
An integrative sustainability assessment of the Tsuji–Trost reaction simulating allylic amination under non-conventional (vs. conventional) conditions†
Sangita Dattatray
Shinde‡
,
Gargi Nikhil
Vaidya‡
 ,
Shyam Kumar
Lokhande‡
,
Anil
Shaha
,
Ramesh Hiralal
Choudhary
and
Dinesh
Kumar
*
,
Shyam Kumar
Lokhande‡
,
Anil
Shaha
,
Ramesh Hiralal
Choudhary
and
Dinesh
Kumar
*
Department of Medicinal Chemistry, National Institute of Pharmaceutical Education and Research (NIPER), Ahmedabad, Palaj, Gandhinagar-382355, Gujarat, India. E-mail: dkchem79@gmail.com; dineshk@niperahm.res.in
First published on 7th February 2024
Abstract
With allyl compounds being a valuable chemical commodity, it is important to understand the sustainability aspects of the Tsuji–Trost reaction in terms of productivity (reactivity-coupled utility), environmental impact (including health hazards), and economic burden and benefits of commercially available allylic precursors under varying conditions. However, comprehensive literature searches reveal that no such study is publicly accessible; the majority of the literature focuses only on the synthetic utility of a few allylic precursors. In this context, we undertook a study for sustainability assessment of 26 allyl precursors (with diverse ionisable partners) by simulating an allylic amination reaction in an alternative reaction medium, water (vs. an organic solvent, OS), under Earth-abundant nickel (vs. traditional palladium) catalysis. The study was accomplished through: (1) conducting reactivity-utility profiling of allylic precursors; (2) identifying the major side reactions occurring with allylic precursors (water vs. OS); and (3) determining green metrics (E-factor, atom economy, atom efficiency, RME, and EcoScale) of the amination process linked with allylic precursors. The key highlights of the study include the following: (a) water is as diverse as an organic solvent in facilitating the Tsuji–Trost allylic amination; (b) diverse allylic precursors could be employed in water as well as in an organic solvent to realize allylic amination; (c) nickel catalysis is a viable alternative to palladium for allylic amination with distinct superiority in the water medium while using an allyl alcohol and allyl amine (trans-N-allylation). Further advancing sustainability and catalysis, we demonstrated for the first time a fully catalytic ‘in-water’ allylic amination reaction using an allylic alcohol and a Ni(II)-catalyst with features like scale-up synthesis (up to 10 g), catalytic usage of a reductant, etc. The authors propose this work as a sustainability guide to steer further research and developments in academia and industry related to allylation chemistries.
Introduction
Sustainable development in chemical processes and technology is essential for tackling global issues and creating a more resilient and eco-friendly future.1–4 It benefits present and future generations by reducing the environmental impact (including carbon footprints), preserving resources, promoting economic success, and increasing health and well-being. One crucial step towards achieving this goal is to advance and embrace green (sustainable) chemistry in the ongoing chemical process, considering the negative impacts of the manufacturing of fine chemicals, agrochemicals, and pharmaceuticals on health and environmental wellness.5–15The presence of an allylic functionality in an organic framework is very diverse owing to its direct utility (as pharmaceuticals, bioactive compounds, and fine chemicals) and indirect applications (further derivatization through vinylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and/or allylic C–H bond manipulations).16–26 As a consequence, the direct installation of an allyl functionality is a hot area of research, and substantial progress has been made to introduce it using diverse methods and strategies.27–33 In this context, the Tsuji–Trost reaction remains the prime tool considering its process robustness, broad scope, functional group compatibility, and adaptation to asymmetric variations. Over time, promising growth has been realised in this field, including the use of different allylic precursors, the use of alternative reaction media, and the inclusion of Earth-abundant metal catalysis.34–52
C and/or allylic C–H bond manipulations).16–26 As a consequence, the direct installation of an allyl functionality is a hot area of research, and substantial progress has been made to introduce it using diverse methods and strategies.27–33 In this context, the Tsuji–Trost reaction remains the prime tool considering its process robustness, broad scope, functional group compatibility, and adaptation to asymmetric variations. Over time, promising growth has been realised in this field, including the use of different allylic precursors, the use of alternative reaction media, and the inclusion of Earth-abundant metal catalysis.34–52
Given the rapid expansion of the Tsuji–Trost reaction,34–52 it is important to have an integrative (and holistic) sustainability assessment of the Tsuji–Trost reaction in the context of productivity (reactivity-coupled utility), environmental impact (including health hazards), and economic burden of commercially available allylic precursors under varying conditions to fulfil the sustainability goal of an important chemical transformation. It is to be noted that no such information (or study) is publicly available or accessible; the majority of the literature deals with the synthetic utility of a few allylic precursors. In this context, we looked into an integrative sustainability assessment of 26 allyl precursors (with diverse ionisable partners) by simulating an allylic amination reaction in the most preferred green solvent water (vs. a traditional organic solvent) under nickel (vs. traditional palladium) catalysis. The key findings indicate: (a) the suitability and compatibility of water in facilitating the allylic amination reaction with diverse allylic partners; (b) the superior catalysis of nickel over palladium in water, particularly using an allylic alcohol; (c) the possibility of trans-N-allylation in water under nickel catalysis; and (d) elucidating the role of H-bond networking in activating allylic precursors. In a further sustainable development, we demonstrated for the first time a fully catalytic ‘in-water’ allylic amination using an allylic alcohol and a Ni(II) catalyst, featuring properties such as scale-up synthesis (up to 10 g), catalytic usage of a reductant, etc., representing further advancement in catalysis and sustainability of the allylic amination reaction.
Results and discussion
Background information and methodology employed
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving the reaction of a nucleophile with a substrate having a leaving group at an allylic position. Originally discovered as an allylic alkylation reaction, the scope of the reaction has been expanded significantly to different carbon, nitrogen, and oxygen-based nucleophiles.53–55 Among the adopted nucleophiles, nitrogen-based nucleophiles are very well adapted owing to the diverse applications of allylamines ranging from materials to medicines.17,56–59 Logically, it was decided to adhere to an amine as a model nucleophile for the target study as described in the Fig. 1. | ||
| Fig. 1 List of allylic precursors (2a–2z) under examination and adopted methodology (model reaction conditions). | ||
Our lab is actively engaged in the development of sustainable organic reactions for diverse applications, including clean pharmaceutical synthesis.60–66 In this context, we recently described an elegant Ni(cod)2-catalyzed ‘in-water’ allylic amination protocol with a diverse substrate scope.61 To the best of our knowledge, there is no existing method that demonstrates a better diversity and adaptability of the substrate scope than this protocol in an aqueous environment, and hence we adopted this protocol as a model reaction and reference method (Method A) for this study. Further variations were done relative to Method A to constitute Method B (allylic amination in organic media under nickel catalysis) and Method C (allylic amination in water under palladium catalysis), as described in Fig. 1.
Assessment of the reactivity-utility profile of allyl precursors in water under nickel catalysis (Method A)
Using the model reaction conditions, the reactivity-coupled utility of 26 different allyl precursors (2a–2z) featuring C–X (X = O/N/S/P/C/B/Si) bond ionisation with phenyl piperazine 1a in aqueous TPGS media under Ni-catalysis was assessed (Method A, Fig. 1). Allyl halides (chloro 2a, bromo 2b, and iodo 2c) underwent amination to yield 3a in 15, 68, and 69%, respectively; the corresponding yields may be correlated to intrinsic halide ionisation properties. Allyl alcohol 2d and their derivatives (2e–2p) were found to be effective in producing 3a, the noted exceptions were allyl trifluoroacetate 2f, allyl oxalyl chloride 2l, and allyl methyl ether 2n. Alcohol 2d and their derivatives, acetate 2e, cinnamate 2g, carbonates (2h, 2i), carbamate 2j, phosphate 2k, and phenyl ether 2o, were superior to give 3a with yields ranging from 81–89% owing to their ease of ionisation of the C–O bond. Allyl trifluoroacetate 2f underwent competitive N-acylation (vs. π-allyl-Ni-complexation) to yield the corresponding amide 4a (38%) as the exclusive product. Similarly, 2l did not yield any desired product 3a; the obtained product corresponds to amide 4b (41%). Allyl glycidyl ether 2p gave 3a (59%) as the major product, along with the epoxide aminolysis product 4c (15%). Allyl methyl ether 2n was found to be non-reactive under the reaction conditions.Allylic amination (trans-N-allylation) following C–N bond ionisation was insignificant (urea 2r, isocyanate 2s, isothiocyanate 2t) except for 2q (N-methylallylamine) wherein the formation of the desired product 3a was observed with an excellent yield (76%). In the case of isocyanate 2s, the major product corresponds to urea 4c (22%).
The 3a formation following C–S bond ionization was observed in sulfone 2v (64%) only. No allylic amination was noticed following C–P (2w), C–Si (2x), C–B (2y), or C–C (2Z) cleavage. Thus, out of 26 allylic precursors (2a–2z) tested, 16 were found to be reactive under the reaction conditions, yielding 3a with variable yields, validating the wider catalytic profile of nickel in the aqueous system (Fig. 2 & 3).
 | ||
| Fig. 2 Assessment of allylic precursors towards allylic amination yielding 3a under Ni(0)-catalysis (aqueous media vs. OS). | ||
 | ||
| Fig. 3 Identification of major side reactions occurring with allylic precursors. | ||
Assessment of the reactivity-utility profile of allyl precursors in an organic solvent (OS) under nickel catalysis (Method B)
Next, we examined the reactivity-coupled-utility of these 26 precursors in an organic solvent (1,4-dioxane)67 under similar conditions but in the absence of aq. TPGS (Method B, Fig. 1). The reactivity-utility profile of 2e, 2g, 2h, 2j, and 2o in dioxane nearly parallels that under aqueous conditions with comparable yields. The striking difference was in the yields of 3a employing allyl alcohol 2d (48% in OS vs. 89% in aq. TPGS) and N-allylmethylamine 2q (28% in OS vs. 76% in aq. TPGS). Such significant yield variations (OS vs. H2O) in turn provide insights into the distinct role of water (H-bond networking) in the activation of 2d (and 2q) towards the π-allyl-Ni-complexation. Similarly, the yields of 3a employing 2k and 2m could be explained on the basis of the H-bond-assisted activation of allyl precursors (Fig. 2 & 3). The other noted differences were in the case of allyl trifluoroacetate 2f, which formed 35% of 3a (vs. 0% in aq. TPGS). The halide precursors (2a, 2b, 2c) also resulted in an inferior 3a yield (vs. aq. TPGS) (Fig. 2).Assessment of the reactivity-utility profile of allyl precursors in water under palladium catalysis (Method C)
The analogous Pd(0)-catalysed reactions were performed to measure the relative scope, reactivity, and utility of allyl precursors in water under palladium catalysis (Method C, Fig. 1). As indicated, in many cases, palladium offers a similar reactivity-utility profile to allyl precursors as nickel; however, surprisingly, allylic amination was distinctly inferior using allyl alcohol 2d under the optimized conditions (Ni vs. Pd: 89 and 0). Such contrasting yields of 3a could be due to the smaller size of Ni, resulting in a higher nucleophilic nature, and variable oxidation states (0, +2 as well as +1, +3) making them more effective to harness C–O bond manipulations compared to Pd.68 The other noted difference with respect to the formation of 3a was observed in the cases of allyl trifluoroacetate 2f (Ni vs. Pd: 0 and 44), allyl oxalyl chloride 2l (Ni vs. Pd: 0 and 25), allyl isocyanate 2s (Ni vs. Pd: 21 and 48), and allyl isothiocyanate 2t (Ni vs. Pd: 0 and 41). The comparative yields corresponding to each allylic precursor (Ni vs. Pd) are depicted in Fig. 4. | ||
| Fig. 4 Allylic cross-amination employing different allylic precursors in aqueous micelles: Ni vs. Pd. | ||
Role of water
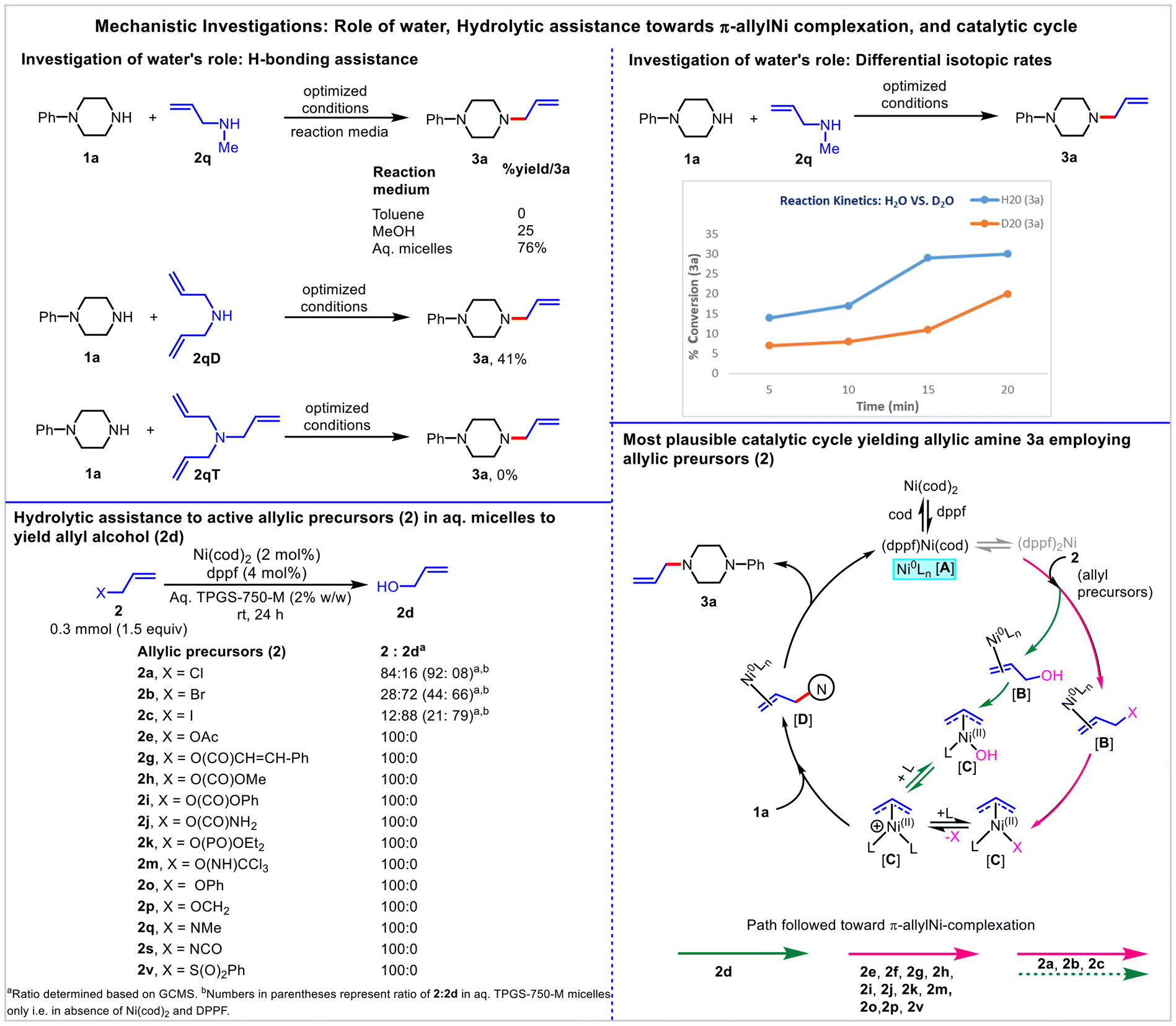
Two substantial issues were brought to light by the results of the current investigation. First, how important is water, especially considering that some allyl precursors, such as N-methylallylamine 2q, show excellent reactivity and yield (3a) when dissolved in aqueous micelles (vs. organic solvent)? Second, in the case of allyl precursors (where X ≠ OH), whether the ‘in-water’ allylic amination follows the direct ionization of allylic C–X or it is combined, including the ionization of allyl alcohol 2d resulting from the hydration of allylic precursors.To access the specific role of water, analogous reactions employing 2q were performed in toluene and MeOH under optimized conditions. The reaction was non-productive in toluene (3a, 0%), while MeOH was found to be conducive to yield 3a, albeit in poor yield (25%). Furthermore, the 3a (41%) formation was observed in the case of diallylamine 2qD (having free NH) while no such amination was noticed using triallylamine 2qT (lacking free NH). These results suggested the definitive role of H-bond networking and insights into the water-enabled hydrogen bond-assisted activation of allyl amine 2q towards the π-allyl-Ni complexation.46,69 To investigate the role of the HB effect in the activation of allyl amine 2q further, a kinetic (yield vs. time) study of the model reaction was planned (D2O vs. H2O-derived micelles).70,71 Treatment of 1a with 2q separately in H2O-micelles and D2O-micelles under the optimized conditions, and the formation of 3a were monitored for 20 minutes at a 5-minute interval. On each occasion, a better yield of 3a was obtained for the reactions performed in H2O compared to the D2O system. This decrease in 3a yield is indicative of the lesser HB affinity of N–D2O vs. N–H2O. This agrees with the reported results of weaker hydrogen bonding by deuterium with oxygen and nitrogen acceptors.72,73
Thus, water not only provides sustainability by eliminating the need for VOS as reaction media, but it also activates otherwise poorly reactive (or non-reactive) allyl amine 2qvia H-bond networking and stabilization of the resulting amine anion by a strong solvation effect (Fig. 5).
 | ||
| Fig. 5 Differential ionization pathways delivering π-allyl-Ni-complex to yield 3a employing different allylic precursors in aqueous micelles under Ni(0)-catalysis. | ||
Subsequently, to determine whether the ‘in-water’ allylic amination follows the direct ionization of allyl precursors (where X ≠ OH) or it is combined, including the ionization of 2d resulting from the hydration of allyl precursors (where X ≠ OH), the different allyl precursors (where X ≠ OH) were subjected to the optimized conditions but in the absence of 1a. A variable amount of 2d (GCMS) was observed in 2a (16%), 2b (72%), and 2c (88%) indicating the possible hydrolytic assistance towards the formation of 3a in the case of allylic halides. In other cases, π-allyl complexation resulted directly via ionization of allyl precursors, as no formation of 2d was observed (Fig. 5). The above study also justifies the higher 3a yield in aq. micelles (vs. organic solvent) employing an allylic halide. In many cases, in addition to the C–X bond dissociation energy (ESI†),74 the effects of water's atypical chemical and physical properties cannot be ruled out as contributing factors to the observed reactivity or selectivity of allylic precursors.
Based on these studies, the proposed Ni(0)/Ni(II) catalytic cycle that accounts for product formation is shown in Fig. 5. First, ligand exchange between Ni(cod)2 and dppf generates the catalytically active Ni-complex [(dppf)Ni(cod)] A.75–77 Because of the greater steric demand of dppf, the favored state involves a single dppf ligand in the Ni sphere with additional stabilization by the relatively small cod ligand. The formed Ni-complex A coordinates with the vinylic CC double bond of 2 to generate the complex B. Subsequent oxidative addition (facilitated by water-assisted H-bond networking) results in the formation of the electrophilic η3 π-allyl-NiII complex C. This was followed by the nucleophilic attack to generate the penultimate intermediate D, which ultimately led to product formation following the detachment of nickel from alkene. The formation of a π-allyl-Ni-complex employing different allylic precursors in aqueous micelles is presented in Fig. 5. It is to be noted that the formation of (dppf)2Ni cannot be ruled out; however, it suffers from an initial high-energy dissociation of one ligand prior to oxidative addition, rendering the system poorly active. Whereas in the case of [(dppf)Ni(cod)], a weaker-binding cod ligand displaced easily to enable a facile oxidative addition.75 Furthermore, the role of water in the stabilization of cationic nickel allylic complexes with a hydroxyl group cannot be ruled out.
Estimation of green metrics and environmental, health, and safety (EHS) hazards assessment of allyl precursors (2a–2z)
The environmental and eco-toxicological impact of allyl precursors, as well as the relative green metrics of the associated process, were subsequently established.78–81 To calculate green metrics (E-factor, atom economy, atom efficiency, RME, and EcoScale), we chose allyl precursors (2a, 2b, 2c, 2d, 2e, 2g, 2h, 2i, 2j, 2k, 2m, 2o, 2p, 2q, 2s, 2v) that were found to be active in aqueous micelles under nickel catalysis, as shown in Table 1. It should be emphasised that the EcoScale has been presented as a relative EcoScale due to shared factors such as technical setup, temperature/time, workup, and purification. The EHS risks of the allyl precursors (2a–2z), specifically in terms of flammability, mutagenicity, carcinogenicity, aquatic hazard, biodegradability, and the LD50 (oral) value, were analysed based on the information retrieved from the MSDS data of the allyl precursors (2a–2z) (Table 2).| Allylic pre. (2) | E-Factor | % atom economy | %Atom efficiency | % RME | Rel. EcoScalea |
|---|---|---|---|---|---|
| a The EcoScale has been represented as relative EcoScale wherein the common parameters (technical setup, temperature/time, workup and purification) have been excluded while calculating the penalty points as they will be the essentially same in all the cases. | |||||
| 2a | 8.15 | 84.66 | 12.69 | 10.92 | 42.5 |
| 2b | 1.50 | 71.37 | 48.53 | 39.93 | 69 |
| 2c | 2.80 | 61.21 | 33.05 | 26.30 | 62 |
| 2d | 0.38 | 91.75 | 81.65 | 72.04 | 79.5 |
| 2e | 0.79 | 77.04 | 66.25 | 55.56 | 78 |
| 2f | N/A | N/A | N/A | N/A | N/A |
| 2g | 1.68 | 57.68 | 47.29 | 37.23 | 81 |
| 2h | 1.05 | 72.62 | 58.82 | 48.59 | 75.5 |
| 2i | 1.53 | 59.38 | 49.87 | 39.48 | 82 |
| 2j | 0.82 | 76.76 | 65.24 | 54.69 | 82.5 |
| 2k | 1.6 | 56.71 | 47.63 | 37.40 | 82 |
| 2l | N/A | N/A | N/A | N/A | N/A |
| 2m | 2.9 | 55.42 | 32.69 | 30.93 | 59.5 |
| 2n | N/A | N/A | N/A | N/A | N/A |
| 2o | 1.2 | 68.19 | 55.23 | 44.99 | 70.5 |
| 2p | 1.8 | 73.14 | 41.68 | 34.52 | 63.5 |
| 2q | 0.7 | 86.60 | 65.81 | 57.10 | 86.5 |
| 2r | N/A | N/A | N/A | N/A | N/A |
| 2s | 5.7 | 82.39 | 17.30 | 14.77 | 45.5 |
| 2t | N/A | N/A | N/A | N/A | N/A |
| 2u | N/A | N/A | N/A | N/A | N/A |
| 2v | 2.3 | 58.68 | 37.55 | 29.66 | 72 |
| 2w | N/A | N/A | N/A | N/A | N/A |
| 2x | N/A | N/A | N/A | N/A | N/A |
| 2y | N/A | N/A | N/A | N/A | N/A |
| 2z | N/A | N/A | N/A | N/A | N/A |
| Allylic precursors | Code | Flammability | Mutagenicity | Carcinogenicity | Aquatic hazard | Biodegradability | LD50 oral (rat) |
|---|---|---|---|---|---|---|---|
| a No data available. b Carcinogenicity, no ingredient of this product present at levels greater than or equal to 0.1% is identified as probable, possible, or confirmed human carcinogen by IARC. | |||||||
| Allyl chloride | 2a | Flammable (Cat. 2) | Suspected (Cat. 2) | Suspected (Cat. 2) | Very toxic | Readily | 419 mg kg−1 |
| Allyl bromide | 2b | Flammable (Cat. 2) | Suspected (Cat. 1B) | Suspected (Cat. 1B) | Very toxic | Readily | 200 mg kg−1 |
| Allyl iodide | 2c | Flammable (Cat. 2) | Suspected (Cat. 1B) | Suspected (Cat. 1B) | Very toxic | Readily | 200 mg kg−1 |
| Allyl alcohol | 2d | Flammable (Cat. 2) | — | NOb | Very toxic | Readily | 105 mg kg−1 |
| Allyl acetate | 2e | Flammable (Cat. 2) | NDAa | NOb | NDAa | NDAa | 130 mg kg−1 |
| Allyl trifluoroacetate | 2f | Flammable (Cat. 2) | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl cinnamate | 2g | NDAa | NDAa | NOb | NDAa | NDAa | 1.52 mg kg−1 |
| Allyl methyl carbonate | 2h | Flammable (Cat. 3) | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl phenyl carbonate | 2i | NDAa | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl carbamate | 2j | NDAa | NDAa | NOb | Very toxic | NDAa | NDAa |
| Allyl diethyl phosphate | 2k | NDAa | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl oxalyl chloride | 2l | Flammable (Cat. 3) | NDAa | NDAa | Toxic | NDAa | NDAa |
| Allyl imidate | 2m | NDAa | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl methyl ether | 2n | Flammable (Cat. 2) | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl phenyl ether | 2o | NDAa | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl glycidyl ether | 2p | Flammable (Cat. 3) | Suspected (Cat. 2) | Suspected (Cat. 2) | Harmful | Not readily | 1.60 mg kg−1 |
| N-Allylmethylamine | 2q | Flammable (Cat. 2) | NDAa | NDAa | NDAa | NDAa | 100 mg kg−1 |
| Allyl urea | 2r | NDAa | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl isocyanate | 2s | Flammable (Cat. 3) | NDAa | NDAa | NDAa | NDAa | 168 mg kg−1 |
| Allyl isothiocyanate | 2t | Flammable (Cat. 3) | NDAa | NOb | Very toxic | Not readily | 425.4 mg kg−1 |
| Allyl methyl sulfide | 2u | Flammable (Cat. 2) | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl phenyl sulfone | 2v | NDAa | NDAa | NDAa | NDAa | NDAa | NDAa |
| Diethyl allyl phosphonate | 2w | NDAa | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl(chloro)dimethylsilane | 2x | Flammable (Cat. 2) | NDAa | NDAa | NDAa | NDAa | NDAa |
| Allyl pinacol boronate | 2y | Flammable (Cat. 3) | NDAa | NOb | NDAa | NDAa | NDAa |
| Allyl benzene | 2z | Flammable (Cat. 3) | NDAa | NOb | NDAa | NDAa | 5.54 mg kg−1 |
Practical demonstration of true catalytic ‘in-water’ allylic amination using a Ni(0) pre-catalyst
In the context of allylic amination reactions, Ni(cod)2-catalyzed allylic amination reactions were found to be highly promising with diverse substrate scope, selectivity, and sustainability.46,47,52 However, the air-sensitive nature of Ni(cod)2 poses a handling issue and limits their catalytic applications.82 This implies looking out for an alternative Ni(0) pre-catalytic system aiming to address the challenges of using reductants in an aqueous environment. The intense interest in this area led to the development of an elegant Ni(0) pre-catalyst system with a wide substrate scope.50,51 Despite notable development, these protocols require organic solvents as reaction media, including the use of dimethylacetamide (DMA), a solvent classified as a Substance of Very High Concern (SVHC) for its reproductive toxicity and teratogenicity.83,84 Therefore, we planned to investigate a genuine catalytic ‘in-water’ allylic amination employing alcohol directly using a Ni(0)-pre-catalyst.The study began with a systematic evaluation of different Ni(0) pre-catalysts in the presence of the dppf ligand and stoichiometric Zn powder (reductant) for the model reaction involving treatment of 1a with 2a at room temperature in aq. TPGS-750-M micelles. Promising results were obtained in the case of NiBr2 (3a, 42%) followed by NiCl2 glyme (3a, 29%). We next explored the opportunity to improve the 3a yield with a catalytic nano-Zn reductant. It was delightful to note that the stoichiometric replacement of Zn powder (2 equiv.) with catalytic nano-Zn (20 mol%) not only minimizes the waste footprint of the process but also improves the product yield significantly (3a, 42 → 67%). Replacement of aq. TPGS-750 M with aq. PTS significantly improved the product yield further (3a, 67 → 81%). Keeping these parameters intact, a detailed optimization study revealed that the use of 2 mol% of NiBr2, 4 mol% of dppf, and nano-Zn (20 mol%) at rt is optimal with an excellent yield of 3a (81%). The use of other ligands (phosphines, bisphosphines, bipyridines, and NHCs) was not found to be superior to that of dppf under the optimized conditions (Fig. 6). The key highlight of this protocol is its adaptability to scale up reactions. Reactions at the scale of 0.5 g, 1.0 g, 5.0 g, and 10.0 g were found to be promising, with yields ≥78% in each case. Another important aspect of this process concerns the amount of Ni that is carried through and into the product. ICP-MS analysis of 3c, prepared and isolated using standard chromatography, afforded product 3c with 0.0402 ppm nickel (ESI†). The recyclability of the spent micelles was studied. Aq. PTS micelles were found to be recyclable up to five times without compromising the 3a yield. The comparative analysis of the micellar composition (1stvs. 6th run) indicated that although there is a significant change in the average particle size and particle distribution index (PDI) of the micelles, these ranges are conducive to performing micellar catalysis, justifying the multiple recycling and reuse of aq. PTS (ESI†). Towards the end, the calculation of commonly exercised green metrics (atom economy, atom efficiency, RME, E-factor, and EcoScale) indicated promising values (well within the acceptable range) considering the making of fine chemicals.
 | ||
| Fig. 6 Demonstration of the Ni(0)-precatalyst for the ‘in-water’ allylic amination. | ||
Conclusions
The study provides for the first time an integrated sustainability assessment of the widely used Tsuji–Trost reaction modelling allylic amination reaction, taking into account the reactivity-coupled utility, environmental impact (including health hazards), and economic burden (and benefits) of various allylic precursors in alternative solvents (water) under alternative Earth-abundant metal catalysis. The finding suggested the remarkable adaptability of water as a reaction medium for the Tsuzi–Trost allylic aminations with a variety of allylic precursors and the superiority of nickel catalysis (over palladium) in a water medium, particularly using an allyl alcohol and non-reactive allyl amine (trans-N-allylation). We believe this study will work as a guide to stimulate further research and developments in academia and industry related to allylation chemistries in an environmentally friendly fashion.Conflicts of interest
There is no conflict of interest.Acknowledgements
The authors thank the Department of Pharmaceuticals (DoP), the Ministry of Chemical and Fertilizers, and NIPER-Ahmedabad for financial support. DK gratefully acknowledges the DST-SERB for the award of the Ramanujan Fellowship (File No. SB/S2/RJN-135/2017), Start-up Research Grant (File No. SRG/2020/000658/CS), and Core Research Grant CRG/2022/004057). DK also thanks CSIR-HRDG for CSIR Research Grants (File No. 02/0456/21/EMR-II).References
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, 2000 Search PubMed.
- F. Gallou, Chimia, 2020, 74, 538–548 CrossRef CAS PubMed.
- I. T. Horváth, Chem. Rev., 2018, 118, 369–371 CrossRef PubMed.
- S. Lems, H. J. Van der Kooi and J. d. Swaan Arons, Green Chem., 2002, 4, 308–313 RSC.
- V. G. Zuin, I. Eilks, M. Elschami and K. Kümmerer, Green Chem., 2021, 23, 1594–1608 RSC.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- F. Ferlin, G. Brufani, G. Rossini and L. Vaccaro, Green Chem., 2023, 25, 7916–7933 RSC.
- W. J. W. Watson, Green Chem., 2012, 14, 251–259 RSC.
- P. T. Anastas, Green Chem., 2003, 5, G29–G34 RSC.
- M. Poliakoff and P. Licence, Nature, 2007, 450, 810–812 CrossRef CAS PubMed.
- G. T. Whiteker, Org. Process Res. Dev., 2019, 23, 2109–2121 CrossRef CAS.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- D. E. Fitzpatrick, C. Battilocchio and S. V. Ley, ACS Cent. Sci., 2016, 2, 131–138 CrossRef CAS PubMed.
- C. M. Friend and B. Xu, Acc. Chem. Res., 2017, 50, 517–521 CrossRef CAS PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- L. E. Overman, Acc. Chem. Res., 1980, 13, 218–224 CrossRef CAS.
- E. M. Skoda, G. C. Davis and P. Wipf, Org. Process Res. Dev., 2012, 16, 26–34 CrossRef CAS PubMed.
- T. Itoh, K. Jitsukawa, K. Kaneda and S. Teranishi, J. Am. Chem. Soc., 1979, 101, 159–169 CrossRef CAS.
- I. Strambeanu and M. C. White, J. Am. Chem. Soc., 2013, 135, 12032–12037 CrossRef CAS PubMed.
- N. Astrain-Redin, C. Sanmartin, A. K. Sharma and D. Plano, J. Med. Chem., 2023, 66, 3703–3731 CrossRef CAS PubMed.
- J. Fong, M. Yuan, T. H. Jakobsen, K. T. Mortensen, M. M. S. Delos Santos, S. L. Chua, L. Yang, C. H. Tan, T. E. Nielsen and M. Givskov, J. Med. Chem., 2017, 60, 215–227 CrossRef CAS PubMed.
- H. Zhao, J. Brånalt, M. Perry and C. Tyrchan, J. Med. Chem., 2023, 66, 7730–7755 CrossRef CAS PubMed.
- I. V. Overmeire, S. A. Boldin, K. Venkataraman, R. Zisling, S. D. Jonghe, S. V. Calenbergh, D. D. Keukeleire, A. H. Futerman and P. Herdewijn, J. Med. Chem., 2000, 43, 4189–4199 CrossRef PubMed.
- H. Kuramochi, J. Med. Chem., 1996, 39, 2877–2886 CrossRef CAS PubMed.
- W. K. Anderson, R. H. Dewey and B. Mulumba, J. Med. Chem., 1979, 22, 1270–1272 CrossRef CAS PubMed.
- M. Rahman, A. Ali, E. Sjöholm, S. Soindinsalo, C.-E. Wilén, K. K. Bansal and J. M. Rosenholm, Pharmaceutics, 2022, 14, 798 CrossRef CAS PubMed.
- B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed.
- N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 7, 2821–2847 CrossRef CAS.
- R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed.
- C. J. Lu, D. K. Chen, H. Chen, H. Wang, H. Jin, X. Huang and J. Gao, Org. Biomol. Chem., 2017, 15, 5756–5763 RSC.
- M. Albert-Soriano, L. Hernández-Martínez and I. M. Pastor, ACS Sustainable Chem. Eng., 2018, 6, 14063–14070 CrossRef CAS.
- W. P. Teh, D. C. Obenschain, B. M. Black and F. E. Michael, J. Am. Chem. Soc., 2020, 142, 16716–16722 CrossRef CAS PubMed.
- H. Huang and J. Y. Kang, J. Org. Chem., 2017, 82, 6604–6614 CrossRef CAS PubMed.
- S.-C. Sha, J. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602–17609 CrossRef CAS PubMed.
- W.-B. Xu, M. Sun, M. Shu and C. Li, J. Am. Chem. Soc., 2021, 143, 8255–8260 CrossRef CAS PubMed.
- P. Fang, M. R. Chaulagain and Z. D. Aron, Org. Lett., 2012, 14, 2130–2133 CrossRef CAS PubMed.
- F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968–10969 CrossRef CAS PubMed.
- T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317–14328 CrossRef CAS PubMed.
- D. Banerjee, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 11556–11560 CrossRef CAS PubMed.
- D. Kumar, S. R. Vemula and G. R. Cook, Green Chem., 2015, 17, 4300–4306 RSC.
- J. Jing, X. Huo, J. Shen, J. Fu, Q. Meng and W. Zhang, Chem. Commun., 2017, 53, 5151–5154 RSC.
- G. Hirata, H. Satomura, H. Kumagae, A. Shimizu, G. Onodera and M. Kimura, Org. Lett., 2017, 19, 6148–6151 CrossRef CAS PubMed.
- C. Qiao, M. M. Belmonte, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2019, 12, 3152–3158 CrossRef PubMed.
- H. Kinoshita, H. Shinokubo and K. Oshima, Org. Lett., 2004, 6, 4085–4088 CrossRef CAS PubMed.
- T. Nishikata and B. H. Lipshutz, Org. Lett., 2009, 11, 2377–2379 CrossRef CAS PubMed.
- Y. Kita, H. Sakaguchi, Y. Hoshimoto, D. Nakauchi, Y. Nakahara, J.-F. Carpentier, S. Ogoshi and K. Mashima, Chem. – Eur. J., 2015, 21, 14571–14578 CrossRef CAS PubMed.
- M. S. Azizi, Y. Edder, A. Karim and M. Sauthier, Eur. J. Org. Chem., 2016, 2016, 3796–3803 CrossRef CAS.
- M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33–79 CrossRef CAS.
- N. R. Lee, F. A. Moghadam, F. C. Braga, D. J. Lippincott, B. Zhu, F. Gallou and B. H. Lipshutz, Org. Lett., 2020, 22, 4949–4954 CrossRef CAS PubMed.
- J. B. Sweeney, A. K. Ball, P. A. Lawrence, M. C. Sinclair and L. J. Smith, Angew. Chem., Int. Ed., 2018, 57, 10202–10206 CrossRef CAS PubMed.
- G. N. Vaidya, M. Nagpure and D. Kumar, ACS Sustainable Chem. Eng., 2021, 9, 1846–1855 CrossRef CAS.
- G. N. Vaidya, R. H. Choudhary, M. Nagpure, S. K. Lokhande, P. Rana and D. Kumar, Green Chem., 2022, 24, 3977–3984 RSC.
- J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387–4388 CrossRef.
- B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J. E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed.
- A. Stütz, Angew. Chem., Int. Ed., 1987, 26, 320–328 CrossRef.
- D. H. Halat, S. Younes, N. Mourad and M. Rahal, Membrane, 2022, 12, 1171 CrossRef PubMed.
- W. G. Xiao, B. Xuan, L. J. Xiao and Q. L. Zhou, Chem. Sci., 2023, 14, 8644–8650 RSC.
- J. Al Ameri, A. Alsuraifi, A. Curtis and C. Hoskins, J. Pharm. Sci., 2020, 109, 3125–3133 CrossRef CAS PubMed.
- G. N. Vaidya, M. Nagpure and D. Kumar, ACS Sustainable Chem. Eng., 2021, 9, 1846–1855 CrossRef CAS.
- G. N. Vaidya, R. H. Choudhary, M. Nagpure, S. K. Lokhande, P. Rana and D. Kumar, Green Chem., 2022, 24, 3977–3984 RSC.
- G. N. Vaidya, S. K. Lokhande, S. D. Shinde, D. P. Satpute, G. Narang and D. Kumar, Green Chem., 2022, 24, 4921–4927 RSC.
- G. N. Vaidya, S. Fiske, H. Verma, S. K. Lokhande and D. Kumar, Green Chem., 2019, 21, 1448–1454 RSC.
- S. Kumar, D. P. Satpute, G. N. Vaidya, M. Nagpure, S. K. Lokhande, D. Meena and D. Kumar, Tetrahedron Lett., 2020, 61, 152017 CrossRef CAS.
- S. K. Lokhande, G. N. Vaidya, D. P. Satpute, A. Venkatesh, S. Kumar and D. Kumar, Adv. Synth. Catal., 2020, 362, 2857–2863 CrossRef CAS.
- D. P. Satpute, G. N. Vaidya, S. K. Lokhande, S. D. Shinde, S. M. Bhujbal, D. R. Chatterjee, P. Rana, A. Venkatesh, M. Nagpure and D. Kumar, Green Chem., 2021, 23, 6273–6300 RSC.
- Evaluation of organic solvents (toluene, 1,4-dioxane, THF, DMF, acetonitrile) concluded 1,4-dioxane is optimal.
- B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed.
- Y.-N. Wang, B.-C. Wang, M.-M. Zhang, X.-W. Gao, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2017, 19, 4094–4097 CrossRef CAS PubMed.
- M. Calvin, J. Hermans and H. A. Scheraga, J. Am. Chem. Soc., 1959, 81, 5048–5050 CrossRef CAS.
- D. N. Kommi, P. S. Jadhavar, D. Kumar and A. K. Chakraborti, Green Chem., 2013, 15, 798 RSC.
- G. Dahlgren and F. A. Long, J. Am. Chem. Soc., 1960, 82, 1303–1308 CrossRef CAS.
- A. Grimison, J. Phys. Chem., 1963, 67, 962–964 CrossRef CAS.
- P. C. St John, Y. Guan, Y. Kim, S. Kim and R. S. Paton, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed.
- A. B. Dürr, G. Yin, I. Kalvet, F. Napoly and F. Schoenebeck, Chem. Sci., 2016, 7, 1076–1081 RSC.
- M. E. Greaves, T. O. Ronson, F. Maseras and D. J. Nelson, Organometallics, 2021, 40, 1997–2007 CrossRef CAS PubMed.
- J. Dewulf, H. Van Langenhove, J. Mulder, M. M. D. Van Den Berg, H. J. Van Der Kooi and J. De Swaan Arons, Green Chem., 2000, 2, 108–114 RSC.
- T. Collins, Green Chem., 2003, 5, G51–G52 RSC.
- I. Yasui, Green Chem., 2003, 5, G70–G73 RSC.
- S. M. Mercer, J. Andraos and P. G. Jessop, J. Chem. Educ., 2012, 89, 215–220 CrossRef CAS.
- S. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7245–7256 RSC.
- Agreement of the Member State Committee on the Identification of N,N-Dimethylacetamide (DMAC) as a Substance of Very High Concern − Adopted on 24 November 2011 https://echa.europa.eu/documents/10162/8b76202f-f4e0-5909-ee10-0ceb4861f3f2.
- Committee for Risk Assessment RAC Opinion on N,N-Dimethylacetamide (DMAC), European Chemicals Agency, 12 September 2014 https://www.echa.europa.eu/documents/10162/a435d3fca05f-b558-3f51-9aff166f2de0.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04216b |
| ‡ Equal first authorship. |
| This journal is © The Royal Society of Chemistry 2024 |