
Recent advances in nitrogen-doped graphene-based heterostructures and composites: mechanism and active sites for electrochemical ORR and HER
Reena
Saini
a,
Farha
Naaz
 b,
Ali H.
Bashal
c,
Ashiq Hussain
Pandit
d and
Umar
Farooq
*a
b,
Ali H.
Bashal
c,
Ashiq Hussain
Pandit
d and
Umar
Farooq
*a
aDepartment of Chemistry, School of Basic Science, Galgotais University, Greater Noida, India. E-mail: darumer27@gmail.com
bIndependent Researcher, Greater Noida, India
cDepartment of Chemistry, Faculty of Science, Taibah University, Yanbu, 46423, Saudi Arabia
dDepartment of Chemistry, Jamia Millia Islamia, New Delhi, 110025, India
First published on 7th December 2023
Abstract
The energy crisis, dependence on non-renewable energy resources and environmental pollution pose a great threat to the ecosystem. Consequently, the generation and utilization of hydrogen as a renewable, pollution-free, sustainable energy resource has attracted significant attention. In this case, different approaches have been explored, among which the electrochemical production of hydrogen through HER provides an efficient, green and clean approach for the mass production of H2. Besides HER, other reactions, such as OER and ORR, are core reactions involved in different electrochemical devices that are being developed to produce green energy technologies. In all these reactions, the development of low-cost materials with high electrochemical activity compared to the state-of-the-art noble metal electrocatalysts is important. Over the last few decades, different catalyst materials have been developed for HER, OER and ORR. Among them, graphene-based materials have been widely explored either as a support material or active catalyst for the above-mentioned electrochemical reactions. Specifically, the intrinsic electrochemical activity of graphene (G) is negligible, and therefore, different strategies, including heteroatom doping, composite development and development of heterojunctions, have been used to improve its electrochemical characteristics for HER/OER and ORR. However, although great advancements have been made using N-doped graphene (NG)-based heterostructures and composites as active electrocatalysts, there is still a lack of understanding of their reaction mechanism and the active sites responsible for the enhanced electrochemical performances are still under debate. Hence, considering the research interest over the past few years on NG-based electrocatalysts, herein we attempt to present a holistic summary of the recent advances made in the synthesis strategies and understanding of the role played by the interface, composite development and active sites in HER/OER and ORR over NG-based electrocatalysts.
Reena Saini has completed her bachelor's from Chaudhary Charan Singh (CCS) University Uttar Pradesh, India. She got her master's in chemistry from CCS University. Currently she is doing her Ph.D. under the supervision of Dr Umar Farooq Department of Chemistry, School of Basic Sciences at Galgotias University, India. |
Farha Naaz has pursued her master's degree in chemistry from Hemwati Nandan Bahuguna Garhwal University, Srinagar. She has recently completed Ph.D. under the supervision of Prof. Tokeer Ahmad from the Department of Chemistry, Jamia Millia Islamia, New Delhi. Her research interest is to development nanodimensional functional materials for highly selective and efficient organic transformation reactions and electrochemical water splitting for hydrogen generation. Currently, she is working as an independent researcher towards development of efficient catalysts for sustainable future. |
Ali H. Bashal is currently working as an associate professor at department of chemistry, college of science at Taibah University (Yanbu Branch). He got his master's degree from USA (university of northern Colorado) and his doctoral degree from UK (University of Liverpool). His research interest in developing a nano metal oxide for catalytic reaction and electro-optical applications. |
Aashiq Hussian pandit is currently working as a Postdoctoral fellow IIT Kanpur, India. He pursued his Doctorate degree in chemistry from Jamia Millia Islamia, New Delhi, India. A. H. P. has completed his bachelor's from Government Boys Degree College, Pulwama, affiliated to University of Kashmir. His research interest is development of novel and efficient functional materials like Hydrogels for Biomedical and Sustainable energy applications. |
Umar Farooq is currently working as an assistant professor in department of chemistry, School of Basic Sciences at Galgotias University, India. He pursued his Doctorate and master's degree in chemistry from Jamia Millia Islamia, New Delhi, India. U. F. has completed his bachelor's from Government Boys Degree College, Pulwama, affiliated to University of Kashmir. His research interest is development of novel and efficient functional materials like metal oxide and carbon based heterostructures and MOF's for sustainable energy and environmental remediation applications. He is a lifetime member of the Society of Material Chemistry (SMC), BARC India. |
1. Introduction
Presently, civilization is facing various health, industrial, energy and environmental-related problems, most of which are interrelated. For example, the growth and development of various industries depend on energy as a basic and fundamental requirement. In this case, currently most of the energy comes from fossil fuels, which are formed due to the decomposition of animal and plants. Therefore, environment issues lead to energy, industrial and health crises and vice versa. Hence, for the smooth functioning of human civilization, it is imperative to develop suitable clean, nontoxic and renewable energy resources. Presently, the energy demand is satisfied by fossil fuels. In this case, the formation, characteristics and usage of fossil fuels have attracted significant attention from researchers and environmentalists. The data on population growth, fossil fuel consumption, and production of greenhouse gases is depicted in Fig. 1, showing the direct relation between population growth and environmental pollution.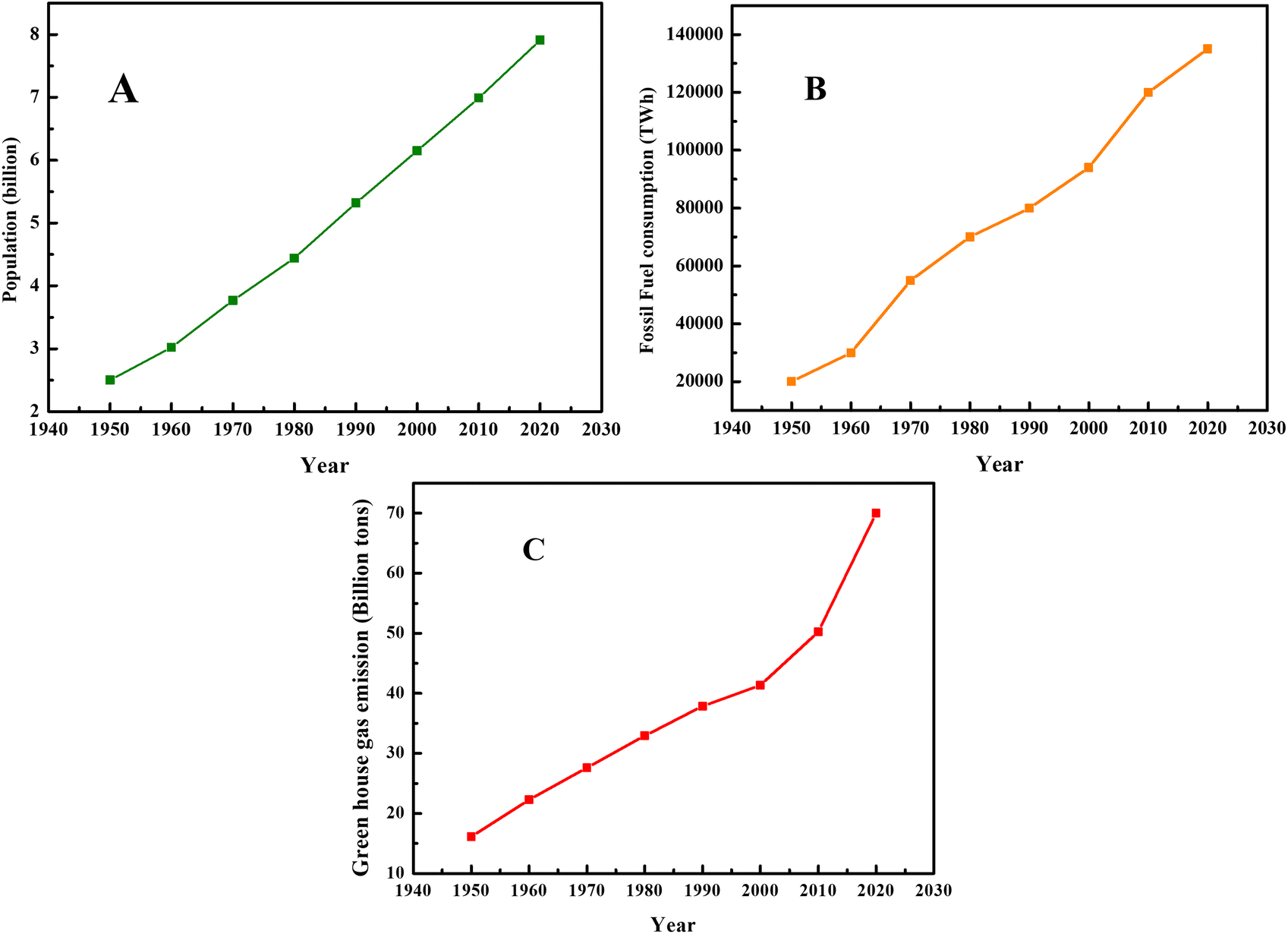 | ||
| Fig. 1 (A) Population growth, (B) fossil fuel consumption and (C) greenhouse gas emissions since 1950. Data obtained from our world in data website. | ||
To meet the ever-increasing energy demands of the growing population without sacrificing the ecosystem/environment, the development and exploration of green, sustainable and clean energy sources have become a hot research topic attracting immense attention.1–9 To date, various alternative energy sources have been explored, among which electrochemical energy is envisioned as a suitable candidate with potential to solve the current energy and environmental crises. Furthermore, it is expected that electrochemical energy devices, such as solar cells, supercapacitors, fuel cells and batteries, will have a high rate of commercialization depending on the energy requirements of different regions across the world.10,11 However, the main problem or concern is the development and supply of a huge amount of green and sustainable energy, which can possibly replace fossil fuels for industrial applications. In this case, different chemical compounds, such as hydrogen, methanol, ammonia and methyl cyclohexane, are considered promising candidates.12–16 Regarding sustainable energy, the electrochemical splitting of water using fuel cells is important technology to address issues related to energy. Based on the natural abundance of water and high energy density of hydrogen, electrochemical water splitting has attracted significant interest.17,18 To carry out water splitting reactions at minimum energy input, suitable, highly efficient, durable catalysts are highly important. Presently, for the HER, OER and ORR, the state-of-the-art catalysts including noble metal-based catalysts such as Pt, IrO2, and Pd are considered benchmark catalysts.19–21 However, these state-of-the-art catalysts are associated with certain drawbacks including high cost, limited reservoirs, dissolution during use, and CO poisoning, hindering their practical application on the industrial scale.20
Thus, to overcome the limitations associated with the state-of-the-art HER, OER and ORR catalysts, it is necessary to explore low-cost active catalysts that are highly stable, abundant and durable. Presently, a large library of less expensive, earth abundant, active catalyst materials has been developed to investigate their possible application in the HER, OER and ORR.22–25 These low-cost materials include metal oxides, phosphides, nitrides, dichalcogenides, and carbides. Recently, carbon-based materials such as carbon dots, carbon nanotubes, carbon cloths, and graphene have been explored as fascinating perspective electrocatalytic materials for water splitting reactions.26–30 Considerable and persistent research efforts have been devoted to the application of carbon-based 2D graphene sheets possessing hexagonal lattices with a single layer of carbon atoms in electrochemical energy devices. Although graphene is an electrochemically inert material, its possible chemical modifications offer great scope to use its π conjugated carbon network as a potential alternative to noble metal-based catalysts for the HER, OER and ORR.31 Graphene offers various opportunities to realize enhanced electrochemical activity including doping with heteroatoms, and it also acts as an efficient substrate material for different electrochemically active catalysts (like metals), thus improving their exposed electrochemical active sites.32 In addition, 2D graphene plays an important role in controlling the morphology and microstructure of electroactive materials together with a strong synergistic coupling effect for improved electrochemical HER, OER and ORR performances.33
Considering that graphene is an intrinsically inert electrochemical material, different strategies have been explored and utilized to improve its application of in electrochemical water splitting reactions.34 The development of heterostructures and composites are important strategies used to develop efficient graphene-based electrochemically active materials. The development of heterostructures offers significant advantages compared to their single pristine components. As an efficient catalyst, heterostructures provide a possible strategy to tune their crystal structure and electronic structures, together with a synergistic coupling effect.35 Compared to 3D materials, the large active surface area of their 2D counterparts make them excellent candidates for the development of heterostructures. Among the 2D materials, graphene is an excellent and promising material with interesting characteristics such as large number of reactive sites on its surface, high conductivity and structural flexibility.36 All these characteristic properties of graphene provide fast electrolyte active site interactions, fast kinetics in catalytic reactions and high stability under harsh electrochemical conditions. Thus, due to these fascinating properties, graphene can easily form heterostructures with other materials such as metals, metal oxides, nitrides, and dichalcogenides.37 Furthermore, the diverse properties of graphene help in the construction and design of hybrid multi-component systems as catalysts, in which the merits of each component are combined, resulting in an improved efficient catalytic performance.
However, the presence of unstable intrinsic bonding states in heterostructures results in a high activated Fermi energy state, which limits their practical.38 Therefore, to further improve their practical application, another important strategy is heteroatom doping of graphene, which not only improves the catalytic activity but also controls the size and shape of graphene, thus creating proper room for the manipulation of its active sites and surface area density. The heteroatoms used for doping include electronegative elements such as B, F, N and S.39,40 The electronegative characteristic of dopant elements improves the water adsorption activity of the materials, which is highly beneficial for hydrogen evolution catalytic activity and stability of the energy states of the modified material. Alternatively, in the ORR, the difference in the electronegativity of the dopant element and carbon atoms of graphene promotes the adsorption of O2, thus weakening the O–O bond to enhance the ORR. However, for easy and meaningful control of the local active sites of graphene, the use of different dopants depends on the knowledge of the local structure of each active site. To analyse the local structure of pure and doped graphene, different studies and investigations have been carried, out, as discussed later in this article. Further, to prepare electrochemically active, efficient and stable graphene-based materials, the synergistic coupling of heteroatom doping, incorporation of electroactive materials and heterostructures has been evaluated.41,42 Moreover, according to theoretical and experimental investigations, it has been observed that the activity and lifespan of electrochemical-active materials for the OER, HER and ORR are strongly dependent on their structure, composition and morphology.43–45 Therefore, the construction of graphene-based materials with rational control of their size, shape, morphology, composition and structure is highly desirable for electrochemical water splitting applications.
A large number of review articles focusing on the use of heterostructures based on metals, metal oxides, metal phosphides, metal carbides and dichalcogenides has been published to date.45–50 In addition, comprehensive and critical reviews regarding the application of either pure graphene or doped graphene in electrochemical HER or ORR have been published.51,52 However, to the best of our knowledge, there is a lack of a comprehensive reviews regarding N-doped graphene-based heterostructures and composite materials for HER, OER and ORR electrocatalytic processes. Thus, in this review article (Section 2), we aim to present an overview of the different synthesis strategies used to develop both doped and hetero-structured graphene-based catalyst materials. Then, in Section 3, we systematically explore and summarize the progress made in the last few years on N-doped graphene-based hetero-structured materials and electroactive-incorporating composite materials for electrochemical water splitting reactions including the HER/OER and ORR.
2. Advantages and types of N-doped graphene-based heterostructures for electrochemical applications
Considering the limitations (including low activity/performance, poor conductivity, low charge transfer activity, small number of active sites, and low stability) of pristine electrochemical catalysts, the development of a heterojunction between two or more different materials has emerged as a new strategy to develop different novel catalysts, which offer better activity, stability, and large number of active sites for the HER/OER and ORR. In a typical heterostructure, two or more single components with different properties and characteristics are coupled, and consequently they possess unique properties compared to their pristine counterparts. From the perspective of electrocatalysis, heterostructure catalysts allow the rearrangement of electrons at their interface, thus modifying the number of active sites. In addition, the pristine components are chosen in a manner to have a synergistic effect between the active sites present on different single components, which can promote the reaction kinetics for faster electrochemical reactions.53–55 The main advantages of hetero-structured catalysts include (i) large number of active sites produced due to lattice strain present at the interface of the heterojunction, (ii) morphological diversity of heterostructures, (iii) design privilege, providing high surface active catalytic sites and high specific surface area and (iv) better charge carrier and charge transfer ability due to complementary redox properties between different components of the heterostructure, promoting the activity and efficiency of heterostructure catalyst materials due to their synergistic effect.56–60 Regarding graphene-based heterostructures, different types of hybrid materials, as shown in Fig. 2, have been developed to beneficially utilize the characteristics of both single components for enhanced electrocatalysis.61–63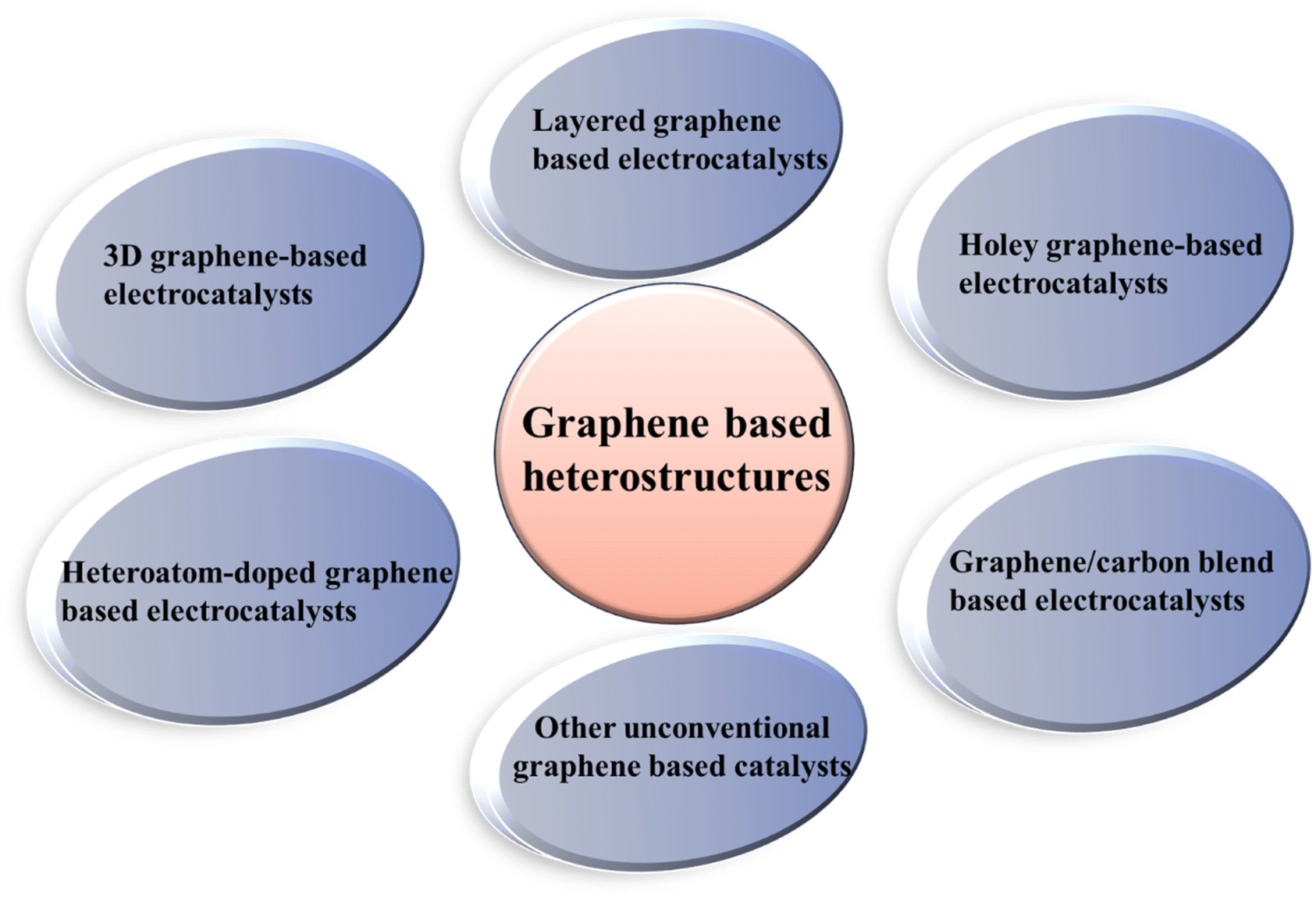 | ||
| Fig. 2 Different types of hybrid heterostructures based on graphene. | ||
3. Recent advances in the synthesis of N-doped graphene heterostructure-based catalysts
All catalytic reactions exhibit a notable difference in their mechanism, which offers different construction and design principles for the development of the corresponding active catalyst materials. Similarly, for the HER, OER and ORR, different design strategies and principles are followed. Recently, graphene-based materials have been explored for the HER, OER or ORR and great success and advancements have been achieved. For the development of N-doped graphene, different approaches have been employed with the main aim to not only achieve unique physicochemical properties but also incorporate desirable active sites for different electrochemical reactions. For the synthesis of pure graphene, five different categories of synthesis approaches have been employed, which include solvothermal/hydrothermal approach, electrochemical, chemical vapour deposition, thermal treatment and physical approaches.63–68 In this section, we discuss the advances made in the design and synthesis of N-doped graphene-based heterostructure/composites for different applications.3.1. Hydrothermal synthesis approach
Numerous researchers have certified that graphene can be either used as a substrate for active catalysts or employed as a non-metal ORR catalyst. For example, N-doped graphene was successfully synthesized by Qu et al. using the CVD approach and an excellent ORR performance was observed.69 Later, most graphene-based materials were mainly synthesized using chemical exfoliation or CVD method.70,71 Based on recent investigations, it has been observed that together with N-doping, the development of heterostructures is an important approach to improve electrochemical performances based on the effect of synergistic coupling.72 Regarding the development of N-doped graphene-based heterostructures for improved electrochemical performances, different approaches have been employed. For example, the hydrothermal approach was used to successfully synthesize N-TiO2 nanowires (NW) and N-doped graphene (NG)-based heterostructures.73,74 The hydrothermal approach has been developed to overcome limitations such as 100 °C as the upper limit temperature range for reactions taking place in the aqueous phase, where this low upper limit temperature range limits the use of aqueous-phase reactions to synthesize materials that are produced at a relatively high temperature. Using the hydrothermal/solvothermal approach, high-quality control of morphology, shape and size can be achieved. Liu et al. have employed N-doped graphene and N-doped TiO2 for the development of a heterostructure.75 Both TiO2 and graphene were doped with nitrogen, followed by the development of the heterostructure to exploit the benefits of both doping and heterostructure for efficient photocatalytic reactions. During the synthesis process, urea was used as the source of N for both TiO2 NW and graphene. In a typical synthesis approach, a hybrid N-TiO2/NG catalyst was synthesized using a self-assembly-assisted hydrothermal reaction by employing urea as a source of N. During the synthesis, 0.3 g TiO2 NW (synthesized using hydrothermal approach in this report) and 15 g urea (as N source for both TiO2 and GO) were added to a homogenised graphene oxide (GO) (prepared by modified Hammers process in this report) aqueous solution. This reaction mixture was stirred for 15 min, and then transferred to a Teflon-lined autoclave and heated at 200 °C for 10 h. After hydrothermal treatment, the reaction mixture was cooled to room temperature and black cylinders of the hybrid photocatalyst were obtained. The obtained sample was analysed using different characterization techniques and the successful N-doping in TiO2 NW and GO was confirmed using Raman and X-ray photoelectron spectroscopy. Another report demonstrated the use of the hydrothermal approach for the synthesis of a layered 3D architecture of MoS2/N-doped graphene hybrid heterostructure using graphene oxide (GO), sodium molybdate, urea and L-cysteine as precursors for the final hybrid structure.76 During the synthesis, GO was synthesized using graphite powder produced via the modified Hummers’ method. During a typical synthesis process, 0.1 g of GO was added to 60 mL water, followed by the addition of sodium molybdate (0.5 g) and L-cysteine (1 g) at pH 6.5 (maintained by adding 0.1 M NaOH). After sonication, 10 g urea was added to the reaction mixture and transferred to a Teflon-lined reactor and heated at 180 °C for 36 h to get the final hybrid material. According to the investigation, it was observed that layered MoS2 was assembled face-to-face on graphene sheets, followed by doping with N. Different analogues were prepared with different ratios of precursors. The typical process for the formation of hybrid MoS2/GNs and MoS2/NG is depicted in Fig. 3a. During the synthesis of MoS2/NG, the microstructure was tuned by changing the ratio of precursor for Mo and GO (Na2MoO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GO) (1.5:1, 3:1 and 5:1). The microstructure at different ratios was analysed by SEM, as shown in Fig. 3b–d. The formation of the 3D architecture was due to the flexible nature of graphene, which showed a coalescing process during hydrothermal treatment. Also, the high surface energy of the 2D material resulted in the formation of a 3D architecture. The sample obtained at 180 °C corresponding to a 1.5:1 ratio showed a petal-like morphology, as observed in the TEM and HETEM analysis shown in Fig. 3e and f, respectively. A similar morphology was also reported by Chang et al., where MoS2/G was hydrothermally synthesized at a 1:2 M ratio and 240 °C.77
:GO) (1.5:1, 3:1 and 5:1). The microstructure at different ratios was analysed by SEM, as shown in Fig. 3b–d. The formation of the 3D architecture was due to the flexible nature of graphene, which showed a coalescing process during hydrothermal treatment. Also, the high surface energy of the 2D material resulted in the formation of a 3D architecture. The sample obtained at 180 °C corresponding to a 1.5:1 ratio showed a petal-like morphology, as observed in the TEM and HETEM analysis shown in Fig. 3e and f, respectively. A similar morphology was also reported by Chang et al., where MoS2/G was hydrothermally synthesized at a 1:2 M ratio and 240 °C.77
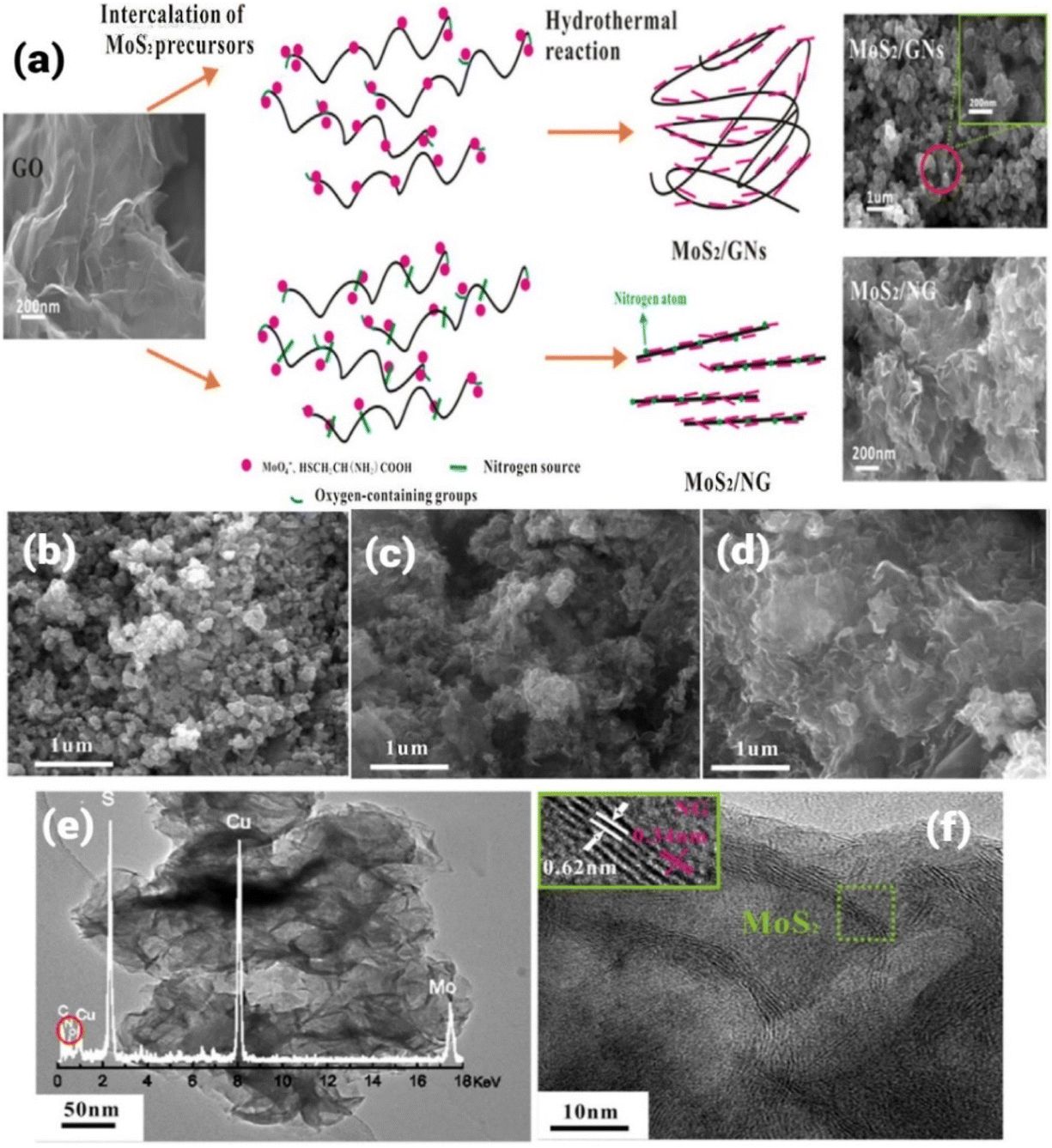 | ||
| Fig. 3 (a) Schematic representation of the synthesis of MoS2/NG and MoS2/graphene nanosheet hybrids using typical hydrothermal approach, (b–d) SEM micrographs at 1.5:1, 3:1 and 5:1 ratio of Na2MoO4:GO respectively, and (e) TEM and (f) HRTEM micrographs of MoS2/NG at 1.5:1 Na2MoO4:GO ratio. Reprinted with a permission from Reference (76). Copyright 2016, Elsevier. | ||
Pie et al. reported the synthesis of a heterostructure between N-doped graphene quantum dots and cadmium tungstate nanorods using the solvothermal/hydrothermal approach.78 The as-developed heterostructure was further analysed as a photocatalyst and photo-electrocatalyst for water splitting reactions. In a typical synthesis procedure, firstly graphene oxide (GO) and N-doped graphene (NG) were synthesized using the Hummer's process and solvothermal process, respectively. During the synthesis of NG quantum dots, GO (50 mg) was dissolved in N,N-dimethylformamide (DMF, 20 mL), followed by sonication for 30 min. Then, the as-obtained solution was transferred to a Teflon-lined autoclave and heated at 200 °C for 6 h. After cooling, the black precipitate from the suspension was filtered using a 0.22 μm membrane and the filtrate containing N-GQD having a brown-yellow colour was obtained. The final powdered product containing N-GQD was obtained using a rotatory evaporator. The concentration of aqueous solution of N-GQD was 0.1 mg mL−1. Finally, the heterostructure was prepared using the hydrothermal approach. During a typical process, different amounts (5 mL, 10 mL, 20 mL and 40 mL) of as-developed N-GQD were employed. Briefly, 0.44 g of Cd(OAc)2·2H2O was dissolved in different amounts of aqueous solution of N-GQD, followed by vigorous stirring for 2 h and addition of 25 mL aqueous solution of Na2WO4·2H2O with constant stirring. The final reaction mixture was heated in a 100 mL Teflon-lined autoclave at 160 °C for 24 h. After natural cooling, the final product was obtained and washed several times with water. Different heterostructures were obtained by changing the amount of N-GQD solution and denoted as N-GQDs-CdWO4-1 to N-GQDs-CdWO4-4. The FESEM micrograph showed that nanorods of CdWO4 were formed with scattered N-GQD, as shown in Fig. 4a and b.
 | ||
| Fig. 4 (a and b) SEM micrographs of N-GQD/CdWO4 at different scales. Reprinted with permission from ref. 78. Copyright 2017. Elsevier. | ||
Similarly, Zn0.1Cd0.9S-incorporated N-GQD and graphene heterostructure for enhanced photoelectric performance was prepared by Jiang et al. using the hydrothermal approach. During the synthesis of the heterostructure, the required amount of cadmium acetate, zinc acetate and thiourea was dissolved in water as the source of Cd, Zn and S, respectively, and ultrasonicated for 15 min. Subsequently, 1 mL solution of N-GQD and 2 mg of GO were added to the solution, followed by ultrasonication and hydrothermal treatment in a 100 mL autoclave at 140 °C for 4 h.79 Kang et al. designed a ternary heterostructure comprised of 1D hollow nanofibers of g-C3N4 (HGCNF) having S/N-doped graphene and MoS2 decorated on it.80 The synthesis of HGCNF was carried out using the electrospinning and sintering approach. Alternatively, the heterostructure was synthesis using an in situ hydrothermal method, in which 0.1 g of HGCNF was added and ultrasonically well dispersed in an aqueous solution of GO, followed by the addition of thiourea as a reducing agent and source of S/N for GO. After 1 h stirring, 0.4 g of Na2MoO4 was added to the reaction mixture as the source of Mo, followed by stirring for 1 h and addition of 0.8 g of thiourea. Finally, the obtained reaction mixture was heated under hydrothermal conditions at 200 °C for 24 h. Similarly, a binary heterostructure of HGCNF was synthesized using the same approach without the addition of GO. Recently, Zhou et al. reported the synthesis of ultrasmall NiS/NiS2 heteronanoparticles monodispersed on N-doped graphene nanosheets via an in situ hydrothermal approach.81 During a typical synthesis, 40 mg GO was dispersed in water by the ultrasonication process, after which 1.5 mmol Ni(NO3)2·6H2O was added and kept under vigorous stirring for 30 min. Then, 3 mmol of CH4N2S was added and after 30 min stirring, the solution was finally treated under hydrothermal condition in a 50 mL autoclave at 180 °C for 24 h. The schematic formation of the ultra-fine monodispersed NiS/NiS2 on NG nanosheets is illustrated in Fig. 5. Recently, Wang et al. reported the synthesis of vertically arrayed MoS2 nanosheets on N-doped reduced graphene oxide using a microwave-assisted hydrothermal/solvothermal approach followed by heating at 600 °C.82 In this report, 1 mg mL−1 GO together with 162.6 mg (NH4)2MoS4 and 15 mg ascorbic acid was dissolved in 25 mL DMF and transferred to a 35 mL vial and heated at 200 °C for 10 h under microwave conditions. Finally, the obtained powder after washing and drying at 60 °C was heated at 600 °C under an NH3 atmosphere for 2 h to get N-doped GO. A schematic representation of the synthesis process and micrographs of the as-prepared samples are shown in Fig. 6a. The SEM and TEM micrographs show that N-rGO was uniformly modified by the MoS2 nanosheets, as observed in Fig. 6b–h. Similarly, many other reports have recently employed the hydrothermal approach for the synthesis of N-doped graphene-based heterostructures for electrocatalytic energy generation applications. Table 1 presents a summary of the reports in the past five years using the hydrothermal/solvothermal approach for the preparation of N-doped graphene (NG)-based heterostructure hybrid materials. Table 1 also includes the reaction conditions such as temperature, pH, concentration and precursors used during the synthesis of NG-based heterostructures.
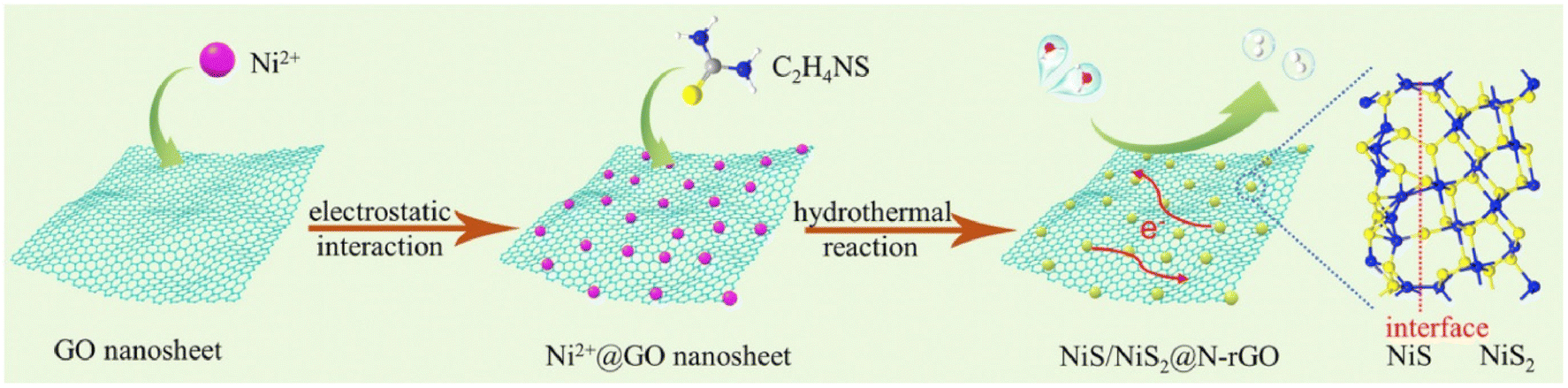 | ||
| Fig. 5 Schematic reaction process for the synthesis of NiS/NiS2@N-rGO. Reprinted with permission from ref. 81. Copyright 2019, Elsevier. | ||
 | ||
| Fig. 6 (a) Schematic reactions steps involved in the synthesis of MoS2/NGO vertical heterostructure, (b and c) SEM micrographs, (d and e) TEM micrographs, and (f and g) HRTEM images of MoS2/NGO vertical heterostructure. Reprinted with permission from ref. 82. Copyright 2021, Elsevier. | ||
| Type of heterostructure electrocatalyst | Precursors | Reaction condition | Ref. |
|---|---|---|---|
| Hydrothermal method | |||
| MoSe2/NG | GO, N2H4·H2O, Na2MoO4·2H2O, hydrazine hydrate | 220 °C, 24 h | 105 |
| Carbon rich C3N4/NG | Melamine, glucose, NG, Isopropanol, HCl | 180 °C, 12 h | 106 |
| N/Ti3+-TiO2/Graphene | GO, TiN | 200 °C, 36 h | 107 |
| NG/BiFeO3 | GO, KOH, BiFeO3 (Precursor for BiFeO3 prepared via reverse co-precipitation) | 180 °C, 12 h | 108 |
| NG/Co0.85Se nanospheres | GO, sodium stearate, CoCl2·6H2O | 180 °C, 15 h | 109 |
| NG/SnS2/TiO2 | Poly(diallyldimethylammonium chloride), GO, thioacetamide, ethylenediamine, titanium glycolates, SnCl4·5H2O | 180 °C, 20 h | 110 |
| NiCo2O4/NG | Co(NO3)2·6H2O, Ni(NO3)2. 6H2O, hexamethylenetetramine | 90 °C, 6 h | 111 |
| Annealing (300 °C, 2 h) | |||
| N-RGO/TiO2 | Ti(SO)4, GO-NH2, GO, HF | 180 °C, 12 h | 112 |
| MoS2/NG/Ionic Liquid | GO, 1-butyl-3-methylimidazolium tetrafluoroborate, (NH4)2MoS4, N2H4·H2O and NH3·H2O | 240 °C, 24 h | 113 |
| MoS2/Sb/NG | Antimony(III) fluoride, sodium molybdate dehydrate, polyvinylpyrrolidone, GO | 200 °C, 24 h | 114 |
| NG-Co3O4 | CoCl2·6H2O, GO | 120 °C, 16 h | 115 |
| MnSnO3/N-RGO | GO, Na2SnO3·3H2O, MnCl2·4H2O, NH3 | 180 °C, 24 h | 116 |
| Heating at 400 °C, 200 min | |||
| SnS/NG | Carbon disulphide, dodecanethiol, SnCl2·5H2O, ethylenediamine, | 180 °C, 48 h (SnS) | 117 |
| 200 °C, 24 h (SnS/NG) | |||
| N-doped graphene/Alloys (NiCo, CoFe) | GO, NiCo-NiOCoO, Ni(NO3)2·6H2O, | 150 °C, 12 h | 118 |
| CVD approach | |||
| N-doped graphene | MgO as template, HCl | Ar (up to 950 °C), CH4 (10 min), NH3, H2 | 119 |
| Co3O4/NGF | NG, Co(NO3)2·H2O, Ni foam | 250 °C, 2 h | 120 |
| Co3O4/CNT | NG, Co(NO3)2·6H2O, ethanol, Ni Foam, DI | 60 °C, 12 h | 121 |
| Annealing (350 °C, 2 h) | |||
| Co/Co3O4/N-doped graphene | Cobalt acetate, GO | NH3, Ar (800 °C, 2 h) | 122 |
| N-CoS2@Co3O4 | Co(NO3)2·6H2O, Isopropanol, Glycerol, sulfur powder, ethanol, NH3.H2O | N2 (700 °C, 2 h) | 123 |
| Fe3O4−rgo | Fe(NO3)3·9H2O, glucose, NaCl | 80 °C, 24 h, Ar, CH4 (800 °C,30 min) | 124 |
| CoMn2O4 N-doped graphene | NCNTs, Co(NO3)2·6H2O, Mn(NO3)2·4H2O, urea, ethanol | 150 °C, 10 h, Ar, H2 (1000 °C, 15 min) | 125 |
| CH4 (90 min) | |||
| N-doped CNT/ZnO | NCNTs, diethyl zinc, Si-wafers | N2 gas | 126 |
| LaNiO3-NCNTs | La(NO3)3·6H2O, C4H6O4Ni·4H2O, diethylene glycol, DW, melamine | 60 °C 3 h, annealing (700 °C, 12 h) Ar flow | 127 |
| Ni0.9Co0.1Fe2O4/NCNTs | Cobalt(II)-nitrate hexahydrate, nickel(II)-nitrate hexahydrate, iron(III)-nitrate nonahydrate, diethylene glycol, NaOH, hexadecanoic acid, IPA, urea, SiO2 Aerosil | 100 °C, 12 h | 128 |
| 600 °C, 3 h, Ar, H2 | |||
| RuO2/NCNTs | (Ru3(CO)12, CNTs) | 120–300 °C, 2 h | 129 |
| N-doped NiCo2O4 | Ni(NO3)2·6H2O, Co(NO3)2·6H2O,HCl, urea, Ni Foam | Ar, NH3 | 130 |
| Co/CoS/Co3O4-SNC | Co(NO3)2·6H2O, 2-methylimidazole, methanol | 80 °C, 24 h | 131 |
| Ar (750 °C, 1 h) | |||
| MnOx/CoOx /NG | Mn, Co, GO | −55 °C, 48 h | 132 |
| NH3, Ar (800 °C, 2 h) | |||
| N-doped CNTs/Ni/NiO | PVP, Ni(AC)2·4H2O, ethanol, pyridine, Al foil | 660 °C, 2 h, N2 (800 °C, 30 min) | 133 |
| Co@CoOx/HNCNTs | Co(NO3)2·6H2O, urea, DI, carbon fibre, melamine | 60 °C, 8h | 134 |
| Ar, C2H4 (900 °C, 30 min) | |||
| N-RGO/CNT-MnO2 | CNT, PEG, KMnO4, GO | NH3, 900 °C | 135 |
| γ-Fe2O3/CNTs | Ferrocene, thiophene, toluene, ethylene | — | 136 |
| Al2O3/BiVO4/WO3 | bismuth oxide, vanadium oxide, butyl acetate, tungsten hexachloride, DMF, trimethylaluminum | 773 K, 30 min | 137 |
| ZnFe2O4/NG/ZnO | SiO2/PDA, dopamine, Zn(CH3COO)2·2H2O, NH3·H2O, PEG,EG, FeCl3·6H2O, CH3COONa, Pyridine | 600 °C, 12 h | 86 |
| N2, annealing 500 °C, 3 h | |||
| Thermal annealing | |||
| Co3O4/MnO2/NGO | Co(NO3).6H2O, Mn (CH3COO)2·4H2O, KMnO4, urea, 30% aqueous ammonia solution | 90 °C, 12 h (under vacuum) | 138 |
| Annealing (650 °C, 8 h) | |||
| PdeCeO2-N-doped graphene | Hydrazine, uric acid, graphite flakes, Al powder, PdCl6(NH4), CeO2-NR, NaBH4 | 180 °C, 24 h | 139 |
| 200 °C, 2 h | |||
| MnO/CoMn/N-doped graphitic composites | Co(NO3)2·6H2O, Mn(NO3)2·4H2O, K3Co(CN)6 | N2 (900 °C, 2 h) | 140 |
| Co/Co3O4/NC | Co(NO3)2·6H2O, trimesic acid, 4,4-bipyridine, DMF, DEG | 80°, 48 h | 141 |
| N2, O2 (30 min), 900 °C, 2 h | |||
| Fe3O4/NHCSs | ZnO, DMF, FeCl3, 2-aminoterephthalic acid | 600 °C, 12 h | 142 |
| Ar (900 °C, 2 h), | |||
| CoO/Co/N-rGO | Cobalt acetate tetrahydrate, GO ethylene glycol, melamine, acetone | N2, 600 °C 3 h | 143 |
| Fe/Co/CoO/N doped carbon frameworks | Zn(NO3)2·6H2O and 2-methylimidazole, methanol, Co(NO3)2·6H2O, FeCl3·6H2O,melamine | 40 °C, 12 h | 144 |
| Annealing N2 (700, 800 °C, 900 °C, 3 h) | |||
| Co3O4/CN HNPs | Co(OAc)2·4H2O, PANI | 70 °C, 12 h | 145 |
| Annealing (300 °C, 4 h) | |||
| Co/CoO-NGA | Co(NO)3·6H2O, urea, GO | 800 °C, 2 h | 146 |
| MnO/N-rGO | KMnO4, N-rGO, sucrose | 70 °C, 12 h | 147 |
| Annealing N2 (800 °C, 1 h) | |||
| Co/CoFe2O4/N-graphene | GO, Tris-buffer, FeCl3, Co (NO3)2·6H2O, dopamine | Ar (800 °C, 3 h) | 148 |
| N-GQDs/Co3O4 | CoCl2, N-GQDs, NaBH4 | 60 °C, 10 h | 149 |
| Annealed Ar (600 °C, 2 h) | |||
| N-doped graphene-wrapped carbon nanoparticles | CCl4, urea, metallic potassium | 80 °C, 12 h | 150 |
| Annealed Ar (1000 °C, 2 h) | |||
| Fe/Co3O4/N-GCFs | Co (OAc)2·4H2O, Fe(acac)3, PAN, DMF | 250 °C, 4 h | 151 |
| Annealed H2, N2 (800 °C, 3 h) | |||
| N-doped graphene/TiO2 nanocomposite | SDS, H2O2, Na2SO4, titanium trichloride | 90 °C, 12 h | 152 |
| N-doped graphene/Si3N4/SiC | SiC, N2, Ar | Annealed at 800 °C to 1300 °C 2 × 10–5 to 2 × 10–4 Torr | 153 |
| Quenched to 500 °C | |||
| MoO2/NG composite | MoO2(acac)2, urea, GO | Ar (400–600 °C, 2 h) | 154 |
| Ni/NiO/NCSs | NCSs, urea, Ni(CH3COO)2·4H2O | 600 °C, annealed Ar (500 °C, 2 h) | 155 |
| ZnCo2O4@nitrogen doped graphene oxide/PANI | Aniline, ferric chloride, H2O2, PANI, N-GO, zinc nitrate, cobalt nitrate | 90 °C, 12 h | 156 |
| 550 °C, 8 h | |||
Based on the above discussion, it can be observed that the hydrothermal/solvothermal method has been successfully used for the synthesis of a variety of N-doped graphene-based hetero-structured materials at low temperature compared to other reported approaches such as CVD, thermal treatment, and physical approaches.
3.2. Chemical vapour deposition (CVD)
For practical applications, new approaches are being adopted for the scalable, low-cost production of new pristine and heterostructure materials with novel properties, exotic physical phenomena and high-quality applications. In this quest, CVD is considered a powerful approach to meet the practical applications for electrochemical phenomena. In the case of graphene, as a 2D material, several correlations have been observed and demonstrated between developed graphene-based materials and reaction parameters used in CVD. In a typical CVD approach, materials are developed either by carrying out vapour phase reaction of gaseous materials or reaction on the surface of a substrate. Subsequently, the deposited vapour phase is transformed into the desired solid material. The main advantage of CVD is in the fact that the morphology, orientation of planes, number of layers, size, defects and dopants can be easily controlled by varying the reaction parameters, including temperature, distance between source and substrate, substrate concentration, and flow rate of carrier gas. Recently, traditional and modified CVD approaches have been widely used for the development and fabrication of 2D graphene-based materials and their heterostructures. Shi et al. developed an MoS2/graphene heterostructure by employing an all-CVD synthesis approach.83 During the typical synthesis process, a monolayer of graphene was grown on Au foil under ambient pressure CVD at 970 °C together with gases such as CH4, H and Ar. For the growth of MoS2, MoO3 and S were used as the reaction precursors at low-pressure CVD and the growth of all the samples was carried out in a three-zone furnace having a quartz tube with a diameter equal to 1 inch. During the synthesis, the temperature in the three-zone furnace was equal to 680 °C, 530 °C and 102 °C for Gr/Au, MoO3 and S powder, respectively. After the successful growth, a monolayer heterostructure of MoS2/Gr was detached from the substrate surface and during the whole process, H2 and Ar were used as carrier gases. A schematic illustration of the development of the MoS2/Gr vertical heterostructure is presented in Fig. 7. Xia et al. also employed the CVD approach for the synthesis of N-doped graphene/ReSe2/Ti3C2 MXene.84 During the synthesis, the first step was to obtain a template in the form of MXene. Prior to the synthesis of the heterostructure, Ti3C2Tx nanosheets and polymethyl methacrylate (PMMA) microspheres were prepared, followed by the synthesis of a 3D porous network of MXene. The ReSe2/MXene heterostructure was prepared by mixing 100 mg as-prepared 3D MXene and 10 mg of NH4ReO4 in a quartz boat placed at the centre of a CVD furnace with three temperature zones. At the edge near the gas inlet of the furnace, another quartz boat containing 1 g of Se was placed. After 30 min Ar purging, the temperature in the regions of the boats containing Se and NH4ReO4 was increased to 400 °C and 550 °C, respectively, and the process was left to continue for 15 min under an Ar/H atmosphere. Finally, the NG/ReSe2/MXene heterostructure was synthesized using PECVD (Plasma enhanced CVD), during which the as-synthesized hybrid ReSe2/MXene was placed in another furnace near the plasma generator at 3 Pa, and for the growth of NG/ReSe2/MXene, pyridine was used as both the nitrogen and carbon source at the growth temperature of 550 °C for 30 min. This process was carried out under an Ar atmosphere. A schematic representation of the synthesis of NG/ReSe2/MXene together with SEM micrographs of pure MXene, ReSe2/MXene and NG/ReSe2/MXene is illustrated in Fig. 8a and b–d, respectively.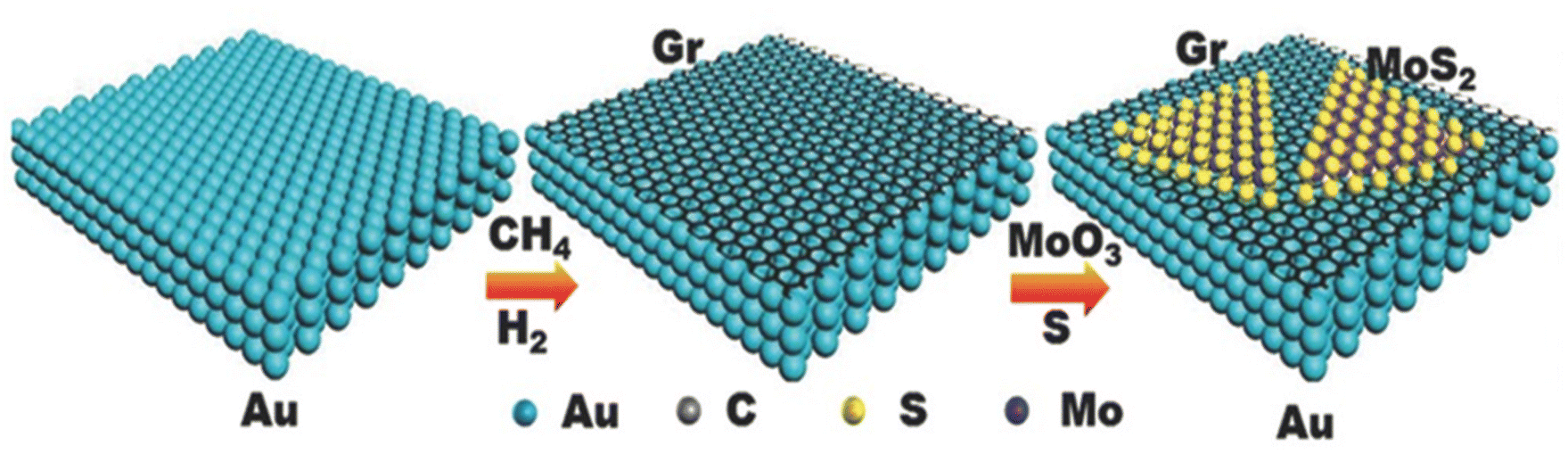 | ||
| Fig. 7 Schematic illustration of reaction steps involved in the synthesis of MoS2/Gr heterostructure using CVD approach. Reprinted with permission from ref. 83. Copyright 2015, Wiley. | ||
 | ||
| Fig. 8 (a) Schematic illustration of CVD synthesis of NG/ReSe2/MXene and (b–d) SEM images of MXene, ReSe2/MXene and NG/ReSe2/MXene heterostructure, respectively. Reprinted with permission from ref. 84. Copyright 2015, Wiley. | ||
An important modified approach using CVD to develop a vertically stacked monolayer of WS2 heterostructure with graphene was reported by Tan et al.85 In this study, the CVD approach was used to separately synthesize WS2 and graphene. For the synthesis of the WS2 monolayer, SiO2/Si was used as the template substrate. The typical synthesis was carried out in a double-temperature zone furnace, in which two precursors of WO3 and S were separately placed in a double-walled quartz tube and positioned at the external and internal tubes, respectively. Both precursors were heated to their respective vaporization temperature and carried to the substrate surface for sulfurization reaction of WO3 to give WS2 as the final product. Monolayer graphene was synthesised using W as the substrate and Cu as the catalyst. During the synthesis process, 1 cm2 W and Cu pieces were rinsed with iso-propyl alcohol (IPA) and acetone, and before rinsing Cu, it was treated with HCl to remove the oxide layer. Then, W with mounted Cu on it was placed in a furnace and annealed at 1090 °C for 30 min under an H and Ar atmosphere at 100 sccm and 200 sccm, respectively, before the growth of the graphene sample. Finally, graphene was grown on molten Cu under a flow of mixed carrier gas containing Ar, H and CH4 for 90 min, followed by a second growth process at 1060 °C for 30 min. Subsequently, the sample was moved out of the hot zone to cool to room temperature. For the development of a G/WS2 heterostructure, the as-developed CVD samples were spin-coated and cured at 150 °C for 15 min using a PMMA scaffold at 4500 rpm. The W and SiO2/Si substrates used during CVD synthesis were etched using the electrochemical approach in NaOH solution and KOH at 60 °C, respectively. The Cu substrate was removed by etching with 0.1 M (NH4)2S2O8. Finally, the floating PMMA/WS2 and PMMA/graphene were transferred carefully to deionised water for washing to remove/dilute the residual contaminants and the as-developed films were deposited on an Si chip, followed by baking at 150 °C for 30 min.
Wang et al. developed hollow spheres of monolayer N-doped graphene and ZnO on ZnFe2O4.86 In this report first hollow spheres of ZnFe2O4/ZnO were synthesized using dual template and self-dissolution approach. Then in situ CVD approach was employed to grow N doped graphene on ZnFe2O4/ZnO. Firstly, nanospheres of SiO2/PDA/ZnFe2O4 were prepared using SiO2 as a hard template, as shown in Scheme 1. During the synthesis of NG/ZnFe2O4/ZnO, 0.2 g of as-prepared SiO2/PDA/ZnFe2O4 was placed in a combustion boat and transferred to a tubular furnace and heated at 700 °C under an N2 atmosphere. After 30 min, using a syringe pump, 0.02 mL pyridine was introduced at a flow rate of 400 sccm. After 30 min incubation, the reaction system was allowed to cool to room temperature to obtain SiO2/PDA/ZnFe2O4/NG. Subsequently, the SiO2/PDA template was removed via an etching process by employing KOH solution having a pH of 13.5 at 90 °C for 3 h. Simultaneously, during the etching process, the Zn precursor was added in the form of (CH3COO)2·2H2O, followed by refluxing at 80 °C for 2 h, resulting in the formation of Zn-hydroxide precipitate. The obtained precipitate was annealed at 500 °C for 3 h under an N2 atmosphere. During the etching process, Zn(II) in ZnFe2O4 reacted with alkali, resulting in the formation of oxide, and during calcination, porous ZnFe2O4 was formed, which finally resulted in the formation of a heterostructure of ZnFe2O4/ZnO/NG.
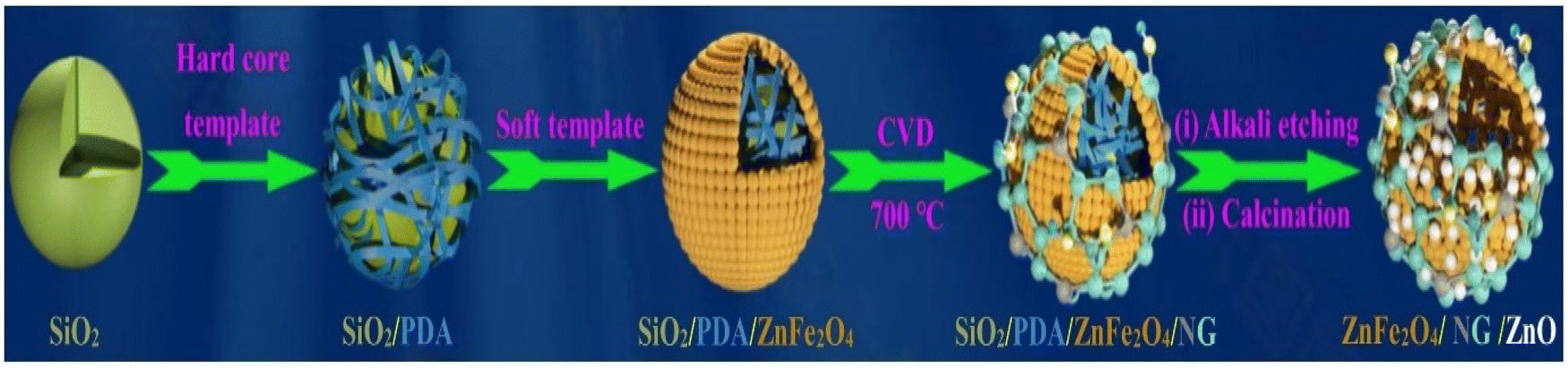 | ||
| Scheme 1 Reaction steps involved in the synthesis of ZnFe2O4/NG/ZnO heterostructure. Reprinted with permission from ref. 86. Copyright 2021, Elsevier. | ||
Wei et al. developed an ultralight ReS2@NG hybrid heterostructure interlayer using an all-CVD approach.87 During the typical synthesis procedure, a two-zone-temperature CVD furnace was employed to grow vertically aligned ReS2 nanosheets on a clean glass substrate. For the synthesis of ReS2 nanosheets, S powder and NH4ReO4 were placed in the centre of the two heating zones with a glass substrate placed over the NH4ReO4 powder under 1000 sccm Ar purging for 10 min. Subsequently, the furnace with two temperature zones was heated up to 300 °C and 700 °C for 30 min under a constant flow of Ar 50 sccm throughout the process. For NG at the growth temperature, plasma-assisted evaporation of the pyridine precursor was carried out and incorporated in the system. During the CVD synthesis reaction, the system was purged with H and Ar carrier gases at 30 sccm and 1000 sccm, respectively, under a base pressure of 1 Pa. Then, after CVD treatment, the as-prepared sample was spin-coated with PMMA at 2000 rpm for 1 min and dried at 160 °C for 5 min. The as-developed film was etched using HF to remove the glass substrate and the final PMMA/ReS2@NG film was obtained and attached to commercial polypropylene. The removal of PMMA from the film could be easily achieved during electrochemical analysis in an electrolyte solution. A schematic illustration of the CVD and PECVD approach used for the synthesis of ReS2@NG is shown in Fig. 9a. Fig. 9b presents an FESEM micrograph of the as-developed vertically aligned ReS2 and ReS2@NG.
 | ||
| Fig. 9 (a) Synthesis steps involved in the two-step CVD synthesis of ReS2@NG, (b and c) SEM micrographs of ReS2 and ReS2@NG nanosheets, (d and e) cross-sectional SEM micrograph of ReS2 nanosheets and ReS2@NG nanosheets with corresponding elemental mapping, and (f–i) corresponding TEM and HRTEM micrographs of ReS2 and ReS2@NG nanosheets. Reprinted with permission from ref. 87. Copyright 2019, Elsevier. | ||
In addition to N doping, other metal- and non-metal-doped graphene materials have been developed using the CVD approach for different electrochemical applications. For example, Zhang et al. developed an Si-doped graphene/GaAs heterojunction using the CVD approach for a better performance in G/GaAs solar cells.88 The synthesis of pure SiG was accomplished using a Cu substrate during CVD synthesis. During the synthesis, annealing of Cu was carried out in a reactor heated at 1015 °C under an H2 and Ar atmosphere for 30 min. Then, a precursor mixture of SiH4 and CH4 was introduced in the reactor for the synthesis of SiG. For sample preparation, different flux ratios of SiH4 and CH4 was employed to obtain different doping concentrations of Si in graphene. However, under a high flux of precursors, SiG was not successfully prepared in this report. Finally, for the preparation of GaAs/SiG, the as-prepared SiG film was transferred to SiO2/Si, germanium or CaAs substrate using PMMA and baking process at 120 °C for 2 min, followed by etching process to remove the Cu substrate using Cu2SO4/HCl/H2O solution. Feng et al. employed Ni foam (NF) as a substrate during the synthesis of N-doped graphene.89 In a typical synthesis, high-purity methane was introduced in a preheated furnace containing NF in a quartz tube at 1000 °C under a 12 sccm H2 flow. After cooling under a flow of mixed gases, the obtained graphene was added to HCl solution for 12 h and washed several times with water. N doping of the as-obtained graphene sample was carried out using the hydrothermal approach, during which the graphene sample was placed in urea solution and heated at 180 °C for 12 h, followed by washing and annealing at 800 °C under an Ar atmosphere. Similarly, for the synthesis of the heterostructure, the hydrothermal approach was employed, during which different precursors such as NiCl2 (0.064 g), Na2MoO4·2H2O (0.1 g), and urea (0.1 g) were added to 40 mL water, followed by the addition of as-prepared NG to a Teflon-lined autoclave and heated at 160 °C for 3 h in a furnace. Finally, the cooled samples were collected, washed and dried to get the NiMoO4/NG heterostructure. The whole synthesis process is schematically presented in Fig. 10.
 | ||
| Fig. 10 Illustration of the fabrication process of NG/NiMoO4. Reprinted with permission from ref. 89. Copyright 2020, Elsevier. | ||
Recently, Aydin et al. developed an N- and S-doped graphene 2D heterostructure on MoOx for efficient hydrogen generation.90 In this report, firstly graphene and MoOx were synthesized separately. For the synthesis of graphene, the thermal CVD approach was used by employing Cu foil as the growth substrate. After proper cleaning in water, HF, IPA, and acetone using ultrasonication, the Cu foil was dried using N2 gas and inserted in the quartz tube. The temperature of the tube was increased to 1050 °C under Ar:H2 at 200:30 sccm and the Cu foil was annealed at this temperature for 2 h. Using a mixture of Ar:CH4:H2 at100:5:100 sccm, the growth of graphene was completed. MoOx was prepared by plasma processing using Mo pellets deposited via electron beam evaporation. The Mo seed layer was oxidised in 13.5 MHz inductively coupled plasma (ICP). The residual cleansing of the ICP-developed layer was carried out by introducing several gases including Ar at 20 sccm for 10 min and H2 plasma at 20 sccm for 15 min for the removal of the native oxide from the metal surface. Final oxidation of Mo metal was carried out at 150 °C under O2 plasma at 10 sccm and a pressure and power of 50 mTorr and 550 W, respectively. Then, in the next step, CVD-developed graphene was transferred to MoOx using PMMA via spin coating. The as-developed PMMA/G/Cu was cured at 90 °C for 10 min, followed by vacuum treatment for several hours for proper interface formation. Finally, the Cu substrate was separated by an etching process and the remaining PMMA/G was transferred to the as-synthesized MoOx sample with the removal of PMMA by employing acetone to obtain G/MoOx/GCE. In the last step, S and N were doped in G/MoOx/GCE using the plasma doping process under Ar:H2S and N2 plasma. A schematic representation of all the steps involved in the synthesis process are depicted in Fig. 11.
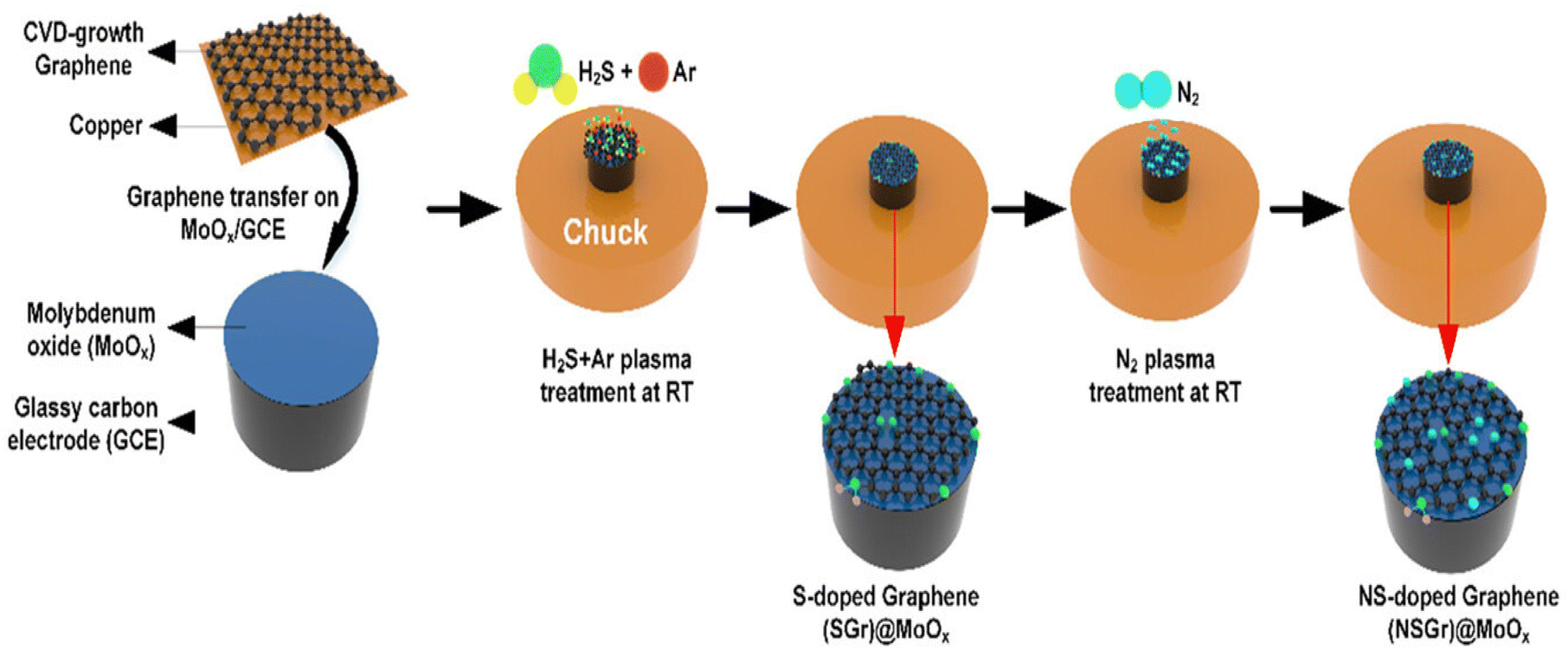 | ||
| Fig. 11 Schematic illustration of reaction steps used to synthesize NSGr@MoOx thin film heterostructure by ICP-assisted CVD approach. Reprinted with permission from ref. 90. Copyright 2023, Wiley. | ||
Bie et al. used an alternative method to the traditional approach for developing a catalyst on NG. In this report, the in situ development of NG over CdS hollow spheres was carried out using a three-step strategy, as shown in Fig. 12a.91 During the first step, spherical SiO2 was employed as a template for the development of the CdS/SiO2 composite. Then, the as-developed composite was placed in a tube furnace, followed by the introduction of pyridine feedstock for the synthesis of NG over the CdS/SiO2 composite. During the synthesis, pyridine was transformed into carbonaceous residue via pyrolysis and dehydrogenation reactions and was deposited on the surface of CdS due to the coordination between the N and Cd atoms. During the cooling process, compared to ordered carbon, the disordered carbon was transformed into graphene due to the energy difference between them. By using the alkali-assisted etching process, the SiO2 template was removed to obtain the final CdS/NG heterostructure with a large contact area between CdS and NG, as is clear from the SEM and TEM micrographs in Fig. 12b–j.
 | ||
| Fig. 12 (a) Schematic illustration of reaction steps involved in the synthesis of NG@CdS hollow spheres, (b–e) FESEM of sequential appearance of material at each step from left to right in (a), (f and g) FESEM images of sample represented as CdG2, where 2 represents concentration of pyridine used, (h and i) TEM and HRTEM micrographs of CdG2 sample, and (j) STEM images of CdG2 with corresponding elemental mapping, respectively. Reprinted with permission from ref. 91. Copyright 2019, Wiley. | ||
It has been observed that it is very difficult to synthesize a uniform monolayer of graphene via the CVD approach. Thus, to obtain a monolayer of graphene, Pang et al. employed a simple confinement method.92 During the synthesis process, SiO2/Si was employed as the substrate with three different configurations, as shown in Fig. 13. The three configurations included (a) Si/SiOx with the oxide face exposed and facing up, (b) Si/SiOx with the oxide phase masked and facing down and (c) sandwiched oxide face using two wafers of Si/SiOx. During the CVD synthesis in a horizontal tube furnace, Si/SiOx was first cleansed up by sonication in acetone and dried in an N2 atmosphere. Under the flux of a mixture of Ar/H2/CH4 gases at flow rates of 120/30/1.5 sccm, respectively, samples with different growth times were heated at 1185 °C under ambient pressure. Before passing the mixed gas of Ar/H2/CH4, the sample was heated under an Ar/H2 flow until a stable reaction temperature was achieved. In this report, the cleanest graphene monolayer was obtained when the sample was post-treated with Ar/H2 before cooling. According to the investigation, it was observed that using the sandwiched configuration, high-quality graphene with almost the same properties as that grown on other metal substrates such as Cu was produced. Also, it was observed that the formation of hexagonal faceted graphene starts from a round nucleating island. The faceting phenomenon occurs to minimize the energy, thus resulting in the formation of hexagonal facets similar to that present in beehives. Thorough investigations have shown that once the whole surface of a substrate is covered with a graphene layer, the formation of graphene stops, and thus this synthesis approach is considered as self-terminating, resulting in the formation of monolayer graphene. Table 1 presents the precursors and reaction conditions employed by different researchers for the synthesis of NG-based heterostructures.
 | ||
| Fig. 13 (a) Schematic illustration of direct synthesis of graphene over Si/SiOx employing CVD approach with different configurations of substrate: (a) SiOx facing up, (b) SiOx facing down, and (c) Si/SiOx–SiOx/Si sandwiched configuration. Reprinted with permission from ref. 92. Copyright 2019, Wiley. | ||
3.3. Thermal-assisted approach
In addition to the above-mentioned method, another common and the most widely used method for the synthesis of NG-based heterostructure catalyst materials is the thermal treatment approach. The high reaction temperature employed during the synthesis process improves the degree of graphitization. Considering the limitation of the CVD approach, thermal treatment is considered an effective and less expensive method to synthesize graphene-based heterostructures. Chen et al. employed a facile, inexpensive, thermal decomposition reduction approach for the synthesis of NG/MoS2/NG.93 During the synthesis process, as-purchased GO was ultrasonicated in deionised water for 2 h, followed by the addition of (NH4)6Mo7O24·4H2O (1.12 g) and CH4N2S (1.16 g) with constant stirring. Subsequently, after freeze drying at −20 °C, the as-obtained precursor (GO-(NH4)6Mo7O24-CH4N2S) was annealed at 750 °C for 2 h in a tube furnace under an Ar atmosphere at 100 sccm to obtain the final product in the form of NDG/MoS2/NDG. The same strategy was employed for the synthesis of NG/MoS2 and MoS2 samples. The reaction mechanism for the synthesis of the NG/MoS2/NG heterostructure was elucidated using the same reaction conditions in the absence of (NH4)6Mo7O24·4H2O, CH4N2S and CH4N2S-(NH4)6Mo7O24·4H2O, respectively. A schematic representation of the mechanistic steps involved in the synthesis process is presented in Fig. 14. During the initial step of the reaction, the well-dispersed GO adsorbs Mo7O246− ions on its surface, followed by the adsorption of CH2N2S around the Mo7O246− anions. Freeze drying of the sample resulted in the in situ layer-by-layer arrangement of the GO/(NH4)6Mo7O24-CH4N2S/GO precursor with an exposed GO layer. Under an Ar atmosphere, the annealing process at 750 °C resulted in the thermal decomposition of CH4N2S and (NH4)6Mo7O24 into H2S/NH3 and MoOx/NH3, respectively. Finally, MoOx was in situ reduced to MoS2 by H2S and GO was doped by N with the help of NH3 to obtain the final product.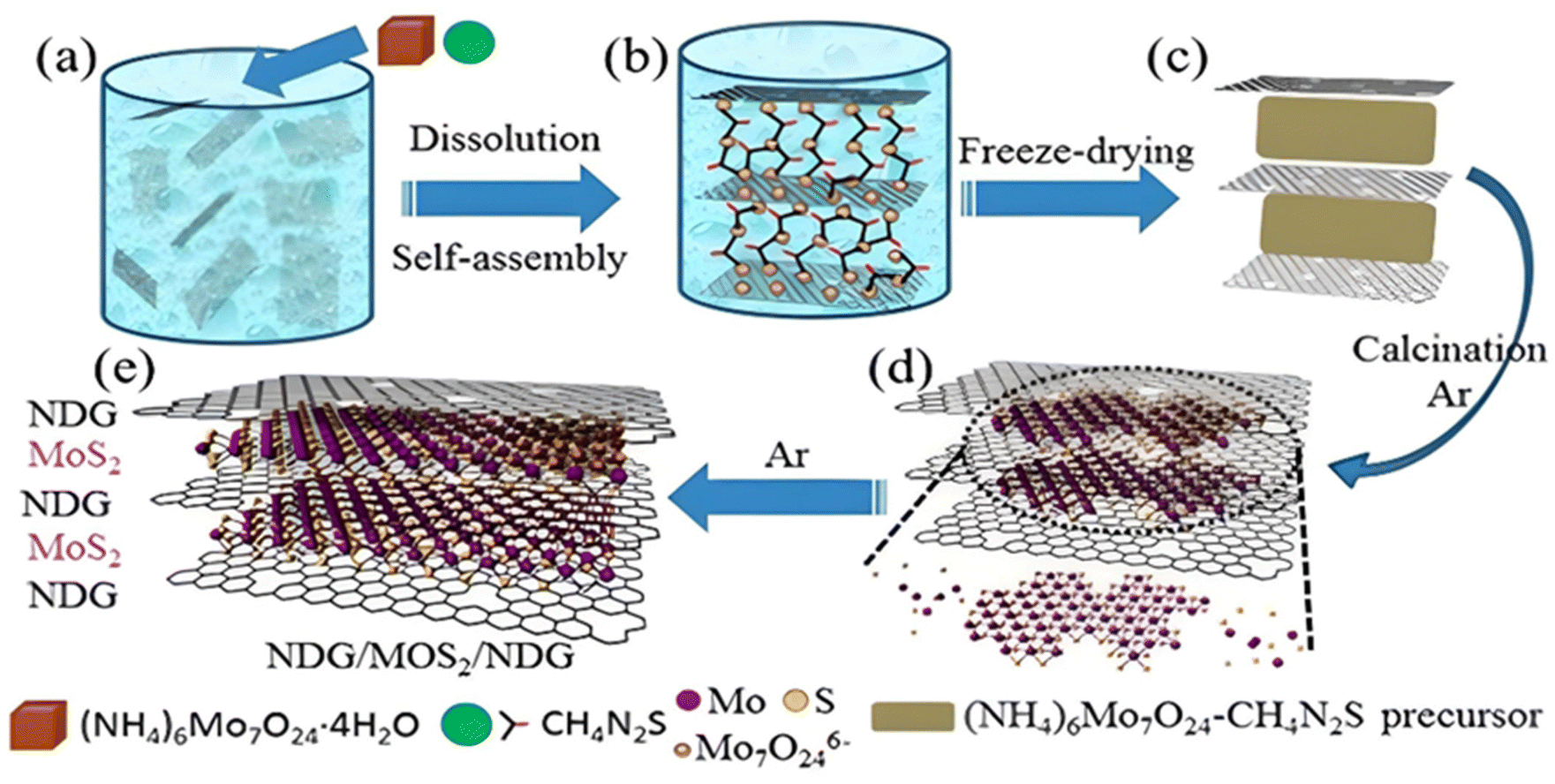 | ||
| Fig. 14 (a–e) Schematic representation of different steps involved in the synthesis of layer-by-layer NG/MoS2/NG stacking heterostructure. Reprinted with permission from ref. 93. Copyright 2017, Elsevier. | ||
Another important catalyst hybrid material consisting of Ni–Fe nitride coupled with NG was designed and developed by Fan et al. using the thermal annealing approach. During the synthesis, an ex situ approach was employed to prepare Ni–Fe hydroxide nanosheets by the exfoliation of Ni–Fe double-layered hydroxide.94 The as-obtained Ni–Fe hydroxide nanosheet colloidal suspension was added to GO colloid in 100 mL formamide. After centrifugation, the sample was lyophilised, and the obtained powder was thermally treated at 700 °C in ammonia for 2 h to obtain the final Ni–Fe nitride/NG hybrid material. The ex situ approach was employed to avoid Ni–Fe nitride phase separation in the hybrid material.
During the synthesis of N-doped based heterostructures, GO is one of the main precursors used; however, the synthesis of GO or RGO is a highly complex, time-consuming and laborious process. Therefore, the direct synthesis of graphene-based heterostructures without the use of GO or RGO is highly desirable. The use of thermal treatment has been observed to directly produce graphene-based materials using some other precursors besides GO. For instance, Huang et al. designed and synthesized MoP nanoflake-intercalated NG nanobelts by employing MoO3-amine hybrid.95 During the synthesis, a one-step thermal approach was used for the preparation of MoP/NG using phosphiding organic inorganic hybrid dodecylamine (DDA)-incorporated MoO3 nanobelts. During a typical synthesis process, 30 mL alcoholic solution of hydrothermally synthesized MoO3 nanobelts and DDA was placed in a 50 mL Teflon-lined autoclave and heated at 100 °C for 24 h. The as-obtained white precursor MoO3/DDA was placed in a furnace downstream to NaH2PO2 at a distance 20 cm. Subsequently, the furnace temperature was increased to 700 °C for 2 h under an Ar atmosphere to get the hybrid MoP/NG catalyst. During the reaction, the intercalated DDA molecules were carbonised to NG and sandwiched MoO3 was converted to MoP to produce the alternately stacked MoP/NG hybrid. Similarly, an important investigation was carried out by Cai et al., in which graphene nanoribbons (GNR) and their heterojunction/heterostructure were synthesized using different non-GO precursors as the starting material.96 During the synthesis, Ar-ion bombarded a highly clean single crystal of Au(111) annealed at 470 °C as the substrate for the growth of GNR. For the synthesis of GNR, the deposition of the monomer 6,11-dibromo-1,2,3,4-tetraphenyltriphenylene on the substrate surface was done using the sublimation process at a rate of 2 Å min−1. The substrate with deposited monomer was kept at 200 °C for dehalogenation and radical addition reactions for the synthesis of N-GNR, and finally annealed at 400 °C for 10 min to carry out cyclodehydrogenation of the polymers for GNR formation. For the pristine GNR, a similar approach was used with a slight change in the substrate temperature, i.e., 250 °C for monomer deposition and 450 °C for the post-annealing process. For the synthesis of the heterostructure, two precursor species were deposited on the Au (111) substrate in a layer-wise fashion one after another and heated at 200 °C with subsequent annealing at 400 °C. The two substrates employed included 1,2-di(pyrimidin-5-yl)ethyne and 6,11-dibromo-1,2,3,4-tetraphenyltriphenylene. Similarly, Yan et al. developed NGQD-BiVO4/g-C3N4 Z scheme photocatalysts using a non-GO precursor of nitrogen-doped graphene quantum dots (NGQD). Here, g-C3N4 was synthesized using the thermal treatment approach, during which urea was heated in a crucible at 550 °C for 4 h. Subsequently, the BiVO4 and NGQD samples were synthesized using the hydrothermal and pyrolysis approach, respectively, followed by the development of the hybrid heterostructure. For NGQD, C6H17N3O7 was used as the precursor. For the preparation of the NGQDs-BiVO4/g-C3N4 sample, the as-prepared BiVO4 was added to a mortar and pestle together with g-C3N4 and ground and mixed, followed by calcination at 400 °C for 4 h to obtain the BiVO4/g-C3N4 sample. To prepare NGQDs-BiVO4/g-C3N4, different amounts of NGQD were dissolved in 25 mL water and the NGQDs-BiVO4 sample added and kept for 24 h, and finally the sample was dried at 80 °C to obtain the NGQDs-BiVO4/g-C3N4 Z-scheme catalyst material.
Similarly, Feng et al. used the coprecipitation method, followed by post-synthesis annealing to synthesize an NG-encapsulated NiFe alloy by employing a non-GO precursor.97 During a typical synthesis process, Prussian blue (PB) analogue gels of NiFe were prepared. By controlling the amount and changing the precursors, a wide range of NiFe-PB analogues was synthesized. During the synthesis process, firstly, two solutions containing 143 mg and 132 mg of NiCl2·xH2O and K3[Fe(CN)6], respectively, were prepared in 20 mL water, followed by mixing of the two solutions under stirring for 1 min. After mixing, the solution was aged for 24 h, followed by the collection of the precipitate by centrifugation. For the synthesis of nanocubes of NiFe-PBA, a solution of 20 mL water containing 143 mg of NiCl2·xH2O and 180 or 330 mg of sodium citrate was prepared, together with an aqueous solution of K3[Fe(CN)6]. The as-prepared solutions were mixed in a similar fashion as discussed above. Similarly, nanoplates were synthesized using a similar approach except that different amounts of NiCl2·xH2O (95 mg) and aqueous solution of potassium tetracyanonickelate(II) (96 mg) in 20 mL water were used. Finally, the as-prepared NiFe-PBA and NiNi-PBA were annealed at 600 °C, 700 °C and 800 °C for 4 h at a heating rate of 5 °C min−1 using an Ar atmosphere. The N-doped graphene was developed by carrying out the etching process in 1 M H2SO4 for 24 h for the sample annealed at 700 °C.
During thermal treatment, it has been observed that high temperature for a much longer time is required for the synthesis of different NG-based hybrid materials. Thus, to reduce the thermal treatment reaction temperature, energy and reaction time, different hybrid approaches have been employed, for instance, the use of hydrothermal-assisted thermal treatment and microwave-assisted thermal treatment approach, which are considered suitable for the easy synthesis of NG catalyst materials. For example, Zhang et al. reported the preparation of an efficient hybrid electrode material based on hierarchal ordered 1D MnO/graphene scroll (GS) core sheath together with 2D nitrogen-doped ribbons (NGR) using a hydrothermally developed precursor, followed by thermal annealing, as shown in Fig. 15.98 During the synthesis process, firstly ultralong nanowires of MnO2 were prepared via a modified hydrothermal approach, followed by the synthesis of MnO/N-GS/GR. For the synthesis of MnO/N-GS/GR, a dispersion of as-developed MnO nanowires (80 mg) in aqueous PDDA was added to an aqueous suspension of GO (20 mL) with constant magnetic stirring for 4 h. During the stirring process, an electrostatic interaction took place between the cationic PDDA-modified MnO and anionic GO sheets. Then, the MnO/GO sample was collected by centrifugation, followed by washing and freeze drying. Subsequently, the MnO/GO hybrid was employed to develop MnO/N-GS/GR hybrid materials using the hydrothermal approach. During the hydrothermal reaction, an aqueous dispersion of MnO/GO was prepared in urea, and an additional 2 mL aqueous dispersion of GO was added to it, followed by hydrothermal treatment at 120 °C for 3 h in a Teflon-lined autoclave. After cooling, a 3D macroscopic hydrogel was obtained, followed by freeze drying and annealing at 600 °C for 2 h under Ar conditions to get the final 3D MnO/N-GS/GR hierarchal sample.
 | ||
| Fig. 15 Schematic representation of the synthesis of hierarchical 3D MnO@NGS, which involves two steps, i.e., self-scrolling and self-assembly process between GO sheets and MnO2 nanowires, followed by annealing treatment. Reprinted with permission from ref. 98. Copyright 2017, Elsevier. | ||
3.4. Other synthesis approaches
Based on elevated temperature in the thermal treatment approach and tedious CVD approach, new approaches or methodologies are being explored and used to develop NG-based heterostructures. In this quest, other approaches such as electrochemical approach, microwave-assisted, biomass-assisted method and chemical precipitation approaches have attracted significant attention.99–101 Given that herein we are focused on the electrochemical application of NG-based hetero-structured catalysts, the electrochemical synthesis approach offers certain advantages compared to the above-discussed methods. Using the electrochemical approach, post-synthesis transfer of the synthesised material on the electrode surface is not required, which proves to be helpful to preserve the original characteristic of the catalyst material without any degradation in its catalytic properties. Using and controlling electrochemical parameters, the final characteristics such as morphology, particle shape and size can be easily modulated. Wang et al. employed the electrochemical deposition technique to synthesize N-doped TiO2/graphene hybrid catalysts.102 In this report, firstly, pure and N-doped TiO2 nanotubes were synthesized using the electrochemical approach. For the synthesis of N-TiO2, TiO2 nanotubes were prepared using Ti foil as the anode and graphite as the cathode in a two-electrode system during the electrochemical synthesis reaction. During the synthesis, 0.138 M NaF, 0.5 M H3PO4 and 10 mL ethylene glycol were used as the electrolyte under a potential of 20 V for 30 min. The as-developed TiO2 nanotubes were used for the development of N-doped TiO2 by carrying out NH4+ enrichment on TiO2 nanotubes using NH4Cl at 5 V for 1 h. Subsequently, after washing, the as-obtained sample was annealed for 2 h. Finally, the as-developed N-doped TiO2 was electrophoretically deposited with graphene from an electrolyte solution containing 1 mg mL−1 GO solution at 2 V for 30 min. During the final step, N-doped TiO2 was employed as the cathode and graphite was used as the anode. Similarly, a solution-phase self-assembling approach was employed by Xing et al. to develop a heterostructure between a non-equilibrium rock salt solid solution of MnS0.5Se0.5 nanocubes and NG.103 In this report, initially, MnS0.5Se0.5 and NG were synthesized separately using a one-pot single-step simple route and hydrothermal approach, respectively. During the typical synthesis of MnS0.5Se0.5, 0.2 mmol, 0.05 mmol and 0.05 mmol of manganese acetylacetonate, dibenzyl disulfide, and dibenzyl diselenide were added to a 100 mL three-neck flask, together with oleylamine and 1-octadecene as the ligand and solvent, respectively. The obtained solution was heated and stirred at 150 °C for 30 min under an Ar atmosphere to remove possible impurities and moisture content. Then, to get the final product, the sample was heated at 320 °C for 10 min, followed by washing with toluene and IPA. The next step involved the synthesis of NG using GO (20 mg) and urea (400 mg) precursors under hydrothermal conditions at 160 °C for 3 h. Finally, using the as-synthesized NG and MnS0.5Se0.5, a hybrid material was developed by employing the self-assembling strategy. During the synthesis, the as-prepared MnS0.5Se0.5 and NG were dispersed in toluene and mixed, followed by sonication for 1 h to obtain a hybrid material. The as-prepared sample was dried under vacuum and heated under an Ar/H2 flow at 250 °C for 2 h.Bu et al. employed another important strategy, in which the ultrafast microwave-assisted thermal conversion approach was used to synthesize FeNiP/graphene composites, which could be transformed into adjustable heterostructures such as FeNiP/PG and FeNiP/GC/PG. During the synthesis, different precursors such as GO and different PBA analogues of NiFe/GO were initially synthesized.104 For instance, the synthesis of GO was carried out by adding 3.0 g of graphite to a concentrated H2SO4 (70 mL), followed by the addition of sodium nitrate (1.5 g) and cooling of the reaction mixture to 0 °C. In the next step, potassium permanganate was added slowly to keep the temperature of the reaction mixture to below 20 °C. Then, the reaction mixture was put in a water bath at 35–40 °C, resulting in the formation of a thick paste. To this paste, 150 mL water was added for 15 min, followed by the further addition of 500 mL water and 20 mL H2O2, which turns the colour from brown to yellow. Finally, the as-prepared sample was washed with HCl solution, followed by centrifugation and dialysis filtration to get the pure GO sample. Simultaneously, in this report, the synthesis of Ni–Fe-PBA/GO composite analogues such as Ni–Fe PBA/GO sponge and Ni–Fe PBA/GO/NaH2PO2 sponges were synthesized. In general, 20 μL 0.5 M Na4Fe(CN)6 was added to 1 mL GO (4 mg mL−1) dispersion and sonicated for 10 min. Then, 0.2 mL 0.5 M NiCl2 solution was added to this mixture, followed by centrifugation to obtain the Ni–Fe-PBA/GO composite. Similarly, metal organic framework/GO composites were synthesized using the same approach as that employed for the Ni–Fe-PBA/GO composites except the metal ions and ligands were changed. Finally, for the synthesis of transition metal phosphide (TMP) phosphorous-doped graphene (PG) heterostructure, as-prepared MOF/GO/NaH2PO2 sponges were loaded in a glass vial containing carbon cloth under an Ar atmosphere and subjected to microwave irradiation at 1000 W for 20 s. Similarly, for FeNiP/PG, 0.0625 mmol of NaH2PO2 containing Ni–Fe PBA/GO/NaH2PO2 sponge was employed, while for FeNiP/GC/PG, 0.25 mmol NaH2PO2 containing Ni–Fe PBA/GO/NaH2PO2 sponge was employed. From this investigation, it was observed that the ultrafast preparation of NG-based heterostructures could be easily achieved using the microwave-assisted approach.
3.5. Advantages and disadvantages of different synthesis approaches
As discussed above, several approaches are being used to synthesize different NG-based heterostructure materials. Based on the final application of the synthesized NG catalyst materials, each method has its own advantages and disadvantages. Some of the advantages and disadvantages of the different methods used for the synthesis of NG-based hybrid materials are presented in Table 2.| Synthesis method | Advantages | Disadvantages |
|---|---|---|
| Hydrothermal/solvothermal | Good control over crystallinity, phase purity and morphology | Difficult optimization the reaction condition and reaction parameters such as concentration of precursors, pH, optimum temperature and pressure for the desired morphology and active plane. |
| Needs detailed knowledge of phase diagrams of catalytic systems. | ||
| Use of organic moieties for control over shape, size and morphology adds extra effort for synthesis and reduces activity. | ||
| Thermal treatment method | Rapid reaction process, Simple to use. | High temperature, no control over shape, size and morphology |
| CVD | Significant control over shape, size and morphology | Highly expensive, limited substrates explored, Substrate and reaction chamber size limitation for large scale synthesis. Use of gases during synthesis may be toxic. |
| Electrochemical approach | Good control over composition, scalable for bulk synthesis, cost effective, controlled thickness and morphology, and solution processing compatibility. | Limited substrate options, difficult to develop specific heterostructure, and requirement of post-synthesis thermal treatments. |
4. Advances in ORR applications of N-doped graphene (NG) heterostructures
In sustainable and renewable energy devices such as metal air batteries, fuel cells and water electrolysis, the ORR is one of the important reactions.The reaction mechanism involved in the ORR includes the following steps:
(1) O2 + H2O + 2e− → HO2− + OH−
(2) HO2− + H2O + 2e− → 3OH−
Overall ORR reaction involved:
| O2 + 2H2O + 4e− → 4OH− |
As is known, the industrial-scale application of these devices is dependent on the development of highly active, low-cost, stable electrochemical catalysts that have potential to replace the most active scarce noble metal catalysts. Among the different already explored alternative catalyst materials, carbon-based electrochemical catalysts are considered as potential alternatives to noble metal electrocatalysts. However, their practical application is hindered either by their low activity or poor stability. Thus, to improve their ORR catalytic efficiencies, different strategies have been adopted, including the incorporation of foreign elements as dopants and development of heterostructures. In the case of doping, N is considered as an important desirable heteroatom, which not only improves the ORR activity but also improves the active site density in graphene. In addition to N, Qiao et al. employed other heteroatoms such as B, O, P and S to investigate the effect of heteroatom doping on the ORR electrochemical activity of graphene.157 Similarly, Dai et al. also developed carbon foam co-doped with N and P.158 In these investigations, it was observed that the B, C and P atoms either bonded to N or near to the N heteroatoms on the graphitic sheet act as an active sites for the ORR or OER. Based on the configuration of N in the carbon framework of graphene, N exists in different configurations including pyridinic-N, pyrrolic-N, graphitic-N, oxidized-N and interstitial-N in NG.159 The different configurations of N in NG are illustrated in Fig. 16. The activity of NG toward the ORR or HER is attributed to these different N configurations. In the case of ORR activity, the role of these N configurations is still ambiguous given that many researchers attributed the superior ORR activity to pyridinic-N or pyrrolic-N, while as many researchers believe that graphitic-N is mainly responsible for the enhanced ORR activity. Further, the catalytic efficiencies were observed to be dependent on the valence orbital levels, spin density and charge density of the C atoms in the graphene sheet. However, despite the improved ORR activity due heteroatom doping, the activity still does not match the state-of-the-art noble metal-based ORR catalysts. Hence, the development of heterostructures based on NG has been explored to investigate and understand how the synergistic effect arises from the atomic level and what is the correlation between the electronic band structure and catalytic activities of hybrid structures? According to recent investigations, it has been observed that the synergistic effect of different metal-based compounds with carbon-based nanodimensional materials has demonstrated enhanced electrocatalytic activity for both the ORR and OER. To utilize the different properties (beneficial for ORR activity) of NG at their full potential, different heterostructure strategies have been employed. Either NG has been utilised as an active catalyst material for the ORR or a substrate material for the dispersion of different catalytic active materials on it.
 | ||
| Fig. 16 Different configurations of N in nitrogen-doped graphene. | ||
Among the different non-precious heterostructures or composites, NG and less expensive non-noble earth abundant transition metals have shown remarkable synergism for both the electrochemical ORR and OER. The interaction of the π electrons of NG and d electrons of the metal leads to the favourable adsorption of O2 and other intermediates formed during the ORR or OER, thus leading to the enhanced activity of heterostructures. Luque-Centeno et al. reported a simple one-pot method for the synthesis of nanocomposites of Ti and Co with NG for electrochemical ORR and OER applications.160 From the investigations, it was observed that both Ti/NG and Co/NG composites were electrochemically active and their activity in this report was compared with Pt/C. According to the electrochemical analysis, Pt/C showed a more positive onset potential and the onset potential of Co/NG was 100 mV more positive compared to that of Ti/NG. The enhanced electrocatalytic ORR activity of the Co/NG composites was attributed to the improved kinetics on the active sites present due to Co species. The lower net-limiting diffusion current density of both composites compared to that of the Pt/C-based electrode was attributed to the lower number of electrons transferred, as deduced using the Levich equation in this report. The mechanistic investigation showed that the number of electrons transferred for the ORR in both composites was the same for Co/NG and Ti/NG (3.6 and 3.5), respectively. In addition, the number of electrons produced showed an insignificant change with the applied potential, thus indicating that the reduction mechanism closely follows a complete four-electron reduction process of oxygen. The significant difference in ORR activity was found in the potential at which the reduction reaction occurs rather than in the mechanism involved in the reaction. According to the already reported investigations based on use of NG as an active catalyst material for ORR application, the role of N doping is under investigation and two types of active N species, i.e., pyridinic and quaternary N, are considered to play a crucial role. It has been observed that the difference in electronegativities between N and C plays a crucial role in the improved ORR activity. The quaternary and pyridinic N are beneficial for the promotion of electron transfer between the C pz orbitals adjacent to N and low onset potential, respectively. However, in this report, using Co/NG and Ti/NG, the role of both pyridinic and quaternary N was different compared to NG. It was observed that the percentage of different N in the as-prepared sample primarily depends on the transition metal employed. The improved ORR activity of Co/NG compared to Ti/NG was attributed to the higher concentration of pyridinic N in Co/NG and possible beneficial Co–N interactions, as reported by Liang et al. for a Co3O4 reduced graphene oxide heterostructure.161
Liu et al. developed NG nanosheets co-decorated with Cu and Co as a bifunctional electrocatalyst for the OER and ORR.162 Different composites consisting of CuCo@NG, Co@NG, Cu@NG and NG were developed to analyse their comparative electrochemical activity towards the ORR and OER. According to the observed results, it was deduced that the CuCo@NG-based composites showed efficient ORR activity compared to the other developed catalysts with an onset potential of 0.92 V and half-wave potential of 0.81 V. According to the kinetic process investigations, the electron transfer number was found to be 3.89 for CoCu@NG, which suggests that compared to the other developed materials in this report, a four-electron transfer process occurred on the surface of the catalyst, reducing the generation of peroxide to a negligible amount. The same mechanism of the ORR was verified using a rotating ring disk electrode (RRDE), which showed that the electron transfer number was equal to 3.89 and the percentage yield of peroxide formation was less than 3.9% for the CoCu@NG electrode, suggesting that Cu and Co co-decorated NG is an efficient (better than Pt/C electrode used in this study) ORR electrocatalyst. The efficient enhanced ORR and OER activity of CoCu@NG was attributed to the strong boosted electron exchange between Cu and Co decorated in the NG nanosheets, improved conductivity and increase in the number of catalytic active sites. Another important investigation was carried out by Liu et al., in which a trifunctional FeNi3N (110 exposed active site)/NG electrocatalyst was developed.163 Prior investigations showed the better electrochemical performance of FeNi for the HER and OER with negligible activity towards the ORR. However, together with HER and OER activities, the as-developed ultrasmall (7.6 nm) FeNi3N nanoparticles deposited on NG showed unprecedently enhanced ORR activity. The reason for this unprecedented ORR activity was investigated using theoretical investigations. According to the experimental HRTEM results, it was demonstrated that both Ni4N and FeNi3N have (110) exposed facets and the DFT investigations over both catalysts showed that the affinity of the FeNi3N (110) surface towards O2 adsorption is very high compared to Ni4N, and based on theoretical observations, the cleavage of O2 on the FeNi3N surface is much easier compared to Ni4N. Similarly, according to the DOS analysis, the electron transfer from the Fe active site to the half-filled π orbitals of O2 takes place more conveniently compared to from Ni 3d. Thus, easy and more filling of the O2 π* orbital leads to easier cleavage of the O–O bond, which thoroughly explains why FeNi3N shows better ORR activity compared to Ni4N. Another interesting study was carried out by Wang et al., in which trifunctionality (HER, OER and ORR activity) in a defect-rich NG-encapsulated Co nanostructure was investigated.164 During the investigation, six different samples were prepared and investigated for their ORR and HER/OER activity, and regarding the ORR, sample consisting of Co/defect-induced NG prepared at 900 °C (CoDNG900) outperformed all the other catalysts including Pt/C. The as-prepared CoDNG900 showed an onset potential of 0.943 V and half-wave potential of 0.864 V, which was comparable to that of the state-of-the-art Pt catalyst. In addition, the reaction dynamics results also showed that CoDNG900 acts a better catalyst with smallest Tafel slope (47 mV dec−1), which was even smaller than that of Pt catalysts (56 mV dec−1), as can clearly be observed in Fig. 17I(a–f). The K–L plot investigation showed that for the reduction of one O2 molecule, the electron transfer number is 3.97, and thus the ORR follows the 4e− pathway. The enhanced ORR activity of Co incorporated in defected NG was attributed to the higher concentration of N and defect sites induced in NG due to Zn evaporation, as observed from the HRTEM results depicted in Fig. 17II(a–f). Thus, it was concluded that by incorporating Co nanoparticles, the formation of sufficient M–N moieties and constructive defective sites in NG had a considerable positive influence on the ORR activity. Another important way to improve the ORR activity is to develop single active isolated atoms attached to a support material. These hybrid catalysts show high selectivity, high activity, excellent durability and optimal utilization of active metal atoms. However, the synthesis of single-atom-based active ORR hybrid catalysts is quite challenging because of their agglomeration and easy dissolution. Thus, an interesting, novel highly active and stable dual-deposited Fe single atom/Fe nanoparticle-incorporated NG was investigated by Nguyen et al. for ORR applications.165 According to this report, important results were inferred, which showed that the as developed hybrid catalysts outperformed commercial Pt/C catalysts with a positive onset (0.94 V) and halfwave potential (0.81 V). The developed hybrid ORR catalysts showed only 10% loss of activity after 15000 s with improved methanol tolerance. These improved catalytic characteristics on FeSA–FeNPs/NG were attributed to the formation of a unique nanoarchitecture with improved conductivity and specific surface area. Therefore, a high surface area provides a large number of active sites and conductivity results in fast charge transfer, resulting in better, effective adsorption and catalytic reduction of O2 to the final product. According to the detailed investigation, the main reasons responsible for the enhanced ORR activity were evaluated. The enhanced catalytic activity was attributed to the formation of positively charged Fe ion centres, which promote the strong adsorption of O2. The further formation of FeSA–FeNPs centres alters the surface properties and electronic configuration, producing more electroactive sites for the reduction of O–O bond strength in O2 molecule for fast reduction reaction.
 | ||
| Fig. 17 I (a) represents ORR LSV polarization plot of CoDNG900, CoNG900, DNG900, NG900 catalysts, (b) shows Tafel plot of as prepared samples, (c) represents half-wave potential, onset potential and number of electrons (n) for as-synthesized catalysts, (d) showing comparative i–t curves for Pt/C and CoDNG900 at 0.6 V (e) showing methanol resistance curves of Pt/C and CoDNG900 and (f) represents Δj = ja–jc at 1.15 V at different scan rates for as-prepared samples II (a–n) XRD patterns of all the catalysts, TEM and HRTEM micrographs of CoDNG900, STEM images and corresponding elemental mapping of different atoms, respectively. Reprinted with permission from ref. 164. Copyright 2021, Elsevier. | ||
Barman et al. developed NG tubes with highly dispersed ultrathin Ru nanocrystals as a bifunctional catalyst for the OER or HER.166 RDE and RRDE in 0.1 M KOH were employed to analyse the ORR activity of the as-developed Ru@NG catalyst. The ORR activity was monitored under O2 saturation showing an onset potential of 0.97 V, 1 V and 0.92 V for Ru@NG, Pt/C and NG, respectively. In addition, Ru@NG showed an enhanced limiting current density compared to that of Pt/C and NG. Further, fuel intolerance, which is the main disadvantage of Pt/C-based electrodes, was investigated, showing that the Ru@NG-based cathode showed better methanol tolerance compared to the Pt/C-based cathode. The catalytic poisoning analysis showed that in the case of pure NG, there is a small change in current density compared to Ru@NG. This was attributed to the fact that in the case of NG, surface poisoning of Co-Nx occurred, and thus Co as an active site plays little role in the ORR. Alternatively, in the case of Ru@NG 40%, a reduction in the limiting current was observed during the surface poisoning analysis due to the poisoning of the Ru-Nx or N-O6-Ru/O-O6-Ru sites, thus clearly confirming the role of these sites in the ORR. After extensive investigations, the enhanced OER activity of Ru@NG was attributed to the formation of the Ru-oxo-N4 centre. Due to the formation of the Ru-oxo-N4 centre, Ru has a high oxidation state, which results in the favourable shifting of the free energy of different intermediates formed during the ORR, thus finally resulting in the favourable ORR activity of Ru@NG. The kinetic analysis of ORR activity during the investigation using RRDE showed that the reaction follows the four-electron reduction pathway. Chuong et al. developed a ternary heterostructure consisting of hierarchal-arranged α-Fe2O3 homogenously dispersed and entrapped in MoS2/NG nanosheets as an effective electrocatalyst for the ORR.167 The development of ternary heterostructures was aimed to overcome the limitations of each pristine counterpart. For comparative analysis, different composites were synthesized such as MoS2, NG, α-Fe2O3, α-Fe2O3@MoS2/NGNS, and α-Fe2O3@MoS2/NG. During the ORR activity analysis, it was observed that the development of a heterostructure of MoS2 well dispersed in graphene nanosheets (GNS) showed better electrochemical ORR activity. This was attributed to the presence of a large number of active sites and better charge transfer between the two components. Further, the incorporation of α-Fe2O3 in the MOS@GNS heterostructure significantly shifted the onset and half-wave potential towards a positive potential. Among the developed heterostructures, α-Fe2O3@MoS2/NGNS showed the highest impressive ORR activity with a low onset potential of 0.947 V, small half-wave potential of 0.854 V and low Tafel slope of 73 mV dec−1. The enhanced ORR activity of α-Fe2O3@MoS2/NG was attributed to the low charge resistance due to the uniform dispersion of ultrasmall α-Fe2O3 in MoS2 embedded in a highly conductive NGNS hierarchal hybrid structure. The stability analysis showed significantly high stability even after 1000 cycles without any change in onset and half-wave potential. The enhanced overall ORR activity was attributed to the synergistic effect of the physicochemical properties of homogenously dispersed α-Fe2O3 entrapped in the hierarchical heterostructure of MoS2/NGNS. The structural similarities between MoS2 and NGNS resulted in the formation of an ideal interface with beneficial modification of electronic and structural characteristics, thus providing improved active adsorption sites and enhanced charge transfer for the ORR. The presence of S and N on the MoS2/NGNS core shell heterostructure effectively anchors and disperses the α-Fe2O3 nanoparticles with unique electronic interactions between the different components of the heterostructure. Thus, these interactions lead to an alteration in the electronic states of the nanoparticles with amplified electrochemical active surface for better ORR activity. In the comparative analysis, it was observed that α-Fe2O3@MoS2/NG outperformed Pt/C in terms of efficiency, cost effectiveness, durability and methanol tolerance. In addition to the formation of enhanced active sites, the development of the hybrid α-Fe2O3@MoS2/NG heterostructure resulted in low resistance to the flow of charge compared to the other samples. Further, the mechanistic investigations indicated that the ORR over α-Fe2O3@MoS2/NG follows the four-electron reaction pathway under alkaline conditions, as follows.
Another important intriguing investigation was carried out by Jeong et al., who developed an NG-encapsulated alloy of CoFe/mesoporous NG foam and used it as a bifunctional electrocatalyst for the OER and ORR.168 The linear sweep voltammogram (LSC) measurements revealed that the as-developed CoFe@mesoporous NG showed strikingly enhanced ORR activity with a low onset potential and Tafel slope compared to the Pt/C-based electrode. The kinetics analysis using the Koutecky–Levich (K–L) equation showed that the electron transfer number changes from 2.1 to 3.6 at different potentials ranging from 0.15 V to 0.35 V, indicating that with an increase in potential, there is change in the mechanism pathway for the ORR. The change in mechanism pathway was attributed to the presence of an NG shell on the CoFe alloy nanoparticles and mesoporous NG as a substrate during the ORR analysis.
As discussed and previously reported, heteroatom-doped graphene/carbon materials show significantly enhanced electrochemical activity. However, their aggregation in terms of bundle formation and restacking in the case of carbon nanotubes and graphene, respectively, significantly hinder the exposure of active sites and mass transfer process, thus limiting their potential as ORR catalysts. Therefore, considerable attention and research are needed in this direction to develop efficient NG-based ORR catalysts without aggregation. In this direction, Cheng et al. developed an NG/carbon nanotube heterostructure with enhanced inter-molecular charge transfer for improved ORR activity.169 The as-developed NG/CNT-900 (900 represents the thermolysis temperature at which the sample was prepared) showed enhanced ORR activity with considerable methanol tolerance. The efficient ORR activity was attributed to the highly porous architecture, abundant defective sites and high specific surface area. All these characteristics impart efficient hetero-interlayer charge transfer and accumulation of electrons on the surface layer carbon atoms, thus improving the adsorption and catalysis of O2 molecules on the surface. The enhanced ORR activity was also attributed to the reduced aggregation of the NG layers due to the incorporation of CNT, which improved the accessibility of the electrolyte to more active sites present in the bulk of the material. The origin of the enhanced electrocatalytic activity was evaluated using first principal simulations and DFT. According to the DFT investigations, the charge transfer at the 2D/1D interface was investigated as the main contributor to the ORR activity using the NG/CNT hybrid structures. Three different hybrid structures (NPyri–G/P-CNT, NPyri–G/NG-CNT, and NPyri–G/NPyri-CNT) were constructed and analysed, where NPyri and NG represent pyridinic N and graphitic N, respectively, and P-CNT represents pristine CNT. It was observed that the incorporation of Npyri in the carbon network induced Lewis basic character in the carbon atoms next to Npyri, which distinctly enhanced the ORR activity of the hybrid structure. For the comparative analysis, a single layer of NPyri–G layer was also established. According to the observed results, as shown in Fig. 18, the developed NPyri–G/CNT showed excess charge transfer at the interface from the bottom CNT to the top NPyri–G layer, which resulted in the accumulation of more electrons on the NPyri–G surface, especially in NPyri–G/NG-CNT, as shown in Fig. 18b and c. The obtained results showed that all the NPyri–G/CNT heterostructures possess a large density of states near the Fermi level, resulting in the easy transfer of electrons and enhanced conductivity compared to the single NPyri–G layer. All the theoretical results were found to be consistent with the experimental results. Thus, according to the simulation studies, it was concluded that the more negatively charged carbon atom on the top of the heterostructure favours O2 activation, and consequently results in a 4e- transfer process for the ORR. An optimized reaction mechanism atomic model was developed, as shown in Fig. 18d. All these investigations showed that the interaction of M–Nx significantly modifies the electronic interactions due to the difference in the electronegativities of the metal, N and C present in NG-based hybrid catalysts. Hence, the different metals present in the NG heterostructure behave differently, as is clear from the above discussion. However, in addition to the metal, N also plays a significant role. Therefore, the role of N has been under extensive investigation since the last decade. Jia et al. carried out an important investigation, in which metal carbide wrapped with an NG layer was synthesized using a self-sacrificing dual N source strategy.170 Graphitic carbon nitride and dopamine were employed as the source of N during the synthesis. During the investigation, different samples with different metal precursor concentrations and different heat treatment temperatures were prepared. The detailed characterization of all the prepared samples showed that in the sample heated at 900 °C, the g-C3N4 structure was completely preserved despite the addition of dopamine. According to the Raman analysis, the highest ratio of the intensity of the D band and G-band was found in Fe3C@NG40-900, depicting the presence of more Sp2 hybridized C and defect sites, which is responsible for the rapid charge transfer during the ORR. Thus, among the synthesized samples, the highly porous Fe3C@NG40-900 showed enhanced ORR activity with a positive onset potential, limiting current density of 6.3 mA cm−2 half-wave potential of 0.87 V and low peroxide yield, which was less than 2.5%. This enhanced ORR activity of the reported catalyst was attributed to more than one factor such as highly porous hierarchical arrangement of graphitic layers, offering highly exposed active sites, enhanced charge and mass transfer from the surface to the bulk of the catalyst material, use of dual-source strategy, which enriched the graphitic layers with N, thus modifying the electronic structure for better ORR activity, and synergism between Fe3C and NG. According to the experimental characterizations and ORR activity analysis, it was observed that more N is immobilised due to the incorporation of Fe. In addition, among the prepared samples, Fe3C@NG40-900 showed the highest N content, thus confirming that the dual N source enabled more N to be present in the catalyst. According to the X-ray photoelectron spectroscopic (XPS) analysis, five different N configuration (pyridinic-N, Fe-N, pyrrolic-N, graphitic-N, and oxidized-N) with different binding energies were present in the samples. Also, the content of the different configurations was found to be temperature dependent. Among these configurations, pyridinic N was beneficial for stabilising the metal centres, while the four-electron mechanism was strongly dependent on graphitic-N and additional active sites for enhanced activity were provided by the Fe–N centres. All these factors were considered to play vital role in controlling the mechanism and enhanced ORR activity of the Fe3C@NG40-900 sample. Another strategy for enhanced ORR activity was adopted by Roy Chowdhury et al., in which a hybrid catalyst was developed by employing CuCo2S4 and dual heteroatom (B,N)-doped rGO.171 The electrocatalytic investigations showed that the as-developed hybrid catalyst possessed enhanced bifunctional ORR and OER catalytic activity. In the case of the ORR, the as-synthesized CuCo2S4@B,N rGO exhibited significantly enhanced activity compared to CuCo2S4 without B,N-doped rGO and Pt/C with a positive onset potential of 0.88 V, half-wave potential of 0.73 V and improved methanol tolerance. The electron transfer number during the ORR was nearly equal to 4, showing that the reaction follows the typical four-electron pathway for the reduction of O2. The enhanced catalytic activity in this report was attributed to the synergistic effect between the two pristine materials and dual heteroatom doping. The improved ORR activity was further supported by the theoretical DFT investigations in previous studies. In these reports, it was theoretically determined that for the ORR activity, N doping facilitates bond dissociation in O2 due to the formation of the superoxide ion, thus improving the catalytic efficiency. Similarly, other investigations revealed that the formation of B–C–N is highly effective and crucial for the ORR. This bond formation first results in the polarization of C due to the bonded N atom, which further induces charge transfer from C to B, thus activating B as an active site for the ORR. On the contrary, it has been reported that the B–N configuration without C bridging is highly determinantal for ORR activity. Therefore, the formation of the B–C–N configuration in this report is considered one of the possible reasons for the better ORR activity. Similarly, several studies have been reported in which the properties of NG such as introduction of defects on adjacent catalysts in the hybrid material, which alters the charge density and asymmetric spin density for improved superoxide ion formation to weaken the O–O bond for enhanced ORR activity, formation of a bond between the metal and highly polar C–N group for enhanced stability, prevention of recrystallization, agglomeration and dissolution of active materials are utilized. In this quest, Gautam et al. developed a proper hybridization between crystalline spinel Zn2Mn3O8 nanocrystals stabilized in NG nanosheets.172 The as-developed shape, size and morphology-controlled hybrid catalysts showed enhanced ORR activity with a positive onset, half-wave potential and enhanced methanol tolerance. The enhanced electrocatalytic ORR activity was attributed to the synergistic effect, efficient charge and mass transfer tendency, better conductivity, surface area and diffusion channels. According to the ORR investigations on the as-developed hybrid structure, it was observed that the reaction follows the four-electron reaction pathway under alkaline electrolyte conditions. Regarding the enhanced electrochemical activity, the formation of more defect sites in graphene due to N doping and development of hybrid structures were considered as the primary reason according to the Raman analysis.
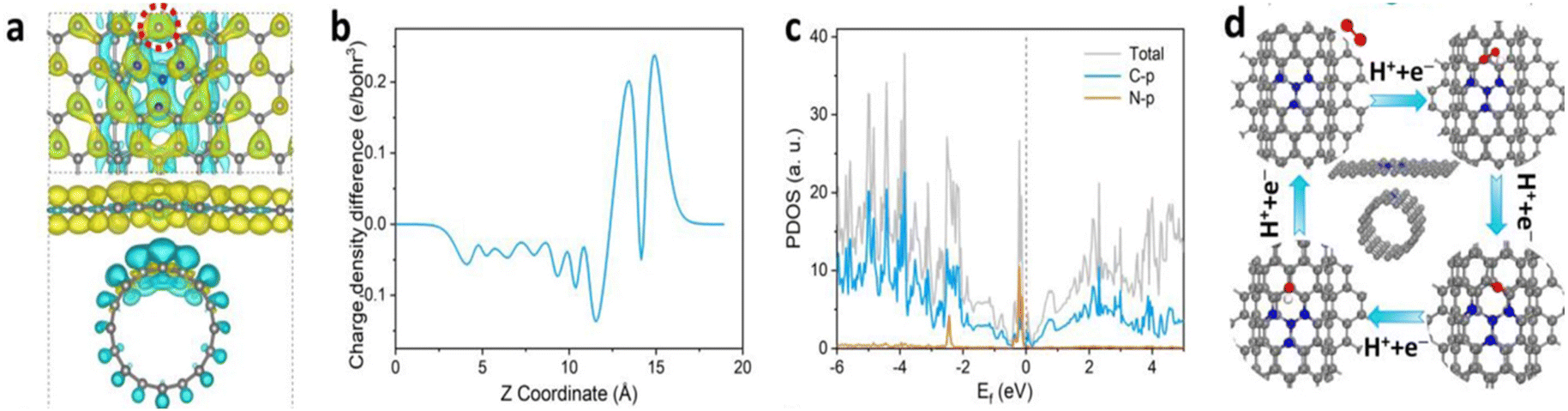 | ||
| Fig. 18 (a) Charge density difference between NG-CNT and NPyri–G in NPyri–G/NG-CNT with top and side view having augmented and reduced charge marked by yellow and cyan colours, respectively, active sites are represented by red circles, C atom demonstrated by grey balls and N atoms by blue balls. (b) 1-D charge difference between NG-CNT and NPyri–G in NPyri–G/NG-CN, (c) partial density of states with Fermi level shown by black line and (d) mechanistic steps of ORR over NPyri–G/NG-CNT catalyst. | ||
Considering the construction of hybrid NG-based electrocatalysts, two of the commonly developed and used hybrid structures include (a) core shell structure (which includes the formation of a coating of NG on another pristine material) and (b) adhering of different particles on NG surface. However, both approaches have certain obvious drawbacks such as the loss of active sites due to the formation of the NG coating and agglomeration of the particles adhered on NG. Therefore, efforts have been devoted to developing possible novel strategies to overcome these limitations. For instance, Bai et al. developed a confined Co9S8@NG catalyst derived from an intercalated Co(OH)2 precursor for enhanced ORR catalytic activity.173 Co9S8@NG with a flower-like morphology was obtained through the interlayer-confined carbonization of metanilic intercalated Co(OH)2 together with sulfidation of the host layers. The investigation showed that Co9S8 was covered by a few layers of NG, thus preventing its agglomeration and producing better ORR activity with a half-wave potential of 0.861 vs. RHE. It was observed that pristine NG showed lower ORR activity, and therefore in the hybrid catalysts, Co9S8 was considered the main active site with additional synergism offered by NG. The electron transfer number was calculated to be equal to 4, which indicates that the ORR on the as-synthesized catalyst occurs in a single step with only 2.5% production of H2O2 as a product of the 2e− process. During the ORR investigation and post-ORR characterization, it was observed that the surface cobalt sulphide is partially oxidised into the corresponding CoOx during the initial stage of the ORR. It was observed that this conversion is beneficially self-activating for ORR activity of the as-prepared sample. The increased ORR activity due to surface oxidation was attributed to the increase in electrochemical surface area, thus providing more active sites for the ORR. The role of surface oxidation was further elucidated using DFT analysis. Using DFT investigations, two models of non-oxidised surface (100 (crystal plane)) and oxidised surface were employed to determine the energy of adsorption of different ORR intermediates. Using the non-oxidised and oxidised surface for different intermediates, the corresponding adsorption models were developed, as shown in Fig. 19. According to the free energy measurements for the rate-determining step at equilibrium potential, it was observed that the energy barrier for the non-oxidised surface was 0.88 eV, whereas that for the oxidised surface was only 0.35 eV. These theoretical calculations revealed how the formation of an oxide layer on the surface of a material enhances its ORR activity. The observed results and theoretical results were consistent. The reaction steps involved in ORR were evaluated using both the non-oxidised and oxidised models, as follows:
| O2 + H2O + 4e− + * → OOH* + OH− + 3e− |
| OOH* + 3e− → O* + OH− + 2e− |
| O* + H2O + 2e− → OH* + OH− + e− |
| OH* + e− → OH− + * |
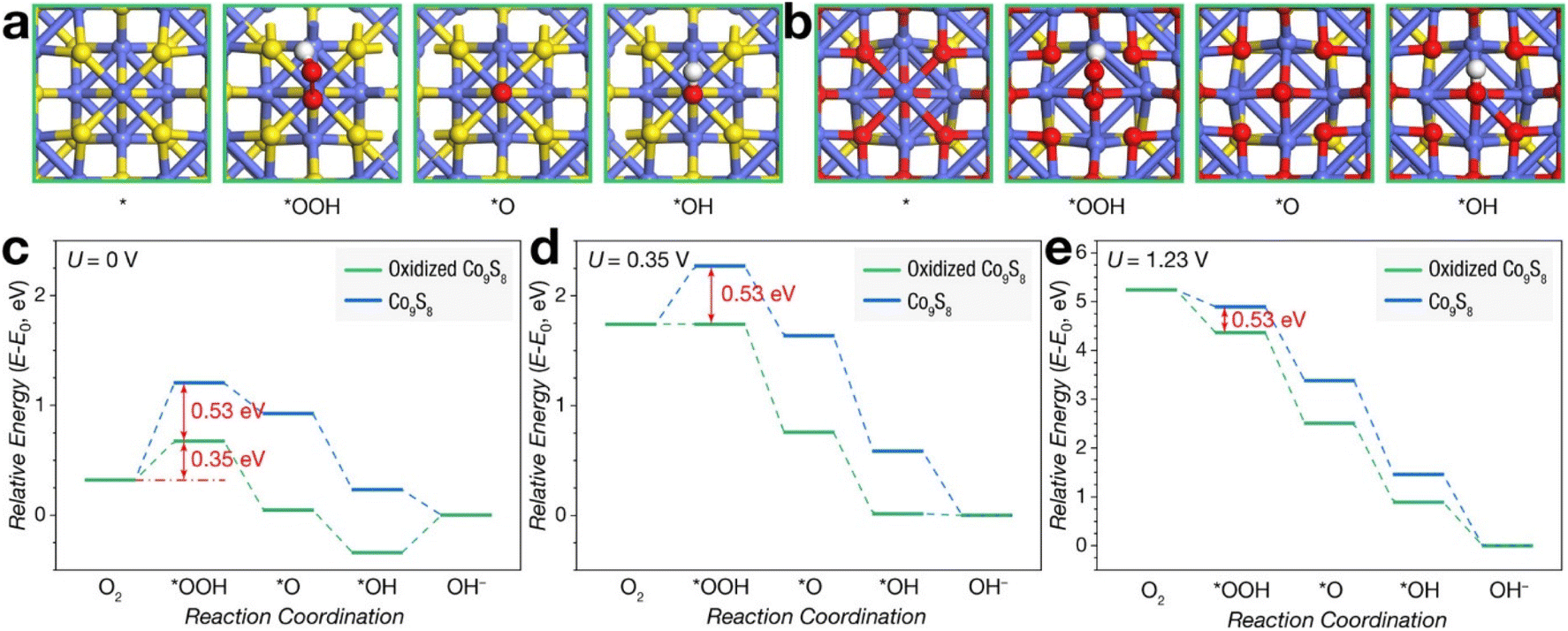 | ||
| Fig. 19 Optimised structure of intermediates adsorbed on (a and b) Co9S8 and surface-oxidised Co9S8 for ORR process and (c–e) energy landscapes calculated at U = 0 V, 0.35 V and 1.23 V for Co9S8 and surface-oxidised Co9S8, respectively. Reprinted with permission from ref. 173. Copyright 2020, the American Chemical Society. | ||
Another important device used for sustainable energy is sea water batteries (SWB). SWB have attracted tremendous attention due to their beneficial and superior characteristics. However, the performance of SWB and durability of their cathode are limited by the presence of chloride ions. Thus, to develop a Cl-resistant cathode electrocatalyst, Kim et al. developed a bifunctional ORR/OER hybrid catalyst consisting of a pyridine N-rich graphene layer encapsulating Co.174 During the investigation, the authors for the first time employed the interface dipole moment to reduce the poisoning caused by Cl ions due to coulombic repulsion. The theoretical DFT investigation revealed that doping of pyridinic N significantly enhanced the catalytic performance of the NG-encapsulated Co nanoparticles together with the strengthening of the electric field of the hybrid catalyst surface. According to both the theoretical and experimental investigations, the overall ORR activity was enhanced in the case of the heterostructure due to the large difference in work function between the two components, which improved the charge transfer capacity for the ORR. The theoretical induced electric field developed at the surface of Co@G and Co@NG is depicted in Fig. 20, which showed that a strong negative electric field developed (more in case of Co@NG), due to which Cl ions may be repelled, thus improving the durability of cathode electrocatalysts in SWB toward Cl poisoning. Further, the experimentally improved electrocatalytic ORR activity was attributed to the efficient charge transfer and synergistic effect between Co nanoparticle and NG.
 | ||
| Fig. 20 (a) Electric field calculated for (a) Co(fcc)Gr and Co (fcc)NGr in z-direction as a function of position with different charge density and (b) graphical representation of charge density difference along z-axis with monolayer and multilayer N-Gr over Co(fcc), where the numerical values present in the different graphs represent the dipole moment. Blue and yellow regions represent the electron accumulation and depletion regions, respectively. Reprinted with permission from ref. 174. Copyright 2022, Elsevier. | ||
As discussed above, different factors such as heteroatom doping, pyridinic N, graphitic N, and hetero-structured development play a vital role in improving the overall ORR activity of carbon-based materials and non-noble metal-based electrocatalysts. In the case of M–N–C (where M is a metal and NC represents N-doped carbon material), single atomic site loading on a matrix having different nanostructures has been explored; however, the synthesis of these materials will a high concentration and low agglomeration is still challenging. Thus, to overcome these challenges, different researchers have suggested that the development of nanoclusters/particles in NG also act as promising electrocatalysts for ORR with simple, cost-effective and easy synthesis. Several nanodimensional particles embedded in NG have been developed, for example, Fe3C@NG has been considered an effective ORR electrocatalyst. For instance, Yang et al. developed Fe3C quantum dots embedded in Fe and N-codoped hollow carbon nanospheres by employing a triblock copolymer as the C source and N-rich melamine formaldehyde as the N source.175 The ORR activity measurements showed that the as-develop Fe3C-based catalysts have great potential for practical applications in place of the commercially used Pt/C electrocatalyst. However, the exact mechanism of how the electronic structure, electron transfer and O2 adsorption reduction capacity of NG are modified due to the addition of Fe3C is still far from being understood. In addition, the understanding of the degradation of Fe3C@NG during the ORR is still not clear. Therefore, to investigate and understand all these issues, Fe3C quantum dots well dispersed in NG mesoporous spheres were developed by Yang et al. using in situ-produced silica as a template. Based on experimental and theoretical investigations, the formation of a Mott–Schottky heterojunction between Fe3C and NG was proposed. It was observed that the as-prepared Fe3C quantum dot@NG Mott–Schottky heterojunction showed enhanced ORR activity and extreme stability under corrosive electrolyte conditions. The enhanced ORR activity was explored both experimentally and theoretically, revealing that the fast electron transfer from Fe3C to the valence band of NG contributed to the improved ORR activity. The highly porous nano hierarchal structure of NG provides a large number of active sites and rapid mass transfer dynamics. In addition, graphene provides a shielding effect for Fe3C in the highly corrosive environment. The as-developed Fe3C@NG together with Zn/G as the anode using a solid-state electrolyte was assembled as a battery and optimised, which delivered an open circuit voltage of 1.506 V together with an energy density of 706.4 W h kg−1 and stability for about 1000 h. A schematic representation of the Fe3C@NG Mott–Schottky heterojunction before and after contact, energy profile of different intermediates, energy profile at different potentials and molecular dynamic simulations of the as-prepared ORR catalyst are presented in Fig. 21. Similarly, other studies investigated the role of hybrid NG/oxide-based heterostructures for ORR application, as presented in Table 3.
 | ||
| Fig. 21 Mott–Schottky heterojunction in Fe3C@NG (a) before and (b) after contact, (c) mechanism of the ORR, (d and e) charge distribution model of Fe3C@NG, (f) ORR on Fe3C@NG model, (g) energy profiles and (h) energy change in ORR process on different active sites over G, NG, Fe3C, and Fe3C@NG materials, (i) energy profile diagram at different potentials for Fe3C@NG, and (j) molecular dynamics simulation of Fe3C@NG in H2SO4 solution (0.5 M). Reprinted with permission from ref. 175. Copyright 2022, Wiley. | ||
| Type of electrocatalyst | Type of electrolyte | Onset potential | Half-wave potential | ORR mechanism (4e/2e) | Ref. |
|---|---|---|---|---|---|
| N-doped CNT/Co2P-CoN | 0.1 M KOH | ≈0.96 V | 0.85 V | 4e− | 176 |
| GC@COF-NC | 0.1 M KOH | 0.923 V | 0.841 V | 4e− | 177 |
| Co1−xZnxSe@NCF | 0.1 M KOH | 913 mV | 0.83 V/829 mV | 4e− | 178 |
| NC/BiOCl-CNTs | 0.1 M KOH | 0.98V | 0.85 V | 4e− | 179 |
| Co/Co3O4@NHCS | 0.1M KOH | 0.95V | 0.82V | 4e− | 180 |
| Co3ZnC/Co@CN | 1 M KOH | 0.912V | 725 mV | 2e−/4e− | 181 |
| Fe2O3-MoO3/NG | 0.1 M KOH | — | ∼0.82 V | 4e− | 182 |
| Co–B–Nx | 0.1 m KOH | 1.05 V | 0.94 V | 4e− | 183 |
| PdxSy-MoS2/N-GR | 0.1 M KOH | −0.141 V | −0.214 V | 4e− | 184 |
| N-doped porous C/CoO/MoC | 0.1 M KOH | 0.941 V | 0.841 V | 4e− | 185 |
| Fe3C/Fe2O3/N-doped graphene | 0.1 M KOH/0.1 M HClO4 | 80 mV | 0.86 V | 4e− | 186 |
| Co3O4−x/N–C | 0.1 M KOH | 0.936 V | 0.845 V | 4e− | 187 |
| Fe2O3/Fe3O4/hNCNC | 0.1 M KOH | 1.03 V | 0.838 V | 4e− | 188 |
| MoS2/Co/CNT | 0.1 M KOH | 931 mV | 792 mV | 189 | |
| PBA/PPy/N-doped/FeCo | 0.1 M KOH | — | 0.83V | 4e− | 190 |
| Co9S8/CeO2/Co/NCNTs | 0.1 M KOH | — | 0.875 V | 4e− | 191 |
| Ag/Ce/Mn3O4 | 0.1 M KOH | ∼0.96 V | ∼0.88 V | 4e− | 192 |
| MnO-Co@NC | 0.1 M KOH | 0.9 V | 0.75 V | 4e− | 193 |
| fct-PdFe/Pd/NG | 0.1 M KOH | ∼0.97 V | ∼0.83 V | 4e− | 194 |
| MoS2/CoP/NPC | 0.1 M KOH | — | 0.790 V | 4e− | 195 |
| NCNS/Fe2O3/Fe3C | 0.1 M KOH/1 M Na2SO4 | 0.90 V | 0.82 V | 4e− | 196 |
| NiCo/NC | 0.1 M KOH | 0.94 V | 0.84 V | 4e− | 197 |
| Mo2C/Fe5C2/NC | 0.1 M KOH | 0.92 V | 0.78 V | 4e− | 198 |
5. Advances in HER applications of N-doped graphene (NG) heterostructures
For sustainable electrocatalytic water splitting, the HER and OER are the two half-cell reactions involved. Considering future green energy, hydrogen is considered to play a significant role as an alternative to the finite, pollution-creating fossil fuels. To produce hydrogen from water or any other source, both the HER and OER occur simultaneously. Regarding the HER and OER, the basic chemistry involved was discussed in detail in a previous report. From an energy perspective, with respect to the standard hydrogen electrode (SHE), the HER requires an external potential of 0.0 V, while that for OER with respect to the SHE is 1.23 V. From a mechanistic viewpoint, the HER and OER are 2e− and 4e− processes, respectively, and thus the OER requires more external energy compared to the HER. Thermodynamically and kinetically, both these reactions are non-spontaneous and do not take place in the absence of external energy and catalysts.199 Regarding external energy, different sources (light, electric potential, electric field, etc.) are employed, while as the catalyst, different materials have been developed and investigated for application as photocatalysts, electrocatalysts, photo-electrocatalysts, etc. The main steps involved in the HER are presented below.The HER is an important two-electron reaction taking place at the cathode. The mechanistic reaction steps involved in the HER under acidic electrolyte conditions are as follows:
(1) Step one involves the adsorption of an H+ ion on the catalyst surface.
| H+ + e− ⇌ H(adsorbed) (Volmer step) |
(2) In the second step, another H+ is adsorbed and reacts with the already adsorbed H+ ion as follows:
| H(adsorbed) + H+ + e− ⇌ H2 (Heyrovsky reaction/step) |
(3) Finally, the adsorbed H2 molecule gets desorbed from the catalyst surface.
| H2(adsorbed) ⇌ H2 (Tafel step) |
The HER in acidic medium takes place as follows:
| 2H(aq) + 2e− ⇌ H2(gas) |
Alternatively, under alkaline conditions, the reaction follows:
1. 2H2O + 2e− ⇌ 2H(adsorbed) + 2OH− (Volmer step)
2. H2O + H(adsorbed) + e− ⇌ H2 + OH−(Heyrovsky reaction/step)
3. 2Hadsorbed ⇌ H2 (Tafel step)
The HER in alkaline medium takes place as follows:
| 2H2O + 2e− ⇌ H2(gas) + 2OH(aq)− |
For electrocatalytic HER, currently the state-of-the-art catalysts displaying the highest activity is noble metal-based Pt electrocatalysts. However, the practical application of Pt is significantly hindered by its high cost and limited resources. Similarly, for the OER, the state-of-the-art catalysts are based on IrO2, which also belongs to the noble family. Therefore, it is necessary to develop alternative efficient catalyst that can overcome these limitation with comparable activity and stability of the state-of-the-art HER and OER electrocatalysts. In this quest, different catalysts have already been developed, which show promising results, but still more efforts are needed to completely replace the state-of-the-art noble metal-based electrocatalysts. In this quest heteroatom doped graphene has evolved one of an important material, which can either function as pristine electrocatalysts for the HER/OER or function as a substrate material to enhance the activity of other active materials (including metal oxides, metal sulphides, metal phosphides, and MXenes). Here, in this section, we discuss the recent advances in improving the catalytic activity of N-doped graphene-based heterostructures for the HER and OER. Further, the role of different active sites such as pyridinic N, quaternary N, metals and metal oxides dispersed uniformly in NG for enhanced HER/OER activity will be discussed. Most of the single-component electrocatalysts investigated to date do not produce the expected activity to replace noble metal-based electrocatalysts. Therefore, doping and development of multicomponent heterostructures have been explored and are expected to attain higher HER activity compared to the state-of-the-art electrocatalysts. Among the different non-noble metal-based electrocatalysts, graphene shows great potential compared to other carbon-based electrocatalysts due to its several advantageous structural characteristics such as large active surface area, efficient electron transport channels, abundant catalytic active sites and impressive stability. Utilising these favourable characteristics for the HER, the development of NG-based heterostructures has shown great potential as HER catalysts.
As an alternative HER catalyst, different single-component non-noble metal-based electrocatalysts such as Co, Ni, and Fe have been explored. Alternatively, multicomponent HER catalysts are associated with a series of merits such as better conductivity, large number of active sites, and synergism between two or more components in multicomponent heterostructures. Utilizing these beneficial characteristics, an enormous number of NG@metal heterostructures have been developed for the HER. For instance, Jiao et al. developed a multicomponent NG-based heterostructure for the HER.199 In this report, a Co@Zn-based heterostructure (Co@Zn@NG) was successfully synthesized and investigated for the HER. The HER investigations showed that the Co@Zn@NG sample heated at 850 °C displayed considerable enhanced HER activity (such as lower overpotential η10 (34 mV), smaller Tafel slope (36 mV dec−1)) as compared to bare NG, Co3O4@ZnO@NG and commercial Pt/C. The enhanced HER activity was attributed to the faster reaction kinetics and low impedance of Co@Zn@NG, as observed during the analysis. Hence, due to the lower impedance of the as-developed Co@Zn@NG catalyst, it showed efficient charge transfer ability with a high density of active sites and no change in morphology, thus confirming its stability. From a mechanism viewpoint, the role of carbon-based materials has been divided into three categories including (a) substrate, (b) main catalyst and (c) auxiliary catalysts. According to this investigation, it was concluded that the C atom attached to N acts as an active site for the HER, which was also illustrated by the XPS analysis before and after the catalytic investigation. Further, the role of the developed heterostructure in the enhanced HER activity was investigated and it was confirmed that after heterostructure development, electron transfer from NG occurs rapidly due to the more electron-deficient Co (Co3+), thus promoting the HER activity of Co@Zn@NG. From a mechanistic view point, a kinetic analysis was carried out and it was observed that in the case of the most active sample, Co@Zn@NG prepared at oxidation temperature of 800 °C has a low Tafel slope. Thus, it was concluded that the Tafel step is the main rate-controlling step for the HER, while the sample prepared at 750 °C had the highest Tafel slope, and thus the Volmer step is considered the main HER-controlling step. Further, the EIS analysis demonstrated that the Co@Zn@NG sample showed a low resistance to electron flow, thus contributing to the enhanced HER activity.
Another interesting study was carried out by Wang et al., in which Ni2P@FeP (NFP) hybrid nanoparticles were incorporated in NG nanosheets and investigated as multifunction electrode materials for overall water splitting reactions.200 For the HER, the as-developed heterostructure hybrid catalyst showed considerably improved activity with low onset potentials (−110 and −144 mV) and low overpotential (−250 and 240 mV at 10 mA cm−2) both in alkaline and acidic electrolyte, respectively. The as-synthesized NFP@NG was simultaneously investigated as an anode and cathode, which revealed long-term stability and a current density of 10 mA cm−2 at a low cell voltage of 1.69 V for overall water splitting. The superior HER activity of NFP@NG was attributed to the presence of a high electrochemical specific surface area and large number of active sites, as observed from the ECSA studies. In addition, the synergistic effect between bimetallic phosphide and NG resulted in faster charge transfer due to the enhanced conductivity. Another important transition metal phosphide heterostructure was developed by Suryawanshi et al., in which Ni2P (0D) was encapsulated in 2D N and S-doped graphene (NSG) nanosheets. The as-developed NP@NSG sample was analysed for overall water splitting activity and better HER activity was observed in 1.0 M KOH electrolyte solution.201 In the comparative analysis, other electrodes were utilised including Ni2P@NG, Ni2P@SG, Ni2P, NG, Ni2P@G, Ni2P and Pt materials as the catalyst. The linear sweep voltammetry (LSV) measurements showed that the overpotential required for the HER to obtain a current of 10 mA cm−2 for Ni2P@NSG was lower (110 mV) compared the other catalysts except Pt/C. This enhanced and effective HER activity was ascribed to the synergistic effect of defective NSG and Ni2P. In addition, the use of the Ni2P (001) plane comprised of highly exposed P and Ni sites possessed both hydride acceptor Ni site and proton acceptor P site, which resulted in enhanced HER activity. Rather than showing pure metal character, Ni2P (001) acts as hydrogenase due to the presence of a P atom on its surface. In addition, the slight positive and negative charges (observed from XPS analysis) on the Ni and P atoms promote the easy adsorption and desorption of the reactant and products molecules, respectively. Further, the encapsulation of Ni2P in the defective NSG significantly enhances the HER activity due to the possible synergistic effect, large number of surface-active sites and rapid transfer of charge carriers. To analyse the reaction mechanism and rate-determining step for the HER over the Ni2P@NSG electrocatalysts, a Tafel investigation was carried out. The Tafel measurements showed that the HER follows the Volmer–Heyrovsky pathway, and hydrogen adsorption and desorption are considered as the possible rate-determining step. According to the different investigations on this type of composite hybrid catalyst, it was established that graphene or doped graphene consists of most of the electrocatalytic active sites responsible for the HER. Specifically, pyridinic N and graphitic N are considered more active for the HER compared to pyrrolic N. Moreover, the formation of surface defects, lattice distortions and structural edge defects due to heteroatom doping are considered as the main reasons responsible for better HER activity. In the case of the Ni2P@NSG hybrid electrocatalyst, the strong interaction between 0D Ni2P and 2D NSG alters the electronic structure of NSG, which significantly tunes the binding energy of the different reaction intermediates formed during the HER. Also, further insight into the mechanism or reasons responsible for the enhanced HER activity in the Ni2P@NSG sample was gained by carrying out DFT studies of the different developed samples in this report. The DFT analysis indicated that the interaction of N and S dopant with Ni2P reduces ΔGH2O* to −0.15 eV and ΔGH* to −0.22 eV, which is close to optimum ΔGH2O* of 0 eV. Therefore, the as-developed Ni2P@NSG hybrid catalysts benefit from the coordinated dual interaction of N and S doping together with optimized adsorption energies for better HER activity.
Another important class of metal-based catalysts includes transition metal oxides (TMOs). Owing to their semiconducting properties, low cost, natural abundance and catalytic activities, TMOs act as important functional materials. Among the different TMOs, 2D MoOx have attracted significant attention and considered as non-metallic catalysts compared to FeOx, NiOx catalysts for the HER and considered a suitable candidate for the design of non-metal-based electrocatalysts. However, under acidic conditions, MoOx-based materials undergo corrosion and do not possess hydrogen adsorption sites, making these materials ineffective for the HER. Thus, to overcome these limitations, Aydin et al. developed a hybrid catalyst consisting of N and S-doped graphene and MoOx, and then analysed its activity towards the HER.90 In this report, layered MoOx was synthesized, and then a thin film of graphene was transferred to it, followed by doping with N and S. The HER analysis showed that the as-developed hybrid catalyst showed a significantly improved catalytic performance with significantly low overpotential of 359 mV at 10 mA cm−2 compared to pure graphene and MoOx. The observed enhanced activity was attributed to the presence of oxygen in graphene, which resulted in the better electrical contact and interfacial interactions between graphene and MoOx. The activation energy analysis showed that the main active sites for the HER come from the superficially deposited N and S-doped graphene. In addition, the better HER catalytic performance of the MoOx@NSG hybrid catalyst was attributed to the synergistic effect of co-doped atoms present at the interface between NSG and MoOx and the presence of more pyrrolic N atoms, which provide more active sites for catalytic action. The pyrrolic N–C configurations have a lower hydrogen binding free energy compared to the graphitic N and pyridinic N, and thus promote efficient H adsorption, enhancing the catalytic performance of the hybrid catalyst. CV activation of the hybrid catalysts was carried out and it was observed that after 1000 cycles of CV activation, the hybrid catalysts showed the optimum HER activity, which was reduced after further cycles due to the possible degradation of the catalyst in terms of cyclic potentiometric analysis. The HER mechanism was evaluated using different characterization techniques. According to the Raman analysis, a schematic of the material surface was developed, as shown in Fig. 22. The Raman investigation of undoped graphene on an MoOx/glass calomel electrode (GCE) structure showed that the absence of defective graphene prevented the adsorption of H+ ions. Hence, the coupling of non-adsorbed H+ ions only with the electrons present on graphene surface takes place without MoOx playing any role besides transferring electrons from the GCE to graphene. In the case of graphene doped with N and S, the formation of defects in graphene resulted in the adsorption of H+ ions. The adsorbed H+ ions are transferred to the MoOx materials, where the reduction process occurs, thus resulting in the evolution of H2 in addition to the H2 produced on the graphene surface due to the reduction of non-adsorbed H+ ions, as shown in Fig. 22. Therefore, this synergistic effect due to doping and heterostructure development resulted in an enhanced HER performance by the MoOx@NSG hybrid catalyst.
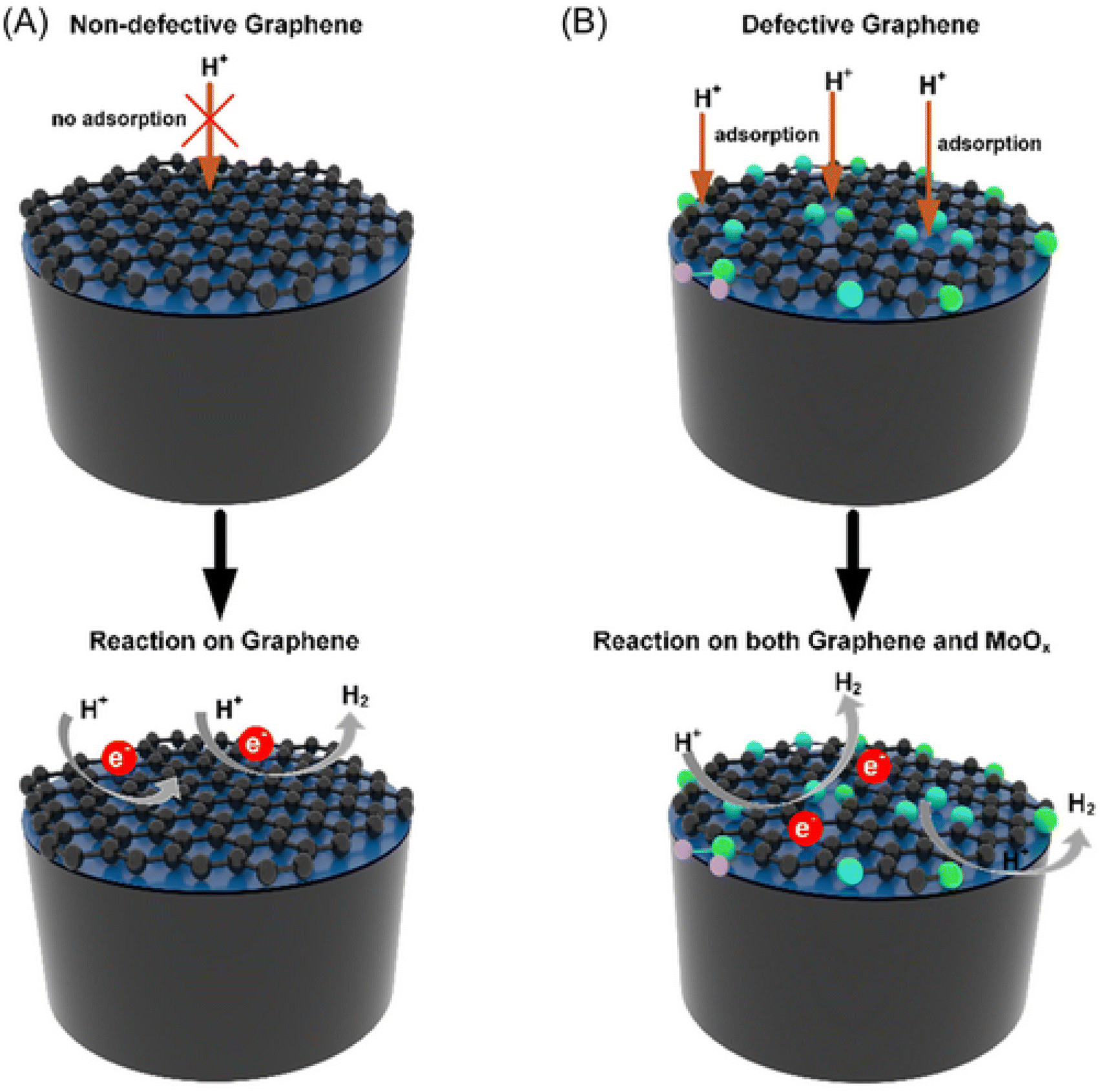 | ||
| Fig. 22 Schematic illustration of the reaction steps involved in the HER over (A) undoped graphene and (B) N and S-co-doped graphene@MoOx/GCE hybrid electrode. Reprinted with permission from ref. 90. Copyright 2023, Wiley. | ||
Another important study was carried out by Gao et al., in which Ru/RuS2 nanoparticles were composited with NG as an electrocatalyst for the HER in alkaline medium.202 As observed from different investigations, the adsorption and desorption of H on the catalyst surface play a vital role in the HER. The theoretical investigations indicated that Ru offers strong binding towards H, which is unfavourable for the HER, whereas RuS2 shows a moderate HER performance due to its similar binding energy for H as that of Pt. Subsequently, Gao et al. developed a hybrid catalyst to utilise the characteristics of both Ru and RuS2 for moderate H adsorption and desorption. The Ru/RuS2@NG hybrid catalyst was utilised for the HER under alkaline electrolyte conditions and the effect of the annealing temperature on its HER activity was investigated. The observed HER results depicted that the as-developed hybrid catalyst annealed at 500 °C showed significantly enhanced activity with a low overpotential of 18 mV at 10 mA cm−2 compared to the samples annealed at higher temperatures. According to the results, it was inferred that the crystallinity of the as-synthesized catalyst plays significant role in its HER activity, and hence the material synthesized at 500 °C with low crystallinity demonstrated higher HER activity. The kinetic investigations and double-layer capacitance measurements showed that the Ru/RuS2@NG sample annealed at 500 °C offer fast reaction kinetics together with a high electrochemically active surface area (ECSA), thus providing more active sites for efficient HER activity. The enhanced HER activity was attributed to the easy dissociation and adsorption of H2O and H on Ru and RuS2, respectively. According to the comparative investigation, the sample without N doping offers higher resistance to charge transfer compared to the sample having NG as one of the main components. Therefore, it was concluded that N doping significantly accelerates the charge transfer between the different components of the heterostructure catalyst. Similarly, Wang et al. engineered Rh2P/Rh heterostructure nanoparticles on P,N-co-doped graphene to investigate the effect of heterojunction and degree of phosphidation on HER activity under both alkaline and acidic conditions.203 It was observed that phosphides of Pt group metals such as Ru, Ir and Rh show interesting electrochemical HER activity and the dosage of P in these materials has a considerable influence on the overall activity of the catalyst. However, during the synthesis process, it is exceedingly difficult to control the degree of phosphidation. In this report, the successful synthesis of Rh2P was conducted with a highly controlled P dosage and its influence on HER activity was investigated. Subsequently, it was inferred that the P-deficient Rh2P/Rh heterostructure with a mixed metallic and phosphide phase outperformed both pure Rh2P and metallic Rh nanoparticles. Then, the as-developed heterostructure was coupled with P,N-co-doped graphene (PNG) and employed as a catalyst for the HER in alkaline and acidic electrolyte solution. According to the HER analysis of Rh2P/Rh@PNG, it exhibited remarkable activity with a significantly small overpotential of 19 and 17 mV at 10 mA cm−2 current density in acidic and alkaline solution, respectively. The activity of the as-developed catalyst in this report surpasses the benchmark Pt/C- and Rh-based competitive catalysts. This enhanced HER activity of the as-developed Rh2P/Rh@PNG electrocatalysts was attributed to the near to zero adsorption energy of H, which results in accelerated water dissociation and boosted H evolution activity. It was also observed that most of the active sites in this hybrid catalyst were present at the interface of Rh2P and Rh, thus promoting efficient charge transfer, while PNG provides a highly conductive skeleton for better charge carrier conductivity. Another important new family of materials consisting of van der Waals (vdW) heterostructures has emerged via the layer-by-layer assembly of 2D materials. For practical catalytic applications, vdW 3D heterostructures offer molecular-level design and precision. Utilising a plurality of 2D materials with different electronic affinities and bandgaps, Duan et al. developed a 3D vdW hybrid catalyst consisting of artificially assembled 2D WS2 nanolayers with 100% surface-exposed atoms coupled with N, P and O-doped G.204 The as-developed 3D material was employed as a self-supported electrode catalyst for the HER. According to the HER analysis, the WS2@P,N,OG catalysts showed superior HER activity close to the Pt/C electrode with efficient favourable kinetics and durability. The superior HER activity was attributed to the hierarchical porous structure, continuous conducting network, expanded exposed surface-active site and synergistic effect between different components of the catalyst. The results obtained during the HER analysis are shown in Fig. 23. The Tafel analysis showed that the electrochemical desorption Heyrovsky step is the rate-determining step of the HER over the WS2@P,N,OG catalyst. Thus, in the HER process, the Volmer–Heyrovsky mechanism was considered as a possible mechanism over the WS2@P,N,OG catalyst surface. The Volmer–Heyrovsky mechanism followed by HER in acidic medium consisted of two steps, which proceeds via two proton–electron-coupled steps, as follows:
In step 1, the Volmer reaction takes place, during which electro-sorption of the solvated proton takes place, as follows:
| [(H2O)n-H] + e− ⇌ H* + (H2O)n |
The second step involves the Heyrovsky step, in which Eley–Rideal-type recombination takes place between the adsorbed H* and other solvated protons to produce molecular hydrogen, as follows:
| [(H2O)n-H] + H* ⇌ H2 + (H2O)n |
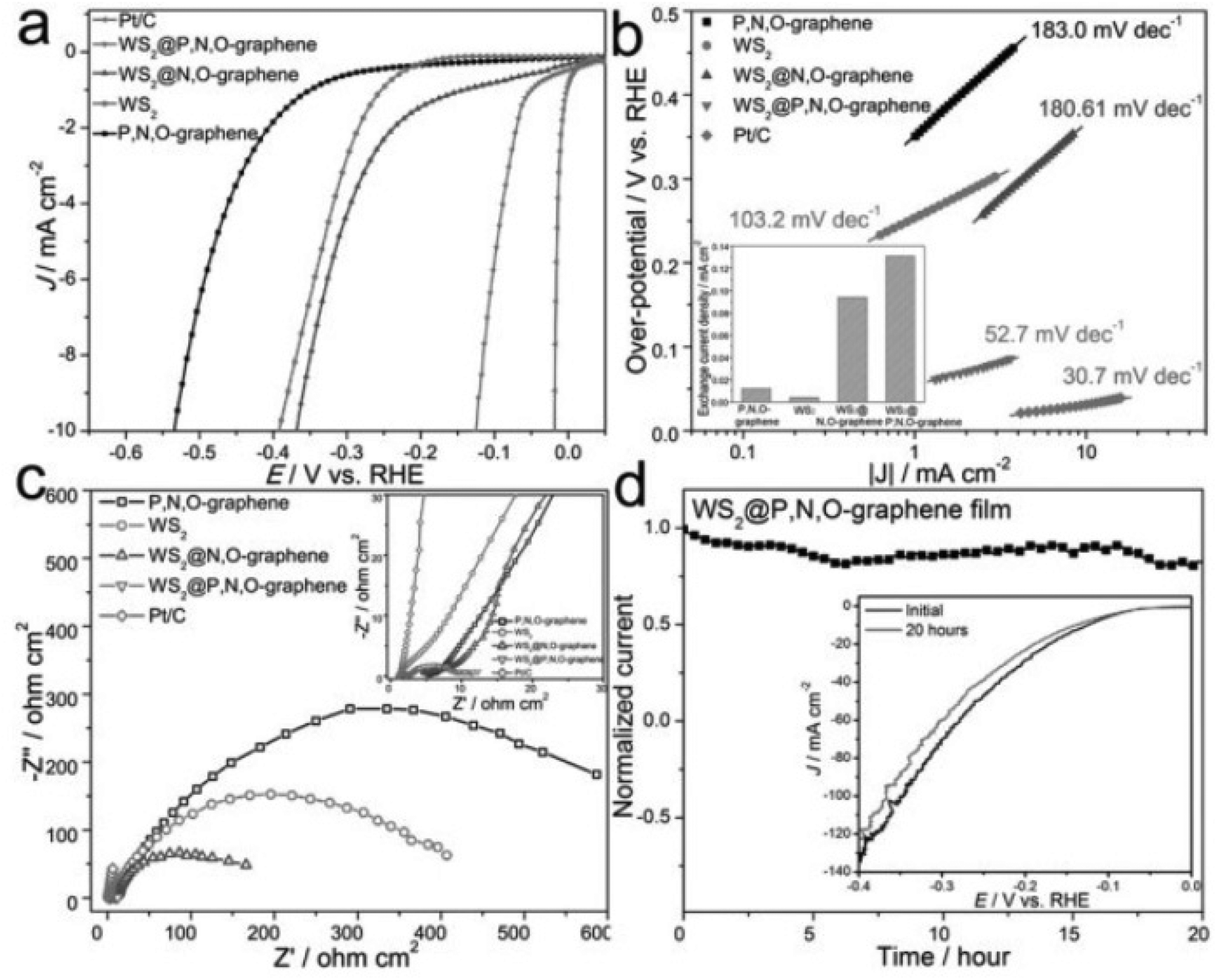 | ||
| Fig. 23 (a) LSV polarization curves below 10 mA cm−2, (b) Tafel plots, (c) EIS spectra with enlarged spectra of as-prepared catalysts in inset, and (d) chronoamperometric response plot of WS2@P,N,O-doped graphene film, with LSV polarization curves before and after 20 h of operation shown in inset. Reprinted with permission from ref. 203. Copyright 2017, Elsevier. | ||
According to the HER mechanism, the dual active sites (first arising due to heteroatom doping and second type arises due to strained metallic edges in 1T structure of WS2 compared to 2H structure) were considered responsible for superior activity.
Another intriguing study was conducted by Song et al., in which doping and heterostructure strategies were coupled to efficiently utilize all the active sites and all components of the heterostructure for better electrocatalytic application.205 In this report, V-doped Ni3FeN was integrated with Ni, and then anchored on NG. The HER investigation showed that the as-developed hybrid catalyst demonstrated better HER and OER activity with an overpotential of 66 mV and 252 mV, respectively, at 10 mA cm−2 current density. The enhanced electrocatalytic activity was attributed to the d-band modulation of the host Ni and Fe due to V doping, change in local charge distribution at the interface of the V-Ni3FeN/Ni heterojunction and better transfer of charge carriers due to the introduction of highly conductive NG skeleton. All these factors contributed to the efficient HER and OER activity of the hybrid V-Ni3FeN/Ni@NG heterojunction electrocatalyst.
Based on the HER/OER analysis of metal-based electrocatalysts, it has been observed that these catalysts face certain limitations such as poor charge carrier and charge transfer ability. Therefore, metal-free electrocatalysts are being explored to overcome these limitations. In this quest, different metal-free electrocatalysts such as black phosphorous (BP), exfoliated BP (EBP), and carbon nitride have been developed. Using metal-free ultrathin black phosphorous, Yuan et al. developed a hybrid catalyst on NG for overall water splitting.206 In this report, solution-processible EBP was coupled with NG having higher Fermi energy levels to form a unique novel metal-free 2D/2D heterostructure with unique electronic configuration and well-modulated interface for HER activity. By coupling the intrinsic activity and stability of pristine EBP and NG, the HER activity was significantly enhanced. The electrocatalytic investigations showed that among the different as-developed heterostructures with different ratios, 2D/2D EBP@NG (1:4) showed efficient HER activity with a low onset potential (191 mV) at 10 mA cm−2, which was significantly lower than that of pure EBP (370 mV) and NG (445 mV). In addition, the kinetic analysis showed that the as-developed EBP@NG(1:4) has a smaller Tafel slope of 76 mV dec−1 with lower charge transfer impedance compared to that of pristine EBP and NG of 135 mV dec−1 and 102 mV dec−1, respectively. The electrochemical double-layer capacitance investigations showed that EBP@NG(1:4) possess significantly improved electrochemical surface area, thus contributing to the significant improvement in electrochemical HER activity. It was observed that using different mass ratios, the electrochemical activity could be easily modulated because different sites are responsible for HER activity. During the investigation, it was observed that EBP is mainly responsible for the HER activity, whereas NG showed better charge transport activity compared to EBP. However, the electronic interactions occurring in the hybrid catalyst are responsible for the enhanced HER activity. The development of a heterostructure between the lower Fermi energy EBP and higher Fermi level NG results in directional charge transfer at the interface, and consequently electronic enrichment of the EBP surface with the optimized adsorption/desorption of H, thus enhancing the HER activity of the hybrid catalyst material. The redistribution of charge after the formation of the heterojunction, differential charge density of EBP and NG and theoretical free energy for the HER at 1.23 V are shown in Fig. 24. Many other novel electrocatalysts, as presented in Table 4, have been explored for the HER and significant advancements have been achieved to meet the current expectations of a material that can replace the state-of-art noble metal-based electrocatalysts.
 | ||
| Fig. 24 (a) P 2p XPS spectra of reference and EBP@NG samples, (b) work function plots of NG and EBP samples, (c) redistribution of interfacial charge between EBP and NG, (d) differential charge density of EBP and NG with yellow and blue for hole- and electron-rich area, respectively, and free energy diagrams for (e) HER on BP-NG and BP, (f) OER on BP-NG and BP at 1.23 V. Reprinted with permission from ref. 205. Copyright 2019, the American Chemical Society. | ||
| Types of electrocatalyst | Current density | Overpotential at 10 mA cm−2 | Onset potential | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|---|
| N-doped MoS2/Ti3C2 | 140 | 225 | — | 100 | 207 |
| Co2P/CoN/NCNTs | 10 | 98 | ≈0.96 V | 57 | 172 |
| CoP(MoP)-CoMoO3/CN | 10 | 198 | — | 95 | 208 |
| CoP/NiCoP/NC | 10 | 60 | — | 58 | 209 |
| Mo2C/MoC/MoP/NC | 0.398 | 109 | — | 52 | 210 |
| N-NiS/NiS2 | 10 | 185 | — | 106 | 211 |
| (Ni/NiO/NCW) | 10 | 105.3 | 55.2 | 212 | |
| N-doped Ni3S2/VS2 | 10 | 151 | — | 107.5 | 213 |
| Ni/MoN/NCNT/CC | 10 | 207 | — | 93.0 | 214 |
| N-doped NiMoO4/Ni3N | 100 | 112 | — | 45.47 | 215 |
| Co/Mo2C-NCNTs | 10 | 170 | — | 79.5 | 216 |
| Co/Mo2C/NC | 50 | 146 | — | 218.06 | 217 |
| Ni/C-V2O3/NF | 500 | 254 | — | 58.10 | 218 |
| MoC-MoP/BCNC | 10 | 158 | 114 | 58 | 219 |
| Mo2C/NC/Pt | 10 | 27 | — | 28 | 220 |
| Fe–Co–oxide/Co/NC-mNS | 10 | 112 | — | 96 | 221 |
| MoS2/Mo2/NC | 10 | 129 | 28 | 78 | 222 |
| MoS2 NSs/NGR | 10 | 208 | — | 79 | 223 |
| MoC1−xMoP/C | 10 | 173 | — | 57 | 224 |
| Au/CoP/NC | 10 | 118.2 | — | 57.75 | 225 |
| Ru/RuO2/N-rGO | 10 | 11 | — | 44.2 | 226 |
| NC/Vo-WON | 10 | 16 | — | 33 | 227 |
| Fe3C-Mo2C/NC | −10 | 116 | 42 | 43 | 228 |
| HGCNF/SNG/MoS2 | 10 | −282 | −0.16 | 83 | 80 |
| CoMoS/CoMoO4/NRGO | 10 | 80 | 35 | 58 | 229 |
| Mo–N/C/MoS2 | 10 | ≈117 | ≈0.90V | ≈64.3 | 230 |
| Mo2C/HNCPs | 10 | 89 | — | 39 | 231 |
6. Theoretical investigations
6.1 Theoretical DFT investigation of ORR activity of NG based heterostructures
Presently, theoretical DFT investigation is considered a crucial tool to theoretically investigate the actual reason responsible for the enhanced properties of NG-based hetero-structured catalysts. Using DFT analysis, different models are employed to investigate the energetics of different intermediates formed during the ORR to elucidate the possible mechanism involved in the reaction. For example, Reda et al. performed a DFT investigation for the ORR on Fe and Fe3C coated with carbon.232 Based on materials such as NG and graphene (G) supported on Fe (110) and Fe3C (010), different models were used to conduct the computational investigation. The computational analysis results evidenced that the Fe3C@NG model has higher ORR activity compared to that of the Fe-supported model, which agreed with the experimental results. During the calculation, the solvation layer on the surface of a hybrid catalyst was also modelled given that the ORR is always carried out in aqueous medium. For a comparison, the free energy diagrams for each model employed were determined at a fixed potential of U = 0.8 VRHE. The Gibbs free energy of adsorption obtained are depicted in Fig. 25(I–IV). According to this figure, all the ORR intermediates (*OOH, *O and *OH) are significantly stabilised over both Fe3C and Fe-supported NG. However, the stabilization is even stronger on the Fe-supported model and O* is more stabilised compared to the other intermediates. This strong binding of O* was correlated with the charge on the oxygen atom. As evidenced by the DFT calculations, the Fe3C@NG heterostructure shows superior ORR activity due to the accumulation of O* on its surface and the ORR occurs when oxygen coverage is between 0.5 < T0 < 1.0. However, in the case of Fe-supported NG, the binding of O* is so strong that the surface of the catalyst becomes effectively blocked for the ORR. The effect of the NG layers also showed that although only two layers of NG converge, the ORR activity of the supported system was superior to that on the unsupported NG. The effect of electron donation by the substrate was also investigated. According to this investigation, it was reported that the stronger electron donation, which is a key parameter for the ORR, may result in the suppression of ORR activity, as observed in the case of the Fe-supported catalyst. Therefore, it was concluded that any substrate material have electron-donating affinity as strong as that of Fe3C but lower than that of Fe can have similar or enhanced activity as that of the Pt (111) surface.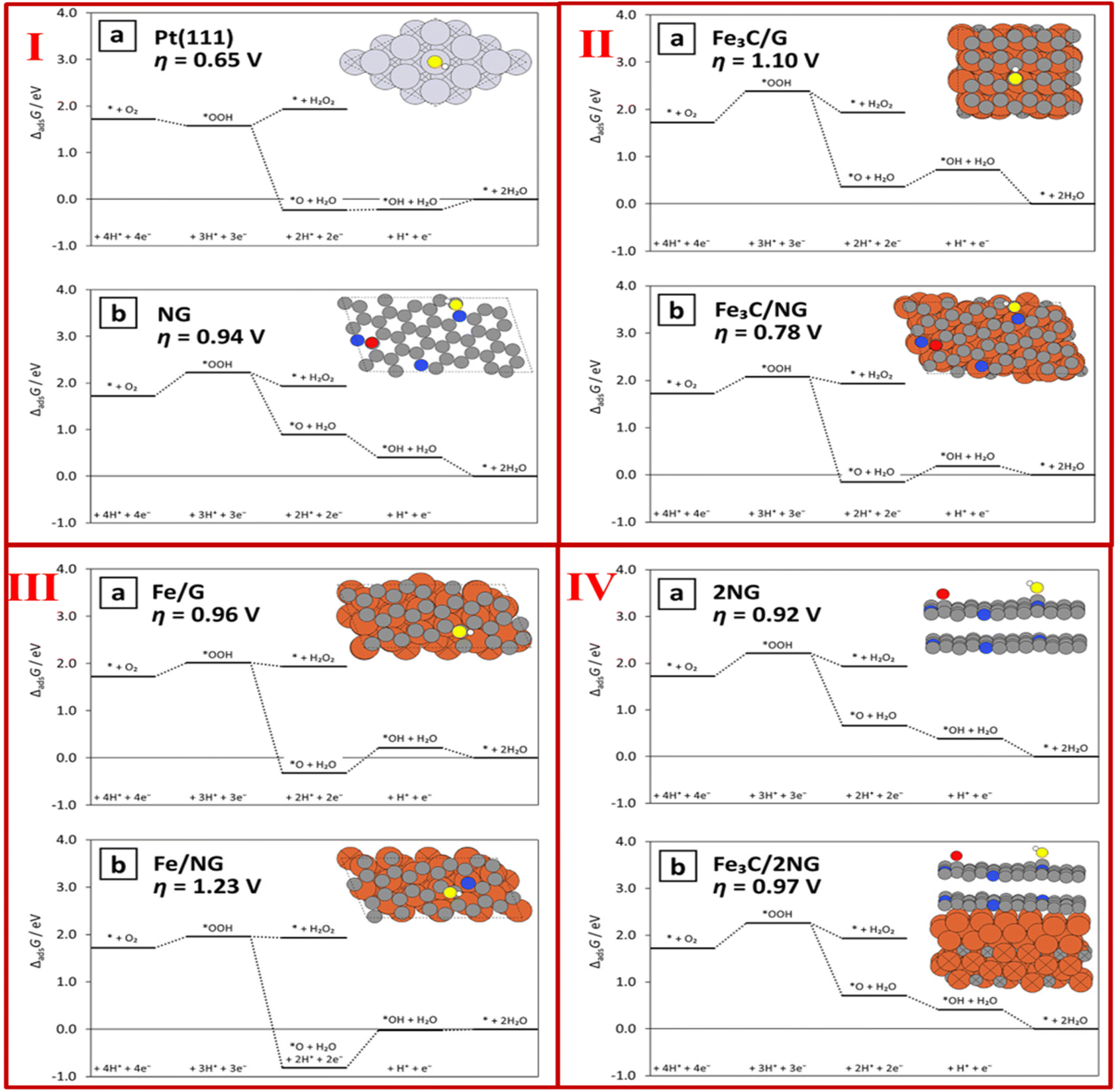 | ||
| Fig. 25 Free energy diagrams for ORR at U = 0.8 V RHE for: (I) (a) Pt (111) and (b) NG, (II) (a) Fe3C/G, (b) Fe3C/NG, and (III) (a) Fe/G, (b) Fe/NG, (IV) (a) NG with two NG layer, and (b) Fe3C/NG with two NG layers with simulation cells shown in inset with OH intermediates bound to active site with different colour codes for different atom simulation cells shown in the inset. Reprinted with permission from ref. 231. Copyright 2018, the American Chemical Society. | ||
Similarly, another study regarding the DFT investigation of an MXene-based NG heterostructure was carried out by Chen et al. In this report, the M-N4-Gr/V2C heterostructure was employed as the model for the DFT investigation of the ORR.233 Eight different metals (M = Ti, Cr, Mn, Fe, Co, Ni, Cu, and Zn), together with NG and V2C were explored. All the samples except Zn-N4-Gr/V2C were found to be thermodynamically stable. Among the 8 samples, the Ni- and Co-based heterostructures showed potential better ORR activity with a theoretical onset potential of 0.32 and 0.45 V, respectively. In this report, four N atoms were used for graphene modification and the metal atom was placed at the centre of the N atoms, as depicted in Fig. 26I. The free energy of adsorption of the different intermediates formed during the mechanistic reduction steps on the different MXene/NG/V2C hybrid catalysts at zero electrode potential, equilibrium potential and working potential determined by DFT is depicted in Fig. 26II. It was observed that the strong binding of O hinders the further reaction. According to the active volcano map, heterostructure stability, electronic structure of the transition metals, electrical conductivity and adsorption energy of the intermediates, it was observed that Ni-N4G/V2C has the highest potential to act as an ORR catalyst with an overpotential of 0.32 V and good volcanic curve of d-band centre with potential. The incorporation of V2C was considered to be responsible for the improved conductivity due to the strong electron jump at the Fermi energy level. Thus, the DFT investigations showed that the free energies of adsorption of the different intermediates play a vital role in predicting the efficiency of ORR catalysts. The higher the interaction and adsorption energy of the different intermediates, the lower the ORR activity.
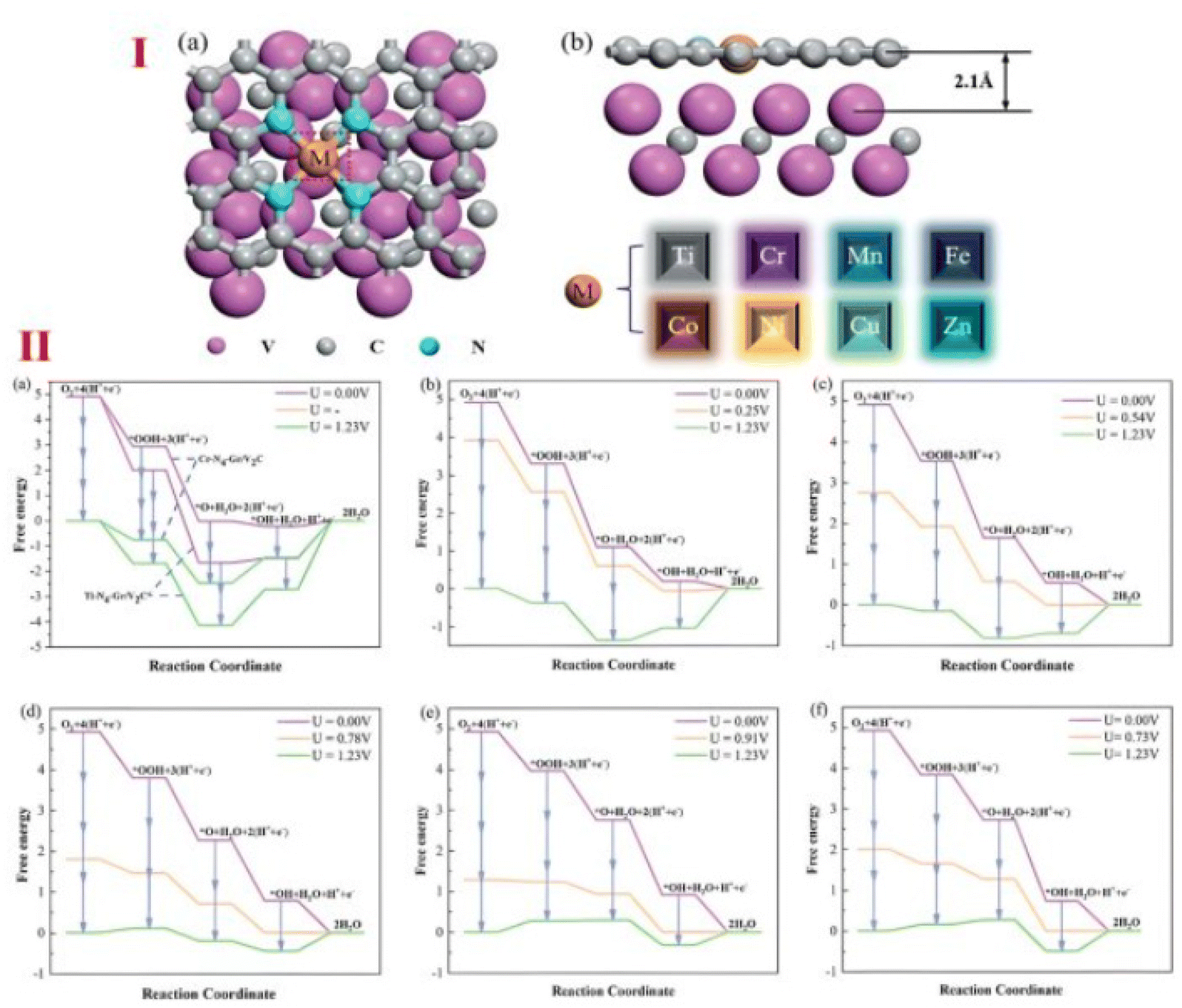 | ||
| Fig. 26 I (a) Top view and (b) side view of M-doped N-coordinated graphene supported by V2C/MXene and interlayer distances, where pink, grey and cyan balls represent V, C and N atoms, respectively. ORR intermediate free energy diagram on II (a) Ti-N4-Gr/V2C, Cr-N4-Gr/V2C, (b) Mn-N4-Gr/V2C, (c) Fe-N4-Gr/V2C, (d) Co-N4-Gr/V2C, (e) Ni-N4-Gr/V2C and (f) Cu-N4-Gr/V2C surfaces at (zero, working and equilibrium electrode potential represented by purple, orange and green lines, respectively). Reprinted with permission from ref. 232. Copyright 2022, The Royal Society of Chemistry. | ||
In another report, a rational design approach was proposed for enhancing the electrocatalytic sites and electron-transport behaviours of CuMo2ON by encasing it in NC shells and anchoring it on an NG matrix.234 The catalytic activity of CuMo2ON@NG was attributed to the synergistic electron transfer observed between the active CuMo2ON nanoparticles, doped N species, and graphitic carbon, as demonstrated by first-principles DFT calculations. The generation of O* intermediates on the CuMo2ON lattice in the core–shell CuMo2ON@NG nanohybrid was shown to be an important and energetically demanding step that governs the rate of achieving both ORR/OER activities. Initially, the structure optimization was carried out to achieve the optimal catalyst. During the process of pyrolysis, it was observed that the nitrogen (N) atom exhibited partial substitution of oxygen (O) atoms. This substitution led to the emergence of a delocalized electron environment, which was characterized by the density of states (DOS) in the vicinity of the Fermi level. The electron distribution of the metal and basic structure modified by elemental nitrogen insertion simplified the adsorption of reactive hydrogen. The introduction of nitrogen in the Cu/Mo catalyst at the ideal molar ratio resulted in a wide DOS peak in close proximity to the Fermi level. This phenomenon has the potential to improve the electronic conductivity of CuMo2ON@NG. Additionally, it is worth noting that the DOS of CuMo2ON@NG exhibited a significantly elevated charge density around the Fermi level, resulting in a substantial abundance of charge carriers. The DOS distribution of the CuMo2ON@NG material around the Fermi surface is beneficial for enhancing the conductivity. However, superior catalytic activity is not necessarily correlated with superior conductivity. The high conductivity and strong hydrogen adsorption capacity of oxynitride have been found to increase its HER activities. Moreover, it has been proposed that the presence of CuMo2ON@NG may lead to an improvement in OER and ORR activities, which can be attributed to the increased affinity of CuMo2ON@NG towards OH* reactants, particularly during the development of a highly porous network. Similarly, Pham et al. used DFT to investigate the electrocatalytic activity of the graphitic edge and in-plane defects in pyridinic-N-doped single and bilayer graphene for the ORR in alkaline media.235 Compared to the N-doped single-layer graphene, the N-doped bilayer graphene demonstrated superior ORR activity. The 4e− associative ORR mechanism was facilitated by graphitic-N-doped multilayer graphene, with OOH* production serving as the rate-limiting step. The interlayer covalent π–π bonding between the graphene layers in N-doped Bernal bilayer graphene structures strengthened the binding of the intermediate species of the ORR (OOH*, O*, and OH*) in comparison to the N-doped single-layer graphene. Moreover, the Bernal stacking in bilayer graphene can significantly increase the ORR and stability. To study Cu-nitrogen-doped fuel cell cathode catalysis, a novel Cu–N-doped graphene nanocomposite catalyst was developed by Balasooriya et al.236 They utilized the Gaussian software for the DFT computations to examine the ORR on the developed catalyst in low-temperature fuel cells. To explore the properties of the fuel cell, three distinct nanocomposite structures including Cu2–N6/Gr, Cu2–N8/Gr, and Cu–N4/Gr were considered in acidic medium under standard conditions (298.15 K, 1 atm). Based on the findings, each structure was found to be stable in the potential range of 0 and 5.87 V. According to the HOMO–LUMO and Mulliken charge energy calculations, Cu2–N6/Gr and Cu2–N8/Gr are structurally more stable. However, based on the free energy calculations, the Cu2–N8/Gr and Cu–N4/Gr structures enable spontaneous ORR.
Similarly, Bhatt et al.237 investigated the ORR on the surface of M-N2-G (M = Ru, Ti, Mo, Cu, and Nb,) electrocatalysts through DFT computations. The authors examined the adsorption qualities of six intermediates (O2, O, OOH, OH, H2O and H2O2) to provide a clearer explanation for the catalytic efficiency of the M-N2-G electrocatalyst. Moreover, they also calculated the adsorption property of the ORR intermediates on Cu-N2-G in the presence of H2O, OH−, and H3O+. Based on the findings, it was apparent that the most stable adsorption site for the ORR intermediates in the presence of H2O, acid, and base is the top. Furthermore, it was also observed that the adsorption characteristics of the ORR intermediates compared to acid are quantitatively more stable than base. Tian et al.238 developed a heterogeneous CoS/CoO nanocrystalline material with well-defined interfacial structure assisted with N-doped graphene via rational interfacial regulation. Based on their DFT measurements, it can be assumed that the boundary interface between CoO and CoS induces a charge transfer effect, which significantly modulates the electronic structure and improved their electronic conductivity. The DFT investigation showed that compared to pure CoS (100) and CoO (111), a greater density of states was present near the Fermi energy level in the CoS/CoO heterostructure interface, thus inducing metallic characteristic or improved intrinsic conductivity, which can be highly beneficial for improved electrocatalytic performance. In this study, the role of NG as a substrate was investigated and it was observed that the graphene layer acts as a stabilising agent for CoS/CoO heterostructures with improved electrocatalytic performances. Also, the robust interaction between CoS/CoO and N-doped graphene further increases the transfer of electrons and enhances the stability of the catalyst. The synthesized noble-metal free bifunctional CoS/CoO@NGNs as an electrocatalyst offers outstanding electrocatalytic activity and improved stability toward the ORR.
In another study, Zhao et al.239 designed a metal-free heterostructure via the in situ coupling of N-SMCTs with N-rGO nanosheets by controlling the N configurations in the respective component. Their results revealed that the efficient redistribution of electrons at the interface of the heterojunction results in the rapid transfer of electrons, which results in the generation of abundant positive charge on N-SMCTs and increasing electron density on N-rGO. Due to this fact, N-SMCTs@N-rGO presents outstanding ORR/OER. The DFT calculations demonstrated a well organised dual-site mechanism, in which the carbon adjacent to pyridinic-N in N-rGO benefits the adsorption of *O/*OH. However, the carbon adjacent to graphitic-N in N-SMCTs facilitates the adsorption of *OOH, thus achieving the lowest possible calculated overpotential values of 0.28 eV and 0.47 eV for the OER and ORR, respectively.
The catalytic activity and stability of N-doped graphene reinforced on a metal-iron carbide heterostructure, i.e., Co–Zn, Ta–W, M = Cr–Mn and Nb–Mo, towards the ORR was studied by Patniboon et al. with the help of DFT calculations.240 The metal-iron carbide heterostructure was developed by encapsulating iron carbide on N-doped graphene by the substitution of the Fe atoms in Fe3C near the interface of NG/Fe3C with the metal. The DFT calculations revealed that the incorporation of the metal atom significantly affected the work function of the overlayer of N-doped graphene, which is largely correlated with the binding strength of the ORR intermediate. The NG/Fe3C heterostructure displayed enhanced ORR activity by the introduction of Co or Ni atoms around the interface, thus stabilizing the heterostructure. In this report, the authors focused on the energetically more favourable associative mechanism (given below) compared to the dissociative mechanism. As in the case of the dissociative mechanism, the dissociative barrier for O2 dissociation at the NG active sites is very high (1.20 eV), which is unlikely to occur at room temperature.
| * + O2 + H+ + e− → *OOH |
| *OOH + H+ + e− → *O + H2O |
| *O + H+ + e− → *OH |
| *OH + H+ + e− → * + H2O |
The DFT investigation showed that the presence of a new metal near the NG/Fe3C interface significantly alters the free energy of adsorption of the different ORR intermediates, thus resulting in different activities corresponding to a particular metal. Among the different metals used in place of Fe near the NG/Fe3C interface, it was observed that the Co and Ni atoms show more prominent enhanced ORR activity compared to other metals. The effect of an increase in metal concentration was also evaluated and it was observed that with an increase in concentration, the binding energy of different intermediates of the ORR can be tuned, thus reducing the overall overpotential of the ORR. Therefore, based on all these investigations, it can be concluded that DFT investigation plays a vital role in predicting the mechanism of the ORR by measuring the adsorption free energies of the different intermediates formed during the reaction. However, for further accurate results, many other reaction parameters (free N in the case of NG or presence of free metal ions) affecting the reaction pathway or interaction of activation sites with reactants need to be included during the measurements.
6.2. Theoretical DFT investigation of HER activity of NG-based heterostructures
To improve the development of clean hydrogen energy, it is important to investigate the composition and structures of electrocatalysts with high performance for the HER. Recently, Zhang et al. presented a report based on first-principles study over W2C and N-doped graphene heterostructures for improved HER performance.241 Several catalytic hetero-structure models were proposed using DFT, which included combined W2C and graphene-like models, with particular focus on N-doped graphene-like models (GN, P3N, and P2V4N). Initially, to compare the HER properties on bulk W2C (W2C (001) surface) with G-like models (G, GN, P3N, and P2V4N), theoretical calculations were conducted. Then, the HER performance of small W2C (W2C and W4C2) NPs on G-like structures was determined. The HER characteristics of W2C and W4C2 NPs on G-like models were estimated to understand the comparatively small W2C with a diameter of 0.2 to 0.5 nm. Moreover, the bulk W2C combined with G-like structures also investigated to estimate their high HER performance mechanisms. Finally, the HER characteristics of the above-mentioned models under the entire pH range (0–14) were evaluated, providing a theoretical basis for W2C-based catalysts under varying experimental conditions. To deduce the mechanism of the HER, the formation energies (Eformation) of all the heterogenous models were calculated, in which the Eformation of the W4C2@G-like model group (W4C2@G, W4C2@GN, W4C2@P3N, W4C2@P2V4N-1 and W4C2@P2V4N-2) was more stable than the other groups of models (W2C@G-like and G-like@W2C groups). On comparing the Eformation of the W2C@G-like and W4C2@G-like groups, it appears that the bigger W2C NPs exhibit a higher affinity for binding to the surfaces of the G-like models (G, GN, P3N, and P2V4N). To investigate the potential HER mechanism, the hetero-structure models W2C@GN, W4C2@P2V4N-2, P3N@W2C-2, and P2V4N@W2C-2 are energetically preferable. The adsorption of a second hydrogen atom on the surface of W2C@GN exhibits a high degree of ease, suggesting that the HER process on this surface likely follows the Volmer-Tafel mechanism. Alternatively, second H adsorption on the surface of W4C2@P2V4N-2, P3N@W2C-2, and P2V4N@W2C-2 shows greater difficulty, indicating that the HER process on these surfaces probably complies with the Vomer–Heyrovsky mechanism. The total density of states (TDOS) of all the proposed individuals and heterostructures models were calculated. The incorporation of N atoms and vacancies in graphene can lead to an increase in the TDOS in close proximity to the Fermi level (Ef). Consequently, this modification has potential to enhance the conductivity of graphene. The hybridization of electron orbitals C-p, W-d, and N-p is a significant factor in governing the variations in ΔGH in the proposed W2C and G-like coupled heterostructures, particularly in relation to the active sites. Also, the impact of pH on ΔGH in the vicinity of the electrode/electrolyte interface has also been examined for these heterostructures. The impact of pH on the HER performance of all the sites included in the proposed models is displayed in Fig. 27. The results revealed that the heterostructure consisting of N-modified graphene-like structures and W2C includes catalytically active sites with ΔGH = 0 eV, regardless of the acidity level (acidic, neutral or alkaline). This suggest that the introduction of nitrogen doping allows these heterostructures to maintain a high level of catalytic activity through a broader pH range. Thus, this report offers a theoretical basis for the design and generation of catalysts that demonstrate superior performances in the HER process.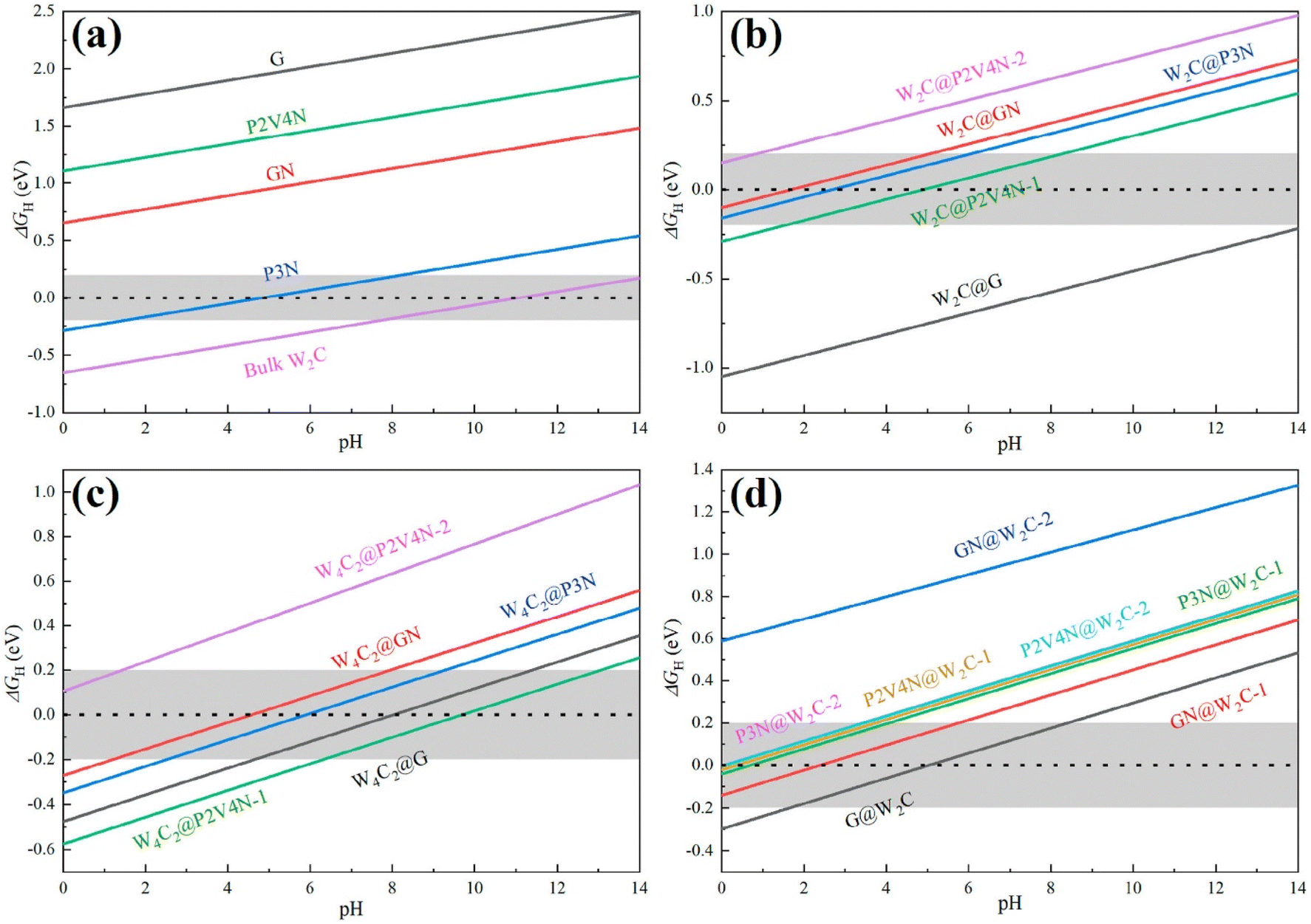 | ||
| Fig. 27 pH-dependent H adsorption Gibbs free energy ΔGH. (a) Bulk W2C and G-like models, (b) W2C@G-like models, (c) W4C2@G-like models, and (d) G-like@W2C models. Area in grey (HER preferable area) −0.1 to 0.2 eV. Reprinted with permission from ref. 236. Copyright 2021, Elsevier. | ||
Several researchers also investigated MXenes with versatile chemistry and high electrical conductivity and found that they are breakthrough materials for energy storage and catalysis. In a recent report, Zhou et al. proposed heterostructures of N-doped graphene supported by MXene monolayers as bifunctional electrocatalysts for the ORR and HER, inspired by recent hybridization experiments of MXenes with carbon materials.242 The first principles study showed that the graphitic sheet on V2C and Mo2C MXenes is very active, with an ORR overpotential as low as 0.36 V and reaction free energies for the HER that are close to zero and low kinetic barriers. The electrical coupling between the graphitic sheet and MXene, pz band center of the carbon atoms on the surface, and the work function of the heterostructures are responsible for the excellent catalytic properties. The heterostructures formed by combining graphene with MXene exhibit interlayer distances ranging from 2.13 to 2.40 Å, as well as binding energies ranging from −0.42 to −0.17 per carbon atom in graphene. Due to the presence of robust interfacial coupling, the significant transfer of electrons was observed from MXene to the graphitic sheet. This transfer of electrons would occupy C pz orbitals, hence causing a disruption in the conjugation of graphene. The total strength of oxygen binding was observed to follow the sequence of G/V2C > G/Mo2C > G/Nb2C > G/Ti2C. The binding energies and ORR overpotentials of the graphene/MXene hybrids are influenced by the structural environment of the surface C atoms. The graphitic sheet of heterostructures has a non-uniform electron density distribution due to electron transport from MXene to the surface C atoms and C to N atoms. The carbon atoms with lower electron density bind oxygen intermediates efficiently. In contrast, MXene C atoms on the top site for chemical bonds with the metal atoms below them and gain more electrons than that on the hollow site, binding the oxygen intermediates weakly. The hollow-site C atoms in G/V2C and G/Mo2C creating many active sites and C atoms near the pyridinic N dopants had the lowest overpotentials, i.e., 0.36 and 0.39 V for G/V2C and G/Mo2C, respectively. The G/Nb2C and G/Ti2C systems show weaker binding strengths and overpotentials of above 0.54 and 0.64 V, respectively. The increased surface reactivity of graphene/MXene heterostructures is also advantageous for HER catalysis. The strong binding of C atoms with oxygen intermediates also promotes the adsorption of H* species, as indicated by the linear relationships between the binding energies of the different reaction intermediates. The heterostructures G/V2C and G/Mo2C exhibit moderate binding strength for H* adsorption, with the adsorption free energy being approximately in equilibrium with that of gaseous H2. The C atoms near the hollow-site of the V2C substrate displayed ΔGH* ranging from −0.04 to 0.17 eV, making them suitable for catalysing the HER. The hollow-site C atoms near the pyridinic and graphitic N dopants showed the highest activity with ΔGH* values of −0.04 and 0.04 eV, respectively. These values are comparable to the activity of Pt, which has a ΔGH* value of −0.10 eV. In the case of G/Mo2C, which is less reactive than G/V2C, only a few hollow-site C atoms proximal to the N dopants can offer appropriate binding strength for HER catalysis with ΔGH* 0.05–0.26 eV. The H* binding energy is too weak for HER catalysis with ΔGH* greater than 0.2 eV in the case of G/Nb2C and G/Ti2C. Further, the kinetic process for the HER was also studied for the above-mentioned heterostructures through the Tafel mechanism, which includes the desorption of two H* molecules and formation of an H2 molecule. The reaction barriers for G/V2C and G/Mo2C are 1.33 and 1.56, respectively, which are higher than Pt and comparable to the values of the MoS2 edges.
In comparison to the tremendous advances in electrical and optoelectrical applications, there are very few papers on electrocatalysis by metal-free exfoliated black phosphorus (EBP)s. For example, Zhonge et al. fabricated processable ultrathin EBP nanosheets with a higher Fermi level of N-doped graphene into a new metal-free 2D/2D heterostructure (EBP@NG) with well-designed interfaces and a unique electronic configuration as efficient and durable bifunctional catalysts towards the HER and OER. The synergy of EPG and NG was completely exploited by rational interface engineering, which not only increased the stability of EBP but also efficiently adjusted the electrical structures of each component to boost their intrinsic activities. Because EBP has a lower Fermi level than NG, their electronic interaction caused directional interfacial electron transfer, which not only enriched the electron density on EBP and optimized the H adsorption/desorption for the HER, but also incorporated abundant positively charged carbon sites on NG and favoured the formation of key OER intermediates (OOH*) to improve the OER energetics. Thus, although pure EBP or NG alone has weak or insignificant activity, EBP@NG exhibited significantly improved HER/OER activities.243
Similarly, in another report, N-doped graphene-encapsulated Ni NPs combined with MoO2 nanosheets self-supported on 3D nickel foam were investigated theoretically and experimentally to confirm the electron transfer from Ni to NG at the interfaces, which further enhanced the HER performance.244 The DFT calculations were performed by constructing a correlative theoretical model of Ni(111)@NC with an N-doped carbon layer covering the Ni (111) surface. It is important to note that the configurations where H is adsorbed at the bridge or hollow site could not be achieved, as H readily moves to the corresponding top site. Four primary sites were identified including ortho-, meta-, and para-carbon atoms, which were referred to as o-TC, m-TC, and p-TC, respectively, together with a nitrogen atom denoted as TN, as shown in Fig. 28(a). The o-TC and p-TC sites displayed the lowest ΔGH* value, indicating their superior HER catalytic activity. The main reason for this is the positioning of the ortho- and para-carbon atoms on the Ni–Ni–Ni hollow sites. The m-TC site is positioned above the Ni atom on the Ni(111) surface. This arrangement decreases the interatomic distance, leading to a more robust bonding interaction between the Ni and C atoms in m-TC compared to o-TC and p-TC. Consequently, during H attack, the ortho- and para-carbon atoms exhibit a greater tendency to deviate from the NC plane to facilitate their reaction with H. The computed ΔGH* results predict that this leads to an improvement in the HER activity. Hence, the ortho- and para-carbon atoms exhibit high reactivity, making them ideal sites for the HER and enhancing the catalytic activity. As depicted in Fig. 28(b), the o-TC and p-TC sites can also have substantially lower ΔGH* values than pure NC (0.615 eV) or Ni(111) (0.390 eV), indicating that the hybrid material has stronger catalytic activity for the HER. The charge density difference (Δρ) for the Ni(111)@NC model was also subjected to correlative computation. Fig. 28(c) demonstrates that the Ni(111) surface can undergo a complex electron transfer process (Ni → C → N) when the N-doped carbon layer is applied. In this process, the Ni atoms act as electron donors, N atoms act as electron acceptors, and the C atoms are in closer proximity to the N atom. This electron transfer process effectively activates the corresponding carbon atoms in the NC layer, resulting in hybrid systems that exhibit excellent catalytic activity for the hydrogen evolution reaction (HER).
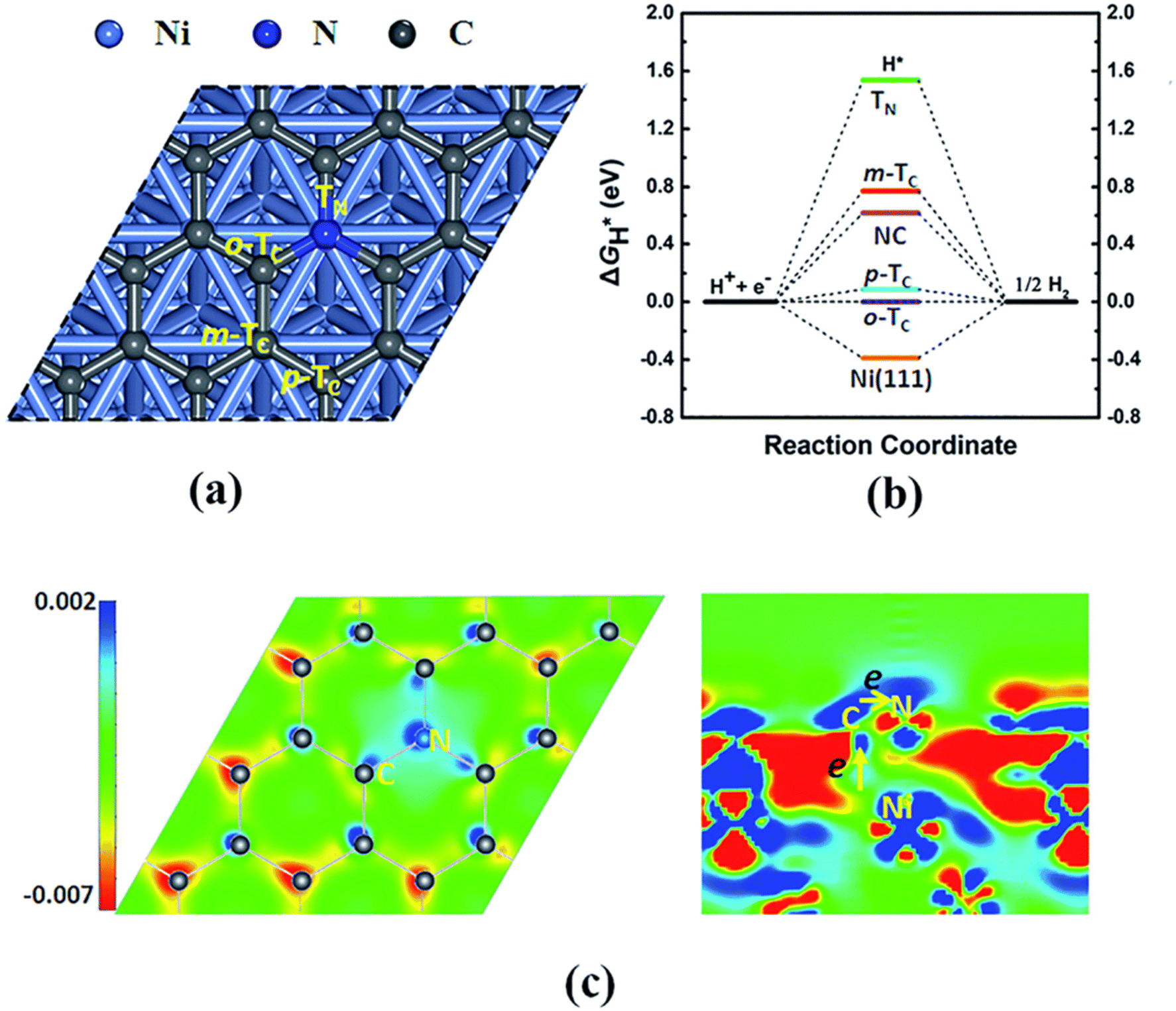 | ||
| Fig. 28 Typical electron transfer process Ni → C → N. (a) Theoretical model used in DFT calculations and obtained adsorption site of H* on model surface. (b) Computed free energies for adsorption sites. (c) Charge density difference of the Ni(111)@NC model, where red and blue areas signify low and high electron density, respectively. Reprinted with permission from ref. 239. Copyright 2020, The Royal Society of Chemistry. | ||
7. Conclusions, perspectives and challenges
For the development of sustainable green energy electrochemical devices such as fuels cells, metal–air batteries and electrolytic cells have attracted tremendous attention. In all electrochemical devices, different reaction such as the HER/OER and ORR under different electrolytic conditions occur in the presence of an electrocatalysts. For each reaction, different set of characteristic electrocatalysts have been developed. However, the most active and efficient electrocatalysts developed to date for these processes are noble metal-based catalyst materials, which are very expensive to employ in practical applications. Therefore, carbon-based NG has been explored to overcome the limitations (high cost, poor long-term stability, methanol intolerance for the ORR, etc.) of noble metal-based electrocatalysts. Due to their important characteristics such as superior conductivity, high stability, and high surface area, graphene-based materials have been extensity explored by different researchers as HER, OER and ORR catalyst materials. Herein, we mainly focused on heteroatom-doped graphene-based hetero-structured catalysts and discussed the recent advancements in synthesis strategies (such as hydrothermal, CVD, thermal treatment and other methods) to develop a universal, simple, cost-effective, energy-efficient and environmentally benign approach for the synthesis of NG heterostructures. Then, we attempted to provide a comprehensive, focused understanding of the effect of heteroatom doping and heterostructure development of active sites and activity of graphene-based electrocatalysts. In addition, the multiple functionalities of graphene to promote the ORR and HER/OER activity of hetero-structured electrocatalysts were discussed. The underlying mechanism and active sites responsible for the better performance of NG-based heterostructures was discussed in detail, which showed that the pyridinic N and C atoms adjacent to it play a significant role in rapid electron transport due to the difference in their electronegativity, which is further enhanced by the development heterojunctions. Meanwhile, further investigations are being carried out to improve the density of catalytic active sites to obtain electrocatalysts that can replace noble metal-based catalysts for practical applications.Although significant advancements have been achieved in the application of graphene either as a support material for active catalysts or an active catalyst for the OER and HER/ORR, there are still different challenges that need to be addressed. Firstly, the real catalytic active sites responsible for the boosted electrocatalytic activity are still unclear. During electrochemical investigation under different electrolytic conditions, the as-developed NG-based heterostructures may undergo significant structural, morphological and electronic changes, which may result in a change in the nature of the catalytic active sites, and therefore the understanding of the exact behaviour of the active sites under this condition is highly difficult. To avoid these difficulties, the development of in situ operando characterization techniques together with electrochemical workstation-coupled electron microscopy is required to investigate and unravel the evolution of the structural changes of the catalytic active sites, which can be highly beneficial for the design of efficient electrocatalysts. Secondly, for the ORR, the catalysts suffer from methanol poisoning, while for the HER/OER, the graphene-based network undergoes a performance degradation during the electrochemical investigation. Due to the harsh electrolyte conditions, the carbon atoms undergo oxidation, resulting in the formation of CO2 and the structural collapse of the carbon network and deterioration of the catalytic performances. This deterioration is usually caused by the presence of defects, and therefore it is imperative to design catalyst systems with a controlled defect density and preserved catalytic active sites and long-term stability. Thirdly, another challenge associated with graphene-based hetero-structured catalysts is the absence of a universal cost-effective synthesis approach. Given that the different methods used for the synthesis of graphene-based materials produce different physicochemical characteristics, during the application analysis, different graphene samples pose a barrier to reproduce high performances by HER/ORR electrocatalysts. Therefore, it is necessary to have a unified standard to analyse the properties of graphene-based samples, which can result in the development of an approach for the continuous measurement and monitoring of electrochemical activity. Fourthly, the important challenge faced by NG-based heterostructure catalysts is the discrepancy between theoretically and experimentally measured HER/ORR activities. The deviation from theoretically measured values is still not clear, and therefore further investigations are needed to achieve close to theoretical performance by NG-based catalysts. DFT measurements have been used to investigate the effect of active sites on the mechanistic steps of different electrocatalytic reactions; however, in the case of hetero-structured NG catalyst materials, multiple active sites are responsible for the better performances, and hence it is very difficult to deduce the exact mechanism of the reaction steps taking place on heteroatom-doped graphene heterostructure catalysts.
However, despite all the above-mentioned challenges, NG based hetero-structured catalysts have attracted significant attention from researchers worldwide for the development of a better combination of NG and other active catalysts for enhanced HER/OER/ORR performances. These continuous efforts have resulted in the development of NG heterostructures that outperform the ORR activity of Pt/C in terms of activity and methanol tolerance stability. However, we believe that a better understanding of the mechanism and electron transfer process between the different components of NG heterostructures can help in further improving the electrocatalytic efficiencies to achieve the dream of cost-effective sustainable energy. Further, the detailed understanding of the structural and electronic properties and electronic interactions taking place between the different components present in the heterostructure will be highly beneficial to provide an enhanced output performance by developing favourable multiple interfaces. Moreover, these multiple interfaces may provide multifunctionalities, which can aid in the development of multifunctional catalyst materials that can simultaneously act as an efficient electrode material for the HER, OER and ORR. We believe that there are many other fascinating features of graphene-based materials that need to be explored and utilised for efficient catalytic performances. Thus, we consider that this article will trigger interest among researchers and readers to further explore the effect of heteroatom doping and heterostructure development on the catalytic performance of graphene-based materials and to understand the role of the active sites in determining the final mechanism of the reaction steps involved in the electrocatalytic processes.
Author contributions
Reena Saini (R. S.), Farha Naaz (F. N.) and Umar Farooq (U. F.) have contributed equally to this manuscript. Reena Siani and Farha Naaz contributed to writing of original draft which includes data collection and writing, the conceptualization and writing of the article was done by U. F. Ali H. Bashal and Ashiq Hussain Pandit have contributed to final editing and preparation of revised manuscript.Conflicts of interest
The authors declare no financial competing and no conflict of interest.Acknowledgements
Authors Reena Saini, Umar Farooq of this review article highly acknowledge School of Basic Sciences, Chemistry Division, Galgotais University, Greater Noida-India, for providing a platform and facilities to carryout research. Author Farha Naaz acknowledges Nanochemistry Laboratory for energy applications, Department of Chemistry, Jamia Millia Islamia, New Delhi-India. Ali H. Bashal is highly thankful to Department of Chemistry, Taibah University, Yanbu for providing the research facilities and fundings for research activities. Ashiq Hussain pandit is highly thankful to department of Chemistry Jamia Millia Islamia, New Delhi for providing research facilities to carryout his research.References
- W. Li, M. Sohail, U. Anwar, T. A. Taha, A. G. Al-Sehemi, S. Muhammad and Z. Ajmal, Int. J. Hydrogen Energy, 2022, 47, 21067–21118 CrossRef CAS.
- A. Hayat, M. Sohail, J. Ali Shah Syed, A. G. Al-Sehemi, M. H. Mohammed, A. A. Al-Ghamdi, T. A. Taha, H. S. AlSalem, A. M. Alenad, M. A. Amin, A. Palamanit, C. Liu, W. I. Nawawi, M. T. S. Chani and M. M. Rahman, Chem. Rec., 2022, 22, 202100310 CrossRef PubMed.
- U. Farooq, R. Phul, S. M. Alshehri, J. Ahmed and T. Ahmad, Sci. Rep., 2019, 9, 4488 CrossRef.
- C. S. Blázquez, D. Borge-Diez, I. M. Nieto, A. F. Martín and D. González-Aguilera, Renewable Energy, 2021, 163, 1682–1691 CrossRef.
- G. K. Karayel, N. Javani and I. Dincer, Energy, 2022, 249, 123597 CrossRef CAS.
- G. A. Kumar and Shivashankar, Int. J. Energy Environ. Eng., 2022, 13, 77–103 CrossRef.
- W. Dong, G. Zhao, S. Yüksel, H. Dinçer and G. G. Ubay, Renewable Energy, 2022, 85, 321–337 CrossRef.
- M. Usman and M. Radulescu, Sci. Total Environ., 2022, 841, 156662 CrossRef CAS.
- U. K. Pata and A. Samour, Prog. Nucl. Energy, 2022, 149, 104249 CrossRef.
- R. Lin, J. Xu, M. Wei, Y. Wang, Z. Qin, Z. Liu and H. Tan, Nature, 2022, 603, 73–78 CrossRef CAS.
- A. G. Olabi, Q. Abbas, A. Al Makk and M. A. Abdelkareem, Energy, 2022, 248, 123617 CrossRef CAS.
- R. Dickson, M. S. Akhtar, A. Abbas, E. D. Park and J. Liu, Green Chem., 2022, 24, 8484–8493 RSC.
- S. Y. Obara, Energy, 2019, 174, 848–860 CrossRef CAS.
- O. Oner and K. Khalilpour, Renewable Sustainable Energy Rev., 2022, 168, 112764 CrossRef CAS.
- J. S. Lee, A. Cherif, H. J. Yoon, S. K. Seo, J. E. Bae, H. J. Shin and C. J. Lee, Renewable Sustainable Energy Rev., 2022, 165, 112556 CrossRef CAS.
- J. Scharf, M. Kübler, V. Gridin, W. D. Z. Wallace, L. Ni, S. D. Paul and U. I. Kramm, SusMat, 2022, 2, 630–645 CrossRef CAS.
- H. Z. He, Y. Zhang, Y. Li and P. Wang, Int. J. Hydrogen Energy, 2021, 46, 7848–7865 CrossRef CAS.
- B. Devi, R. R. Koner and A. Halder, ACS Sustainable Chem. Eng., 2018, 7, 2187–2199 CrossRef.
- F. Gao, Y. Zhang, Z. Wu, H. You and Y. Du, Coord. Chem. Rev., 2021, 436, 213825 CrossRef CAS.
- S. D. Bhoyate, J. Kim, F. M. de Souza, J. Lin, E. Lee, A. Kumar and R. K. Gupta, Coord. Chem. Rev., 2023, 474, 214854 CrossRef CAS.
- T. Noor, L. Yaqoob and N. Iqbal, ChemElectroChem, 2021, 8, 447–483 CrossRef CAS.
- U. Farooq, T. Ahmad, F. Naaz and S. U. Islam, Energy Fuels, 2023, 37, 1577–1632 CrossRef CAS.
- U. Farooq, A. H. Pandit and R. Phul, Environ. Nanotechnol. Water Purif., 2020, 187–216 CAS.
- U. Farooq, J. Ahmed, S. M. Alshehri, Y. Mao and T. Ahmad, ACS Omega, 2022, 7, 16952–16967 CrossRef CAS.
- U. Farooq, F. Naz, R. Phul, N. A. Pandit, S. K. Jain and T. Ahmad, J. Nanosci. Nanotechnol., 2020, 20, 3770–3779 CrossRef CAS PubMed.
- Z. Li, Y. Li, L. Wang, L. Cao, X. Liu, Z. Chen and M. Wu, Electrochim. Acta, 2017, 235, 561–569 CrossRef CAS.
- K. Sheoran, V. K. Thakur and S. S. Siwal, Mater. Today: Proc., 2022, 56, 9–17 Search PubMed.
- R. Tjandra, W. Liu, M. Zhang and A. Yu, J. Power Sources, 2019, 438, 227009 CrossRef CAS.
- Y. C. Hsiao, J. L. Hung, S. Kubendhiran, S. Yougbaré, L. Y. Lin and Y. F. Wu, J. Energy Storage, 2022, 56, 105902 CrossRef.
- I. Sadiq, S. A. Ali and T. Ahmad, Catalyst, 2023, 13, 109 CrossRef CAS.
- S. K. Singh, K. Takeyasu and J. Nakamura, Adv. Mater., 2019, 31, 1804297 CrossRef.
- K. Baruah and P. Deb, Nanoscale Adv., 2021, 3, 3681–3707 RSC.
- S. Choi, C. Kim, J. M. Suh and H. W. Jang, Carbon Energy, 2019, 1, 85–108 CrossRef CAS.
- J. Li, Z. Zhao, Y. Ma and Y. Qu, ChemCatChem, 2017, 9, 1554–1568 CrossRef CAS.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS.
- D. Geng and H. Y. Yang, Adv. Mater., 2018, 30, 1800865 CrossRef.
- H. Huang, M. Yan, C. Yang, H. He, Q. Jiang, L. Yang and Y. Yamauchi, Adv. Mater., 2019, 31, 1903415 CrossRef CAS.
- J. Jiang, F. Meng, Q. Cheng, A. Wang, Y. Chen, J. Qiao and L. Han, Small Methods, 2020, 4, 2000238 CrossRef CAS.
- T. Z. Hou, X. Chen, H. J. Peng, J. Q. Huang, B. Q. Li, Q. Zhang and B. Li, Small, 2016, 12, 3283–3291 CrossRef CAS.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- Y. Tang, C. Yang, M. Sheng, X. Yin and W. Que, ACS Sustainable Chem. Eng., 2020, 8, 12990–12998 CrossRef CAS.
- J. Linnemann, K. Kanokkanchana and K. Tschulik, ACS Catal., 2021, 11, 5318–5346 CrossRef CAS.
- R. Jiang, S. O. Tung, Z. Tang, L. Li, L. Ding, X. Xi, Y. Liu, L. Zhang and J. Zhang, Energy Storage Mater., 2018, 12, 260–276 CrossRef.
- S. Zhao, L. Yan, H. Luo, W. Mustain and H. Xu, Nano Energy, 2018, 47, 172–198 CrossRef CAS.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., 2021, 133, 17976–17996 CrossRef.
- T. Ahmad, U. Farooq and R. Phul, Ind. Eng. Chem. Res., 2018, 57, 18–41 CrossRef CAS.
- S. T. Oyama, T. Gott, H. Zhao and Y. K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- X. Zhang, J. Shao, C. Yan, R. Qin, Z. Lu, H. Geng and L. Ju, Mater. Des., 2021, 200, 109452 CrossRef CAS.
- S. Manzeli, D. Ovchinnikov, D. Pasquier, O. V. Yazyev and A. Kis, Nat. Rev. Mater., 2017, 2, 1–15 Search PubMed.
- S. Bolisetty, M. Peydayesh and R. Mezzenga, Chem. Soc. Rev., 2019, 48, 463–487 RSC.
- D. Higgins, P. Zamani, A. Yu and Z. Chen, Energy Environ. Sci., 2016, 9, 357–390 RSC.
- N. Talukder, Y. Wang, B. B. Nunna and E. S. Lee, Carbon, 2021, 185, 198–214 CrossRef CAS.
- A. Wu, Y. Xie, H. Ma, C. Tian, Y. Gu, H. Yan and H. Fu, Nano Energy, 2018, 44, 353–363 CrossRef CAS.
- J. Liang, Q. Yu, X. Yang, T. Zhang and J. Li, Nano Res., 2018, 11, 1599–1611 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- P. Krauspe, D. Tsokkou, E. Buchaca-Domingo, Z. Fei, M. Heeney, N. Stingelin and N. Banerji, J. Mater. Chem. A, 2018, 6, 22301–22309 RSC.
- L. Najafi, S. Bellani, B. Martín-García, R. Oropesa-Nuñez, A. E. Del Rio Castillo, M. Prato and F. Bonaccorso, Chem. Mater., 2017, 29, 5782–5786 CrossRef CAS.
- B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 RSC.
- T. A. Shifa, F. Wang, Z. Cheng, X. Zhan, Z. Wang, K. Liu and J. He, Nanoscale, 2015, 7, 14760–14765 RSC.
- Y. J. Tang, M. R. Gao, C. H. Liu, S. L. Li, H. L. Jiang, Y. Q. Lan and S. H. Yu, Porous molybdenum–based hybrid catalysts for highly efficient hydrogen evolution, Angew. Chem., 2015, 127, 13120–13124 CrossRef.
- H. Zhou, F. Yu, J. Sun, R. He, Y. Wang, C. F. Guo and S. Chen, J. Mater. Chem. A, 2016, 4, 9472–9476 RSC.
- S. Zhang, X. Yu, F. Yan, C. Li, X. Zhang and Y. Chen, J. Mater. Chem. A, 2016, 4, 12046–12053 RSC.
- D. M. Fernandes, M. P. Araujo, A. Haider, A. S. Mougharbel, A. J. Fernandes, U. Kortz and C. Freire, ChemElectroChem, 2018, 5, 273–283 CrossRef CAS.
- R. Ye, D. K. James and J. M. Tour, Adv. Mater., 2019, 31, 1803621 CrossRef PubMed.
- L. Qu, Y. Liu, J. B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS.
- X. Li, C. W. Magnuson, A. Venugopal, R. M. Tromp, J. B. Hannon, E. M. Vogel and R. S. Ruoff, J. Am. Chem. Soc., 2011, 133, 2816–2819 CrossRef CAS.
- H. Huang, L. Ma, C. S. Tiwary, Q. Jiang, K. Yin, W. Zhou and P. M. Ajayan, Small, 2017, 13, 1603013 CrossRef.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- A. Nasir, S. Khalid, T. Yasin and A. A. Mazare, Energies, 2022, 15, 6248 CrossRef CAS.
- T. San Nwe, Doctoral dissertation, Prince of Songkla University, 2019.
- C. Liu, L. Zhang, R. Liu, Z. Gao, X. Yang, Z. Tu and Y. Li, J. Alloys Compd., 2016, 656, 24–32 CrossRef CAS.
- B. Xie, Y. Chen, M. Yu, T. Sun, L. Lu, T. Xie and Y. Wu, Carbon, 2016, 99, 35–42 CrossRef CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS.
- Y. Pei, T. Fan, H. Chu, Y. Ge, Y. Yang, P. Dong and J. Shen, J. Alloys Compd, 2017, 724, 1014–1022 CrossRef CAS.
- Z. Jiang, Y. Lei, Y. Lin, J. Hu and Z. Ouyang, Ceram. Int., 2020, 46, 15801–15811 CrossRef CAS.
- S. Kang, J. Jang, S. H. Ahn and C. S. Lee, Dalton Trans., 2019, 48, 229 Search PubMed.
- G. Zhou, Y. Chen, H. Dong, L. Xu, X. Liu, C. Ge and Y. Tang, Int. J. Hydrogen Energy, 2019, 44, 26338–26346 CrossRef CAS.
- L. Wang, B. Li, Z. You, A. Wang, X. Chen, G. Song and C. Chen, Anal. Chem., 2021, 93, 11123–11132 CrossRef CAS.
- J. Shi, M. Liu, J. Wen, X. Ren, X. Zhou, Q. Ji and Y. Zhang, Adv. Mater., 2015, 27, 7086–7092 CrossRef CAS.
- Z. Xia, X. Chen, H. Ci, Z. Fan, Y. Yi, W. Yin and J. Sun, J. Energy Chem., 2021, 53, 155–162 CrossRef CAS.
- H. Tan, Y. Fan, Y. Rong, B. Porter, C. S. Lau, Y. Zhou and J. H. Warner, ACS Appl. Mater. Interfaces, 2016, 8, 1644–1652 CrossRef CAS.
- K. Wang, S. Zhan, D. Zhang, H. Sun, X. Jin and J. Wang, Colloids Surf., A, 2021, 618, 126362 CrossRef CAS.
- N. Wei, J. Cai, R. Wang, M. Wang, W. Lv, H. Ci and Z. Liu, Nano Energy, 2019, 66, 104190 CrossRef CAS.
- S. J. Zhang, S. S. Lin, X. Q. Li, X. Y. Liu, H. A. Wu, W. L. Xu and Z. J. Xu, Nanoscale, 2016, 8, 226–232 RSC.
- X. Feng, J. Ning, D. Wang, J. Zhang, M. Xia, Y. Wang and Y. Hao, J. Alloys Compd., 2020, 816, 152625 CrossRef CAS.
- K. Aydin, S. Woo, V. K. Kanade, S. Choi, C. Ahn, B. Lim and T. Kim, Carbon Energy, 2023, 340 CrossRef.
- C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1902868 CrossRef CAS PubMed.
- J. Pang, R. G. Mendes, P. S. Wrobel, M. D. Wlodarski, H. Q. Ta, L. Zhao and M. H. Rümmeli, ACS Nano, 2017, 11, 1946–1956 CrossRef CAS PubMed.
- B. Chen, Y. Meng, F. He, E. Liu, C. Shi, C. He and N. Zhao, Nano Energy, 2017, 41, 154–163 CrossRef CAS.
- Y. Fan, S. Ida, A. Staykov, T. Akbay, H. Hagiwara, J. Matsuda and T. Ishihara, Small, 2017, 13, 1700099 CrossRef.
- C. Huang, C. Pi, X. Zhang, K. Ding, P. Qin, J. Fu and K. Huo, Small, 2018, 14, 1800667 CrossRef.
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang and R. Fasel, Nat. Nanotechnol., 2014, 9, 896–900 CrossRef CAS.
- Y. Feng, X. Y. Yu and U. Paik, Sci. Rep., 2016, 6, 34004 CrossRef CAS PubMed.
- Y. Zhang, P. Chen, X. Gao, B. Wang, H. Liu, H. Wu and S. Dou, Adv. Funct. Mater., 2016, 26, 7754–7765 CrossRef CAS.
- X. Li, H. Zhang, Q. Hu, W. Zhou, J. Shao, X. Jiang and C. He, Angew. Chem., Int. Ed., 2023, 62, 202300478 CrossRef.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- C. Feng, M. Lv, J. Shao, H. Wu, W. Zhou, S. Qi and C. He, Adv. Mater., 2023, 35, 2305598 CrossRef CAS.
- A. Wang, W. Chen, N. Geng, X. Lan, M. Liu and X. Wu, Sustainability, 2022, 14, 10614 CrossRef CAS.
- S. Xing, J. Yang, M. Muska, H. Li and Q. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 22608–22620 CrossRef CAS PubMed.
- F. Bu, W. Chen, M. F. A. Aboud, I. Shakir, J. Gu and Y. Xu, J. Mater. Chem. A, 2019, 7, 14526–14535 RSC.
- L. Zhang, L. Sun, Y. Huang, Y. Sun, T. Hu, K. Xu and F. Ma, J. Mater. Sci., 2017, 52, 13561–13571 CrossRef CAS.
- Y. Yang, B. Hu, W. Zhao, Q. Yang, F. Yang, J. Ren and Q. Pan, Electrochim. Acta, 2019, 317, 25–33 CrossRef CAS.
- X. Lin, M. Sun, S. Sun and Z. Zhang, J. Alloys Compd., 2019, 785, 732–741 CrossRef CAS.
- P. Li, Q. Chen, Y. Lin, G. Chang and Y. He, J. Alloys Compd., 2016, 672, 497–504 CrossRef CAS.
- H. Wang, X. Wang, D. Yang, B. Zheng and Y. Chen, J. Power Sources, 2018, 400, 232–241 CrossRef CAS.
- X. Li, G. Guo, N. Qin, Z. Deng, Z. Lu, D. Shen and H. E. Wang, Nanoscale, 2018, 10, 15505–15512 RSC.
- Y. Chen, T. Liu, L. Zhang and J. Yu, Appl. Surf. Sci., 2019, 484, 135–143 CrossRef CAS.
- W. Y. Yan, Q. Zhou, X. Chen, X. J. Huang and Y. C. Wu, Sens. Actuators, B, 2016, 230, 761–772 CrossRef CAS.
- J. Ye, Z. Yu, W. Chen, Q. Chen, S. Xu and R. Liu, Carbon, 2016, 107, 711–722 CrossRef CAS.
- L. Cao, B. Zhang, H. Xia, C. Wang, B. Luo, X. Fan and X. Ou, J. Chem. Eng., 2020, 387, 124060 CrossRef CAS.
- X. Yao, G. Guo, Y. Zhao, Y. Zhang, S. Y. Tan, Y. Zeng and Y. Zhao, Small, 2016, 12, 3849–3860 CrossRef CAS.
- P. Liu, Q. Hao, X. Xia, W. Lei, H. Xia, Z. Chen and X. Wang, Electrochim. Acta, 2016, 214, 1–10 CrossRef CAS.
- L. Wang, X. Li, Z. Jin, Z. Liang, X. Peng, X. Ren and K. Huo, Mater. Chem. A, 2019, 7, 27475–27483 RSC.
- Z. Wu, P. Li, Q. Qin, Z. Li and X. Liu, Carbon, 2018, 139, 35–44 CrossRef CAS.
- H. F. Wang, C. Tang and Q. Zhang, Catal. Today, 2018, 301, 25–31 CrossRef CAS.
- Y. Zou, I. A. Kinloch and R. A. Dryfe, ACS Appl. Mater. Interfaces, 2015, 7, 22831–22838 CrossRef CAS.
- J. Wu, Y. Liu, D. Geng, H. Liu and X. Meng, Int. J. Energy Res., 2018, 42, 853–862 CrossRef CAS.
- B. He, X. Chen, J. Lu, S. Yao, J. Wei, Q. Zhao and T. Wang, Electroanalysis, 2016, 28, 2435–2443 CrossRef CAS.
- X. Bai, Z. Liu, H. Lv, J. Chen, M. Khan, J. Wang and K. Shi, J. Hazard. Mater., 2022, 423, 127120 CrossRef CAS.
- V. Harnchana, S. Chaiyachad, S. Pimanpang, C. Saiyasombat, P. Srepusharawoot and V. Amornkitbamrung, Sci. Rep., 2019, 9, 1494 CrossRef.
- Y. Liu, F. Zhan, B. Wang, B. Xie, Q. Sun, H. Jiang and X. Sun, ACS Appl. Mater. Interfaces, 2019, 11, 21526–21535 CrossRef CAS.
- R. M. Silva, M. C. Ferro, J. R. Araujo, C. A. Achete, G. Clavel, R. F. Silva and N. Pinna, Langmuir, 2016, 32, 7038–7044 CrossRef CAS.
- J. Chen, J. Wu, Y. Liu, X. Hu and D. Geng, Phys. Status Solidi A, 2018, 215, 1800380 CrossRef.
- G. Seiffarth, M. Steimecke, T. Walther, M. Kühhirt, S. Rümmler and M. Bron, Electroanalysis, 2016, 28, 2335–2345 CrossRef CAS.
- K. Xie, W. Xia, J. Masa, F. Yang, P. Weide, W. Schuhmann and M. Muhler, J. Energy Chem., 2016, 25, 282–288 CrossRef.
- K. Choi, I. K. Moon and J. Oh, J. Mater. Chem. A, 2019, 7, 1468–1478 RSC.
- H. Pan, X. Huang, Z. Lu, Z. Zhang, B. An, D. Wu and F. Cheng, J. Chem. Eng., 2021, 419, 129619 CrossRef CAS.
- Z. Lu, X. Chen, P. Liu, X. Huang, J. Wei, Z. Ren and J. Masa, J. Electrochem. Soc., 2018, 165, 580 CrossRef.
- Y. Luo, M. Weng, J. Zheng, Q. Zhang, B. Xu, S. Song and C. Nan, J. Alloys Compd., 2018, 750, 17–22 CrossRef CAS.
- Y. Liu, B. Wang, Q. Sun, Q. Pan, N. Zhao, Z. Li and X. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 16512–16520 CrossRef CAS PubMed.
- C. Xiong, M. Li, W. Zhao, C. Duan and Y. Ni, J. Mater., 2020, 6, 523–531 Search PubMed.
- M. Tavakkoli, T. Kallio, O. Reynaud, A. G. Nasibulin, J. Sainio, H. Jiang and K. Laasonen, J. Mater. Chem. A, 2016, 4, 5216–5222 RSC.
- Y. Miyase, S. Takasugi, S. Iguchi, Y. Miseki, T. Gunji, K. Sasaki and K. Sayama, Sustainable Energy Fuels, 2018, 2, 1621–1629 RSC.
- S. Ramesh, K. Karuppasamy, H. S. Kim, H. S. Kim and J. H. Kim, Sci. Rep., 2018, 8, 16543 CrossRef.
- J. C. Carrillo-Rodríguez, S. García-Mayagoitia, R. Pérez-Hernández, M. T. Ochoa-Lara, F. Espinosa-Magaña, F. Fernández-Luqueño and F. J. Rodríguez-Varela, J. Power Sources, 2019, 414, 103–114 CrossRef.
- C. Deng, K. H. Wu, J. Scott, S. Zhu, R. Amal and D. W. Wang, J. Mater. Chem. A, 2019, 7, 20649–20657 RSC.
- A. Kumar, A. Sethi, R. M. Lawrence and V. M. Dhavale, Int. J. Hydrogen Energy, 2021, 46, 34701–34712 CrossRef.
- Y. Li, H. Huang, S. Chen, X. Yu, C. Wang and T. Ma, Nano Res., 2019, 12, 2774–2780 CrossRef CAS.
- X. X. Liu, J. B. Zang, L. Chen, L. B. Chen, X. Chen, P. Wu and Y. H. Wang, J. Mater. Chem. A, 2017, 5, 5865–5872 RSC.
- V. Jose, A. Jayakumar and J. M. Lee, ChemElectroChem, 2019, 6, 1485–1491 CrossRef CAS.
- X. Chen, Y. Chen, X. Luo, H. Guo, N. Wang, D. Su and L. Cui, Appl. Surf. Sci., 2020, 526, 146626 CrossRef CAS.
- M. Wang, Y. Hou, R. C. Slade, J. Wang, D. Shi, D. Wexler and J. Chen, Front. Chem., 2016, 4, 36 Search PubMed.
- I. J. R. Sarkar, S. G. Peera and R. J. Chetty, Appl. Electrochem., 2018, 48, 849–865 CrossRef CAS.
- Y. Niu, X. Huang, L. Zhao, W. Hu and C. M. Li, ACS Sustainable Chem. Eng., 2018, 6, 3556–3564 CrossRef CAS.
- A. Muthurasu, S. S. Mers and V. Ganesh, Int. J. Hydrogen Energy, 2018, 43, 4726–4737 CrossRef CAS.
- R. Ma, Y. Zhou, P. Li, Y. Chen, J. Wang and Q. Liu, Electrochim. Acta, 2016, 216, 347–354 CrossRef CAS.
- H. Tang, W. Chen, J. Wang, T. Dugger, L. Cruz and D. Kisailus, Small, 2018, 14(11), 1703459 CrossRef PubMed.
- S. Ni, F. Han, W. Wang, D. Han, Y. Bao, D. Han, H. Wang and L. Niu, Sens. Actuators, B, 2018, 259, 963–971 CrossRef CAS.
- E. Vélez-Fort, E. Pallecchi, M. G. Silly, M. Bahri, G. Patriarche, A. Shukla, F. Sirotti and A. Ouerghi, SSRN, 2014, 7, 835–843 Search PubMed.
- P. Li, X. Yin, Y. Yan, K. Zhan, J. Yang, B. Zhao and J. Li, J. Mater. Sci., 2018, 53, 6124–6134 CrossRef CAS.
- J. Zhu, H. Yin, J. Gong, M. S. H. Al-Furjan and Q. Nie, J. Alloys Compd., 2018, 748, 145–153 CrossRef CAS.
- A. Kathalingam, S. Ramesh, H. M. Yadav, J.-H. Choi, H. S. Kim and H.-S. Kim, J. Alloys Compd., 2020, 830, 154734 CrossRef CAS.
- Y. Jiao, Y. Zheng, K. Davey and S. Z. Qiao, Nat. Energy, 2016, 1(10), 1–9 Search PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10(5), 444–452 CrossRef CAS PubMed.
- H. Wu and P. H. L. Sit, Comput. Theor. Chem., 2021, 1201, 113292 CrossRef CAS.
- J. M. Luque-Centeno, M. V. Martínez-Huerta, D. Sebastián, G. Lemes, E. Pastor and M. J. Lázaro, Renewable Energy, 2018, 125, 182–192 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10(10), 780–786 CrossRef CAS.
- P. Liu, Y. Hu, X. Liu, T. Wang, P. Xi, S. Xi, D. Gao and J. Wang, J. Mater. Chem. A, 2019, 7(20), 12851–12858 RSC.
- L. Liu, F. Yan, K. Li, C. Zhu, Y. Xie, X. Zhang and Y. Chen, J. Mater. Chem. A, 2019, 7(3), 1083–1091 RSC.
- A. Wang, C. Zhao, M. Yu and W. Wang, Appl. Catal., B, 2021, 281, 119514 CrossRef CAS.
- T. H. Nguyen, D. T. Tran, N. H. Kim and J. H. Lee, Int. J. Hydrogen Energy, 2023, 48, 32294–32303 CrossRef CAS.
- B. K. Barman, B. Sarkar, P. Ghosh, M. Ghosh, G. M. Rao and K. K. Nanda, ACS Appl. Energy Mater., 2019, 2(10), 7330–7339 CrossRef CAS.
- N. D. Chuong, T. D. Thanh, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10(29), 24523–24532 CrossRef CAS PubMed.
- D. I. Jeong, H. W. Choi, S. Woo, J. H. Yoo, M. Kumar, Y. H. Song and D. H. Yoon, Appl. Surf. Sci., 2021, 559, 149077 CrossRef CAS.
- C. Cheng, Z. Sun, B. Li, Y. Li, C. Jin, T. Xiang, W. Wang, Z. Wu, H. Xue, Y. Cao and J. Yang, SSRN, 2022, preprint, DOI:10.2139/ssrn.4235928.
- W. Jia, J. Zhang, F. Le, X. Wang, Y. Lv, Y. Cao and D. Jia, J. Colloid Interface Sci., 2020, 567, 165–170 CrossRef CAS.
- S. Roy Chowdhury and T. Maiyalagan, ACS Omega, 2022, 7(23), 19183–19192 CrossRef CAS PubMed.
- J. Gautam, D. T. Tran, N. H. Kim and J. H. Lee, J. Colloid Interface Sci., 2019, 545, 43–53 CrossRef CAS.
- F. Bai, X. Qu, J. Wang, X. Chen and W. Yang, ACS Appl. Mater. Interfaces, 2020, 12(30), 33740–33750 CrossRef CAS PubMed.
- S. Kim, S. Ji, H. Yang, H. Son, H. Choi, J. Kang and L. O. Li, Appl. Catal., B, 2020, 310, 121361 CrossRef.
- X. Yang, X. Zheng, H. Li, B. Luo, Y. He, Y. Yao, H. Zhou, Z. Yan, Y. Kuang and Z. Huang, Adv. Funct. Mater., 2022, 32(31), 2200397 CrossRef CAS.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28(51), 1805641 CrossRef.
- S. Zhang, W. Xia, Q. Yang, Y. V. Kaneti, X. Xu, S. M. Alshehri, T. Ahamad and Y. Yamauchi, Chem. Eng. J., 2020, 396, 125154 CrossRef CAS.
- L. P. Lv, P. Du, P. Liu, X. Li and Y. Wang, ACS Sustainable Chem. Eng., 2020, 8(22), 8391–8401 CrossRef CAS.
- X. Shao, Y. Yang, Y. Liu, P. Yan, S. Zhou, T. T. Isimjan and X. Yang, J. Colloid Interface Sci., 2022, 607, 826–835 CrossRef CAS PubMed.
- X. Wang, H. Gai, Z. Chen, Y. Liu, J. Zhang, B. Zhao, A. Toghan and M. Huang, Mater. Today Energy, 2020, 18, 100497 CrossRef CAS.
- J. Su, G. Xia, R. Li, Y. Yang, J. Chen, R. Shi, P. Jiang and Q. Chen, J. Mater. Chem. A, 2016, 4(23), 9204–9212 RSC.
- K. Maiti, N. H. Kim and J. H. Lee, Chem. Eng. J., 2021, 410, 128358 CrossRef CAS.
- V. Jose, J. M. V. Nsanzimana, H. Hu, J. Choi, X. Wang and J. M. Lee, Adv. Funct. Mater., 2021, 11(17), 2100157 CAS.
- L. G. Bach, M. L. N. Thi, Q. B. Bui and H. T. Nhac-Vu, Synth. Met., 2019, 254, 172–179 CrossRef CAS.
- R. Yin, S. Ma, J. Ying, Z. Lu, X. Niu, J. Feng, F. Xu, Y. Zheng, W. Liu and X. Cao, Batteries, 2023, 9(6), 306 CrossRef CAS.
- Y. Tian, L. Xu, J. Qian, J. Bao, C. Yan, H. Li, H. Li and S. Zhang, Carbon, 2019, 146, 763–771 CrossRef CAS.
- Y. Wang, R. Gan, Z. Ai, H. Liu, C. Wei, Y. Song and M. Dirican, Carbon, 2021, 181, 87–98 CrossRef CAS.
- H. Fan, K. Mao, M. Liu, O. Zhuo, J. Zhao, T. Sun, Y. Jiang, X. Du, X. Zhang, Q. Wu, R. Che, L. Yang, Q. Wu, X. Wang and Z. Hu, J. Mater. Chem. A, 2018, 6(43), 21313–21319 RSC.
- Y. Song and P. Yang, Int. J. Hydrogen Energy, 2021, 46(68), 33801–33808 CrossRef CAS.
- Y. Li, Y. Feng, L. Li, K. Wu, X. Bo, J. Jia and L. Zhu, J. Alloys Compd., 2020, 823, 153892 CrossRef CAS.
- Y. Sun, Y. Guan, X. Wu, W. Li, Y. Li, L. Sun, H. Mi, Q. Zhang, C. He and X. Ren, Nanoscale, 2021, 13(5), 3227–3236 RSC.
- W. Wang, J. Q. Chen, Y. R. Tao, S. N. Zhu, Y. X. Zhang and X. C. Wu, ACS Catal., 2019, 9(4), 3498–3510 CrossRef CAS.
- A. Samanta, B. K. Barman, S. Mallick and C. R. Raj, ACS Appl. Energy Mater., 2020, 3(10), 10108–10118 CrossRef CAS.
- K. Maiti, J. Balamurugan, S. G. Peera, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10(22), 18734–18745 CrossRef CAS PubMed.
- J. Cai, X. Zhang, Y. Pan, Y. Kong and S. Lin, Int. J. Hydrogen Energy, 2021, 46(69), 34252–34263 CrossRef CAS.
- J. Cheng, D. Wu and T. Wang, Inorg. Chem. Commun., 2020, 117, 107952 CrossRef CAS.
- W. Gou, J. Bian, M. Zhang, Z. Xia, Y. Liu, Y. Yang, Q. Dong, J. Li and Y. Qu, Carbon, 2019, 155, 545–552 CrossRef CAS.
- P. Li, Y. Qiu, S. Liu, H. Li, S. Zhao, J. Diao and X. Guo, Eur. J. Inorg. Chem., 2019, 27, 3235–3241 CrossRef.
- M. Jiao, Z. Chen, X. Zhang, K. Mou and L. Liu, Int. J. Hydrogen Energy, 2020, 45(33), 16326–16336 CrossRef CAS.
- H. Wang, X. Wang, B. Zheng, D. Yang, W. Zhang and Y. Chen, Electrochim. Acta, 2019, 318, 449–459 CrossRef CAS.
- U. P. Suryawanshi, U. V. Ghorpade, D. M. Lee, M. He, S. W. Shin, P. V. Kumar, J. S. Jang, H. R. Jung, M. P. Suryawanshi and J. H. Kim, Chem. Mater., 2021, 33(1), 234–245 CrossRef CAS.
- X. Gao, B. Li, X. Sun, B. Wu, Y. Hu, Z. Ning, J. Li and N. Wang, Chin. Chem. Lett., 2021, 32(11), 3591–3595 CrossRef CAS.
- R. Wang, X. Wang, M. Cheng, Y. Wei, J. Xia, H. Lin, W. Sun and W. Hu, Int. J. Hydrogen Energy, 2022, 47(59), 24669–24679 CrossRef CAS.
- J. Duan, S. Chen, B. A. Chambers, G. G. Andersson and S. Z. Qiao, Adv. Mater., 2015, 27(28), 4234–4241 CrossRef CAS PubMed.
- G. Song, S. Luo, Q. Zhou, J. Zou, Y. Lin, L. Wang, G. Li, A. Meng and Z. Li, J. Mater. Chem. A, 2022, 10(36), 18877–18888 RSC.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141(12), 4972–4979 CrossRef CAS PubMed.
- D. Liu, Z. Lv, J. Dang, W. Ma, K. Jian, M. Wang, D. Huang and W. Tian, Inorg. Chem., 2021, 60(13), 9932–9940 CrossRef CAS PubMed.
- L. Yu, Y. Xiao, C. Luan, J. Yang, H. Qiao, Y. Wang, X. Zhang, X. Dia, Y. Yang and H. Zhao, ACS Appl. Mater. Interfaces, 2019, 11(7), 6890–6899 CrossRef CAS PubMed.
- R. Boppella, J. Tan, W. Yang and J. Moon, Adv. Funct. Mater., 2019, 29(6), 1807976 CrossRef.
- H. Q. Chang, G. H. Zhang and K. C. Chou, Electrochim. Acta, 2021, 394, 139119 CrossRef CAS.
- H. Liu, Z. Liu, F. Wang and L. Feng, Chem. Eng. J., 2020, 397, 125507 CrossRef CAS.
- H. Han, S. Park, D. Jang and W. B. Kim, J. Alloys Compd., 2021, 853, 157338 CrossRef CAS.
- X. Zhong, J. Tang, J. Wang, M. Shao, J. Chai, S. Wang, M. Yang, Y. Yang, N. Wang, S. Wang, B. Xu and H. Pan, Electrochim. Acta, 2018, 269, 55–61 CrossRef CAS.
- P. Wang, J. Qi, C. Li, X. Chen, T. Wang and C. Liang, ChemElectroChem, 2020, 7(3), 745–752 CrossRef CAS.
- X. Liu, Y. Guo, P. Wang, Q. Wu, Q. Zhang, E. A. Rozhkova, Z. Wang, Y. Liu, Z. Zheng, Y. Dai and B. Huang, ACS Appl. Energy Mater., 2020, 3(3), 2440–2449 CrossRef CAS.
- J. Zhang, X. P. Sun, P. Wei, G. Lu, S. X. Sun, Y. Xu, C. Fang, Q. Li and J. T. Han, ChemCatChem, 2020, 12(14), 3737–3745 CrossRef CAS.
- G. Liu, K. Wang, L. Wang, B. Wang, Z. Lin, X. Chen, Y. Hua, W. Zhu, H. Li and J. Xia, J. Colloid Interface Sci., 2021, 583, 614–625 CrossRef CAS PubMed.
- G. Qian, J. Chen, L. Luo, H. Zhang, W. Chen, Z. Gao, S. Yin and P. Tsiakaras, ACS Appl. Mater. Interfaces, 2020, 12(34), 38061–38069 CrossRef CAS PubMed.
- N. N. Chen, Q. Mo, L. He, X. Huang, L. Yang, J. Zeng and Q. Gao, Electrochim. Acta, 2019, 299, 708–716 CrossRef CAS.
- J. Q. Chi, J. Y. Xie, W. W. Zhang, B. Dong, J. F. Qin, X. Y. Zhang, J. H. Lin, Y. M. Chai and C. G. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 4047–4056 CrossRef CAS PubMed.
- T. I. Singh, G. Rajeshkhanna, U. N. Pan, T. Kshetri, H. Lin, N. H. Kim and J. H. Lee, Small, 2021, 17(29), 2101312 CrossRef CAS PubMed.
- L. Jia, B. Liu, Y. Zhao, W. Chen, D. Mou, J. Fu, Y. Wang, W. Xin and L. Zhao, J. Mater. Sci., 2020, 55, 16197–16210 CrossRef CAS.
- D. M. Nguyen, P. D. H. Anh, L. G. Bach and Q. B. Bui, Mater. Res. Bull., 2019, 115, 201–210 CrossRef CAS.
- T. Liu, X. Zhang, T. Guo, Z. Wu and D. Wang, Electrochim. Acta, 2020, 334, 135624 CrossRef CAS.
- X. Wang, Y. Fei, W. Li, L. Yi, B. Feng, Y. Pan, W. Hu and C. M. Li, ACS Appl. Mater. Interfaces, 2020, 12(14), 16548–16556 CrossRef CAS PubMed.
- X. Gao, J. Chen, X. Sun, B. Wu, B. Li, Z. Ning, J. Li and N. Wang, ACS Appl. Nano Mater., 2020, 3(12), 12269–12277 CrossRef CAS.
- B. Zhang, J. Hou, Y. Wu, S. Cao, Z. Li, X. Nie, Z. Gao and L. Sun, Adv. Energy Mater., 2019, 9(12), 1803693 CrossRef.
- H. Lin, W. Zhang, Z. Shi, M. Che, X. Yu, Y. Tang and Q. Gao, ChemSusChem, 2017, 10(12), 2597–2604 CrossRef CAS PubMed.
- Y. R. Liu, X. Shang, W. K. Gao, B. Dong, X. Li, X. H. Li, J. C. Zhao, Y. M. Chai, Y. Q. Liu and C. G. Liu, J. Mater. Chem. A, 2017, 5(6), 2885–2896 RSC.
- I. S. Amiinu, Z. Pu, X. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. Zhang and S. Mu, Adv. Funct. Mater., 2017, 27(44), 1702300 CrossRef.
- L. Chai, L. Zhang, X. Wang, Z. Ma, T. T. Li, H. Li, Y. Hu, J. Qian and S. Huang, Electrochim. Acta, 2019, 321, 134680 CrossRef CAS.
- M. Reda, H. A. Hansen and T. Vegge, ACS Catal., 2018, 8(11), 10521–10529 CrossRef CAS.
- Y. Chen, Q. Jiang, X. Bai, P. Shan, T. Liu, Y. Wang, H. Cui and H. Yuan, RSC Adv., 2022, 12(23), 14368–14376 RSC.
- J. Balamurugan, T. T. Nguyen, N. H. Kim, D. H. Kim and J. H. Lee, Nano Energy, 2021, 85, 105987 CrossRef CAS.
- N. N. Pham, K. H. Kim, B. Han and S. G. Lee, J. Phys. Chem. C, 2022, 126, 5863–5872 CrossRef CAS.
- Y. Balasooriya, P. Samarasekara, C. M. Lim, Y. F. C. Chau, M. R. R. Kooh and R. Thotagamuge, Heliyon, 2023, 9, 15989 CrossRef PubMed.
- M. D. Bhatt, G. Lee and J. S. Lee, Electrochim. Acta, 2017, 228, 619–627 CrossRef CAS.
- Y. Tian, L. Xu, M. Li, D. Yuan, X. Liu, J. Qian and S. Zhang, Nanomicro Lett., 2021, 13, 1–15 CAS.
- J. Zhao, Q. Li, Q. Zhang and R. Liu, Chem. Eng. J., 2022, 431, 133730 CrossRef CAS.
- T. Patniboon and H. A. Hansen, ChemSusChem, 2020, 13, 996–1005 CrossRef CAS PubMed.
- B. Zhang, X. Fu, L. Song and X. Wu, Carbon, 2021, 172, 122–131 CrossRef CAS.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10(23), 10876–10883 RSC.
- Z. Yuan, J. Li, M. Yang, Z. Fang, J. Jian, D. Yu, X. Chen and L. Dai, J. Am. Chem. Soc., 2019, 141, 4972–4979 CrossRef CAS PubMed.
- G. Qian, G. Yu, J. Lu, L. Luo, T. Wang, C. Zhang, R. Ku, S. Yin, W. Chen and S. Mu, J. Mater. Chem. A, 2020, 8, 14545–14554 RSC.
| This journal is © The Royal Society of Chemistry 2024 |