
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparative regio- and stereoselective α-hydroxylation of medium chain mono- and dicarboxylic fatty acids†
Klara
Bangert
 a,
Alexander
Swoboda
b,
Stephan
Vrabl
a,
Haris
Rudalija
c,
Mattia
Lazzarotto
c,
Stefan
Payer
c,
Anton
Glieder
d,
Christian A. M. R.
van Slagmaat
e,
Stefaan M. A.
De Wildeman
e and
Wolfgang
Kroutil
*afg
a,
Alexander
Swoboda
b,
Stephan
Vrabl
a,
Haris
Rudalija
c,
Mattia
Lazzarotto
c,
Stefan
Payer
c,
Anton
Glieder
d,
Christian A. M. R.
van Slagmaat
e,
Stefaan M. A.
De Wildeman
e and
Wolfgang
Kroutil
*afg
aDepartment of Chemistry, University of Graz, Heinrichstraße 28, 8010 Graz, Austria. E-mail: Wolfgang.Kroutil@uni-graz.at
bAustrian Centre of Industrial Biotechnology, c/o University of Graz, Heinrichstraße 28, 8010 Graz, Austria
cEnzyan Biocatalysis GmbH, Stiftingtalstrasse 14, 8010 Graz, Austria
dBisy GmbH, Wünschendorf 292, 8200 Hofstätten an der Raab, Austria
eB4Plastics BV, IQ-Parklaan2 A, 3650 Dilsen-Stokkem, Belgium
fBioTechMed Graz, 8010 Graz, Austria
gField of Excellence BioHealth, University of Graz, 8010 Graz, Austria
First published on 5th January 2024
Abstract
Regio- and stereoselective functionalisation reactions like C–H oxidation are of high importance for instance for the valorization of renewables like fatty acids by α-hydroxylation. Here, peroxygenases were envisioned to be of high interest as they require common hydrogen peroxide as the only oxidant generating water as the sole side product. As the unspecific peroxygenase from Hypoxylon sp. (HspUPO) turned out to be not selective for α-hydroxylation, various bacterial peroxygenases from the CYP152 family were tested for the stereoselective α-hydroxylation of medium chain fatty acids (C6, C8, C10). The enzyme P450Exα proved to be highly suitable for the conversion of caproic acid (C6) (95% conv.) and showed high regioselectivity to give the α-hydroxylated product (α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-selectivity = 14:1). Additionally, P450Exα successfully converted the dicarboxylic acids azelaic acid (C9) and sebacic acid (C10) exclusively to the corresponding α-monohydroxylated product (up to >99% conversion). P450Spα hydroxylated the fatty acids C6, C8 and C10 preferentially in α-position giving the optically pure or optically enriched (S)-enantiomer [ee 95–>99% (S)] with up to 99% conversion. Both enzymes were used for preparative synthesis of α-hydroxylated fatty acids at up to 150 mM substrate concentration on 50 mL scale giving for instance 2-hydroxyoctanoic acid with 87% yield on gram scale (1260 mg) reaching TONs up to 42000.
:β-selectivity = 14:1). Additionally, P450Exα successfully converted the dicarboxylic acids azelaic acid (C9) and sebacic acid (C10) exclusively to the corresponding α-monohydroxylated product (up to >99% conversion). P450Spα hydroxylated the fatty acids C6, C8 and C10 preferentially in α-position giving the optically pure or optically enriched (S)-enantiomer [ee 95–>99% (S)] with up to 99% conversion. Both enzymes were used for preparative synthesis of α-hydroxylated fatty acids at up to 150 mM substrate concentration on 50 mL scale giving for instance 2-hydroxyoctanoic acid with 87% yield on gram scale (1260 mg) reaching TONs up to 42000.
Introduction
Due to the need to change our fossil-based economy to a renewable based one,1–6 methods to transform renewables with high selectivity are in demand. Thereby fatty acids can be considered as a broadly available raw material and selected methods for their transformation have been reported.7–9 Although functionalised derivatives like α-hydroxylated fatty acids serve as intermediates for the synthesis of α-keto acids,10 α-amino acids,11 as building blocks for bio-polyester12,13 and to produce pharmaceuticals,14 as anti-microbial agents and natural products,15 their preparation is still challenging when starting from the non-functionalised fatty acid.Using traditional chemical routes, functionalising saturated fatty acids bearing only a carboxylic acid group and many C–H bonds requires rather harsh conditions and is only moderately regioselective and not stereoselective, thus leading to the racemic product.16,17 Although enzymes/biocatalysts are known to work under mild conditions in buffer and display high regio- and stereoselectivity,18–21 C–H functionalisation is still a challenge. Typical enzymes for hydroxylation are P450 monooxygenases mediating e.g. the oxidation of aliphatic C–H bonds, aryl C–H bonds and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds with a high degree of chemo-, regio- and stereoselectivity.22–25 P450 peroxygenases and unspecific peroxygenases (UPOs) allow comparable oxy-functionalisation to P450 monooxygenases but are independent of reduced nicotinamide cofactors and electron transport chains, and rely on hydrogen peroxide as the only oxidant and source of oxygen.26–31 Hydrogen peroxide can be considered as a green reagent,32 as it is a stable oxidant and gives water as the only side product. The challenges with H2O2-driven biocatalysis are the inactivation of an enzyme at elevated peroxide concentration as amino acids may be oxidized,33,34 or the heme group itself gets degraded.35–37 Another challenge is the over-oxidation of desired products, like recently observed for the α-hydroxylation of fatty acids leading to α-keto acids,38 which are sensitive to decarboxylation in the presence of H2O2. Consequently, various approaches have been developed to generate the H2O2in situ to keep the H2O2 concentration low.39–45
C double bonds with a high degree of chemo-, regio- and stereoselectivity.22–25 P450 peroxygenases and unspecific peroxygenases (UPOs) allow comparable oxy-functionalisation to P450 monooxygenases but are independent of reduced nicotinamide cofactors and electron transport chains, and rely on hydrogen peroxide as the only oxidant and source of oxygen.26–31 Hydrogen peroxide can be considered as a green reagent,32 as it is a stable oxidant and gives water as the only side product. The challenges with H2O2-driven biocatalysis are the inactivation of an enzyme at elevated peroxide concentration as amino acids may be oxidized,33,34 or the heme group itself gets degraded.35–37 Another challenge is the over-oxidation of desired products, like recently observed for the α-hydroxylation of fatty acids leading to α-keto acids,38 which are sensitive to decarboxylation in the presence of H2O2. Consequently, various approaches have been developed to generate the H2O2in situ to keep the H2O2 concentration low.39–45
Peroxygenases were mostly used for the hydroxylation of long chain fatty acids, and only few reports investigated medium chain fatty acids.10,11,35,38,46,47 Nevertheless, the global supply of medium chain fatty acids like caproic acid, caprylic acid and capric acid relies on oils like coconut, corn and palm oil48 and the global market of these medium chain fatty acids is expected to reach USD 2.03 Billion by 2028.49 Consequently, our focus here is the regioselective hydroxylation of medium chain fatty acids (C6:0, C8:0 and C10:0) to identify a suitable enzyme and a reaction protocol which can then be used on preparative scale and also for dicarboxylic acids.
Results and discussion
Medium chain fatty acid hydroxylation by an UPO (HspUPO)
In a first approach, the unspecific peroxygenase from Hypoxylon sp. (HspUPO)50 was investigated for the possible regioselective hydroxylation of the fatty acids caproic acid (C6:0, 1a), caprylic acid (C8:0, 1b) and capric acid (C10:0, 1c). The biotransformations were performed in phosphate buffer using acetonitrile (MeCN) as co-solvent and H2O2 as oxidant. The enzyme had been produced by excretion using Pichia pastoris and was therefore a catalase free preparation. In contrast to methods for in situ generation of H2O2 and also in contrast to batchwise addition of H2O2,11,46 here the H2O2 was supplied by continuous feeding of an H2O2-solution to supplement the reaction overall with (at least) stoichiometric amounts of oxidant. Using this approach, we expected to avoid elevated concentration of H2O2. Furthermore, the local H2O2 concentration at the inlet was expected to be kept to a minimum by vigorous shaking or stirring.Under the conditions employed, HspUPO converted caproic acid 1a with 39% conversion (Scheme 1). Thereby the (ω-1)-hydroxylated (ω-1)-2a and the β-hydroxylated product β-2a were detected in a ratio of 1.8:1. Alpha-hydroxylation was not observed at all. The same is true for octanoic acid 1b, for which hydroxylation was primarily observed for the (ω-1)-position but also detected in the β-, γ-, and δ-positions. Also for capric acid 1c the main mono-hydroxylation product had the alcohol-moiety in (ω-1)-position, whereby hydroxylation was also observed in β- and α-position in a ratio of (ω-1):β:α = 1.7:1.5:1. The incomplete mass balance detected for all three fatty acids by GC also indicated, that most likely also decarboxylation occurred, which fits to the observation, that the enzyme performed also β-hydroxylation.51 The observed preferred (ω-1)-hydroxylation for the UPO is also in line with a very recent report describing the improvement of the (ω-1)-preference by enzyme engineering of an UPO from another fungus (AaeUPO).27
 | ||
| Scheme 1 Oxidative biotransformation of medium chain saturated fatty acids with HspUPO. Reactions were performed in 1.5 mL glass crimp vials. Reactions were analysed after derivatisation (BSTFA with 1% TMCS). Conversions [%] refer to the consumption of substrate and were determined by GC-MS using internal standard (ISD) in comparison to the sample at 0 hours. Ratios for mono-hydroxylated products were calculated based on GC-area (Fig. S9–S11†). Red circles illustrate that no α-hydroxylation was observed. | ||
Consequently, as the unspecific peroxygenase HspUPO was found to be indeed unspecific with respect to the position of hydroxylation of the fatty acids investigated and did not give α-hydroxylation in significant amounts, we turned our attention to peroxygenases from the CYP152 family, expecting them to show higher specificity for α-hydroxylation. Furthermore, we also considered to change the co-solvent MeCN, which is sometimes rated as not ideal in the solvent selection guides,52–54 due to supply volatility, difficulties in dealing with MeCN waste and poor scoring with regard to life cycle management.55 Consequently, biotransformations were performed using ethanol as an environmentally more preferable co-solvent.56
Fatty acid hydroxylation by members of the CYP152 family
Since the UPO turned out to be not suitable for hydroxylation in α-position, six α-hydroxylases from the CYP152 family (P450Spα,57 P450CLA,35 P450Bsβ F79L/G290F,58 P450Exα,59 CYP152K660 and P450Jα47) were selected from literature. Some of them (CYP152K6, P450Bsβ F79L/G290F, P450Exα) have not been investigated for the transformation of medium chain fatty acids before, while for the others various challenges like over-oxidation or expression issues have been reported.38 While CYP152s have often been reported to be difficult to express in soluble form or in sufficient quantities in E. coli, all the potential enzyme candidates were successfully expressed (ESI Fig. S1–S6†), whereby the choice of an appropriate expression construct was essential, especially in the case of P450Spα (see ESI Plasmids†). Since E. coli possesses catalase activity, which would disproportionate the H2O2 reagent, the enzymes had to be purified. After purification, P450Exα was obtained with the highest concentration of active enzyme as determined by CO titration [up to 228 μM (10.9 mg mL−1)] (Table S5†).
Biotransformations with P450CLA, P450Spα, P450Exα, CYP152K6 and P450Bsβ F79L/G290F showed successful conversion using 10 mM of substrate to the corresponding α-hydroxylated fatty acids (Table 1). Remarkably, P450Exα was found to be an excellent candidate for the conversion of 1a and exhibited good regioselectivity (14:1) for the α-position over the β-position, giving 7 mM of α-2a and only 0.5 mM of β-2a. The natural substrate for P450Exα has been described to be myristic acid.59
|
|
|||||
|---|---|---|---|---|---|
| Biocatalyst | FA | Conv.a [%] | Productsb [mM] | eeαc [%] |
|
| CYP152 | α-OH | β-OH | |||
| a Conversion [%] (i.e., consumption of substrate 1a–c) was determined by GC using an int. standard (lauric acid) after derivatisation (for GC traces see Fig. S12–14†). b Product concentrations were determined via calibration curves with an internal standard (Fig. S8†). The response factor for β-OH was assumed to be the same as for α-OH. n.d. not detected. c Optical purity was measured after derivatisation on GC using a chiral phase. d ee values in literature are 36% ee (S)-α-2b and 71% ee (S)-α-2c.35 e n.a. = not applicable due to no product formation. f (Z)-2-Hydroxyoct-2-enoic acid (7% and 8% GC-area) was detected by GC-MS. | |||||
| P450CLA | 1a | 44 | 3.0 | n.d. | 8 (S) |
| P450CLA | 1b | >99 | 6.5 | 1.5 | 48 (S)d |
| P450CLA | 1c | 75 | 6.8 | n.d. | 62 (S)d |
| P450Spα | 1a | 35 | 2.1 | n.d. | >99 (S) |
| P450Spα | 1b | >99 | 8.9 | 0.4 | >99 (S) |
| P450Spα | 1c | >99 | 8.5 | 0.6 | >99 (S) |
| P450Exα | 1a | 95 | 7.0 | 0.5 | 81 (S) |
| P450Exα | 1b | >99 | 4.0 | 3.6 | >99 (S) |
| P450Exα | 1c | 96 | 2.9 | 4.0 | >99 (S) |
| P450Jα | 1a | 11 | n.d. | n.d. | n.a.e |
| P450Jα | 1b | 11 | n.d. | n.d. | n.a.e |
| P450Jα | 1c | 24 | 0.5 | n.d. | n.a.e |
| CYP152K6 | 1a | 73 | 5.3 | n.d. | 93 (S) |
| CYP152K6f | 1b | >99 | 6.3 | 1.3 | >99 (S) |
| CYP152K6 | 1c | 92 | 5.9 | 2.2 | >99 (S) |
| P450Bsβ F79L/G290F | 1a | 50 | 2.8 | n.d. | 58 (S) |
| P450Bsβ F79L/G290Ff |
1b | >99 | 5.6 | 1.6 | 79 (S) |
| P450Bsβ F79L/G290F | 1c | 38 | n.d. | 1.3 | n.a.e |

Furthermore, P450Spα reached completion for the conversion of 1b and 1c (>99% conv.) giving the corresponding α-hydroxylated products α-2b and α-2c with 8.9 mM and 8.5 mM, respectively. Only small amounts of β-hydroxylation product were detectable (β-2b and β-2c with 0.4 and 0.6 mM, respectively) corresponding to an α:β ratio of 22:1 and 14:1, respectively. Incomplete mass balances are most likely due to undesired oxidative decarboxylation, which was in general not significant, except for P450Jα for which decarboxylation to the alkene has been reported before.47 Furthermore 1-alkene formation has been observed for P450CLA, P450Spα and P450Exα at elevated H2O2 concentrations.47,59 The enzyme CYP152K6 has been described as α-hydroxylase for substrates like lauric acid, tetradecanoic acid and palmitic acid.60 Nevertheless, our study showed that this biocatalyst exhibits also good regioselectivity for medium chain fatty acids leading in general to the α-hydroxylated product, whereby for 1a the α-product was detected exclusively without any β-product and in case of 1b and 1c, the ratios of α:β-hydroxylation were 4.8:1 and 2.7:1, respectively. The variant P450Bsβ F79L/G290F produced actually only α-2a, although at moderate conversion and low recovery, while the same enzyme gave only the β-product when transforming the C10 fatty acid 1c.
It is worth to mention, that for most enzymes and transformations no over-oxidation e.g. to the corresponding carbonyl compound, thus the α-keto acid, was detected which has been identified as a major issue in previous work.27,38 Only in the case of 1b with CYP52K6 and P450Bsβ F79L/G290F some α-keto acid was detected. The successful minimization/avoidance of the over-oxidation product can most likely be attributed to the continuous addition of the oxidant circumventing sudden high concentrations.
Analysing the chirality and optical purity of the α-hydroxylated products revealed that all six CYP152 enzymes investigated formed preferentially the (S)-enantiomer (Fig. S15–17†). Thereby the biocatalysts P450Spα, P450Exα and CYP152K6 gave access to optically pure (S)-products (>99% ee) in most cases. In contrast, α-hydroxylation by P450CLA was in general poorly enantioselective with ee values in the range of 8–62%.
From these results, P450Spα and P450Exα were chosen as the most suitable enzymes for α-hydroxylation, due to the high conversion, regio- and stereoselectivity and amounts of products achieved.
Conversion of dicarboxylic acids with P450Spα and P450Exα
Since the hydroxylation of mono-carboxylic acids was successful, the two best performing α-hydroxylating enzymes P450Spα and P450Exα were investigated for the hydroxylation of selected dicarboxylic acids. While the dicarboxylic acids azelaic acid 3a (C9) and sebacic acid 3b (C10) were accepted by P450Exα (Scheme 2 and Fig. S18†), P450Spα did not transform these substrates. | ||
| Scheme 2 Mono-hydroxylation of dicarboxylic acids 3a–b with P450Exα. Reactions were performed in 1.5 mL glass crimp vials in a final volume of 1 mL. Conversion [%] (i.e., consumption of substrate) was determined by GC-MS using an int. standard (ISD, lauric acid) after derivatisation (BSTFA, 1% TMCS). | ||
The substrate azelaic acid is produced by ozonolysis of oleic acid61,62 and sebacic acid is obtained by the alkaline cleavage of castor oil.63 The market for azelaic acid is expected to reach USD 1.6 billion by 202564 and USD 313 million by 2026 for sebacic acid.65 Both are building blocks of high interest for industrial applications66 due to their bio-based nature67 and biodegradability.68
Although one might expect that the dicarboxylic acids could be hydroxylated twice in the α-positions of the two carboxylic acid moieties, P450Exα successfully converted 3a and 3b exclusively to the corresponding mono-hydroxylated products α-4a and α-4b, the latter with >99% conv. (Scheme 2).
As shorter chain dicarboxylic acids like succinic acid 3c (C4) or adipic acid 3d (C6) were not converted by P450Exα and P450Spα (Fig. S21–24†), but the longer dicarboxylic acid 3a (C9) and 3b (C10) were converted, we initiated structural investigations in search of a potential explanation. Docking of the dicarboxylic acids C6, C9, C10 and the natural substrate myristic acid (C14) into the active site of P450Exα revealed the following (Fig. 1 and Fig. S24†).
 | ||
| Fig. 1 Docking of dicarboxylic acids C6 (A), C9 (B), C10 (C) and carboxylic acid C14 (D) into the active site of P450Exα (PDB: 5YHJ) Docking studies were performed using YASARA and visualized with PyMOL. The docked substrates are visualized in pink in their lowest energy state. The simulation cell was defined at 4 Å around the iron atom of the heme. | ||
The computational modelling studies revealed that adipic acid (C6) (Fig. 1A) as well as succinic acid (C4) would be completely buried in the hydrophobic access channel. In contrast, the length of the hydrocarbon chains of 3a (C9) (Fig. 1B) and 3b (C10) (Fig. 1C) allowed for one of their carboxylic acid moieties to remain on the outside while they were tightly coordinated to the reaction centre like the natural substrate of P450Exα, myristic acid (C14) (Fig. 1D). Hence, it was presumed that the second carboxyl group of the longer chain dicarboxylic acids 3a and 3b did not lead to unfavourable interactions with the hydrophobic substrate tunnel and therefore to be converted. In contrast, for a transformation of e.g. succinic acid 3c (C4) or 3d (C6) also the second carboxylic acid moiety would need to be in the hydrophobic channel causing non-favoured interaction and therefore these substrates were not converted.
Preparative scale reactions
For the preparative transformations the focus was put first on P450Spα and 1b as high regio- and stereoselectivity was achieved on analytical scale. Running the reactions on a 50 mL scale with 10 and 50 mM 1b substrate concentration, respectively, both experiments resulted in >99% conv. (Table 2, entries 1 and 2) of which the higher substrate concentration (50 mM) reached a TON of 16667. By increasing the substrate concentration further to 100 mM (100 mL scale), the reaction still went to completion. In this experiment, the wildtype enzyme tolerated a total H2O2 concentration of 100 mM giving 1260 mg of product (S)-α-2b (87%, Table 2, entry 3) corresponding to a TON of 33333. It was possible to purify the final product and remove any residual substrate by utilizing heptane as a solvent (adapted procedure from literature69 see ESI “Purification of α-2b after re-extraction in heptane”†). Increasing the substrate concentration even further to 150 mM and thereby also the amount/concentration of H2O2 still allowed to reach 90% conversion, corresponding to a TON of 42000 (Table 2, entry 4). Interestingly, increasing the substrate concentration for P450Spα from 10 mM to 50 mM led to a decrease in ee from >99% to 97%. At 100 and 150 mM the ee was 90–96%.
| Entry | Substrate | Substrate [mM] | Enzyme | Total H2O2 [mM] | GC-MS conv.a [%] | Isolated yield [%]/purityb [%] | TONc |
|---|---|---|---|---|---|---|---|
| Reaction conditions: reactions were performed in a 250 mL reaction flask containing reaction buffer (100 mM KPi buffer, pH 7.4), EtOH (5% v/v), fatty acid (10, 50, 100 or 150 mM) and purified enzyme (3 μM), in a final volume of 50 mL (100 mL for entry 3 and 25 mL for entry 5). H2O2 was added continuously via a syringe pump [entries 1, 5 and 6: 1.6 mM h−1 over 12 h to a final concentration of 20 mM (stock: 320 mM); entries 2 and 7: 8.3 mM h−1 over 12 h to a final concentration of 100 mM (stock: 320 mM), entries 3 and 4: 12.5 mM h−1 over 12 h to a final concentration of 150 mM (stock: 400 mM). N.d. not determined.a Conversion was determined by GC-MS using lauric acid (5 mM) as ISD by comparison with a sample at t = 0.b Isolated yields [%] were calculated based on the measured mass [mg] of isolated and dried product and the maximum theoretical yields [mg]. Purity [%] was calculated based on GC-MS data.c TON = turnover number which is defined as mmol substrate converted per mmol catalyst.d Yield after second purification step.e α-2c 61% GC-area; β-2c and γ-2c product 26% GC-area and 9% GC-area, respectively. | |||||||
| 1 | 1b | 10 | P450Spα | 20 | >99 | 99 (80 mg)/>99 | 3333 |
| 2 | 1b | 50 | P450Spα | 100 | >99 | 89 (401 mg)/94 (6% β-2b) | 16667 |
| 3 | 1b | 100 | P450Spα | 150 | >99 | 87 (1260 mg)/>99d | 33333 |
| 4 | 1b | 150 | P450Spα | 150 | 90 | n.d. | 42000 |
| 5 | 1c | 10 | P450Spα | 20 | 97e | n.d. | 3233 |
| 6 | 3b | 10 | P450Exα | 20 | >99 | 29 (26 mg)/>99 | 3333 |
| 7 | 3b | 50 | P450Exα | 100 | 25 | n.d. | 833 |
The hydroxylation of 1c on 25 mL scale resulted in α-2c as the main product (GC-MS yield of 61%) but also the β- and γ-products were detected via GC-MS (Table 2, entry 5).
Finally, dicarboxylic acid 3b was successfully converted to the mono-hydroxylated product α-4b (Table 2, entry 6) reaching a TON of 3333 under the conditions used. The lower yield (29%) was a result of the higher water solubility of the dicarboxylic acid and using the same work up procedure as for the mono-carboxylic acids.
Increasing the substrate concentration to 50 mM and using 100 mM of H2O2 indicated the limits of the used enzyme reaching 25% conv. (Table 2, entry 7).
Comparison of chemical and biocatalytic methods
Comparing this study with established chemical and other biocatalytic strategies from literature (Table 3), two chemical options for the synthesis of α-2b have been reported using lithium diisopropylamide (LDA), hexamethyl-phosphoramide (HMPA) and either bis(trimethylsilyl)-peroxide (TMSOOTMS) or molecular oxygen (O2) as oxygen source (Table 3, route 1 and 2).16,17 Although the substrate concentration was high (200–250 mM), these routes have the lowest atom economy and use toxic and carcinogenic (HMPA) reagents. Additionally, the products were obtained in low yields and in the racemic form only.| Entry | Catalyst/reagents | Solvent/workup | Conc. 1b/scale | Yield [%] | Conv. [%] (ee) | TONe | Atom economyf [%] |
|---|---|---|---|---|---|---|---|
a Purification by preparative thin-layer chromatography (SiO2, 20% ethyl acetate in hexane).
b Hydroxyperoxide product as side product.
c Intermediate in a cascade reaction.
d Over-oxidation to α-keto acid (2.89 mM).
e TON = turnover number which is defined as mmol substrate converted per mmol catalyst.
f 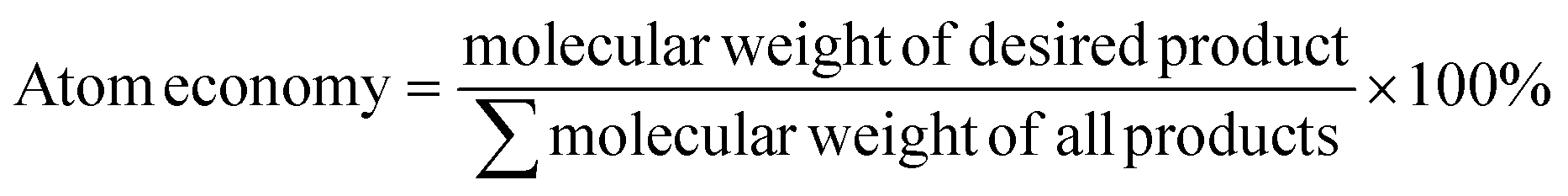 .
g Reduced atom economy due to side product formation (α-keto acid). n.a. = not applicable. .
g Reduced atom economy due to side product formation (α-keto acid). n.a. = not applicable. |
|||||||
| 116 | LDA TMSOOTMS, H+ | THF/extraction with ethyl acetatea | 250 mM/10 mL | 44 | rac | n.a. | 34.4 |
| 217 | O2/LDA, HMPA | THF/crystallization | 200 mM/100 mL | 63b | rac | n.a. | 35.9 |
| 370 | Homogenate of peas/O2 | 0.2 M phosphate buffer | 0.2 mM/150 mL | Not isolated | <10 (99% R) | n.a. | n.a. |
| 438 | P450CLA/H2O2 (24 mM) | Kpi (pH 7.5, 100 mM) | 10 mM 1a/500 μL | Not isolated | 78.4 (36% S) | 1568 | 47.1g |
| EtOH (5% v/v) | 5 mM α-2ad | ||||||
| 546 | P450CLA/H2O2 (20 mM) | Tris-HCl (pH 7.5, 100 mM) | 10 mM/1 mL | Not isolated | >99 (36% S) | 3333 | 89.9 |
| EtOH (5% v/v) | >9.9 mM α-2b | ||||||
| 611 | P450CLA/H2O2 (20 mM) | Kpi (pH 7, 20 mM) | 10 mM/50 mL | >95 | >99 (36% S) | 1666 | 89.9 |
| EtOH (5% v/v) | |||||||
| 710 | P450Spα/H2O2 (20 mM) | KPi (pH 7.4, 100 mM) | 10 mM/1 mL | Not isolatedc | 23 (>99% S) | 767 | 89.9 |
| EtOH (10% v/v) | 2.3 mM α-2b | ||||||
| 8This work | P450Spα/H2O2 (up to 150 mM) | KPi (pH 7, 100 mM) | Up to 150 mM/50 mL | Up to >99% | Up to >99 (90->99% S) | Up to 42000 |
89.9 |
| EtOH (5% v/v)/acidification, extraction with ethyl acetate | |||||||
On the other hand, all biocatalytic approaches are performed in aqueous media requiring H2O2 as oxidation reagent, except for entry 3 (Table 3) where molecular oxygen was used with a homogenate of peas, representing a less well-defined reagent.
The CYP152 family offers high potential for regio- and stereoselective hydroxylation on medium chain fatty acids on preparative scale.
When comparing the two biocatalysts from the CYP152 family used for the α-hydroxylation of 1b (P450CLA and P450Spα) concerning stereoselectivity, P450Spα is superior as highly optically enriched product α-2b was obtained (96–99% ee), while P450CLA gave the α-hydroxy acid with an ee of 36% only (Table 3, entries 6, 7 versus 4–6).
Comparing the approach of this study P450Spα (Table 3, route 8) with all previous studies involving peroxygenases, it becomes clear that P450Spα can be used also at elevated substrate concentration (up to 150 mM), while previous studies used 2a at a maximum substrate concentration of 10 mM only. In comparison to route 7 where the hydroxy acid was not the main target, in our study α-2b was obtained with up to >99% conversion allowing to reach an outstanding TON and the product was isolated from a preparative scale experiment. An important key to success was the method of applying stoichiometric amounts of H2O2 continuously via a syringe pump. This turned out to be clearly superior to the previous approaches, where H2O2 was added at the beginning or was produced during the reaction.
Consequently, high conversions of 1b with P450Spα on 50 mL scale were achieved reaching TONs of up to 42000. So far, the highest turnover number (TON) for CYP152s in literature did not exceed a value of 3333.46 On small scale a TON of 3333 was reached for the conversion of 1b (10 mM) by P450CLA (3 μM). For pelargonic acid (C9:0) the reaction was conducted in 20 mM KPi buffer and yielded in 0.49 g product (>99% conv., 95% α-hydroxylated nonanoic acid, TON 3330) on 150 mL scale.11 Thus, the TON achieved here are one order of magnitude higher than reported before and are therefore the highest reported for a peroxygenase from the CYP152 family.
Conclusions
In this study an atom economical process for the stereoselective biocatalytic α-hydroxylation of medium chain fatty acids was developed, requiring hydrogen peroxide as the only stoichiometric reagent. The enzyme P450Exα showed high conversions, regio- and stereoselectivity for the substrates 1a [conv. 95%; 70% α-2a; ee 81% (S)] and was active towards longer chain dicarboxylic acids 3a (conv. 68%) and 3b (conv. 99%) with a remarkable chemoselectivity for mono-hydroxylation. P450Spα allowed to achieve excellent conversion for the fatty acids C8:0 (1b) and C10:0 (1c) (>99%) with up to >99% (S) ee. These results allowed an efficient and scalable process to produce α-hydroxylated fatty acids where the amount of required chemicals and waste produced were kept at a minimum. In summary, TONs of up to 42000 were achieved for the conversion of 1b on preparative scale using P450Spα as catalyst at substrate concentrations up to 150 mM giving the desired product in gram quantities.
Author contributions
K. B., A. S., S. V., H. R., M. L. and S. P. performed the experiments. K. B., S. W. and W. K. planned experiments. All authors analysed data. M. L., S. P., S. W., W. K., C. S. provided scientific feedback. K. B. and W. K. wrote the manuscript. W. K. and S. W. conceived the study. W. K. raised funding and supervised the study. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under Marie Skolodowska-Curie grant agreement ID: 956621. AS received funding via the COMET center: acib: Next Generation Bioproduction is funded by BMVIT, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency in the framework of COMET – Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency FFG. The University of Graz and BioHealth Graz are acknowledged for financial support.References
- G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS PubMed.
- I. Vural Gursel, B. Elbersen, K. P. H. Meesters and M. van Leeuwen, Sustainability, 2022, 14, 12780 CrossRef.
- X. Yue and Y. Queneau, ChemSusChem, 2022, 15, e202102660 CrossRef CAS PubMed.
- B. Duchemin, Green Chem., 2022, 24, 2653–2679 RSC.
- P. Intasian, K. Prakinee, A. Phintha, D. Trisrivirat, N. Weeranoppanant, T. Wongnate and P. Chaiyen, Chem. Rev., 2021, 121, 10367–10451 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- J. García, J. Fraile, C. Herrerías and E. Pires, Synthesis, 2017, 1444–1460 CrossRef.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS PubMed.
- A. Enferadi Kerenkan, F. Béland and T.-O. Do, Catal. Sci. Technol., 2016, 6, 971–987 RSC.
- S. Gandomkar, A. Dennig, A. Dordic, L. Hammerer, M. Pickl, T. Haas, M. Hall and K. Faber, Angew. Chem., Int. Ed., 2018, 57, 427–430 CrossRef CAS PubMed.
- A. Dennig, F. Blaschke, S. Gandomkar, E. Tassano and B. Nidetzky, Adv. Synth. Catal., 2019, 361, 1348–1358 CrossRef CAS.
- E. Gabirondo, A. Sangroniz, A. Etxeberria, S. Torres-Giner and H. Sardon, Polym. Chem., 2020, 11, 4861–4874 RSC.
- K. M. Nampoothiri, N. R. Nair and R. P. John, Bioresour. Technol., 2010, 101, 8493–8501 CrossRef PubMed.
- N. M. Carballeira, M. Cartagena, F. Li, Z. Chen, C. F. Prada, E. Calvo-Alvarez, R. M. Reguera and R. Balaña-Fouce, Pure Appl. Chem., 2012, 84, 1867–1875 CrossRef CAS PubMed.
- N. M. Carballeira, H. Cruz, C. D. Kwong, B. Wan and S. Franzblau, Lipids, 2004, 39, 675–680 CrossRef CAS PubMed.
- M. Pohmakotr and C. Winotai, Synth. Commun., 1988, 18, 2141–2146 CrossRef CAS.
- D. Konen, L. Silbert and P. Pfeffer, J. Org. Chem., 1975, 40, 3253–3258 CrossRef CAS.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- E. Romero, B. S. Jones, B. N. Hogg, A. Rue Casamajo, M. A. Hayes, S. L. Flitsch, N. J. Turner and C. Schnepel, Angew. Chem., Int. Ed., 2021, 60, 16824–16855 CrossRef CAS PubMed.
- G. Grogan, Curr. Opin. Chem. Biol., 2011, 15, 241–248 CrossRef CAS PubMed.
- M. T. Lundemo and J. M. Woodley, Appl. Microbiol. Biotechnol., 2015, 99, 2465–2483 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- D. Permana, T. Kitaoka and H. Ichinose, Biotechnol. Bioeng., 2023, 120, 1725–1745 CrossRef CAS PubMed.
- J. Akter, T. P. Stockdale, S. A. Child, J. H. Z. Lee, J. J. De Voss and S. G. Bell, J. Inorg. Biochem., 2023, 244, 112209 CrossRef CAS PubMed.
- P. Gomez de Santos, A. Gonzalez-Benjumea, A. Fernandez-Garcia, C. Aranda, Y. Wu, A. But, P. Molina-Espeja, D. M. Mate, D. Gonzalez-Perez, W. Zhang, J. Kiebist, K. Scheibner, M. Hofrichter, K. Swiderek, V. Moliner, J. Sanz-Aparicio, F. Hollmann, A. Gutierrez and M. Alcalde, Angew. Chem., Int. Ed., 2023, 62, e202217372 CrossRef CAS PubMed.
- A. González-Benjumea, J. Carro, C. Renau-Mínguez, D. Linde, E. Fernández-Fueyo, A. Gutiérrez and A. T. Martínez, Catal. Sci. Technol., 2020, 10, 717–725 RSC.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- O. Shoji and Y. Watanabe, J. Biol. Inorg. Chem., 2014, 19, 529–539 CrossRef CAS PubMed.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- K. Hernandez, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Org. Chem., 2012, 16, 2652–2672 CrossRef CAS.
- E. Stadtman and R. Levine, Amino Acids, 2003, 25, 207–218 CrossRef CAS PubMed.
- M. Girhard, S. Schuster, M. Dietrich, P. Dürre and V. B. Urlacher, Biochem. Biophys. Res. Commun., 2007, 362, 114–119 CrossRef CAS PubMed.
- R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS.
- A. W. Munro, H. M. Girvan, A. E. Mason, A. J. Dunford and K. J. McLean, Trends Biochem. Sci., 2013, 38, 140–150 CrossRef CAS PubMed.
- X. Yu, X.-Y. Chen, H.-L. Yu, J.-H. Xu and Z.-J. Zhang, Green Chem., 2023, 25, 3469–3474 RSC.
- S. Lütz, E. Steckhan and A. Liese, Electrochem. Commun., 2004, 6, 583–587 CrossRef.
- Y. Ni, E. Fernández-Fueyo, A. G. Baraibar, R. Ullrich, M. Hofrichter, H. Yanase, M. Alcalde, W. J. van Berkel and F. Hollmann, Angew. Chem., Int. Ed., 2016, 55, 798–801 CrossRef CAS PubMed.
- C. E. Paul, E. Churakova, E. Maurits, M. Girhard, V. B. Urlacher and F. Hollmann, Bioorg. Med. Chem., 2014, 22, 5692–5696 CrossRef CAS PubMed.
- D. I. Perez, M. M. Grau, I. W. Arends and F. Hollmann, Chem. Commun., 2009, 6848–6850 RSC.
- E. Churakova, M. Kluge, R. Ullrich, I. Arends, M. Hofrichter and F. Hollmann, Angew. Chem., Int. Ed., 2011, 50, 10716–10719 CrossRef CAS PubMed.
- D. S. Choi, Y. Ni, E. Fernández-Fueyo, M. Lee, F. Hollmann and C. B. Park, ACS Catal., 2017, 7, 1563–1567 CrossRef CAS.
- L. Schmermund, S. Reischauer, S. Bierbaumer, C. K. Winkler, A. Diaz-Rodriguez, L. J. Edwards, S. Kara, T. Mielke, J. Cartwright, G. Grogan, B. Pieber and W. Kroutil, Angew. Chem., Int. Ed., 2021, 60, 6965–6969 CrossRef CAS PubMed.
- A. Dennig, S. Gandomkar, E. Cigan, T. C. Reiter, T. Haas, M. Hall and K. Faber, Org. Biomol. Chem., 2018, 16, 8030–8033 RSC.
- J. Armbruster, M. Steinmassl, C. A. Müller Bogotá, G. Berg, B. Nidetzky and A. Dennig, Chem. – Eur. J., 2020, 26, 15910–15921 CrossRef CAS PubMed.
- V. Kostik, S. Memeti and B. Bauer, J. Hyg. Eng. Des., 2013, 4, 112–116 Search PubMed.
- https://www.zionmarketresearch.com/report/medium-chain-triglycerides-market , published 2021; accessed 14.08.2023.
- L. Rotilio, A. Swoboda, K. Ebner, C. Rinnofner, A. Glieder, W. Kroutil and A. Mattevi, ACS Catal., 2021, 11, 11511–11525 CrossRef CAS PubMed.
- M. Pickl, S. Kurakin, F. G. Cantu Reinhard, P. Schmid, A. Pocheim, C. K. Winkler, W. Kroutil, S. P. de Visser and K. Faber, ACS Catal., 2019, 9, 565–577 CrossRef CAS PubMed.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- I. F. McConvey, D. Woods, M. Lewis, Q. Gan and P. Nancarrow, Org. Process Res. Dev., 2012, 16, 612–624 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC.
- I. Matsunaga, E. Kusunose, I. Yano and K. Ichihara, Biochem. Biophys. Res. Commun., 1994, 201, 1554–1560 CrossRef CAS PubMed.
- T. Fujishiro, O. Shoji, S. Nagano, H. Sugimoto, Y. Shiro and Y. Watanabe, J. Biol. Chem., 2011, 286, 29941–29950 CrossRef CAS PubMed.
- H. Onoda, O. Shoji, K. Suzuki, H. Sugimoto, Y. Shiro and Y. Watanabe, Catal. Sci. Technol., 2018, 8, 434–442 RSC.
- H. M. Girvan, H. Poddar, K. J. McLean, D. R. Nelson, K. A. Hollywood, C. W. Levy, D. Leys and A. W. Munro, J. Inorg. Biochem., 2018, 188, 18–28 CrossRef CAS PubMed.
- R. G. Ackman, M. Retson, L. Gallay and F. Vandenheuvel, Can. J. Chem., 1961, 39, 1956–1963 CrossRef CAS.
- A. Köckritz and A. Martin, Eur. J. Lipid Sci. Technol., 2010, 113, 83–91 CrossRef.
- J. L. Dubois, in Chemicals and Fuels from Bio–Based Building Blocks, 2016, pp. 535–548 Search PubMed.
- https://markwideresearch.com/azelaic-acid-market/ , published 2023; accessed 13.06.2023.
- https://www.expertmarketresearch.com/reports/sebacic-acid-market , published 2022; accessed 13.06.2023.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- E. Brenna, D. Colombo, G. Di Lecce, F. G. Gatti, M. C. Ghezzi, F. Tentori, D. Tessaro and M. Viola, Molecules, 2020, 25, 1882 CrossRef CAS PubMed.
- G. Z. Papageorgiou, D. N. Bikiaris, D. S. Achilias, E. Papastergiadis and A. Docoslis, Thermochim. Acta, 2011, 515, 13–23 CrossRef CAS.
- E. Busto, N. Richter, B. Grischek and W. Kroutil, Chem. – Eur. J., 2014, 20, 11225–11228 CrossRef CAS PubMed.
- W. Adam, M. Lazarus, C. R. Saha-Mǒller and P. Schreier, Tetrahedron: Asymmetry, 1996, 7, 2287–2292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04593e |
| This journal is © The Royal Society of Chemistry 2024 |