhyay, P. Nockemann, S. F. Parker, S. Rudić, S. M. Stana, J. Tomkinson, D. J. Yeadon, K. R. Seddon and N. V. Plechkova, J. Phys. Chem. C, 2021, 125, 24463–24476 CrossRef PubMed
- S. E. Goodwin, D. E. Smith, J. S. Gibson, R. G. Jones and D. A. Walsh, Langmuir, 2017, 33, 8436–8446 CrossRef CAS PubMed
- J. P. Belieres and C. A. Angell, J. Phys. Chem. B, 2007, 111, 4926–4937 CrossRef CAS PubMed
- G. L. Burrell, I. M. Burgar, F. Separovic and N. F. Dunlop, Phys. Chem. Chem. Phys., 2010, 12, 1571–1577 RSC
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161–5178 RSC
- A. Brzęczek-Szafran, J. Więcławik, N. Barteczko, A. Szelwicka, E. Byrne, A. Kolanowska, M. Swadźba Kwaśny and A. Chrobok, Green Chem., 2021, 23, 4421–4429 RSC
- D. J. Yeadon, J. Jacquemin, N. V. Plechkova, M. Maréchal and K. R. Seddon, ChemPhysChem, 2020, 21, 1369–1374 CrossRef CAS PubMed
- M. Thomas, M. Brehm, O. Hollóczki and B. Kirchner, Chem. – Eur. J., 2014, 20, 1622–1629 CrossRef CAS PubMed
- M. Wild, F. Stolz, S. Naumov and B. Abel, Mol. Phys., 2021, 119, 17–18 CrossRef
- I. Chiarotto, L. Mattiello, F. Pandolfi, D. Rocco and M. Feroci, Front. Chem., 2018, 6, 355 CrossRef PubMed
- T. Fukuyama, M. T. Rahman, H. Mashima, H. Takahashi and I. Ryu, Org. Chem. Front., 2017, 4, 1863–1866 RSC
- F. C. Binks, G. Cavalli, M. Henningsen, B. J. Howlin and I. Hamerton, Polymer, 2018, 139, 163–176 CrossRef CAS
- M. Swadzba-Kwasny, L. Chancelier, S. Ng, H. G. Manyar, C. Hardacre and P. Nockemann, Dalton Trans., 2012, 41, 219–227 RSC
- T. R. Gohndrone, T. Bum Lee, M. A. Desilva, M. Quiroz-Guzman, W. F. Schneider and J. F. Brennecke, ChemSusChem, 2014, 7, 1970–1975 CrossRef CAS PubMed
- T. R. Gohndrone, T. Song, M. Aruni DeSilva and J. F. Brennecke, J. Phys. Chem. B, 2021, 125, 6649–6657 CrossRef CAS PubMed
- T. B. Lee, S. Oh, T. R. Gohndrone, O. Morales-Collazo, S. Seo, J. F. Brennecke and W. F. Schneider, J. Phys. Chem. B, 2015, 120, 1509–1517 CrossRef PubMed
- S. Oh, O. Morales-Collazo, A. N. Keller and J. F. Brennecke, J. Phys. Chem., 2020, 124, 8877–8887 CrossRef CAS PubMed
- B. H. R. Suryanto, K. Matuszek, J. Choi, R. Y. Hodgetts, H. L. Du, J. M. Bakker, C. S. M. Kang, P. V. Cherepanov, A. N. Simonov and D. R. MacFarlane, Science, 2021, 372, 1187–1191 CrossRef CAS PubMed
- D. R. MacFarlane and K. R. Seddon, Aust. J. Chem., 2007, 60, 3–5 CrossRef CAS
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178–5186 RSC
- Y. Deng, J. Yao and H. Li, AIChE J., 2020, 66, e16982 CrossRef CAS
- J. E. S. J. Reid, C. E. S. Bernardes, F. Agapito, F. Martins, S. Shimizu, M. E. Minas Da Piedade and A. J. Walker, Phys. Chem. Chem. Phys., 2017, 19, 28133–28138 RSC
- A. Mariani, M. Bonomo, X. Gao, B. Centrella, A. Nucara, R. Buscaino, A. Barge, N. Barbero, L. Gontrani and S. Passerini, J. Mol. Liq., 2021, 324, 115069 CrossRef CAS
- S. K. Davidowski, F. Thompson, W. Huang, M. Hasani, S. A. Amin, C. A. Angell and J. L. Yarger, J. Phys. Chem. B, 2016, 120, 4279–4285 CrossRef CAS PubMed
- K. R. Harris, J. Phys. Chem. B, 2019, 123, 7014–7023 CrossRef CAS PubMed
- G. Huang, L. Porcarelli, M. Forsyth and H. Zhu, J. Phys. Chem. Lett., 2021, 12, 5552–5557 CrossRef CAS PubMed
- H. Doi, X. Song, B. Minofar, R. Kanzaki, T. Takamuku and Y. Umebayashi, Chem. – Eur. J., 2013, 19, 11522–11526 CrossRef CAS PubMed
- H. Watanabe, N. Arai, Y. Kameda, R. Buchner and Y. Umebayashi, J. Phys. Chem. B, 2020, 124, 11157–11164 CrossRef CAS PubMed
- H. Watanabe, T. Umecky, N. Arai, A. Nazet, T. Takamuku, K. R. Harris, Y. Kameda, R. Buchner and Y. Umebayashi, J. Phys. Chem. B, 2019, 123, 6244–6252 CrossRef CAS PubMed
- H. Watanabe, N. Arai, H. Jihae, Y. Kawana and Y. Umebayashi, J. Mol. Liq., 2022, 352, 118705 CrossRef CAS
- R. Jacobi, F. Joerg, O. Steinhauser and C. Schröder, Phys. Chem. Chem. Phys., 2022, 24, 9277 RSC
- C. H. C. Janssen, J. Mol. Liq., 2020, 304, 112738 CrossRef CAS
- F. Joerg and C. Schröeder, Phys. Chem. Chem. Phys., 2022, 24, 15245 RSC
- J. Ingenmey, S. Gehrke and B. Kirchner, ChemSusChem, 2018, 11, 1900–1910 CrossRef CAS PubMed
- J. E. S. J. Reid, S. Shimizu and A. J. Walker, J. Mol. Liq., 2020, 297, 111746 CrossRef CAS
- C. Karlsson, C. Strietzel, H. Huang, M. Sjödin and P. Jannasch, ACS Appl. Energy Mater., 2018, 1, 6451–6462 CrossRef CAS
- P. Jannasch, J. Rehmen, D. Evans and C. Karlsson, J. Phys. Chem. C, 2019, 123, 23427–23432 CrossRef CAS
- M. A. R. Martins, P. J. Carvalho, L. M. N. B. F. Santos, S. P. Pinho and J. A. P. Coutinho, Fluid Phase Equilib., 2021, 531, 112919 CrossRef CAS
- V. K. Venkatesan and C. V. Suryanarayana, J. Phys. Chem., 1956, 60, 777–779 CrossRef CAS
- G. M. Barrow, J. Am. Chem. Soc., 1956, 78, 5802–5805 CrossRef CAS
- V. K. Venkatesan and C. V. Suryanarayana, J. Phys. Chem., 1956, 60, 777–779 CrossRef CAS
- K. M. Johansson, E. I. Izgorodina, M. Forsyth, D. R. Macfarlane and K. R. Seddon, Phys. Chem. Chem. Phys., 2008, 10, 2972–2978 RSC
- J. A. McCune, A. H. Turner, F. Coleman, C. M. White, S. K. Callear, T. G. A. Youngs, M. Swadźba-Kwaśny and J. D. Holbrey, Phys. Chem. Chem. Phys., 2015, 17, 6767–6777 RSC
- A. McGrogan, E. L. Byrne, R. Guiney, T. F. Headen, T. G. A. Youngs, A. Chrobok, J. D. Holbrey and M. Swadźba-Kwaśny, Phys. Chem. Chem. Phys., 2023, 25, 9785 RSC
- N. P. Aravindakshan, K. E. Gemmell, K. E. Johnson and A. L. L. East, J. Chem. Phys., 2018, 149, 094505 CrossRef PubMed
- J. Zhang, J. Yao and H. Li, Ind. Eng. Chem. Res., 2021, 60, 13719–13726 CrossRef CAS
- D. R. MacFarlane, A. L. Chong, M. Forsyth, M. Kar, R. Vijayaraghavan, A. Somers and J. M. Pringle, Faraday Discuss., 2018, 206, 9–28 RSC
- Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119–7130 RSC
- T. Stettner, S. Gehrke, P. Ray, B. Kirchner and A. Balducci, ChemSusChem, 2019, 12, 3827–3836 CrossRef CAS PubMed
- S. Zahn, K. Wendler, L. Delle Site and B. Kirchner, Phys. Chem. Chem. Phys., 2011, 13, 15083–15093 RSC
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2012, 51, 7468–7471 CrossRef CAS PubMed
- D. Yalcin, C. J. Drummond and T. L. Greaves, Phys. Chem. Chem. Phys., 2019, 21, 6810–6827 RSC
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed
- H. P. Steinrück and P. Wasserscheid, Catal. Lett., 2015, 145, 380–397 CrossRef
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed
- S. Zhao, X. Wang and L. Zhang, RSC Adv., 2013, 3, 11691–11696 RSC
- A. G. Ying, L. Liu, G. F. Wu, G. Chen, X. Z. Chen and W. D. Ye, Tetrahedron Lett., 2009, 50, 1653–1657 CrossRef CAS
- A. Brzęczek-Szafran, K. Erfurt, M. Swadźba-Kwaśny, T. Piotrowski and A. Chrobok, ACS Sustainable Chem. Eng., 2022, 10, 13568–13575 CrossRef
- K. Matuszek, A. Brzeczek-Szafran, D. Kobus, D. R. Macfarlane, M. Swadźba-Kwaśny and A. Chrobok, Aust. J. Chem., 2019, 72, 130–138 CrossRef CAS
- T. Wang, C. Shen, G. Yu and X. Chen, Polym. Degrad. Stab., 2022, 203, 110050 CrossRef CAS
- I. A. Andreev, N. K. Ratmanova, A. U. Augustin, O. A. Ivanova, I. I. Levina, V. N. Khrustalev, D. B. Werz and I. v. Trushkov, Angew. Chem., Int. Ed., 2021, 60, 7927–7934 CrossRef CAS PubMed
- P. Zhang, H. Liu, M. Fan, Y. Liu and J. Huang, Curr. Org. Chem., 2016, 20, 752–760 CrossRef CAS
- Z. Ullah, A. S. Khan, N. Muhammad, R. Ullah, A. S. Alqahtani, S. N. Shah, O. ben Ghanem, M. A. Bustam and Z. Man, J. Mol. Liq., 2018, 266, 673–686 CrossRef CAS
- C. Chiappe, S. Rajamani and F. D'Andrea, Green Chem., 2013, 15, 137–143 RSC
- C. Chiappe, S. Rajamani and F. D'Andrea, Green Chem., 2013, 15, 137–143 RSC
- Z. Man, Y. A. Elsheikh, M. A. Bustam, S. Yusup, M. I. A. Mutalib and N. Muhammad, Ind. Crops Prod., 2013, 41, 144–149 CrossRef CAS
- B. S. Caldas, C. S. Nunes, P. R. Souza, F. A. Rosa, J. v. Visentainer, O. de O. S. Júnior and E. C. Muniz, Appl. Catal., B, 2016, 181, 289–297 CrossRef CAS
- Z. S. Qureshi, K. M. Deshmukh, M. D. Bhor and B. M. Bhanage, Catal. Commun., 2009, 10, 833–837 CrossRef CAS
- H. Luo, H. Yin, R. Wang, W. Fan and G. Nan, Catalysts, 2017, 7, 102 CrossRef
- S. Das, A. J. Thakur and D. Deka, Sci. World J., 2014, 2014, 180983 Search PubMed
- A. G. Khiratkar, K. R. Balinge, D. S. Patle, M. Krishnamurthy, K. K. Cheralathan and P. R. Bhagat, Fuel, 2018, 231, 458–467 CrossRef CAS
- A. N. Masri, M. I. Abdul Mutalib, N. F. Aminuddin and J. M. Lévêque, Sep. Purif. Technol., 2018, 196, 106–114 CrossRef CAS
- L. Zhang, M. Xian, Y. He, L. Li, J. Yang, S. Yu and X. Xu, Bioresour. Technol., 2009, 100, 4368–4373 CrossRef CAS PubMed
- L. Zhang, M. Xian, Y. He, L. Li, J. Yang, S. Yu and X. Xu, Bioresour. Technol., 2009, 100, 4368–4373 CrossRef CAS PubMed
- M. B. Martini, C. G. Adam and J. L. Fernández, Mol. Catal., 2021, 513, 111821 CrossRef CAS
- M. Przypis, K. Matuszek, A. Chrobok, M. Swadźba-Kwaśny and D. Gillner, J. Mol. Liq., 2020, 308, 113166 CrossRef CAS
- A. Brzęczek-Szafran, J. Więcławik, N. Barteczko, A. Szelwicka, E. Byrne, A. Kolanowska, M. Swadźba Kwaśny and A. Chrobok, Green Chem., 2021, 23, 4421–4429 RSC
- T. M. Ukarde, J. S. Mahale, P. H. Pandey, A. Vasishta, A. M. J. C. Harrish and H. S. Pawar, ChemistrySelect, 2021, 6, 9616–9624 CrossRef CAS
- F. Rajabi and R. Luque, Mol. Catal., 2020, 498, 111238 CrossRef CAS
- A. G. Khiratkar, K. R. Balinge, M. Krishnamurthy, K. K. Cheralathan, D. S. Patle, V. Singh, S. Arora and P. R. Bhagat, Catal. Lett., 2018, 148, 680–690 CrossRef CAS
- Y. Chen and T. Mu, Green Chem., 2019, 21, 2544–2574 RSC
- D. Wei-Li, J. Bi, L. Sheng-Lian, L. Xu-Biao, T. Xin-Man and A. Chak-Tong, J. Mol. Catal. A: Chem., 2013, 378, 326–332 CrossRef
- W. Dai, W. Yang, Y. Zhang, D. Wang, X. Luo and X. Tu, J. CO2 Util., 2017, 17, 256–262 CrossRef CAS
- L. Han, S. J. Choi, M. S. Park, S. M. Lee, Y. J. Kim, M. il Kim, B. Liu and D. W. Park, React. Kinet., Mech. Catal., 2012, 106, 25–35 CrossRef CAS
- J. Qiu, Y. Zhao, Z. Li, H. Wang, M. Fan and J. Wang, ChemSusChem, 2017, 10, 1120–1127 CrossRef CAS PubMed
- S. G. Khokarale and J. P. Mikkola, Green Chem., 2019, 21, 2138–2147 RSC
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS
- G. Xu, Z. Tu, X. Hu, M. Li, X. Zhang and Y. Wu, Fuel, 2023, 339, 127334 CrossRef CAS
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC
- T. Wang, C. Shen, G. Yu and X. Chen, Polym. Degrad. Stab., 2022, 203, 110050 CrossRef CAS
- K. Kumar, F. Parveen, T. Patra and S. Upadhyayula, New J. Chem., 2018, 42, 228–236 RSC
- C. Chiappe, M. J. Rodriguez Douton, A. Mezzetta, L. Guazzelli, C. S. Pomelli, G. Assanelli and A. R. De Angelis, New J. Chem., 2018, 42, 1845–1852 RSC
- P. Liu, K. Cai, M. Liu, F. Liu, P. Chen and T. Zhao, AIChE J., 2023, 69, e17944 CrossRef CAS
- X. Zhang, W. Xiong, M. Shi, Y. Wu and X. Hu, Chem. Eng. J., 2021, 408, 127866 CrossRef CAS
- P. Y. S. Nakasu, P. Verdía Barbará, A. E. J. Firth and J. P. Hallett, Trends Chem., 2022, 4, 175–178 CrossRef CAS
- A. R. Abouelela, A. al Ghatta, P. Verdía, M. Shan Koo, J. Lemus and J. P. Hallett, ACS Sustainable Chem. Eng., 2021, 9, 10524–10536 CrossRef CAS
- S. Geleynse, K. Brandt, M. Garcia-Perez, M. Wolcott and X. Zhang, ChemSusChem, 2018, 11, 3728–3741 CrossRef CAS PubMed
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS
- D. Gabriëls, W. Y. Hernández, B. F. Sels, P. van der Voort and A. Verberckmoes, Catal. Sci. Technol., 2015, 5, 3876–3902 RSC
- K. K. Ramasamy, M. Gray, H. Job, D. Santosa, X. S. Li, A. Devaraj, A. Karkamkar and Y. Wang, Top. Catal., 2016, 59, 46–54 CrossRef CAS
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- L. P. Manker, G. R. Dick, A. Demongeot, M. A. Hedou, C. Rayroud, T. Rambert, M. J. Jones, I. Sulaeva, M. Vieli, Y. Leterrier, A. Potthast, F. Maréchal, V. Michaud, H. A. Klok and J. S. Luterbacher, Nat. Chem., 2022, 14, 976–984 CrossRef CAS PubMed
- M. Pointner, P. Kuttner, T. Obrlik, A. Jäger and H. Kahr, Agron. Res., 2014, 12, 391–396 Search PubMed
- J. P. Reddy and J. W. Rhim, J. Nat. Fibers, 2018, 15, 465–473 CrossRef CAS
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed
- C. Liu and C. E. Wyman, Ind. Eng. Chem. Res., 2004, 43(11), 2781–2788 CrossRef CAS
- U. Avci, X. Zhou, S. Pattathil, L. da Costa Sousa, M. G. Hahn, B. Dale, Y. Xu and V. Balan, Front. Energy Res., 2019, 7, 85 CrossRef
- T. A. Lloyd and C. E. Wyman, Bioresour. Technol., 2005, 96, 1967–1977 CrossRef CAS PubMed
-
G. A. Smook, Handbook for Pulp and Paper Technologists, Angus Wilde Publications, Vancouver, 1992, vol. 11 Search PubMed
- G. Gellerstedt, Ind. Crops Prod., 2015, 77, 845–854 CrossRef CAS
-
J. Fernández-Rodríguez, X. Erdocia, F. Hernández-Ramos, M. G. Alriols and J. Labid, in Separation of Functional Molecules in Food by Membrane Technology, Elsevier, 2018, pp. 229–265 Search PubMed
- A. George, A. Brandt, K. Tran, S. M. S. N. S. Zahari, D. Klein-Marcuschamer, N. Sun, N. Sathitsuksanoh, J. Shi, V. Stavila, R. Parthasarathi, S. Singh, B. M. Holmes, T. Welton, B. A. Simmons and J. P. Hallett, Green Chem., 2015, 17, 1728–1734 RSC
- C. L. Chambon, T. Y. Mkhize, P. Reddy, A. Brandt-Talbot, N. Deenadayalu, P. S. Fennell and J. P. Hallett, Biotechnol. Biofuels, 2018, 11, 1–16 CrossRef PubMed
- A. Brandt-Talbot, F. J. V. Gschwend, P. S. Fennell, T. M. Lammens, B. Tan, J. Weale and J. P. Hallett, Green Chem., 2017, 19, 3078–3102 RSC
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC
-
J. Viell, Doctoral Thesis, RWTH Aachen, 2014
- P. Verdía, A. Brandt, J. P. Hallett, M. J. Ray and T. Welton, Green Chem., 2014, 16, 1617–1627 RSC
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489–2499 RSC
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 690–695 CrossRef PubMed
-
W. Schutyser, T. Renders, G. van den Bossche, S. van den Bosch, S.-F. Koelewijn, T. Ennaert and B. F. Sels, in Nanotechnology in Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2017, pp. 537–584 Search PubMed
- D. Gao, C. Haarmeyer, V. Balan, T. A. Whitehead, B. E. Dale and S. P. S. Chundawat, Biotechnol. Biofuels, 2014, 7, 175 CrossRef PubMed
- E. Palmqvist, B. Arbel and H.-H. Agerdal, Bioresour. Technol., 2000, 74, 25–33 CrossRef CAS
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344 Search PubMed
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed
- F. J. V. Gschwend, C. L. Chambon, M. Biedka, A. Brandt-Talbot, P. S. Fennell and J. P. Hallett, Green Chem., 2019, 21, 692–703 RSC
- A. R. Abouelela and J. P. Hallett, ACS Sustainable Chem. Eng., 2021, 9, 704–716 CrossRef CAS
- L. M. Hennequin, S. yin Tan, E. Jensen, P. Fennell and J. P. Hallett, Ind. Crops Prod., 2022, 176, 114259 CrossRef CAS
- L. M. Hennequin, K. Polizzi, P. S. Fennell and J. P. Hallett, RSC Adv., 2021, 11, 18395–18403 RSC
- M. Graglia, N. Kanna and D. Esposito, ChemBioEng Rev., 2015, 2, 377–392 CrossRef
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS
-
G. Gellerstedt and G. Henriksson, Monomers, Polymers and Composites from Renewable Resources, Elsevier, Amsterdam, 2008 Search PubMed
- F. J. V. Gschwend, F. Malaret, S. Shinde, A. Brandt-Talbot and J. P. Hallett, Green Chem., 2018, 20, 3486–3498 RSC
- A. M. da Costa Lopes and R. M. Łukasik, ChemSusChem, 2018, 11, 1099–1107 CrossRef CAS PubMed
- A. Bukowski, D. Esau, A. A. Rafat Said, A. Brandt-Talbot and J. Albert, ChemPlusChem, 2020, 85, 373–386 CrossRef CAS PubMed
-
D. di Maio and D. Turley, Lignocellulosic feedstock in the UK, NNFCC Report, York, 2014 Search PubMed
- K. Shimada, S. Hosoya and T. Ikeda, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS
- J. L. Wen, T. Q. Yuan, S. L. Sun, F. Xu and R. C. Sun, Green Chem., 2014, 16, 181–190 RSC
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed
- A. Ovejero-Pérez, V. Rigual, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Int. J. Biol. Macromol., 2022, 197, 131–140 CrossRef PubMed
- M. Chen, F. Malaret, A. E. J. Firth, P. Verdía, A. R. Abouelela, Y. Chen and J. P. Hallett, Green Chem., 2020, 22, 5161–5178 RSC
- A. Wufuer, Y. Wang and L. Dai, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01846-7
- L. L. Reys, S. S. Silva, J. M. Oliveira, S. G. Caridade, J. F. Mano, T. H. Silva and R. L. Reis, Biomed. Mater., 2013, 8, 1–11 Search PubMed
- T. H. Silva, A. Alves, B. M. Ferreira, J. M. Oliveira, L. L. Reys, R. J. F. Ferreira, R. A. Sousa, S. S. Silva, J. F. Mano and R. L. Reis, Int. Mater. Rev., 2012, 57, 276–307 CrossRef CAS
- S. S. Silva, J. F. Mano and R. L. Reis, Green Chem., 2017, 19, 1208–1220 RSC
- M. Li, H. Zang, J. Feng, Q. Yan, N. Yu, X. Shi and B. Cheng, Polym. Degrad. Stab., 2015, 121, 331–339 CrossRef CAS
- H. Zang, S. Yu, P. Yu, H. Ding, Y. Du, Y. Yang and Y. Zhang, Carbohydr. Res., 2017, 442, 1–8 CrossRef CAS PubMed
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC
- A. Blakers, M. Stocks, B. Lu and C. Cheng, Prog. Energy, 2021, 3, 022003 CrossRef
- U.S. Department of Energy, Energy Storage Grand Challenge: Energy Storage Market Report, 2020.
- International Energy Agency, Renewables, 2021 Search PubMed
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed
- H. Zhang, L. Qiao, H. Kühnle, E. Figgemeier, M. Armand and G. G. Eshetu, Energy Environ. Sci., 2023, 16, 11–52 RSC
- J. Zhang, X. Yao, R. K. Misra, Q. Cai and Y. Zhao, J. Mater. Sci. Technol., 2020, 44, 237–257 CrossRef CAS
- D. D. Patel and J. M. Lee, Chem. Rec., 2012, 12, 329–355 CrossRef CAS PubMed
- A. Balducci, Top. Curr. Chem., 2017, 375, 1–27 CrossRef CAS PubMed
- N. Böckenfeld, M. Willeke, J. Pires, M. Anouti and A. Balducci, J. Electrochem. Soc., 2013, 160, A559–A563 CrossRef
- A. T. Nasrabadi and V. Ganesan, J. Phys. Chem. B, 2019, 123, 5588–5600 CrossRef CAS PubMed
- T. Stettner and A. Balducci, Energy Storage Mater., 2021, 40, 402–414 CrossRef
- S. Menne, J. Pires, M. Anouti and A. Balducci, Electrochem. Commun., 2013, 31, 39–41 CrossRef CAS
- T. Vogl, S. Menne, R. S. Kühnel and A. Balducci, J. Mater. Chem. A, 2014, 2, 8258–8265 RSC
- T. Méndez-Morales, J. Carrete, Ó. Cabeza, O. Russina, A. Triolo, L. J. Gallego and L. M. Varela, J. Phys. Chem. B, 2014, 118, 761–770 CrossRef PubMed
- T. Vogl, S. Menne and A. Balducci, Phys. Chem. Chem. Phys., 2014, 16, 25014–25023 RSC
- P. Ray, A. Balducci and B. Kirchner, J. Phys. Chem. B, 2018, 122, 10535–10547 CrossRef CAS PubMed
- Y. Lu, Y. Xu, L. Lu, Z. Xu and H. Liu, Phys. Chem. Chem. Phys., 2021, 23, 18338–18348 RSC
- S. Menne, M. Schroeder, T. Vogl and A. Balducci, J. Power Sources, 2014, 266, 208–212 CrossRef CAS
- S. Menne, T. Vogl and A. Balducci, Chem. Commun., 2015, 51, 3656–3659 RSC
- T. Vogl, P. Goodrich, J. Jacquemin, S. Passerini and A. Balducci, J. Phys. Chem. C, 2016, 120, 8525–8533 CrossRef CAS
- T. Vogl, S. Passerini and A. Balducci, Electrochem. Commun., 2017, 78, 47–50 CrossRef CAS
- T. Stettner, F. C. Walter and A. Balducci, Batteries Supercaps, 2019, 2, 55–59 CrossRef CAS
- T. Stettner, R. Dugas, A. Ponrouch and A. Balducci, J. Electrochem. Soc., 2020, 167, 100544 CrossRef CAS
- M. Arnaiz, P. Huang, J. Ajuria, T. Rojo, E. Goikolea and A. Balducci, Batteries Supercaps, 2018, 1, 204–208 CrossRef CAS
- J. Xu, Y. Dou, Z. Wei, J. Ma, Y. Deng, Y. Li, H. Liu and S. Dou, Adv. Sci., 2017, 4, 1700146 CrossRef PubMed
- P. Ray, T. Vogl, A. Balducci and B. Kirchner, J. Phys. Chem. B, 2017, 121, 5279–5292 CrossRef CAS PubMed
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271 CrossRef CAS
- T. Vogl, C. Vaalma, D. Buchholz, M. Secchiaroli, R. Marassi, S. Passerini and A. Balducci, J. Mater. Chem. A, 2016, 4, 10472–10478 RSC
- X. Min, J. Xiao, M. Fang, W. Wang, Y. Zhao, Y. Liu, A. M. Abdelkader, K. Xi, R. V. Kumar and Z. Huang, Energy Environ. Sci., 2021, 14, 2186–2243 RSC
- X. Zhang, R. S. Kühnel, H. Hu, D. Eder and A. Balducci, Nano Energy, 2015, 12, 207–214 CrossRef CAS
- M. Arnaiz, A. Bothe, S. Dsoke, A. Balducci and J. Ajuria, J. Electrochem. Soc., 2019, 166, A3504–A3510 CrossRef CAS
- S. Hosseini, S. Masoudi Soltani and Y. Y. Li, Chem. Eng. J., 2021, 408, 127241 CrossRef CAS
- Y. Guo, S. Wu, Y.-B. He, F. Kang, L. Chen, H. Li and Q.-H. Yang, eScience, 2022, 2, 138–163 CrossRef
- M. Zhao, B. Q. Li, X. Q. Zhang, J. Q. Huang and Q. Zhang, ACS Cent. Sci., 2020, 6, 1095–1104 CrossRef CAS PubMed
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS PubMed
- Z. Liu, S. Z. El Abedin and F. Endres, Electrochem. Commun., 2015, 58, 46–50 CrossRef CAS
- P. Jankowski, K. Matuszek, M. Treskow, M. Armand, D. MacFarlane and P. Johansson, Chem. Commun., 2019, 55, 12523–12526 RSC
- T. Stettner, G. Lingua, M. Falco, A. Balducci and C. Gerbaldi, Energy Technol., 2020, 8, 2000742 CrossRef CAS
- G. Lingua, M. Falco, T. Stettner, C. Gerbaldi and A. Balducci, J. Power Sources, 2021, 481, 228979 CrossRef CAS
- J. Wang, S. Y. Chew, Z. W. Zhao, S. Ashraf, D. Wexler, J. Chen, S. H. Ng, S. L. Chou and H. K. Liu, Carbon, 2008, 46, 229–235 CrossRef CAS
- K. Kopczyński, A. Gabryelczyk, M. Baraniak, B. Łęgosz, J. Pernak, E. Jankowska, W. Rzeszutek, P. Kędzior and G. Lota, J. Energy Storage, 2019, 26, 100996 CrossRef
- Z. Li, S. Gadipelli, H. Li, C. A. Howard, D. J. L. Brett, P. R. Shearing, Z. Guo, I. P. Parkin and F. Li, Nat. Energy, 2020, 5, 160–168 CrossRef CAS
- S. Satpathy, N. Kumar, V. Goyal, B. K. Bhattacharyya and C. Shekhar, J. Energy Storage, 2023, 57, 106198 CrossRef
-
S. Banerjee, B. De, P. Sinha, J. Cherusseri and K. K. Kar, Applications of Supercapacitors, 2020, vol. 300 Search PubMed
- L. Miao, D. Zhu, M. Liu, H. Duan, Z. Wang, Y. Lv, W. Xiong, Q. Zhu, L. Li, X. Chai and L. Gan, Chem. Eng. J., 2018, 347, 233–242 CrossRef CAS
- A. B. McEwen, H. L. Ngo, K. LeCompte and J. L. Goldman, J. Electrochem. Soc., 1999, 146, 1687–1695 CrossRef CAS
- K. C. Lethesh, M. O. Bamgbopa and R. A. Susantyoko, Front. Energy Res., 2021, 9, 1–16 Search PubMed
- L. Yu and G. Z. Chen, Front. Chem., 2019, 7, 1–15 CrossRef PubMed
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC
- T. Stettner, S. Gehrke, P. Ray, B. Kirchner and A. Balducci, ChemSusChem, 2019, 12, 3827–3836 CrossRef CAS PubMed
- S. Sultana, K. Ahmed, P. K. Jiwanti, B. Y. Wardhana and M. N. I. Shiblee, Gels, 2022, 8, 2 CrossRef CAS PubMed
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS
- A. Eftekhari, Energy Storage Mater., 2017, 9, 47–69 CrossRef
- S. Zhang, A. Ikoma, K. Ueno, Z. Chen, K. Dokko and M. Watanabe, ChemSusChem, 2015, 8, 1608–1617 CrossRef CAS PubMed
- E. Kim, J. Han, S. Ryu, Y. Choi and J. Yoo, Materials, 2021, 14, 400 CrossRef PubMed
- S. Zhang, K. Dokko and M. Watanabe, Chem. Mater., 2014, 26, 2915–2926 CrossRef CAS
- N. Fechler, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2013, 1, 14097–14102 RSC
- L. Sun, Y. Yao, Y. Zhou, L. Li, H. Zhou, M. Guo, S. Liu, C. Feng, Z. Qi and B. Gao, ACS Sustainable Chem. Eng., 2018, 6, 13494–13503 CrossRef CAS
- L. Sun, H. Zhou, Y. Li, F. Yu, C. Zhang, X. Liu and Y. Zhou, Mater. Lett., 2017, 189, 107–109 CrossRef CAS
- L. Sun, H. Zhou, L. Li, Y. Yao, H. Qu, C. Zhang, S. Liu and Y. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 26088–26095 CrossRef CAS PubMed
- L. Sun, Y. Zhou, L. Li, H. Zhou, X. Liu, Q. Zhang, B. Gao, Z. Meng, D. Zhou and Y. Ma, Appl. Surf. Sci., 2019, 467–468, 382–390 CrossRef CAS
- H. Zhou, Y. Zhou, L. Li, Y. Li, X. Liu, P. Zhao and B. Gao, ACS Sustainable Chem. Eng., 2019, 7, 9281–9290 CrossRef CAS
- H. Wang, H. Zhou, S. Wu, Z. Li, B. Fan, Y. Li and Y. Zhou, Electrochim. Acta, 2021, 380, 138230 CrossRef CAS
- H. Zhou, S. Wu, H. Wang, Y. Li, X. Liu and Y. Zhou, J. Hazard. Mater., 2021, 402, 124023 CrossRef CAS PubMed
- S. Wu, C. Feng, B. Fan, Y. Li, H. Wang and Y. Zhou, J. Alloys Compd., 2022, 899, 163282 CrossRef CAS
- F. Al-Zohbi, F. Ghamouss, B. Schmaltz, M. Abarbri, M. Zaghrioui and F. Tran-Van, Materials, 2021, 14, 4275 CrossRef CAS PubMed
- A. Bouzina, H. Perrot, C. Debiemme-Chouvy and O. Sel, ACS Appl. Energy Mater., 2022, 5, 14934–14944 CrossRef CAS
- R. M. Darling, Curr. Opin. Chem. Eng., 2022, 37, 100855 CrossRef
- L. Li, S. Kim, W. Wang, M. Vijayakumar, Z. Nie, B. Chen, J. Zhang, G. Xia, J. Hu, G. Graff, J. Liu and Z. Yang, Adv. Energy Mater., 2011, 1, 394–400 CrossRef CAS
- V. M. Ortiz-Martínez, L. Gómez-Coma, G. Pérez, A. Ortiz and I. Ortiz, Sep. Purif. Technol., 2020, 252, 117436 CrossRef
- A. Joseph, J. Sobczak, G. Żyła and S. Mathew, Energies, 2022, 15, 4545 CrossRef CAS
- G. Nikiforidis, A. Belhcen and M. Anouti, J. Energy Chem., 2021, 57, 238–246 CrossRef CAS
- G. Nikiforidis and M. Anouti, Le Studium Multidisciplinary J., 2021, 5, 1–5 Search PubMed
- X. Zhou, R. Xue, Y. Zhong, Y. Zhang and F. Jiang, J. Membr. Sci., 2020, 595, 117614 CrossRef CAS
- A. L. O. Smith and D. L. Crittenden, Chem. – Asian J., 2023, 18, e202201296 CrossRef PubMed
- P. Navalpotro, J. Palma, M. Anderson and R. Marcilla, Angew. Chem., Int. Ed., 2017, 56, 12460–12465 CrossRef CAS PubMed
- C. Karlsson and P. Jannasch, ACS Appl. Energy Mater., 2019, 2, 6841–6850 CrossRef CAS
- K. Mishra, S. A. Hashmi and D. K. Rai, J. Solid State Electrochem., 2014, 18, 2255–2266 CrossRef CAS
- H. Wang, R. Emanuelsson, C. Karlsson, P. Jannasch, M. Strømme and M. Sjödin, ACS Appl. Mater. Interfaces, 2021, 13, 19099–19108 CrossRef CAS PubMed
- M. Liao, X. Ji, Y. Cao, J. Xu, X. Qiu, Y. Xie, F. Wang, C. Wang and Y. Xia, Nat. Commun., 2022, 13, 1–9 Search PubMed
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed
- P. Jannasch, J. Rehmen, D. Evans and C. Karlsson, J. Phys. Chem. C, 2019, 123, 23427–23432 CrossRef CAS
- C. Karlsson, C. Strietzel, H. Huang, M. Sjödin and P. Jannasch, ACS Appl. Energy Mater., 2018, 1, 6451–6462 CrossRef CAS
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
- X. Tian, X. F. Lu, B. Y. Xia and X. W. D. Lou, Joule, 2020, 4, 45–68 CrossRef CAS
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 Search PubMed
- Y. Yang, M. Luo, W. Zhang, Y. Sun, X. Chen and S. Guo, Chem, 2018, 4, 2054–2083 CAS
- J. Snyder, K. Livi and J. Erlebacher, Adv. Funct. Mater., 2013, 23, 5494–5501 CrossRef CAS
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904–907 CrossRef CAS PubMed
- Y. Li, J. Hart, L. Profitt, S. Intikhab, S. Chatterjee, M. Taheri and J. Snyder, ACS Catal., 2019, 9, 9311–9316 CrossRef CAS
- M. George, G. R. Zhang, N. Schmitt, K. Brunnengräber, D. J. S. Sandbeck, K. J. J. Mayrhofer, S. Cherevko and B. J. M. Etzold, ACS Catal., 2019, 9, 8682–8692 CrossRef CAS PubMed
- G. R. Zhang, M. Munoz and B. J. M. Etzold, ACS Appl. Mater. Interfaces, 2015, 7, 3562–3570 CrossRef CAS PubMed
- M. Qiao, G. A. Ferrero, L. Fernández Velasco, W. Vern Hor, Y. Yang, H. Luo, P. Lodewyckx, A. B. Fuertes, M. Sevilla and M. M. Titirici, ACS Appl. Mater. Interfaces, 2019, 11, 11298–11305 CrossRef CAS PubMed
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S. G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC
- A. Alashkar, A. Al-Othman, M. Tawalbeh and M. Qasim, Membranes, 2022, 12, 1–30 CrossRef PubMed
- L. K. Seng, M. S. Masdar and L. K. Shyuan, Membranes, 2021, 11, 728 CrossRef CAS PubMed
- A. L. L. East, C. M. Nguyen and R. Hempelmann, Front. Energy Res., 2023, 11, 728 Search PubMed
- M. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 3, 938–939 RSC
- A. Noda, M. A. Bin Hasan Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS
- H. Nakamoto and M. Watanabe, Chem. Commun., 2007, 2539–2541 RSC
- H. Nakamoto, A. Noda, K. Hayamizu, S. Hayashi, H. O. Hamaguchi and M. Watanabe, J. Phys. Chem. C, 2007, 111, 1541–1548 CrossRef CAS
- S. Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed
- Z. Zhu, X. Yan, H. Tang, H. Cai, M. Pan, H. Zhang and J. Luo, J. Power Sources, 2017, 351, 138–144 CrossRef CAS
- D. E. Smith and D. A. Walsh, Adv. Energy Mater., 2019, 9, 1–6 Search PubMed
- G. De Araujo Lima E Souza, M. E. Di Pietro, F. Castiglione, P. H. Marques Mezencio, P. Fazzio Martins Martinez, A. Mariani, H. M. Schütz, S. Passerini, M. Middendorf, M. Schönhoff, A. Triolo, G. B. Appetecchi and A. Mele, J. Phys. Chem. B, 2022, 126, 7006–7014 CrossRef CAS PubMed
- O. Danyliv and A. Martinelli, J. Phys. Chem. C, 2019, 123, 14813–14824 CrossRef CAS
- S. Maity, S. Singha and T. Jana, Polymer, 2015, 66, 76–85 CrossRef CAS
- A. K. Mishra, T. Kuila, D. Y. Kim, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 24366–24372 RSC
- A. Eguizábal, J. Lemus and M. P. Pina, J. Power Sources, 2013, 222, 483–492 CrossRef
- G. Skorikova, D. Rauber, D. Aili, S. Martin, Q. Li, D. Henkensmeier and R. Hempelmann, J. Membr. Sci., 2020, 608, 118188 CrossRef CAS
- T. Yasuda, S. Nakamura, Y. Honda, K. Kinugawa, S.-Y. Lee and M. Watanabe, ACS Appl. Mater. Interfaces, 2012, 4, 1783–1790 CrossRef CAS PubMed
- J. A. F. L. Batalha, K. Dahmouche, R. B. Sampaio and A. de Souza Gomes, Macromol. Mater. Eng., 2017, 302, 1–11 CrossRef
- X. Wang, Y. Rong, F. Wang, C. Zhang and Q. Wang, Microporous Mesoporous Mater., 2022, 346, 112314 CrossRef CAS
- Z. Guan, Y. Jin, S. Shi, B. Jin, M. Zhang and L. Zhao, Polymer, 2022, 254, 125011 CrossRef CAS
- J. Rao, X. Wang, R. Yunis, V. Ranganathan, P. C. Howlett, D. R. MacFarlane, M. Forsyth and H. Zhu, Electrochim. Acta, 2020, 346, 136224 CrossRef CAS
- G. Huang, L. Porcarelli, Y. Liang, M. Forsyth and H. Zhu, ACS Appl. Energy Mater., 2021, 4, 10593–10602 CrossRef CAS
- G. Huang, H. Zhu, L. Porcarelli, Y. García, L. A. O'Dell and M. Forsyth, Macromol. Chem. Phys., 2022, 223, 2200124 CrossRef CAS
- S. Sen, S. E. Goodwin, P. V. Barbará, G. A. Rance, D. Wales, J. M. Cameron, V. Sans, M. Mamlouk, K. Scott and D. A. Walsh, ACS Appl. Polym. Mater., 2021, 3, 200–208 CrossRef CAS
- A. Fernicola, S. Panero and B. Scrosati, J. Power Sources, 2008, 178, 591–595 CrossRef CAS
- V. Di Noto, M. Piga, G. A. Giffin, S. Lavina, E. S. Smotkin, J. Y. Sanchez and C. Iojoiu, J. Phys. Chem. C, 2012, 116, 1370–1379 CrossRef CAS
- Y. Zhang, R. Xue, Y. Zhong, F. Jiang, M. Hu and Q. Yu, Fuel Cells, 2018, 18, 389–396 CrossRef CAS
- R. Sood, C. Iojoiu, E. Espuche, F. Gouanve, G. Gebel, H. Mendil-Jakani, S. Lyonnard and J. Jestin, J. Phys. Chem. C, 2014, 118, 14157–14168 CrossRef CAS
- Y. Li, Y. Shi, N. Mehio, M. Tan, Z. Wang, X. Hu, G. Z. Chen, S. Dai and X. Jin, Appl. Energy, 2016, 175, 451–458 CrossRef CAS
- M. Díaz, A. Ortiz, M. Isik, D. Mecerreyes and I. Ortiz, Int. J. Hydrogen Energy, 2014, 40, 11294–11302 CrossRef
- L. Zanchet, L. G. da Trindade, W. Bariviera, K. M. Nobre Borba, R. D. M. Santos, V. A. Paganin, C. P. de Oliveira, E. A. Ticianelli, E. M. A. Martini and M. O. de Souza, J. Mater. Sci., 2020, 55, 6928–6941 CrossRef CAS
- C. Xu, X. Liu, J. Cheng and K. Scott, J. Power Sources, 2015, 274, 922–927 CrossRef CAS
- L. G. da Trindade, L. Zanchet, P. C. Martins, K. M. N. Borba, R. D. M. Santos, R. da S. Paiva, L. A. F. Vermeersch, E. A. Ticianelli, M. O. de Souza and E. M. A. Martini, Polymer, 2019, 179, 121723 CrossRef CAS
- K. Huang, T. Song, O. Morales-Collazo, H. Jia and J. F. Brennecke, J. Electrochem. Soc., 2017, 164, F1448–F1459 CrossRef CAS
- R. Izumi, Y. Yao, T. Tsuda, T. Torimoto and S. Kuwabata, Adv. Mater. Interfaces, 2018, 5, 1–5 CAS
- R. Izumi, Y. Yao, T. Tsuda, T. Torimoto and S. Kuwabata, J. Mater. Chem. A, 2018, 6, 11853–11862 RSC
- H. Zhang, J. Liang, B. Xia, Y. Li and S. Du, Front. Chem. Sci. Eng., 2019, 13, 695–701 CrossRef CAS
- X. Yan, F. Zhang, H. Zhang, H. Tang, M. Pan and P. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 6111–6117 CrossRef CAS PubMed
- T. Sasaki, S. Inoue and S. Kuwabata, Electrochemistry, 2021, 89, 83–86 CrossRef CAS
- J. Guo, A. Wang, W. Ji, T. Zhang, H. Tang and H. Zhang, Polym. Test., 2021, 96, 107066 CrossRef CAS
- J. Guo, A. Wang, W. Ji, T. Zhang, H. Tang and H. Zhang, Polym. Test., 2021, 96, 107066 CrossRef CAS
- R. Thimmappa, K. Scott and M. Mamlouk, Int. J. Hydrogen Energy, 2020, 45, 28303–28312 CrossRef CAS
- R. Thimmappa, D. Walsh, K. Scott and M. Mamlouk, J. Power Sources, 2020, 449, 227602 CrossRef CAS
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC
- C. Zhao, G. Burrell, A. A. J. Torriero, F. Separovic, N. F. Dunlop, D. R. MacFarlane and A. M. Bond, J. Phys. Chem. B, 2008, 112, 6923–6936 CrossRef CAS PubMed
- X. Lu, G. Burrell, F. Separovic and C. Zhao, J. Phys. Chem. B, 2012, 116, 9160–9170 CrossRef CAS PubMed
- R. Y. Kannah, S. Kavitha, O. P. Karthikeyan, G. Kumar, N. V. Dai-Viet and J. R. Banu, Bioresour. Technol., 2021, 319, 124175 CrossRef PubMed
- K. Chen, B. Xu, L. Shen, D. Shen, M. Li and L.-H. Guo, RSC Adv., 2022, 12, 19452–19469 RSC
- R. F. De Souza, J. C. Padilha, R. S. Gonçalves and J. Rault-Berthelot, Electrochem. Commun., 2006, 8, 211–216 CrossRef CAS
- F. Fiegenbaum, E. M. Martini, M. O. de Souza, M. R. Becker and R. F. de Souza, J. Power Sources, 2013, 243, 822–825 CrossRef CAS
- M. G. Freire, C. M. S. S. Neves, I. M. Marrucho, J. A. P. Coutinho and A. M. Fernandes, J. Phys. Chem. A, 2010, 114, 3744–3749 CrossRef CAS PubMed
- K. Saihara, Y. Yoshimura, H. Fujimoto and A. Shimizu, J. Mol. Liq., 2016, 219, 493–396 CrossRef CAS
- T. Li, X. Wang, W. Yuan and C. M. Li, ChemElectroChem, 2016, 3, 204–208 CrossRef CAS
- R. Thimmappa, D. Walsh, K. Scott and M. Mamlouk, J. Power Sources, 2020, 449, 227602 CrossRef CAS
- E. Reichert, R. Wintringer, D. A. Volmer and R. Hempelmann, Phys. Chem. Chem. Phys., 2012, 14, 5214–5221 RSC
- T. K. F. Dier, D. Rauber, D. Durneata, R. Hempelmann and D. A. Volmer, Sci. Rep., 2017, 7, 1–12 CrossRef CAS PubMed
- W. Ma, G. Liu, Q. Wang, J. Liu, X. Yuan, J. Xin, S. Wang and H. He, J. Mol. Liq., 2022, 367, 120407 CrossRef CAS
- C. J. M. Van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, A. N. Simonov, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC
- C. S. M. Kang, X. Zhang and D. R. MacFarlane, J. Phys. Chem. C, 2018, 122, 24550–24558 CrossRef CAS
- M. Alvarez-Guerra, J. Albo, E. Alvarez-Guerra and A. Irabien, Energy
Environ. Sci., 2015, 8, 2574–2599 RSC
- D. Yang, Q. Zhu and B. Han, The Innovation, 2020, 1, 100016 CrossRef CAS PubMed
- A. Atifi, D. W. Boyce, J. L. DiMeglio and J. Rosenthal, ACS Catal., 2018, 8, 2857–2863 CrossRef CAS PubMed
- T. Kunene, A. Atifi and J. Rosenthal, ACS Appl. Energy Mater., 2021, 4, 13605–13616 CrossRef CAS
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed
- D. H. Pool, M. P. Stewart, M. O'Hagan, W. J. Shaw, J. A. S. Roberts, R. M. Bullock and D. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15634–15639 CrossRef CAS PubMed
- E. S. Wiedner, A. M. Appel, S. Raugei, W. J. Shaw and R. M. Bullock, Chem. Rev., 2022, 122, 12427–12474 CrossRef CAS PubMed
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S.-G. Sun and Y. Shao-Horn, Nat. Catal., 2021, 4, 753–762 CrossRef CAS
- K. Zhao, X. Chang, H. Su, Y. Nie, Q. Lu and B. Xu, Angew. Chem., 2022, 134, e202207197 CrossRef
- T. L. Greaves, D. Dharmadana, D. Yalcin, J. Clarke-Hannaford, A. J. Christofferson, B. J. Murdoch, Q. Han, S. J. Brown, C. C. Weber and M. J. S. Spencer, Langmuir, 2022, 38, 4633–4644 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |