
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of depolymerizable polymers toward a circular economy
Julian F.
Highmoore
 a,
Lasith S.
Kariyawasam
a,
Scott R.
Trenor
b and
Ying
Yang
*a
a,
Lasith S.
Kariyawasam
a,
Scott R.
Trenor
b and
Ying
Yang
*a
aUniversity of Nevada, Reno, Department of Chemistry, Reno, Nevada, USA. E-mail: yingy@unr.edu
bThe Association of Plastic Recyclers (APR), Washington, DC, USA
First published on 24th January 2024
Abstract
Transition from the traditional linear economy to a circular economy is the key to reducing the amount of plastic that ends up in the environment and recovering value from the waste. However, energy-efficient, environmentally friendly, and economical technologies to recycle the mounting volume of low-grade plastic waste are lacking when compared to the low cost of primary fossil-based plastic production. This led to extensive efforts to identify methods that mitigate the waste that already exists and develop new polymers to eliminate the poor recyclability problem caused by current materials. Polymers capable of depolymerizing back to their own monomers provide a promising solution. This chemical recycling process has the potential to eliminate contaminants, selectively depolymerize a particular polymer from a waste stream, and recover high-purity monomers from contaminated wastes; however, the process must be energy-efficient and economical. This review discusses recent advances in chemical design of polymers that can depolymerize back to their monomers with an emphasis on strategies that shift the monomer–polymer equilibrium to enable reversible depolymerization and polymerization. Novel approaches developed for ring-opening polymerization and ring-closing depolymerization, reversing free radical polymerization, and reversible reactions that enable depolymerization of highly crosslinked thermosets to small molecular monomers are reviewed. Self-immolative polymers, which have a long history of chemistry innovation to realize the balance between stability and rapid depolymerization, are briefly discussed to serve as an inspiration for new polymer designs. Any new materials designed should have at least matching properties and stabilities compared to existing materials. Thus, this review reflects the status of current research that not only demonstrates the reversible polymerization chemistry but also achieves outstanding material properties.
 Julian F. Highmoore | Julian F. Highmoore is a PhD student in the group of Prof. Ying Yang at the University of Nevada, Reno. They are primarily working on chemically recyclable thermoset polymers. Julian earned their B.S in Chemistry from Clarkson University (New York) in 2021 under the guidance of Prof. Devon Shipp. |
 Lasith S. Kariyawasam | Dr Lasith S. Kariyawasam is a postdoctoral scholar in the group of Prof. Ying Yang at University of Nevada, Reno. He is primarily working on chemically recyclable dithioacetal polymers. Lasith earned his Ph.D. in Organic Chemistry from Miami University (Ohio) in 2020 under the supervision of Prof. C. Scott Hartley. His work focused on the use of common reagents, specifically carbodiimides, to fuel the formation of transient aqueous anhydrides from carboxylic acid precursors. |
 Scott Trenor | Dr Scott Trenor is Technical Director at The Association of Plastics Recyclers (APR). He is leading efforts on APR programs in design for recycling, recyclability testing protocols and global harmonization of packaging design protocols. Prior to joining APR, he was involved in R&D and Sustainability at Milliken & Company and Kraton Polymers giving him over 18 years of experience in the polymer industry. His functional experience includes the discovery, development and commercialization of novel polymers and polymer additives as well as in technology scouting, acquisition, and licensing. Scott earned his Ph.D. in Macromolecular Science and Engineering from Virginia Tech in 2004. |
 Ying Yang | Dr Ying Yang completed her PhD studies in Materials Science and Engineering at Clemson University under the supervision of Prof. Marek Urban. She then started a postdoc appointment in Prof. Urban's group working on self-healing materials. She joined the Department of Chemistry at the University of Nevada, Reno (UNR) as an assistant professor in 2019. Her lab works on designing molecular platforms for highly recyclable polymers that can depolymerize back to their feedstock. Another research focus is bioinspired materials. Her lab is involved in broad collaborations in various disciplines including geothermal energy, 3D printing, and the study of natural products. |
1. Introduction
The world is drowning in plastic. This is a result of the traditional linear economic model of taking raw materials from nature, making the product, using it, and then discarding the materials as waste. The desirable qualities that make plastic usage so prolific – durability and stability – are the same qualities causing long-lasting plastic pollution problems. Strategies to improve the sustainability of plastics involve but are not limited to four important aspects: finding renewable feedstocks,1,2 extending the service life of traditional polymers,3,4 mitigation of the waste that already exists,5–7 and the development of new materials to eliminate the poor recyclability problem caused by current materials.8–12 The production of plastics is in fact more energy efficient than non-plastic alternatives such as glass, aluminum, steel, or paper, requiring roughly half the energy, a fraction of the water consumption, and produces a fraction of the smog.13 Thus, the issue lies in how plastics are handled post-consumption. One approach to address the end-of-life management is transitioning into a circular economy. A circular economy keeps materials, products, and services in circulation for as long possible, minimizing environmental waste. It reduces material use, redesigns materials, products, and services to be less resource intensive, and recaptures “waste” as a resource to manufacture new materials and products.14Plastic recycling strategies that turn wastes into products are being pursued with intensive efforts from both the industry and the scientific community.15 The recycling strategies can be categorized into three different cycles (Fig. 1):16 the polymer loop, the monomer loop, and the molecular loop. The polymer loop refers to the most commonly practiced mechanical recycling where the polymer backbone remains unchanged in chemical composition. This is currently the most common method of recycling well-known post-consumer plastics such as polyethylene terephthalate (PET), high density polyethylene (HDPE), low density polyethylene (LDPE), and polypropylene (PP). This approach is the most technologically mature and most energy-efficient and should be considered as the first choice. However, mechanical recycling is not suitable for low-grade wastes (highly contaminated wastes, mixed waste streams, products that have been downgraded over many mechanical recycling cycles), thermosets, and applications with high requirements for color, odor, and other properties. In these cases, the monomer and molecular loops offer viable solutions. The monomer loop refers to depolymerizing the polymer backbone into monomers. The monomers are purified and reused to make new polymers. The molecular loop refers to breaking the polymers down into small molecules that require additional steps to form monomers before repolymerization. Processes such as pyrolysis and gasification fall into the molecular loop and are the most energy-intensive processes. Advances in the monomer and molecular loop, have the potential to offer scalable and energy efficient solutions to traditionally non-recyclable wastes and have been the focus of recent research.
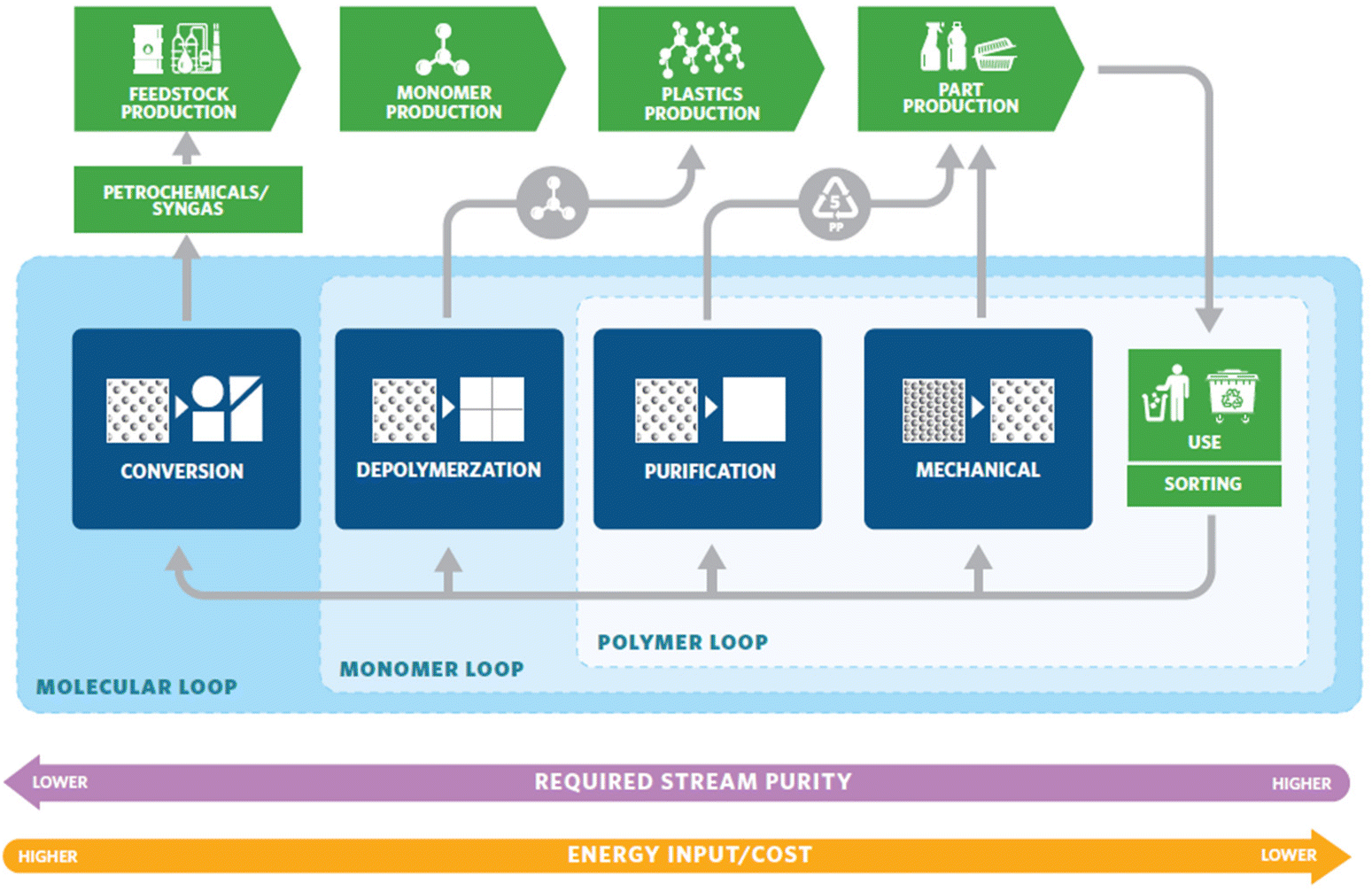 | ||
| Fig. 1 Diagram for a circular plastic economy. Reproduced from ref. 16 with permission from American Chemical Society, copyright 2020. | ||
Over the last two decades, the pursuit of backbone degradable polymers has expanded significantly from biodegradation17 and partial degradation,18 to full backbone depolymerization.19 The field can be split into two broad categories: (1) chemically recycling of preexisting commodity polymers and (2) development of new polymers to replace current commodity polymers that are more easily recycled. Traditional methods such as hydrolysis, glycolysis, alcoholysis, and aminolysis to recover valuable monomers from commercial polyesters, polyurethanes, polyamides, and polycarbonates have been extensively studied and are seeing increasing success on industrial scale.20–23 Chemical recycling often faces solvent and energy constrains. Thus, new catalytic systems are being developed to improve the conversion and product selectivity under mild conditions.24,25 Catalysis and processes such as temperature-gradient thermolysis are paving the way for upcycling of polyolefins.26–28 Investigations into different solvents,29,30 and conditions to depolymerize vinyl polymers31 are also among the forefront of recent progresses. Developing new polymers to replace current ones also have many challenges to be addressed, chief among them is finding materials that have comparable stability and mechanical performances to existing commodity polymers and achieving economic feasibility of scaling up. As mentioned in numerous reviews, we are at an early stage of tackling the plastic waste problem, and therefore multipronged approach should be taken to accelerate the technological advances.6,10,32
This review comes at a time when the urge to develop a sustainable polymer economy is at its highest. We will focus our discussion on the redesign of polymer structures capable of the monomer loop via depolymerization. Unfortunately, depolymerization is difficult to realize with many existing plastic wastes, especially the widely used polyolefins. New polymers with inherent depolymerizability, repolymerizability, and with performances comparable to various commodity polymers, along with recycling processes that are economically feasible, represent the design goal for the next-generation plastics. This review aims to comprehensively evaluate recent synthetic innovations of depolymerizable polymers focusing on the polymerization and depolymerization chemistries as well as the mechanical performances of the new materials in comparison to commodity plastics. We will first dive into the design methodologies including ring-chain equilibrium, reversing free radical polymers, and depolymerizable crosslinked thermosets. For the ring-chain equilibrium enabled recycling, in addition to the enthalpy-driven ring-opening polymerization (ROP) of strained cyclic monomers, we will highlight entropy-driven ring-opening polymerization (ED-ROP) as an emerging method of chemical recycling, such as polycarbonates and polydithioacetals, which have not been discussed in extent in prior reviews. We also include a brief discussion of self-immolative polymers (SIPs) considering the rich chemistries that have been developed in the field of SIP since its introduction in 200833 that offer inspirations for recyclable polymer design. Finishing out this review are design parameters that should be considered moving forward. Due to the rapid progress in polymer recycling in the last five years, we limit the scope of this review to examples of degradable polymers that recycle back to monomers with high yields and exclude the ones that degrade to oligomers.
2. Depolymerizable polymers based-on ring-chain equilibrium
In recent years, there has been significant focus on using reversible ring-opening polymerization (ROP) to develop recyclable polymers. Cyclic monomers and their corresponding polymers have the same types of bonding. It follows then that the thermodynamics can be tuned by the ring-strain energy (RSE) and the entropy change of polymerization with high sensitivity to reaction conditions, including temperature and concentration. To understand how the thermodynamics can be tuned, we can start by viewing polymerization as a reversible reaction: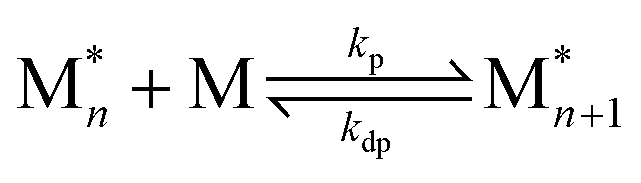 | (1) |
When the rate of polymerization equals the rate of depolymerization, an equilibrium is reached. For most monomers, the equilibrium greatly favors the product due to the large enthalpic driving force. At higher temperatures, the rate constant of depolymerization (kdp) increases more rapidly than polymerization (kp), which shifts the equilibrium to the monomer side. This usually occurs above the polymer's decomposition temperature. Therefore, in pursuit of depolymerizable polymers, the strategy is to identify monomer–polymer systems that are near their thermodynamic equilibrium.
The ROP offers several advantages. The ring-closing depolymerization (RCD) and ROP cycles do not generate any side products. Polymers with a variety of heteroatom backbones similar to step-growth polymers can be prepared, but with much higher molecular weights. The ring-opening metathesis polymerization (ROMP) of cycloalkenes form polymers with all hydrocarbon backbones, which are promising replacements for polyolefins. Even being near equilibrium, the ROP polymers sustain high thermal stability in the absence of catalysts. Although we used the term “ring-chain equilibrium” here, the polymer “chain” can also be a cyclic polymer. Cyclic polymers have even higher stabilities than their linear counterparts due to the absence of end groups.34 Functional groups can be conveniently introduced via ring modifications. Thus, ROP offers high control over both the polymer structure and polymerization–depolymerization equilibrium.
2.1 Tuning the thermodynamics of ring-chain equilibrium
There are two types of ROP, enthalpy-driven ROP for small to medium sized rings with ring-strains, and entropy-driven ROP (ED-ROP) of strainless macrocyclic rings (Fig. 2). This section will discuss how to control the equilibrium positions between the monomer and the polymer during the polymerization and depolymerization cycles. For these ROPs, both the enthalpy and entropy terms play a role in determining the equilibrium position.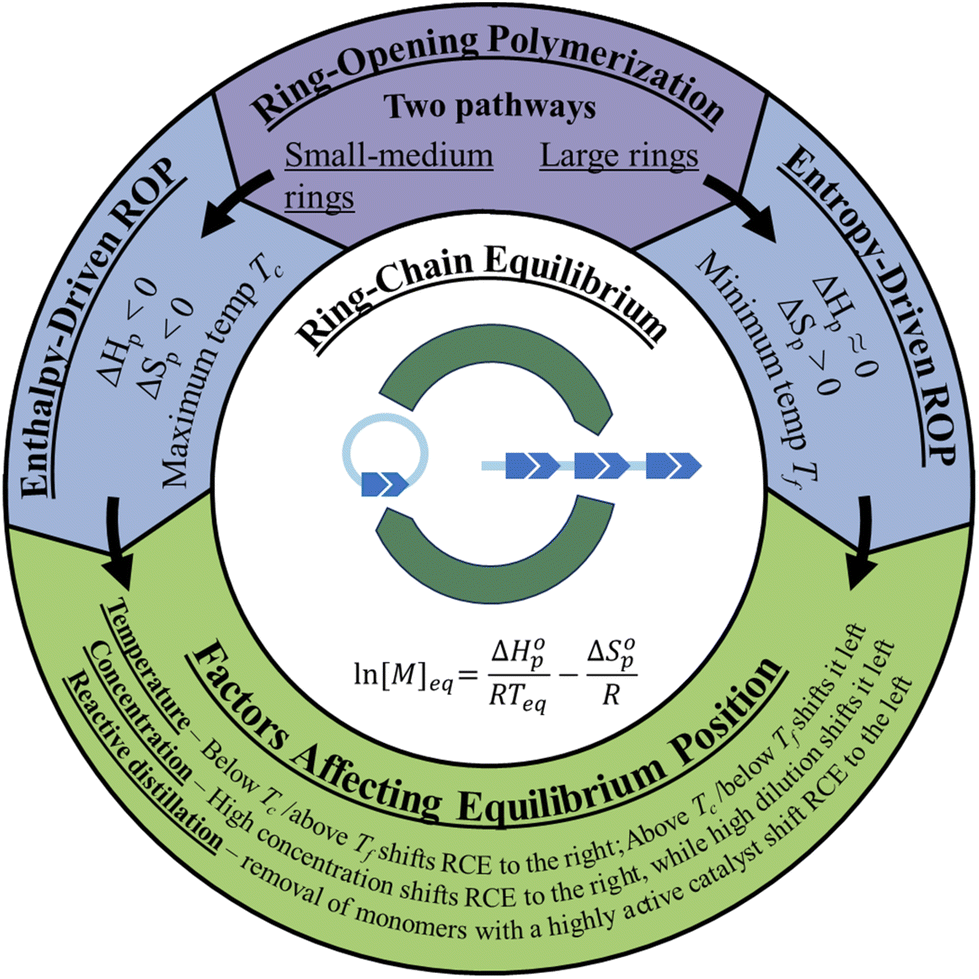 | ||
| Fig. 2 Graphic depiction of the two pathways of ROP and the factors affecting the ring-chain equilibrium. | ||
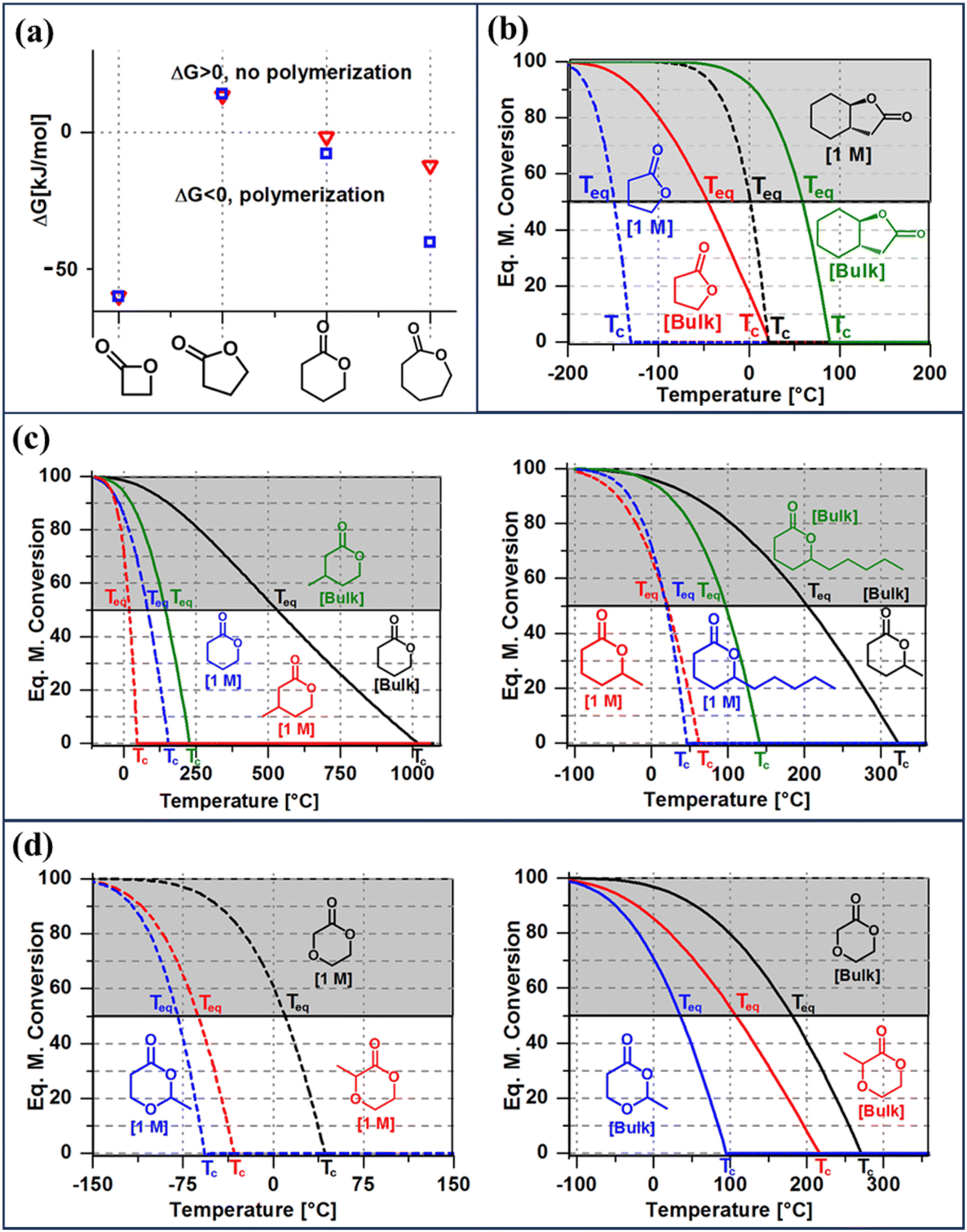 | ||
| Fig. 3 Thermodynamic considerations for ring opening polymerization. (a) The difference in ΔGp as a function of ring-size at normal pressure and 25 °C. (b) The effect of fused-ring on the equilibrium monomer conversion as a function of temperature of γ-lactones. (c) The effect of ring-substituents of various lengths on the equilibrium monomer conversion of δ-lactones. (d) The effect of substituent position on the equilibrium monomer conversion of ether-δ-lactones. For each monomer shown in figures (b)–(d), the equilibrium monomer conversion over temperature curves are shown for two starting monomer concentrations, bulk and 1 M. Reproduced from ref. 35 with permission from American Chemical Society, copyright 2016. | ||
Fig. 3a shows the differences in ΔGp as a function of ring size. Compared to the 4-membered ring, 5- and 6-members rings are promising candidates for depolymerizable polymers as they are closer to their equilibrium (ΔGp = 0) at 25 °C. With near equilibrium systems, they tend to yield a mixture of monomers and low molecular weight polymers. Thus, the questions are, how to push the equilibrium to the polymer side to obtain high monomer conversion and high molecular weight polymers, and conversely how to shift the equilibrium to the monomer side quantitively.
The equilibrium control is best described by the Dainton and Ivin equation:36
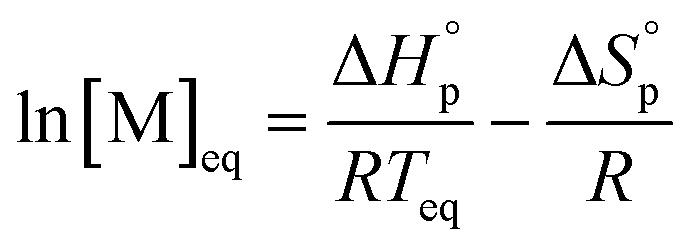 | (2) |
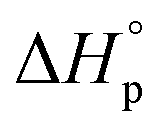 , and
, and 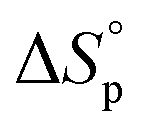 . This relationship is derived from the equilibrium condition. At equilibrium,
. This relationship is derived from the equilibrium condition. At equilibrium, 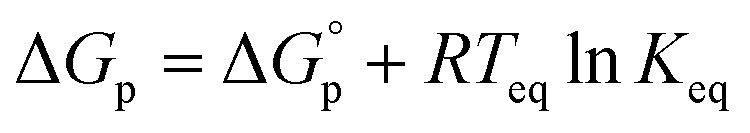 . When ΔGp = 0, the equilibrium constant
. When ΔGp = 0, the equilibrium constant 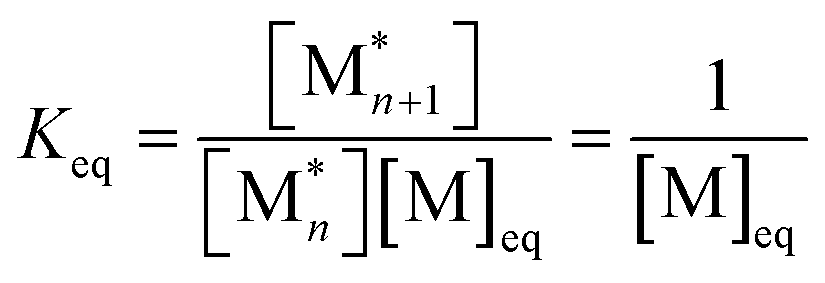 . Combining these equations gives eqn (2), which can be rewritten as
. Combining these equations gives eqn (2), which can be rewritten as | (3) |
Thus, a ceiling temperature (Tc) is defined to describe a scenario where the equilibrium lies on the very left side. It is the temperature at or above which the concentration of monomer in equilibrium with its polymer becomes essentially equal to the initial monomer concentration ([M]0).
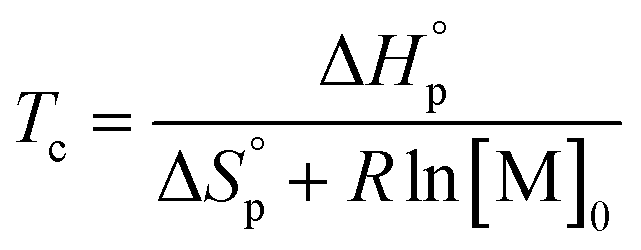 | (4) |
Usually, Tc is reported for [M]0 at unit molarity or bulk state; however, one can see that Tc is a function of [M]0 and that it decreases with dilution. 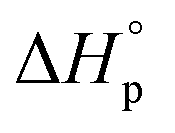 can be adjusted by RSE and
can be adjusted by RSE and  can be fine-tuned by the size of the monomer.
can be fine-tuned by the size of the monomer.
Thus, eqn (2) and (4) give us a general guideline to control the equilibrium, an example of which is illustrated in Fig. 3b–d for lactones with different ring-size and substituents. The characteristic behavior of enthalpy-driven ROP is shown in equilibrium conversion vs. temperature curves, where increasing the temperature shifts the equilibrium to the left, promoting depolymerization. Conversely, lowering the temperature induces propagation due to the negative 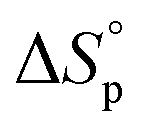 . The Teq shown in Fig. 3 is defined as when [M]eq is 50% of the starting monomer concentration, [M]0. As the temperature continues to increase, Tc is reached and the monomer conversion drops to zero. The concentration dependency of Tc modeled in eqn (4) is shown in Fig. 3 as Tc drops going from bulk polymerization to a starting [M]0 of 1 M.
. The Teq shown in Fig. 3 is defined as when [M]eq is 50% of the starting monomer concentration, [M]0. As the temperature continues to increase, Tc is reached and the monomer conversion drops to zero. The concentration dependency of Tc modeled in eqn (4) is shown in Fig. 3 as Tc drops going from bulk polymerization to a starting [M]0 of 1 M.
Equilibrium control that relies exclusively on temperature and concentration is quite limited. For example, γ-butyrolactone is difficult to polymerize as it has an extremely low Tc even in bulk. Thus, design tools that change the  and
and 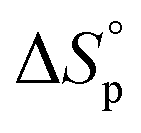 values are extremely useful. A fused-ring approach is commonly used to increase the RSE, and thus the Tc, to make the monomer more polymerizable. This is shown in Fig. 3b, where Tc increases by over 100 °C when γ-butyrolactone is fused with a cyclohexane ring. In certain cases a fused-ring strategy can also decrease the ring-strain energy.37 Ring-substitutions that do not create a fused ring usually decrease the Tc. As shown in Fig. 3c, six-membered δ-lactones have quite high Tc's even though its ΔGp is slightly below zero; however, a large drop of Tc is observed in ring-substitutions with aliphatic groups of increasing length. The trend can be explained by the Thorpe–Ingold effect: as the steric hinderance of a substituent increase, the molecule will favor ring closure and intramolecular reactions.38 This effect is more profound at high concentrations but become negligible in certain cases at 1 M monomer concentration (Fig. 3c). The position of the substitute also plays an important role on the thermodynamics as demonstrated by Fig. 3d. Hillmyer et al. showed that polymerization rate depends strongly on substituent position and exhibits a more modest dependence on alkyl length (from –CH3 to –(CH2)8CH3) under bulk room temperature conditions. However, the physical properties of the resulting aliphatic polyesters depend much more on substituent length than on substituent position.39
values are extremely useful. A fused-ring approach is commonly used to increase the RSE, and thus the Tc, to make the monomer more polymerizable. This is shown in Fig. 3b, where Tc increases by over 100 °C when γ-butyrolactone is fused with a cyclohexane ring. In certain cases a fused-ring strategy can also decrease the ring-strain energy.37 Ring-substitutions that do not create a fused ring usually decrease the Tc. As shown in Fig. 3c, six-membered δ-lactones have quite high Tc's even though its ΔGp is slightly below zero; however, a large drop of Tc is observed in ring-substitutions with aliphatic groups of increasing length. The trend can be explained by the Thorpe–Ingold effect: as the steric hinderance of a substituent increase, the molecule will favor ring closure and intramolecular reactions.38 This effect is more profound at high concentrations but become negligible in certain cases at 1 M monomer concentration (Fig. 3c). The position of the substitute also plays an important role on the thermodynamics as demonstrated by Fig. 3d. Hillmyer et al. showed that polymerization rate depends strongly on substituent position and exhibits a more modest dependence on alkyl length (from –CH3 to –(CH2)8CH3) under bulk room temperature conditions. However, the physical properties of the resulting aliphatic polyesters depend much more on substituent length than on substituent position.39
With a negligible ΔHp, ED-ROP exhibits high sensitivity to temperature and concentration which originates from the TΔSp contribution to the overall ΔGp. Opposite of the enthalpy-driven ROP, ED-ROP is favored at higher temperatures due to the positive ΔSp. Reversible ED-ROP systems with near-quantitative depolymerization have been sporadically reported.40,41,51 There remains an unexplored design space for ED-ROP-based chemically recyclable polymers. Comparatively, enthalpy and entropy driven ROP are similar in that they both utilize the unique chemical environments present in cyclic monomers, albeit of different sizes. They share many attractive features, such as the perfect stoichiometry and no emission of small molecules or volatiles, making them eco-friendly. ED-ROP also exhibits unique advantages.41 Virtually no heat is evolved during ED-ROP because the mixtures of homologous macrocycles are often strainless. In addition, the macrocyclic monomers are usually solids which are easy to store and transport. Thermal stability is less of a concern even in the presence of catalyst because polymerization is favored at high temperatures. As such, strict requirements for catalyst removal, which can be difficult such as when making thermosets, becomes unnecessary. In comparison, low Tc polymers derived from strained cyclic monomers need strict catalyst removal to avoid depolymerization that can be activated at higher temperatures.
The concentration dependency of the equilibrium is illustrated in Fig. 4. Monomers have higher translational entropy, whereas polymers exhibit higher conformational entropy. Under concentrated conditions, the conformational entropy dominates, shifting the equilibrium to the polymers. At low concentrations, translational entropy dominates and the equilibrium shifts to the monomer side. Thus, high dilution significantly decreases the depolymerization temperature.
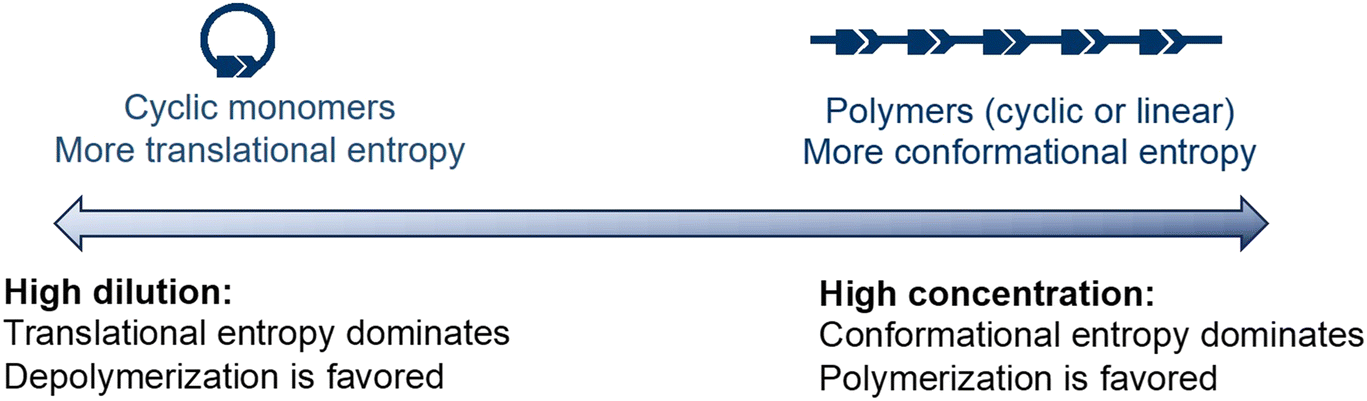 | ||
| Fig. 4 Graphical depiction of the effect of concentration on the ring-chain equilibrium. | ||
The use of solvent can be avoided by reaction distillation, although the trade-off is the higher processing temperature required. Ideally, depolymerization should be carried out at temperatures well below Tc to obtain depolymerization in high yields. However, for polymers with intermediate to high Tc's, depolymerization is possible above the Tc under reactive distillation. Monomers are removed as soon as they are formed, which shifts the reaction out of equilibrium and toward the monomer formation. This process requires a highly reactive catalyst to activate the exchange reactions that enable monomer formation via ring-closing depolymerization. Byers group found that ZnCl2, when combined with poly(ethylene glycol), forms a particularly active catalyst that works universally for RCD of a variety of polyesters and polycarbonates.52 The catalyst induces random chain scissions, allowing monomer recovery under distillation at 160 °C, which is near or below the Tc of the polymers (153 °C < Tc < 261 °C). The use of more reactive catalysts is expected to further lower the distillation temperature.
In the following sections 2.2–2.6, we will introduce specific examples of depolymerizable polymers synthesized from enthalpy- and entropy-driven ROPs.
2.2 ROP of lactones and thiolactones
Polyesters are the world's fourth-most produced synthetic polymer, after polyethylene, polypropylene, and polyvinylchloride, making them the most used heterochain polymer in plastic bottles and fabrics. Polyester is also the most recyclable commodity polymer. Recycled polyethylene terephthalate (PET) bottles are shredded, washed, and then used for remaking the bottles or spinning into fibers. However, low-quality PET waste, such as polyester carpeting wastes, rely on chemical recycling, including hydrolysis, glycolysis, methanolysis, and enzymatic recycling to recover valued products, or otherwise be downcycled. Chemical recycling approaches are being actively investigated to identify energy efficient and environmentally benign processes53–58 with commercialization efforts from industry on a global scale.59,60Commercial polyesters are typically synthesized from step-growth polymerization (Fig. 5a). However, new advances in the ROP of lactones have led to high molecular weight polyesters with thermal and mechanical properties that are outperforming polyolefins, while still maintaining their recyclability via RCD. The cyclic monomers can be derived from biomass through simple steps, thereby reducing the need for petrochemical starting materials. Two approaches are used to tackle the recyclability. One is to develop new synthetic strategies for cyclic monomers that are usually considered as non-polymerizable due to low RSEs, such as 5-membered γ-butyrolactone (γBL). The corresponding polymers have low thermodynamic stability (low Tc) but can be kinetically stabilized. They easily depolymerize under ambient conditions with a catalyst. The other approach is to depolymerize high Tc polymers, which are traditionally considered as non-depolymerizable. Reaction distillation has thus far been extensively investigated and Fig. 5 summarizes the recent progress, ranging from 4- to 6-membered cyclic esters.
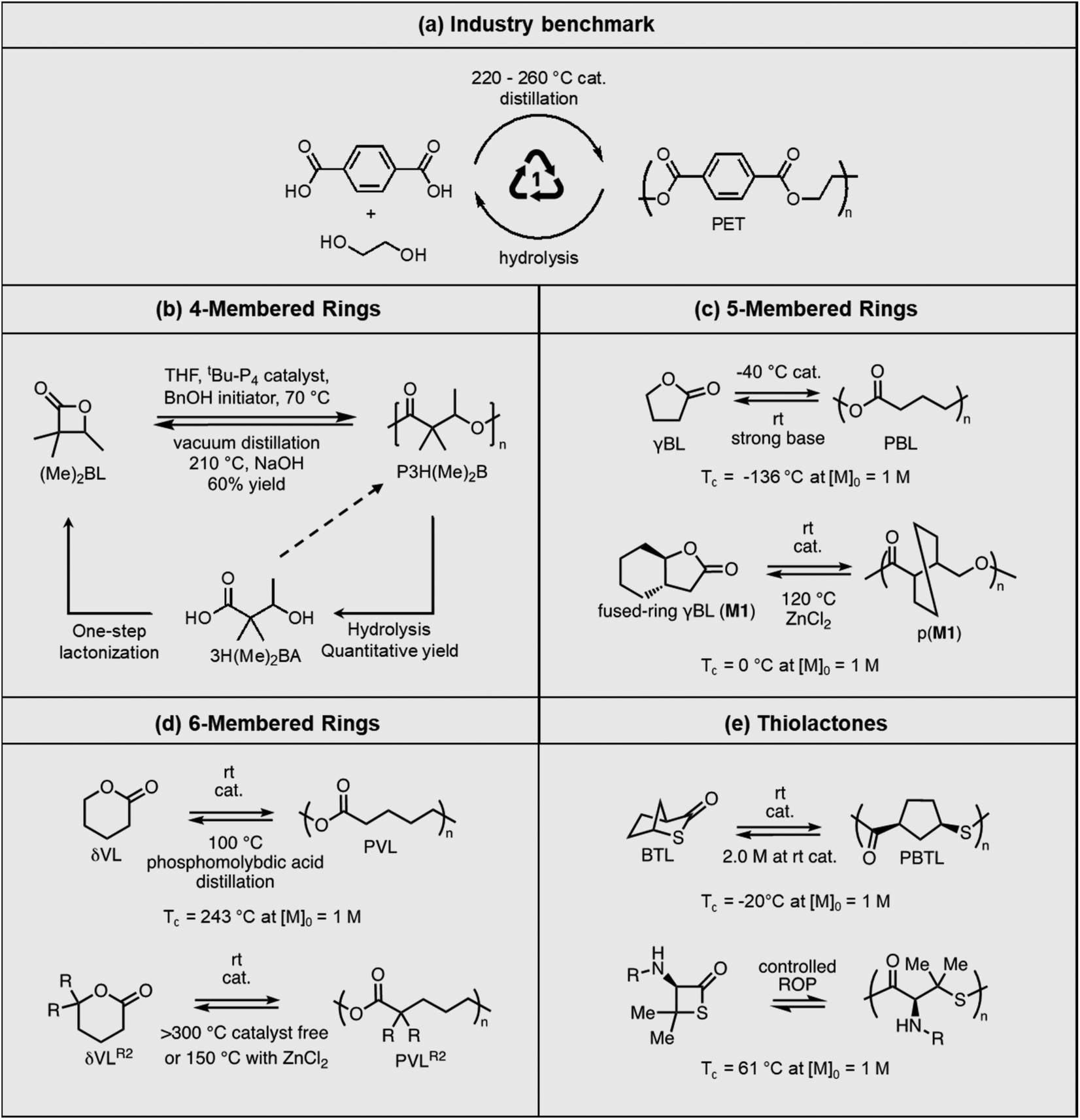 | ||
| Fig. 5 Polymerization and depolymerization of various polyesters. (a) Chemical recycling of PET currently used in industry. (b) Recycling between α,α-dimethyl-β-butyrolactone ((Me)2BL) and its polymer (P3H((Me)2B) (ductile; Tm = 243 °C; Td = 335 °C). c) Top: recycling between γ-butyrolactone (γBL) and its polymer (PBL) (brittle; Tg ∼ −52 °C; Tm ∼ 60 °C; Tc = −9 °C at [M]0 = 10 M); bottom: recycling between fused-ring-γBL (M1) and its polymer (pM1) (slightly ductile; Tg ∼ 50 °C; Tm ∼ 188 °C; Tc = 88 °C at [M]0 = 8 M). (d) Top: recycling between δ-valerolactone (δVL) and its polymer (PVL) (ductile; Tg ∼ −53 °C; Tm ∼ 57 °C; Td > 300 °C); bottom: recycling between gem-dialkyl-substituted δVL (δVLR2) and its polymer (PVLR2) (highly variable thermal and mechanical properties depending on the position and length of the R groups). (e) Top: recycling between 2-thiabicyclo[2.2.1]heptan-3-one (BTL) and its polymer (PBTL) (ductile; Tm = 166–213 °C; Td > 320 °C); bottom: geminal dimethyl substituted penicillamine-derived β-thiolactone (NR-PenTL) and its polymer (PNR-PenTL) (brittle at rt; Tg = 50 °C; Tm = 100 °C; Td = −192 °C). | ||
The first example shown in Fig. 5b is a new polyhydroxyalkanoate (PHA) synthesized from substituted four-membered β-lactone. PHAs have long been of interest as sustainable plastics because of their biorenewability, biocompatibility, and biodegradability in the ambient environment. They are typically biosynthesized from microorganisms, or abiotically via polycondensation of hydroxyacids. ROP of 4-membered β-lactones or 8-membered diolides are also commonly used. However, despite the extensive research,61,62 industrial applications of PHAs are limited by the long-standing challenges of lack of melt processability, mechanical brittleness, and unrealized closed-loop recyclability. The Chen group addressed these challenges using α,α-dialkylated β-butyrolactone ((Me)2BL) as the monomer, which eliminates the α-hydrogens in PHA.63 The backbone α-hydrogen causes the facile cis-elimination responsible for PHA's thermal instability. This causes the molecular weight to decrease during melt-processing conditions as often observed for unsubstituted PHA at only 180 °C. ROP of (Me)2BL proceeds with a superbase catalyst tBu-P4 {1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)phosphoranyliden-amino]2λ5,4λ5-catenadi(phosphazene)}, affording well-controlled molecular weight ranging from 43.0 to 554 kg mol−1. Compared to unsubstituted PHA, this new polymer exhibited 56 to 85 °C enhancement of thermal stability, enabling melt-processing. The polymer showed a rare case of tacticity-independent crystallinity with Tm of 167 and 176 °C for the atactic polymer, and it increased to 243 °C for the isotactic counterpart. The α,α-dialkyl substitution along with the high molecular weights also overcame the brittleness (Fig. 6a). Degradation was most efficient via hydrolysis in aqueous lithium hydroxide, affording the hydroxy acid (3H(Me)2BA). The 4-membered monomer can reform from the degradation product via a one-step cyclization. Although 3H(Me)2BA can directly polymerize via step-growth polymerization, it only yielded oligomers or low molecular weight polymers. Synthesis of the virgin (Me)2BL monomer was demonstrated on a 200 g scale, and the starting compounds can be bio sourced.
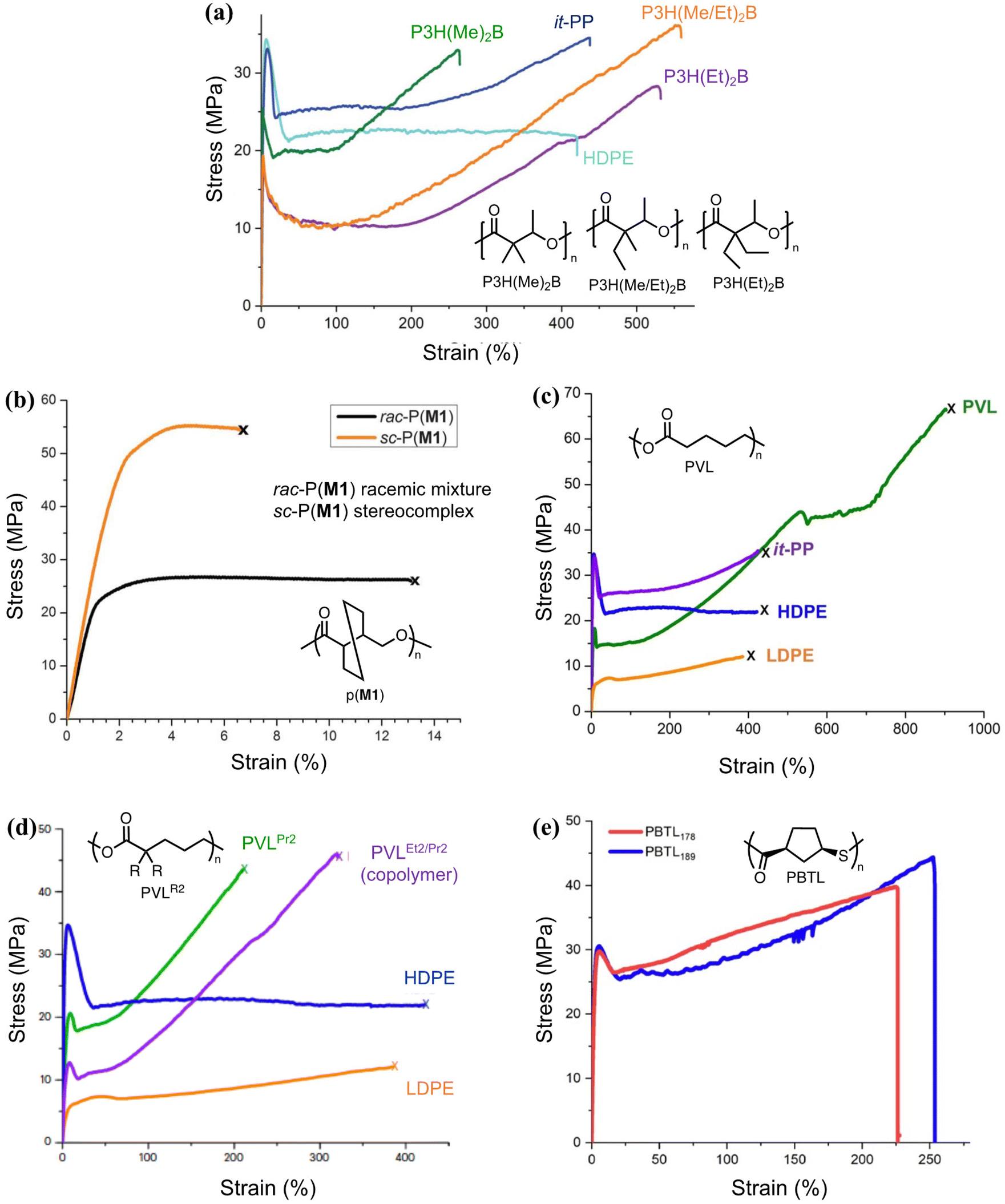 | ||
| Fig. 6 A side-by-side comparison of the tensile curves of various circular polyesters, polythioester, in comparison to isotactic polypropylene (it-PP), high-density polyethylene (HDPE), and low-density polyethylene (LDPE). Details about their syntheses and recycling properties are shown in Fig. 5. (a) is reproduced from ref. 63 with permission from AAAS, copyright 2023. (b) is reproduced from ref. 64 from AAAS, copyright 2018. (c) is reproduced from ref. 65 with permission from Wiley, copyright 2023. (d) is reproduced from ref. 66 with permission from Springer Nature, Copyright 2023. (e) is reproduced from ref. 67 with permission from AAAS, copyright 2020. | ||
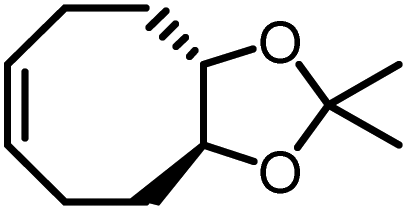
Opposite of the 4-membered β-lactones, 5-membered γ-butyrolactones (γBL) have less RSEs and were considered non-polymerizable. This limitation was tackled by polymerizing γBL at −40 °C.34 The low temperature compensates for the entropy loss. Solvent was selected so that the polymers formed precipitates from the solution in a crystalline state to further shift the equilibrium toward propagation. Under ambient pressure with a proper catalyst, this approach yielded 90% monomer conversion and polymer molecular weights up to 30 kg mol−1.34 Although Tc of the poly(γ-butyrolactone) (PBL) is low (−9 and −136 °C for [M]0 = 10 M and 1 M, respectively), the polymer was shown to be kinetically stable up to 300 °C without catalysts. Catalyzed by a strong base such as DBU, depolymerization proceeded rapidly within minutes at just room temperature due to the low Tc. Despite the success in polymerization, PBL suffers from low mechanical robustness and impractically low synthetic temperature. This was addressed by trans-ring fusion at the β and γ positions as shown in Fig. 5c, monomer M1, which effective increases the ring-strain energy.64 As a result, the Tc was enhanced by over 100 °C compared to γBL and polymerization of M1 proceeded at room temperature, reaching a much higher molecular weight of 875 kg mol−1 for the resulting polymer. The presence of cyclohexanyl groups in the backbone increased both the Tg and Tm by over 100 °C as well. The mechanical strength was improved; however, the maximum strain did not surpass 20% (Fig. 6b). With a higher Tc, the depolymerization temperature increased to 120 °C catalyzed by ZnCl2, which provides enhanced room temperature stability. It should be noted that the γBL monomer can be produced from succinic acid, a fermentation product from glucose.
The 6-membered lactone, δ-valerolactone (δVL) has a higher RSE compared to the 5-memberd γBL. It leads to a tendency towards ROP; however, with a high Tc of 298 °C at [M]0 = 1 M, the polymer is difficult to recycle. In this case, a kinetic approach that shifts the equilibrium to depolymerization by removing the monomer via solvent-free vacuum distillation proved to be feasible (Fig. 5d).65 Its success relies on highly active catalysts. With 2.0 mol% of ZnCl2 under 0.07 Torr pressure at 150 °C, which is below the Tc, the monomer was recovered in 99% yield. When catalyzed by an inorganic polyacid, phosphomolybdic acid (PMA), at 2.0 wt%, the depolymerization preceded at only 100 °C, recovering 98% of the monomer in 3.5 h. The use of more active catalysts allows for vacuum distillation at lower temperatures and improved energy efficiency.
The properties of PVL are tunable via ring substitutions. Monoalkyl-substituted δVL gives amorphous polymers, allowing for the use in soft elastomers.39 Racemic carbomethoxy substitution at the γ-position surprisingly gave a semi-crystalline polymer (Tm = −7–95 °C varied by the length of substituents).68,69 A strategy to further increase the melting transition temperature was demonstrated using gem-dialkyl-substitution that eliminated the stereogenic center. As shown in Fig. 5d, polymers formed from δVLR2 monomer exhibited a wide range of Tc from 67–115 °C and Tm from 44–140 °C depending on the length of the alkyl substituents, making it a highly tunable circular platform with mechanical properties comparable to polyolefins (Fig. 6c and d).66
The success of recyclable polyesters inspired the studies on polythioesters. Replacing oxygen with sulfur changes the monomer RSE. The 4- and 7-membered thiolactones readily polymerize via anionic70,71 or free radical initiated ROP,72 but 5- or 6-membered rings are difficult to polymerize.73 Thioester is more reactive than ester toward nucleophiles and makes a promising candidate for use in dynamic polymers.74 Although the 5-memberd γ-thiolactone is reluctant to polymerize, its bridged bicyclic form, 2-thiabicyclo[2.2.1]heptan-3-one (BTL), polymerizes at room temperature to high molecular weight poly(2-thiabicyclo[2.2.1]heptan-3-one) (PBTL) (Fig. 5e, top).67 With a low Tc of −20 °C at [M]0 = 1 M, PBTL depolymerizes easily at room temperature. PBTL also exhibits unique intrinsic crystallinity. The atactic PBTL has a Tm of 166 °C which increases to 213 °C with 100% tacticity, and the degree of crystallinity and tacticity can be fine-tuned within this range through catalyst control. The tensile properties of PBTL closely resemble polyolefins (Fig. 6e). Fully recyclable polythioesters have also been demonstrated for γ-thiolactone bridged with pyrrolidine, giving an amorphous polymer with relatively low dispersity (Đ = 1.32) even when reaching a molecular weight of 226 kg mol−1.75 β-Thiolactone (4-membered ring, Fig. 5e, bottom) modified with geminal dimethyl group and a secondary amine group also shown to be easily recyclable although the semicrystalline polymer is brittle.76 The ultimate advantage of the last two systems is that the amine groups allow for post-polymerization modifications.
There is one more interesting avenue of synthesis using lactones by employing a hybrid monomer. The Chen group developed a bicyclic lactone/olefin hybrid of γ-butyrolactone (γ-BL) and cyclohexene.77 This hybrid monomer can produce two different polymers depending on the catalyst employed: polyester via ROP (P(BiL![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) )ROP) and functionalized poly(cyclic olefin) via ROMP (P(BiL)ROMP) (Scheme 1). The polyester synthesized is semicrystalline with a high Tg (103–115 °C) and a Tm in the 141–239 °C range, while the poly(cyclic olefin) is amorphous with a Tg of 111–113 °C. The tacticity of polymers can be controlled via catalyst selection. Both polymers are thermally robust and depolymerize under mild conditions (25–40 °C) with the appropriate depolymerization catalyst. The hybrid monomer can then be cleanly recovered via chain unzipping (ROP) or scission (ROMP). The functional groups remain intact post polymerization (double bond in ROP, lactone in ROMP) allowing for further tuning of thermal and mechanical characteristics. The versatility of the hybrid monomer shows promise and demonstrates that it is possible to have a closed polymer system with the application of an orthogonal polymerization/depolymerization strategy and prompts the investigation into other hybrid monomer systems.
)ROP) and functionalized poly(cyclic olefin) via ROMP (P(BiL)ROMP) (Scheme 1). The polyester synthesized is semicrystalline with a high Tg (103–115 °C) and a Tm in the 141–239 °C range, while the poly(cyclic olefin) is amorphous with a Tg of 111–113 °C. The tacticity of polymers can be controlled via catalyst selection. Both polymers are thermally robust and depolymerize under mild conditions (25–40 °C) with the appropriate depolymerization catalyst. The hybrid monomer can then be cleanly recovered via chain unzipping (ROP) or scission (ROMP). The functional groups remain intact post polymerization (double bond in ROP, lactone in ROMP) allowing for further tuning of thermal and mechanical characteristics. The versatility of the hybrid monomer shows promise and demonstrates that it is possible to have a closed polymer system with the application of an orthogonal polymerization/depolymerization strategy and prompts the investigation into other hybrid monomer systems.
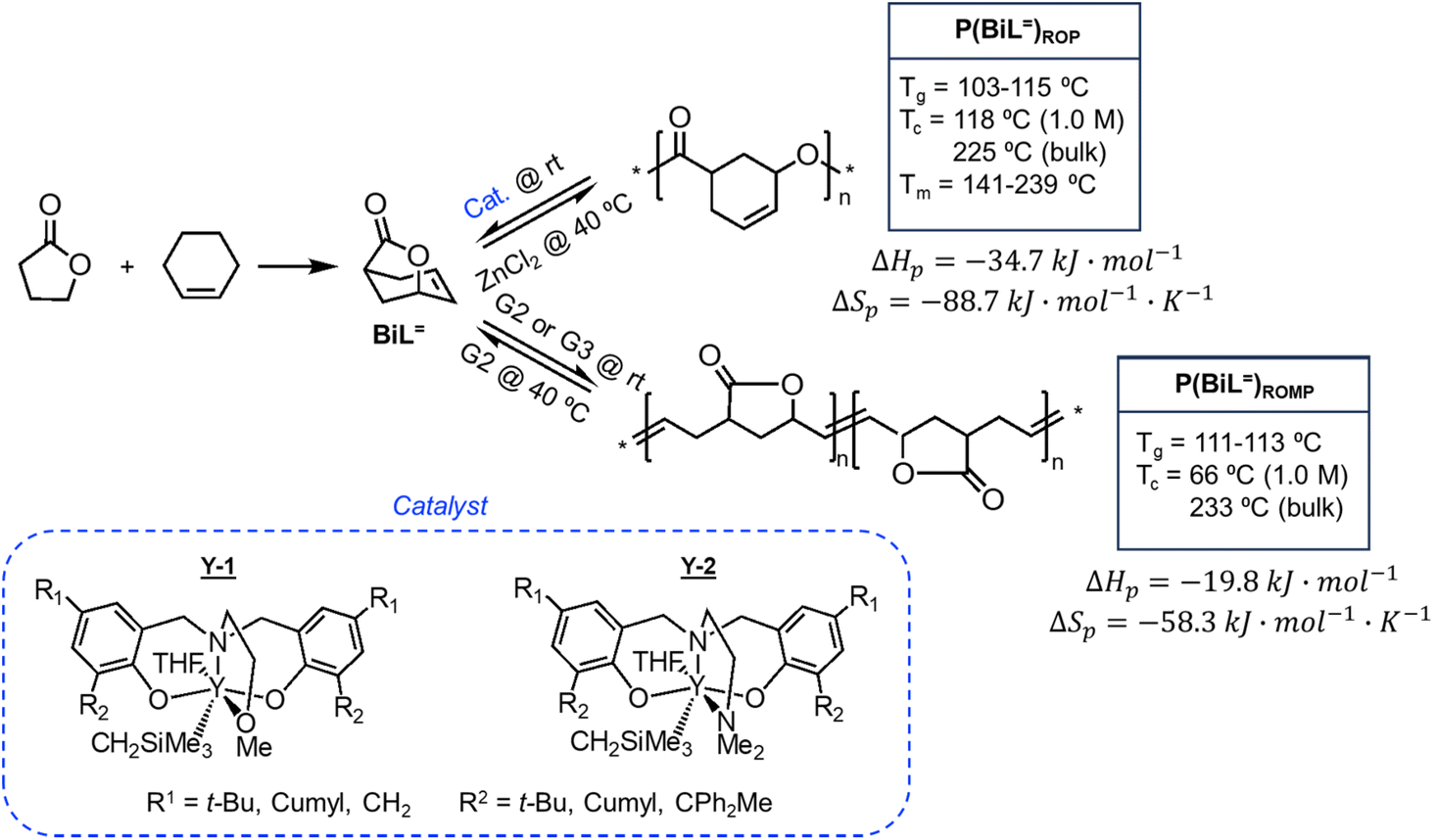 | ||
| Scheme 1 Hybrid monomer design and polymerization. Reproduced from ref. 77 with permission from American Chemical Society, copyright 2022. | ||
The studies discussed above showed similar characteristics for polymers derived from various lactones and thiolactones. They are strong, semicrystalline polymers with high melting transition temperatures. The Tm is adjustable via stereo-control and substitutions. Amorphous polymers form with large substituents. Fig. 6 which summarizes the tensile properties of the various polymers discussed in Fig. 5, shows that their Young's moduli, yield stress, and ductility are comparable or even surpass those of polyolefins, showcasing their potential as polyolefin replacements. The p(M1) is not particularly ductile but exhibits high tensile stress under low strain. It is noteworthy that the high mechanical performance is realized through not only the monomer design, but also synthetic techniques that involve the design of the catalyst, initiator, solvent, and reaction conditions to enable well-controlled polymerization leading to high molecular weight polymers. Their low molecular weight counterparts are usually brittle. Chemistry innovation is the key in such material designs even though details about the reaction systems are not discussed extensively in this review.
2.3 Ring-opening metathesis polymerization (ROMP) of cycloalkenes
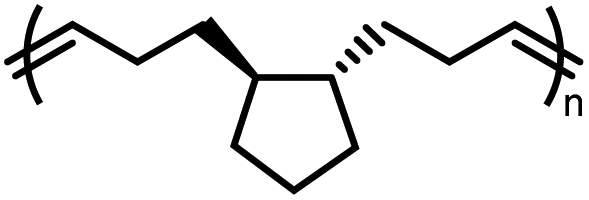
ROMP of cycloalkenes is particularly attractive for the design of recyclable polymers as it produces all-hydrocarbon backbones without heteroatoms. Upon complete removal of the catalysts, these polymers are suitable for applications where greater hydrolytic and thermal stability are necessary. ROMP is based on the mechanism of olefin metathesis of strained cycloalkenes catalyzed by a transition metal alkylidene complex, most famously the ruthenium-carbene Grubbs catalysts. This chain-growth polymerization produces stereoregular polymers with low dispersity, high molecular weight, and consequently outstanding mechanical performance being either rigid or flexible. ROMP has been used for industrial production of trans-polyoctenamer (a reactive plasticizer used in the rubber industry), polynorbornene elastomer, and poly(dicyclopentadiene) (for reaction injection molding of large engineering parts). Traditionally, ROMP is irreversible due to the release of significant RSE; however, by lowering the ring-strain, a near-equilibrium system that undergoes ring-closing metathesis (RCM) to regenerate the monomer can be realized.Rings such as cyclopropene and cyclobutene have RSEs that are too high to allow for realistic depolymerization, and the RSE of cyclohexene is too low to enable polymerization at all. With the relative rarity of cycloheptenes, that leaves cyclopentenes and cyclooctenes as promising candidates with the appropriate amount of RSE to enable recyclability.78
Polypentenamers from cyclopentenes have been shown to depolymerize since 1972.79 The early work on ROMP of cyclopentenes took place in industry driven by their similarities to natural rubber but was limited by synthetic challenges. Recent advances in synthetic approaches and understanding of the fundamental mechanisms allow the synthesis of polypentenamers with high molar mass, low dispersity, and precise microstructures.80–82 These polymers exhibit interesting elastomeric properties which strongly depend on trans/cis content and pendent groups. trans-Polypentenamers showed a strain-induced crystallization which provides self-reinforcement effect similar to natural rubber.83 Precise placement of phenyl pendent groups produced an ethylene–styrene copolymer structure that behaves as a strong elastomer.84 Polypentenamers synthesized from cyclopentene can depolymerize in solution.85,86 They have also found their way into bottlebrush copolymers that depolymerize via end-to-end unzipping instead of random scission like unmodified polypentenamers.87 Since polypentenamers are soft and amorphous, they are suggested as recyclable tire additives.88
The Xia group discovered that the five-membered enol ether ring, 2,3-dihydrofuran, can undergo ROMP with Grubbs catalysts,89 even though enol ethers have been used as quenching agents for Grubbs catalysts and thus viewed as terminating agents for ring-opening reactions as opposed to monomers. Thermodynamics for the ROP of 2,3-dihydrofuran is like cyclopentene and rubbery polymers with a Mn up to 127.7 kg mol−1 can be obtained at room temperature with a Tg = −50.5 °C and a subzero Tc. The deactivation of the catalyst following the metathesis reaction was done by cleaving the catalyst through peroxide oxidation and provides thermal stability up to 320 °C. Depolymerization in the bulk state at 60 °C by 0.01 mol% of Grubbs second-generation catalyst (G2) recovered >90% pure monomer after only 2.5 h. Its polymerizability is promising for a compound previously considered as only a quenching agent.
The ring strain energy of cyclopentene prevents bulk depolymerization of polycyclopentene under ambient conditions. The Moore group addressed this limitation by attaching bulky substituents directly on the cyclopentene ring.90 The substituents significantly lowered the Tc, with more pronounced decrease for bulker substituents, which can be attributed to a smaller loss of entropy (ΔSp). Depolymerization in the bulk was achieved in the 50–100 °C temperature range with different substituents. Crosslinked networks were formed with bifunctional and trifunctional monomers which proved to be thermodynamically stable and mechanically robust at room temperature, but readily depolymerizing above bulk Tc to give liquid monomers that can be repolymerized through cooling to room temperature again. Furthermore, degradation temperature can be fine-tuned by copolymerization of monomers that exhibit different Tc's.
Given the importance of substituents on changing the thermodynamics, the Kennemur group sought computational approaches to predict the RSE for a large set of substituted cyclopentene as a means to guide the monomer design.91,92 The DFT calculations were based on homodesmotic equations that capture the conformational changes upon ring-opening and side-chain rearrangements, providing the ability to calculate the effects of substituents on the RSE of a monomer. Fig. 7a shows an example of a homodesmotic reaction where the reactants and products contain equal numbers of carbon atoms and have the same hybridization state before and after, as well as the same substituents. By matching the hybridization and substituent groups, the enthalpy of the equation is representative of the ring strain energy in the cyclic monomer. Some of the screened cyclopentenes are shown in Fig. 7b. In general, it was found that the homoallylic (R3) substitution resultes in slightly larger RSEs than for alkenyl (R1) or allylic substitution (R2) across the board. R3 substituents experience higher repulsions from eclipsing interactions with the hydrogens on C2 and C4 positions. In addition, the RSE is most sensitive to the first atom of the substituent. Increasing atom size increases the bond length, thus decreases the RSE. Electronegativity generally increases the RSE in the R1 position, while the R2 and R3 substituents do not appear to follow a trend. Thus, in addition to developing and solidifying the reliability of a predictive method of RSE, the parameters that need to be considered when selecting monomers would be the size and substitution of the first atom primarily considered, and position (allylic vs. homoallylic) of the substituent secondarily considered.92
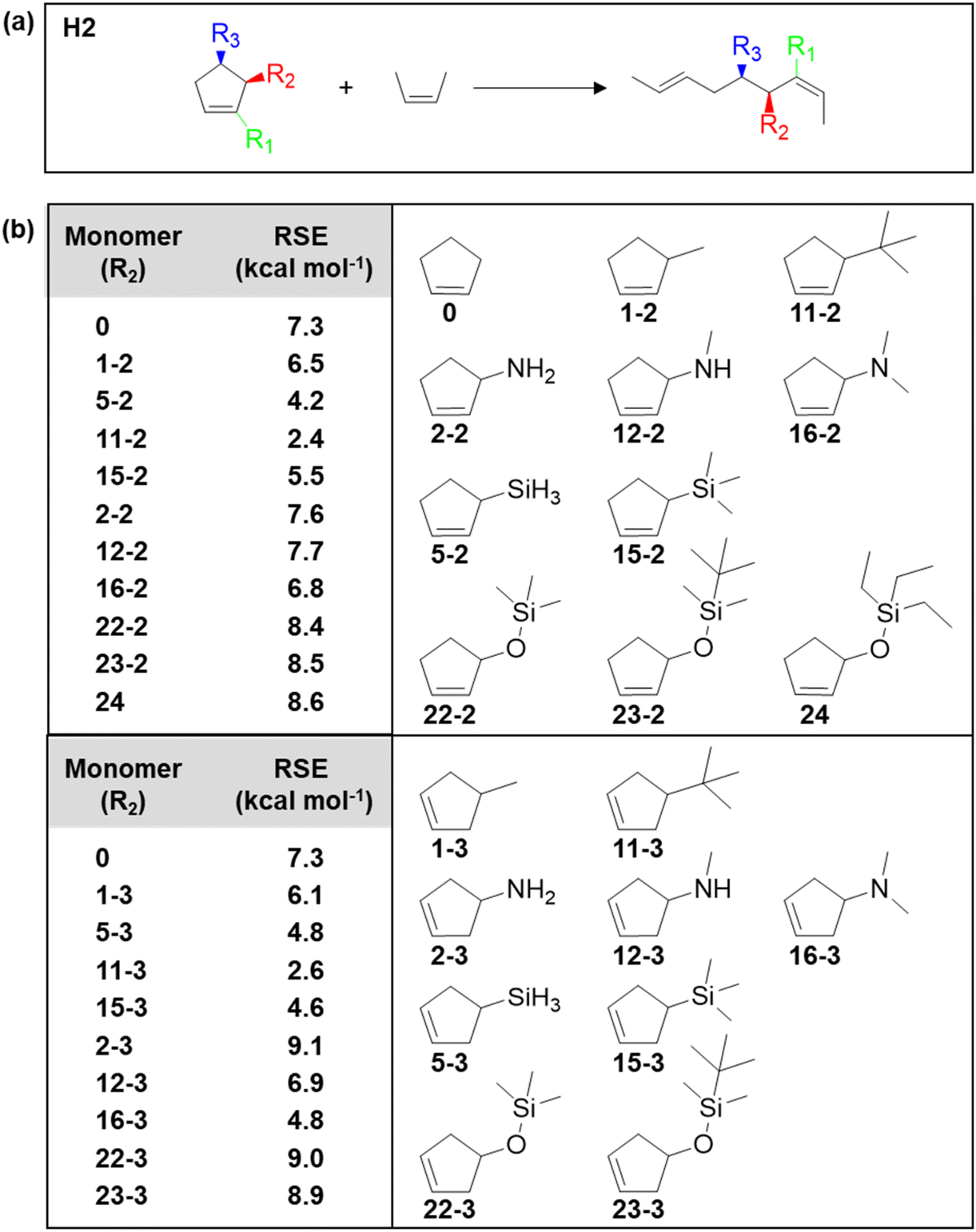 | ||
| Fig. 7 (a) Homodesmotic equations used to calculate RSEs. Substituent locations (R) are denoted as alkenyl (R1), allylic (R2), or homoallylic (R3). (b) Calculated RSEs of allylic (R2) and homoallylic (R3) monomers. The monomer structures correspond to the monomer RSEs in the table. Reproduced from ref. 92 with permission from American Chemical Society, copyright 2023. | ||
Different from polypentanemers which can serve as strong rubbers, ROMP of fused-ring cyclooctene produces rigid polymers with physical properties comparable to commodity plastics such as polystyrene.37,93,94 The ring-strain of unmodified cyclooctene is too high to enable efficient depolymerization; however, based on the hypothesis that the ring-strain of cyclooctene can be lowered by a fused-ring, the Wang group calculated RSEs for cyclooctene with 3-, 4-, 5- and 6-membered rings both cis- and trans-fused at the 5- and 6-positions using density function theory (DFT).93 The results identified that trans-cyclobutane (CB)-fused cyclooctene (Fig. 8a) has the lowest RSE, which was verified experimentally.
 | ||
| Fig. 8 Dihedral angles of substituted and non-substituted cyclooctene. Reproduced from ref. 93 with permission from Springer Nature, copyright 2021. | ||
The main contribution for the low RSE of trans-cyclobutane-fused cyclooctene is the change of tortional strain going from the cyclic to the ring-opened linear form. As shown in Fig. 8b, the non-substituted cyclooctene shows a significant change of dihedral angles H5–C5–C6–C7, H6–C6–C5–C4 and H5′–C5–C6–H6′ after ring-opening. In comparison, for trans-cyclobutane fused cyclooctenes (trans-CB), since the tortional strains are locked in the acyclic diene, the dihedral angles remain identical between the cyclic and acyclic forms. This leads to an overall reduction in RSE; however, for cis-cyclobutane fused cyclooctenes (cis-CB) the reduction in tortional strain is offset by the large distortion of the cycloalkane ring, which is released upon the opening of the cycloalkene ring.
A particularly valuable finding from this study is the importance of entropy change in the equilibrium of an enthalpy-driven ROMP. As shown in Table 1, the Wang group studied the structure–polymerization thermodynamics relationships for nine fused-ring cyclooctenes with varying fused-ring sizes and substituents.94M1ester (5.0 kcal mol−1) and M1imide (5.1 kcal mol−1) have lower RSEs than cyclopentene (5.2 kcal mol−1) and thus, lower ΔHp.93 One would usually expect them to have lower Tc's and be difficult to polymerize. But their Tc's are much higher than cyclopentene and can polymerize more easily. This is because of their less negative ΔSp values. The lower decrease in entropy upon polymerization can be attributed to the lower loss in translational entropy for the larger-sized monomers and the higher gain in rotational entropy due to the higher number of freed bonds in the eight-membered ring that were attained through the ring-opening reaction. Over 90% depolymerization into the corresponding monomers was achieved by heating these polymers (25 mM) at 50 °C with G2 (1 mol%) for 2 h. Increasing the fused-ring size from 4 to 5 increases the ΔHp, which is unfavorable for depolymerization. Interestingly, this can be compensated by substituents, which largely vary the ΔSp without significant effects on ΔHp. This is shown in Table 1, M2 and M3a–e. The gem-disubstitution of M3c and M3d reduced the Tc by ∼300 °C than the mono-substituted M3b, making them more depolymerizable. This Thorpe–Ingold effect which promotes cyclization is entropic in origin as the gem-disubstituted M3c and M3d have more negative ΔSp. The germinal substituents constrain the conformational freedom of the polymer, meaning less gain of rotational freedom upon polymerization, thus a more negative ΔSp dominated by the loss of translational entropy.
| Entry | Structure | ΔH (kcal mol−1) | ΔS (cal mol−1 K−1) | T c at 1.0 M (°C) | Entry | Structure | M n (kDa) | Đ | T d (°C) | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| M1 |
|
−2.1 ± 0.1 | −3.4 ± 0.3 | 335 | P1 |

|
58 | 1.61 | 391 | −55 |
M1ester![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |

|
−1.7 ± 0.3 | −3.6 ± 0.9 | 199 |
P1ester |
|
142 | 1.55 | 401 | 18 |
|
P1ether |
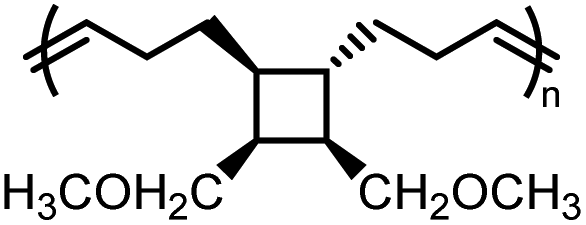
|
293 | 1.91 | 372 | −34 | |||||
|
M1imide |

|
−2.8 ± 0.5 | −4.9 ± 1.6 | 295 |
P1imide |
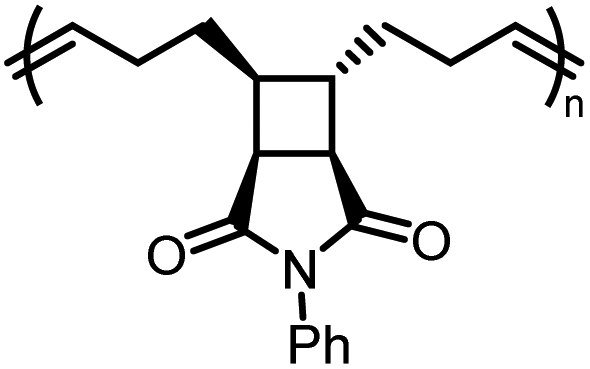
|
198 | 1.96 | 409 | 100 |
| M2 |
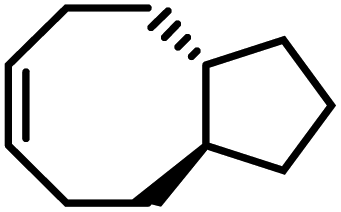
|
−2.9 ± 0.3 | −3.2 ± 0.8 | 614 | P2 |
|
82 | 1.64 | 405 | −33 |
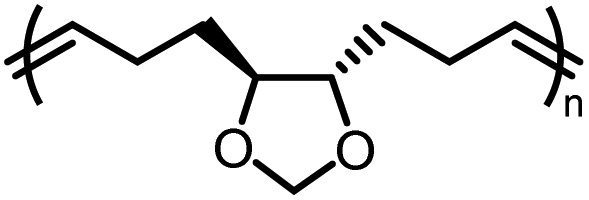
| M3a |
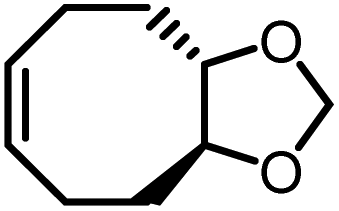
|
−2.7 ± 0.2 | −2.9 ± 0.7 | 646 | P3a |
|
171 | 1.71 | 360 | −30 |
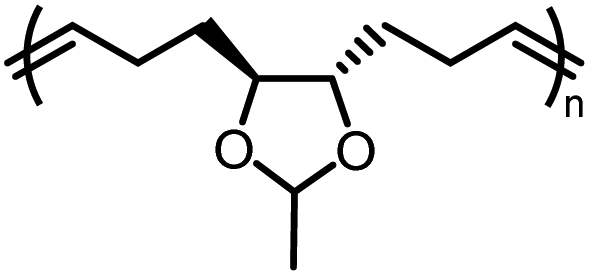
| M3b |
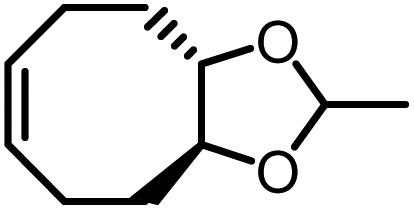
|
−2.6 ± 0.2 | −2.7 ± 0.7 | 675 | P3b |
|
102 | 1.78 | 367 | −14 |
| M3c |
|
−3.2 ± 0.1 | −4.9 ± 0.3 | 376 | P3c |

|
247 | 1.89 | 342 | −4 |
| M3d |
|
−3.3 ± 0.2 | −5.0 ± 0.6 | 380 | P3d |
|
164 | 20.8 | 327 | −27 |
| M3e |

|
−2.8 ± 0.1 | −3.3 ± 0.5 | 571 | P3e |

|
258 | 1.86 | 547 | 8 |
These polymers showed good thermal stability (Table 1) and strong mechanical properties in addition to their improved recyclability. Varying the substituents from flexible pendant groups such as butyl ester to rigid imide rings, the Tg can be tuned by a range of 155 °C from −55 to 100 °C.37 The crosslinked polymers made from the low Tg fused-ring cyclooctenes showed tough elastomeric characteristics with a tensile strain upwards of 200% and the tensile stress in the megapascal range. The imide functionalized ring resulted in a glassy polymer with tensile characteristics comparable to polystyrene.93
2.4 SNArROP
Nucleophilic aromatic substitution (SNAr) is a well-known dynamic covalent chemistry.95–99 In small molecules, SNAr is the displacement of a good leaving group, typically a halide, by a nucleophile. This forms an intermediate Meisenheimer complex100 that facilitates the switch between leaving group and nucleophile. SNAr has been utilized to synthesize high-performance industrial materials; however, there are few reported uses of its inherent reversibility.101,102 Gutekunst and coworkers have taken this idea of SNAr reversibility and combined it with ROP to facilitate depolymerizability using a cyclic aromatic thioether denoted as SNArROP (Scheme 2).103 The resulting polythioether has a Tg of 79.5 °C and expressed mechanical properties comparable to commonly used thermoplastics such as polyethylene, poly vinyl acetate, and poly vinyl chloride. The depolymerization proceeded through backbone cleavage with the addition of thiol and DBU followed by back-biting, showing a successful isolated yield of 85% at 90 °C and a concentration of 10 mg mL−1. This was confirmed by depolymerizing iodoacetamide end-capped polythioether, resulting in a 70% isolated yield. The polymers can be tailored with different R groups, possess high recyclability and impressive mechanical characteristics, ultimately showing promise for industrial applications.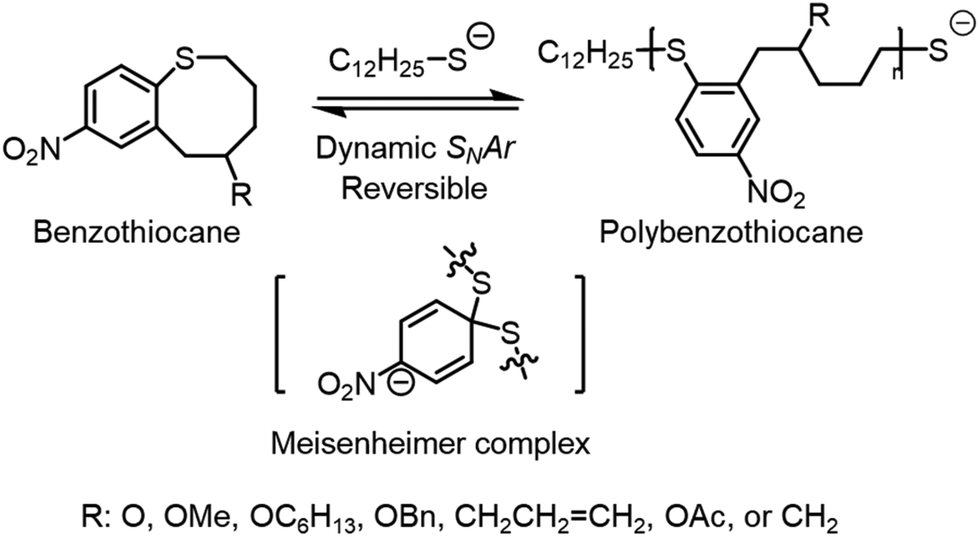 | ||
| Scheme 2 Nucleophilic aromatic substitution of cyclic aromatic thioether. Reproduced from ref. 103 with permission from American Chemical Society, copyright 2023. | ||
2.5 Polyacetals
Polyacetals are promising candidates for depolymerizable polymers due to the dynamics of acetal bonds in the backbones.104,105 Acetals undergo rapid exchange reactions under acidic conditions. Hydorlysis of non-cyclic acetal bonds in polymers readily proceeds under a pH of 7.4, leading to numerous biomedical applications.106 Cyclic acetal units are more resistant toward hydrolysis and provide strong mechanical stability but are still hydrolyzable with a strong acid.107 Due to their tunable degradability, polyacetals have attracted interest lately in advancing their synthetic techniques;106 however, a major limitation is the lack of molecular weight control. For example, poly(1,3-dioxolane) (PDXL) can be synthesized via ROP of 1,3-dioxolane cyclic monomer catalyzed by either Brønsted or Lewis acids, but its applications as bulk materials were limited by the low molecular weights (up to 44 kg mol−1).108A breakthrough over molecular weight control was made by the Coates group via reversible-deactivation cationic polymerization.109 The reaction uses halomethyl ether as the initiator (MOMX), a Lewis acid catalyst, and 2,4-di-tert-butyl pyridine as the acid trap. As shown in Fig. 9a, propagation is mediated by reversible activation of the dormant halide-terminated chain ends by a Lewis acid such as InX3 or ZnX2. In choosing the Lewis acid, selectivity toward chain-end activation relative to binding oxygens in the monomer or polymer is critical. GaCl3, SbCl5, SnCl4, and FeCl3 directly activated the monomer even in the presence of the initiator, and thus did not provide any control over the polymerization. The more labile Br leaving group increased the polymerization rate by 25-fold compared to Cl, giving high molecular weight semi-crystalline polymers in minutes at room temperature (Fig. 9b). Material properties improved significantly from brittle to tough as molecular weight increased from 37.9 kg mol−1 to 180 kg mol−1, with tensile properties comparable to isotactic polypropylene and high-density polyethylene (Fig. 9c).110 Dispersity values varied between 1.51–1.74 due to transacetalization, but it did not hinder the living nature of the polymerization or physical strengths of the resulting material. Even though acetals are susceptible to acid hydrolysis, the commercial success of polyoxymethylene (POM) as a highly crystalline rigid plastic demonstrates that they sustain sufficient chemical and thermal stability for real world applications. With complete removal of the catalyst, the polymer remained stable up to 350 °C.
 | ||
| Fig. 9 Poly(1,3-dioxolane) (PDXL) circular system (Tc = 13 °C). (a) Proposed mechanism for the reversible-deactivation polymerization of cyclic acetals. (b) Reaction rate increases when increasing the [Br]:[Cl] ratio in InX3/MOMX catalyst/initiator system. (c) Tensile properties of PDXL with molecular weight of 37.9 kg mol−1 (top) and 182 kg mol−1 (bottom). (d) Recovery of the monomer from a mixed commodity plastics feedstock. Recycling condition: PDXL 80–180 kg mol−1, acid resin (Dowex-50, 5 wt%, nonvolatile), mechanically stirred at 150 ± 5 °C, distilled under ambient pressure, monomer recovered in 96% yield and >99% purity. Reproduced from ref. 109 with permission from AAAS, copyright 2021. | ||
Depolymerization took place via RCD catalyzed by strong acids such as camphorsulfonic acid and diphenylphosphoric acid. The H+ activates oxygen in the backbone and initiates the RCD. A representative degradation procedure is shown in Fig. 9d. Monomer of high purity was recovered by distillation from a mixture of different plastics containing various dyes and additives due to the low boiling point of the 1,3-dioxolane monomer. Availability of the monomer, mechanical strength of the bulk polymer, high thermal and chemical stability, and the energy-efficient depolymerization to recover pure monomers from complex mixed waste, make PDXL an outstanding circular system.
2.6 Polydithiolanes
First discovered in the 1950s, ROP of 5-membered 1,2-dithiolanes provides unique opportunities as the polymerization and depolymerization can be conducted in water.111,112 Lipoic acid, also known as thioctic acid, serves as the monomer (Fig. 10) and it is widely available as a low-cost over-the-counter nutritional supplement. Deprotonation of the carboxylic acid with a base allows the monomer to dissolve in water, and ROP proceeds as water evaporates, producing a polymer with disulfide in the backbone and ionic carboxylate pendant groups, which assembles into a mechanically tough and strong material.111 The ROP can also be carried out by simple solvent-free melt polymerization at 70 °C and retain the carboxylic acid groups.112 Diluting the polymer to a lower concentration in basic aqueous solution lowers the Tc and induces RCD. Acidification of the solution changes the monomer from hydrophilic to hydrophobic, enabling monomer recovery by precipitation or solvent extraction. The study also demonstrated that depolymerization in water allows for the separation of additives and contaminants such as dyes, reinforcing agents, and flame retardants by simple filtration before the monomer recovery step since they are usually water insoluble. The wide availability of the monomer feed and robust polymerization conditions are promising for scale-up and commercialization.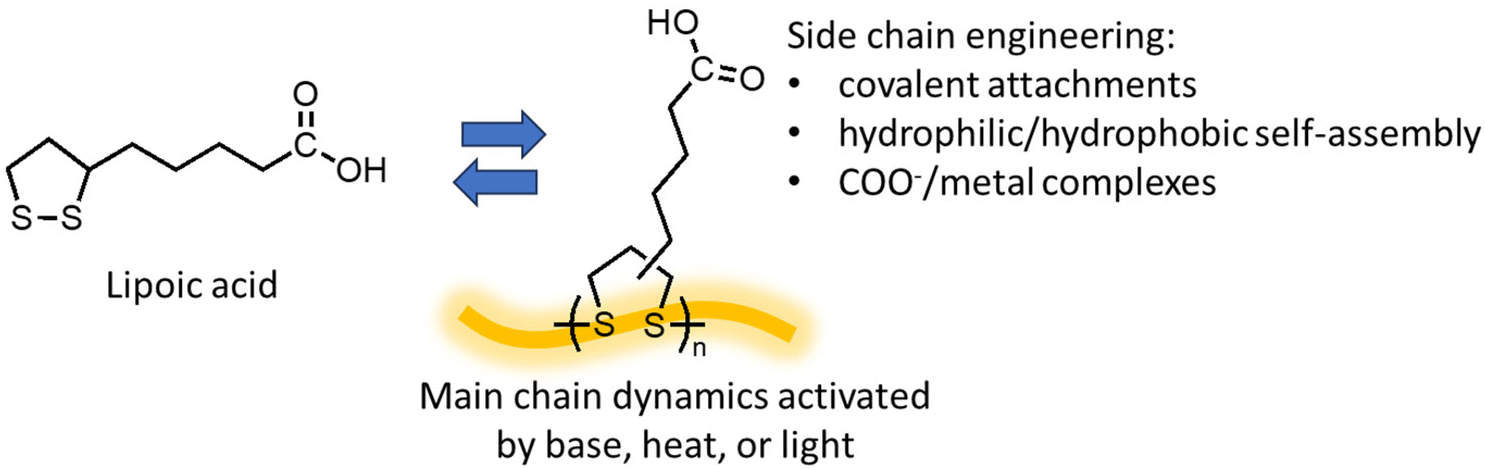 | ||
| Fig. 10 Graphical depiction of the polymerization of lipoic acid. | ||
Newly designed polymers should be intrinsically recyclable while serving as a platform for functionalization. 1,2-dithiolane polymers are such an example. The dynamics of the disulfide bond in the backbone in combination with substitutions via the carboxylic acid groups opens opportunities for material applications. Opportunities arise from the dynamic chemistry of 1,2-dithiolane polymers for the design of dynamic smart materials have been discussed in detail in a perspective by Tian.113 Both light and heat can activate the disulfide bond and induce polymerization, leading to photo and thermally responsive materials.114 Disulfide metathesis enables efficient self-healing that prolongs the lifespan of materials.114,115 With reducing agents, disulfide breaks and forms two thiols, providing another approach to depolymerize poly(1,2-dithiolane)116 and is applicable to removable adhesives.115,117 The side group modifications produce liquid crystalline elastomers,118 bottlebrush polymer networks,114 and cell-penetrating polymers.119 Covalent and carboxylate-metal noncovalent crosslinks can conveniently tune the polymer viscoelasticity.120 Synthetic methodologies that provide the control over polymer dispersity and topology (linear vs. cyclic) have also been developed.121 Meanwhile, the labile nature of the disulfide also challenges the durability of the polymers. Reaching a balance between durability and dynamic functionality requires new perspectives and approaches about the materials lifecycle.
2.7 Polyamides and polycarbonates
Due to the popularity of polyamides in consumer products, it is pertinent to briefly discuss research into potential application of polyamides in a circular economy. However, since many other reviews have covered polyamide synthesis122–124 and recyclability,125 this section will be kept brief. In the realm of polyamides, the current research is heavily geared towards depolymerization of post-consumer nylon-6 through RCD and development of its replacement. Several groups over the past few years have taken to finding and developing efficient catalysis to recover ε-caprolactam.126,127 Recovery yields have been improved through the use of lanthanide catalysts126 over ruthenium-based catalysts127 though both provide a method of chemically recycling post-consumer nylon-6. As for development of materials that are easier to chemically recycle, hybrid nylon has become a popular focus. Synthesized from bicyclic lactam with the properties of high Tc ε-caprolactam and low Tc pyrrolidone, hybrid nylon 4/6 shows promising characteristics. A copolymer made of nylon 4/6 and nylon 4 is more thermally stable than nylon 6 with Tg values 40–100 °C higher with the added benefit of accessible chemical recyclability.128Chemical recycling to monomers of polycarbonates has also been more extensively reviewed elsewhere,10,129,130 and thus much like our discussion on polyamides this will be kept brief. There has been a heavy focus on using renewable sources as feedstocks for polycarbonates made through ROP. Seven-membered fused-ring monomers derived from maleic anhydride, furan, and α-terpinene can undergo ROP to give aliphatic polycarbonates or ROMP to give poly(cyclic olefins) with a wide range of Tg values (80 °C to 200 °C).131 There has also been investigation into tandem ROP of imidazole carboxylate bearing cyclic carbonate with renewable alcohols of glucose tetraacetate, acetylated isosorbide, lauryl alcohol, and ethanol in the presence of an organocatalyst to generate recyclable functional polycarbonates.132 The chemical recyclability of these polycarbonates shows extreme promise, as many methods exist and are still being developed to push polycarbonates through RCD back to their cyclic monomers. Recent methods include using seven membered ring carbonates with trans-cyclohexyl fused ring.131 These monomers yielded high molecular weight polymers (Mn upwards of 650 kg mol−1), high thermal stability with degradation temperatures greater than 300 °C, complete depolymerization into dimers that can be repolymerized in moderate yields, and they can be functionalized post polymerization through crosslinking and UV induced “click” reactions. N-Heterocyclic carbene-mediated ring-expansion of cyclic monomers were also used to generate cyclic polymers through zwitterionic ROP. They gave high yield (>90%) and high molecular weight (110–150 kg mol−1) recyclable polymers via a “ring to ring” circular approach.133 Varying catalysts have been a target of method improvement as well with the goal of creating alternatives to toxic metal catalysts. An example of this is the synthesis of poly(cyclopentene carbonate) (PCPC) from copolymerization of CO2 and cyclopentene oxide (CPO). It is promising but ultimately was a challenging chemically recyclable polymer to make. The Wu group found a work around using metal-free catalysis via organoboron catalysts. Their method cuts out low reactivity toxic metal catalysts and increases catalytic efficiency (1 kg PCPC per g of catalyst). In addition, depolymerization returns CPO via a combination pathway of random chain scission and chain unzipping in near quantitative yield (>99%).134 As mentioned earlier, this is a sampling of recent research and there is much more research happening in the realm of polycarbonates that has been extensively covered elsewhere.
2.8 Polyoxalate
Polyoxalate synthesis has been reported since the 1930s by Carothers who found that the poly(ethylene oxalate) (PEOx) can be prepared from ethyl oxalate and ethylene glycol, which under distillation, depolymerizes into 1,4-dioxane-2,3-dione cyclic monomers along with other thermolysis products. The system was described as “the monomer polymerizes spontaneously, and purified polymers are partially depolymerized”. Other polyoxalates were synthesized by Carothers but all the polyoxalates synthesized at the time held no practical value as their molecular weights were low with low hydrolytic stability.135 Later work by Alksnis and McNeill around 1980s confirmed that the equilibrium relationship existed between the cyclic monomer and the polymer.136,137 Polyoxalates are known to have renewable feedstocks derived from biomass or CO2, their syntheses have been advanced to reach a molecular weight of ∼70 kg mol−1, a broad range of thermal properties can be obtained using different diol feedstocks, and hydrolysis produces nontoxic low molecular weight compounds suitable for biomedical applications.138 Driven by these advantages, Wei and coworkers investigated the feasibility of scaling up the synthesis of PEOx from dimethyl oxalate (DMO) and ethylene glycol (EG) in a melt polycondensation reaction (Scheme 3) to a 5 L scale, while retaining the chemical recyclability and material characteristics shown on benchtop experiments.139 The resulting PEOx has Tm up to 176 °C, Td,5% up to 227 °C, and elongation of break at 2.5%. The PEOx showed degradability via hydrolysis in simulated marine environments. After 24 days, 27% of PEOx was hydrolyzed, a characteristic of which is unachievable with most polyesters. Closed-loop recycling was initiated through high temperature thermolysis followed by pyrolysis and produced a white powder of the cyclic monomer 1,4-dioxane-2,3-dione. The recovered monomer polymerized spontaneously without the need for catalyst due to the high activity of the cyclic monomer. The material ultimately shows promise in not only CRM, but also in reducing the environmental impact of polymers that escape from a circular economy. | ||
| Scheme 3 Synthesis and closed-looped recycling of PEOx.139 DEG stands for diethylene glycol. Reproduced from ref. 139 with permission from American Chemical Society, copyright 2023. | ||
2.9 Entropy-driven ROP polymers
As discussed in section 2.1, ED-ROP of large strainless cyclic monomers has a unique set of design parameters that allows for the development of novel recyclable materials. Studies of ED-ROP are still in its infancy with regards to chemical recycling. Only a limited number of ED-ROP polymers have been reported so far40,41,51 and fewer studies on reversible ED-ROP. One such example is ED-ROP of macrocyclic carbonates for a fast and simple method to synthesize and recycle polycarbonates developed by the Odelius group.49 As shown in Scheme 4, the polycarbonates were first synthesized via polycondensation of various diols with diethyl carbonate catalyzed by sodium hydride. Interestingly, distilling the polycarbonate at 240 °C catalyzed by sodium hydride (NaH) formed dimeric macrocyclic carbonates as the primary products which can be collected using a sublimation apparatus in high yield. Monomeric cycles were formed simultaneously in smaller amounts as vapor which did not interfere with the crystallization of dimeric products. To assess the ROP behavior and recyclability of the macrocyclic carbonates, ED-ROP was done in tetrahydrofuran with benzyl alcohol initiator and the catalysts in Scheme 4 at ambient temperatures. The fastest polymerization was observed with sodium tert-butoxide (t-BuONa) catalyst, giving high monomer conversion (98%) within 3 seconds in ambient temperatures. In order of decreasing conversion, t-Bu-P4 and potassium tert-butoxide (t-BuOK) showed high monomer conversion within 10 seconds, where lithium tert-butoxide (t-BuOLi) showed no conversion at all. Thus, the recyclability of the polycarbonates back to monomers was established giving a promising method that can be scaled up for industrial use with relative efficiency.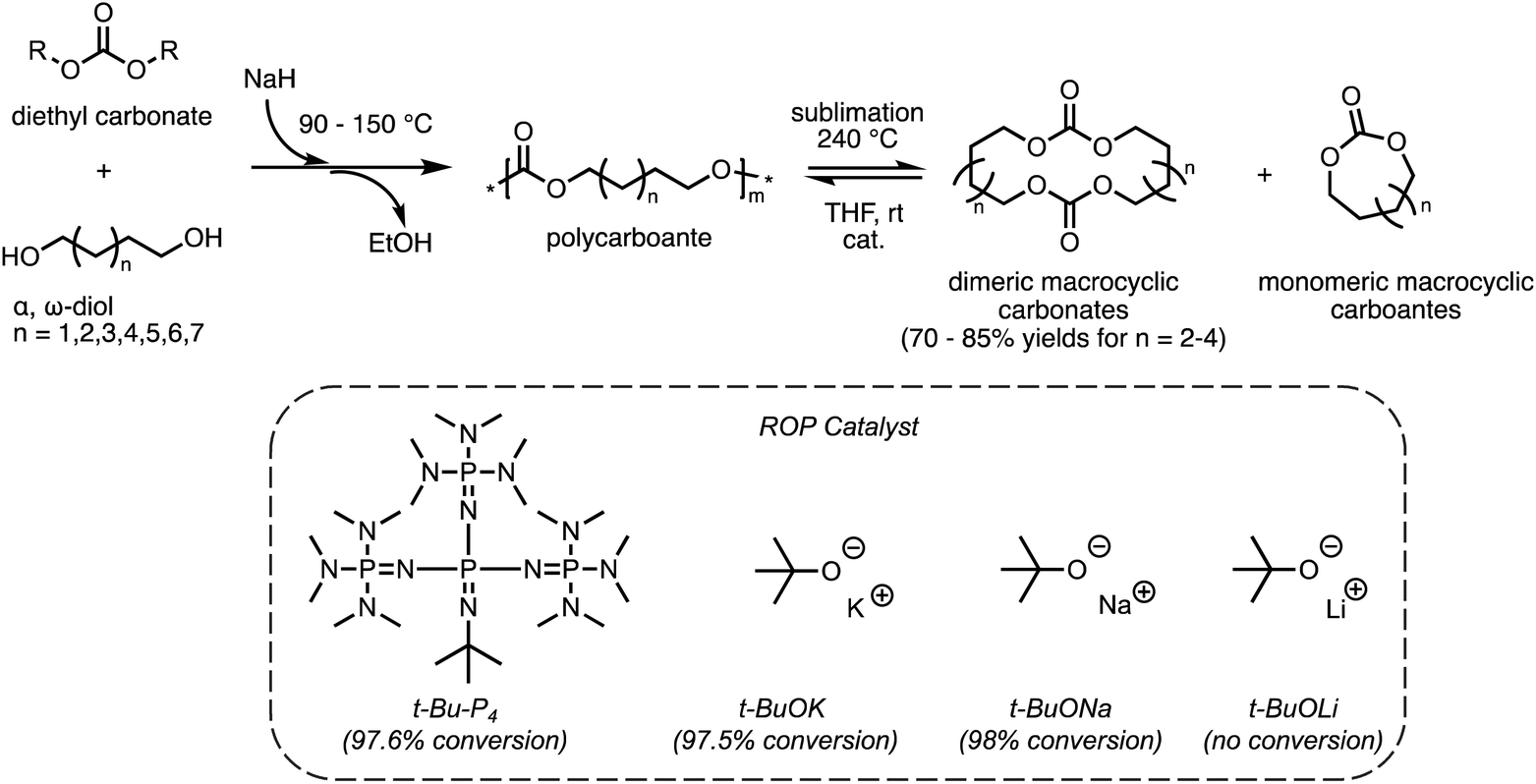 | ||
| Scheme 4 Polymerization pathway of polycarbonates from α,ω-diol and diethyl carbonate and its recycling via RCD and ED-ROP. Reproduced from ref. 49 with permission from American Chemical Society, copyright 2022. | ||
Polysilylethers can be made via ED-ROMP by bifunctional silyl ether-based monomers using a Grubbs 3rd generation bipyridyl complex (G3). Previously, the Johnson group had success with this method making copolymers of cyclic bifunctional silyl ether olefins with a variety of ROMP-active norbornene derivatives.140–142 Most recently they have taken an interest in the facile deconstruction of the homopolymer, for which the thermal properties are comparable to polydimethylsiloxane.43 Homopolymerization with 7 and 8-membered bifunctional silyl ether olefins were tested. The 7-membered olefin formed a cyclic dimer without polymerization; however, the 8-membered silyl ether polymerized with a molecular weight upwards of 150 kg mol−1 in 2 h (Fig. 11a). The homopolymerization is concentration dependent as demonstrated in Fig. 11b, indicating the sensitivity of the entropy-driven ring-chain equilibrium to concentration. In the depolymerization studies, a solution of the polymer treated with G3 showed a decrease in the chain size by an order of magnitude. The treatment led to the reformation of the monomers with a ∼28% yield with this method. The design of the polymer allows for an alternative pathway for depolymerization. When exposed to a Brønsted acid or fluoride ions, the polymer rapidly deconstructed via Si–O cleavage into cis- and trans-isomers of butene, pentene, and hexene. Repolymerizability of the recovered deconstruction products is theoretically possible, enabling them to be reused elsewhere.
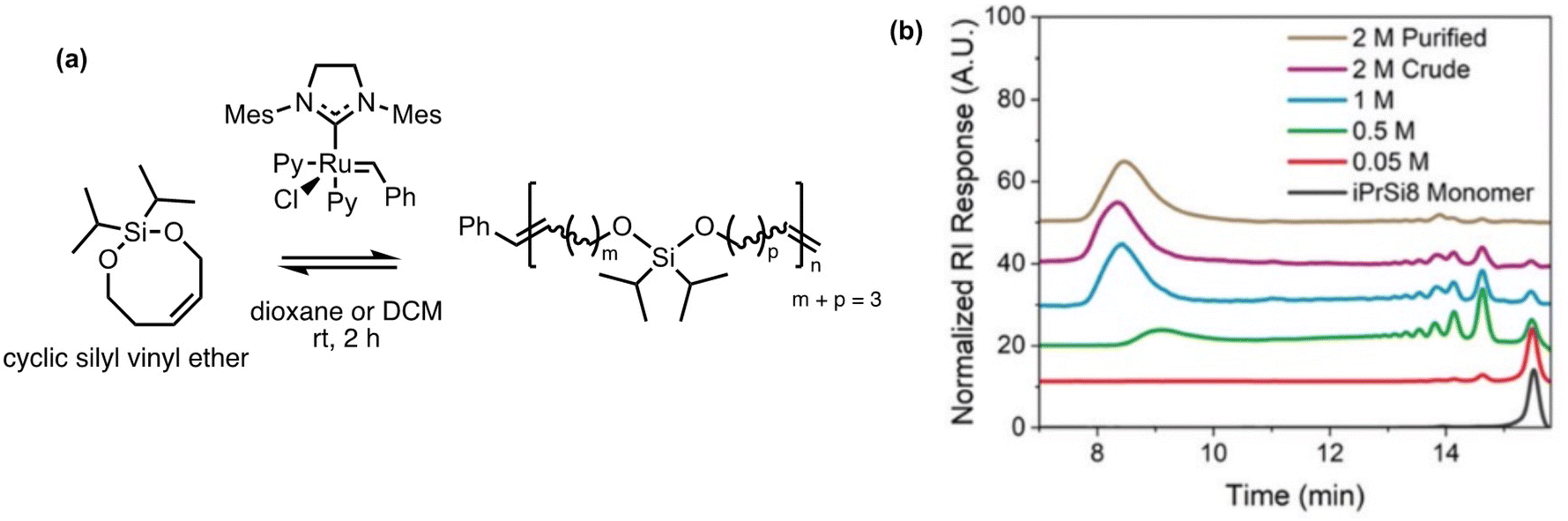 | ||
| Fig. 11 Homopolymerization of bifunctional 8-membered silyl ethers via ROMP.43 (a) Scheme of 8-membered silyl ether forming high molecular weight polysilylether. (b) Size exclusion chromatography of polysilylether prepared at increasing monomer concentration in dioxane. Reproduced from ref. 43 with permission from Royal Society of Chemistry, copyright 2022. | ||
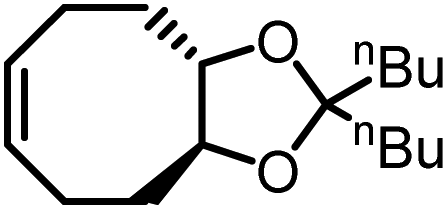
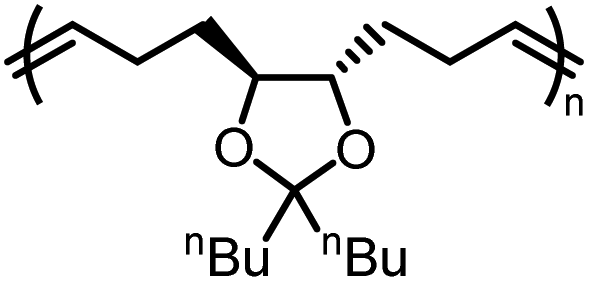
Our group demonstrated a new class of polymer, polydithioacetal (PDTA), that undergoes highly reversible ED-ROP cultivated by dithioacetal exchanges.143 Dithioacetals have been widely used in synthetic organic chemistry as a protective group but has received only limited attention in polymer chemistry. Dithioacetal is formed from the reaction of an aldehyde with two thiol groups. It exhibits higher hydrolytic stability compared to the O,O-acetal.144 Although dithioacetals are relatively stable, they can be activated by strong acid catalysts. Given their straightforward synthesis, tunability, and dynamics, we investigated dithioacetal as a reversible bond in developing recyclable polymers. We synthesized PDTAs using 3,4,5-trimethoxybenzaldehyde and alkyl dithiols catalyzed by p-toluenesulfonic acid (PTSA) via a step-growth mechanism. Chemical recycling is achieved through RCD of the PDTA to generate cyclic monomers which can then be repolymerized via ED-ROP (Fig. 12a). RCD takes place by diluting the polymer and refluxing it in toluene for 24 h catalyzed by zinc(II) triflate. The result is a mixture of macrocycles of various sizes, which is characteristic of an entropy-driven ring-chain equilibrium, in the form of a sticky white-yellow powder (Fig. 12b–d). The macrocycles could then be repolymerized back into the polymer at room temperature upon the addition of an acid catalyst. The recyclable PDTA system represents several advantages: (1) easy to synthesize – the polymers are formed via a one-step reaction between commercially available aldehyde and dithiol monomers, which then undergo closed-loop recycle via RCD and ED-ROP; the macrocycles can be repolymerized at ambient temperatures without isolating specific ring sizes and yield high molecular weight polymers; and the cationic ED-ROP can be conducted conveniently in the fume hood with all starting compounds and solvents used as purchased without further purifications; (2) functionalizable – the benzaldehyde and dithiol starting compounds provide chemical handles for versatile backbone and side-chain engineering, which opens opportunities for functionalization; (3) stability – a unique feature of ED-ROP is that polymerization is favored with increasing temperature and concentration, thus depolymerization at high temperature for bulk materials is not a concern, even in the presence of the catalyst which activates the dithioacetal exchanges. Due to the unique duality of structural stability and local bond dynamics, PDTA can be integrated into crosslinked networks to create recyclable thermosets which will be discussed in section 5.
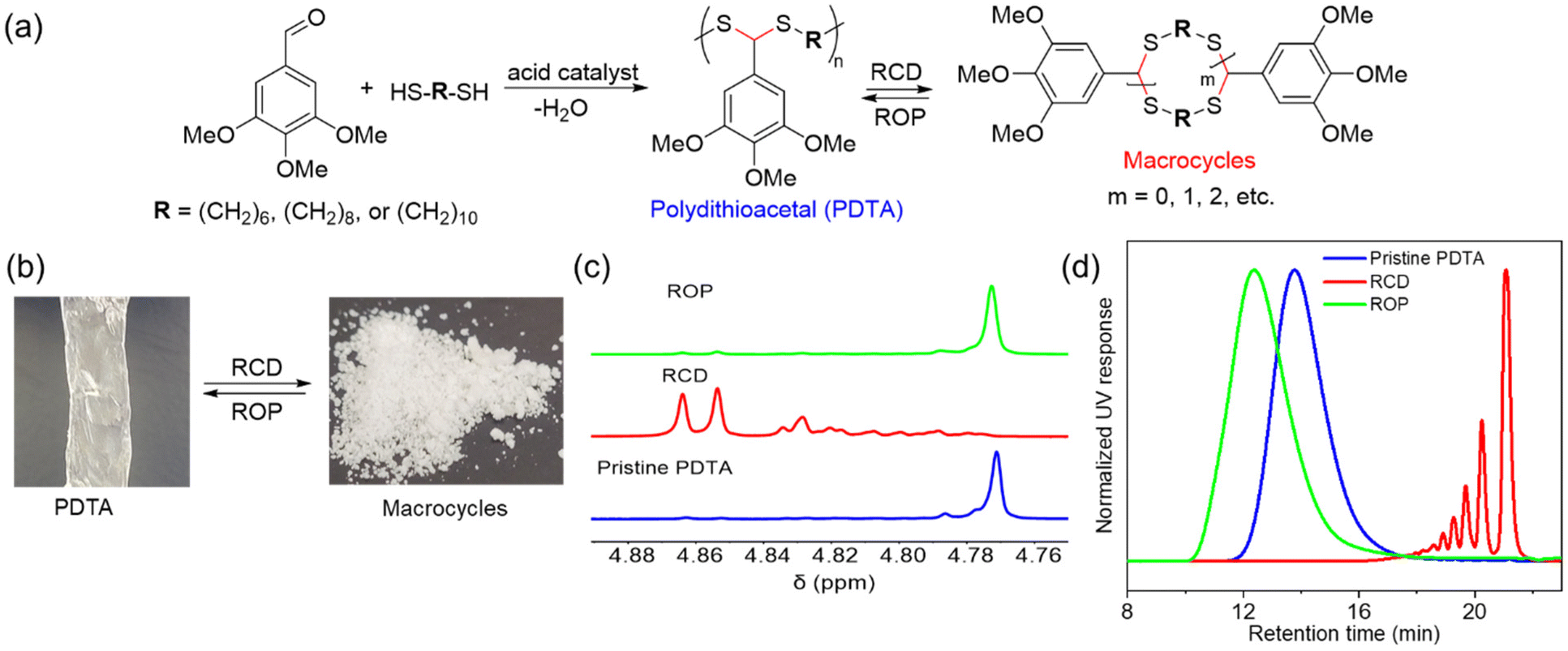 | ||
| Fig. 12 Chemical recycling of PDTA. (a) Scheme for the formation of pristine PDTA and its recycling via reversible RCD and ED-ROP of macrocycles. (b) Representative pictures of the pristine PDTA and macrocyclic mixtures formed after RCD. (c) Partial 1H spectra of pristine PDTA (blue), the crude macrocyclic mixture of various sized rings (red), and after ROP of the RCD mixtures (green). (d) GPC traces showing the normalized UV (254 nm) response as a function of retention time. The color coding is the same as for 1H NMRs. Reproduced from ref. 143 with permission from Wiley, copyright 2023. | ||
3. Reversing free radical polymerization
Vinyl polymers are known to be difficult to depolymerize due to their high Tc's that exceed the decomposition temperature, and the difficulty in activating the highly stable carbon–carbon backbone. In fact, for some, their Tc's are within reach for depolymerization such as disubstituted monomers and monomers with large substituents. For example, poly(methyl methacrylate) (PMMA), a common commodity polymer known as plexiglass, has a Tc of only ∼200 °C for [M]0 = 1 M.145 Thus, there has been a recent focus on developing methods to reverse the radical polymerization of methacrylic polymers to recover the vinyl monomers.One may question that with such a low Tc, why large-scale recycling of PMMA is still absent. In fact, PMMA synthesized by free radical polymerization must be heated ∼200 °C above its Tc to depolymerize. This is due to the high energy barrier to break the carbon–carbon bond to generate a backbone free radical that induces depolymerization.146 Once the free radical is formed, depolymerization occurs in an end-to-end fashion. An earlier study reported depolymerization of PMMA at lower temperatures during radical addition-fragmentation chain-transfer (RAFT) polymerization147 and a similar observation was made for atom transfer radical polymerization (ATRP).148 These depolymerization behaviors were observed under dilute conditions via re-activation of their labile end-groups – C–S homolytic cleavage and halogen abstraction.
A near-quantitative depolymerization was reported by Anastasaki group based on the hypothesis that a depolymerizable vinyl polymer should meet two pre-requisites: (1) a relatively low Tc that favors depolymerization thermodynamically, and (2) a labile bond, usually at the chain end, that generates a backbone free radical under mild conditions, to induce the depolymerization.149 The Anastasaki group addressed these two pre-requisites via (1) using dilute conditions to shift the reaction in favor of depropagation and (2) creating weak C–S bond at the chain end. The latter was achieved by synthesizing PMMA and PMMA-derivatives via RAFT polymerization with 2-cyano-2-propyl dithiobenzoate as the chain transfer agent (CTA) (Scheme 5). When heated, chain-end radicals are produced and trigger the depolymerization without any catalyst. The catalyst free RAFT reversal depolymerization strategy can be applied to linear, bulk, crosslinked, and functionalized polymethacrylic materials synthesized through RAFT with near-quantitative yield.
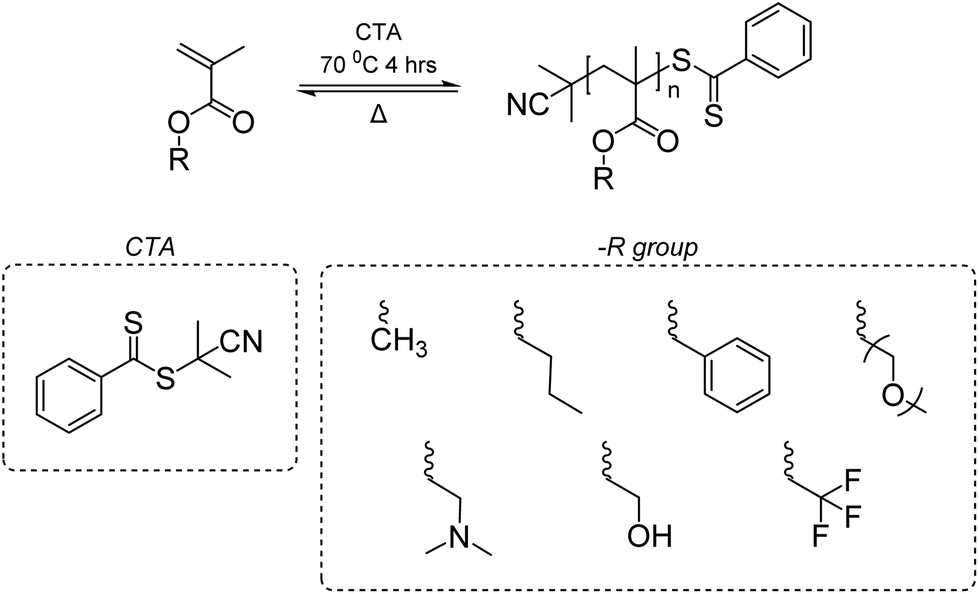 | ||
| Scheme 5 Polymerization and depolymerization of polymethacrylates via RAFT. Reproduced from ref. 149 with permission from American Chemical Society, copyright 2022. | ||
While the Anastasaki group demonstrated the feasibility of the depolymerization in dilution, solvent-free and catalyst-free depolymerization of PMMA can be done in bulk reactive distillation enabled by the integration of thermolytically labile end-groups. The Sumerlin group accomplished this using a combination of α-end functionalization with N-hydroxyphthalimide esters (Phth) and ω-end functionalization with trithiocarbonates.150 It was first discovered that PMMA functionalized with only one end group gives only partial depolymerization. Specifically, as shown in Fig. 13, PMMA terminated with Phth at the α-end was synthesized with an ATRP initiator. 65% depolymerization occurred via a mechanism of thermally activated decarboxylation of the Phth ester and subsequent loss of isobutylene from the α-chain end. ω-end terminated PMMA with trithiocarbonates was synthesized via RAFT polymerization, but only 42% depolymerization was reached from the cleavage of C–S bond at the ω-chain end. Based on these observations, a Phth-modified RAFT agent was synthesized to install thermally labile groups on both chain ends. Activation at both the α- and ω-chain ends significantly enhanced the extent of depolymerization to 92%. Depolymerization was performed at 210–220 °C, 250 °C lower than current industrial methods. This method can be extended to depolymerize ultra-high molecular weight difunctional PMMA (980 kg mol−1) at 41% depolymerization. The lower yield is consistent with highly dilute conditions where it is attributed to the loss of end-group fidelity when reaching ultra-high molecular weight.149
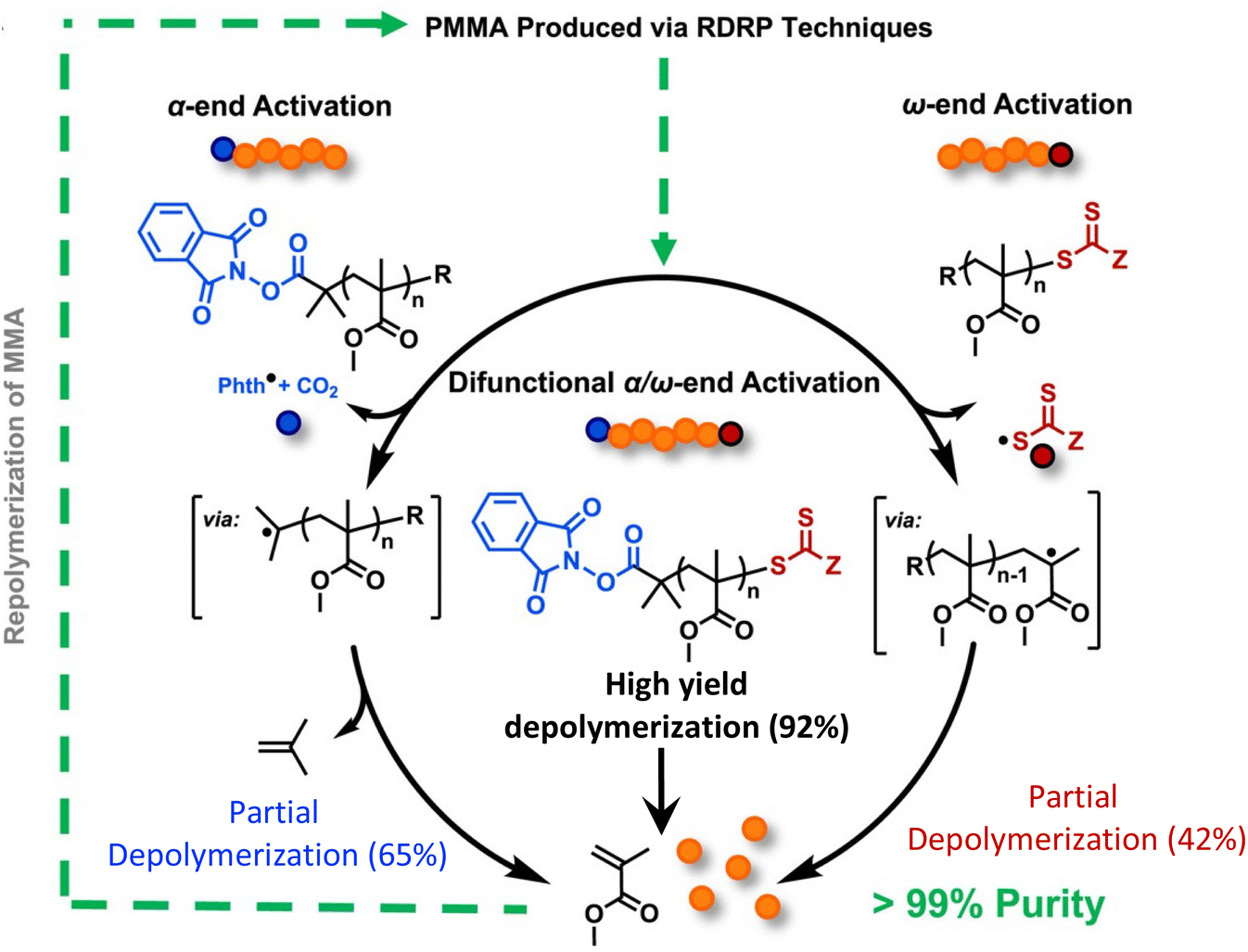 | ||
| Fig. 13 A scheme depicting the chemical recycling of PMMA to MMA via activation of the α-chain ends, ω-chain ends, or a combination of both. Reproduced from ref. 150 with permission from Elsevier, copyright 2023. | ||
Photo assisted RAFT depolymerization can be used to depolymerize polymers with thiocarbonylthio end-groups by leveraging photolysis to drastically increase the radical concentration compared to thermolysis on its own. Within 1 h, depolymerization can reach 87%.151 Light can be used to accelerate the RAFT depolymerization in the presence of Eosin Y, using a wide range of wavelengths as well.152
Depolymerization of ATRP polymers can be accomplished through activation of terminal groups by halogen abstraction. A reaction using an indenyl-based ruthenium catalyst depolymerized a narrowly distributed chlorine capped PMMA.148 The reaction, performed at 100 °C in solution, allowed the molecular weight to decrease without changing the distribution. Using a copper(II) chloride/tris(2-pyridylmethyl)-amine catalyst (CuCl2/TPMA), chlorine-capped poly(poly(dimethylsiloxane) methacrylate) (P(PDMS11MA-Cl)) bottle brush polymers153 and poly(n-butyl methacrylate) (PBMA)154 can also depolymerize into monomers. The efficient atom transfer between the alkyl halide and catalyst enabled 80% monomer recovery of P(PDMS11MA-Cl) and ∼70% n-butyl methacrylate within minutes. Increasing the amount of the ligand present during experiments with high catalyst loadings improved the yield and selectivity of depolymerization of P(PDMS11MA-Cl). When the catalyst loading was decreased, there was better depolymerization selectivity of the monomer over oligomers but this was at the expense of significantly reduced depolymerization rates. By the intrinsic virtue of halogen abstraction, only chains containing chlorine functionalities could be depolymerized enabling selective depolymerization in a mixture of functionalized and unfunctionalized polymers. The depolymerization can also be mediated by iron salts and iron powder, resulting in 75% monomer recovery with a more cost-effective reagent.155 CuCl2/TPMA mediated depolymerization also showed the ability to quickly depolymerize PMMA and poly(n-butyl methacrylate) in under 20 minutes using reactive distillation.156
4. Self-immolative polymers
Self-immolative polymers (SIPs) are a unique class of materials that are programmed to depolymerize in a controlled fashion through the removal of a triggering end group upon interaction with external stimuli. Removal of the stabilizing end groups results in domino-like cascade depolymerization. Since their introduction in 2008, the methods of depolymerization of SIPs have expanded from just quinone-methide elimination to include β-elimination of 4-hydroxy-2-butanone, 1,6-eliminations in polyanilines, and pinacol-borate head triggers in the presence of hydrogen peroxide just to name a few.157 They have been used as stimuli responsive materials and sacrificial layers for surface regeneration in materials sciences. The early developments centered on the concept of self-immolation around room temperature and volatile degradation products. With the latest growing interests in depolymerizable polymers for the purpose of recycling, the two fields certainly merge with a common principle of controlling the monomer–polymer equilibrium.158 Some of the polymers we have discussed so far depolymerize in an end-to-end fashion, such as the polymethacrylates, but at higher temperatures.There are two broad categories of SIP: irreversible and reversible.33 Irreversible SIPs depolymerize into monomers that are different than their starting monomers. They are prepared in a step-wise fashion and depolymerized through elimination or cyclization reactions. The most widely used irreversible SIP is poly(benzyl carbamate)s (PBC) derived from 4-aminobenzyl alcohol.159 Polycarbonates and variations have also been introduced to the field.33 Irreversible depolymerization of SIPs has found applications mostly as sensors and in drug delivery where recovery of the initial monomers is not required and sometimes undesirable. Reversible SIPs depolymerize into their original monomers, enabling repolymerization and chemical recycling, much like circular monomers used in reversible ROP reactions. They tend to have low Tc but are kinetically stabilized through their end groups to operate above their Tc. Their Tc's are usually below room temperature to realize rapid depolymerization under ambient conditions. SIPs have been reviewed in several papers.33,157,160,161 Although SIPs are usually synthesized at very low temperatures due to low Tc and are not all investigated for bulk material applications, the rich chemistry developed in this field over the years can potentially shine light on the design of next generation recycling polymers. Thus, we highlight a few selected examples of reversible SIPs.
4.1 Poly(phthalaldehyde)
Poly(phthalaldehyde)s (PPA)s are high Tg and low Tc (−40 °C) polymers. There are two paths to the synthesis of PPA from phthalaldehyde monomers: anionic polymerization giving linear PPA, and cationic polymerization generating cyclic PPA (cPPA).162 cPPA depolymerizes using thermally mediated cleavage of the acetal backbone to initiate ring-opening depolymerization (Fig. 14A(a)), thus lowering the kinetic barrier of the thermodynamically unstable polymer chain with a Tc of −42 °C.163 The low Tc favors rapid depolymerization to the monomer o-phthalaldehyde (o-PA). The monomer is volatile at the conditions required for depolymerization enabling the recovery of the monomer via a cold trap. This allows for cPPA to depolymerize and be separated from mixed waste streams including dyes (Fig. 14A(b)). The mechanical properties of cPPA is characterized with a tensile strength in the 40–50 MPa region and Youngs Moduli around 3.5 GPa; however, it is brittle on its own suffering break around 1.2% strain. Brittleness aside, cPPA has comparable tensile properties to industry standard thermoplastics such as polystyrene.163 Considering the problems facing the recyclability of fiber-reinforced polymer composites, the White group used cPPA as the bonding polymer matrix to create the composites. This addresses a major issue facing composite recycling, namely that the methods of recovering the fibers from the polymer matrix they are integrated into, such as pyrolysis or solvolysis, require high temperatures (>200 °C) that damage the fibers and alternative mild degradation conditions do not completely remove the polymer matrix, leaving fibers encased in residual resin. With the use of cPPA as the polymer matrix, the matrix can be removed without damaging the fibers or leaving residual polymer matrix on the fibers themselves (Fig. 14A(c)). | ||
| Fig. 14 (A) a. Thermally reversible polymerization of cPPA. b. The solvent cast cPPA films and the monomer (oPA) collected following depolymerization of the dyed films. Blue arrows indicate polymerization and red arrows indicate depolymerization. c. Photographs of a cPPA/carbon nano fiber composite film (left) and sublimation apparatus after heating the composite film to 120 °C for 1 h (right). Reproduced from ref. 163 with permission from American Chemical Society, copyright 2019. (B) Polymerization of a functionalized poly(phthalaldehyde) copolymer and subsequent cross-linking of the self-immolative backbone via thiol–ene photopolymerization. The green lines indicate pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) cross-linkers and the red stars indicate removable end-caps. Reproduced from ref. 162 with permission from Wiley, copyright 2021. (C) Polymerization and depolymerization of poly(ethyl glyoxylate). Reproduced from ref. 164 with permission from American Chemical Society, copyright 2016. (D) The solid-state synthesis of pDTT end-capped with pyridinic disulfide. Reproduced from ref. 165 with permission from American Chemical Society, copyright 2020. | ||
Another means to improve the mechanical strength of PPA is crosslinking. One such method is to make a copolymer of phthalaldehyde (PA) and allylated phthalaldehyde (Fig. 14B).162 The allylated comonomer enables photo induced crosslinking via thiol–ene click chemistry. The network showed the same trends of depolymerization with the caveat of showing oligomers once depolymerized which is attributed to the presence of thiol–ene crosslinks. This study also showed that the crosslinked networks can depolymerize with tunable stimuli dependent on the end-cap identity. Using tert-butyldimethylsilyl group (TBDMS) as the labile end group, the polymer is sensitive to acid and base, and depolymerized in under 10 minutes when added to either acidic (trifluoroacetic acid) or basic tetrabutylammonium fluoride (TBAF) solutions. Using 2-(2-nitrophenyl)propyl chloroformate (NPPOC) as a photosensitive labile group, the network depolymerized in a few hours after irradiation in a solution of dichloromethane/pyridine mixture.
4.2 Polyglyoxylates
Polyglyoxylates are soft polymers with low ceiling temperatures that can undergo depolymerization through chain end cleavage. The monomers used to make polyglyoxylates are commercially available or can be readily prepared from easy to obtain starting materials such as maleic or fumaric acid. They are recyclable via photodegradation of the end cap and the volatile ethyl glyoxylate monomers can be collected and repolymerized (Fig. 14C).164 It was demonstrated that the mildly acidic conditions used to cleave the backbones of polyacetals instead slow the depolymerization process as the hemiacetals cleave more readily under neutral conditions. In addition, the time to depolymerize was predictably dependent upon temperature and coating thickness and showed no dependency on the presence of water.4.3 Polydisulfides
A major drawback of SIPs is the low temperature required for their syntheses. Pedersen and Daasbjerg developed a method of synthesizing poly(DL-dithiotheritol) (pDTT) in the solid state at room temperature with high yield.165 Cyclic DL-dithiotheritol (cDTT) is difficult to polymerize via ROP due to its low ring strain. Inspired by the step-growth polymerization of aliphatic dithiol to form polydisulfides developed by Ghosh et al.,166 the solid-state synthesis was realized through a sequence of thiol exchanges (Fig. 14D). These exchanges occur at room temperature in the solid state and the molecular weight of these polymers is directly dependent on the excess of the end-cap moiety, 2,2′-dithiodipyridine (DTDP) (ζ) (Fig. 14D(a)). pDTT showed good chemical and thermal stabilities with a shelf life of over 300 days in bulk. Uncapping using DTT itself via thiol-disulfide exchange induced depolymerization via backbiting to generate the stable cyclic compound cDTT within minutes (Fig. 14D(b and c)). Depolymerization can be quenched at any given time by adding DTDP to reinstall the pyridinic disulfide end-caps. Treating the depolymerization product cDTT with reducing agents recovers the linear DTT monomers, which can be repolymerized to generate a close-looped system (Fig. 14D(d)).5. Design methodologies for thermoset polymers capable of depolymerization to monomers
The thermoset market, including mainly unsaturated polyester, epoxy, polyurethane, phenolic resin, and vinyl ester resin, accounts for 15% of the global plastic production.6 Different from thermoplastics thermosets in the form of composites or foams, serve as long-lasting structural materials used in construction, automotive, aerospace, and wind turbine industries. Their recycling is incredibly difficult due to the highly crosslinked structure. Wind turbine blades, for example, with an average length of 35 meters, are being landfilled or incinerated at their end-of-life. Technologies that rely on chemical recycling that break down thermosets into repolymerizable liquid products are being developed, including acidolysis, solvolysis, and enzymolysis to produce a mixture of monomers and other small molecules. Usually, monomers can only be partially recovered after separation and purification. Recently developed approach is to incorporate cleavable bonds into the network structure, so that crosslinks can be dissociated, and later being reformed, or degrade polymers into oligomers that can be repolymerized.162,167 Alternatively, chemically recycling to the same monomers offers its own advantage. Production of thermosets relies heavily on the processing, such as liquid molding and foaming. Recycled monomers with the same structures as the virgin ones ensure the same rheological properties suitable for production using the same infrastructure without reformulating or changing the properties of the final products. Herein, we summarize the chemistries that are currently being investigated for fully depolymerizable thermosets.5.1 Densely crosslinked polycyanurate network
Depolymerizable polycyanurate network developed by Lei group pioneered the idea of redesigning the synthetic route for the same polymer but with reversible chemistry.99 This approach uses retrosynthetic analysis and it pursues alternative synthetic routes for the existing thermosets. If the alternative route involves reversible chemistry, a traditionally un-recyclable polymer thus becomes recyclable. Scheme 6a shows the synthesis used in industry for the most commercially produced polycyanurate thermoset. The starting compound, bisphenol A, reacts with cyanogen chloride in the presence of a base to produce the cyanate ester, which then trimerize to form a cyanurate ring structure at a higher temperature in the presence of metal salts to form a cross-linked network.168 The high crosslink density and high aromatic content gives a Tg value that exceeds 250 °C, along with high thermal stability, low water uptake, small dielectric constant, and low dissipation factor, making it suitable for aerospace and electronics industries. However, the trimerization step is irreversible, limiting the polymer's recyclability. The Lei group redesigned the synthetic route by forming the triazine carbon and oxygen single bond via reversible nucleophilic aromatic substitution (SNAr) between alkoxy triazine and alcohol (Scheme 6b). The monomer, 2,4,6-tiethoxy-1,3,5-triazine (TETA) can be synthesized from industrially available cyanuric chloride. Polymerization proceeds via the SNAr reaction between TETA and diol catalyzed by a strong base, triazabicyclodecene (TBD). The use of different aliphatic and aromatic diols provides access for a broad range of glass transition temperatures. For example, when using 1,4-butanediol as the monomer, the network has a Tg of 65.5 °C, exhibiting properties of a tough and strong material with an elongation at break of over 45%, a tensile strength of 45 MPa, and a Young's modulus of 1.1 GPa. At their end-of-life, closed-loop-recycling can be achieved by refluxing in ethanol catalyzed by potassium carbonate (Scheme 6b). Within 16 hours, both monomers are recovered in ∼90% isolated yield. Alkyl polycyanurate has not been synthesized before due to side reactions involved in the traditional cyanate ester cyclotrimerization reactions. Thus, in addition to providing a sustainability solution, the chemistry innovation here expands to the library of polycyanurate networks to access more diverse material properties. | ||
| Scheme 6 Synthetic pathways for polycyanurate networks. (A) Network formation by cyclotrimerization of cyanate ester which is used for industrial productions but is irreversible. (B) Reversible network formation by nucleophilic aromatic substitution of TETA by a diol and removal of ethanol. Adapted from ref. 99 with permission from Springer Nature, copyright 2022. | ||
5.2 Polydiketoenamines and polytriketoenamines
This group of polymers represents the effort in designing new polymer networks with outstanding mechanical performance and recyclability. Leveraging on the reversible formation of diketoenamine, a series of polydiketoenamines (PDK) capable of depolymerization were developed, allowing recovery of the monomers in their pure states.169 Diketoenamine bond forms spontaneously from condensation of triketone and amines, while reverse reaction occurs by acidolysis. The polymerization and depolymerization chemistry along with the processes involved in separating the two monomers from the degradation products are shown in Fig. 15a.170 Highly crosslinked PDK networks can be conveniently formed by ball milling of a bifunctional triketone and a trifunctional amine crosslinker at room temperature without any solvents or catalysts, producing water as the sole byproduct. The crosslinked polymers in the powder form are then compression-molded into bars and films. This is enabled by the bond exchanges of diketoenamine, allowing it to be thermally reprocessed as a vitrimer. At their end-of-life, treatment with strong acids, such as concentrated HCl or H2SO4 aqueous solutions, induces complete depolymerization, producing insoluble triketone monomer which can be separated by filtration with the soluble amine remaining in the acidic solution. The ammonium monomer is then recovered by ion exchange.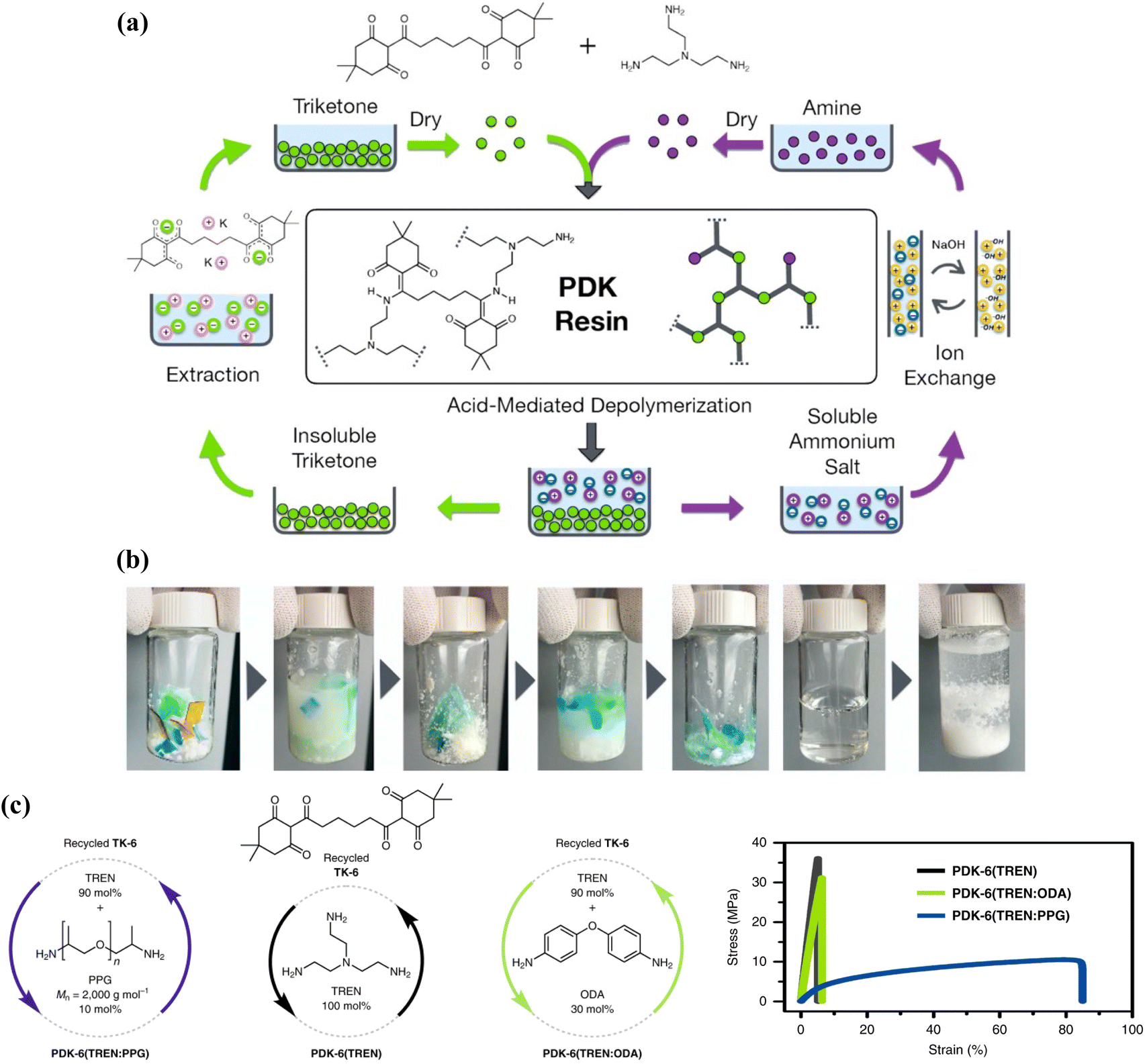 | ||
| Fig. 15 (a) Schematic illustration for the synthesis of PDK networks from a ditopic triketone (TK-6) and an amine crosslinker (TREN). The polymer depolymerizes back to the monomers via hydrolysis in strong acid. The two monomers can be separated based on their different solubilities. (b) Photographs showing orthogonal depolymerization of PDKs and triketone monomer recovery from mixed plastic waste containing poly(ethylene terephthalate) (PET), nylon-6,6 (PA), polyethylene (PE), poly(vinyl chloride) (PVC) and polycarbonate (PC). (c) Reformulation of PEK network using recycled TK-6 and different amines, and their corresponding tensile curves. Reproduced from ref. 170 with permission from Springer Nature, copyright 2019. | ||
The unique processing conditions for monomer recovery, owing to the pH-dependent solubility of the triketone in water, enables PDK recycling in the presence of any water-insoluble contaminants such as dyes, pigments, flame retardant, reinforcing agents, and mixed waste plastics. Specifically, these additives are mostly hydrophobic, so that after depolymerization, they end up in the filtrate along with insoluble triketone monomers (Fig. 15b). Extraction of the triketone into aqueous base allows removal of the contaminants, and subsequent acidification of the basic extract precipitates the pure triketone monomer.
PDK network is a versatile platform with tunable Tgs, mechanical properties, and degradation rates, due to the broad structural design space. Densely crosslinked PDK as shown in Fig. 15a has a high Tg of 125 °C. Crosslink density can be controlled by a combination of diamine monomer and triamine crosslinker, where rigidity of the diamine also dictates Tg and mechanical performances. The use of macromonomers with amine end groups is anticipated to open the door to even more variety of polymers. This is like polyurethanes, for which a broad range of products for different applications, from coatings, rubbers, adhesives, to foams, are developed relying on different polyols. For example, replacing 10 mol% of the triamine crosslinker with poly(propylene glycol) bis(2-aminopropyl ether) (PPG, Mn = 2000 g mol−1) yielded a ductile polymer (Fig. 15c). This showcases the design space in varying the mechanical characteristics based on the PDK platform. Polymers made from recycled monomers exhibit identical mechanical properties compared to the virgin one.
The structural variations also have dramatic impacts on the kinetics of degradation. With heteroatom substitutions on the triketone monomers, diketoenamines formed from TK1 and TK2 hydrolyzes >150 and >25 times faster than the one from TK4, respectively (Fig. 16a).171 The large rate difference allowed them to be recycled at different temperatures. Increasing the carbon spacing of the amine crosslinker from 2 to 3 also significantly decreases the depolymerization rate (Fig. 16b), which is surprising since the spacer is far from the diketoenamine reaction center.171 When a triketoenamine is formed from secondary amines and 2,4,6-triformylphloroglucinol, the absence of intramolecular hydrogen bonds further increase the rate of hydrolysis, and depolymerization completes within two hours under otherwise similar conditions (Fig. 16c).172 NaOH solution also induces complete depolymerization of polytriketoenamine to the monomers at an even faster rate. The underlying mechanisms determining the rate of hydrolysis for these systems were studied in detail using DFT calculations.
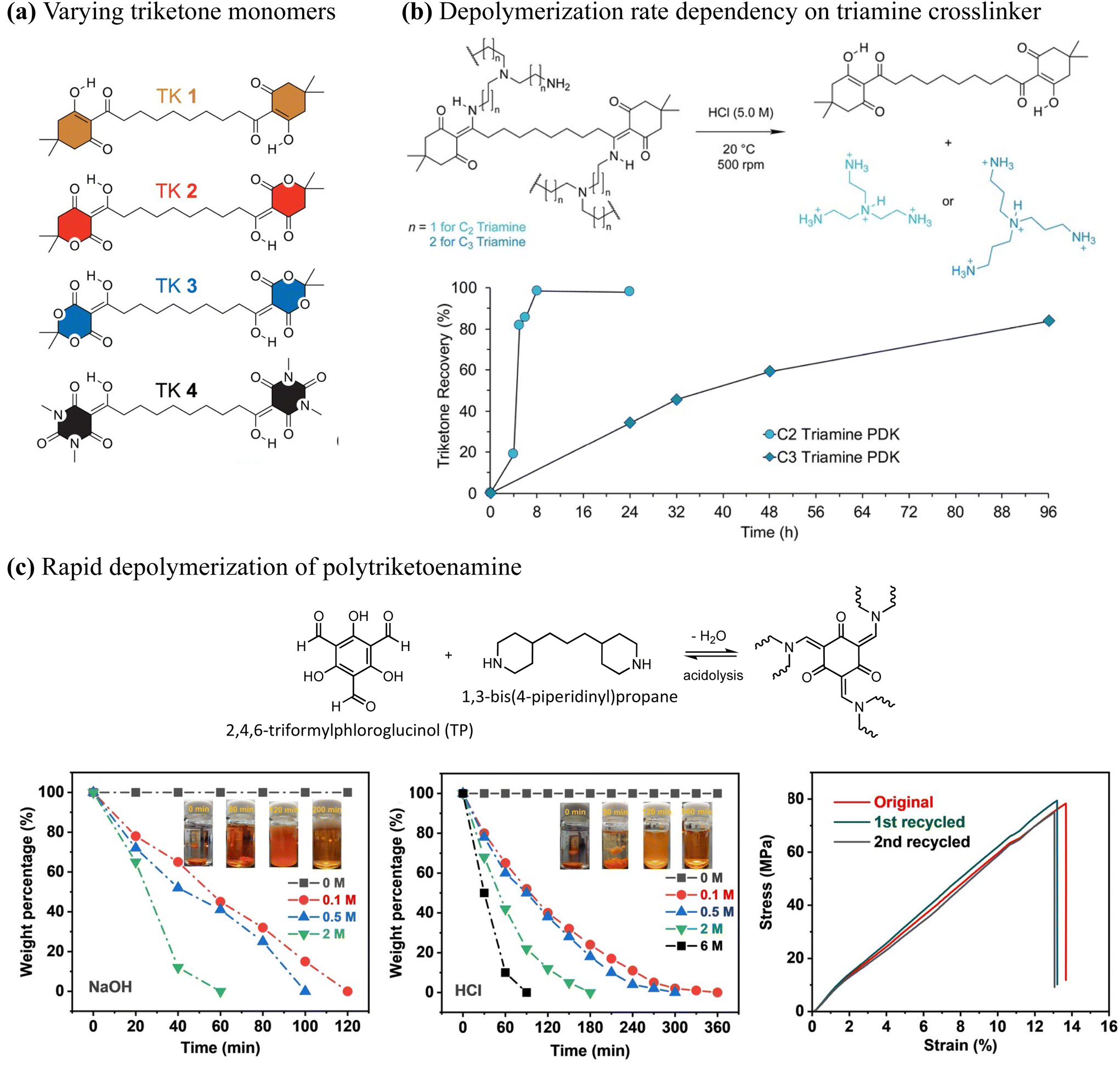 | ||
| Fig. 16 (a) Structures of triketone monomers with heteroatoms. Adapted from ref. 173 with permssion from AAAS, copyright 2022. (b) Depolymerization scheme with varying structures of the amine crosslinker (top) and their corresponding depolymerization over time (bottom). Adapted from ref. 171 with permission from American Chemical Society, copyright 2023. (c) Synthesis and hydrolysis of polytriketoenamine (top). Their degradation in basic solution (bottom left) and acidic solution (bottom middle). Tensile properties of original and recycled polytriketoenamine (bottom right). Adapted from ref. 172 with permission from American Chemical Society, copyright 2023. | ||
5.3 Depolymerization of covalently adaptable networks
Covalently adaptable networks (CANs) are materials that combine the durability and stability of crosslinked thermosets and the reprocessability of thermoplastics. They contain exchangeable bonds that respond to various stimuli (heat, pH, light, etc.) and induce inter-chain exchanges, effectively enabling plasticity and reshaping on a molecular level.99,170,174–183 This can be accomplished in one of two ways: dissociative mechanisms (i.e., elimination–addition mechanism) or associative mechanisms (i.e., addition–elimination mechanism). Dissociative CANs are known as reversible networks and can show a decrease in viscosity while stimuli are applied due to the drop in network connectivity. Associative CANs are known as permanent networks or vitrimers. These preserve their network integrity while stimuli are applied and do not experience a drop in viscosity or gel-to-sol transitions.184 Since some of the dynamic exchangeable bonds used in CANs are also reversible, complete depolymerization of CANs into the monomers is feasible. As such, thermoset wastes can be mechanically reprocessed and remolded for multiple cycles till significant deterioration of properties occur, they can then be depolymerized to monomers to remake high-quality products. In fact, the depolymerizable networks that as discussed above, including diketoenamine polymers developed by Helms group and the polycyanurate network from the Zhang group, all exhibit such properties. Several studies on vitrimers also reported complete depolymerizability.175,176,179,185 However, investigations on close-loop recycling of vitrimers, and CANs in general, are lacking. With this consideration, the Weder group selected vinylogous urethane vitrimers as a model study to demonstrate the potential of closed-loop recycling of vitrimers.185 Vinylogous urethanes are formed from the reaction of bis(acetoacetate)-terminated polyol building block with amine-based crosslinker (Scheme 7a). This material was first developed by the De Prez group186 and was selected in this model study for its ease of synthesis, high processibility, and broad applicability which have been demonstrated in prior studies.187–192 Transamination in the presence of excess amines at high temperatures (60–150 °C) enables the thermal reprocessing without de-crosslinking (Scheme 7b, left). Depolymerization to monomers is realized via hydrolysis at room temperature (Scheme 7b, right). If vinylogous urethanes are synthesized from hydrophilic polyol like poly(ethylene glycol), depolymerization proceeds in simply water at rt, allowing 97–99% recovery of the polyol and 88% recovery of the amine crosslinker of high purity after treatment with acidic ion exchange resin. When hydrophobic polyol such as poly(tetrahydrofuran), the polymer remains stable in neutral or basic water, but depolymerizes in 1 M HCl aqueous solution at rt.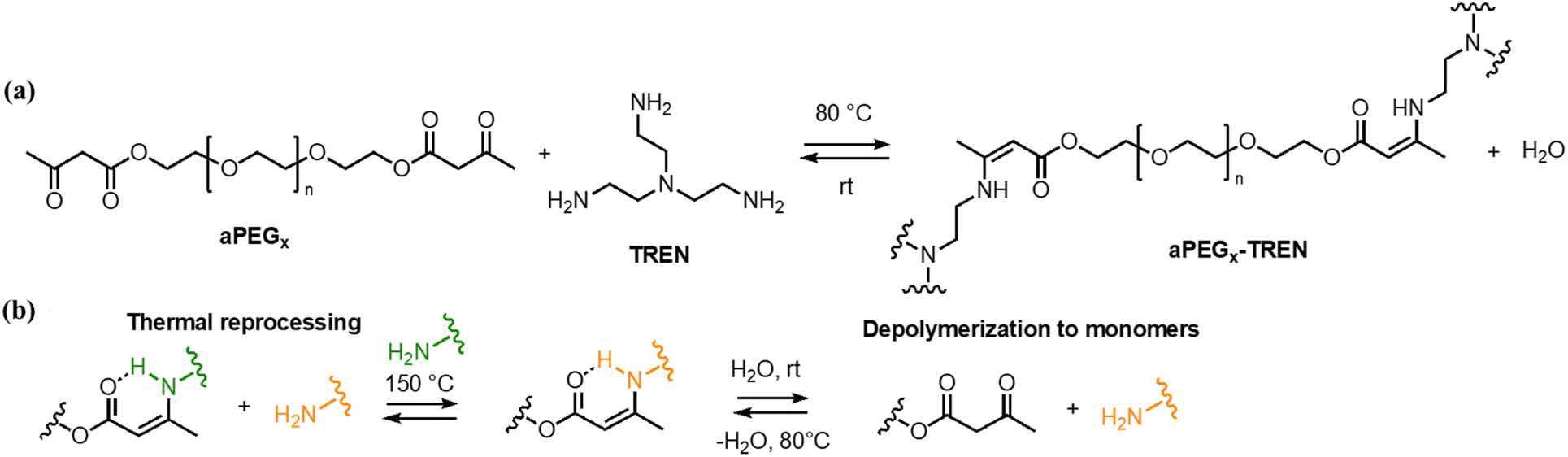 | ||
| Scheme 7 (a) Synthesis of the vinylogous urethane network from bis(acetoacetate)-terminated poly(ethylene glycol) (aPEGx) and tris(2-aminoethyl) amine (TREN) as the crosslinker. (b) Transamination between an amine and a vinylogous urethane (left) and the reversible hydrolytic depolymerization to the parent monomers (right). Reproduced from ref. 185 with permission from Wiley, copyright 2023. | ||
5.4 Depolymerization via ring-closing depolymerization
As discussed in section 2, numerous depolymerizable polymers have been developed based on reversible ROP and RCD reactions. Hypothetically, crosslinked networks should retain their ability of RCD. This was first demonstrated by the Moore group with the reversible ROMP of cyclopentene derivatives in the bulk state which was discussed in section 2.3.90 The network reversibly changes between solid crosslinked state at low temperature and liquid depolymerized state at temperatures above Tc. Key findings to note is that compared to monofunctional cyclopentene derivatives which form linear polymers, bifunctional and trifunctional monomers that contain two and three cyclopentene rings linked by a spacer are more polymerizable characterized by higher monomer conversions under the same conditions. This also results in higher depolymerization temperatures. The polymerizability is highly sensitive to the anchoring groups on the rings but not so to the overall length of the spacer.For entropy-driven systems with a Tf, the crosslinked network behaves differently since polymerization is favored at high temperatures. It forms a different kind of CANs. Activating the bond exchanges without depolymerization should enable thermal reprocessability. The network depolymerizes when needed through RCD via dilution or reactive distillation. This was recently demonstrated by our group via crosslinked polydithioacetals.143 Entropy-driven ROPs of the monofunctional macrocyclic dithioacetal rings and their subsequent RCDs are discussed in section 2.9. As shown in Fig. 17a, crosslinked polydithioacetal (cPDTA) was synthesized with terephthalaldehyde crosslinker to form a soft rubbery material. The concept of depolymerizable crosslinked network was demonstrated as illustrated in Fig. 17b,143 and we expect it to be applicable for many other depolymerizable ROP polymers. The polydithioacetal system is particularly suitable for this application since its polymerization is favored at high temperatures in bulk by virtue of being an ED-ROP polymer. The network will not depolymerize even when dithioacetal exchanges are activated. The network underwent ten cycles of heat reprocessing under pressure (5 tons of pressure at 100 °C) without loss of desirable properties (Fig. 17c). Dithioacetal bonds enabled CANs have also been reported in several other studies.174,179,193 Complete depolymerization of the cPDTA network into large, strainless macrocyclic monomers via RCD was achieved by refluxing in toluene under dilute conditions. The recovered macrocyclic monomers are then able to reform the crosslinked network through ED-ROP in mild conditions, requiring a minimum amount of solvent and 2 h at room temperature, much like their linear counterparts discussed in section 2.9. The chemically recycled network can subsequently be heat reprocessed again. It should be noted that all the chemical recycling and reprocessing processes were catalyzed by zinc(II) triflate which was added to catalyze the synthesis of virgin crosslinked network. It remained in the crosslinked network to activate the dithioacetal exchanges. Purification of the macrocyclic monomers to remove the catalyst or isolation of certain macrocycles based on their sizes is not required for the system to repolymerize back into the crosslinked network. Macrocycles in the solid state do not polymerize at room temperature even in the presence of catalyst. Change of the tensile properties through multiple cycles was observed as shown in Fig. 17d, but we expect that it can be mitigated via optimization of the synthetic and reprocessing conditions.
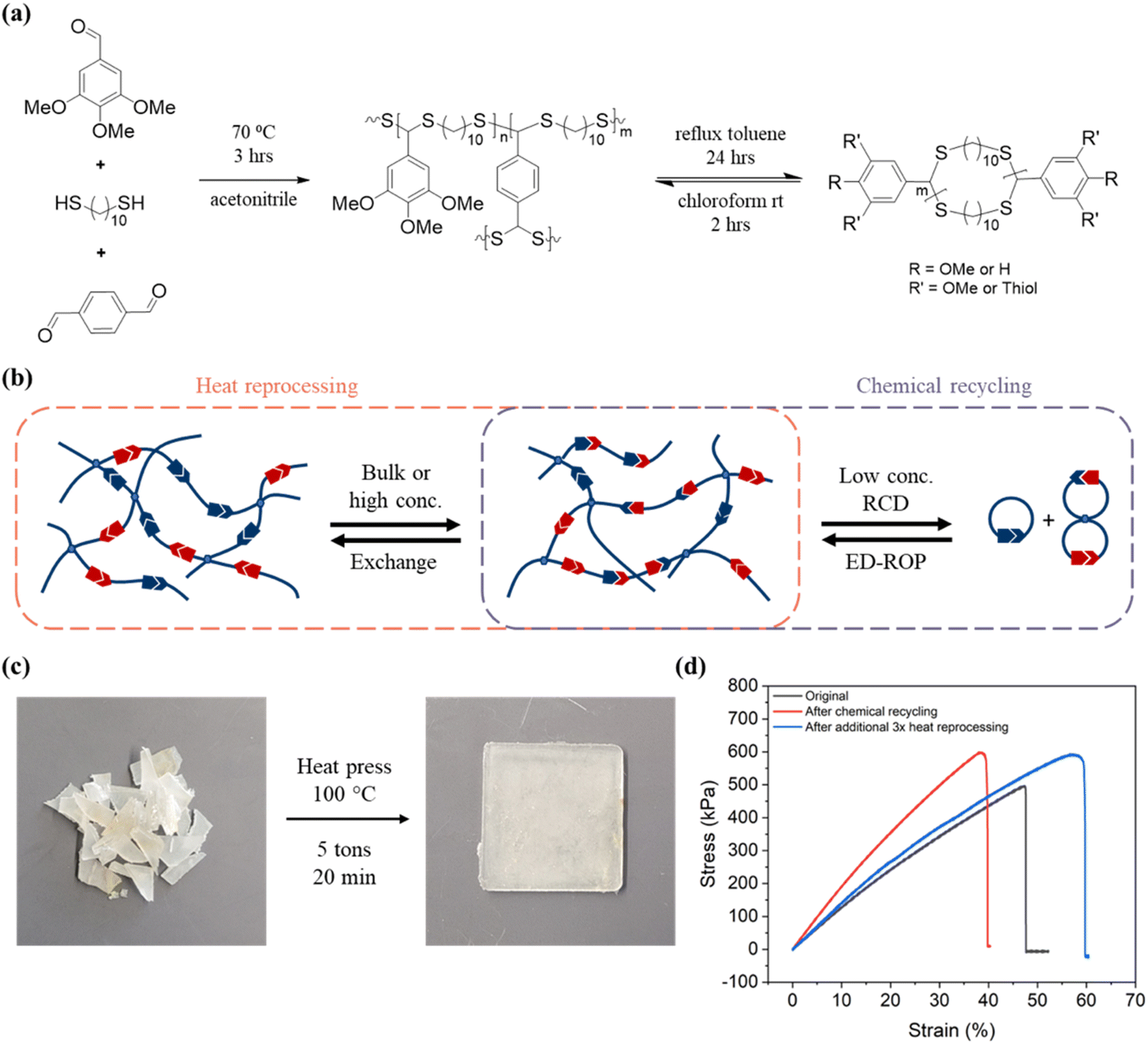 | ||
| Fig. 17 Reprocessability and mechanical properties of the crosslinked PDTA network. (a) Synthesis of cPDTA and recycling process. (b) Schematic illustration of the reprocessing and recycling mechanisms. (c) Pictures of heat pressing processing. (d) Stress–strain curves of the original network (black), immediately post-chemical reprocessing (red), and 3× mechanical recycling post-chemical reprocessing (blue). Reproduced from ref. 143 with permission from Wiley, copyright 2023. | ||
6. Closing remarks
Realizing the circularity is a complex task that requires the engagement of academia, industry, government, and the society at large. Within the scope of research, intensive efforts from the polymer science society have significantly expanded the scope of material design, which is showcased in this review. If the polymer is to be considered for practical applications, its widespread adoption rests heavily on finding polymers that depolymerize and have comparable material characteristics to current commodity polymers and economics. For years the concern has been that the incorporation of degradable bond compromises material stability and performance. As highlighted in this review, recent advancements demonstrated that new recyclable materials can maintain thermal mechanical performance and stability matching those of the existing materials such as polyolefins. Moving forward, application-specific requirements, such as gas permeability for packaging materials, toxicity for food containers, should be met.While the physical properties and methods are crucial to the polymer design, so is the projected life cycle. Replacing current commodity polymers with recyclable counterparts is one step towards increased sustainability for the industry. However, several questions need to be asked: where are the monomers coming from? Can they be obtained from renewable feedstocks or is it a more efficient use of current petroleum feedstocks? Can the replacements be reused effectively? Or is the recycling of said polymers more detrimental in terms of the energy input needed to recycle them? Do they require high amounts of heat or reaction time? How would that be accomplished on an industrial scale? Replacing current polymers means nothing if what is needed to recycle them puts more pressure on the environment than what we have now.
Many of the chemical recycling processes rely on solvents. The use of large amounts of solvents creates a thermodynamically favorable condition that makes depolymerization possible at lower temperatures. Studies showed that solvents that interact strongly with the monomer can reduce the depolymerization temperature.194 New types of solvents offer new opportunities, such as ionic liquids whose chemical design can be tailored to target catalytic performance, decomposition temperatures, and polymer solubilities for specific deconstruction applications.30 These studies confirmed that solvent effects on depolymerization are an important factor, which requires more in-depth studies with green solvents being prioritized. The high costs and challenges in handling high dilution needs to be addressed. Reactive distillation, on the other hand, avoids the use of large amounts of solvents which makes the process greener, but the high depolymerization temperature makes it more energy intensive. It is anticipated that future catalyst development can lower the depolymerization temperatures, as Cantat and coworkers have already shown the feasibility of doing so with the application of hydrosilanes195 along with numerous other studies.24,29 Reactive distillation can be more easily adopted with the current industrial infrastructure. To top it off, the distilled products exhibit a high purity.
Another important consideration in material and processing design is the tolerance for contaminants. The depolymerization needs to be selective and efficient so that there is not more energy going into the process than the polymer is worth and selectivity of the depolymerization is extremely important as it is unreasonable to expect that used plastic recycling will be perfectly sorted should the polymer be put to practical use. It then follows that the methods of purification for both the polymer and the depolymerization products also need to be efficient because of the use of additives, specifically modifiers, dyes, plasticizers, catalysts, stabilizers, flame retardants, etc. need to be easily removed upon depolymerization. New systems must address these issues to be considered for industrial applications. A lucrative approach to this is using hyper-selective depolymerization conditions that are unique to the polymer, enabling mixed plastic waste to be run through various stimuli to recover monomers in stages. Ideally this separation would be selective enough to remove contaminants in as few steps as possible and there are polymers that have demonstrated this ability at a bench-top scale as described in this review.
Optimization is key to convert research into industry. When compared to research, the industry error tolerances can be much narrower. This includes time, materials, and projected loss or error. For example, a reaction that takes 16 h to complete goes from a reasonable time frame in the lab to an eternity in an industrial setting. Production that requires large amounts of solvent, in the hundreds of liters when scaled up, with a 0.5% solvent loss in recycling said solvent becomes an exponentially large amount compared to what 0.5% solvent loss looks like inside a research lab. If there is any hope to transfer an experimental material from a bench top into industry, speed and minimizing loss is crucial. With these things in mind, the path toward a circular economy becomes clearer cut but also faces challenges.
The commonly diagramed circular economy in Fig. 1 is grossly oversimplified with respects to what is typically summarized by the use/sorting aspect. Regardless of the material or application, the circular economy is a complex system that requires coordination between every level of the supply chain, a properly designed regulatory system as well as involvement at the consumer/end user to be successful. A pinch point or failure in any one of these groups can lead to failure of the entire system. For example, in Norway, Infinitum achieved a return rate of 92.8% and 91.5% for PET and aluminum containers, respectively for recycling.196 These return rates were due to a combination of strong regulatory policy including a deposit return system, infrastructure, collaboration between the grocery and beverage industry and consumer involvement.197 The collection rates outpaces PET container collection rates for European (average of 61%)198 and the US (26.6%)199 where nationwide collection policies are nonexistent and regional policies are fragmented at best. In both regions as well as globally, developing policies such as the Intergovernmental Negotiating Committee on Plastic Pollution,200 European Union's Plastics Strategy201 and Packaging and Packaging Waste Directive,202 and US state-based approach with California's Truth in Labeling (SB343) and EPR Program (SB54), Oregon's Plastic Pollution and Recycling Modernization Act, and Colorado's Producer Responsibility Program for Statewide Recycling Act aim at tackling some of the hurdles of circularity from the regulatory perspective. Similarly, regardless of material and application, once collected, items must be properly designed for sorting through existing and developing infrastructure to reach the proper mechanical, physical, or chemical recycling process. For plastic packaging, the Association of Plastics Recyclers203 in the US and RecyClass204 in Europe developed designs for recycling guidelines to ensure that packaging is sortable at Material Recovery Facilities (MRFs) that operate at volumes as high as 300000 metric tons per year.205,206 Regardless of recycling technology and tolerance to contamination, effective separation early on ensures the item enters the correct stream, improves quality, reduces yield loss and improves economic viability. Additionally, as the range of incoming streams expands to textiles, automotive, appliance, construction and other durable goods, infrastructure similar to the MRFs that exist for curbside recycling streams will need to be developed and scaled to broaden the range of material available for mechanical and chemical recycling processes.
Lastly, life-cycle analysis and techno-economic analysis that provide preliminary evaluations on the carbon footprints and cost effectiveness associated with new feedstocks, recycling processes such as the use of catalyst, solvent, temperature, pressure, infrastructures for polymer production and recycling, storage, and transportation, must be well-integrated to guide the material and process designs.207
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Science Foundation under Award CHE-2305045.References
- Y. Zhu, C. Romain and C. K. Williams, Sustainable Polymers from Renewable Resources, Nature, 2016, 540(7633), 354–362 CrossRef CAS PubMed.
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a Circular Economy, Nat. Rev. Mater., 2022, 7(2), 117–137 CrossRef PubMed.
- S. Wang, M. Urban and W. Self-Healing Polymers, Nat. Rev. Mater., 2020, 5(8), 562–583 CrossRef CAS.
- T. H. Epps, L. T. J. Korley, T. Yan, K. L. Beers and T. M. Burt, Sustainability of Synthetic Plastics: Considerations in Materials Life-Cycle Management, JACS Au, 2022, 2(1), 3–11 CrossRef PubMed.
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer and S. R. Nicholson, Technical, Economic, and Environmental Comparison of Closed-Loop Recycling Technologies for Common Plastics, ACS Sustainable Chem. Eng., 2023, 11(3), 965–978 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Critical Advances and Future Opportunities in Upcycling Commodity Polymers, Nature, 2022, 603(7903), 803–814 CrossRef CAS PubMed.
- A. H. Westlie, E. Y. X. Chen, C. M. Holland, S. S. Stahl, M. Doyle, S. R. Trenor and K. M. Knauer, Polyolefin Innovations toward Circularity and Sustainable Alternatives, Macromol. Rapid Commun., 2022, 43(24), 2200492 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, 50th Anniversary Perspective: There Is a Great Future in Sustainable Polymers, Macromolecules, 2017, 50(10), 3733–3749 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Chemically Recyclable Polymers: A Circular Economy Approach to Sustainability. Green Chemistry, Royal Society of Chemistry, 2017, pp. 3692–3706 Search PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical Recycling to Monomer for an Ideal, Circular Polymer Economy, Nat. Rev. Mater., 2020, 5(7), 501–516 CrossRef CAS.
- G. Xu and Q. Wang, Chemically Recyclable Polymer Materials: Polymerization and Depolymerization Cycles. Green Chemistry, Royal Society of Chemistry, 2022, pp. 2321–2346 Search PubMed.
- J. Zhou, T. Hsu and J. Wang, Mechanochemical Degradation and Recycling of Synthetic Polymers, Angew. Chem., Int. Ed., 2023, 62(27), e202300768 CrossRef CAS PubMed.
- M. Levy, C. Cirko, F. Edgecombe, A. Carlson and E. Tipaldo, Life Cycle Impacts of Plastic Packaging Compared to Substitutes in the United States and Canada Theoretical Substitution Analysis Prepared for The Plastics Division of the American Chemistry Council (ACC), 2018 Search PubMed.
- What is a Circular Economy? https://www.epa.gov/circulareconomy/what-circular-economy (accessed Oct 29, 2023).
- F. M. Haque, J. S. A. Ishibashi, C. A. L. Lidston, H. Shao, F. S. Bates, A. B. Chang, G. W. Coates, C. J. Cramer, P. J. Dauenhauer, W. R. Dichtel, C. J. Ellison, E. A. Gormong, L. S. Hamachi, T. R. Hoye, M. Jin, J. A. Kalow, H. J. Kim, G. Kumar, C. J. Lasalle, S. Liffland, B. M. Lipinski, Y. Pang, R. Parveen, X. Peng, Y. Popowski, E. A. Prebihalo, Y. Reddi, T. M. Reineke, D. T. Sheppard, J. L. Swartz, W. B. Tolman, B. Vlaisavljevich, J. Wissinger, S. Xu and M. A. Hillmyer, Defining the Macromolecules of Tomorrow through Synergistic Sustainable Polymer Research, Chem. Rev., 2022, 122(6), 6322–6373 CrossRef CAS PubMed.
- S. Billiet and S. R. Trenor, 100th Anniversary of Macromolecular Science Viewpoint: Needs for Plastics Packaging Circularity, ACS Macro Lett., 2020, 9(9), 1376–1390 CrossRef CAS PubMed.
- M. Vert, Aliphatic Polyesters: Great Degradable Polymers That Cannot Do Everything, Biomacromolecules, 2005, 6(2), 538–546 CrossRef CAS PubMed.
- V. Delplace and J. Nicolas, Degradable Vinyl Polymers for Biomedical Applications, Nat. Chem., 2015, 7(10), 771–784 CrossRef CAS PubMed.
- J. M. Fishman and L. L. Kiessling, Synthesis of Functionalizable and Degradable Polymers by Ring–Opening Metathesis Polymerization, Angew. Chem., 2013, 125(19), 5165–5168 CrossRef.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Towards Closed-Loop Recycling of Multilayer and Coloured PET Plastic Waste by Alkaline Hydrolysis, Green Chem., 2020, 22(16), 5376–5394 RSC.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Recycling of Polyurethanes from Laboratory to Industry, a Journey towards the Sustainability, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- A. J. Minor, R. Goldhahn, L. Rihko-Struckmann and K. Sundmacher, Chemical Recycling Processes of Nylon 6 to Caprolactam: Review and Techno-Economic Assessment, Chem. Eng. J., 2023, 474, 145333 CrossRef CAS.
- J. G. Kim, Chemical Recycling of Poly(Bisphenol A Carbonate), Polym. Chem., 2020, 11(30), 4830–4849 RSC.
- K. V. Khopade, S. H. Chikkali and N. Barsu, Metal-Catalyzed Plastic Depolymerization, Cell Rep. Phys. Sci., 2023, 4(5), 101341 CrossRef CAS.
- S. Zhang, M. Li, Z. Zuo and Z. Niu, Recent Advances in Plastic Recycling and Upgrading under Mild Conditions, Green Chem., 2023, 25(18), 6949–6970 RSC.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Catalytic Deconstruction of Waste Polyethylene with Ethylene to Form Propylene, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- Z. Xu, N. Eric Munyaneza, Q. Zhang, M. Sun, C. Posada, P. Venturo, N. A. Rorrer, J. Miscall, B. G. Sumpter and G. Liu, Chemical Upcycling of Polyethylene, Polypropylene, and Mixtures to High-Value Surfactants, Science, 2023, 381, 666–671 CrossRef CAS PubMed.
- H. Li, J. Wu, Z. Jiang, J. Ma, V. M. Zavala, C. R. Landis, M. Mavrikakis and G. W. Huber, Hydroformylation of Pyrolysis Oils to Aldehydes and Alcohols from Polyolefin Waste, Science, 2023, 381, 660–666 CrossRef CAS PubMed.
- A. Chaudhary and R. Srivastava, Ionic Liquid–Assisted Depolymerization of Condensation Polymers: A Review, ChemistrySelect, 2023, 8(30), e202301709 CrossRef CAS.
- T. Christoff-Tempesta and T. H. Epps, Ionic-Liquid-Mediated Deconstruction of Polymers for Advanced Recycling and Upcycling, ACS Macro Lett., 2023, 12(8), 1058–1070 CrossRef CAS PubMed.
- G. R. Jones, H. S. Wang, K. Parkatzidis, R. Whitfield, N. P. Truong and A. Anastasaki, Reversed Controlled Polymerization (RCP): Depolymerization from Well-Defined Polymers to Monomers, J. Am. Chem. Soc., 2023, 145(18), 9898–9915 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Toward Polymer Upcycling-Adding Value and Tackling Circularity, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- R. E. Yardley, A. R. Kenaree and E. R. Gillies, Triggering Depolymerization: Progress and Opportunities for Self-Immolative Polymers, Macromolecules, 2019, 52(17), 6342–6360 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Completely Recyclable Biopolymers with Linear and Cyclic Topologies via Ring-Opening Polymerization of γ-Butyrolactone, Nat. Chem., 2016, 8(1), 42–49 CrossRef CAS PubMed.
- P. Olsén, K. Odelius and A. C. Albertsson, Thermodynamic Presynthetic Considerations for Ring-Opening Polymerization, Biomacromolecules, 2016, 17(3), 699–709 CrossRef PubMed.
- F. S. Dainton and K. J. Ivin, Changes of Entropy and Heat Content During Polymerization, Trans. Faraday Soc., 1950, 46, 331–348 RSC.
- D. Sathe, H. Chen and J. Wang, Regulating the Thermodynamics and Thermal Properties of Depolymerizable Polycyclooctenes through Substituent Effects, Macromol. Rapid Commun., 2023, 44(1), 2200304 CrossRef CAS PubMed.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, CXIX.—The Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane, J. Chem. Soc., Dalton Trans., 1915, 107(0), 1080–1106 RSC.
- D. K. Schneiderman and M. A. Hillmyer, Aliphatic Polyester Block Polymer Design, Macromolecules, 2016, 49(7), 2419–2428 CrossRef CAS.
- S. Strandman, J. E. Gautrot and X. X. Zhu, Recent Advances in Entropy-Driven Ring-Opening Polymerizations, Polym. Chem., 2011, 2(4), 791–799 RSC.
- P. Hodge, Entropically Driven Ring-Opening Polymerization of Strainless Organic Macrocycles, Chem. Rev., 2014, 114(4), 2278–2312 CrossRef CAS PubMed.
- G. Odian, Radical Chain Polymerization, in Principles of Polymerization, John Wiley& Sons, Inc., Hoboken, 2004, pp. 282 Search PubMed.
- A. M. Johnson, K. E. L. Husted, L. J. Kilgallon and J. A. Johnson, Orthogonally Deconstructable and Depolymerizable Polysilylethers via Entropy-Driven Ring-Opening Metathesis Polymerization, Chem. Commun., 2022, 58(61), 8496–8499 RSC.
- R. Ogawa and M. A. Hillmyer, High Molar Mass Poly(Ricinoleic Acid): Via Entropy-Driven Ring-Opening Metathesis Polymerization, Polym. Chem., 2021, 12(15), 2253–2257 RSC.
- A. G. Amador, A. Watts, A. E. Neitzel and M. A. Hillmyer, Entropically Driven Macrolide Polymerizations for the Synthesis of Aliphatic Polyester Copolymers Using Titanium Isopropoxide, Macromolecules, 2019, 52(6), 2371–2383 CrossRef CAS.
- J. A. Nowalk, C. Fang, A. L. Short, R. M. Weiss, J. H. Swisher, P. Liu and T. Y. Meyer, Sequence-Controlled Polymers Through Entropy-Driven Ring-Opening Metathesis Polymerization: Theory, Molecular Weight Control, and Monomer Design, J. Am. Chem. Soc., 2019, 141(14), 5741–5752 CrossRef CAS PubMed.
- D. Myers, T. Witt, A. Cyriac, M. Bown, S. Mecking and C. K. Williams, Ring Opening Polymerization of Macrolactones: High Conversions and Activities Using an Yttrium Catalyst, Polym. Chem., 2017, 8(37), 5780–5785 RSC.
- F. N. Behrendt, A. Hess, M. Lehmann, B. Schmidt and H. Schlaad, Polymerization of Cystine-Derived Monomers, Polym. Chem., 2019, 10(13), 1636–1641 RSC.
- J. Huang, P. Olsén, E. Svensson Grape, A. K. Inge and K. Odelius, Simple Approach to Macrocyclic Carbonates with Fast Polymerization Rates and Their Polymer-to-Monomer Regeneration, Macromolecules, 2022, 55(2), 608–614 CrossRef CAS.
- T. Matsuhira and H. Sakai, Entropy-Driven Supramolecular Ring-Opening Polymerization of a Cyclic Hemoglobin Monomer for Constructing a Hemoglobin-PEG Alternating Polymer with Structural Regularity, Biomacromolecules, 2021, 22(5), 1944–1954 CrossRef CAS PubMed.
- H. Huang, W. Wang, Z. Zhou, B. Sun, M. An, F. Haeffner and J. Niu, Radical Ring-Closing/Ring-Opening Cascade Polymerization, J. Am. Chem. Soc., 2019, 141(32), 12493–12497 CrossRef CAS PubMed.
- C. F. Gallin, W. Lee and J. A. Byers, A Simple, Selective, and General Catalyst for Ring Closing Depolymerization of Polyesters and Polycarbonates for Chemical Recycling, Angew. Chem., 2023, 135(25), e202303762 CrossRef.
- J. J. R. Arias and W. Thielemans, Instantaneous Hydrolysis of PET Bottles: An Efficient Pathway for the Chemical Recycling of Condensation Polymers, Green Chem., 2021, 23(24), 9945–9956 RSC.
- P. Pereira, P. E. Savage and C. W. Pester, Neutral Hydrolysis of Post-Consumer Polyethylene Terephthalate Waste in Different Phases, ACS Sustainable Chem. Eng., 2023, 11(18), 7203–7209 CrossRef CAS.
- S. Kaabel, J. P. D. Therien, C. E. Deschênes, D. Duncan, T. Friščić and K. Auclair, Enzymatic Depolymerization of Highly Crystalline Polyethylene Terephthalate Enabled in Moist-Solid Reaction Mixtures, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(29), e2026452118 CrossRef CAS PubMed.
- T. Uekert, J. S. DesVeaux, A. Singh, S. R. Nicholson, P. Lamers, T. Ghosh, J. E. McGeehan, A. C. Carpenter and G. T. Beckham, Life Cycle Assessment of Enzymatic Poly (Ethylene Terephthalate) Recycling, Green Chem., 2022, 24(17), 6531–6543 RSC.
- N. M. Wang, G. Strong, V. DaSilva, L. Gao, R. Huacuja, I. A. Konstantinov, M. S. Rosen, A. J. Nett, S. Ewart, R. Geyer, S. L. Scott and D. Guironnet, Chemical Recycling of Polyethylene by Tandem Catalytic Conversion to Propylene, J. Am. Chem. Soc., 2022, 144(40), 18526–18531 CrossRef CAS PubMed.
- G. Xi, M. Lu and C. Sun, Study on Depolymerization of Waste Polyethylene Terephthalate into Monomer of Bis(2-Hydroxyethyl Terephthalate), Polym. Degrad. Stab., 2005, 87(1), 117–120 CrossRef CAS.
- J. Payne and M. D. Jones, The Chemical Recycling of Polyesters for a Circular Plastics Economy: Challenges and Emerging Opportunities, ChemSusChem, 2021, 14(19), 4041–4070 CrossRef CAS PubMed.
- E. Barnard, J. J. R. Arias and W. Thielemans, Chemolytic Depolymerisation of PET: A Review, Green Chem., 2021, 23(11), 3765–3789 RSC.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J. Carpentier, Recent Advances in Metal–mediated Stereoselective Ring–opening Polymerization of Functional Cyclic Esters towards Well–defined Poly (Hydroxy Acid) s: From Stereoselectivity to Sequence–control, Chem. – Eur. J., 2020, 26(1), 128–138 CrossRef CAS PubMed.
- D. Tan, Y. Wang, Y. Tong and G.-Q. Chen, Grand Challenges for Industrializing Polyhydroxyalkanoates (PHAs), Trends Biotechnol., 2021, 39(9), 953–963 CrossRef CAS PubMed.
- L. Zhou, Z. Zhang, C. Shi, M. Scoti, D. K. Barange, R. R. Gowda and E. Y.-X. Chen, Chemically Circular, Mechanically Tough, and Melt-Processable Polyhydroxyalkanoates, Science, 2023, 380(6640), 64–69 CrossRef CAS PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, A Synthetic Polymer System with Repeatable Chemical Recyclability, Science, 2018, 360(6387), 398–403 CrossRef CAS PubMed.
- X.-L. Li, R. W. Clarke, H.-Y. An, R. R. Gowda, J.-Y. Jiang, T.-Q. Xu and E. Y.-X. Chen, Dual Recycling of Depolymerization Catalyst and Biodegradable Polyester That Markedly Outperforms Polyolefins, Angew. Chem., Int. Ed., 2023, e202303791 CAS.
- X.-L. Li, R. W. Clarke, J.-Y. Jiang, T.-Q. Xu and E. Y.-X. Chen, A Circular Polyester Platform Based on Simple Gem-Disubstituted Valerolactones, Nat. Chem., 2023, 15(2), 278–285 CrossRef CAS PubMed.
- C. Shi, M. L. McGraw, Z. C. Li, L. Cavallo, L. Falivene and E. Y. X. Chen, High-Performance Pan-Tactic Polythioesters with Intrinsic Crystallinity and Chemical Recyclability, Sci. Adv., 2020, 6(34), 1–12 Search PubMed.
- G. W. Fahnhorst and T. R. Hoye, A Carbomethoxylated Polyvalerolactone from Malic Acid: Synthesis and Divergent Chemical Recycling, ACS Macro Lett., 2018, 7(2), 143–147 CrossRef CAS PubMed.
- G. W. Fahnhorst, G. X. De Hoe, M. A. Hillmyer and T. R. Hoye, 4-Carboalkoxylated Polyvalerolactones from Malic Acid: Tough and Degradable Polyesters, Macromolecules, 2020, 53(8), 3194–3201 CrossRef CAS PubMed.
- T. J. Bannin and M. K. Kiesewetter, Poly (Thioester) by Organocatalytic Ring-Opening Polymerization, Macromolecules, 2015, 48(16), 5481–5486 CrossRef CAS PubMed.
- M. Suzuki, K. Makimura and S. Matsuoka, Thiol-Mediated Controlled Ring-Opening Polymerization of Cysteine-Derived β-Thiolactone and Unique Features of Product Polythioester, Biomacromolecules, 2016, 17(3), 1135–1141 CrossRef CAS PubMed.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, Radical Approach to Thioester-Containing Polymers, J. Am. Chem. Soc., 2019, 141(4), 1446–1451 CrossRef CAS PubMed.
- C. G. Overberger and J. K. Weise, Anionic Ring-Opening Polymerization of Thiolactones, J. Am. Chem. Soc., 1968, 90(13), 3533–3537 CrossRef CAS.
- S. Mavila, B. T. Worrell, H. R. Culver, T. M. Goldman, C. Wang, C.-H. Lim, D. W. Domaille, S. Pattanayak, M. K. McBride and C. B. Musgrave, Dynamic and Responsive DNA-like Polymers, J. Am. Chem. Soc., 2018, 140(42), 13594–13598 CrossRef CAS PubMed.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, 4-Hydroxyproline-Derived Sustainable Polythioesters: Controlled Ring-Opening Polymerization, Complete Recyclability, and Facile Functionalization, J. Am. Chem. Soc., 2019, 141(12), 4928–4935 CrossRef CAS PubMed.
- W. Xiong, W. Chang, D. Shi, L. Yang, Z. Tian, H. Wang, Z. Zhang, X. Zhou, E. Q. Chen and H. Lu, Geminal Dimethyl Substitution Enables Controlled Polymerization of Penicillamine-Derived β-Thiolactones and Reversed Depolymerization, Chem, 2020, 6(7), 1831–1843 CAS.
- C. Shi, R. W. Clarke, M. L. McGraw and E. Y. X. Chen, Closing the “One Monomer-Two Polymers-One Monomer” Loop via Orthogonal (De)Polymerization of a Lactone/Olefin Hybrid, J. Am. Chem. Soc., 2022, 144(5), 2264–2275 CrossRef CAS PubMed.
- P. v. R. Schleyer, J. E. Williams and K. R. Blanchard, Evaluation of Strain in Hydrocarbons. The Strain in Adamantane and Its Origin, J. Am. Chem. Soc., 2002, 92(8), 2377–2386 CrossRef.
- E. A. Ofstead and N. Calderon, Equilibrium Ring–opening Polymerization of Mono–and Multicyclic Unsaturated Monomers, Makromol. Chem., 1972, 154(1), 21–34 CrossRef CAS.
- W. D. Mulhearn and R. A. Register, Synthesis of Narrow-Distribution, High-Molecular-Weight ROMP Polycyclopentene via Suppression of Acyclic Metathesis Side Reactions, ACS Macro Lett., 2017, 6(2), 112–116 CrossRef CAS PubMed.
- W. J. Neary and J. G. Kennemur, Variable Temperature ROMP: Leveraging Low Ring Strain Thermodynamics to Achieve Well-Defined Polypentenamers, Macromolecules, 2017, 50(13), 4935–4941 CrossRef CAS.
- W. J. Neary and J. G. Kennemur, Polypentenamer Renaissance: Challenges and Opportunities, ACS Macro Lett., 2019, 8(1), 46–56 CrossRef CAS PubMed.
- C. R. López-Barrón, B. Rohde, A. V. Zabula, J. J. Schaefer and J. A. Throckmorton, Molecular Orientation and Strain-Induced Crystallization in Trans-Polypentenamer, Macromolecules, 2020, 53(4), 1356–1367 CrossRef.
- R. J. Kieber, W. J. Neary and J. G. Kennemur, Viscoelastic, Mechanical, and Glasstomeric Properties of Precision Polyolefins Containing a Phenyl Branch at Every Five Carbons, Ind. Eng. Chem. Res., 2018, 57(14), 4916–4922 CrossRef CAS.
- R. Tuba and R. H. Grubbs, Ruthenium Catalyzed Equilibrium Ring-Opening Metathesis Polymerization of Cyclopentene, Polym. Chem., 2013, 4(14), 3959–3962 RSC.
- R. Tuba, M. Al-Hashimi, H. S. Bazzi and R. H. Grubbs, One-Pot Synthesis of Poly(Vinyl Alcohol) (PVA) Copolymers via Ruthenium Catalyzed Equilibrium Ring-Opening Metathesis Polymerization of Hydroxyl Functionalized Cyclopentene, Macromolecules, 2014, 47(23), 8190–8195 CrossRef CAS.
- W. J. Neary, T. A. Isais and J. G. Kennemur, Depolymerization of Bottlebrush Polypentenamers and Their Macromolecular Metamorphosis, J. Am. Chem. Soc., 2019, 141(36), 14220–14229 CrossRef CAS PubMed.
- R. Tuba, J. Balogh, A. Hlil, M. Barłóg, M. Al-Hashimi and H. S. Bazzi, Synthesis of Recyclable Tire Additives via Equilibrium Ring-Opening Metathesis Polymerization, ACS Sustainable Chem. Eng., 2016, 4(11), 6090–6094 CrossRef CAS.
- J. D. Feist and Y. Xia, Enol Ethers Are Effective Monomers for Ring-Opening Metathesis Polymerization: Synthesis of Degradable and Depolymerizable Poly(2,3-Dihydrofuran), J. Am. Chem. Soc., 2020, 142(3), 1186–1189 CrossRef CAS PubMed.
- H. Liu, A. Z. Nelson, Y. Ren, K. Yang, R. H. Ewoldt and J. S. Moore, Dynamic Remodeling of Covalent Networks via Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2018, 7(8), 933–937 CrossRef CAS PubMed.
- B. M. Coia, S. E. Werner and J. G. Kennemur, Conformational Bias in Density Functional Theory Ring Strain Energy Calculations of Cyclopentene Derivatives: Towards Predictive Design of Chemically Recyclable Elastomers, J. Polym. Sci., 2022, 60(24), 3391–3403 CrossRef CAS.
- B. M. Coia, L. A. Hudson, A. J. Specht and J. G. Kennemur, Substituent Effects on Torsional Strain in Cyclopentene Derivatives: A Computational Study, J. Phys. Chem. A, 2023, 127(23), 5005–5017 CrossRef CAS PubMed.
- D. Sathe, J. Zhou, H. Chen, H.-W. Su, W. Xie, T.-G. Hsu, B. R. Schrage, T. Smith, C. J. Ziegler and J. Wang, Olefin Metathesis-Based Chemically Recyclable Polymers Enabled by Fused-Ring Monomers, Nat. Chem., 2021, 13(8), 743–750 CrossRef CAS PubMed.
- J. Zhou, D. Sathe and J. Wang, Understanding the Structure–Polymerization Thermodynamics Relationships of Fused-Ring Cyclooctenes for Developing Chemically Recyclable Polymers, J. Am. Chem. Soc., 2022, 144(2), 928–934 CrossRef CAS PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Dynamic Covalent Chemistry Approaches Toward Macrocycles, Molecular Cages, and Polymers, Acc. Chem. Res., 2014, 47(5), 1575–1586 CrossRef CAS PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations, Adv. Mater., 2017, 29(14), 1606100 CrossRef PubMed.
- A. G. Orrillo and R. L. E. Furlan, Sulfur in Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2022, 61(26), e202201168 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41(6), 898–952 CrossRef PubMed.
- Z. Lei, H. Chen, C. Luo, Y. Rong, Y. Hu, Y. Jin, R. Long, K. Yu and W. Zhang, Recyclable and Malleable Thermosets Enabled by Activating Dormant Dynamic Linkages, Nat. Chem., 2022, 14(12), 1399–1404 CrossRef CAS PubMed.
- J. Meisenheimer, Ueber Reactionen Aromatischer Nitrokorper, Justus Liebigs Ann. Chem., 1902, 323(2), 205–246 CrossRef CAS.
- M. J. Robb and D. M. Knauss, Poly(Arylene Sulfide)s by Nucleophilic Aromatic Substitution Polymerization of 2,7-difluorothianthrene, J. Polym. Sci., Part A: Polym. Chem., 2009, 47(9), 2453–2461 CrossRef CAS.
- H. Jiang, T. Chen, Y. Q1 and J. Xu, Macrocyclic Oligomeric Arylene Ether Ketones: Synthesis and Polymerization, Polym. J., 1998, 30(4), 300–303 CrossRef CAS.
- Y.-L. Su, L. Yue, H. Tran, M. Xu, A. Engler, R. Ramprasad, H. J. Qi and W. R. Gutekunst, Chemically Recyclable Polymer System Based on Nucleophilic Aromatic Ring-Opening Polymerization, J. Am. Chem. Soc., 2023, 145(25), 13950–13956 CrossRef CAS PubMed.
- P. Ortmann, I. Heckler and S. Mecking, Physical Properties and Hydrolytic Degradability of Polyethylene-like Polyacetals and Polycarbonates, Green Chem., 2014, 16(4), 1816 RSC.
- B. S. Rajput, U. Chander, K. Arole, F. Stempfle, S. Menon, S. Mecking and S. H. Chikkali, Synthesis of Renewable Copolyacetals with Tunable Degradation, Macromol. Chem. Phys., 2016, 217(12), 1396–1410 CrossRef CAS.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Thermoplastic, Polyacetals: Chemistry from the Past for a Sustainable Future?, Polym. Chem., 2019, 10(1), 9–33 RSC.
- M. Shen, S. Vijjamarri, H. Cao, K. Solis and M. L. Robertson, Degradability, Thermal Stability, and High Thermal Properties in Spiro Polycycloacetals Partially Derived from Lignin, Polym. Chem., 2021, 12(41), 5986–5998 RSC.
- H. Qiu, Z. Yang, M. Köhler, J. Ling and F. H. Schacher, Synthesis and Solution Self-Assembly of Poly (1, 3-Dioxolane), Macromolecules, 2019, 52(9), 3359–3366 CrossRef CAS.
- B. A. Abel, R. L. Snyder and G. W. Coates, Chemically Recyclable Thermoplastics from Reversible-Deactivation Polymerization of Cyclic Acetals, Science, 2021, 373(6556), 783–789 CrossRef CAS PubMed.
- H. G. Hester, B. A. Abel and G. W. Coates, Ultra-High-Molecular-Weight Poly(Dioxolane): Enhancing the Mechanical Performance of a Chemically Recyclable Polymer, J. Am. Chem. Soc., 2023, 145(16), 8800–8804 CrossRef CAS PubMed.
- Q. Zhang, Y.-X. Deng, H.-X. Luo, C.-Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D.-H. Qu, Assembling a Natural Small Molecule into a Supramolecular Network with High Structural Order and Dynamic Functions, J. Am. Chem. Soc., 2019, 141(32), 12804–12814 CrossRef CAS PubMed.
- Q. Zhang, Y. Deng, C.-Y. Shi, B. L. Feringa, H. Tian and D.-H. Qu, Dual Closed-Loop Chemical Recycling of Synthetic Polymers by Intrinsically Reconfigurable Poly (Disulfides), Matter, 2021, 4(4), 1352–1364 CrossRef CAS.
- Q. Zhang, D.-H. Qu, B. L. Feringa and H. Tian, Disulfide-Mediated Reversible Polymerization toward Intrinsically Dynamic Smart Materials, J. Am. Chem. Soc., 2022, 144(5), 2022–2033 CrossRef CAS PubMed.
- C. Choi, J. L. Self, Y. Okayama, A. E. Levi, M. Gerst, J. C. Speros, C. J. Hawker, J. Read De Alaniz and C. M. Bates, Light-Mediated Synthesis and Reprocessing of Dynamic Bottlebrush Elastomers under Ambient Conditions, J. Am. Chem. Soc., 2021, 143(26), 9866–9871 CrossRef CAS PubMed.
- Q. Zhang, C.-Y. Shi, D.-H. Qu, Y.-T. Long, B. L. Feringa and H. Tian, Exploring a Naturally Tailored Small Molecule for Stretchable, Self-Healing, and Adhesive Supramolecular Polymers, Sci. Adv., 2018, 4(7), 8192–8219 CrossRef PubMed.
- K. R. Albanese, P. T. Morris, J. Read de Alaniz, C. M. Bates and C. J. Hawker, Controlled-Radical Polymerization of α-Lipoic Acid: A General Route to Degradable Vinyl Copolymers, J. Am. Chem. Soc., 2023, 145(41), 22728–22734 CrossRef CAS PubMed.
- K. R. Albanese, Y. Okayama, P. T. Morris, M. Gerst, R. Gupta, J. C. Speros, C. J. Hawker, C. Choi, J. R. de Alaniz and C. M. Bates, Building Tunable Degradation into High-Performance Poly (Acrylate) Pressure-Sensitive Adhesives, ACS Macro Lett., 2023, 12, 787–793 CrossRef CAS PubMed.
- S. Huang, Y. Shen, H. K. Bisoyi, Y. Tao, Z. Liu, M. Wang, H. Yang and Q. Li, Covalent Adaptable Liquid Crystal Networks Enabled by Reversible Ring-Opening Cascades of Cyclic Disulfides, J. Am. Chem. Soc., 2021, 143(32), 12543–12551 CrossRef CAS PubMed.
- E. K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, Substrate-Initiated Synthesis of Cell-Penetrating Poly(Disulfide)s, J. Am. Chem. Soc., 2013, 135(6), 2088–2091 CrossRef CAS PubMed.
- Q. Zhang, Y. Deng, C. Y. Shi, B. L. Feringa, H. Tian and D. H. Qu, Dual Closed-Loop Chemical Recycling of Synthetic Polymers by Intrinsically Reconfigurable Poly(Disulfides), Matter, 2021, 4(4), 1352–1364 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, Architecture-Controlled Ring-Opening Polymerization for Dynamic Covalent Poly(Disulfide)s, J. Am. Chem. Soc., 2019, 141(43), 17075–17080 CrossRef CAS PubMed.
- M. Varghese and M. W. Grinstaff, Beyond Nylon 6: Polyamides via Ring Opening Polymerization of Designer Lactam Monomers for Biomedical Applications, Chem. Soc. Rev., 2022, 51(19), 8258–8275 RSC.
- K. Yan, J. Wang, Z. Wang and L. Yuan, Bio-Based Monomers for Amide-Containing Sustainable Polymers, Chem. Commun., 2022, 59(4), 382–400 RSC.
- M. Winnacker, Polyamides Derived from Terpenes: Advances in Their Synthesis, Characterization and Applications, Eur. J. Lipid Sci. Technol., 2023, 125(5), 2300014 CrossRef CAS.
- V. Hirschberg and D. Rodrigue, Recycling of Polyamides: Processes and Conditions, J. Polym. Sci., 2023, 61(17), 1937–1958 CrossRef CAS.
- L. Wursthorn, K. Beckett, J. O. Rothbaum, R. M. Cywar, C. Lincoln, Y. Kratish and T. J. Marks, Selective Lanthanide–Organic Catalyzed Depolymerization of Nylon–6 to E–Caprolactam, Angew. Chem., Int. Ed., 2023, 62(4), e202212543 CrossRef CAS PubMed.
- C. Alberti, R. Figueira, M. Hofmann, S. Koschke and S. Enthaler, Chemical Recycling of End-of-Life Polyamide 6 via Ring Closing Depolymerization, ChemistrySelect, 2019, 4(43), 12638–12642 CrossRef CAS.
- R. M. Cywar, N. A. Rorrer, H. B. Mayes, A. K. Maurya, C. J. Tassone, G. T. Beckham and E. Y. X. Chen, Redesigned Hybrid Nylons with Optical Clarity and Chemical Recyclability, J. Am. Chem. Soc., 2022, 144(12), 5366–5376 CrossRef CAS PubMed.
- F. Siragusa, C. Detrembleur and B. Grignard, The Advent of Recyclable CO2-Based Polycarbonates, Polym. Chem., 2023, 14(11), 1164–1183 RSC.
- Y. Liu and X. B. Lu, Chemical Recycling to Monomers: Industrial Bisphenol-A-Polycarbonates to Novel Aliphatic Polycarbonate Materials, J. Polym. Sci., 2022, 60(24), 3256–3268 CrossRef CAS.
- W. Zhang, J. Dai, Y. C. Wu, J. X. Chen, S. Y. Shan, Z. Cai and J. B. Zhu, Highly Reactive Cyclic Carbonates with a Fused Ring toward Functionalizable and Recyclable Polycarbonates, ACS Macro Lett., 2022, 11(2), 173–178 CrossRef CAS PubMed.
- D. J. Saxon, E. A. Gormong, V. M. Shah and T. M. Reineke, Rapid Synthesis of Chemically Recyclable Polycarbonates from Renewable Feedstocks, ACS Macro Lett., 2021, 10(1), 98–103 CrossRef CAS PubMed.
- J. Huang, R. Yan, Y. Ni, N. Shi, Z. Li, C. Ma and K. Guo, Cyclic Polycarbonates by N-Heterocyclic Carbene-Mediated Ring-Expansion Polymerization and Their Selective Depolymerization to Monomers, ACS Sustainable Chem. Eng., 2022, 10(46), 15007–15016 CrossRef CAS.
- G. Yang, Y. Wang, H. Qi, Y. Zhang, X. Zhu, C. Lu, L. Yang and G. Wu, Highly Selective Preparation and Depolymerization of Chemically Recyclable Poly(Cyclopentene Carbonate) Enabled by Organoboron Catalysts, Angew. Chem., Int. Ed., 2022, 61(46), e202210243 CrossRef CAS PubMed.
- W. H. Carothers, J. A. Arvin and G. L. Dorough, Studies on Polymerization and Ring Formation V. Glycol Esters of Oxalic Acid, J. Am. Chem. Soc., 1930, 52(8), 3292–3300 CrossRef CAS.
- A. Alksnis and D. Deme, Investigations on Equilibrium of Poly(Ethylene Oxalate) and 1,4-dioxane-2,3-dione, J. Polym. Sci., Polym. Chem. Ed., 1979, 17(9), 2701–2706 CrossRef CAS.
- I. C. McNeill and M.-L. Pereira Leão, Degradation Studies of Some Polyesters and Polycarbonates: Part 4—Poly(Ethylene Oxalate), Polym. Degrad. Stab., 1986, 15(4), 345–356 CrossRef CAS.
- J. J. Garcia and S. A. Miller, Polyoxalates from Biorenewable Diols via Oxalate Metathesis Polymerization, Polym. Chem., 2014, 5(3), 955–961 RSC.
- Z. Tu, Y. Lu, L. Sang, Y. Zhang, Y. Li and Z. Wei, Kilogram-Scale Preparation of Poly(Ethylene Oxalate) toward Marine-Degradable Plastics, Macromolecules, 2023, 56(8), 3149–3159 CrossRef CAS.
- P. Shieh, H. V. T. Nguyen and J. A. Johnson, Tailored Silyl Ether Monomers Enable Backbone-Degradable Polynorbornene-Based Linear, Bottlebrush and Star Copolymers through ROMP, Nat. Chem., 2019, 11(12), 1124–1132 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Cleavable Comonomers Enable Degradable, Recyclable Thermoset Plastics, Nature, 2020, 583(7817), 542–547 CrossRef CAS PubMed.
- K. E. L. Husted, P. Shieh, D. J. Lundberg, S. L. Kristufek and J. A. Johnson, Molecularly Designed Additives for Chemically Deconstructable Thermosets without Compromised Thermomechanical Properties, ACS Macro Lett., 2021, 10(7), 805–810 CrossRef CAS PubMed.
- L. S. Kariyawasam, J. Rolsma and Y. Yang, Chemically Recyclable Dithioacetal Polymers via Reversible Entropy–Driven Ring–Opening Polymerization, Angew. Chem., Int. Ed., 2023, 62(26), e202305900 CrossRef.
- D. P. N. Satchell and R. S. Satchell, Mechanisms of Hydrolysis of Thioacetals, Chem. Soc. Rev., 1990, 19(1), 55 RSC.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley, New York City, 4th edn, 1999 Search PubMed.
- C. B. Godiya, S. Gabrielli, S. Materazzi, M. S. Pianesi, N. Stefanini and E. Marcantoni, Depolymerization of Waste Poly(Methyl Methacrylate) Scraps and Purification of Depolymerized Products, J. Environ. Manage., 2019, 231, 1012–1020 CrossRef CAS PubMed.
- M. J. Flanders and W. M. Gramlich, Reversible-Addition Fragmentation Chain Transfer (RAFT) Mediated Depolymerization of Brush Polymers, Polym. Chem., 2018, 9(17), 2328–2335 RSC.
- Y. Sano, T. Konishi, M. Sawamoto and M. Ouchi, Controlled Radical Depolymerization of Chlorine-Capped PMMA via Reversible Activation of the Terminal Group by Ruthenium Catalyst, Eur. Polym. J., 2019, 120, 109181 CrossRef CAS.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, Reversing RAFT Polymerization: Near-Quantitative Monomer Generation Via a Catalyst-Free Depolymerization Approach, J. Am. Chem. Soc., 2022, 144(10), 4678–4684 CrossRef CAS PubMed.
- J. B. Young, R. W. Hughes, A. M. Tamura, L. S. Bailey, K. A. Stewart and B. S. Sumerlin, Bulk Depolymerization of Poly(Methyl Methacrylate) via Chain-End Initiation for Catalyst-Free Reversion to Monomer, Chem, 2023, 9(9), 2669–2682 CAS.
- J. B. Young, J. I. Bowman, C. B. Eades, A. J. Wong and B. S. Sumerlin, Photoassisted Radical Depolymerization, ACS Macro Lett., 2022, 11(12), 1390–1395 CrossRef CAS PubMed.
- V. Bellotti, K. Parkatzidis, H. S. Wang, N. De Alwis Watuthanthrige, M. Orfano, A. Monguzzi, N. P. Truong, R. Simonutti and A. Anastasaki, Light-Accelerated Depolymerization Catalyzed by Eosin Y, Polym. Chem., 2022, 14(3), 253–258 RSC.
- M. R. Martinez, S. Dadashi-Silab, F. Lorandi, Y. Zhao and K. Matyjaszewski, Depolymerization of P(PDMS11MA) Bottlebrushes via Atom Transfer Radical Polymerization with Activator Regeneration, Macromolecules, 2021, 54(12), 5526–5538 CrossRef CAS.
- M. R. Martinez, F. De Luca Bossa, M. Olszewski and K. Matyjaszewski, Copper(II) Chloride/Tris(2-Pyridylmethyl)Amine-Catalyzed Depolymerization of Poly(n-Butyl Methacrylate), Macromolecules, 2022, 55(1), 78–87 CrossRef CAS.
- M. R. Martinez, D. Schild, F. De Luca Bossa and K. Matyjaszewski, Depolymerization of Polymethacrylates by Iron ATRP, Macromolecules, 2022, 55(23), 10590–10599 CrossRef CAS.
- F. De Luca Bossa, G. Yilmaz and K. Matyjaszewski, Fast Bulk Depolymerization of Polymethacrylates by ATRP, ACS Macro Lett., 2023, 12(8), 1173–1178 CrossRef CAS PubMed.
- O. Shelef, S. Gnaim and D. Shabat, Self-Immolative Polymers: An Emerging Class of Degradable Materials with Distinct Disassembly Profiles, J. Am. Chem. Soc., 2021, 143(50), 21177–21188 CrossRef CAS PubMed.
- Z. Deng and E. R. Gillies, Emerging Trends in the Chemistry of End-to-End Depolymerization, JACS Au, 2023, 3(9), 2436–2450 CrossRef CAS PubMed.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, Self-Immolative Polymers., J. Am. Chem. Soc., 2008, 130(16), 5434–5435 CrossRef CAS PubMed.
- J. A. Kaitz, O. P. Lee and J. S. Moore, Depolymerizable Polymers: Preparation, Applications, and Future Outlook, MRS Commun., 2015, 5(2), 191–204 CrossRef CAS.
- F. Wang and C. E. Diesendruck, Polyphthalaldehyde: Synthesis, Derivatives, and Applications, Macromol. Rapid Commun., 2018, 39(2), 1700519 CrossRef PubMed.
- S. Soars, J. Kamps, B. Fairbanks and C. Bowman, Stimuli-Responsive Depolymerization of Poly(Phthalaldehyde) Copolymers and Networks, Macromol. Chem. Phys., 2021, 222(17), 2100111 CrossRef CAS.
- E. M. Lloyd, H. Lopez Hernandez, A. M. Feinberg, M. Yourdkhani, E. K. Zen, E. B. Mejia, N. R. Sottos, J. S. Moore and S. R. White, Fully Recyclable Metastable Polymers and Composites, Chem. Mater., 2019, 31(2), 398–406 CrossRef CAS.
- B. Fan, J. F. Trant, R. E. Yardley, A. J. Pickering, F. Lagugné-Labarthet and E. R. Gillies, Photocontrolled Degradation of Stimuli-Responsive Poly(Ethyl Glyoxylate): Differentiating Features and Traceless Ambient Depolymerization, Macromolecules, 2016, 49(19), 7196–7203 CrossRef CAS.
- S. Pal, A. Sommerfeldt, M. B. Davidsen, M. Hinge, S. U. Pedersen and K. Daasbjerg, Synthesis and Closed-Loop Recycling of Self-Immolative Poly(Dithiothreitol), Macromolecules, 2020, 53(12), 4685–4691 CrossRef CAS.
- D. Basak, R. Kumar and S. Ghosh, Telechelic Poly(Disulfide)s and Related Block Copolymer, Macromol. Rapid Commun., 2014, 35(15), 1340–1344 CrossRef CAS PubMed.
- Y. Liu, Z. Yu, B. Wang, P. Li, J. Zhu and S. Ma, Closed-Loop Chemical Recycling of Thermosetting Polymers and Their Applications: A Review. Green Chemistry, Royal Society of Chemistry, 2022, pp. 5691–5708 Search PubMed.
- N. Ramdani, H. Zaimeche and M. Derradji, Biobased Thermally-Stable Aromatic Cyanate Ester Thermosets: A Review, React. Funct. Polym., 2021, 168, 105037 CrossRef CAS.
- B. A. Helms, Polydiketoenamines for a Circular Plastics Economy, Acc. Chem. Res., 2022, 55(19), 2753–2765 CrossRef CAS PubMed.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Closed-Loop Recycling of Plastics Enabled by Dynamic Covalent Diketoenamine Bonds, Nat. Chem., 2019, 11(5), 442–448 CrossRef CAS PubMed.
- A. R. Epstein, J. Demarteau, B. A. Helms and K. A. Persson, Variable Amine Spacing Determines Depolymerization Rate in Polydiketoenamines, J. Am. Chem. Soc., 2023, 145(14), 8082–8089 CrossRef CAS PubMed.
- Z. Hu, F. Hu, L. Deng, Y. Yang, Q. Xie, Z. Gao, C. Pan, Y. Jin, J. Tang and G. Yu, Reprocessible Triketoenamine Based Vitrimers with Closed–loop Recyclability, Angew. Chem., Int. Ed., 2023, 62(34), e202306039 CrossRef CAS PubMed.
- J. Demarteau, A. R. Epstein, P. R. Christensen, M. Abubekerov, H. Wang, S. J. Teat, T. J. Seguin, C. W. Chan, C. D. Scown, T. P. Russell, J. D. Keasling, K. A. Persson and B. A. Helms, Circularity in Mixed-Plastic Chemical Recycling Enabled by Variable Rates of Polydiketoenamine Hydrolysis, Sci. Adv., 2022, 8, 8823 CrossRef PubMed.
- Q. Li, S. Ma, N. Lu, J. Qiu, J. Ye, Y. Liu, S. Wang, Y. Han, B. Wang, X. Xu, H. Feng and J. Zhu, Concurrent Thiol-Ene Competitive Reactions Provide Reprocessable, Degradable and Creep-Resistant Dynamic-Permanent Hybrid Covalent Networks, Green Chem., 2020, 22(22), 7769–7777 RSC.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Exploring How Vitrimer-like Properties Can Be Achieved from Dissociative Exchange in Anilinium Salts, Macromolecules, 2020, 53(4), 1233–1244 CrossRef CAS.
- K. Saito, F. Eisenreich, T. Türel and Ž Tomović, Closed–Loop Recycling of Poly(Imine–Carbonate) Derived from Plastic Waste and Bio–based Resources, Angew. Chem., Int. Ed., 2022, 61(43), e202211806 CrossRef CAS PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Vitrimers Designed Both to Strongly Suppress Creep and to Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments, Macromolecules, 2018, 51(15), 5537–5546 CrossRef CAS.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Catalyst-Free Vitrimers from Vinyl Polymers, Macromolecules, 2019, 52(5), 2105–2111 CrossRef CAS.
- J. S. A. Ishibashi and J. A. Kalow, Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum's Acid-Derived Cross-Links, ACS Macro Lett., 2018, 7(4), 482–486 CrossRef CAS PubMed.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, Poly(Oxime-Ester) Vitrimers with Catalyst-Free Bond Exchange, J. Am. Chem. Soc., 2019, 141(35), 13753–13757 CrossRef CAS PubMed.
- H. Zeng, Z. Tang, Y. Duan, S. Wu and B. Guo, Recyclable Crosslinked Elastomer Based on Dynamic Dithioacetals, Polymer, 2021, 229, 124007 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–Yne Cross–Linking, Angew. Chem., 2020, 132(9), 3637–3646 CrossRef.
- C. A. Tretbar, J. A. Neal and Z. Guan, Direct Silyl Ether Metathesis for Vitrimers with Exceptional Thermal Stability, J. Am. Chem. Soc., 2019, 141(42), 16595–16599 CrossRef CAS PubMed.
- J. Luo, Z. Demchuk, X. Zhao, T. Saito, M. Tian, A. P. Sokolov and P. F. Cao, Elastic Vitrimers: Beyond Thermoplastic and Thermoset Elastomers, Matter, 2022, 5(5), 1391–1422 CrossRef CAS.
- Y. Ma, X. Jiang, Z. Shi, J. A. Berrocal and C. Weder, Closed–Loop Recycling of Vinylogous Urethane Vitrimers, Angew. Chem., Int. Ed., 2023, 62(36), e202306188 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Vinylogous, Urethane Vitrimers, Adv. Funct. Mater., 2015, 25(16), 2451–2457 CrossRef CAS.
- S. Efstathiou, C. Ma, D. Coursari, G. Patias, L. Al-Shok, A. M. Eissa and D. M. Haddleton, Functional PH-Responsive Polymers Containing Dynamic Enaminone Linkages for the Release of Active Organic Amines, Polym. Chem., 2022, 13, 2362–2374 RSC.
- F. Chen, Q. Cheng, F. Gao, J. Zhong, L. Shen, C. Lin and Y. Lin, The Effect of Latent Plasticity on the Shape Recovery of a Shape Memory Vitrimer, Eur. Polym. J., 2021, 147, 110304 CrossRef CAS.
- F. Wang, K. Huang, Z. Xu, F. Shi and C. Chen, Self-Healable Nanocellulose Composite Hydrogels Combining Multiple Dynamic Bonds for Drug Delivery, Int. J. Biol. Macromol., 2022, 203, 143–152 CrossRef CAS PubMed.
- W. Niu, Z. Zhang, Q. Chen, P. F. Cao and R. C. Advincula, Highly Recyclable, Mechanically Isotropic and Healable 3D-Printed Elastomers via Polyurea Vitrimers, ACS Mater. Lett., 2021, 3(8), 1095–1103 CrossRef CAS.
- Z. Shen, Y. Wu, S. Qiu, H. Deng, R. Hou and Y. Zhu, UV-Thermal, Dual-Cured Polymers with Degradable and Anti-Bacterial Function, Prog. Org. Coat., 2020, 148, 105783 CrossRef CAS.
- Y. Lin, Y. Chen, Z. Yu, Z. Huang, J. C. Lai, J. B. H. Tok, Y. Cui and Z. Bao, Reprocessable and Recyclable Polymer Network Electrolytes via Incorporation of Dynamic Covalent Bonds, Chem. Mater., 2022, 34(5), 2393–2399 CrossRef CAS.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–Yne Cross–Linking, Angew. Chem., 2020, 132(9), 3637–3646 CrossRef.
- L. Cederholm, J. Wohlert, P. Olsén, M. Hakkarainen and K. Odelius, “Like Recycles Like”: Selective Ring-Closing Depolymerization of Poly(L-Lactic Acid) to L-Lactide, Angew. Chem., Int. Ed., 2022, 61(33), e202204531 CrossRef CAS PubMed.
- L. Monsigny, J.-C. Berthet and T. Cantat, Depolymerization of Waste Plastics to Monomers and Chemicals Using a Hydrosilylation Strategy Facilitated by Brookhart's Iridium(III) Catalyst, ACS Sustainable Chem. Eng., 2018, 6(8), 10481–10488 CrossRef CAS.
- Annual Report 2021, Infinitum, 2021.
- Norway's deposit return scheme is world's recycling role model https://www.tomra.com/en/reverse-vending/media-center/feature-articles/norway-deposit-return-scheme (accessed Oct 8, 2023).
- PET MARKET IN EUROPE STATE OF PLAY 2022 PRODUCTION, COLLECTION AND RECYCLING, Eunomia, 2022.
- C. Staub, US PET bottle recycling rate continues to sink https://resource-recycling.com/plastics/2021/11/03/us-pet-bottle-recycling-rate-continues-to-sink/ (accessed Oct 8, 2023).
- Intergovernmental Negotiating Committee on Plastic Pollution https://www.unep.org/inc-plastic-pollution (accessed Oct 8, 2023).
- Plastics Strategy https://environment.ec.europa.eu/strategy/plastics-strategy_en (accessed Oct 8, 2023).
- Packaging Waste https://environment.ec.europa.eu/topics/waste-and-recycling/packaging-waste_en (accessed Oct 8, 2023).
- APR Design Guide https://plasticsrecycling.org/apr-design-guide (accessed Oct 8, 2023).
- Design for Recycling Guidelines https://recyclass.eu/recyclability/design-for-recycling-guidelines/ (accessed Oct 8, 2023).
- M. Heffernan, Largest commingled MRF in North America adds robots https://resource-recycling.com/recycling/2022/12/06/largest-commingled-mrf-in-north-america-adds-robots/#:˜:text=Sims%20Municipal%20Recycling's%20(SMR)%20Sunset,Sanitation%20throughout%20the%20five%20boroughs (accessed Oct 8, 2023).
- Investment Pays off at a Pioneering Material Recovery Facility, 2016.
- D. E. Fagnani, J. L. Tami, G. Copley, M. N. Clemons, Y. D. Y. L. Getzler and A. J. McNeil, 100th Anniversary of Macromolecular Science Viewpoint: Redefining Sustainable Polymers, ACS Macro Lett., 2021, 10(1), 41–53 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |