
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A visible-light-promoted metal-free approach for N–H insertions by using donor/donor diazo precursors†
Yu
Zhang‡
 *a,
Qiannan
Li‡
a,
Ping
Wang‡
a,
Jinxin
Wang
b,
Jingchuan
Lin
a,
Dingding
Xia
a,
Er-Jun
Hao
c,
Xin
Luan
a,
Shoubhik
Das
*d and
Wei-Dong
Zhang
*abe
*a,
Qiannan
Li‡
a,
Ping
Wang‡
a,
Jinxin
Wang
b,
Jingchuan
Lin
a,
Dingding
Xia
a,
Er-Jun
Hao
c,
Xin
Luan
a,
Shoubhik
Das
*d and
Wei-Dong
Zhang
*abe
aShanghai Frontiers Science Center for Chinese Medicine Chemical Biology, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, No. 1200, Cailun Road, Shanghai 201203, China. E-mail: yzhang@shutcm.edu.cn; wdzhangy@hotmail.com
bSchool of Pharmacy, Second Military Medical University, Shanghai 200433, China
cDepartment School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
dDepartment of Chemistry, University of Bayreuth, Bayreuth, Germany. E-mail: Shoubhik.Das@uni-bayreuth.de
eInstitute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100193, China
First published on 28th February 2024
Abstract
The metal-catalyzed carbenoid-based N–H insertions of α-stabilized diazo compounds and relative precursors serve as robust strategies for the formation of C–N bonds. However, carbenoid-based N–H insertions with aminopyridines are usually challenging due to the undesired coordination effect between the pyridine and the metal. Moreover, the coupling of donor/donor diazo compounds with amines remains challenging due to the instability and unavailability of donor/donor diazo compounds. Considering all these existing challenges, herein, a metal-free strategy is reported to achieve N–H insertions via coupling donor/donor diazo precursors (N-tosylhydrazones) with a plethora of amines including aminopyridines, anilines, aliphatic amines, and other nucleophiles such as imidazoles, pyrroles, and indoles. Expediently, β-amino esters are also afforded by using this mild strategy. The utility of this protocol is further demonstrated by the gram-scale synthesis and modifications of pharmaceuticals. Therefore, it is very clear that this metal-free approach uniquely tolerates a wide range of nucleophiles and opens a straightforward synthetic route to synthesize diverse value-added amines.
Introduction
Nitrogen-containing molecules, such as amino acids, alkaloids, and amines, possess various biological and pharmacological properties and more than 85% of FDA-approved pharmaceuticals contain nitrogen atoms (Fig. 1A).1 Inspired by this, chemists strive to develop facile strategies to introduce diversity for the formation of C–N bonds.2–4 In this context, carbenoid-based N–H insertions via the coupling of diazo compounds with amines are highly noteworthy (Fig. 1B).5 In this strategy, the metal carbenoid is initially formed from a diazo compound which subsequently reacts with molecules containing N–H bonds to achieve N–H insertions.4g,6,7 For example, Zhou and co-workers reported enantioselective N–H insertions between α-diazoacetate and aryl amines by the cooperative action of copper complexes and a chiral spiro bisoxazoline ligand.8 Following this work, other catalytic strategies were further developed by the same group and others.9 To realize the direct activation of diazo esters under metal-free conditions, Davies et al. disclosed that aryldiazoacetates could undergo direct photolysis under the irradiation of blue light to form donor/acceptor carbene intermediates and further react with amines to achieve C–N bond formation.10 Later, to achieve the asymmetric transformation of C–N bond formation, the Zhou group disclosed that high-pKa Brønsted acid catalysts enabled N–H insertions to prepare the corresponding chiral α-amino acid derivatives under the irradiation of visible light.6a Despite these advances, donor and donor/donor diazo compounds without electron-withdrawing groups have rarely been applied to N–H insertions due to their instability and unavailability, which certainly results in poor structural diversity of carbenoid based N–H insertions.11 | ||
| Fig. 1 Overview of N–H insertions and our work. | ||
To explore the application of donor/donor diazo compounds (precursors) in N–H insertions, Che and co-workers developed Fe-porphyrin complexes and applied them in diaryl carbene transfer reactions. However, the formation of the corresponding amines required a large excess (10 equiv.) of aminating reagents.12a Later, the Koenig group successfully achieved the N–H insertion of diaryl diazo compounds in the presence of a silver catalyst, but aryl alkyl diazo compounds were still incompatible and only anilines could be used as aminating reagents.12b Recently, Bi reported impressive progress so that diaryl N-triftosylhydrazones could also undergo N–H insertions to provide the desired diaryl methylamines. However, the presence of a precious silver catalyst and high temperature was essential to the reaction. Additionally, this strategy was also infeasible for utilizing the aryl/alkyl diazo precursors.12c Therefore, there is an ongoing need to develop approaches for achieving N–H insertions with a broad substrate scope, including previously challenging aryl/alkyl or alkyl/alkyl diazo compounds or precursors. In addition, developing a metal-free strategy to realize N–H insertions with structural diversity is also certainly appealing since this approach will provide an alternative reactivity principle that allows a conceptual advancement in this domain.
Besides the challenging applications of aryl/alkyl or alkyl/alkyl diazo compounds or precursors in N–H insertions, aminopyridines as aminating sources are also often incompatible in N–H insertions due to the undesired coordination between pyridines and the respective transition metals.12d Davies et al. disclosed the metal-free N–H insertions of donor/acceptor diazo compounds with a few aminopyridines under thermal conditions. However, the scope of aminopyridines was still highly limited (Fig. 1C).13a Inspired by this elegant effort from the Davies group, we became interested in the achievement of N–H insertions from challenging aryl/alkyl diazo precursors with aminopyridines and other heteroaryl amines under metal-free conditions, which are still completely underexplored.12d,13b
In the past decade, light-mediated organic transformations via photoredox catalysis,14 energy transfer,15 the formation of an EDA complex,16 or direct photolysis have emerged as powerful strategies in synthetic chemistry.17a Based on these advances and our continuous efforts in the activation and application of N-tosylhydrazones,17b,c we argued that donor/donor diazo compounds could be formed from N-tosylhydrazones via the formation of non-covalent interactions between the N-tosylhydrazone anion and the organic base. Afterwards, the corresponding N–H insertions were introduced by coupling donor/donor diazo compounds with amines. In particular, this metal-free approach should bypass current limitations to utilize donor/donor diazo compounds and diversify challenging aminating reagents (Fig. 1D).
Results and discussion
At first, N-tosylhydrazone (1a) was synthesized simply from 3-acetylbenzo[b]furan and p-toluenesulfonyl hydrazide as the model donor/donor diazo precursor. After careful and systematic optimization, we finalized the best reaction conditions by using DBN as the base and DCM as the solvent to furnish the desirable product in good yield (Table 1, entry 1). Other organic bases such as DBU and TBD also facilitated N–H insertions, but the yields were slightly low (Table 1, entries 2 and 3), and Cs2CO3 exhibited limited reactivity (Table 1, entry 4). Next, 390 nm and 456 nm Kessil lamps were used to verify the best light source. However, both of them showed less reactivity compared to the 427 nm Kessil lamp (Table 1, entries 5 and 6). Later, lowering the quantity of 1b led to lower reactivity than the standard conditions (Table 1, entry 7). It was also found that increasing the amount of base could not improve the yield (Table 1, entry 8). In addition, control experiments indicated that the presence of both base and light was critical for this reaction (Table 1, entries 9 and 10).|
|
||
|---|---|---|
| Entry | Changes from standard conditions | Yieldb [%] |
| a General reaction conditions: 1a (0.1 M in DCM), 1b (5.0 equiv.), DBN (1.5 equiv.), 427 nm 40 W Kessil lamp, room temperature, 6 h. n.d. = not detected. b Yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 77 |
| 2 | DBU instead of DBN | 67 |
| 3 | TBD instead of DBN | 60 |
| 4 | Cs2CO3 instead of DBN | 20 |
| 5 | 390 nm instead of 427 nm | 28 |
| 6 | 456 nm instead of 427 nm | 46 |
| 7 | 3.0 equiv. of 1b were used | 60 |
| 8 | 3.0 equiv. of DBN were used | 58 |
| 9 | In the dark | n.d. |
| 10 | Without a base | n.d. |
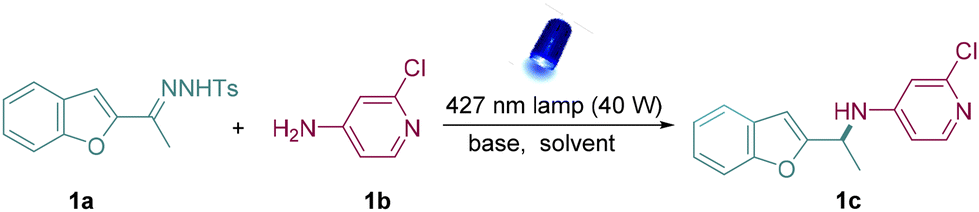
Having established the optimal conditions for the metal-free N–H insertion, the generality of this protocol was further evaluated. First, diverse N-tosylhydrazones obtained from heteroaryl alkyl ketones successfully served as the donor/donor diazo precursors and afforded the corresponding amines in medium to good isolated yields (Table 2). It was found that heteroaromatic N-tosylhydrazones which contain coordinating groups such as benzofuryl (1c, 2c), benzothiophene (3c), indole (4c), and thiophenes (5c, 6c), delivered the corresponding aminated products successfully. Subsequently, N-tosylhydrazones containing other heterocycles including 6,7-dihydro-4-benzothiophen (7a), imidazolpyridyl (8a), and Boc-protected 1,5,6,7-tetrahydro-4H-indol-4-one (9a) also underwent the metal-free N–H insertion successfully, indicating the strong substrate tolerance of our strategy.

Besides the heteroaromatic N-tosylhydrazones, general aromatic N-tosylhydrazones (10a) and other tosylhydrazones derived from monosubstituted acetophenone at the para- or meta-position with methyl-, imidazolyl-, phenoxy-, and cyclohexyl-groups also proved to be feasible in our system and desirable amines were obtained in good to excellent yields (11a–13a). Moreover, the aryl motifs of N-tosylhydrazones bearing two different substituents at the ortho-, meta-, or para-positions also successfully coupled with 1b to afford the corresponding secondary amines (14c, 15c) in moderate to good yields. Naphthyl-substituted N-tosylhydrazone (16a) also proved to be a suitable substrate. Moreover, different alkyl groups in N-tosylhydrazones (17c–20c) proved to be feasible, and these products were obtained in moderate to good yields. The amine product was also obtained from diaryl N-tosylhydrazone (21c) in good isolated yield. Furthermore, N-tosylhydrazones obtained from benzaldehydes bearing electron-rich substituents (22c, 23c), including methoxy and tert-butyl groups, successfully yielded the corresponding amines. N-Tosylhydrazone derived from an aldehyde bearing a chloride group (24c) also reacted smoothly with aminopyridine to generate the corresponding amine in good yield, indicating the stage for subsequent late-stage diversifications.
A range of different amines under the standard conditions was also examined. The N-tosylhydrazone derived from benzofuran was selected as the model substrate since the benzofuran heterocyclic scaffold is frequently found in many pharmaceuticals and natural products.18a At first, anilines with various substituents at the para-position (25c–28c) reacted with N-tosylhydrazone to provide the corresponding aminated products in moderate to good yields. Phenyl-substituted aniline (29c) at the ortho-position was also accommodated in our system and the aminated product was obtained in good yield. Disubstituted anilines were well-tolerated to give aminated products in moderate to good yields (30c–32c). Furthermore, aminopyridines with substitutions in different positions (33c–36c) also successfully underwent N–H insertions to furnish products in moderate to good yields. However, pyrimidine-derived amines (37c) showcased limited reactivity. Next, the range was extended to different secondary amines such as N-methylaniline, 3,4-dihydro-2H-1,4-benzoxazine and 1,2,3,4-tetrahydroquinoline. And desirable tertiary amines were afforded in moderate to good reactivities (38c–41c). Notably, nucleophilic alkylamine reagents (42c–44c) were shown to be tolerable under slightly modified reaction conditions. In addition, diverse nucleophilic reagents such as imidazole, benzimidazole, pyrrole, indole, triazole, tetrahydropyrrole, piperidine, morpholine, and N-Boc-piperazine were also well-tolerated in our system to deliver the aminated products (45c–53c). Thus, this simple and metal-free approach exhibited broad applicability, functional group compatibility, and a wide scope.
β-Amino acids/esters are structural motifs that are frequently found in biologically active molecules, drugs and natural products. Notably, peptides composed of β-amino acids result in more stable helical secondary structures than those composed of original α-amino acids.18b,c,d Therefore, they are broadly utilized in constructing bioactive peptides and peptidomimetics, thanks to their increased metabolic stability.19 With this benign approach, the synthesis of β-amino esters directly via the coupling of N-tosylhydrazones derived from ethyl 3-oxo-3-phenylpropanoate (54b) and anilines has been reported. In this case, β-amino esters were obtained under the standard conditions in medium to good isolated yields (Table 3). First, commercially diverse anilines with different substituents (54c–63c) were engaged in this reaction and the desired β-amino-esters were obtained in medium to good yields. Subsequently, various aminopyridines (64c–68c) bearing substituents in different positions were also applicable and exhibited medium reactivities. In addition to primary amines, the scope was further expanded to secondary amines such as N-allylaniline and indoline which have proven to be suitable aminating reagents (69c–72c). Next, the tolerability of substituted N-tosylhydrazones was also examined and they were found to exhibit good compatibility (73c–78c).
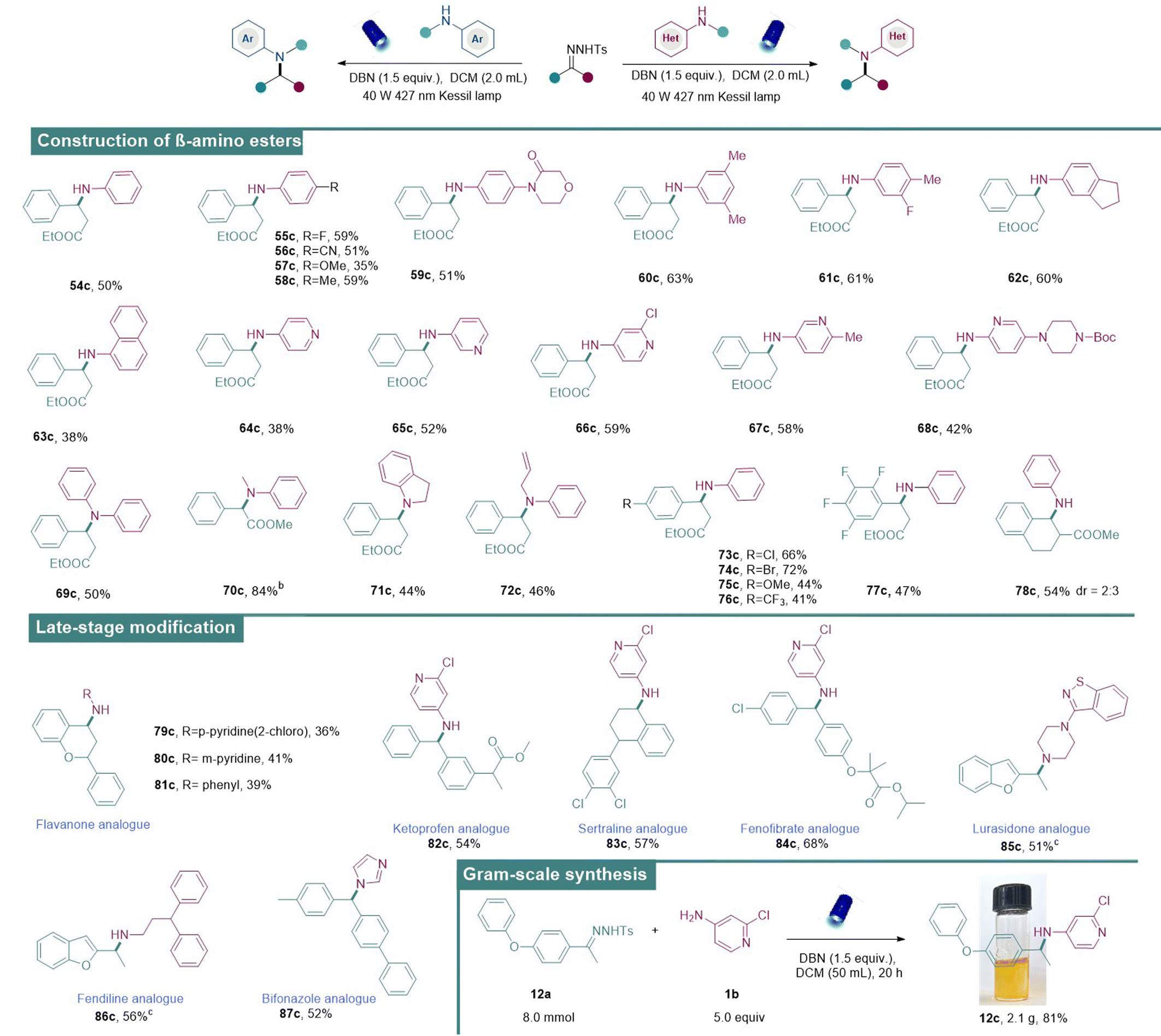
We then became interested in diversifying pharmaceutically important molecules using our approach. For instance, flavanone, a natural product having potential pharmacological activities, underwent N–H insertions successfully with different aminating reagents (79c–81c). Moreover, N–H insertions of N-tosylhydrazones derived from ketoprofen (82c, anti-inflammatory drug), sertraline (83c, anti-depressive drug), fenofibrate (84c, lipid-regulating drug), lurasidone (85c, anti-psychotic drug), fendiline (86c, anti-anginal drug), and bifonazole (87c, anti-fungal drugs) provided aminated drug analogues in medium to good yields. The practical utility of this approach was further demonstrated by carrying out a gram-scale reaction of 12a under the optimal conditions to provide 12c (2.1 g) in 81% yield.
Next, mechanistic investigations were carried out to gain insights into the mechanism of this N–H insertion reaction. First, the UV/Vis absorption spectra showed a clear red (bathochromic) shift between DBN and the N-tosylhydrazone anion, with visible-light absorption tailing at 400–460 nm (Fig. 2A, details in Fig. S2†), which suggested the existence of non-covalent interactions in our system. Afterwards, Job's plot of the complex was also obtained and it was noticed that the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the N-tosylhydrazone anion and DBN in DCM showed maximal absorption, which further indicated the formation of non-covalent interactions (Fig. 2B).17b Meanwhile, NMR titration experiments were performed to examine the interactions of the N-tosylhydrazone anion 10a′ and DBN. An upfield shift of the N-tosylhydrazone anion 10a′ was observed upon increasing the amount of DBN, further indicating that the complexation occurred between the N-tosylhydrazone anion 10a′ and DBN (Fig. 2C).20 Moreover, on–off experiments proved that light was critical to promoting the reaction (Fig. 2D). A deuterium labelling experiment was also performed, indicating a proton transfer from the aminating reagent to the benzylic position (Fig. 2E).
:1 ratio of the N-tosylhydrazone anion and DBN in DCM showed maximal absorption, which further indicated the formation of non-covalent interactions (Fig. 2B).17b Meanwhile, NMR titration experiments were performed to examine the interactions of the N-tosylhydrazone anion 10a′ and DBN. An upfield shift of the N-tosylhydrazone anion 10a′ was observed upon increasing the amount of DBN, further indicating that the complexation occurred between the N-tosylhydrazone anion 10a′ and DBN (Fig. 2C).20 Moreover, on–off experiments proved that light was critical to promoting the reaction (Fig. 2D). A deuterium labelling experiment was also performed, indicating a proton transfer from the aminating reagent to the benzylic position (Fig. 2E).
 | ||
| Fig. 2 Mechanistic investigations of coupling of N-tosylhydrazones with amines. | ||
In addition, a set of control experiments were performed to elucidate possible intermediates (Fig. 2F). First, N-tosylhydrazone 20a reacted under standard conditions without the addition of an aminating reagent and the formation of diazo compounds indicated that the metal-free strategy enabled the formation of donor/donor diazo compounds rather than the carbene species since the carbene coupling product 20aa was not observed.17c Furthermore, a competition reaction was performed to understand if the reaction proceeded through the free carbenoid N–H insertion or the nucleophilic addition of diazo intermediates to induce C–N bond formation. Only a trace amount of cyclopropane was detected, whereas the aminating product was formed with good reactivity. Both observations indicated that N–H insertions were obtained via the nucleophilic addition of diazo intermediates as the major pathway rather than carbenoid N–H insertions.21
Based on the above-mentioned mechanistic experiments and our previous report,17b a plausible mechanism was proposed (Fig. 2G). First, N-tosylhydrazone 1a was deprotonated by DBN, resulting in the generation of N-tosylhydrazone anion 1a′. Afterwards, it was suggested that through the non-covalent interaction, 1f was formed from 1a′ and DBN. Then 1f was subjected to blue light irradiation (427 nm) to generate diazo intermediate 1gvia the release of the sulfonyl anion and DBN. The electrophilic diazo intermediate was further attacked by the aminating reagent to furnish intermediate 1h. Afterwards, the ylide species 1m was formed with the concomitant extrusion of nitrogen gas. Finally, the final product 1c was formed via the 1,2-H shift process.
Conclusions
In summary, a metal-free strategy enabling N–H insertions has been described to afford diverse amines and β-amino esters through the coupling between in situ generated donor/donor diazo intermediates and amines including aminopyridines, anilines, aliphatic amines, and other nucleophiles, such as imidazoles, pyrroles, and indoles. The utility of this protocol is further demonstrated by the synthesis of drug analogues and gram-scale synthesis. This efficient strategy is believed to benefit C–N bond formation and the synthesis of amines and β-amino esters. In addition, considering the further improvement of the sustainability of our system, the green solvent (2-Me-THF) was applied as the reaction medium, and the corresponding amination product was obtained with a yield of 63% after slight modifications (see details in the ESI†). Further studies with other green solvents and the determination of bioactivities are underway in our lab.Conflicts of interest
A Chinese patent on this work was applied for with the number (202310364042.2) on 7 April 2023. The remaining authors declare no competing interests.Acknowledgements
This work was supported by the Chenguang Program of the Shanghai Education Development Foundation and the Shanghai Municipal Education Commission (22CGA51, Y. Z.), the National Natural Science Foundation of China (82141203 and 82003624, W. Z.), the National Key Research and Development Program of China (2022YFC3502000, W. Z.), the Shanghai Municipal Science and Technology Major Project (ZD2021CY001, W. Z.), the Three-year Action Plan for the Shanghai TCM Development and Inheritance Program [ZY(2021-2023)-0401, W. Z. ], and the Innovation Team and Talents Cultivation Program of the National Administration of Traditional Chinese Medicine (ZYYCXTDD-202004, W. Z.). We are also grateful to Dr Lu Lu for carrying out the NMR experiments.References
-
(a) M. Beller, J. Seayad, A. Tillack and H. J. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed
; (b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. B. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed
-
(a) J. Bariwal and E. V. D. Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC
-
(a) Y. Z. Qin, Q. L. Zhu, R. Sun, J. M. Ganley, R. R. Knowles and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 10232–10242 CrossRef CAS PubMed
-
(a) C.-M. Ho, J.-L. Zhang, C.-Y. Zhou, O.-Y. Chan, J. J. Yan, F. Y. Zhang, J. S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS PubMed
-
(a) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed
-
(a) M. L. Li, J. H. Yu, Y. H. Li, S. F. Zhu and Q. L. Zhou, Science, 2019, 366, 990–994 CrossRef CAS PubMed
-
(a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed
- B. Liu, S. F. Zhu, W. Zhang, C. Chen and Q. L. Zhou, J. Am. Chem. Soc., 2007, 129, 5834–5835 CrossRef CAS PubMed
-
(a) Z. R. Hou, J. Wang, P. He, J. Wang, B. Qin, X. H. Liu, L. L. Lin and X. M. Feng, Angew. Chem., 2010, 122, 4873–4876 CrossRef
- I. D. Jurberg and H. M. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC
-
(a) D. Zhu, L. F. Chen, H. L. Fan, Q. L. Yao and S. F. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC
-
(a) H. X. Wang, Q. Wan, K. H. Low, C. Y. Zhou, J. S. Huang, J. L. Zhang and C. M. Che, Chem. Sci., 2020, 11, 2243–2259 RSC
-
(a) S. R. Hansen, J. E. Spangler, J. H. Hansen and H. M. L. Davies, Org. Lett., 2012, 14, 4626–4629 CrossRef CAS PubMed
-
(a) S. Cuadros, G. Goti, G. Barison, A. Raulli, T. Bortolato, G. Pelosi, P. Costa and L. Dell'Amico, Angew. Chem., 2023, 135, e202303585 CrossRef
-
(a) K. Gadde, P. Mampuys, A. Guidetti, H. V. Ching, W. A. Herrebout, S. Van Doorslaer, K. A. Tehrani and B. U. Maes, ACS Catal., 2020, 10, 8765–8779 CrossRef CAS
-
(a) J. Mateos, F. Rigodanza, P. Costa, M. Natali, A. Vega-Peñaloza, E. Fresch, E. Collini, M. Bonchio, A. Sartorel and L. Dell'Amico, Nat. Synth., 2023, 2, 26–36 CrossRef PubMed
-
(a) A. G. Herraiz and M. G. Suero, Chem. Sci., 2019, 10, 9374–9379 RSC
-
(a) G. Khodarahmi, P. Asadi, F. Hassanzadeh and E. Khodarahmi, J. Res. Med. Sci., 2015, 20, 1094–1104 CrossRef CAS PubMed
-
(a) G. Cardillo and C. Tomasini, Chem. Soc. Rev., 1996, 25, 117–128 RSC
-
(a) K. Sun, A. Z. Shi, Y. Liu, X. L. Chen, P. J. Xiang, X. T. Wang, L. B. Qu and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC
-
(a) C. Empel, C. Pei, F. F. He, S. Jana and R. M. Koenigs, Chem. – Eur. J., 2022, 28, e202104397 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and details, mechanistic and optimization results and NMR spectra. See DOI: https://doi.org/10.1039/d3gc04431a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |