
Metal-free upcycling of plastic waste: photo-induced oxidative degradation of polystyrene in air†
Shuoyu
Xu‡
,
Shuxin
Liu‡
,
Wangze
Song
 and
Nan
Zheng
*
and
Nan
Zheng
*
School of Chemical Engineering, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: nzheng@dlut.edu.cn
First published on 9th January 2024
Abstract
Chemical recycling of plastics, as a promising strategy to reduce waste pollution and yield value-added chemicals, is showing great potential in the circular economy. In this work, a photooxidation method was reported to facilitate the degradation of polystyrene (PS) in air using porphyrin-based porous organic polymers (PPOPs) as the photocatalysts, associated with the recovery of benzoic acid in high yield (72%) and with excellent selectivity (97%). From the method aspect, the degradation could happen under very mild conditions using EtOAc as the green solvent and air as the oxidant. From the application perspective, such a method could efficiently facilitate the degradation of numerous PS derivatives and PS-based plastic wastes, with the recovery of benzoic acid. The optimized PPOP structure was screened with the desired degradation efficiency and benzoic acid recovery yield. Detailed mechanisms have been investigated to demonstrate that reactive oxygen species generated by light-triggered PPOPs play crucial roles in plastic waste upcycling.
Plastics have been widely used in all aspects of people's daily lives due to their low cost and wide range of applications. Up to the present, more than 8 billion tons of plastics have been produced, and plastic production is expected to grow at an annual rate of 2–5%, reaching more than 1.1 billion tons per year by 2050.1–4 However, most of the plastics are buried in landfills or leaked into the environment,5 which has caused a global environmental pollution crisis,6–11 and the existing infrastructure cannot solve this crisis,12–14 which has brought great trouble to people's lives and health. This challenge has accelerated research on new chemical recovery technologies. In recent years, some technologies that can convert plastic waste streams into valuable chemicals have emerged.3,15–19 The development of a more efficient and low-cost chemical recovery method has become a research hotspot.
Polystyrene (PS), accounting for about 7% of the current global plastic production,5 is widely used in daily life. Due to its stable structure, PS is difficult to degrade without special treatment. In order to facilitate the chemical degradation of plastics, many methods have been developed for recycling PS through pyrolysis,20–23 but many of these methods produce a mixture of organic products. Although purer pyrolysis products can be obtained by multi-step post-modifications,24–26 higher temperature and pressure with very special reactors are still needed, which will lead to an increase in cost (Scheme 1, method A). In the past 20 years, some advancements in the oxidative degradation of PS to benzoic acid have been achieved,27,28 but these approaches often required harsh or complex conditions (Scheme 1, method B). The use of photocatalytic technology in organic reactions has exploded over the past fifteen years.29 Many light-triggered degradation reactions30,31 and photocatalytic conversion applications32,33 have been discovered and utilized. In recent years, several methods have been able to produce benzoic acid with high selectivity for the photocatalytic oxidative degradation of PS.34–41 However, photocatalytic technology has been limited by low energy conversion rates,42 resulting in low or moderate yields of benzoic acid from PS degradation. Pure oxygen is usually required as an oxidant to further improve the oxidative efficiency (Scheme 1, method C). In addition, some other methods for degrading PS, including electrochemical degradation,43 biodegradation,44,45 and ultrasonic degradation,46 have also been developed recently (Scheme 1, method D). However, methods for degrading PS remain limited. Therefore, it is crucial and urgent to develop a green, clean and simple method for degrading PS to recover high-value chemical products.
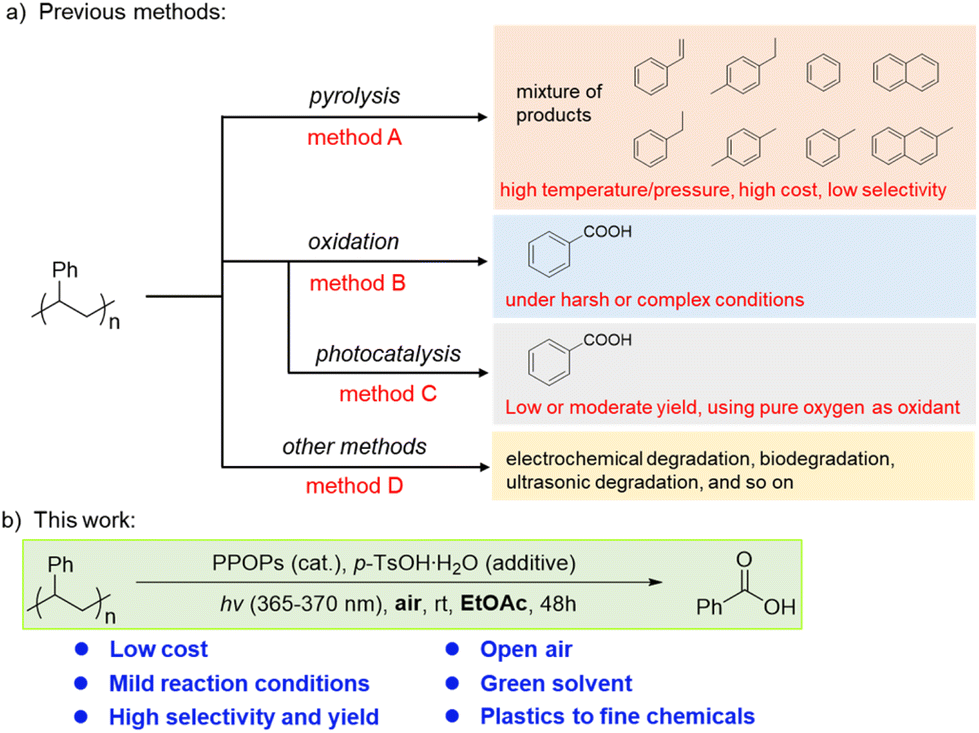 | ||
| Scheme 1 Methods for the chemical degradation of PS. | ||
Previously, we developed a catalytic system based on porphyrin-based porous organic polymers (PPOPs) and applied it to various dehydrogenation reactions.47,48 We found that these PPOPs can be very efficient at generating reactive oxygen species (ROS) in air. Porous organic polymers (POPs) are constructed by rigid monomers through irreversible covalent bonds, which endow them with high structural and chemical stability in various solvents and under harsh conditions.49,50 Porphyrins and their derivatives are a class of photosensitizers, which have a wide range of applications in photocatalytic reactions.47,48 Singlet oxygen (1O2) is a kind of ROS, which can induce the oxidation of various molecules. It is widely used in chemical reactions, biochemical processes and organic pollutant treatment.51–57 Xiao's group has demonstrated that 1O2 can efficiently seize a proton from the benzylic C–H bond of PS by hydrogen atom transfer (HAT), which produces benzyl radicals, further induces the oxidative degradation of PS, and finally obtains benzoic acid.36 The critical role of 1O2 inspired us to consider whether the PPOPs can trigger the degradation of PS efficiently even in the air.
Herein, 7 different PPOPs by copolymerization of bromo-substituted porphyrins and diynes were afforded with different structures. Taking advantage of the porous structures and confined spaces, PPOPs could efficiently produce ROS under air and at room temperature compared to porphyrin monomers and achieve more efficient photocatalytic oxidative degradation of PS. Moreover, PPOPs have high structural and chemical stability. Impressively, controlled experiments have shown that PPOPs exhibit overwhelming advantages over those small-molecule photosensitizers. Therefore, such PPOPs provide an excellent photocatalytic platform for the oxidative degradation of PS (Scheme 1).
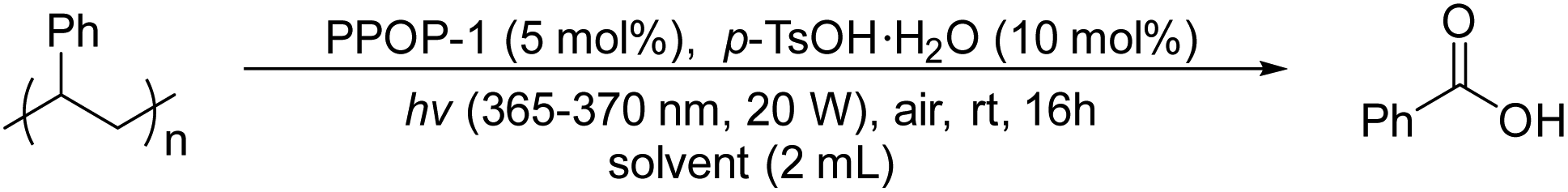
Initially, we synthesized the photocatalyst PPOP-1 by the Sonogashira polycondensation according to a method reported in the literature.47,48 We performed a series of characterization methods for the obtained PPOP-1 to confirm its structure (Fig. S1–S6, in the ESI†). Then we used PS (Mw ∼ 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) as a model substrate to optimize the oxidative degradation conditions. We surprisingly found that PPOP-1 (5 mol%) can effectively catalyze the degradation of PS using p-toluenesulfonic acid monohydrate (p-TsOH·H2O) (10 mol%) as an additive under the irradiation of black light (365–370 nm, 20 W) in ethyl acetate (EtOAc), yielding 48% of benzoic acid (Table 1, entry 1). Light and air were necessary for the degradation since PS could not be degraded without them (Table S1, entries 2 and 3 in the ESI†). Among different light sources, black light (365–370 nm, 20 W) performed best for acquiring benzoic acid (Table S1, entries 1 and 4–6 in the ESI†). In the case of using photosensitizers without additives, 26% yield of benzoic acid could be obtained (Table S1, entry 7 in the ESI†). Similarly, only 18% yield of benzoic acid could be obtained using additives without photosensitizers (Table S1, entry 8 in the ESI†), which corresponded to the literature reporting that acids could catalyze PS degradation to a certain extent.36 Next, different solvents were screened (Table 1, entries 1–5). The results showed that the yield of benzoic acid using 1,2-dichloroethane (DCE) and dimethyl carbonate (DMC) as solvents was slightly lower than that using EtOAC, while no reaction occurred using other solvents. It might be due to the high solubility of PS in EtOAc.37 Then, the amount of photosensitizer was studied and the results indicated that 5 mol% photosensitizer was the optimized loading equivalent (Table 1, entries 1, 6 and 7). The type and amount of additive was next optimized to be 10 mol% p-TsOH·H2O, with the highest yield of benzoic acid (Table 1, entries 1, 8–14, see Table S1† for more details). As the reaction time was extended to 32 hours and 48 hours, the yield of benzoic acid was also gradually increased. Finally, 62% yield of benzoic acid was afforded in 48 hours (Table 1, entries 15 and 16). More optimization details for solvents, additives, amounts of photosensitizers and additives, and light sources are given in the ESI (Table S1†).
000 g mol−1) as a model substrate to optimize the oxidative degradation conditions. We surprisingly found that PPOP-1 (5 mol%) can effectively catalyze the degradation of PS using p-toluenesulfonic acid monohydrate (p-TsOH·H2O) (10 mol%) as an additive under the irradiation of black light (365–370 nm, 20 W) in ethyl acetate (EtOAc), yielding 48% of benzoic acid (Table 1, entry 1). Light and air were necessary for the degradation since PS could not be degraded without them (Table S1, entries 2 and 3 in the ESI†). Among different light sources, black light (365–370 nm, 20 W) performed best for acquiring benzoic acid (Table S1, entries 1 and 4–6 in the ESI†). In the case of using photosensitizers without additives, 26% yield of benzoic acid could be obtained (Table S1, entry 7 in the ESI†). Similarly, only 18% yield of benzoic acid could be obtained using additives without photosensitizers (Table S1, entry 8 in the ESI†), which corresponded to the literature reporting that acids could catalyze PS degradation to a certain extent.36 Next, different solvents were screened (Table 1, entries 1–5). The results showed that the yield of benzoic acid using 1,2-dichloroethane (DCE) and dimethyl carbonate (DMC) as solvents was slightly lower than that using EtOAC, while no reaction occurred using other solvents. It might be due to the high solubility of PS in EtOAc.37 Then, the amount of photosensitizer was studied and the results indicated that 5 mol% photosensitizer was the optimized loading equivalent (Table 1, entries 1, 6 and 7). The type and amount of additive was next optimized to be 10 mol% p-TsOH·H2O, with the highest yield of benzoic acid (Table 1, entries 1, 8–14, see Table S1† for more details). As the reaction time was extended to 32 hours and 48 hours, the yield of benzoic acid was also gradually increased. Finally, 62% yield of benzoic acid was afforded in 48 hours (Table 1, entries 15 and 16). More optimization details for solvents, additives, amounts of photosensitizers and additives, and light sources are given in the ESI (Table S1†).
|
|
||||
|---|---|---|---|---|
| Entry | PPOP-1 (mol%) | Additive (mol%) | Solvent | Yieldc (%) |
| a The reaction was carried out with PS (0.2 mmol based on C8H8 as the repeating unit of PS, 1.0 equiv.) in the presence of 5 mol% PPOP-1 and 10 mol% additive in 2 mL solvent under air, black light (365–370 nm, 20 W) and room temperature for 16 h. b The amounts of PPOP-1, additive and the yield of products were based on the repeating unit of PS. c Yield was determined by the crude 1H NMR spectrum with 1,3,5-trimethoxybenzene as an internal standard; isolated yield in parentheses. d Room temperature for 32 h. e Room temperature for 48 h. | ||||
| 1 | 5 | p-TsOH·H2O (10) | EtOAc | 49 (48) |
| 2 | 5 | p-TsOH·H2O (10) | DCE | 44 |
| 3 | 5 | p-TsOH·H2O (10) | Acetone | 27 |
| 4 | 5 | p-TsOH·H2O (10) | MeCN | Trace |
| 5 | 5 | p-TsOH·H2O (10) | DMC | 45 |
| 6 | 2.5 | p-TsOH·H2O (10) | EtOAc | 44 |
| 7 | 10 | p-TsOH·H2O (10) | EtOAc | 50 |
| 8 | 5 | p-TsOH·H2O (5) | EtOAc | 38 |
| 9 | 5 | p-TsOH·H2O (20) | EtOAc | 50 |
| 10 | 5 | HCl (10) | EtOAc | 35 |
| 11 | 5 | CF3COOH (10) | EtOAc | 26 |
| 12 | 5 | ZnCl2 (10) | EtOAc | 37 |
| 13 | 5 | Yb(OTf)3 (10) | EtOAc | 33 |
| 14 | 5 | Sc(OTf)3 (10) | EtOAc | 36 |
| 15d | 5 | p-TsOH·H2O (10) | EtOAc | 55 |
| 16 | 5 | p-TsOH·H 2 O (10) | EtOAc | 63 (62) |
With the optimized reaction conditions in hand, several photocatalysts were evaluated systematically, including some commercially available small-molecule photosensitizers and different PPOPs (Table 2, entries 1–10; see Fig. 1, Fig. S9 and Table S2† for more details). Among these small-molecule photosensitizers, tetrabutylammonium decatungstate (TBADT) (Table 2, entry 1) and 9-mesityl-10-methylacridinium tetrafluoroborate (Mes-Acr-MeBF4) (Table 2, entry 2) promoted such transformation slightly, while 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) (Table S2, entry 3 in the ESI†), tris(2,2′-bipyridyl)ruthenium(II) dichloride hexahydrate (Ru(bpy)3(Cl)2·6H2O) (Table S2, entry 4 in the ESI†) and bis[2-(2,4-difluorophenyl)-5-trifluoromethylpyridine][2-2′-bipyridyl]iridium hexafluorophosphate ([Ir(dF(CF3)ppy)2(dtbbpy)]PF6) (Table S2, entry 5 in the ESI†) were not effective. The small-molecular porphyrin photosensitizers such as 5,10,15,20-tetraphenylporphyrin (TPP) (Table 2, entry 3) and 5,10,15,20-tetra(4-carboxyphenyl)-porphyrin (TCPP) (Table S2, entry 7 in the ESI†) exhibited a poor oxidative degradation effect on PS. Based on the structure of PPOP-1, we designed another six PPOPs by changing the structure of porphyrins and diynes and evaluated their photocatalytic effects (Table 2, entries 4–10). We were delighted to find that all the PPOPs containing porphyrin components showed much higher photodegradation efficiency of PS than small-molecule photosensitizers, among which PPOP-7 with a reduced tetraphenylporphyrin structure showed the optimal effect (71% yield of benzoic acid) (Fig. S11–S17 in the ESI†). This may be due to the large specific surface area (270.07 m2 g−1) and suitable pore size (2.6 nm) of PPOP-7. Therefore, in the following experiments, PPOP-7 was chosen as a photocatalyst for the degradation of PS-based plastic waste.
 | ||
| Fig. 1 Structures of photocatalysts, PPOPs. | ||
|
|
||
|---|---|---|
| Entry | Photocatalyst | Yieldb (%) |
| a Reaction conditions: PS (0.2 mmol based on C8H8 as the repeating unit of PS, 1.0 equiv.), photocatalyst (0.01 mmol, 5 mol%), p-TsOH·H2O (0.02 mmol, 10 mol%), EtOAc (2.0 mL), black light (365–370 nm, 20 W), open in air, and room temperature for 48 h. b Yield was determined by the crude 1H NMR spectrum with 1,3,5-trimethoxybenzene as an internal standard; isolated yield in parentheses. | ||
| 1 | TBADT | 25 |
| 2 | Mes-Acr-MeBF4 | 36 |
| 3 | TPP | 17 |
| 4 | PPOP-1 | 63 (62) |
| 5 | PPOP-2 | 61 |
| 6 | PPOP-3 | 60 |
| 7 | PPOP-4 | 62 |
| 8 | PPOP-5 | 61 |
| 9 | PPOP-6 | 65 |
| 10 | PPOP-7 | 71 (71) |
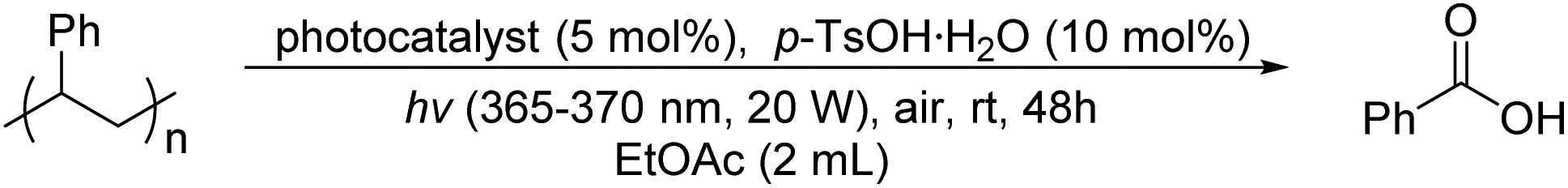
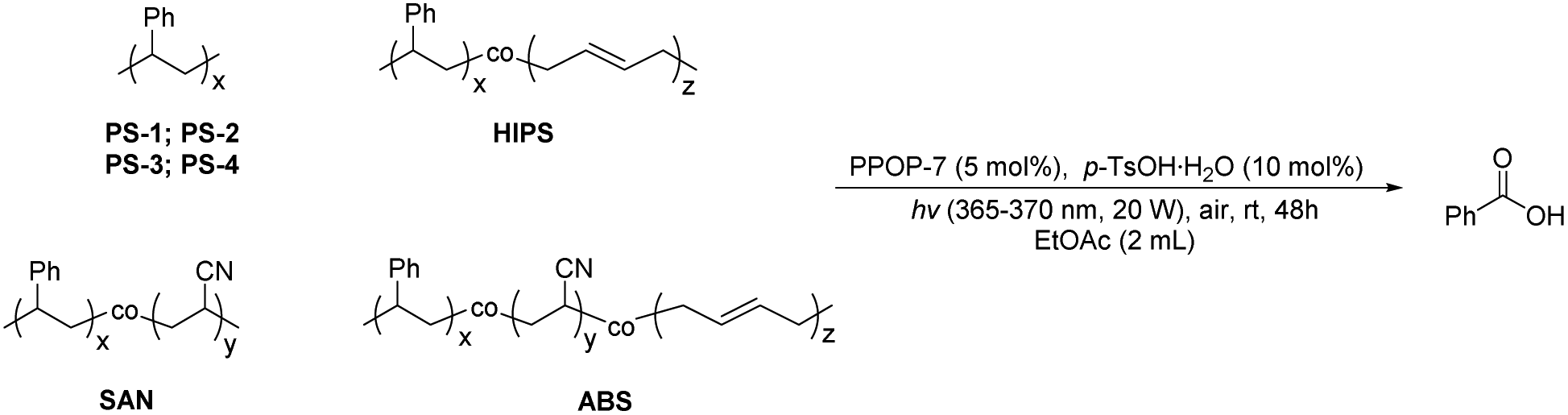
Subsequently, we applied PPOP-7 to the photodegradation of PS with different molecular weights and styrene copolymers with different structural units (Table 3). For the PS with different molecular weights, it could be well aerobically degraded to benzoic acid, and 69–72% yields could be obtained (Table 3, entries 1–4). For the copolymers of styrene, the yields of benzoic acid were not as high as those of PS. HIPS, graft-copolymerized by styrene and butadiene with strong impact strength, could be degraded to benzoic acid in 50% yield (Table 3, entry 5). Styrene-based block copolymers like SAN and ABS possess strong heat resistance and stability due to their acrylonitrile units, which makes their chemical degradation full of challenges.39 Considering that the solubility of SAN and ABS in acetone is much better than that in EtOAc, we used acetone as the solvent with trifluoroacetic acid (TFA) as the additive instead of p-TsOH·H2O to give a desired yield of benzoic acid (40–42% yields) (Table 3, entries 6 and 7). In summary, polymers containing a styrene structural unit can be aerobically degraded efficiently to produce benzoic acid as the only product. The method we disclosed provides a convenient way for the degradation of such plastics under green and mild conditions.
|
|
||
|---|---|---|
| Entry | Polymer | Yield (%) of benzoic acidb |
| a Reaction conditions: polymer (0.2 mmol based on C8H8 as the repeating unit of PS, 1.0 equiv.), PPOP-7 (0.01 mmol, 5 mol%), p-TsOH·H2O (0.02 mmol, 10 mol%), EtOAc (2.0 mL), black light (365–370 nm, 20 W), open air, and room temperature for 48 h. b Isolated yield. c Acetone (2 mL) was used as the solvent and TFA (0.02 mol, 10 mol%) was used as the additive. | ||
| 1 |
PS-1 (Mw ∼ 65000 g mol−1) |
72 |
| 2 |
PS-2 (Mw ∼ 192000 g mol−1) |
71 |
| 3 |
PS-3 (Mw ∼ 350000 g mol−1) |
71 |
| 4 |
PS-4 (Mw ∼ 650000 g mol−1) |
69 |
| 5 |
HIPS (Mw ∼ 140000 g mol−1, styrene 30 wt%) |
50 |
| 6c |
SAN (Mw ∼ 165000 g mol−1, styrene 75 wt%) |
42 |
| 7c |
ABS (Mw ∼ 238000 g mol−1, styrene 72 wt%) |
40 |
According to the above experimental results, we finally investigated the aerobic degradation of waste PS products from daily life. As shown in Table 4, these waste PS products can be aerobically degraded to benzoic acid. PS wastes like these from EPS foams, plastic plates, food boxes, cup lids, yoghurt containers and packaging were all suitable as substrates, providing benzoic acid with 54–65% isolated yields (Table 4, entries 1–6). In addition, it is worth noting that for the degradation of SBS plastic, benzoic acid could also be obtained in a 44% isolated yield (Table 4, entry 7). These results demonstrated the practicality and potential applications of this polymer degradation strategy.

A series of tracking methods were used to reveal the degradation process. By sampling and analyzing the photodegradation process of PS, we could observe that the water contact angle on the PS membrane decreased with the progress of degradation, which indicated that the hydrophilicity of the membrane was enhanced, and a large number of hydrophilic groups, such as hydroxyl and carboxyl groups, were produced during the degradation process (Fig. 2a). An X-ray diffraction (XRD) experiment was further used to track the change of PS at different degradation times, which showed that the original crystal peak of PS gradually decreased upon degradation (Fig. 2b). The XRD experiment was also carried out using the PS degradation product (48 hours of degradation). From the obtained images, obvious diffraction peaks could be seen, indicating that benzoic acid was generated (Fig. S19b in the ESI†). The control experiments of PS in EtOAc with or without light were conducted, and the changes in glass transition temperature (Tg) with degradation time in these two cases were recorded. It could be clearly seen that PS was gradually decomposed during the reaction (Fig. 2c, red line, and Fig. S20 in the ESI†). The degradation process of PS under standard reaction conditions was monitored by gel permeation chromatography (GPC). Through the decrease of molecular weight, we could see that PS was gradually degraded during the reaction (Fig. 2d).
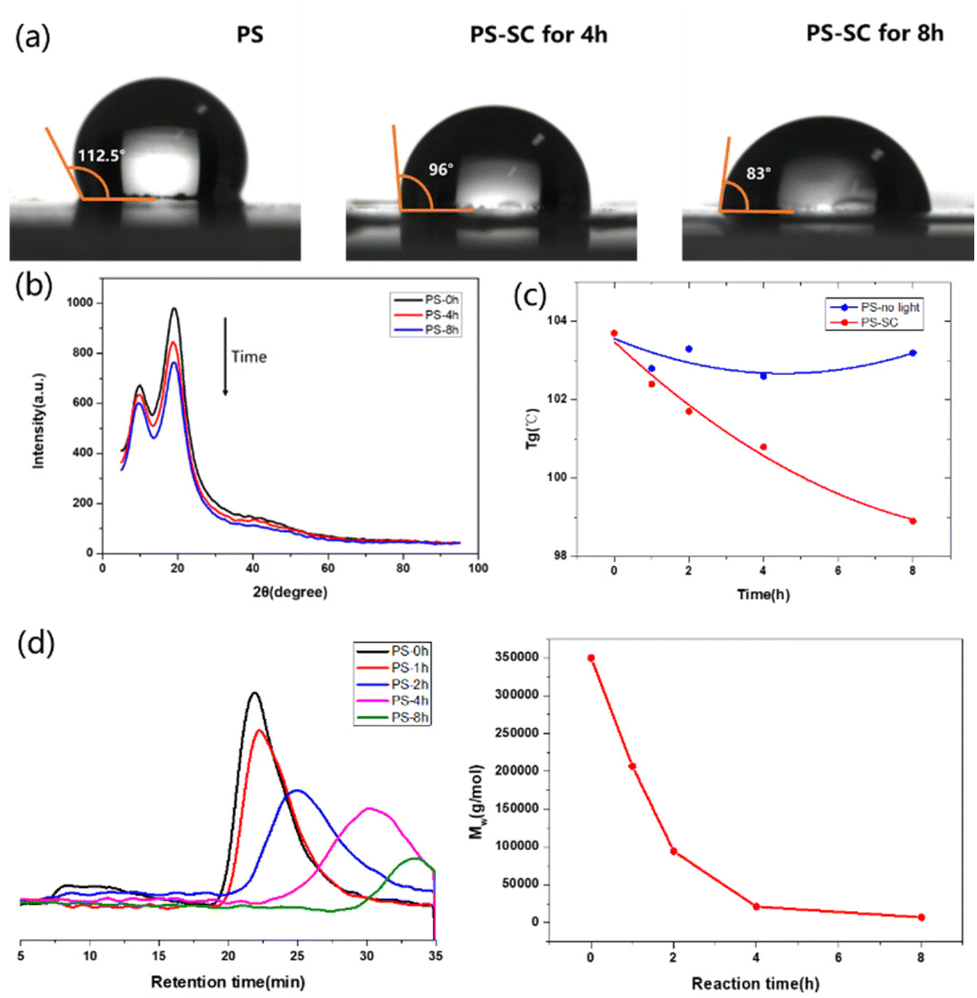 | ||
| Fig. 2 Characterization of polymer degradation (SC: standard conditions). (a) Comparison of water contact angles on the top surface of PS-based membranes. (b) X-ray diffraction of PS depolymerization. (c) Tracing of glass transition temperature for PS degradation when reacting with or without light. (d) GPC tracing and the degradation trend for PS. | ||
In order to elucidate the possible reaction pathways, a series of controlled experiments were carried out. Under standard conditions, the degradation experiments were conducted in the presence of 9,10-diphenylanthracene (DPA) or NaN3 as an 1O2 trap or scavenger;36 however, the expected degradation products were not obtained (Scheme 2, eqn (1)). These results suggested that 1O2 played crucial roles in the aerobic degradation of PS. In addition, a radical trap experiment was designed using 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) as the radical scavenger. No degradation of PS occurred under standard conditions with the addition of TEMPO, indicating that the radical also contributed to the degradation of PS (Scheme 2, eqn (2)).
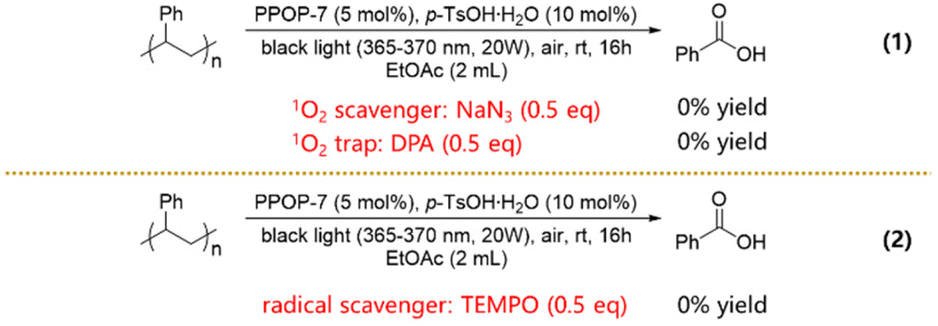 | ||
| Scheme 2 Controlled experiments. | ||
Based on the above experimental results and related literature reports,35–38,57 we proposed a possible reaction mechanism (Fig. 3). 1O2 was first produced by two possible pathways. One way was that the electrophilic protonation of PS initially occurred under acidic reaction conditions to form an aromatic cation species A. Under the irradiation of black light, A was further excited to form an excited state A*. The excited state A* underwent energy transfer with triplet oxygen (3O2), which led to the generation of 1O2. Another way was that the photocatalyst PPOP-7 was irradiated to an excited state PPOP-7*, which could generate 1O2 by energy transfer from 3O2. The 1O2 could react with 1 (PS) via the HAT process to produce benzyl radical 2 and ˙OOH. Next, ˙OOH reacted with benzyl radical 2 to generate the corresponding peroxide intermediate 3. Homolytic O–O bond cleavage under the irradiation of black light yielded the oxygen radical 4 and the hydroxyl radical (˙OH). 4 underwent a β-scission to produce fragments 5 and 6, which with repeated HAT and β-scission processes generated benzoylformic acid 7. Under light irradiation, 7 underwent decarbonylation to form benzoyl radical 8 with CO and ˙OH removal. Benzaldehyde 9 was formed from benzoyl radical 8 by HAT, which was further oxidized to afford benzoic acid 10. Alternatively, 7 underwent a decarboxylation process to directly obtain benzaldehyde 9 with CO2 release, which was then oxidized to obtain benzoic acid 10.
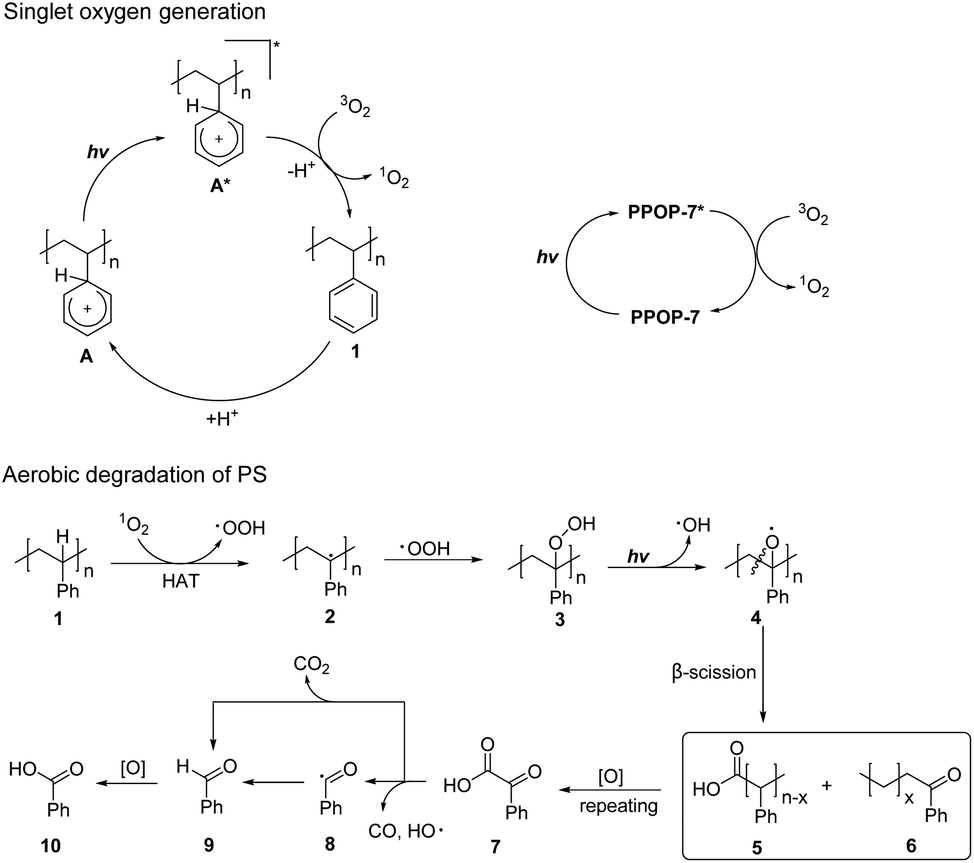 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Conclusions
In summary, the efficient and green degradation of polystyrene under air was realized using a porphyrin-based porous organic polymer (PPOP) as a photocatalyst. Benzoic acid was obtained as the only product with good selectivity and yield (up to 72% yield). This approach for the degradation of plastics features mild conditions, green solvents, simple operation, and air as the oxidant. It can convert a wide range of polystyrene wastes into high-value products by upcycling. This method is also suitable for other polymers containing a styrene structural unit to obtain benzoic acid. Mechanistic studies have demonstrated that both 1O2 and radicals play crucial roles in the degradation process. We believe that this photo-induced oxidative degradation will not only provide a unique method for upcycling real plastic wastes, but also address the increasingly serious environmental crisis in the future.Author contributions
N. Z. designed the study. S. X. and S. L. performed the experiments and analyzed the data. S. X., S. L., W. S. and N. Z. wrote the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (22375027), the Natural Science Foundation of Jiangsu Province (BK20221265 and BK20211100), and the Fundamental Research Funds for the Central Universities (DUT23YG133). The authors acknowledge the assistance of the Dalian University of Technology (DUT) Instrumental Analysis Center.References
- N. Simon, K. Raubenheimer, N. Urho, S. Unger, D. Azoulay, T. Farrelly, J. Sousa, H. van Asselt, G. Carlini, C. Sekomo, M. L. Schulte, P.-O. Busch, N. Wienrich and L. Weiand, Science, 2021, 373, 43–47 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- S. S. Borkar, R. Helmer, F. Mahnaz, W. Majzoub, W. Mahmoud, M. Al-Rawashdeh and M. Shetty, Chem Catal., 2022, 2, 3320–3356 CrossRef CAS.
- A.-C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- L. E. Revell, P. Kuma, E. C. Le Ru, W. R. C. Somerville and S. Gaw, Nature, 2021, 598, 462–467 CrossRef CAS PubMed.
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373, 51–55 CrossRef CAS PubMed.
- R. G. Santos, G. E. Machovsky-Capuska and R. Andrades, Science, 2021, 373, 56–60 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- L. Weiss, W. Ludwig, S. Heussner, M. Canals, J.-F. Ghiglione, C. Estournel, M. Constant and P. Kerhervé, Science, 2021, 373, 107–111 CrossRef CAS PubMed.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- A. Milbrandt, K. Coney, A. Badgett and G. T. Beckham, Resour., Conserv. Recycl., 2022, 183, 106363 CrossRef.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- J. Nisar, G. Ali, A. Shah, M. R. Shah, M. Iqbal, M. N. Ashiq and H. N. Bhatti, Energy Fuels, 2019, 33, 12666–12678 CrossRef CAS.
- J. Wang, J. Jiang, X. Wang, R. Wang, K. Wang, S. Pang, Z. Zhong, Y. Sun, R. Ruan and A. J. Ragauskas, J. Hazard. Mater., 2020, 386, 121970 CrossRef CAS PubMed.
- O. Dogu, A. Eschenbacher, R. John Varghese, M. Dobbelaere, D. R. D'Hooge, P. H. M. Van Steenberge and K. M. Van Geem, Chem. Eng. J., 2023, 455, 140708 CrossRef CAS.
- S. Fan, Y. Zhang, L. Cui, T. Maqsood and S. Nižetić, J. Cleaner Prod., 2023, 390, 136102 CrossRef CAS.
- R. Li, Z. Zhang, X. Liang, J. Shen, J. Wang, W. Sun, D. Wang, J. Jiang and Y. Li, J. Am. Chem. Soc., 2023, 145, 16218–16227 CrossRef CAS PubMed.
- Z. Xu, F. Pan, M. Sun, J. Xu, N. E. Munyaneza, Z. L. Croft, G. G. Cai and G. Liu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203346119 CrossRef CAS PubMed.
- N. E. Munyaneza, C. Posada, Z. Xu, V. De Altin Popiolek, G. Paddock, C. McKee and G. Liu, Angew. Chem., Int. Ed., 2023, 62, e202307042 CrossRef CAS PubMed.
- A. Pifer and A. Sen, Angew. Chem., Int. Ed., 1998, 37, 3306–3308 CrossRef CAS PubMed.
- K. P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Roman-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef CAS PubMed.
- H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- X. Fang, Y. Tang, Y. Ma, G. Xiao, P. Li and D. Yan, Sci. China Mater., 2023, 66, 664–671 CrossRef CAS.
- X. Fang, R. Gao, Y. Yang and D. Yan, iScience, 2019, 16, 22–30 CrossRef CAS PubMed.
- P. Li, T. Zhang, M. A. Mushtaq, S. Wu, X. Xiang and D. Yan, Chem. Rec., 2021, 21, 841–857 CrossRef CAS PubMed.
- P. Li, Y. Liu, M. A. Mushtaq and D. Yan, Inorg. Chem. Front., 2023, 10, 4650–4667 RSC.
- G. Zhang, Z. Zhang and R. Zeng, Chin. J. Chem., 2021, 39, 3225–3230 CrossRef CAS.
- T. Li, A. Vijeta, C. Casadevall, A. S. Gentleman, T. Euser and E. Reisner, ACS Catal., 2022, 12, 8155–8163 CrossRef CAS PubMed.
- Z. Huang, M. Shanmugam, Z. Liu, A. Brookfield, E. L. Bennett, R. Guan, D. E. Vega Herrera, J. A. Lopez-Sanchez, A. G. Slater, E. J. L. McInnes, X. Qi and J. Xiao, J. Am. Chem. Soc., 2022, 144, 6532–6542 CrossRef CAS PubMed.
- Y. Qin, T. Zhang, H. Y. V. Ching, G. S. Raman and S. Das, Chem, 2022, 8, 2472–2484 CAS.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- C. Li, X. Y. Kong, M. Lyu, X. T. Tay, M. Đokić, K. F. Chin, C. T. Yang, E. K. X. Lee, J. Zhang, C. Y. Tham, W. X. Chan, W. J. Lee, T. T. Lim, A. Goto, M. B. Sullivan and H. S. Soo, Chem, 2023, 9, 2683–2700 CAS.
- A. Ong, J. Y. Q. Teo, Z. Feng, T. T. Y. Tan and J. Y. C. Lim, ACS Sustainable Chem. Eng., 2023, 11, 12514–12522 CrossRef CAS.
- W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang and L. C. Sim, Environ. Sci. Pollut. Res., 2020, 27, 2522–2565 CrossRef CAS PubMed.
- J. Lu, R. Hou, Y. Wang, L. Zhou and Y. Yuan, Water Res., 2022, 226, 119277 CrossRef CAS PubMed.
- P. Xiang, Y. Zhang, T. Zhang, Q. Wu, C. Zhao and Q. Li, J. Hazard. Mater., 2023, 458, 131856 CrossRef CAS PubMed.
- Z. Chen, Y. Zhang, R. Xing, C. Rensing, J. Lü, M. Chen, S. Zhong and S. Zhou, Environ. Sci. Technol., 2023, 57, 7867–7874 CrossRef CAS PubMed.
- J. Zimmermann, D. Kruppa, M. Fischlschweiger, S. Beuermann and S. Enders, Chem. Ing. Tech., 2021, 93, 289–296 CrossRef CAS.
- X. Sun, N. Zheng, G. Liu, Q. Wu and W. Song, Chem. Commun., 2022, 58, 13234–13237 RSC.
- J. Bai, X. Xu, Y. Zheng, W. Song and N. Zheng, Green Synth. Catal., 2023 DOI:10.1016/j.gresc.2023.03.001.
- Y. Zhang and S. N. Riduan, Chem. Soc. Rev., 2012, 41, 2083–2094 RSC.
- S. B. Yu, F. Lin, J. Tian, J. Yu, D. W. Zhang and Z. T. Li, Chem. Soc. Rev., 2022, 51, 434–449 RSC.
- A. Sagadevan, K. C. Hwang and M.-D. Su, Nat. Commun., 2017, 8, 1812 CrossRef PubMed.
- S. Ren, H. Li, X. Xu, H. Zhao, W. He, L. Zhang and Z. Cheng, Biomater. Sci., 2023, 11, 509–517 RSC.
- M. Wang, J. Guo, L. Chen and X. Zhao, Biomater. Sci., 2023, 11, 1776–1784 RSC.
- W. Wang, Y. Song, Y. Tian, B. Chen, Y. Liang, Y. Liang, C. Li and Y. Li, Biomater. Sci., 2023, 11, 2828–2844 RSC.
- M. Wang, Y. Zheng, H. He, T. Lv, X. Xu, X. Fang, C. Lu and H. Yang, Biomater. Sci., 2023, 11, 7423–7431 RSC.
- J. Al-Nu'airat, I. Oluwoye, N. Zeinali, M. Altarawneh and B. Z. Dlugogorski, Chem. Rec., 2021, 21, 315–342 CrossRef PubMed.
- J. Wu, J. Chen, L. Wang, H. Zhu, R. Liu, G. Song, C. Feng and Y. Li, Green Chem., 2023, 25, 940–945 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04197b |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |