
Shell waste valorization to chemicals: methods and progress
Lavanya
Korampattu
 ab,
Neha
Ghosh
ab and
Paresh L.
Dhepe
*ab
ab,
Neha
Ghosh
ab and
Paresh L.
Dhepe
*ab
aCatalysis & Inorganic Chemistry Division, CSIR-National Chemical Laboratory, Pune-411008, Maharashtra, India. E-mail: lavanyakunni@gmail.com; nehaghosh.lbc@gmail.com; pl.dhepe@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 19th March 2024
Abstract
In light of growing energy and resource demands, the conversion of biomass materials into diverse chemicals and fuels holds significant importance. This process enables the utilization of biomass as a valuable and renewable resource to meet these escalating needs. The current biomass valorization processes are largely based on plant-derived lignocellulosic biomass materials. Studying new genres of biomass materials and their value addition is highly desirable, and they would complement and expand the existing biorefinery system. Crustacean shell waste represents a highly potential bioresource that is composed of a set of useful chemicals, among which chitin, the amino polysaccharide, has come to the fore as a promising material for a plethora of applications. In terms of the abundance of biopolymers on Earth, cellulose is the most prevalent biopolymer, and chitin ranks as the second. The presence of biologically fixed nitrogen in the structure of chitin opens up new possibilities for making useful chemicals, notably nitrogen-containing chemicals, which are otherwise impossible to produce directly from lignocellulosic biomass. In the last two decades, several attempts have been made by researchers in this area to convert chitin and its derivatives into valuable chemicals. This review comprehensively summarizes the transformation of the chitin fraction from shell waste to various chemicals through different thermo-catalytic processes and appraises the advancements in this area. The effect of various catalytic systems on chitin biomass valorization processes and the challenges and opportunities allied to this are discussed.
 Lavanya Korampattu | Lavanya Korampattu was born in 1995 in Kerala, India. She received her Bachelor's degree in Chemistry from the University of Calicut and Master's in Chemistry from the Central University of Kerala, Kasargod. She is currently pursuing her Ph.D. in the Catalysis and Inorganic Chemistry Division at CSIR-National Chemical Laboratory, Pune, with Dr Paresh L. Dhepe. Her research focuses on the synthesis of biomass-derived carbon materials and exploring their applications in various organic transformations and electrochemistry. |
 Neha Ghosh | Neha Ghosh was born in 1991 in Ludhiana, Punjab. She received her Ph.D. in Chemistry at CSIR-National Chemical Laboratory, Pune, under the supervision of Dr Paresh L. Dhepe. Prior to this, she recieved her B.Sc. in Chemistry from the University of Calcutta and M.Sc. in Chemistry from the Indian Institute of Science Engineering and Technology, Shibpur (formerly BESU, Shibpur). Her research has been focused on understanding the physico-chemical properties of N-containing saccharides through various analytical techniques, their valorization using solid acid catalysts and analytical method developments for the separation and identification of products. |
 Paresh L. Dhepe | Paresh L. Dhepe has been Senior Principal Scientist at CSIR-National Chemical Laboratory, Pune, since 2020. He has worked at the CSIR-National Chemical Laboratory, Pune, since 2007. Prior to this, he completed his post-doctoral studies at Hokkaido University, Japan (2003–2007), with Prof. Atsushi Fukuoka. His Ph.D. is from Hokkaido University Japan (1999–2003). His research focuses on conversion of celluloses into sugars and furfural, valorization of lignin into aromatic monomers and their upgradation, synthesis of platform chemicals from sugars and sugar derivatives, and conversion of chitin-derived biomass into value-added chemicals. He is also working in the area of plastics circularity. |
1 Introduction
The excessive release of carbon dioxide (CO2) has emerged as a significant cause for concern due to its contribution to the global warming phenomenon. The primary culprits behind this surge in CO2 emissions are human activities involved in processing fossil fuel feedstocks to fulfil energy and material demands. Over the past two decades, this has prompted a notable shift in scientific focus towards exploring alternative energy sources and feedstocks. As per current scientific understanding, transitioning from conventional fuels to electricity can substantially mitigate emissions. However, despite progress in the energy sector, there is still a reliance on chemical processes to meet material needs. A potential solution lies in adopting renewable feedstocks, such as waste biomass. This approach not only aids in reducing our carbon footprint but also aligns with the imperative of fulfilling chemical requirements more sustainably.1,2While plant-based biomass is primarily composed of carbon, hydrogen, and oxygen, animal biomass waste, such as shell waste, additionally contains nitrogen in the structure as a result of biological nitrogen fixation. This makes shell waste an appealing alternative feedstock to traditional nitrogen-containing chemicals, which rely heavily on the energy-intensive Haber process for ammonia synthesis. The current biomass utilization is largely focused on lignocellulosic biomass.3–5 In contrast, much less attention is given to the valorization of animal-derived biomass. In this context the horizon of exploring new types of biomass resource will benefit the expanding horizon of biobased products and complement the current lignocellulose-based biorefinery system.
The FAO's 2022 report states that the annual production of crustaceans, including both aquaculture and capture fisheries, reached 17.2 million tons in 2020.6 Most of it comprises crabs, shrimps, lobsters, and prawns. With the increase in the overall production of crustaceans, the shell waste management facilities also have to be improved. The disposal of the waste produced after shellfish processing represents a practical challenge, as in developing and poor countries, often the shellfish waste is discarded back into the ocean, dumped in landfill sites, burned, or left out to spoil, whereas in developed countries like the UK, the disposal cost for landfill sites is about £80.7 Unregulated disposal can be a nuisance and polluting if not controlled. Shell waste contains several useful chemicals, such as chitin (an amino polysaccharide), protein, minerals like calcium carbonate, and pigments (the detailed composition is discussed in the next section). Hence, the valorization of shell waste by separating its components into different fractions, up-grading of each fraction, and converting them into useful chemicals can be a strategy for the management of shell waste.
The chitin fraction derived from shell waste constitutes a potent biopolymer, ranking as the second most abundant biopolymer in nature, following cellulose. Given its origin, chitin is both inexpensive and abundant, deriving from biomass, rendering it carbon-neutral and aligning well with the principles of the circular economy. Its natural nitrogen content further enhances its appeal as a feedstock. Considering the structural homology between chitin and cellulose, researchers have contemplated a parallel synthetic pathway for the production of value-added chemicals and platform compounds, particularly those featuring nitrogen moieties. Notably, several thermo-catalytic methodologies have been developed for the transformation of chitin biomass. These encompass the depolymerization, hydrolysis, hydrogenation, and oxidation of chitin and chito-oligosaccharides, yielding various nitrogen-containing and non-nitrogen-containing sugars, heterocyclic compounds, polyols, and acids etc. These methods serve the purpose of enhancing the value and utility of chitin-derived chemicals through systematic upgradation processes.
Chitin valorization to chemicals was in its budding stage in the initial years of the last decade. In recent times, there has been a discernible surge in interest surrounding the conversion of chitin, resulting in a substantial body of research and publications on the subject. Fig. 1 illustrates the increasing number of publications related to the thermo-catalytic transformation of chitin biomass into various chemicals. This data compilation is derived from the literature results obtained through searches using specific keywords and their combinations, such as ‘valorization of chitin and chitosan’, ‘depolymerization of chitin and chitosan’, ‘catalytic conversion of chitin biomass’, ‘value-added chemicals from chitin’, ‘transformation of chitin’, and ‘nitrogen compounds from chitin’ in the SciFinder database.
 | ||
| Fig. 1 Number of publications on the transformation of chitin and chitin-derived chemicals. | ||
The transformation of chitin into various chemicals and the aspects of shell biorefinery have been previously addressed in a limited number of earlier reviews and perspectives.8–19 While a few reviews have provided detailed descriptions of various aspects of shell waste valorization and offered insights into the potential of chitin valorization, the majority of other reviews have focused on specific areas, such as the synthesis of one particular category of chemicals. Notably, a comprehensive compilation and discussion on recent advancements in catalytic transformations of chitin biomass is conspicuously absent in the existing literature. This gap in knowledge has motivated our exploration into the intricacies of the valorization of chitin from shell waste. The primary objective of this review is to scrutinize the diverse transformations of chitin leading to the production of valuable chemicals. Through an extensive examination of relevant literature, we present a categorization of chitin valorization methods based on the various products synthesized from chitin biomass. The review also delves into the discussion of the different catalytic systems employed and the distinct types of reaction process utilized in the conversion of chitin. In this review, our major focus is on the value addition of only the chitin fraction from shell waste. Given the abundance of detailed reviews on shell waste components10 and fractionation methods,20 we have included only a general and short description of other shell waste components and methods of shell waste fractionation for them in this review.
2 Shell waste components and uses
The exoskeleton of crustaceans such as crabs, shrimps, lobsters, krill, and prawns consist of mainly four components: the amino polysaccharide chitin (15–40%), protein (20–40%), minerals like calcium carbonate (20–50%), and lipids and pigments like astaxanthin (0–14%).21–23 This composition is different and can vary from species to species, depending on the habitat, geographical area, and season as well. An illustration of shell waste composition and the area of application of each component is presented in Fig. 2. | ||
| Fig. 2 Shell waste components and their applications. | ||
Calcium carbonate is the major component of shell waste. It is also commonly found in rocks, shells of snails and mollusks, and eggs. It has three polymorphic forms in nature. The β, λ and μ-CaCO3 forms are named calcite, aragonite, and vaterite, respectively.24 CaCO3 has a wide range of applications in industry. It is mainly used as a construction material in the building industry, and as an extender in the paint industry. Recent reports have highlighted the importance of shell waste-derived CaCO3 as a green additive to commercial cement.25,26 In medicine, CaCO3 is used for health and dietary applications such as gastric antacids, phosphate binders, etc. It also plays a vital role in soil treatment in the agricultural industry.24 About 20–50% of shell waste is composed of CaCO3. This biological occurrence makes shell waste-derived calcium carbonate a reliable component in food and medicinal applications. The extraction of CaCO3 to usable forms is discussed in some of the previous reports.27–29 Briefly, shell waste undergoes washing with water to remove dirt, salt and remaining flesh, drying and calcination at 500 °C to 1000 °C to produce the CaCO3 as the final powder.
Chitin is a nitrogen-containing polysaccharide that is the second major component of the shell waste of crabs, shrimp, prawns etc. It also occurs in the exoskeletons of arthropods and in certain fungal species. The annual global production of chitin exceeds 100 billion tons.30 The principal source of chitin is shellfish waste, which constitutes about 15–40% of the shell waste. Shrimp-processing wastes contain around 14–27% chitin in terms of their dry weight, while crab-processing wastes contain approximately 13–15% chitin. Chitin acts as a protective component in the cell wall, where the chitin filaments are integrated into a mixture of calcium carbonate, phosphate, and protein, forming a matrix. This protein-based matrix undergoes a hardening process known as tanning.31 Chemically, chitin is defined as a linear homopolymer of N-acetyl-D-glucosamine (NAG), or 2-(acetylamino)-2-deoxy-D-glucose linked via β-(1 → 4) glycosidic bonds. The structure of chitin is very comparable to that of cellulose, where the hydroxyl groups at the C-2 position of the glucose units are replaced by an acetamido group. There are three different polymorphic forms of chitin, namely α, β, and γ-chitin; they vary in the packing and polarities of adjacent chains in successive layers.32 The stacking configuration of different polymorphs of chitin is shown in Fig. 3a.
 | ||
| Fig. 3 (a) Stacking of polymer chains in different polymorphs of chitin and (b) intra- and inter-chain hydrogen bonding in chitin. | ||
Thermodynamically, α-chitin is the most stable form of chitin, with an antiparallel arrangement of polymeric chains with alternating sense. The parallel arrangement of chains with the same sense makes the β-chitin whereas γ-chitin has a both antiparallel and parallel stacking of chains, which was also considered as a variant of the α-chitin family by recent studies. The conversion of β-chitin to α-chitin is possible while the reverse cannot be done. The crystallinity of α-chitin is due to the presence of intra-chain (C3′–OH to O5, C6′–OH to acetamido carbonyl O7 and C6–OH to O6′) and interchain (between NH and the acetamido carbonyl O7′ and C6′–OH to O6) hydrogen bonding between the polymeric chains of the chitin.33 The intra- and inter-chain hydrogen bonding in chitin is shown in Fig. 3b.
α-Chitin is the most abundant among all of the chitin polymorphs, due to its thermodynamic stability, which makes it insoluble in most of the common solvents, consequently making it difficult to break into monomers and other chemicals. Chitin can undergo deacetylation of the acetamido group when treated with a base at elevated temperature to form chitosan, where an amino group is present instead of an acetamido group. The monomer unit of chitosan is D-glucosamine (GA) or 2-(amino)-2-deoxy-D-glucose. Chitosan can have a varying degree of deacetylation (DD) according to the deacetylation conditions. Chitin with more than 50% of DD is considered to be chitosan. The transformation of chitosan is easier since it is less crystalline and has reduced rigidity compared with chitin.34 Chitin and chitosan contain nitrogen along with carbon, hydrogen, and oxygen in their structure which makes them an excellent resource to produce various valuable chemicals. The attractive aspect of chitin valorization lies in its ability to produce nitrogen-containing chemicals, which are particularly valuable compared with those derived from its cellulose counterparts. Moreover, the production of N-containing chemicals requires ammonia or ammonia derivatives, and it is typically a laborious and energy intensive process. The Haber–Bosch process for ammonia synthesis requires high temperature and high pressure, and the hydrogen supply for the process is mainly from the reforming of fossil fuel. Hence, chitin can be a potential resource which is inexpensive and sustainable to produce nitrogen-containing chemicals. Section 4 provides a detailed discussion on the application of chitin as a feedstock and its valorization process.
Apart from the fact that chitin is a sustainable feedstock material, in its native form chitin has got so many interesting properties. The biological properties of both chitin and chitosan such as antibacterial, antioxidative, biocompatibility, biodegradability, etc. have made both chitin and chitosan versatile materials for a variety of applications in the biomedical,35 pharmaceutical,36 and food processing37 fields etc. The advantageous feature of chitin's structure lies in its ease of transformation into diverse physical forms, including fibers, membranes, films, spheres, beads, mats, disks, and more. This inherent flexibility facilitates the creation of chitin-based materials for applications such as wastewater treatment38 and innovative biomedical solutions like tissue engineering, drug delivery, and the fabrication of medical implants with notable effectiveness.39–42 Chitin is thermally and chemically stable and it has an abundance of hydroxyl, primary amino, and acetylamino functional groups that can bind to a diverse range of metal ions. Notably, the occurrence of the amino group is uncommon in polysaccharides, and that makes the tuning and functionalization of chitin a feasible process. The chemically modified chitin in the form of various organo-catalysts and metal incorporated catalysts is explored and utilized in many reactions.13,43–46
In addition to fine chemicals, chitin is also used as a feedstock for biobased nitrogen-doped carbon.47 This is a greener alternative to conventional nitrogen-doped carbon, as it reduces the use of expensive and toxic nitrogen sources like NH3, amines such as urea, melamine, dicyanodiamine, DMF, ethylenediamine, N2H4, aniline,48–52 or nitrogen-containing polymers, ionic liquids, and metal–organic frameworks (MOFs).53–55 The synthesis procedure and utilization of these nitrogen-doped carbons as catalysts,56–58 catalyst supports,59–61 adsorbents61,62etc. are discussed in several earlier literature works. The majority of them are being utilized for electrochemical applications63–68 and various organic transformations.58,59,69,70 Readers are redirected to the references for further reading.
Minor fractions of proteins and lipids are also found in the shell waste. The protein hydrolyzate which is found in some shrimp shells can be used in applications such as animal feed, pharmaceuticals, human nutrition, and cosmetics.71 The essential amino acid percentage in protein hydrolyzate determines the food protein nutritional value. Several species of shrimp shells found to have proteins constituting amino acids glutamic acid, lysine, histidine, and valine which represent shell waste can be used as a nutritious protein source as animal feed and a food supplement.72 Lipids are biomolecules which contain hydrocarbons and are vital building blocks for the structure and functioning of cells. Examples of lipids are fats, waxes, oils, hormones, certain vitamins, and cell membrane etc. The lipids in shrimp shells comprise fatty acids as the majority. These fatty acids are composed of 30–50% saturated and 50–70% unsaturated fats. Eicosanoic acid is the prevalent saturated fatty acid found in the shells of shrimps. Additionally, fatty acids such as docosahexaenoic acid and eicosapentaenoic acid are polyunsaturated, and can also be found in crustacean shells.9 These lipids are used in cosmetics, food, and pharmaceutical applications.
Pigments like astaxanthin, astathin, lutein, canthaxanthin and β-carotene etc. are found in crustacean shells.73 When the crustaceans are alive, they appear in different colors or even as transparent, as the pigments in their shells are bound to the protein. However, upon denaturation when the protein unfolds, the pigment is released. This is the reason for the transformation of seafood to a reddish hue when cooked. Astaxanthin has remarkable antioxidant activity, 10-fold higher than carotenoids and 500 times more advanced than vitamin E; hence, it is used as a dietary compound.74 Since astaxanthin shows antioxidant, anti-immunomodulating, and anti-inflammatory effects it is widely used as a coloring agent in pharmaceuticals, food, and the cosmetics industry. The separation of lipids, proteins and pigments is mainly biochemical using enzymes and microorganisms and is briefly discussed in section 3.
3 Shell waste fractionation methods for the extraction of chitin
Fractionation protocols for shell waste have been developed primarily to extract chitin from discarded shells. Several methods exist for extracting chitin from shell waste (as shown in Fig. 4), in which the chemical extraction method is commonly employed industrially (Industrial method), whereas there are biological extraction procedures based on enzymes and microorganisms (Biological methods). Additionally, ionic liquid and deep eutectic solvent-based solvent extraction procedures have emerged recently (Solvent extraction methods). | ||
| Fig. 4 Methods of shell waste fractionation. | ||
3.1 Industrial method
Industrially, chitin extraction from shell waste follows a well-established chemical method starting from demineralization followed by deproteinization, and decolouration steps.75 The demineralization step is conducted after the drying and powdering of waste shells. Mineral acids like hydrochloric acid (HCl), sulphuric acid (H2SO4), nitric acid (HNO3) and even acetic acid or formic acid are used for demineralization. However, commonly, HCl is used for the treatment, in a concentration ranging from 0.2 to 2 N.30 The time and temperature conditions for extracting chitin from shell waste can vary depending on the type of shells and the desired quality of the chitin. It usually takes a few hours to a couple of days, and the temperature can range from room temperature to around 100 °C. During the acid treatment, the calcium carbonate and other minerals are removed: (CaCO3 + 2HCl → CaCl2 + H2O + CO2↑).Deproteinization is typically performed by alkaline treatment using 0.1 to 2 M concentration of NaOH at temperatures of 50 to 100 °C.76 Proteins and lipids are hydrolysed and removed from the shell waste in this step. The process of base treatment not only removes proteins but also causes the chitin to undergo hydrolysis, resulting in a decrease in its molecular weight and partial deacetylation. After the deproteinization process, residual NaOH is removed by repeated washing with water and oven dried to obtain the chitin. Sometimes, to remove the pigments and to get colorless chitin for commercial applications oxidative bleaching is carried out using H2O2 or NaOCl. The depigmentation step can be carried out before the demineralization to extract valuable pigments like astaxanthin. Even though this method is widely used for commercial processes, the procedure is costly, destructive, and not environmentally friendly. Since it involves corrosive mineral acids and bases it requires corrosion-resistant equipment, and a huge amount of wastewater is generated from the neutralization process which causes extra treatment expenses. These factors account for the high price of chitin in the market. The need for a green and cost-effective method for shell fractionation resulted in many novel strategies, namely, solvent-extraction methods and bioprocessing methods.
3.2 Biological method
Biological extraction of chitin involves the use of enzymes and microorganisms for the isolation of chitin. Biological methods for extracting chitin typically involve two processes: deproteination using enzymes and microorganism-mediated fermentation. Proteolytic enzymes like proteases, which are found in plants, microbes, and animals, are used for deproteinization of shell waste.30 These protease enzymes (e.g. pepsin, papain, trypsin, devolvase etc.) can remove proteins from shells with minimal depolymerization and deacetylation during the extraction of chitin.77 Enzymatic deproteinization of shrimp shells after demineralization has yielded chitin and protein hydrolysate.76 Deproteinization can be conducted later or before the demineralization using crude enzymes as well as pure enzymes. Crude proteases can be obtained from bacteria and fish viscera. Even though enzymatic methods offer high-quality products and remain an eco-friendly process, they are costly, and the overall efficiency is lower than that of chemical processes.78 Around 5 to 10% of residual proteins are found associated with isolated chitin. Additional alkali treatment can carry out the purity enhancement of the isolated chitin fraction.The fermentation method can be utilized effectively by introducing specific microbial strains and endogenous microbes for deproteinization instead of using high-cost enzymes.75 The fermentation process combines the processes of deproteinization and demineralization into a single step. As a result of fermentation, a water-soluble liquor enriched with Ca2+ and protein hydrolysates is produced, which can be utilized as a supplement in animal feed. It should be noted that the efficiency of deproteinization and demineralization of shrimp shells can vary substantially based on the specific types of microorganism employed and the conditions of fermentation.9
There are two types of fermentation process, lactic acid-mediated fermentation and non-lactic acid-mediated fermentation. Lactic acid fermentation of shell waste uses Lactobacillus sp. bacterial strains which are capable of producing proteases and lactic acid during the fermentation process.79 Glucose is converted to lactic acid, which causes a lowering of the overall pH of the feed. This acidic environment helps to inhibit the development of harmful microorganisms. Several factors affect the efficiency of the lactic acid fermentation process. These include the amount and composition of the microbial inoculum, initial pH, change in pH throughout fermentation, concentration and source of carbon, duration, and temperature of fermentation.80 In non-lactic acid-based fermentation processes, various fungi and bacterial species such as Bacillus, Pseudomonas, and Aspergillus etc. have been employed. Among these, the protease-producing bacteria Pseudomonas aeruginosa demonstrated the highest deproteinization efficiency (74.8%) and demineralization efficiency (78.5%).81
3.3 Solvent extraction methods
Even though chitin cannot be solubilized in normal solvents, ionic liquids (ILs)82 in the category of alkyl imidazolium halides or acetates or its acid functionalized derivatives and deep eutectic solvents (DES) (e.g., choline chloride-urea (CCU), choline chloride-thiourea and betaine hydrochloride-urea etc.) can dissolve chitin.83 The literature extensively describes the dissolution of various biopolymers, such as cellulose, lignin, keratin, and others, in several ionic liquids. These studies provide detailed information on the capability of ionic liquids to effectively dissolve these biopolymers.84 The strong interaction of anions in the ionic liquids with the hydroxyl group of the chitin helps to reduce the extent of hydrogen bonding in the structure and aids the dissolution of chitin. This dissolution ability of ILs has been utilized for the extraction of chitin in shell waste. The use of 1-ethyl-3-methylimidazolium acetate ([Emim]OAc) was reported for chitin extraction from shrimp shells. For 0.4 g shell waste with 10 g IL, loading under microwave heating for 2 minutes extracted chitin with high purity and molecular weight.85 Along with chitin, the dissolution of minerals and other components in the shell waste was also observed. Introducing a further step of demineralization by employing citric acid was found to be effective in reducing the mineral contamination in chitin extraction by ILs.86 Recently in situ extraction of chitin was reported using 1-butyl-3-methylimidazolium hydrogen sulfate ([Bmim]HSO4) acidic ionic liquid. This [Bmim]HSO4 acidic ionic liquid facilitates the extraction of chitin and effectively separates it from the protein–mineral matrix simultaneously. This innovative approach offers a more streamlined and efficient method for chitin extraction.87 Acidic and natural DES systems have been found to be effective in the dissolution and extraction of superior quality chitin from raw shell waste effectively.88,89 Extraction methods based on mechanochemistry where the solid-state milling of shell waste with solid acids is followed by aging are emerging as novel and sustainable methods nowadays.90As we discussed earlier, the methods intended to extract chitin from shell waste involve demineralization using acids, in which minerals are often decomposed to carbon dioxide and metal oxide or metal hydroxides, which means that they cannot be used as is. Similarly, base-aided deproteination causes the denaturation of proteins and they cannot be isolated successfully. Hence, the traditional chemical methods of shell waste fraction can recover only a single component of it. However, the simultaneous extraction of protein, chitin, minerals, and pigments is highly desirable.
Biological extraction methods using protease enzymes have been successful in recovering the protein part in the form of protein hydrolyzate along with the recovery of chitin, astaxanthin and minerals.91,92Recombinant aspartic proteases P6281 (protease A) and saccharopepsin (protease B) showed high rates of deproteinization and could yield 40–71% of protein portions with an EAA (essential amino acid) content of 45–49%.91 The extraction of minerals, particularly CaCO3, is highly desirable as they can serve as a reliable component in food and pharmaceutical applications because of their biological origin. The extraction of minerals from crustacean shell waste is not as popular as the mineral extraction from eggshells or the shells of mollusks. However similar methods can be adopted for mineral recovery from crustacean shell wastes. A typical method involves acid digestion of the shell waste followed by Na2CO3 treatment to precipitate out the CaCO3.93
4 Conversion of chitin biomass to chemicals
Chitin, being the bearer of naturally occurring nitrogen in its structure along with carbon, hydrogen, and oxygen, holds the potential for the production of biobased chemicals. Chitosan, the deacetylated chitin derivative, is more suitable for chemical transformation since it has enhanced solubility and less crystallinity than chitin. Hence, both chitin and chitosan are considered as substrates for chemical transformations. There are different classes of chemicals derived from the valorization of chitin and its derivatives. Examples of chitin biomass-derived chemicals explored so far are given in Fig. 5. As the monomers of chitin and chitosan are the amino-sugars, viz., N-acetyl-D-glucosamine (NAG) and D-glucosamine (GA), they are the first set of products that can be made from chitin or chitosan. In further cascade reactions, they can be converted to different chemicals. For example, the hydrogenation of amino sugars leads to the formation of amino or amide polyols, and dehydration reactions lead to various nitrogen heterocycles and nitrogen-containing furan derivatives or even furan derivatives without nitrogen. Oxidation leads to the formation of amino acids as well as acids without nitrogen. The following sections provide a comprehensive discussion on each of these transformations.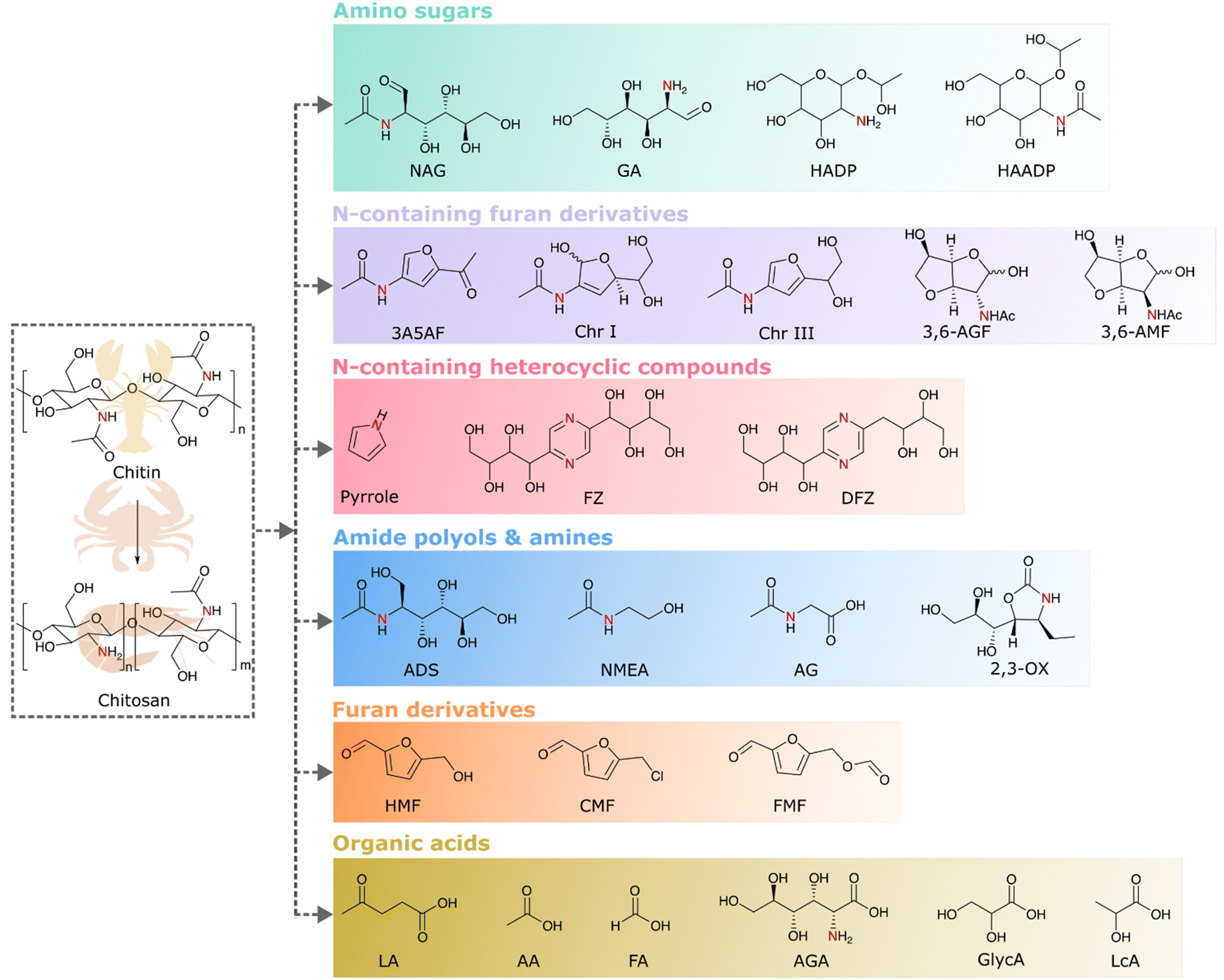 | ||
| Fig. 5 Different classes of chemicals derived from chitin biomass valorization. | ||
4.1 Conversion of chitin to nitrogen-containing chemicals
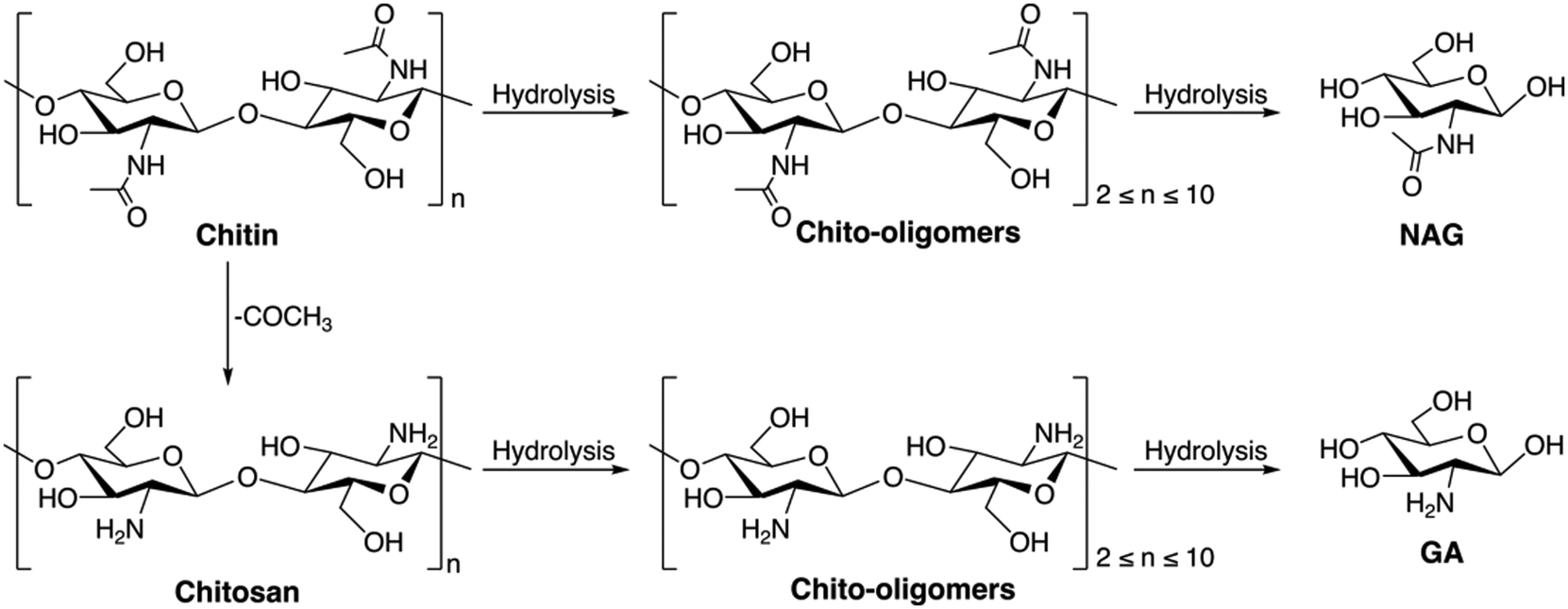 | ||
| Scheme 1 Reaction pathway for chitin biomass hydrolysis to chito-oligomers and amino sugars. | ||
The acid-catalyzed hydrolysis of chitin and chitosan mostly involves the use of homogeneous acid catalysts like mineral acids in either water or ionic liquid media. The hydrolysis process for both chitin and chitosan comprises two important steps: the breakdown of the glycosidic bonds that link the NAG units and the deacetylation of the acetamido group within the NAG units. The rate of acid-catalyzed hydrolysis of the glycosidic linkage in chitin has been observed to be 2–3 times faster than that in chitosan.105,106 The probable reason is that the acetamido group at C2 helps in stabilizing the oxo carbonium ion transition state and accelerates the cleavage of the glycosidic bond. Meanwhile the protonated amino group in the C2 position of the GA unit hinders the proton attack on the glycosidic oxygen atom. The use of HCl (6.0 wt%) promoted the hydrolysis of chitosan in 1-butyl-3-methylimidazolium chloride ([Bmim]Cl) ionic liquid media resulting in a mixture of total reducing sugars (TRS) with a total yield of 60% within 7 hours at 100 °C.107 The H2SO4-catalysed liquefaction of chitin in ethylene glycol (EG) medium has been reported by the same group; at 165 °C within 90 minutes, 75% of chitin was liquefied by using 8 wt% of acid. Hydroxyethyl-2-amino-2-deoxyhexopyranoside (HADP) and hydroxyethyl-2-acetamido-2-deoxyhexopyranoside (HAADP) were obtained as the major products in approximate yields of 30%.108 In an attempt to avoid the use of corrosive mineral acids, sulfonic acid functionalized imidazolium ILs have been used for the microwave-assisted hydrolysis of chitosan109 where the IL 1-(3-sulfonic acid) propylpyridinium hydrogen sulfate ([C3SO3Py]HSO4) showed the best activity in DMSO because of its higher Brønsted acidity. The highest yield of 90% of TRS was obtained within 2 minutes at 640 W microwave power. Although these processes allow direct hydrolysis of chitin under mild reaction conditions, the use of strong mineral acids or other homogeneous alternatives is not desirable as they are corrosive and require a high post-processing cost.
In this context, the ball milling process has been successfully used to reduce the use of large amounts of mineral acids and ionic liquids, as mechanochemical forces can aid the hydrolysis process. Chitin depolymerization using a catalytic amount of H2SO4 was reported by Kobayashi et al. Ball milling of acid-impregnated chitin under solvent-free conditions yielded soluble short-chain oligomers, and further hydrolysis and methanolysis of the ball-milled (BM) sample at 170 °C for 1 hour gave 53% of NAG and 1–70% of O-methyl-N-acetylglucosamine (MeNAG) yield.110 Ning Yan et al. employed a similar strategy for the hydrolysis of chitin, where instead of a water medium, co-solvent systems were used. Various polar aprotic solvents were screened in which with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diethylene glycol diethyl ether (DGDE)/water co-solvent system along with sulfuric acid, an 80% yield of glucosamine was achieved at 175 °C in 1 h from ball-milled chitin.111 The depolymerization and functionalization of ball-milled chitin and chitin from shrimp shells were achieved during formic acid-mediated chitin liquefaction. The total yield of monomeric products obtained was 60% at 100 °C in 12 h.112 Upon longer reaction times, the monomeric products underwent dehydration, resulting in the formation of 5-(formyloxymethyl)furfural (FMF) as the final product.
:1 diethylene glycol diethyl ether (DGDE)/water co-solvent system along with sulfuric acid, an 80% yield of glucosamine was achieved at 175 °C in 1 h from ball-milled chitin.111 The depolymerization and functionalization of ball-milled chitin and chitin from shrimp shells were achieved during formic acid-mediated chitin liquefaction. The total yield of monomeric products obtained was 60% at 100 °C in 12 h.112 Upon longer reaction times, the monomeric products underwent dehydration, resulting in the formation of 5-(formyloxymethyl)furfural (FMF) as the final product.
Another way to reduce the use of mineral acids was using lithium halide molten salt hydrate (MSH) systems due to their greater ability to disrupt the hydrogen bonding network of chitin.113 The use of only 40 mM HCl along with 60 wt% LiBr AMSH resulted in the formation of 71.5% NAG at 120 °C in 30 min. The higher acidity of the catalyst, the higher nucleophilicity of Br− anions and the interaction of Li+ cations with acetamide oxygen helps in breaking the glycosidic bonds without losing the acetamido groups and in turn increases the swelling and dissolution of the chitin. Other successful MSH systems constitute the use of 50% CaCl2 along with 1a 0% ZnBr2 mixture which resulted in a 67% yield of NAG at 120 °C in a 1 h reaction time. However, the chitin was aged in H2SO4 in this case.114
In an attempt to completely get rid of mineral acids, the same group studied use of zeolites in the lithium halide salt hydrate-mediated hydrolysis of chitin.115 Among various zeolites used, both commercial and synthesized SAPO-34 (CHA-type zeolite system) zeolite was found to be a suitable catalyst and gave an NAG yield up to 60%. The authors proposed that the Li+ ions exchange with H+ ions during the reaction and hence the hydrolysis reaction does not happen inside the pores of the catalyst and the released proton hydrolyzed the chitin dissolved in the LiBr molten salt hydrolysate. While the catalytic activity was not related to the zeolite morphology, the higher Brønsted acidity of the SAPO-34 had the key role to play. Meanwhile, the low concentration of Lewis acid sites avoided the formation of undesirable side products.
Carbon-based solid catalysts possess higher stability compared with zeolite catalysts. A glucose-based carbon solid acid catalyst (GCSA) was employed for the hydrolysis of chitosan successfully, with a 98.1% yield of GA at 110 °C in 6 h. The presence of the amino groups in chitosan possesses challenges for solid acid catalysts as they can poison the acid sites. The authors overcame the problem by acidification of the chitosan amino group prior to the reaction. Along with higher activity the catalyst was found to be stable up to 6 catalytic cycles.116 Recently Kobayashi et al. overcame the use of any mineral acids and solvents by employing the mechanochemical hydrolysis of chitin using air-oxidized carbon as a catalyst (AC-Air).117 The weak acid sites induced by air oxidation of carbon in the form of carboxylic acid and hydroxyl groups were found to be responsible for the selective formation (94%) of chito-oligosaccharides. The solid carbon catalyst was able to hydrolyze chitin in a non-aqueous state and when compared with mineral acids and other solid acid catalysts, the selectivity of oligomers was higher in case of carbon catalyst.
Even though acid-catalyzed depolymerization is most favorable for chitin, the base-catalyzed depolymerization of chitin and chitosan has also been explored. Chebotok and co-workers studied the effects of bases on the depolymerization of chitin and chitosan.118 The hydrolysis was performed in various conditions such as in solutions of NaOH, KOH, NaCl, NaI, and KI, and also in air, under O2, and under N2. It was observed that the changes in molecular weight of chitin during its hydrolysis were essentially similar. It was found that neither hydroxide ions nor oxygen initiates the cleavage of the glycosidic bonds, but the excess water present in the system is playing this role. The H+ ions formed by the dissociation of water (even trace amounts) caused the formation of carbenium ions which act as electrophiles in the reaction. Yan et al. studied the base-catalyzed mechano-chemical conversion of chitin and raw shrimp shell waste into low molecular weight chitosan.119 Chitin was ball milled along with different bases such as NaOH, KOH, LiOH, Ba(OH)2, and Ca(OH)2. It was observed that both NaOH and LiOH are effective in depolymerization as since Li+ and Na+ are small, they can easily coordinate to the C1 and C4 oxygens. However, NaOH is more effective in doing both deacetylation and depolymerization. A certain base concentration is necessary, but higher amounts of base cause the absorption of water which hinders the mechanical process. The amount of base used does not significantly affect the conversion of chitin, but notable improvements in conversion have been observed by optimizing ball milling parameters such as the number of balls, mill-cycles, etc. Chitosan obtained from raw shell waste had more than 90% purity and this process enabled a degree of deacetylation and a molecular weight in the range of 40–83% and 1–13 kDa, respectively.
The overall analysis of the conversion of chitin/chitosan to monomeric products shows that conventional acid and base systems are not very selective in forming the monomers, and moreover they often require extreme reaction conditions. At the same time the incorporation of ball milling or aging techniques was found to be very effective along with acid–base catalysts. The use of carbon-based catalysts results in an improved yield and selectivity for monomeric products. The combination of MSH and acid catalyst systems gives better yields of products. Further improvements in the process can be expected when heterogeneous catalyst systems are explored. The literature reports on the conversion of chitin biomass to chito-oligomers and amino sugars are tabulated in Table 1.
| Substrate | Catalyst | Reaction conditions | Product (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | ||||
| a MW: microwave heating. | ||||||
| Chitosan | HCl | [Bmim]Cl | 100 | 7 | TRS (60) | 107 |
| Chitin | H2SO4 | EG | 165 | 1.5 | HADP + HAADP (30) | 108 |
| Chitosan | [C3SO3Py]HSO4 | DMSO | 640 W (MWa) | 0.03 | TRS (90) | 109 |
| Ball milled chitin | H2SO4 | Water | 170 | 1 | NAG (53) | 110 |
| Methanol | 190 | 1 | MeNAG (70) | |||
| Ball milled chitin | H2SO4 | DGDE/water (4:1 v/v) |
175 | 1 | GA (80) | 111 |
| Ball milled chitin | Formic acid | Formic acid | 100 | 12 | Monomeric products (60) | 112 |
| Chitin | LiBr + HCl | Water | 120 | 0.5 | NAG (71.5) | 113 |
| Acid aged chitin | CaCl2 | Water | 120 | 1 | NAG (51) | 114 |
| CaCl2 + ZnBr2 | NAG (66.8) | |||||
| Chitin | LiBr + SAPO-34 | Water | 130 | 2 | NAG (61) | 115 |
| Chitosan | GCSA | HCl | 110 | 6 | GA (98.1) | 116 |
| Ball milled chitin | AC-Air | — | — | 48 | Oligomers (66) NAG (8.3) | 117 |
| Chitin | NaOH (50%) | Water | 150 | 0.25 | Chitosan (DD, 65%) | 118 |
| Ball milled chitin | NaOH | — | — | 3 | Chitosan (DD, 83%) | 119 |
4.1.2.1 Synthesis of 3-acetamido-5-acetylfuran (3A5AF). The utilization of cellulosic biomass for producing furan derivatives such as HMF and furfural has been extensively researched. Similarly, researchers have investigated the chemical conversion of chitin and chitin-derived compounds like GA and NAG to obtain nitrogen-containing furan derivatives, which are more valuable substitutes compared with those derived from cellulose valorization. 3-Acetamido-5-acetylfuran (3A5AF) is the first reported N-containing furan compound from animal biomass sources. It was first obtained as a pyrolysis product of NAG with a yield of 2% along with 3-acetamidofuran (5%) and acetamidoacetaldehyde (3%).120 Later in the thermal degradation of NAG at 200 °C various degradation products were obtained and 3A5AF was identified as the major compound, along with 2-acetylfuran, 3-acetamidofuran, pyrazine, pyridine, ethylpyrazine, methylpyrazine, 2-ethyl-6-methylpyrazine, 2,3-dimethylpyrazine, and acetamide.121
The use of imidazolium ILs for the conversion of glucose and fructose to HMF is known. Drover et al. were pioneers in reporting the chemical conversion of NAG to 3A5AF using ILs. The inherent acidity of imidazolium ionic liquids and the effect of chloride counter-anions aid the dehydration process. They achieved a 25.5% yield of 3A5AF by subjecting NAG to microwave heating at 180 °C for 3 minutes in the presence of [Bmim]Cl.122 The freely bound chloride ions had a positive effect on the reaction and the addition of NaCl could make only a 38.3% yield of 3A5AF. Meanwhile, the use of boric acid (H3BO3) as an additive at 180 °C increased the yield up to 60% during a 1 hour reaction. The Lewis acidity of the H3BO3 along with the formation of a borate–hexose complex accelerated the dehydration process; however due to the presence of the N-acetyl group in NAG, higher amounts of the additive were required.122 Furthermore, they tried the combination of NaCl and boric acid in a ratio 2:1 in the presence of dimethylacetamide (DMA) as the solvent under microwave heating at 220 °C for 15 minutes. The yield of 3A5AF could be maximized to 62% by using of 4 equivalents of NaCl.123 The formation of 3A5AF directly from chitin was reported by Yan et al., where they screened 6 organic solvents and 27 additives, including organic/inorganic acids, metal chlorides, bases, and heteropolyacids for the reaction. At 215 °C for 1 h, a 7.5% yield of 3A5AF was obtained with 50% chitin conversion by using N-methyl-2-pyrrolidone (NMP) as the solvent and H3BO3, LiCl and HCl as additives.124 In addition to 3A5AF, several other products, including levoglucosenone, acetic acid, 4-(acetylamino)-1,3-benzenediol, and humins, were also identified in the reaction mixture, which offers insights into the reaction network. A mechanistic exploration through NMR studies highlights the formation of a boron complex intermediate, explaining boric acid's role. Kinetic analysis underscores the importance of disrupting hydrogen bonding in the chitin crystalline region for improved yields. A probable mechanism for the formation of 3A5AF from chitin is depicted in Scheme 2.
 | ||
| Scheme 2 Plausible pathway for the formation of 3A5AF from chitin. | ||
Subsequently, their research extended to the conversion of chitin and NAG to 3A5AF, involving the testing of 10 different ionic liquids (ILs) and 25 various additives. Replacing NMP with IL resulted in improved yields while lowering the required reaction temperature. In the presence of [Bmim]Cl, along with H3BO3 and HCl as additives, they achieved a 6.2% product yield at 180 °C in just 1 hour.125 This work emphasized the importance of using additives and mineral acids in combination with ILs to enhance chitin conversion. The crystalline nature of chitin posed a challenge in its conversion, prompting the exploration of different pre-treatment methods, including ball milling, steam explosion, alkaline treatment, phosphoric acid treatment, and dissolution/reprecipitation using ionic liquids.126 Ball milling in dry conditions was found to be effective in reducing the crystallinity and H-bonding in chitin; ball-milled chitin yielded 28% 3A5AF, when boric acid and HCl were used as additives and [Bmim]Cl as the solvent at 180 °C and 1 h reaction time.126
While ILs proved to be efficient in this transformation, their higher cost is a notable concern. Therefore, several efforts have been made to render them more cost-effective and tailored to specific tasks. Wang et al. studied the amino acid ionic liquid (AAIL) catalyzed conversion of NAG to 3A5AF. Among them, glycine chloride ionic liquid, without any additive, yielded 43.2% 3A5AF in just 10 minutes at 200 °C. When CaCl2 was introduced as an additive, the yield of 3A5AF increased to 52.6%, with improved conversion.127 The AAIL proved to be reusable, maintaining yields ranging from 43.22% to 36.59% over eight cycles. Zang et al. utilized pyridinium-based ionic liquids to produce 3A5AF from NAG. They achieved a 37.5% yield of 3A5AF with the 1-carboxymethyl pyridinium chloride ([CMPy]Cl) ionic liquid catalyst. This yield was further enhanced to 67.37% at 180 °C in 20 minutes by incorporating B2O3 and CaCl2 as additives. However, this system was applicable to chitin monomer only. Additionally, ethanolamine ionic liquids were employed for NAG conversion, resulting in a 62% yield using triethanolamine hydrochloride (TEA·HCl) at 170 °C within 20 minutes.128 This catalyst proved to be recyclable for five reaction runs. In another study Zang and co-workers tried a series of quaternary ammonium ionic liquids for the conversion of NAG to 3A5AF. Using pyrazine hydrochloride ([Pyz]Cl) in combination with boric acid and CaCl2 as additives in DMA at 190 °C for 60 minutes, they achieved a remarkable (69.5%) yield of 3A5AF.129 The catalyst, [Pyz]Cl, exhibits strong acidity, and when heated, it ionizes to release protons, facilitating the hydrolysis of NAG. This aligns with the known impact of ionic liquid acidity on NAG degradation. The acidic nature of [Pyz]Cl allows H+ and Cl− ions to disrupt the original intramolecular hydrogen bonds in NAG, enhancing the degradation process. In contrast, substituted pyrazinyl ionic liquids show weaker degradation effects, with methyl-substituted ionic liquids exhibiting limited catalytic efficiency due to steric hindrance and hindered interaction with NAG. However, the catalytic activity of [Pyz]Cl was poor for the chitin degradation.
Deep eutectic solvents (DESs), similar to ionic liquids, have gained prominence for their eco-friendly nature. They are formed by mixing hydrogen-bond donors and acceptors, resulting in solutions with lower melting points than their components. DESs offer advantages like easy preparation, low toxicity, and cost-effectiveness. They prove to be effective as catalysts and solvents in biorefining processes for biomass and its derivatives.130,131 Zhang and his team introduced an innovative method for converting NAG and chitin into 3A5AF using a DES based on choline chloride and citric acid (CCCA).132 They explored a range of biobased DESs, incorporating choline chloride and various acids like citric, succinic, malonic, malic, and lactic acids, along with urea and glycerol. By introducing CaCl2·2H2O as an additive along with CCCA, they achieved a 47 mol% yield of 3A5AF from NAG at 210 °C in 20 minutes, with a 1:2 NAG to CCCA ratio. The addition of H3BO3 also improved yields when used as a catalyst. The catalyst proved to be reusable for at least 5 cycles, maintaining a 30 mol% yield in the fifth cycle. Notably, the study also accomplished the direct conversion of chitin to 3A5AF using the same system, achieving a maximum yield of 5 mol% with chitin concentrations of 1 wt% at 210 °C in 120 minutes. Furthermore, they successfully isolated 99% pure 3A5AF from the crude product using a simple extraction followed by concentration, decolorization, crystallization, and drying processes.
Previously mentioned methods have commonly required high temperatures exceeding 180 °C, N-containing aprotic solvents, and chloride additives. However, Padovan et al. recently introduced an alternative approach involving the dehydration of NAG using AlCl3·6H2O and N,N-dimethylformamide (DMF) solvent at a notably lower temperature of 120 °C. This marks a significant reduction in the required temperature for NAG conversion via conventional heating.133 They could achieve a 30% yield of 3A5AF within 30 minutes and also presented a straightforward procedure for obtaining high-purity (98%) 3A5AF through column chromatography with an isolated yield of 18%. Zhang and Chen explored NH4Cl as an economical catalyst for converting NAG to 3A5AF. In the presence of LiCl and DMF, they achieved a 43% product yield at 160 °C in 5 minutes.134 NH4Cl outperformed other chloride compounds tested as a catalyst. Notably, it surpassed a recent study using AlCl3·6H2O. The catalyst maintained 94% residual activity through five reaction runs. Scaling up the reaction to 200 mL demonstrated consistent results, and a purification protocol similar to that of Padovan et al. enabled the isolation of 99% pure 3A5AF with an isolated yield of 30%. Another study using MgCl2·6H2O achieved a 41.6% yield of 3A5AF from NAG, at 180 °C in 1 hour with B2O3 as an additive. They evaluated various N-containing polar aprotic solvents, with NMP demonstrating superior performance. Interestingly, under lower temperatures (120 °C), MoCl5 outperformed MgCl2·6H2O.135
The analysis of the literature highlights the effectiveness of ionic liquids and Lewis acid catalysts in the formation of 3A5AF from NAG. However, these methods typically require higher temperatures and N-containing aprotic solvents. The use of chloride salts and boric acid as additives plays a pivotal role in the reaction. Direct conversion of chitin to 3A5AF remains challenging due to chitin's structural rigidity. Pre-treatment methods like ball-milling have proved effective but necessitate the use of mineral acids. Addressing the commercial feasibility issue with expensive ILs, the utilization of biobased ILs, deep eutectic solvents (DES), and cost-effective Lewis acids shows potential. However, N-containing solvents are often unavoidable. Hence, these studies underscore the need for a heterogeneous catalytic system together with suitable pretreatment methods for chitin conversion. It is significant to tune the catalyst's acidic properties to accommodate chitin's recalcitrant nature and preserve the amino groups during conversion. The successful isolation and purification of 3A5AF on a lab scale holds promise for further valuable transformations into N-containing chemicals. Table 2 provides an overview of literature reports on the conversion of chitin and NAG to 3A5AF.
| Substrate | Catalyst | Reaction conditions | 3A5AF (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (min) | ||||
| a MW: microwave heating. | ||||||
| NAG | [Bmim]Cl | Water | 180 (MWa) | 3 | 25.5 | 122 |
| NAG | H3BO3 + NaCl | DMA | 220 (MW) | 15 | 58 | 123 |
| Chitin | H3BO3 + LiCl + HCl | NMP | 215 | 60 | 7.5 | 124 |
| Chitin (8 wt%) | [Bmim]Cl + H3BO3 + HCl | [Bmim]Cl | 180 | 60 | 6.2 | 125 |
| NAG | [Bmim]Cl + H3BO3 | EtOAc | 180 (MWa) | 3 | 32 | |
| NAG | [Bmim]HSO4 + NaCl | 19.7 | ||||
| Ball milled chitin | H3BO3 + HCl | [Bmim]Cl | 180 | 60 | 28 | 126 |
| NAG | [Gly]Cl + CaCl2 | DMAc | 200 | 10 | 52.6 | 127 |
| NAG | TEA·HCl | 170 | 20 | 62 | 128 | |
| NAG | [Pyz]Cl + B2O3 + CaCl2 | DMAc | 190 | 60 | 69.5 | 129 |
| NAG | CCCA + CaCl2·2H2O | DMAc | 210 | 20 | 47 | 132 |
| Chitin (1 wt%) | 120 | 5 | ||||
| NAG | AlCl3·6H2O | DMF | 120 | 30 | 30 | 133 |
| NAG | NH4Cl + LiCl | DMF | 160 | 5 | 43 | 134 |
| NAG | MgCl2·6H2O + B2O3 | NMP | 180 | 60 | 41.6 | 135 |
| NAG | [CMPy]Cl + B2O3 + CaCl2 | NMP | 180 | 20 | 67.4 | 136 |
The potential of 3A5AF as an important organic compound and building block for the synthesis of other valuable chemicals has been explored by several groups. 3-Aminofuran is a molecule with unique substitution and its chemical transformations provide synthetic scope for novel heterocycles. Moreover, this is the only way of sustainably producing N-heterocycles. The hydrolysis of amido groups and reduction of acetyl groups in 3A5AF have resulted in 2-acetyl-4 aminofuran and 3-acetamido-5-(1-hydroxyethyl)furan.137 Sperry et al. have studied the synthesis scopes for 3-amidofuran from 3A5AF by various ring and keto group functionalizations of 3A5AF.138 Proximicins are excellent chemotherapeutic drug leads. Sperry and co-workers have attempted the synthesis of proximicin A, from chitin-derived 3A5AF. The synthetic steps were found to be greener and more sustainable than the conventional route.139 The oxidative ring expansion of 3A5AF to 2-amino sugars like N-acetyl-L-rednose (RedNAc) was reported with good yield and enantiopurity.140 Deacetylation of RedNAc gives L-rednose. Natural antibiotics such as anthracycline and angucycline contain L-rednose. The compound 3A5AF undergoes Piancatelli rearrangement, resulting in the formation of a 3-aminocyclopentenone. This 3-aminocyclopentenone can be easily transformed into diverse 4-aminocyclopentanones and 4-aminocyclopentene-1,3-diones. These compounds have significant utility in the synthesis of various nitrogen-containing heterocycles.141 The reaction between 3A5AF and aliphatic ketones resulted in the formation of a dihydrodifuropyridine and it is an important heteroaromatic scaffold.142 The chemical transformations of 3A5AF are schematically presented in Scheme 3.
 | ||
| Scheme 3 Potential chemical transformation of 3A5AF. | ||
4.1.2.2 Synthesis of chromogens and bicyclic 3,6-anhydro sugars. Derivatives of NAG such as Chromogen I (2-acetamido-2,3-dideoxy-D-erythro-hex-2-enofuranose), Chromogen III (3-acetamido-5-(1′,2′-dihydroxyethyl)furan), 3,6-AGF (2-acetamido-3,6-anhydro-2-deoxy-D-glucofuranose), and 3,6-AMF (2-acetamido-3,6-anhydro-2-deoxy-D-mannofuranose) are having potent biological applications and could be used in medicine and the food industry as additives.143 Chromogen I (Chr I) was reported as a major product obtained from the Morgan–Elson reaction initially, and later the alkaline and borate treatment of N-acetyl-D-mannosamine (NAM) and NAG has been found to give Chromogen I and III.144 Ogata and co-workers investigated the synthesis of 3,6-anhydrohexofuranose from NAG.145 The reaction was carried out using 100 mM NAG solution taken in 250 mL borate buffer (0.4 M, pH 7) and this solution was incubated for 2 h at 100 °C; the reaction mixture was then analyzed using a charcoal–Celite column with a linear gradient of ethanol. 3,6-AGF and 3,6-AMF were formed at 10.2% and 9.8% respectively. The 3,6-anhydrohexofuranoses were then converted to furanodictines using isovaleryl chloride in a single step. Furanodictines represent the pioneer examples of amino sugars possessing an attractive bicyclic bis-tetrahydrofurofuran framework. They exhibit neuronal differentiation activity and can be used as model structures in the invention of novel nerve-rejuvenation drugs.143 The intricate structure, combined with their potent biological activity, renders them highly desirable targets for total synthesis.146 The formation of 3,6-anhydrohexofuranoses from NAG and the structures of furanodictines are given in Scheme 4.
 | ||
| Scheme 4 Probable reaction pathway for the formation of Chromogen I, Chromogen III, 3A5AF and 3,6-anhyrohexofuranoses form NAG. | ||
High-temperature water was used as a dehydration medium for the conversion of NAG to chromogens, and is noteworthy for being non-catalytic and greener. This approach takes advantage of the higher ion product constant of water in the temperature range of 180–220 °C compared with ambient temperatures, which aids in the efficient dehydration of NAG. Osada and co-workers tried the dehydration of NAG, and they were able to obtain Chr I (23%) and Chromogen III (Chr III) (23.1%) in the presence of high-temperature water at 120–220 °C and 250 bar pressure within 39 seconds.147 In similar conditions the dehydration of a chitin dimer (N,N′-diacetylchitobiose) resulted in a poor yield of chromogens.148 NAG can exist in both its furan ring and open-chain forms when dissolved in water. The epimerisation of NAG to NAM is possible via keto–enol tautomerization. Dehydration predominantly occurs from the open-chain (keto) form, facilitated by the electron-withdrawing properties of the N-acetyl group, which aids in the removal of a proton (H-2). The subsequent ring closure of the dehydrated open chain leads to the formation of Chr I, which can undergo further dehydration to yield Chr III. Finally, the dehydration of Chr III produces 3A5AF. A probable mechanism for the formation of Chr I, Chr III and 3A5AF from NAG and NAM is given in Scheme 4.
An extended work on the non-catalytic conversion of NAG in high-temperature water by Osada, et al. has reported 37% and 34.5% of Chr I and III respectively at 180–280 °C and 250 bar in 5–34 seconds. 3A5AF was also obtained in less than 1% yield from the dehydration of Chr III.149 The dehydration of Chr III proved to be challenging in the absence of a catalyst, resulting in the lower yield of 3A5AF. The effect of acidic and basic catalysts was studied under similar reaction conditions. Acid catalysts were found to suppress the formation of Chr I while promoting the dehydration of Chr I to Chr III. On the other hand, base catalysts exhibited a negative effect on the reactions. The same research group also explored the direct conversion of chitin to Chr I, using a similar approach. The reaction took place at higher temperatures, specifically between 290 and 390 °C, under a pressure of 250 bar, and over a longer duration of 180 minutes.150 Despite their efforts, the yield of Chr I from chitin was only 2.6%, and the conversion of chitin to chromogens was found to be relatively insignificant. This observation suggests that the non-catalytic conversion of chitin and the associated dehydration reactions are sluggish processes.
In summary, chromogens and 3,6-anhydro sugars are rare N-furan derivatives derived from the conversion of chitin or NAG. So far, there are no reported catalytic systems for this conversion, apart from the non-catalytic systems, as mentioned earlier. The mechanism of NAG conversion to these products indicates that designing an effective catalyst poses a significant challenge, as excessive acidity can lead to the formation of 3A5AF or undesirable products. To successfully advance the application of these compounds, proper methods for separation and purification must be devised, especially for Chr I, in order to enable their further transformation into important molecules like furanodictines. A compilation of reports on 3,6-anhydrosugar and chromogen synthesis is given in Table 3.
| Substrate | Reaction conditions | Product (% yield) | Ref. | |||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (s) | Pressure (bar) | |||
| NAG | Borate buffer (pH 7) | 100 | 2 | — | 3,6-AGF (10.2), 3,6-AMF (9.8) | 145 |
| NAG | High-temperature water | 220 | 39 | 250 | Chr I (23), Chr III (23.1) | 147 |
| NAG | High-temperature water | 280 | 34 | 250 | Chr I (37), Chr III (34.5) | 149 |
| Chitin | High-temperature water | 390 | 3600 | 250 | Chr I (2.6) | 150 |
These compounds are naturally produced through the Maillard reaction involving reducing sugars and ammonia-containing compounds. Synthetic pathways for the formation of these compounds from monosaccharides (glucose/fructose) and polysaccharides (cellobiose/inulin) have been established, employing ammonium salts as catalysts in a weak acid aqueous solution. However, these routes require additional nitrogen resources, which does not comply with green chemistry aspects.
There have been few efforts to make these compounds from GA condensation. In the presence of methanol and sodium methoxide at 70 °C, glucosamine hydrochloride (GA·HCl) and mannosamine hydrochloride (MA·HCl) were converted to FZ within 4 hours, yielding 24.8% and 39.4% respectively.153 Candiano et al. tried GA condensation in the presence of lysine in water under simulated physiological conditions [100 mM lysine was incubated with 100 mM GA in phosphate buffer (0.02 M, pH 7.4) for 1 week at 37 °C in the dark] and the condensation product formed was FZ (70% yield).154 This reaction represents the browning process of amino acids and proteins in systems that resemble human blood serum. It also serves as a fundamental tool for studying the circulating and structural proteins in diabetes mellitus and their browning and aging.154 A unique method for producing DFZ was described, involving a condensation reaction of GA·HCl in the presence of phenylboronic acid and NaOH. Remarkably, this approach yielded a 96% of DFZ after 2 hours of reaction. Comparable results were achieved when using disodium tetraborate, sodium metaborate, or a solution of boric acid in NaOH.155
Ionic liquid aiding the conversion of GA·HCl was also studied by several groups. The self-condensation of GA·HCl catalysed by an ethanolic solution of a basic ionic liquid, 1-butyl-3-methylimidazolium hydroxide ([Bmim]OH), was reported. The IL was used as both catalyst and solvent, and a mixture of FZ and DFZ (49%) was obtained at 120 °C in 3 h in a DMSO-IL system.152 Changing the IL to 1-ethyl-3-methylimidazolium acetate ([Emim]OAc) could give a total yield of 37% FZ and DFZ at 100 °C in 1.5 hours.156 The effectiveness of these ionic liquids stemmed from their strong hydrogen bonding capabilities with amino and hydroxyl groups, facilitating the activation of GA and promoting the dehydration reaction to form the desired products. A detailed mechanism for the formation of FZ and DFZ from GA is presented in Scheme 5. The bifunctional nature of the IL has been anticipated in DFT calculations. It was found that the acidity of the cationic component within the IL plays a role in catalyzing the dehydration process, while the basic counter-anion assists in the dehydrogenation step. The formation of an intermediate dihydrofructosazine and the observation of a lower thermodynamic barrier for the formation of DFZ have been corroborated by both computational and experimental findings.157 Furthermore, the impact of various additives on the condensation of GA into FZ and DFZ was investigated. 40.2% of DFZ was obtained via a dehydration process while using the boric acid as additive.158 The dehydrogenation reaction was enhanced when the oxidizing agent used was H2O2 and the major product was FZ (25.3%). Boron coordination helped in a ring-opening and successive dehydration reaction and was favored by both thermodynamic and kinetic factors. Dehydration of the intermediate dihydrofructosazine was fundamental in controlling the complete reaction. A choline chloride (ChCl) based deep eutectic solvent system was also employed for GA condensation as an alternative to ILs. Amino acids were used as cocatalysts, among which arginine was found to be most effective. 13.5% DFZ was obtained at 100 °C and 150 minutes in a choline chloride/urea system.159 The use of arginine enhanced the yield to 30.1% while FZ was found only in traces. The hydrogen bonding interaction of arginine and the presence of the dihydrofructosazine intermediate was observed from in situ NMR studies, suggesting that the mechanism of formation of the products is similar in both cases.
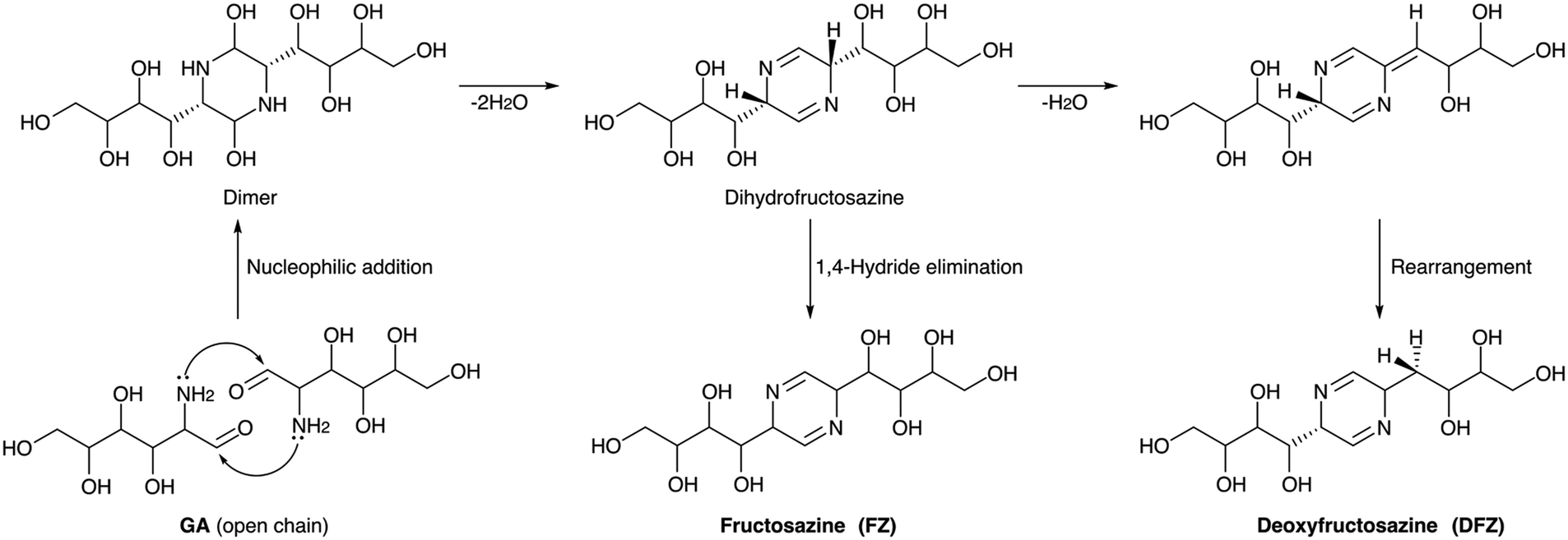 | ||
| Scheme 5 Mechanism of formation of FZ and DFZ from GA condensation. | ||
The synthesis of heterocyclic compounds from chitin is a nascent field requiring in-depth exploration. Investigating the diverse synthetic possibilities for various heterocycles derived from chitin and characterizing them is essential. Compounds like FZ and DFZ hold significant value, and their sustainable synthesis from chitin biomass could be a viable avenue in biomass biorefinery. The use of basic ILs in polar aprotic solvents has demonstrated the ability to produce FZ and DFZ; however challenges remain in addressing their separation and purification. The elevated cost of ILs and concerns about the stability160 of basic ILs add complexity. Separating these compounds from solvents like DMSO is problematic and possesses risks in food industry applications. The use of bio-based DES shows promises due to their tunable properties, cost-effectiveness, and scalability. However, there is a need to explore novel heterogeneous catalytic systems, and effective methods for the separation and purification of these compounds must be developed. Detailed information on the formation of heterocyclic compounds from chitin biomass can be found in Table 4.
| Substrate | Catalyst | Reaction conditions | Product (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (min) | ||||
| Chitin | CuO | NaOH | 325 | 35 | Pyrrole (6) | 151 |
| GA. HCl | NaOMe | MeOH | 70 | 240 | FZ (24.8) | 153 |
| MA·HCl | FZ (39.4) | |||||
| GA | Lysine | Water | 37 | 1 week | FZ (70) | 154 |
| GA | Phenylboronic acid + NaOH | Water | 120 | DFZ (96) | 155 | |
| GA·HCl | [Bmim]OH ethanolic solution | [(Bmim)OH] + DMSO | 120 | 180 | FZ + DFZ (49) | 152 |
| GA | [Emim]OAc | DMSO | 100 | 90 | FZ + DFZ (37) | 156 |
| GA | [Emim]OAc + B(OH)3 | DMSO | 90 | 180 | DFZ (40.2) | 158 |
| [Emim]OAc + H2O2 | DMSO | 80 | 180 | FZ (25.3) | ||
| GA | ChCl–urea + arginine | ChCl–urea | 100 | 150 | FZ (30.1) | 159 |
Yan et al. were the first to report the hydrogenation of NAG using noble metal-supported catalyst (Pt/C, Pd/C, Rh/C and Ru/C) in the presence of hydrogen.166 Ru(1 mol%)/C was found to be effective in converting NAG to 8.7% N-acetylmonoethanolamine (NMEA) under conditions of 180 °C and 40 bar H2 pressure within 1 hour. 6.1% of C4 polyols, and 71.9% of C6-nitrogen-containing polyols were also obtained. 2-Acetamido-2-deoxy-D-sorbitol (ADS) was the major product, and its selectivity was higher at lower temperatures. At high temperature a yield of C2–C4 polyols was increased due to the breaking of C6-polyols. NMEA and C4-polyols were formed in a single step via retro-aldol condensation and the use of NaOH enhanced the yields of the C2 and C4 products. According to the proposed mechanism, four parallel reaction pathways have been elucidated which involve the direct hydrogenation of NAG to ADS, NAG undergoing a retro-aldol reaction followed by hydrogenation to yield NMEA and C4-polyols, deacetylation of NAG to GA and its hydrogenation to form C6-amino sugar alcohols without further breakage to smaller polyols and lastly the dehydration of NAG and its subsequent hydrogenation to form the partially oxygenated C6-amino sugar alcohol. The proposed reaction pathways are outlined in Scheme 6. The catalyst demonstrated recyclability for at least three cycles, with minimal Ru leaching observed at only ppb levels. Reactions using chitin and chitosan as substrates revealed that at elevated temperatures (260 °C), no C6 polyols were detected. Instead, 8% liquid products, including NMEA (from chitin), MEA (from chitosan), ethylene glycol (EG), and acetic acid (AA), were observed, along with 10% gaseous products, primarily methane and carbon dioxide. The generation of gaseous products was attributed to the exceptionally high reaction temperature.
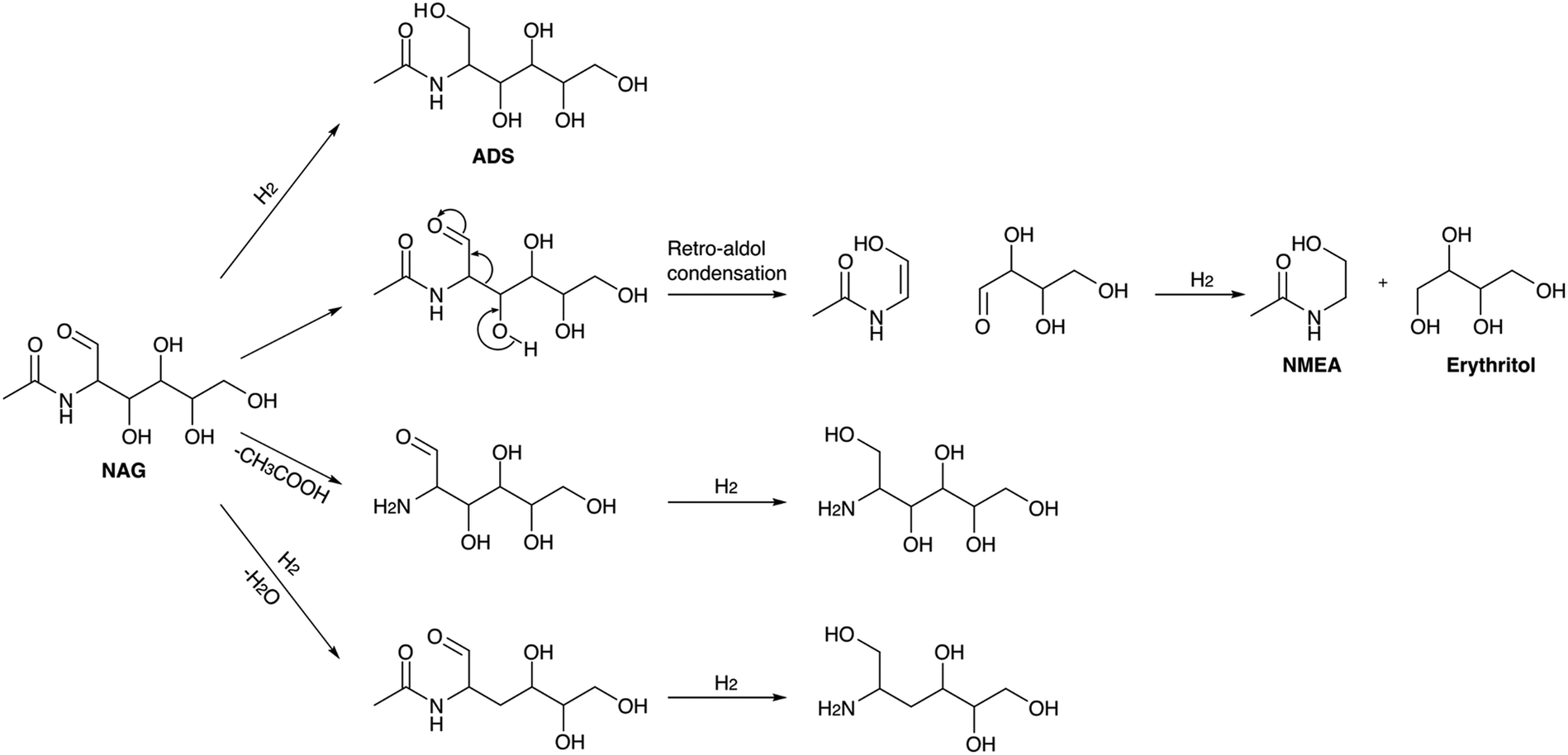 | ||
| Scheme 6 Reaction pathways for the formation of C4, C6 polyols and NMEA from NAG. | ||
Later Kobayashi and co-workers reported a hydrolytic hydrogenation procedure to obtain ADS directly from chitin.167
They ball-milled chitin with H2SO4, and the resulting chito-oligomeric mixture, along with acid, underwent one-pot hydrogenation using 5 wt% Ru/TiO2 as the catalyst at 40 bar H2 pressure. Rapid heating to 200 °C and rapid cooling led to a 25% ADS yield. Improved selectivity and yield were achieved by separating the hydrolysis and hydrogenation steps. The precise control of pH and temperature during hydrolysis (pH 2, 175 °C) and hydrogenation (pH 3–4, 120 °C) was crucial to prevent side reactions under acidic conditions and retro-aldol reactions under basic conditions caused by the Ru catalyst. In a two-step hydrolytic hydrogenation reaction with a maintained pH of 3–4, an overall hydrogenation selectivity of 85% and a 52% ADS yield were obtained. The study indicated ADS stability under the reaction conditions, suggesting that a high-temperature, low pH hydrogenation catalyst holds promise for the rapid and selective conversion of NAG to ADS. Their group also described a method to synthesize acetyl glycine (AG) and NMEA from NAG via oxidation and hydrogenation reactions.168 Using 5 wt% Ru/C as the catalyst with NaHCO3, NAG underwent a base-catalyzed retro-aldol reaction, forming 2-acetamido-acetaldehyde and erythrose. Subsequently, 2-acetamido-acetaldehyde was hydrogenated to NMEA using the metal catalyst at 40 bar H2 pressure and 120 °C in 1 hour, yielding 15% NMEA along with other products such as ADS, 2-acetamido-2-deoxymannitol (ADM), 2-acetamido-2,3-dideoxyhexitols, and acetic acid. The oxidation of the OH group on the NMEA was achieved with the same catalyst in NaHCO3 solution under 10 bar O2 pressure at 120 °C for 1 hour, resulting in 21% AG under optimized conditions. The Ru/C catalyst, with Ru species present on the carbon as hydrous RuO2 with a hydroxo, aqua, μ-hydroxo, and μ-oxo ligand environment, facilitated the oxidation process.
The hydrolysis of NMEA yields ethanolamine (ETA), a versatile component used in CO2 sequestration, dermatological formulations, cleaning agents, and surfactants. Chowdhury et al. explored the hydrogenation of NAG to NMEA using a Ni (10 wt%)/CeO2 catalyst.169 Among the various ceria supports (cubic/rod/polyhedral shaped) employed, cubic CeO2 demonstrated the highest activity under optimized conditions. With NaHCO3 as an additive, 42% NMEA was obtained at 120 °C in 8 hours and 40 bar H2 pressure. Two proposed pathways for NMEA formation involve the retro-aldol condensation of NAG to produce 2-acetamidoacetaldehyde and erythrose, followed by hydrogenation to NMEA over a Ni catalyst. Alternatively, NAG can undergo hydrogenation to ADS, and its subsequent hydrodeoxygenation with C–C cleavage leads to NMEA formation. Control experiments and intermediate compound detection confirm the reaction pathway involving the formation of ADS.
Amine synthesis is crucial for developing pharmaceuticals, materials, and agrochemicals, given the role of amines in biological molecules, drugs, polymers, and pesticides. Amine synthesis from NAG was reported by Lin and co-workers. They have carried out the deoxygenation of NAG using H2 and a Ru/C catalyst under acidic conditions.170 The highest total amine yield (47.5 mol%) was achieved at 250 °C with 33 bar H2 in 2 hours, using a 4.45 molar ratio of H3PO4 to NAG. The identified amine products included n-butylamine, pyrrolidine, 2-methylpyrrolidine, n-amylamine, 2,5-dimethylpyrrolidine, piperidine, 2-methylpiperidine, 3-methylpiperidine, and 1-ethylpiperidine, with cyclic amines prevailing. The hydrogenation environment with Ru/C likely led to the removal of oxygen atoms from the NAG as H2O and/or CO/CO2, generating methane as the primary by-product through the hydrogenation of CO. The in situ formation of acetic acid also resulted in the production of ethane. Additionally, various polyol-based side products were detected in the reaction.
Analogous to the conversion of sorbitol to isosorbide, ADS can be converted to 2-acetamide-2-deoxyisosorbide. Unlike sugar compounds, ADI's solubility is better due to its sole hydroxyl group, and it could be a potential precursor of nitrogen-containing polymers. Kobayashi et al. converted chitin-derived amino sugar alcohol (ADS) to an isosorbide analogue (ADI). NAG to ADS conversion involved a hydrogenation reaction using Ru/C at 1 bar H2 and 150 °C for 3 hours.171 ADS was then transformed into ADI through acid-catalyzed dehydration, utilizing trifluoromethanesulfonic acid (CF3SO3H) under vacuum conditions. CF3SO3H (pKa = −15, S/C = 2) produced the highest ADI yield (33%) under pressures less than 1 bar. Seeking an alternative to the corrosive CF3SO3H, they employed phosphorous acid (H3PO3) for the ADS to ADI conversion through cyclization via phosphite ethers.172 The optimal yield of ADI (25%) was achieved at 130 °C for 3 hours, with H3PO3's reducing property aiding in reducing the humin formation. DFT calculations also supported the reaction proceeding through the formation of phosphite intermediates, lowering the activation energy compared with typical acid-catalyzed dehydration condensation reactions.
Kobayashi et al. explored another possible transformation of ADS. They studied the catalytic conversion of ADS to chiral oxazolidinones namely 4-(1,2,3,4-tetrahydroxybutyl)-1,3-oxazolidin-2-one (1,2-OX) and 4-hydroxymethyl-5-(1,2,3-trihydroxypropyl)-1,3-oxazolidin-2-one (2,3-OX). The reaction was done in methanol and dimethyl carbonate (DMC) at 80 °C for 24 h using KHCO3 and it selectively formed 84% 2,3-OX (5(S)-isomer) which is a potential antibiotic agent.173 DFT calculations revealed that the inner molecular hydrogen bonds in the transition states facilitated 2,3-OX formation by reducing the energy barrier. Interestingly, the direct conversion of NAG to oxazolidinones did not occur under the same conditions. The addition of Na2B4O7 slightly modified the reaction selectivity, increasing the yield of 1,2-OX threefold. Scheme 7 shows the possible transformation of chitin-derived ADS to ADI and oxazolidinones.
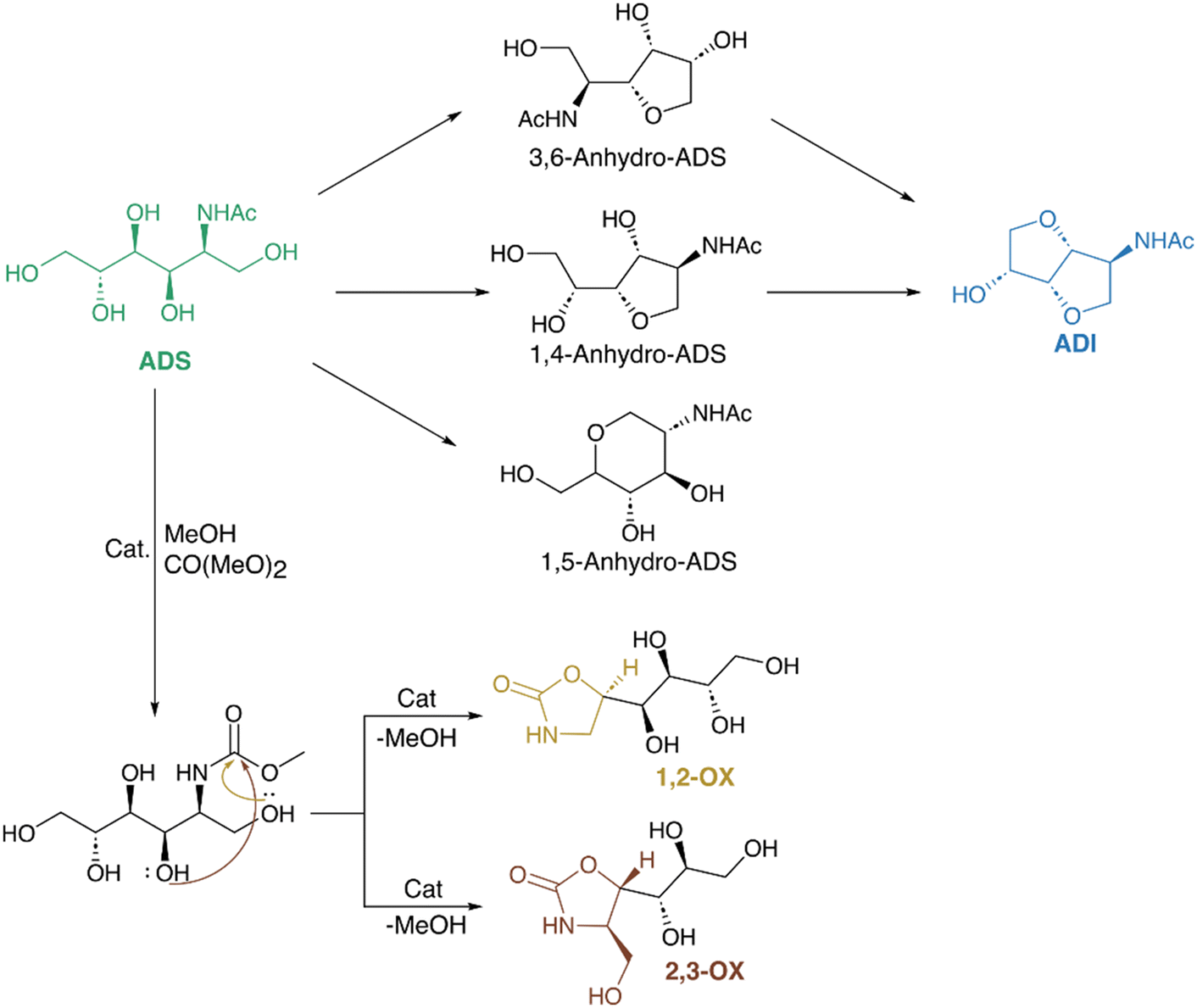 | ||
| Scheme 7 Possible transformation of chitin-derived ADS to ADI and oxazolidinones. | ||
In summary, the hydrogenation of NAG presents a novel avenue for the synthesis of N-containing polyols and amines directly sourced from biomass. While ruthenium-based supported metal catalysts demonstrate superior performance, they all operate under a high hydrogen pressure. Directly hydrogenating chitin proves challenging due to the need for the precise tuning of hydrolysis and hydrogenation conditions to maximize the product yield, making single-step conversion difficult. Exploring low-cost metal catalysts is an alternative, but their selective transformation into specific products remains unexplored. The subsequent upgrading of products, such as ADS to ADI and oxazolidinones, shows potential, with further investigations needed to explore their potential transformations and application. The literature summary of conversion of chitin and chitin-derived chemicals to amide polyols and amines has been listed in Table 5.
| Substrate | Catalyst | Reaction conditions | Product (% yield) | Ref. | |||
|---|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | Pressure (bar) | ||||
| a Chitin ball milled with H2SO4. b Hydrolysis temperature. c Hydrogenation temperature. | |||||||
| NAG | Ru(1)/C | Water | 180 | 1 | H2(40) | C2–C4 polyol (6.1), C6-polyols with N (71.9), NMEA (8.7) | 166 |
| Chitina | Ru(5)/TiO2 | Water | 175b, 120c | 2 | H2(40) | ADS (52) | 167 |
| NAG | Ru(5)/C + NaHCO3 | Water | 120 | 1 | H2(40) | NMEA (15) | 168 |
| 120 | 1 | O2(10) | AG (21) | ||||
| NAG | Ni(10)/CeO2 | NaHCO3 | 120 | 8 | H2(40) | NMEA (42) | 169 |
| NAG | Ru/C | H3PO4 | 250 | 2 | H2(40) | Amines (47.5) | 170 |
| ADS | Ru/C + CF3SO3H | Water | 150 | 3 | H2(1) | ADI (33) | 171 |
| ADS | H3PO3 | 130 | 3 | — | ADI (25) | 172 | |
| ADS | KHCO3 | MeOH + DMC | 80 | 24 | — | 2,3-OX (84), 1,2-OX (6.9) | 173 |
4.2 Conversion of chitin to non-nitrogen chemicals
:2 v/v) at 150 °C in 3 hours, resulting in a yield of 45% for CMF and 29% for LA.175 Cellulose exhibited a more favorable transformation, producing 85% CMF. This discrepancy underscored the inherent challenge associated with chitin conversion, attributed to its structural rigidity. Furthermore, CMF displayed facile conversion into HMF through hydrolysis. When subjected to reactions with ethanol and hydrogen, CMF yielded 5-(ethoxymethyl)furfural and 5-methylfurfural, respectively. In a separate HCl-catalyzed conversion of chitosan, microwave irradiation at 200 °C for 30 minutes resulted in a modest 2.2% HMF yield.176 A low-temperature hydrothermal process involving dilute H2SO4 at 174 °C for 36.9 minutes achieved a more substantial 12.1% HMF yield.177 Notably, under similar conditions, glucosamine (GA) produced only a 1.8% HMF yield in 5 minutes. Levulinic acid (LA), a recurring byproduct of HMF synthesis, was commonly observed in conjunction with HMF formation. Ultrasound-assisted chitosan degradation, utilizing a 0.5% (v/v) acetic acid solution, yielded 12.1% HMF.103 Additionally, sonication of chitosan at 40 °C for 30 minutes resulted in a concentrated soluble fraction of chitosan.
The use of weak acids such as formic acid was also employed for the chitin conversion. During the liquefaction of chitin mediated by formic acid, the formation of 5-(formyloxymethyl)furfural (FMF) was observed with a yield of up to 35% after an extended reaction time of 168 hours at 80 °C.112 The hydroxyl group undergoes formylation, resulting in the formation of soluble polymeric products. These polymers subsequently undergo hydrolysis, leading to the generation of both monomeric products and rehydrated products. Examining GA conversion in the presence of 0.1 M methanesulfonic acid (MSA) at 160 °C for 40 minutes produced a 2.3% HMF yield, accompanied by LA and formic acid (FA).178 A parallel study on chitosan conversion with MSA at 200 °C for 15 minutes resulted in a more substantial 15% HMF yield, employing 2% (w/w) chitosan and 0.2 M MSA as catalyst.179 Mineral Brønsted acids, while providing ease of operation, often cause by-product formation and non-selective transformation, necessitate costly post-treatment, and their inherent corrosiveness that can contribute to reactor part corrosion. In light of these drawbacks, researchers have explored alternative acid catalysts, such as Lewis acidic catalysts including metal chlorides and ionic liquids, for chitin biomass conversion. Table 6 summarizes reports on the synthesis of HMF through mineral and organic acid catalyzed transformations of chitin biomass. Various metal chloride systems were utilized as catalysts, leveraging their Lewis acidity, in the conversion of chitin biomass. Hydrolysis of chitosan using SnCl4·5H2O at 200 °C for 30 minutes under microwave heating was found to produce HMF.176 In this study, metal-containing catalysts were found to be effective among all those screened. Lower SnCl4·5H2O concentrations produced 10 wt% HMF, while higher concentrations yielded 21.6% LA in chitosan hydrolysis. Chitin hydrolysis under similar conditions resulted in 11.5–12.6% LA, and without catalysts or conventional heating, no HMF or LA formed. GA was treated in similar conditions to explain the mechanism of hydrolysis, and under optimised conditions 32 wt% of LA was obtained without HMF formation. The coordination of amino groups with a metal center or proton aided the cleavage of glycosidic bonds in the chitosan to yield glucosamine which on subsequent dehydration and deamination forms HMF.
| Substrate | Catalyst | Reaction conditions | Product (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | ||||
| a MW: microwave heating. b US: ultrasound. | ||||||
| Chitin | HCl | DCE | 150 | 3 | CMF (45) | 175 |
| Chitosan | HCl | Water | 200 (MWa) | 0.5 | HMF (2.2) | 176 |
| Chitosan | H2SO4 | Water | 174 | 0.6 | HMF (12.1) | 177 |
| GA | 0.08 | HMF (1.8) | ||||
| Chitosan | CH3COOH | Water | 40 (USb) | 0.5 | HMF (12.1) | 103 |
| Chitin | HCOOH | HCOOH | 80 | 168 | FMF (35) | 112 |
| GA | 0.1 M MSA | Water | 160 | 0.67 | HMF (2.3) | 178 |
| Chitosan | 0.2 M MSA | Water | 200 | 0.25 | HMF (15) | 179 |
Further rehydration by two molecules of water produces LA according to the proposed mechanism (Scheme 8). SnCl4 often forms SnO2 and HCl in aqueous solution. The reaction carried out using both SnO2 and HCl together produced 27.4 wt% LA from chitosan. They have also compared the effect of microwave heating by keeping the same reactions under conventional heating conditions. The formation of 12.08 wt% LA was found after 9 days of conventional heating whereas there was no HMF formation observed. In another work, a 67 wt% aqueous solution of ZnCl2 was found to catalyse the hydrolysis of chitosan, GA, and NAG to HMF.180 At 120 °C in 90 minutes, 10.1% of HMF was formed from chitosan, and GA and NAG produced 21.6% and 2.8% of HMF respectively. The yield of HMF and conversion of substrate decreased with a decrease in the ZnCl2 concentration. Eight Lewis acidic metal chlorides were screened as cocatalysts, out of which H3BO3 and AlCl3 were having promotional effects in HMF formation. The complex formation of Zn2+ with amino and hydroxyl groups weakened the polymeric backbone and eased the hydrolysis step to form the monomers. The zinc-monomer complex in pyranose form isomerises into open form and then back into cyclic furanose form unlike in the case of tin chloride catalyst, and further dehydration and deamination led to the formation of HMF. HMF formation from NAG was poor, because the presence of the amino group was essential for the HMF formation.
 | ||
| Scheme 8 Mechanism for HMF and LA production from GA. | ||
Zang et al. have screened 16 metal salts in a DMSO–water biphasic system for the hydrothermal conversion of chitin biomass.181 FeCl2·4H2O showed the best catalytic activity in the conversion of GA, NAG, chitosan, and chitin to HMF. The yield of HMF obtained was 24.5, 37.9 (at 180 °C in 5 h), 26.6, and 19.3 mol% (at 190 °C in 6 h) respectively from GA, NAG, chitosan, and chitin. In Lewis acid catalyzed reactions, it has been observed that the coordination of the Cl− anions with –NH2 and –OH groups plays a key role in breaking the hydrogen bonding network present in chitosan and chitin. Also, metal cation interactions weaken the β-glycosidic bond and promote hydrolysis.
Sulfamic acid, also known as amidosulfonic acid, is considered a safer alternative to other strong acids like HCl or H2SO4 in various applications due to its lower corrosivity and higher stability. It possesses several advantageous properties, including being non-volatile, odorless, non-corrosive, moderately acidic (pKa = 1.0), non-hygroscopic, and cost-effective. The catalytic properties of sulfamic acid can be attributed to its inherent zwitterionic nature (H3N+SO3−), which arises from its tautomeric structure.182 Under the conditions of 200 °C and 2 minutes, the conversion of 3% (w/w) chitosan yielded 21.5% HMF in the presence of 0.7 M sulfamic acid.183 This marks a notable improvement over previous studies, demonstrating higher HMF yields in a shorter time frame with sulfamic acid as the catalyst. In a related study by the same group, the conversion of GA to LA was explored. Interestingly, at lower sulfamic acid concentrations, the production of HMF was reduced. Specifically, using 0.3 M sulfamic acid at 200 °C for 15 minutes resulted in 0.14 mol% HMF and 33.76 mol% LA.182
Polyoxometalates (POMs) were another group of catalysts explored for HMF synthesis. They represent a versatile group of anionic metal–oxygen clusters that encompass a broad range of early transition metals. These clusters exhibit diverse chemical properties, including redox potential, acid–base properties, and solubility in both aqueous and organic media. Remarkably, the chemical characteristics of POMs can be readily and precisely adjusted across a wide range by intentionally manipulating their composition and structure. A microwave-assisted chitin conversion using polyoxometalate (POM) clusters as a catalyst was reported recently.184 Using silicotungstic acid (H4[SiW12O40]), a 23.1% HMF yield was obtained at 200 °C within 3 minutes where the solvent system used was 67% DMSO–water. The chitin was first ball milled in the presence of H2SO4, which boosted the reduction in crystallinity and resulted in an improvement in the solubility of the chitin. Table 7 shows the literature reports on Lewis acid-catalyzed transformations of chitin biomass to HMF.
| Substrate | Catalyst | Reaction conditions | HMF (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | ||||
| a MW: microwave heating. | ||||||
| Chitosan | SnCl4·5H2O | Water | 200 (MWa) | 0.5 | 10 | 176 |
| Chitosan | ZnCl2 | Water | 120 | 1.5 | 10.1 | 180 |
| NAG | 2.8 | |||||
| GA | 21.6 | |||||
| Chitin | FeCl2·4H2O | DMSO | 190 | 6 | 19.3 | 181 |
| Chitosan | 180 | 5 | 26.6 | |||
| NAG | 37.9 | |||||
| GA | 24.5 | |||||
| GA | 0.3 M Sulfamic acid | Water | 200 | 0.25 | 0.14 | 182 |
| Chitosan | 0.7 M Sulfamic acid | Water | 200 | 0.03 | 21.5 | 183 |
| Chitin (BM with H2SO4) | H4[SiW12O40] | DMSO + water | 200 | 0.05 (MW) | 23.1 | 184 |
Heterogeneous catalysts offer advantages over homogeneous catalysts, primarily in terms of their potential for separation, recovery, and recyclability. Additionally, they tend to be environmentally friendly and do not corrode the reaction equipment. In the area of biomass conversion reactions, various solid acid–base catalysts have been explored. However, the conversion of chitin biomass to HMF using heterogeneous catalysts is a relatively unexplored area, with only a few recent studies addressing this aspect. Recently, H-β-zeolite was used in combination with acetic acid, to produce HMF from chitosan by Dandekar et al.185 Different catalysts were screened where H-β-zeolite and Amberlyst-15 were found to have the best performance along with 10% acetic acid as additive. Under the optimized reaction conditions (180 °C and 60 minutes in the presence of a 5% catalyst concentration) chitosan with molecular weight 20 kDa, NAG, and GA·HCl yielded 25.16 ± 0.75%, 12.42 ± 0.25%, and 23.78 ± 0.15% of HMF. In this system, the Lewis acidity of the solid catalyst, coupled with the Brønsted acid sites (hydronium ions) from aqueous acetic acid, facilitates the rapid isomerization of the pyranose ring into furan and its subsequent hydrolysis. The addition of acetic acid, at a concentration of 10% v/v, enhances the surface coverage of the solid acid catalyst with the substrate and product. However, using acetic acid alone resulted in a poor yield of HMF. It was observed that higher acid loading resulted in the poisoning of the H-β-zeolite catalyst and the recovered catalyst gave only 7.43% of HMF.
Later Chung et al. reported HMF production from chitosan using Nafion®50 resin in a biphasic solvent system consisting of methyl isobutyl ketone (MIBK) and water (v/v = 2).186 A 32.6 ± 4.2 mol% of reasonable yield of HMF was obtained as in the case of homogeneous acid systems at 180 °C in 2 h. The catalyst's efficacy in converting chitosan can be attributed to the interactions between the chitosan and the perfluorinated surface of the resin, coupled with its highly acidic characteristics. Unfortunately, the recyclability of the catalyst was not demonstrated. A notable challenge in this process involves the deactivation of catalytic sites (acid) due to the release of ammonia from the acetamido/amino group during the reaction. Hence the development of innovative heterogeneous catalytic systems is crucial for efficient HMF production from chitin biomass. The literature studies on the formation of HMF using heterogeneous catalysts are summarized in Table 8.
Ionic liquids are organic salts characterized by a lower melting point (<100 °C) when compared with conventional inorganic salts. In contrast to traditional organic solvents, they possess minimal vapor pressure. Additionally, they demonstrate a broad liquid temperature range, conductivity, broad electrochemical window, and exceptional dissolving power for most organic, inorganic, and organometallic substances.187 The various combinations of cation and anion enable the tuning of their properties desirable for task specific applications.188–190 Over the last two decades, ionic liquids have played a crucial role as both solvents and catalysts in the dissolution and conversion of biomass.191,192 Their extensive use extends to the transformation of lignocellulosic materials, such as cellulose, lignin,193 and hemicellulose,194 into essential platform chemicals. Researchers are actively exploring the use of ILs to depolymerize chitin, leveraging its structural similarity to cellulose. Various attempts have been made to convert chitin biomass specifically into HMF using ILs.
Zang et al. reported the conversion of chitosan to HMF under hydrothermal conditions using 5 wt% of 1-butyl-3-methylimidazolium hydrogen sulfate ([Bmim]HSO4) ionic liquid and AlCl3·6H2O (100 mol%) as co-catalyst. They could achieve 25.2 mol% of HMF in 5 h at 180 °C.195 The catalysts were recyclable, and the activity was found to be constant for the first five cycles. Chitosan was converted into HMF using different acidic ionic liquids via a hydrothermal reaction by Zang and co-workers.196 4 wt% of N-methyl imidazolium hydrogen sulfate ([MIM]HSO4) aqueous solution at 180 °C in 5 h produced 29.5 mol% and 19.3 mol% of HMF from chitosan and chitin, respectively. Among the 9 ILs screened, [MIM]HSO4 was found to be more efficient. The catalyst demonstrated recoverability through straightforward post-processing and remained recyclable for up to five runs. The IL anions’ coordination with hydroxyl and amino groups played a crucial role in disrupting the hydrogen bonding, while the IL cations weakened glycosidic bonding, facilitating monomer formation. The anticipated mechanism involves isomerization, cyclization to furan form, and subsequent dehydration, ultimately leading to HMF formation. Scheme 9 illustrates the anticipated mechanism of HMF formation facilitated by IL. Later, a series of Lewis-Brønsted acidic ionic liquids were employed for the hydrothermal synthesis of HMF from chitosan.197 A 1.25 wt% aqueous solution of the IL 1-hexyl-3-methylimidazolium hydrogen sulfate along with FeCl2 ([Hmim][HSO4]-0.5FeCl2) produced 44.1% of HMF at 180 °C in 4 h. This IL system showed remarkable activity for the first four catalytic cycles. In the DMSO–water system, NAG was successfully converted to HMF through hydrothermal synthesis at 180 °C for 6 hours. Utilizing the [Hmim]HSO4 ionic liquid, a notable 64.6 mol% yield of HMF was achieved.198 Evaluation of various imidazolium and thiazolium cation-based ILs revealed the essential role of the –HSO4− anion for higher HMF yields. Under similar conditions, GA, chitosan, and chitin produced 54.9, 34.7, and 25.7 mol% of HMF, respectively. The decreased accessibility of HSO4− anions to chitosan and chitin resulted in lower HMF yields due to reduced polymer hydrolysis. The catalyst, along with the DMSO, was recovered and reused for up to five catalytic cycles, demonstrating reasonable activity. Benzimidazole and NMP-based ionic liquids were also studied for chitosan degradation by the same group. The catalytic efficiency of the ionic liquid benzimidazolium chloride ([Hbim]Cl) was exceptional, yielding 30.8 mol% HMF in a water medium and 34.9 mol% in a 10% DMSO–water system. The addition of DMSO improved the solubility of the chitosan and minimized side-product formation. The catalyst demonstrated reusable capability for five cycles without compromising its effectiveness.
 | ||
| Scheme 9 Proposed mechanism of IL-promoted conversion of chitosan to HMF. | ||
Table 9 presents a summary of studies on the IL-catalyzed conversion of chitin and its derivatives to HMF. Notably, acidic ionic liquids have demonstrated superior performance compared with conventional acid catalysts in terms of yield, conversion, catalyst recovery, and recyclability for HMF production from chitin biomass. The optimization of ionic liquid properties, either independently or along with co-catalysts, holds potential for enhancing catalytic activity. Future endeavors in this research area should focus on conducting reactions at lower temperatures, with attention given to the specific choice of reaction solvents due to concerns about IL and product separation.
| Substrate | Catalyst | Reaction conditions | HMF (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | ||||
| Chitosan | [BMIM][HSO4] + AlCl3·6H2O | Water | 180 | 5 | 25.2 | 195 |
| Chitosan | [MIM]HSO4 | Water | 180 | 5 | 19.3 | 196 |
| Chitosan | [Hmim][HSO4]-0.5FeCl2 | Water | 180 | 4 | 44.1 | 197 |
| Chitin | [Hmim]HSO4 | Water + DMSO | 180 | 6 | 25.7 | 198 |
| Chitosan | 34.7 | |||||
| NAG | 64.6 | |||||
| GA | 54.9 | |||||
| Chitosan | [Hbim]Cl | Water + DMSO | 180 | 3 | 34.9 | 199 |
In the context of reports on mineral acid-catalyzed chitin conversion to LA, a microwave-assisted hydrothermal process using 2 M H2SO4 at 190 °C for 30 minutes resulted in a yield of 37.8 mol% of LA.201 Similarly, employing HCl under comparable conditions yielded 32.7 mol% of LA. This approach was extended to GA, NAG, and various chitosan samples, where 19.3 to 37 mol% of LA was observed with both HCl and H2SO4 within 10 to 30 minutes. In a separate study, 4 wt% of H2SO4 successfully converted GA into LA (25.3 wt%) at 188 °C in 49 minutes.202 Methanesulfonic acid (MSA) is an environmentally benign and green catalyst. Even though it is a strong acid (pKa = −1.9), it is non-oxidizing, non-foaming, less corrosive and bio-degradable compared with other mineral acids. In a hydrothermal conversion of GA at 200 °C using 0.5 M MSA, 49.9% LA and 50.8% FA was formed in 30 min.178 Under the same reaction conditions 28.2% of LA was formed from 2% chitosan when 0.2 M MSA was used.179
When Lewis acids such as SnCl4·5H2O were used in the hydrothermal conversion of chitosan at 200 °C under microwave irradiation, in 30 minutes 23.9% LA was formed and the conversion of chitin produced 11.5% LA.176 The poor yield of LA from chitin polymer is due to its structural rigidity, intensive network of hydrogen bonding and crystallinity. A very low yield of LA (0.48%) was obtained in a 0.7 M sulfamic acid catalysed hydrothermal degradation of chitosan at 200 °C within 2 min.183 At the same temperature the 0.3 M sulfamic acid catalysed hydrolysis of GA yielded 33.76 mol% of LA within 15 min. 39.03 mol% FA and 26.48 wt% humins (insoluble products) were also detected.182 Zirconium oxychloride (ZrOCl2) was able to catalyse the hydrothermal degradation of chitosan. At 200 °C and 20 min 21.3 mol% of LA was obtained from 5 wt% chitosan using 15 mol% catalyst.203 Solid acid Amberlyst-15 was able to transform GA into 36.9% LA at 180 °C in 60 minutes.204
Sulfonic acid-functionalized imidazolium ionic liquids were used for the transformation of chitosan to LA, and various anion combinations were tried by keeping the cation the same. Among them 1-methyl-3-(3-sulfopropyl)-imidazolium hydrogen sulphate ionic liquid ([C3SO3Hmim]HSO4) showed the best activity. At 170 °C and 5 h, 1 g of the catalyst was able to convert 250 mg of chitosan in water (4 g) to 49% LA.205 The presence of water played a pivotal role in the reaction, as the absence of water led to a decrease in LA yield and the formation of a significant amount of humins. Reducing the substrate amount to 50 mg increased the LA yield to 64%, indicating a dilution effect. The increased selectivity for LA was attributed to the highly acidic nature of the IL. According to the proposed mechanism, chitosan underwent quaternization and depolymerization, forming the glucosammonium salt. This salt then underwent dehydration and deamination to produce HMF. Subsequent rehydration resulted in the formation of LA. However, the catalytic activity of the IL decreased after two cycles due to a reduction in acidity caused by ammonia released during the reaction. The researchers quantified the amount of NH4+ ion in the reaction mixture using ion chromatography analysis. Remarkably, the catalyst was found to be reusable for up to five catalytic cycles when 1 equivalent of H2SO4 was added after recovery.
Liu et al. have recently reported the direct conversion of chitin to LA using the acidic ionic liquid [C3SO3Hmim]HSO4.206 The effect of the structural properties of IL on the chitin conversion and the role of the acetylamino group was investigated in detail. At 180 °C and 5 h, 56.5 mol% of LA was obtained from 3.4 wt% of chitin. A significant increase in LA yield (67 mol%) was observed when 0.7 wt% of chitin was used. [C3SO3Hmim]Cl was found to have remarkable efficiency in LA (54 mol%) formation despite its lower acidity compared with [C3SO3Hmim]HSO4. However, the catalyst's activity decreased in recycling experiments, with the LA yield reaching 34 mol% in the fifth catalytic cycle, likely due to a decline in acidity resulting from an interaction with NH3 released during the reaction. The addition of 1 equivalent of H2SO4 as a supplement maintained a consistent catalytic activity. A two-way approach to the depolymerization and formation of LA was proposed by the investigators (Scheme 10). It can be either via deacetylation followed by depolymerization to GA and LA or by direct depolymerization to NAG and its conversion to GA and LA.
 | ||
| Scheme 10 Proposed mechanism of formation of LA from chitin. | ||
In another study, they presented a highly selective one-step method for synthesizing LA from raw crab shells using [C3SO3Hmim]HSO4.207 They achieved a notable 77.9% LA yield at 180 °C in a 5 hour reaction. The acidity and hydrogen-bonding ability of the IL played a crucial role in the catalytic activity.
Monitoring the degree of acetylation at various time intervals provided insight into the reaction's mechanism, revealing that direct depolymerization and successive deacetylation or depolymerization occurring on the outer surface of the chitin while the inner crystalline structure remained inert were viable pathways. However, the catalytic activity of the IL diminished within the first three cycles due to the deamination of NAG and the CaCO3 component in crab shells. The addition of H2SO4 during recycling proved effective in restoring the activity. Theoretical investigations on chitin to LA conversion also appeared in recent literature. Tian et al. investigated the impact of –SO3H functionalized ionic liquids on chitin conversion to LA through a comprehensive analysis using DFT studies. In their research, the relative catalytic activity of the IL was predicted by calculating the LUMO–HOMO band gap of the most stable complexes of IL-NAG. Imidazolium-based ILs exhibited the highest activity, followed by pyridinium cation- and ammonium cation-based ILs. Through 1H NMR and DFT analysis, [C4SO3Hmim]HSO4 was identified as the catalyst of choice for converting chitin to LA. Under reaction conditions of 190 °C for 5 hours, utilizing 1 g of chitin with a catalyst amount of 1.5 g in 6 g of water, [C4SO3Hmim]HSO4 showed a remarkable LA yield of 74.7%.208
Table 10 summarizes literature findings on converting chitin biomass into LA. These data reveal that for LA synthesis, a catalyst with higher acidity is essential with control of acidity to minimize humin formation. MSA and acid-functionalized ionic liquids exhibited a superior performance among all the catalysts. Although ILs required longer reaction times, they proved to be effective for the direct conversion of raw shell waste. However, challenges arise due to the deactivation of acid catalysts in the presence of minerals in shell waste and the removal of acetamido groups in the form of NH3. Also, the separation and purification of products remain unresolved in the current solvent and catalytic systems. This complexity underscores the need for designing an efficient, recyclable, cost-effective, and environmentally friendly catalyst for the chitin to LA conversion process.
| Substrate | Catalyst | Reaction conditions | LA (% yield) | Ref. | ||
|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | ||||
| a MW: microwave heating. | ||||||
| Chitosan | HCl | Water | 190 (MWa) | 0.5 | 32.7 | 201 |
| H2SO4 | 37.8 | |||||
| GA | H2SO4 | Water | 188 | 0.82 | 25.3 | 202 |
| Chitin | HCl | DCE | 150 | 3 | 29 | |
| GA | 0.5 M MSA | Water | 200 | 0.5 | 49.9 | 178 |
| Chitosan | 0.2 M MSA | Water | 200 | 0.5 | 28.2 | 179 |
| Chitosan | SnCl4·5H2O | Water | 200 (MWa) | 0.5 | 23.9 | 176 |
| Chitin | 11.5 | |||||
| Chitosan | Sulfamic acid | Water | 200 | 0.25 | 33.8 | 182 |
| Chitosan | ZrOCl2 | Water | 200 | 0.3 | 21.3 | 203 |
| GA | Amberlyst-15 | Water | 180 | 1 | 36.9 | 204 |
| Chitosan | [C3SO3Hmim]HSO4 | Water | 170 | 5 | 49 | 205 |
| Chitin | [C3SO3Hmim]HSO4 | Water | 180 | 5 | 56.5 | 206 |
| [C3SO3Hmim]Cl | 54 | |||||
| Crab shell | [C3SO3Hmim]HSO4 | Water | 180 | 5 | 77.9 | 207 |
| Chitin | [C4SO3Hmim]HSO4 | Water | 190 | 5 | 74.7 | 208 |
Acetic acid (AA) is another important organic acid that can be obtained from chitin. The production of acetic acid is more practical from chitin biomass in comparison with lignocellulosic biomass. This is due to the fact that the hydrolysis of the acetamido group in chitin invariably results in the simultaneous removal of acetic acid. Yan and co-workers have demonstrated the hydrothermal oxidation of raw shrimp shells and chitin to produce AA and pyrrole in a single-step transformation.151 100 mg of chitin in 5 mL of 2 M NaOH solution was converted into 23.5% of AA without any oxidant and metal oxide additives at 300 °C in 35 min, while the use of ball milled CuO and 5 bar oxygen enhanced the AA yield up to 38.1% and 47.9% of AA was obtained from crude shrimp shell waste, under similar conditions. Along with AA, other acid products such as LA, FA, oxalic acid (OxA), glycolic acid (GlyA), and various other C4–C6 acids containing hydroxyl groups were also identified and quantified using GC-MS, GC-FID and TOC (total organic carbon) analyses. Using 13C-labeled acetamido groups in NAG, the relative ratio of AA formed from the pyranose ring and the sidechain acetamido group was determined via GC-MS. The analysis revealed that 60% of AA formation resulted from acetamido group hydrolysis, while the remaining 40% originated from pyranose ring decomposition and oxidation. The proposed mechanism involves chitin deacetylation and depolymerization to GA and AA, with subsequent deamination leading to C6-sugars. Parallel reactions were evidenced by the presence of unstable intermediates, ultimately converting into various stable organic acids. Untreated tiger shrimp shell powder, utilized under similar conditions, yielded 47.9% AA, suggesting the protein content in shrimp shells contributes to AA formation.
2-Amino-2-deoxy-D-gluconic acid or glucosaminic acid (AGA) is an α-amino acid which is having potential applications as a starting material in synthetic chemistry and used in various biomedical applications.209 GA can be converted into AGA by the oxidative cleavage of the O–C bond in the pyranose form of GA. While numerous enzymatic and microbial methods exist for this conversion, the literature reports only a limited number of chemo-catalytic approaches for the transformation of GA to AGA. In 1915 oxidation of GA to AGA was tried with HgO and H2S but the process was not well considered due to environmental concern and lack of consistency in the activity, and also the mercuric sulphide left in the reaction was not separable easily.210 Later an active charcoal-supported Pd–Bi bimetallic catalyst in the presence of molecular oxygen was found to oxidize GA to form AGA at 30 °C.211 70% of the AGA was achieved in the reaction in pure form. The mechanism was proposed to be the oxidative dehydrogenation of glucosamine hydrochloride. The role of Bi was as the co-catalyst, and it reduced the over-oxidation of the Pd surface during the reaction. The catalyst was recyclable for eight cycles. Tominaga and colleagues introduced an electrocatalytic method for oxidizing glucosamine (GA) in both basic and neutral pH environments.212 They utilized a carbon electrode modified with 2 nm-sized gold nanoparticles and a gold plate electrode. In an alkaline solution, the conversion to AGA achieved a remarkable current efficiency of 100% at −0.2 V, while in a neutral pH, AGA was detected with an approximate current efficiency of 70% at 0.4 V.
Ebitani et al. investigated the air oxidation of GA·HCl and its derivatives in a base-free environment using gold (Au) particles supported on heterogeneous basic supports.213 Various supports were examined, with Au/HT (hydrotalcite) and Au/MgO catalysts showing the best activity. At 40 °C and 3 hours under O2 flow, Au (2 wt%)/HT and Au (0.9 wt%)/MgO produced 89% and 93% of AGA, respectively. Other amino sugars such as NAG and NAM were also oxidized to yield the corresponding acids at approximately 90% under similar conditions. However, Au/HT exhibited reduced activity in successive cycles, with an agglomeration of Au particles in the spent catalyst. Conversely, the high basicity of MgO prevented Au particle growth, maintaining high catalytic activity during the first and second reuses. Furthermore, Yan et al. developed a two-step process for the transformation of chitosan salts to form AGA using an Au/MgO catalyst sytem.214 Initially, mineral/organic acid-treated chitosan underwent hydrolysis with the solid acid catalyst Amberlyst-15. Subsequently, oxidation using Au/MgO yielded AGA. A monomer yield of 58% was achieved when chitosan treated with H2SO4 was utilized as the substrate at 160 °C in 2 hours. However, the presence of polymeric material and humins during hydrolysis impeded the oxidation catalyst's activity, limiting the AGA yield to 17%. The introduction of a detoxification method using activated carbon for the hydrolyzed product, followed by oxidation, significantly increased the AGA yield to 63% under 5 bar O2 pressure at 40 °C in 3 hours.
Recently Chen et al. reported the conversion of chitin-derived NAG into organic acids. The reaction employed 0.1 M NAG and 0.5 M NaOH at 35 °C for 48 hours, utilizing various oxidants.215 Under ambient air, the total yield of organic acid products reached 72.3%, encompassing acetic acid (AA), formic acid (FA), and glyceric acid (GlycA). Product selectivity was influenced by the choice of oxidants, with a predominant formation of FA (57.1%) observed when hydrogen peroxide (H2O2) was utilized. Conversely, the use of oxygen favored the production of AA and GlycA, constituting 41.2% of the major products. Following this, various metal oxide catalysts were assessed for the direct oxidation of chitin itself, with MgO exhibiting the most efficient catalytic activity, resulting in a significant yield of 10.8% lactic acid (LcA) as the major product at 260 °C in a 6 hour reaction.216 The conversion of GA and NAG under similar reaction conditions showed LcA yields of 23.3% and 7.7%, respectively. While the catalyst demonstrated recyclability for the initial three runs, the first recycling revealed 15% Mg leaching. The accelerated leaching of Mg was attributed to the formation of acidic products.
Table 11 summarizes literature reports on organic acids obtained from chitin biomass oxidation reactions. Given the limited number of studies conducted on the oxidation of chitin biomass, there exists a significant scope for exploring various catalytic systems in this reaction.
| Substrate | Catalyst | Reaction conditions | Product (% yield) | Ref. | |||
|---|---|---|---|---|---|---|---|
| Solvent | Temperature (°C) | Time (h) | O2 (bar) | ||||
| Chitin | CuO | NaOH | 300 | 0.58 | 5 | AA (38.1) | 151 |
| Shrimp shell | AA (47.9) | ||||||
| GA | Pd–Bi/C + KHCO3 | Water | 30 | 6 | Bubbled O2 | AGA (70) | 211 |
| GA | Au on carbon electrode | Alkaline water | — | — | AGA (100) | 212 | |
| GA | Au(0.9)/MgO | Water | 40 | 3 | 1 | AGA (93) | 213 |
| Au(2)/HT | AGA (89) | ||||||
| Chitosan–H2SO4 | Amberlyst-15 + Au/MgO | Water | 40 | 3 | 5 | AGA (63) | 214 |
| NAG | NaOH + H2O2 | Water | 35 | 48 | — | FA (57.1) | 215 |
| NaOH | Water | 35 | 48 | 5 | AA + GlycA (41.2) | ||
| Chitin | MgO | Water | 260 | 6 | — | LcA (10.8) | 216 |
| GA | LcA (23.3) | ||||||
| NAG | LcA (7.7) | ||||||
The incorporation of a base or basic support is crucial in oxidation reactions and designing a suitable catalyst for achieving the selective formation of acids, such as AGA and LcA, is highly desirable. Additionally, addressing the challenge of separating the various acids formed during the reaction underscores the importance of research focused on selectivity.
5 SWOT analysis
A SWOT analysis for the conversion of chitin to value-added chemicals can help to identify the strengths, weaknesses, opportunities, and threats associated with this process. As the drive toward eco-friendly and sustainable solutions gains momentum, understanding the potential and constraints of this process becomes vital. A SWOT analysis diagram for the production of chemicals from chitin is given in Fig. 6. | ||
| Fig. 6 SWOT analysis for the production of chemicals from chitin biomass. | ||
5.1 Strengths
Chitin is the second most abundant biopolymer, making it an excellent resource for the production of renewable chemicals. Chitin is primarily derived from crustacean shells, a byproduct of fisheries and marine aquaculture, ensuring its continued production in the future. The production of nitrogen-containing chemicals from renewable resources, which does not require ammonia synthesized from the energetically inefficient Haber process, is expected to play a vital role in achieving net zero emissions and sustainable development goals. In this context, chitin, as the only nitrogen-containing renewable resource, is invaluable as a feedstock. Furthermore, the use of shell waste would decrease environmental pollution and waste management costs.5.2 Weaknesses
The main weakness is that basic research on chitin as a feedstock is still in its early stages. The major advances in chitin chemistry have been made by drawing parallels with cellulose chemistry due to structural similarities. The general principles of chitin chemistry are not well understood. Furthermore, there is no techno-economic analysis done so far in the field of production of chemicals from the catalytic conversion of chitin biomass to determine the industrial viability of the associated process. Another issue is that commercial chitin extraction methods are not environmentally friendly, as most of the processes focus solely on chitin extraction, while the other components of shell waste are not properly utilized.5.3 Opportunities
The abovementioned weakness creates significant opportunities. More systematic research is needed in the field to determine a chitin extraction process that focuses on utilizing the protein, lipid, and mineral components of shell waste in addition to chitin, as the co-production of all the components would reduce costs. Understanding the reaction mechanism provides ample opportunities for rationally designing catalytic active sites for chitin conversion to chemicals. Because chitin-derived chemicals have a wide range of applications in the polymer and pharmaceutical industries, also the new end products can be compared with existing related products. For example, the properties of polymers that can be synthesized from chitin-derived monomers should be investigated, as this will aid in identifying important monomers and providing researchers with a meaningful goal for the development of catalytic reactions.5.4 Threats
Despite these opportunities, several threats loom over the process. The most serious threat to the process is likely to be the cost competitiveness of the end products. The main competition would come from existing similar products. Furthermore, the industrialization of chitin-based end products may necessitate a different infrastructure than what is currently in place, which could serve as a bottleneck. On the other hand, shell waste supply chain disruptions may occur as a result of rapid climate change, overfishing, or other causes.Overall, the establishment of a chitin-based bio-refinery would require more careful fundamental research and careful governance and policies.
6 Analysis of products in chitin chemistry
As we have discussed in the previous sections there are a reasonable number of studies on the valorization of chitin and chitosan to yield various chemicals with or without nitrogen. However, it is observed from the literature that the reported analytical methods can detect and quantify either oligomer or monomer or furan derivatives. The polarity of amino sugars and sugar derivatives are very close to each other and mostly soluble in water, and that is why they are hard to separate. Moreover, due to lack of optimum analytical conditions, a few products were not separated, and some were undetected. From the literature, it was seen that chitin/chitosan-derived compounds can be separated and quantified by various analytical methods like chromatographic (gas chromatography (GC),111,186,217 column chromatography, high-performance liquid chromatography (HPLC)181,186,218), chemical (Fehling's test, gravimetric analysis)219 and colorimetric methods (DNS method).181,195,218,220In our work, firstly, the properties (e.g., kinetic diameter, melting point, boiling point, degradation temperature, solubility, molecular weight) of chitin-derived compounds (mainly acid-catalysed depolymerization and degradation products of chitin biomass such as GA, NAG and HMF) were examined, which is very essential before starting the analytical method development.221 The study suggested that the compounds may not be separated only on the basis of their polarities since they are mostly polar in nature, which was evidenced from their higher solubility in water compared with methanol. The size-based separation of these compounds is not possible as they have very minimal differences in their size (kinetic diameter of GA, NAG and HMF is 6.954 Å, 7.460 Å, and 6.186 Å respectively). The amino sugar compounds GA and NAG cannot be analyzed by gas chromatography as they degrade at the higher temperatures used to vaporize the samples. A TLC (thin layer chromatography) experiment was conducted to understand the trend in polarity of these compounds and Rf (retention factor) values. It was realized that the polarity trend follows the order GA > NAG > HMF. Considering all these factors, we have presumed that HPLC is the easiest, most convenient, rapid, and sensitive technique to separate chitin-derived chemicals which are both water soluble and organic. Researchers have extensively employed HPLC as a primary analytical technique for chitin/chitosan valorization chemistry. However, there is a lack of prior reports on the simultaneous separation and quantification of all the compounds derived from chitin/chitosan during the reaction. This is primarily due to the limited research conducted in selecting suitable analytical techniques capable of effectively separating and quantifying these compounds.108,186,221
After a thorough literature survey on the analysis of chitin/chitosan-derived products, our group has worked towards the investigation and optimization of various HPLC parameters which can simultaneously separate and quantify various chitin-derived compounds.221 Initially, the mixture of GA, NAG, and HMF was analyzed using a previously reported HPLC method employed in chitin chemistry for HMF analysis. In this analysis, an C18 column (non-polar), a mobile phase consisting of methanol and water (2.3/7.7 v/v), and a UV detector set at a wavelength of 284 nm were used.181 However, only HMF was detected, and no peaks corresponding to GA and NAG were observed. From the UV-Visible absorption study of the compounds, we have observed that the λmax (maximum absorption wavelength) value for amino sugars was at 195 nm. This indicates that the UV wavelength on the detector should be set at 195 nm to effectively detect these compounds. In the previously reported literature, a UV detector with a wavelength of 284 nm was employed, resulting in the detection of only HMF (λmax = 284 nm). As a consequence, compounds that do not exhibit UV activity below 284 nm could not be analyzed using this method.
In our study, setting the UV wavelength to 195 nm allowed the detection of all the compounds, based on the understanding that higher energy is associated with lower wavelengths, effectively exciting electrons in all the compounds. Another challenge that arose was the selection of the VWD wavelength when using a methanol/water mobile phase. It was crucial to set the wavelength above 205 nm due to methanol's UV cut-off at that value. According to the literature, the measurement wavelength should be higher than the UV cut-off of the mobile phase to prevent any contribution from the mobile phase in the measured absorbance.222
To address the limitations posed by methanol's UV cut-off, we replaced it with acetonitrile, which has a lower UV cut-off (<190 nm). The use of acetonitrile led to less difference in the retention times of the compounds compared with methanol, owing to the different polarity index of acetonitrile. Our analysis was conducted with a C18 column, utilizing an acetonitrile/water ratio of 2.3/7.7 v/v and a VWD wavelength of 195 nm. To further enhance the compound separation, we introduced a more polar solvent, specifically water, into the mobile phase. By varying the acetonitrile-to-water ratio, we explored different combinations. After evaluating all the possible ratios, we found that a 1/9 v/v ratio of acetonitrile to water provided the best separation of GA, NAG, and HMF. This optimized condition allowed us to achieve improved resolution and differentiation between the compounds.
During our investigation, we also examined the effect of column length on compound separation. It was determined that longer columns are more effective in separating compounds as they allow for a greater interaction time with the stationary phase, minimizing the chance of peak merging. However, when utilizing a non-polar C18 column for analyzing other sugars and chitin valorized products, the retention times showed only minimal differences, making it difficult to achieve distinct peaks. In an attempt to improve the separation, we opted to use a polar column (amino column) since polar compounds are expected to exhibit strong interactions with a polar stationary phase. However, the chromatogram obtained using the polar column showed peak tailing, suggesting that the compounds were strongly retained on the stationary phase due to strong interactions. This finding led us to conclude that polar columns are not suitable for separating these polar compounds, as they tend to exhibit tailing and peak merging, resulting in undesirable separation. Based on our previous experimental observations, it became clear that the stationary phase should possess an optimum polarity to facilitate the rapid adsorption–desorption of the compounds on the stationary phase. Then we carried out the experiments on an ion-exchange column (monosaccharide lead (Pb2+) column). In this column the stationary phase is composed of sulfonated styrene divinyl benzene polymer with Pb2+ exchanged cations and offering medium polarity. A suitable separation was observed when water was used as the mobile phase (0.5 mL min−1 flow rate), a column temperature of 80 °C, and a VWD wavelength of 195 nm. In addition to this, other sugars and chitin-derived compounds showed clear separation with this column and the abovementioned analysis conditions. A schematic representation of the separation of various amino sugars and chitin-derived chemicals using Pb2+ is given in Fig. 7. Method validation was done by ensuring precision, day to day variations and repeatability and proved to be accurate and sensitive.
 | ||
| Fig. 7 Separation of various chitin-derived chemicals by HPLC on a Pb2+ monosaccharide column (analysis conditions: column temperature: 80 °C, mobile phase: water, flow rate: 0.5 mL min−1, VWD wavelength: 195 nm). | ||
In order to accomplish effective separation of chitin/chitosan-derived compounds, we evaluated various columns with different properties, including polarity, length, and stationary phase, together with mobile phases of varying polarity. Through our experimental study, we discovered that both the C18 (non-polar) column and the cation exchange Pb2+ column (medium polar) was suitable for the detection and separation of amino sugar monomers and dimers, other N-containing compounds, and furan derivatives. Although the non-polar C18 column exhibited better separation compared with the polar amino column, it struggled to separate sugars with similar functionality and polarity, while the cation exchange Pb2+ column was able to effectively separate most of the compounds due to its optimum polarity. A summary of the HPLC analysis parameters and separation of compounds is given in Table 12. With the developed HPLC method, we achieved separation of amino sugars and HMF within 39 minutes and observed a strong linear relationship as indicated by the regression analysis. As a result, we have successfully established a simple, quick, accurate, and precise HPLC method that enables the separation and quantification of chitin/chitosan-derived products.
| Column | Polarity, phase, (length × internal diameter in mm, matrix, particle size in μm, pH range) | Analysis conditions (mobile phase, flowrate, column temperature, VWD wavelength) | Remarks on separation [compounds (RT in minutes)] |
|---|---|---|---|
| C18 | Non-polar, reverse (250 × 4.6, octadecylsilane, 5, 2–9) | Acetonitrile:water (1:9 v/v), 0.5 mL min−1, 25 °C, 195 nm |
Good separation of GA (4.037), NAG (5.582) and HMF (9.735) but retention times of other products were merging [AA (5.983), NAG (5.582)], [GA·HCl dimer (3.994), GA (4.037), AGA (4.126)] |
| Non-polar, reverse (100 × 4.6, octadecylsilane, 5, 2–9) | Acetonitrile:water (1:9 v/v), 0.5 mL min−1, 25 °C, 195 nm |
Peaks of GA and NAG were merged as compounds got less interaction time with the stationary phase | |
| Amino | Polar, normal (250 × 4.6, silica, 2–8) | Acetonitrile:water (8:2 v/v), 0.5 mL min−1, 30 °C, 195 nm |
Tailing of peaks due to strong interaction; peaks of HMF (7.130), acetamide (7.990) and AGA (7.493) were merged due to peak tailing |
| Monosaccharide Pb2+ | Medium polar, reverse (300 × 7.8, sulfonated styrene divinyl benzene, 8, 7) | Water, 0.5 mL min−1, 80 °C, 195 nm | Very good separation of GA (11.517), NAG (15.853) and HMF (36.797). Separations of other products were also good [AGA (9.068), GA·HCl dimer (10.487), LA (19.183), AA (20.883), acetamide (22.173)] |
7 Conclusions
1. In this review, we described various methods of fractionating shell waste into different components. We have described in detail the various products obtained so far from the catalytic conversion of chitin and chitin derivatives to N-containing furan compounds, amino sugars, heterocyclic compounds such as pyrazines and pyrrole, amino acids, amines, amino sugar alcohols, and 5-hydroxymethylfurfural, levulinic acid, acetic acid, and glucosaminic acid, among others.2. Even though shell waste is inexpensive, the current fractionation methods require a higher price for high-purity chitin. New fractionation techniques that are inexpensive, environmentally friendly, and efficient must be developed to obtain chitin without compromising the product purity or resulting in the loss of other valuable components.
3. The direct transformation of chitin is difficult due to the recalcitrant structure of chitin, while the products were easily attainable from low molecular weight chitosan and monomeric compounds.
4. In the hydrolysis and dehydration reactions of chitin biomass, while homogeneous acid and base catalysts exhibit efficiency in achieving reasonable conversion, their drawbacks, including corrosiveness, poor recovery and reusability, and the loss of nitrogen atoms from product molecules pose significant challenges. Conversely, ionic liquid catalytic systems demonstrate promising performance in terms of product selectivity.
5. Due to the significant risk of catalytic site poisoning by ammonia during dehydration reactions, prioritizing the production of nitrogen-containing furan derivatives or other heterocycles over the synthesis of HMF and LA from chitin is recommended. This preference is based on the potential transformations of these nitrogen-containing compounds, underscoring the crucial importance of preserving the amino/acetamido group intact during the catalytic processes.
6. Given the environmentally benign nature and ease of separation, there is a strong inclination towards employing heterogeneous catalysts in chitin biomass conversion. While recent attempts have utilized heterogeneous catalysts for chitin valorization, there remains massive potential for exploration and innovation in the realm of heterogeneous catalytic systems for this purpose.
7. The vast potential of hydrogenation and oxidation reactions remains largely unexplored. There is a need for extensive research into various catalytic systems to selectively produce desired products. Additionally, the exploration of potential upgrades for hydrogenation products represents a promising avenue that has yet to be thoroughly investigated.
8. It is crucial to comprehend both the potential and constraints of chitin valorization to propel this research area into a commercially viable process. Conducting a SWOT analysis enabled us to do a comprehensive exploration of the strengths, weaknesses, opportunities, and threats associated with chitin valorization, thereby enhancing the current scope of this review.
9. Future research in this field is expected to focus on several key directions. First, there will be efforts to develop efficient methods for extracting high-purity chitin from shell waste, optimizing the process to maximize yield and minimize waste and production cost.
10. Another area of interest will be the synthesis of nitrogen-containing chemicals from chitin. Researchers may explore novel approaches and reaction pathways to convert chitin into valuable nitrogen-based compounds, such as amino acids, amines, and other nitrogen-containing derivatives.
11. Designing suitable catalysts for the chemical transformation of chitin biomass will also be a priority. Catalyst development can enable more efficient and selective reactions, leading to improved yields and reduced energy consumption in chitin valorization processes. The designing of the catalysts should also consider the structural rigidity and the features of the target chemicals.
12. Moreover, conducting a thorough examination of reaction mixtures through the utilization of suitable analytical tools should be essential. Employing advanced analytical techniques to characterize reaction intermediates, by-products, and end products will offer profound insights into the intricacies of reaction mechanisms, reaction kinetics, catalyst impact, and the optimization of reaction conditions. This acquired knowledge will play a pivotal role in enhancing the precision and optimization of chitin valorization processes in the future.
13. The separation of the products and their purification is another area which is lacking in the studies of chitin biomass valorization. Devising these protocols is mandatory for commercial viability of the end products. Developing efficient separation techniques, such as chromatography or membrane-based processes, will enable the extraction as well as purification of target compounds from complex reaction mixtures.
14. Detailed investigations are required to make this valorization process economically viable on a large scale in future.
15. Combining the process of chitin extraction from shell waste and its conversion into various value-added products is highly recommended in terms of industrial upscaling of the valorization of raw shell waste.
16. Awareness about the possibilities of shell waste applications should be passed on to the fisheries sector. Also, establishing industries on fisheries sites for the production of chitin and shell waste fractionation could be beneficial in terms of the commercial production of chitin.
17. Research efforts for making cascade processes for the direct conversion of shell waste to value-added chemicals have to be considered and the potential of this conversion process has to be acknowledged and emphasized properly to build a foundation for shell waste-based shell-biorefinery.
Author contributions
Lavanya K: conceptualization, data curation, visualization, writing – original draft, and writing – review & editing, Neha Ghosh: writing – review & editing, Paresh L Dhepe: supervision and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Lavanya K thanks CSIR, New Delhi for the research fellowship.References
- G. Velvizhi, C. Goswami, N. P. Shetti, E. Ahmad, K. Kishore Pant and T. M. Aminabhavi, Fuel, 2022, 313, 122678 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 RSC.
- P. Bhaumik and P. L. Dhepe, Catal. Rev., 2016, 58, 36–112 CrossRef CAS.
- FAO, The State of World Fisheries and Aquaculture 2022, Towards Blue Transformation, Rome, 2022, FAO, DOI:10.4060/cc0461en.
- J. P. Morris, T. Backeljau and G. Chapelle, Rev. Aquac., 2019, 11, 42–57 CrossRef.
- X. Chen and N. Yan, Catal. Surv. Asia, 2014, 18, 164–176 CrossRef CAS.
- X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- M. J. Hülsey, Green Energy Environ., 2018, 3, 318–327 CrossRef.
- A. Fukuoka and H. Kobayashi, J. Jpn. Pet. Inst., 2023, 66, 48–56 CrossRef CAS.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- N. Lucas, A. A. Athawale and C. V. Rode, Chem. Rec., 2019, 19, 1995–2021 CrossRef CAS PubMed.
- C. Schmitz, L. G. Auza, D. Koberidze, S. Rasche, R. Fischer and L. Bortesi, Mar. Drugs, 2019, 17, 452 CrossRef CAS PubMed.
- J. Dai, F. Li and X. Fu, ChemSusChem, 2020, 13, 6498–6508 CrossRef CAS PubMed.
- X. Cai, Z. Wang, Y. Ye, D. Wang, Z. Zhang, Z. Zheng, Y. Liu and S. Li, Renewable Sustainable Energy Rev., 2021, 150, 111452 CrossRef CAS.
- D. Zhou, D. Shen, W. Lu, T. Song, M. Wang, H. Feng, J. Shentu and Y. Long, Molecules, 2020, 25, 541 CrossRef CAS PubMed.
- B. Xu, Z. Du, J. Dai, R. Yang, D. Yang, X. Gu, N. Li and F. Li, Catalysts, 2022, 12, 653 CrossRef CAS.
- X. Shi, X. Ye, H. Zhong, T. Wang and F. Jin, Mol. Catal., 2021, 505, 111517 CrossRef CAS.
- N. Islam, M. Hoque and S. F. Taharat, World J. Microbiol. Biotechnol., 2023, 39, 1–17 CrossRef PubMed.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- V. Šimat, N. B. Rathod, M. Čagalj, I. Hamed and I. Generalić Mekinić, Mar. Drugs, 2022, 20, 206 CrossRef PubMed.
- M. L. Maia, C. Grosso, M. F. Barroso, A. Silva, C. Delerue-Matos and V. F. Domingues, Antioxidants, 2023, 12, 1–12 CrossRef PubMed.
- M. M. H. Al Omari, I. S. Rashid, N. A. Qinna, A. M. Jaber and A. A. Badwan, in Profiles of Drug Substances, Excipients and Related Methodology, ed. H. G. Brittain, Elsevier Inc., 1st edn, 2016, vol. 41, pp. 31–132 Search PubMed.
- G. O. Bamigboye, A. T. Nworgu, A. O. Odetoyan, M. Kareem, D. O. Enabulele and D. E. Bassey, J. Build. Eng., 2021, 34, 101864 CrossRef.
- A. Naqi, S. Siddique, H. K. Kim and J. G. Jang, Constr. Build. Mater., 2020, 230, 116973 CrossRef CAS.
- B. A. Tayeh, M. W. Hasaniyah, A. M. Zeyad and M. O. Yusuf, J. Cleaner Prod., 2019, 237, 117723 CrossRef CAS.
- U. G. Eziefula, J. C. Ezeh and B. I. Eziefula, Constr. Build. Mater., 2018, 192, 287–300 CrossRef CAS.
- Z. Pavlík, M. Záleská, M. Pavlíková, A. Pivák, A. M. Lauermannová, M. Lojka, A. Jiříčková, G. Łagód and O. Jankovský, Constr. Build. Mater., 2023, 394, 132069 CrossRef.
- M. Yadav, P. Goswami, K. Paritosh, M. Kumar, N. Pareek and V. Vivekanand, Bioresour. Bioprocess., 2019, 6, 8 CrossRef.
- D. Sahoo and P. L. Nayak, in Biopolymers, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2011, pp. 129–166 Search PubMed.
- A. C. A. Wan and B. C. U. Tai, Biotechnol. Adv., 2013, 31, 1776–1785 CrossRef CAS PubMed.
- G. Cárdenas, G. Cabrera, E. Taboada and S. P. Miranda, J. Appl. Polym. Sci., 2004, 93, 1876–1885 CrossRef.
- K. M. Varum, M. H. Ottoy and O. Smidsrod, Carbohydr. Polym., 2001, 46, 89–98 CrossRef CAS.
- M. He, X. Wang, Z. Wang, L. Chen, Y. Lu, X. Zhang, M. Li, Z. Liu, Y. Zhang, H. Xia and L. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 9126–9135 CrossRef CAS.
- S. Rejinold N, K. P. Chennazhi, H. Tamura, S. V. Nair and J. Rangasamy, ACS Appl. Mater. Interfaces, 2011, 3, 3654–3665 CrossRef CAS PubMed.
- J. Lv, X. Lv, M. Ma, D. H. Oh, Z. Jiang and X. Fu, Carbohydr. Polym., 2023, 299, 120142 CrossRef CAS PubMed.
- M. Nasrollahzadeh, M. Sajjadi, S. Iravani and R. S. Varma, Carbohydr. Polym., 2021, 251, 116986 CrossRef CAS PubMed.
- J. L. Shamshina, P. Berton and R. D. Rogers, ACS Sustainable Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- P. A. Sandford and A. Steinnes, in Water-Soluble Polymers: Synthesis, Solution Properties, and Applications—Preface, ed. S. W. Shalaby, C. L. McCormick and G. B. Butler, American Chemical Society, 1991, pp. 430–445 Search PubMed.
- N. Kulkarni, S. D. Shinde, G. S. Jadhav, D. R. Adsare, K. Rao, M. Kachhia, M. Maingle, S. P. Patil, N. Arya and B. Sahu, Bioconjugate Chem., 2021, 32, 448–465 CrossRef CAS PubMed.
- D. Sahoo, S. Sahoo, P. Mohanty, S. Sasmal and P. L. Nayak, Des. Monomers Polym., 2009, 12, 377–404 CrossRef CAS.
- Y. Tsutsumi, H. Koga, Z.-D. Qi, T. Saito and A. Isogai, Biomacromolecules, 2014, 15, 4314–4319 CrossRef CAS PubMed.
- M. Nasrollahzadeh, N. Shafiei, Z. Nezafat, N. S. Soheili Bidgoli and F. Soleimani, Carbohydr. Polym., 2020, 241, 116353 CrossRef CAS PubMed.
- Á. Molnár, Coord. Chem. Rev., 2019, 388, 126–171 CrossRef.
- W. Argüelles-Monal, J. Lizardi-Mendoza, D. Fernández-Quiroz, M. Recillas-Mota and M. Montiel-Herrera, Polymers, 2018, 10, 342 CrossRef PubMed.
- R. J. White, M. Antonietti and M. M. Titirici, J. Mater. Chem., 2009, 19, 8645–8650 RSC.
- H. Watanabe, S. Asano, S. I. Fujita, H. Yoshida and M. Arai, ACS Catal., 2015, 5, 2886–2894 CrossRef CAS.
- Z. Li, J. Liu, Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3, 839–845 CrossRef CAS.
- S. Chen, P. Qi, J. Chen and Y. Yuan, RSC Adv., 2015, 5, 31566–31574 RSC.
- J. Luo, F. Peng, H. Wang and H. Yu, Catal. Commun., 2013, 39, 44–49 CrossRef CAS.
- R. P. Rocha, A. G. Gonçalves, L. M. Pastrana-Martínez, B. C. Bordoni, O. S. G. P. Soares, J. J. M. Órfão, J. L. Faria, J. L. Figueiredo, A. M. T. Silva and M. F. R. Pereira, Catal. Today, 2015, 249, 192–198 CrossRef CAS.
- X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- R. V. Jagadeesh, H. Junge, M. Pohl, J. Radnik, A. Bru and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776–10782 CrossRef CAS PubMed.
- K. Shen, L. Chen, J. Long, W. Zhong and Y. Li, ACS Catal., 2015, 5, 5264–5271 CrossRef CAS.
- J. Zhou, L. Bao, S. Wu, W. Yang and H. Wang, Carbohydr. Polym., 2017, 173, 321–329 CrossRef CAS PubMed.
- M. Chu, Y. Zhai, N. Shang, P. Guo, C. Wang and Y. Gao, Appl. Surf. Sci., 2020, 517, 146140 CrossRef CAS.
- Y. Cao, M. Tang, M. Li, J. Deng, F. Xu, L. Xie and Y. Wang, ACS Sustainable Chem. Eng., 2017, 5, 9894–9902 CrossRef CAS.
- X. Xiao, S. H. Lim, W. Chu, Y. Liu, S. Hua Lim, W. Chu and Y. Liu, ACS Sustainable Chem. Eng., 2021, 9, 12655–12662 CrossRef CAS.
- N. Brun, S. A. Wohlgemuth, P. Osiceanu and M. M. Titirici, Green Chem., 2013, 15, 2514–2524 RSC.
- Y. Gao, X. Chen, J. Zhang and N. Yan, ChemPlusChem, 2015, 80, 1556–1564 CrossRef CAS PubMed.
- A. Primo, A. Forneli, A. Corma and H. García, ChemSusChem, 2012, 5, 2207–2214 CrossRef CAS PubMed.
- X. Sun, X. Chen, Z. Wang, X. Ai, Y. Cao and J. Zhou, ACS Appl. Energy Mater., 2022, 5, 11825–11834 CrossRef CAS.
- W. Suginta, P. Khunkaewla and A. Schulte, Chem. Rev., 2013, 113, 5458–5479 CrossRef CAS PubMed.
- R. Vinodh, Y. Sasikumar, H. J. Kim, R. Atchudan and M. Yi, J. Ind. Eng. Chem., 2021, 104, 155–171 CrossRef CAS.
- D. Kasprzak and M. Galiński, J. Power Sources, 2023, 553, 232300 CrossRef CAS.
- Y. Zhai, M. Chu, N. Shang, C. Wang, H. Wang and Y. Gao, Appl. Surf. Sci., 2020, 506, 144681 CrossRef CAS.
- K. Zhang, Q. Meng, H. Wu, T. Yuan, S. Han, J. Zhai, B. Zheng, C. Xu, W. Wu, M. He and B. Han, Green Chem., 2021, 23, 1621–1627 RSC.
- Z. Lin, J. Zou, S. Li, C. Zhang, F. Xie, B. Li and D. Ye, ACS Sustainable Chem. Eng., 2021, 9, 1568–1575 CrossRef CAS.
- D. Polidoro, D. Rodriguez-Padron, A. Perosa, R. Luque and M. Selva, Materials, 2023, 16, 575 CrossRef CAS PubMed.
- S. Ravichandran, G. Rameshkumar and A. Rosario Prince, Am.-Eurasian J. Sci. Res., 2009, 4, 191–194 CAS.
- J. Synowiecki and N. A. A. Q. Al-Khateeb, Food Chem., 2000, 68, 147–152 CrossRef CAS.
- I. Aranaz, M. Mengibar, R. Harris, I. Panos, B. Miralles, N. Acosta, G. Galed and A. Heras, Curr. Chem. Biol., 2009, 3, 203–230 CAS.
- X. Mao, N. Guo, J. Sun and C. Xue, J. Cleaner Prod., 2017, 143, 814–823 CrossRef CAS.
- W. Arbia, L. Arbia, L. Adour and A. Amrane, Food Technol. Biotechnol., 2013, 9862, 12–25 Search PubMed.
- A. Percot, C. Viton and A. Domard, Biomacromolecules, 2003, 4, 1380–1385 CrossRef CAS PubMed.
- M. C. Gortari and R. A. Hours, Electron. J. Biotechnol., 2013, 16, 1–14 Search PubMed.
- S. Kaur and G. S. Dhillon, Crit. Rev. Biotechnol., 2015, 35, 44–61 CrossRef CAS PubMed.
- R. Castro, I. Guerrero-Legarreta and R. Bórquez, Biotechnol. Rep., 2018, 20, e00287 CrossRef PubMed.
- P. Kandra, C. Murali Mohan, P. Smitha and K. P. Hemalatha, Int. J. Appl. Biol. Pharm. Technol., 2010, 1, 903–910 Search PubMed.
- O. Ghorbel-Bellaaj, I. Younes, H. Maâlej, S. Hajji and M. Nasri, Int. J. Biol. Macromol., 2012, 51, 1196–1201 CrossRef CAS PubMed.
- J. L. Shamshina, Green Chem., 2019, 21, 3974–3993 RSC.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 RSC.
- H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913–3929 RSC.
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968–997 RSC.
- T. Setoguchi, T. Kato, K. Yamamoto and J. ichi Kadokawa, Int. J. Biol. Macromol., 2012, 50, 861–864 CrossRef CAS PubMed.
- J. L. Shamshina and N. Abidi, ACS Sustainable Chem. Eng., 2022, 10, 11846–11855 CrossRef CAS.
- M. Bisht, I. P. E. Macário, M. C. Neves, J. L. Pereira, S. Pandey, R. D. Rogers, J. A. P. Coutinho and S. P. M. Ventura, ACS Sustainable Chem. Eng., 2021, 9, 16073–16081 CrossRef CAS.
- W. C. Huang, D. Zhao, N. Guo, C. Xue and X. Mao, J. Agric. Food Chem., 2018, 66, 11897–11901 CrossRef CAS PubMed.
- F. Hajiali, J. Vidal, T. Jin, L. C. De La Garza, M. Santos, G. Yang and A. Moores, ACS Sustainable Chem. Eng., 2022, 10, 11348–11357 CrossRef CAS.
- J. J. Deng, H. H. Mao, W. Fang, Z. Q. Li, D. Shi, Z. W. Li, T. Zhou and X. C. Luo, J. Cleaner Prod., 2020, 271, 122655 CrossRef CAS.
- S. S. Pattanaik, P. B. Sawant, K. A. M. Xavier, K. Dube, P. P. Srivastava, V. Dhanabalan and N. K. Chadha, Aquaculture, 2020, 515, 734594 CrossRef CAS.
- H. B. Kwon, C. W. Lee, B. S. Jun, J. Do Yun, S. Y. Weon and B. Koopman, Resour., Conserv. Recycl., 2004, 41, 75–82 CrossRef.
- L. Liu, Y. Liu, H. Shin, R. Chen, J. Li, G. Du and J. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 6149–6158 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, Cell. Mol. Life Sci., 1997, 53, 131–140 CrossRef CAS PubMed.
- V. K. Mourya, N. N. Inamdar and Y. M. Choudhari, Polym. Sci., Ser. A, 2011, 53, 583–612 CrossRef CAS.
- F. Tian, Y. Liu, K. Hu and B. Zhao, J. Mater., 2003, 8, 4709–4712 Search PubMed.
- S. Tanioka, Y. Matsui, T. Irie, T. Tanigawa, Y. Tanaka, H. Shibata, Y. Sawa and Y. Kono, Biosci. Biotechnol. Biochem., 1996, 60, 2001–2004 CrossRef CAS.
- S. Seo, J. M. King and W. Prinyawiwatkul, J. Food Sci., 2007, 72, 522–526 Search PubMed.
- W. Xia, P. Liu and J. Liu, Bioresour. Technol., 2008, 99, 6751–6762 CrossRef CAS PubMed.
- A. Ajavakom, S. Supsvetson, A. Somboot and M. Sukwattanasinitt, Carbohydr. Polym., 2012, 90, 73–77 CrossRef CAS PubMed.
- E. Savitri, Sumarno and A. Roesyadi, Model. Appl. Sci., 2015, 9, 647–653 Search PubMed.
- E. Savitri, S. R. Juliastuti, A. Handaratri, Sumarno and A. Roesyadi, Polym. Degrad. Stab., 2014, 110, 344–352 CrossRef CAS.
- G. Margoutidis, V. H. Parsons, C. S. Bottaro, N. Yan and F. M. Kerton, ACS Sustainable Chem. Eng., 2018, 6, 1662–1669 CrossRef CAS.
- V. Y. Novikov, Russ. J. Appl. Chem., 2004, 77, 484–487 CrossRef CAS.
- A. Einbu and K. M. Vårum, Biomacromolecules, 2008, 9, 1870–1875 CrossRef CAS PubMed.
- Z. Zhang, C. Li, Q. Wang and Z. K. Zhao, Carbohydr. Polym., 2009, 78, 685–689 CrossRef CAS.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- Q. Chen, W. Xiao, L. Zhou, T. Wu and Y. Wu, Polym. Degrad. Stab., 2012, 97, 49–53 CrossRef CAS.
- M. Yabushita, H. Kobayashi, K. Kuroki, S. Ito and A. Fukuoka, ChemSusChem, 2015, 8, 3760–3763 CrossRef CAS PubMed.
- J. Zhang and N. Yan, ChemCatChem, 2017, 9, 2790–2796 CrossRef CAS.
- J. Zhang and N. Yan, Green Chem., 2016, 18, 5050–5058 RSC.
- G. Gözaydin, S. Song and N. Yan, Green Chem., 2020, 22, 5096–5104 RSC.
- Y. Wang, J. Kou, X. Wang and X. Chen, Green Chem., 2023, 25, 2596–2607 RSC.
- G. Gözaydın, Q. Sun, M. Oh, S. Lee, M. Choi, Y. Liu and N. Yan, ACS Sustainable Chem. Eng., 2023, 11, 2511–2519 CrossRef.
- H. Zhang, Y. Lu, Y. Wang, X. Zhang and T. Wang, RSC Adv., 2018, 8, 5608–5613 RSC.
- H. Kobayashi, Y. Suzuki, T. Sagawa, M. Saito and A. Fukuoka, Angew. Chem., Int. Ed., 2023, 62, e202214229 CrossRef CAS PubMed.
- E. N. Chebotok, V. Y. Novikov and I. N. Konovalova, Russ. J. Appl. Chem., 2006, 79, 1162–1166 CrossRef CAS.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- R. A. Franich, S. J. Goodin and A. L. Wilkins, J. Anal. Appl. Pyrolysis, 1984, 7, 91–100 CrossRef CAS.
- J. Chen, M. Wang and C. T. Ho, J. Agric. Food Chem., 1998, 46, 3207–3209 CrossRef CAS.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- X. Chen, Y. Gao, L. Wang, H. Chen and N. Yan, ChemPlusChem, 2015, 80, 1565–1572 CrossRef CAS PubMed.
- J. Wang, H. Zang, S. Jiao, K. Wang, Z. Shang, H. Li and J. Lou, Sci. Total Environ., 2020, 710, 136293 CrossRef CAS PubMed.
- H. Zang, H. Li, S. Jiao, J. Lou, Y. Du and N. Huang, ChemistrySelect, 2021, 6, 3848–3857 CrossRef CAS.
- Y. Du, H. Zang, Y. Feng, K. Wang, Y. Lv and Z. Liu, J. Mol. Liq., 2022, 347, 117970 CrossRef CAS.
- Y.-L. Loow, E. K. New, G. H. Yang, L. Y. Ang, L. Y. W. Foo and T. Y. Wu, Cellulose, 2017, 24, 3591–3618 CrossRef CAS.
- K. T. T. Amesho, Y.-C. Lin, S. V. Mohan, S. Halder, V. K. Ponnusamy and S.-R. Jhang, Environ. Chem. Lett., 2023, 21, 183–230 CrossRef CAS.
- C. Wu, C. Wang, A. Zhang, K. Chen, F. Cao and P. Ouyang, React. Chem. Eng., 2022, 7, 1742–1749 RSC.
- D. Padovan, H. Kobayashi and A. Fukuoka, ChemSusChem, 2020, 13, 3594–3598 CrossRef CAS PubMed.
- C. Wang, C. Wu, A. Zhang, K. Chen, F. Cao and P. Ouyang, ChemistrySelect, 2022, 7, e202104574 CrossRef CAS.
- H. Zang, Y. Feng, M. Zhang, K. Wang, Y. Du, Y. Lv, Z. Qin and Y. Xiao, Carbohydr. Res., 2022, 522, 108679 CrossRef CAS PubMed.
- H. Zang, J. Lou, S. Jiao, H. Li, Y. Du and J. Wang, J. Mol. Liq., 2021, 330, 115667 CrossRef CAS.
- Y. Liu, C. Stähler, J. N. Murphy, B. J. Furlong and F. M. Kerton, ACS Sustainable Chem. Eng., 2017, 5, 4916–4922 CrossRef CAS.
- T. T. Pham, A. C. Lindsay, S. W. Kim, L. Persello, X. Chen, N. Yan and J. Sperry, ChemistrySelect, 2019, 4, 10097–10099 CrossRef CAS.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- T. T. Pham, G. Gözaydın, T. Söhnel, N. Yan and J. Sperry, Eur. J. Org. Chem., 2019, 2019, 1355–1360 CrossRef CAS.
- T. T. Pham, X. Chen, T. Söhnel, N. Yan and J. Sperry, Green Chem., 2020, 22, 1978–1984 RSC.
- T. T. Pham, X. Chen, N. Yan and J. Sperry, Monatsh. Chem., 2018, 149, 857–861 CrossRef CAS.
- H. Yoda, Y. Suzuki and K. Takabe, Tetrahedron Lett., 2004, 45, 1599–1601 CrossRef CAS.
- R. Kuhn and G. Krüger, Chem. Ber., 1957, 90, 264–277 CrossRef CAS.
- M. Ogata, T. Hattori, R. Takeuchi and T. Usui, Carbohydr. Res., 2010, 345, 230–234 CrossRef CAS PubMed.
- D. Matsuura, T. Mitsui, T. Sengoku, M. Takahashi and H. Yoda, Tetrahedron, 2008, 64, 11686–11696 CrossRef CAS.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, Green Chem., 2013, 15, 2960–2966 RSC.
- M. Osada, K. Kikuta, K. Yoshida, K. Totani, M. Ogata and T. Usui, RSC Adv., 2014, 4, 33651–33657 RSC.
- M. Osada, S. Shoji, S. Suenaga and M. Ogata, Fuel Process. Technol., 2019, 195, 106154 CrossRef CAS.
- M. Osada, H. Kobayashi, T. Miyazawa, S. Suenaga and M. Ogata, Int. J. Biol. Macromol., 2019, 136, 994–999 CrossRef CAS PubMed.
- X. Gao, X. Chen, J. Zhang, W. Guo, F. Jin and N. Yan, ACS Sustainable Chem. Eng., 2016, 4, 3912–3920 CrossRef CAS.
- L. Jia, Y. Wang, Y. Qiao, Y. Qi and X. Hou, RSC Adv., 2014, 4, 44253–44260 RSC.
- S. FujH, R. Kikuchi and H. Kushida, J. Org. Chem., 1966, 31, 2239–2241 CrossRef.
- G. Candiano, G. M. Ghiggeri, R. Gusmano, L. Zetta, E. Benfenati and G. Icardi, Carbohydr. Res., 1988, 184, 67–75 CrossRef CAS PubMed.
- J. Rohovec, J. Kotek, J. A. Peters and T. Maschmeyer, Eur. J. Org. Chem., 2001, 2001, 3899–3901 CrossRef.
- L. Jia, C. M. Pedersen, Y. Qiao, T. Deng, P. Zuo, W. Ge, Z. Qin, X. Hou and Y. Wang, Phys. Chem. Chem. Phys., 2015, 17, 23173–23182 RSC.
- L. Jia, X. Liu, Y. Qiao, C. M. Pedersen, Z. Zhang, H. Ge, Z. Wei, Y. Chen, X. Wen, X. Hou and Y. Wang, Appl. Catal., B, 2017, 202, 420–429 CrossRef CAS.
- L. Jia, Z. Zhang, Y. Qiao, C. M. Pedersen, H. Ge, Z. Wei, T. Deng, J. Ren, X. Liu, Y. Wang and X. Hou, Ind. Eng. Chem. Res., 2017, 56, 2925–2934 CrossRef CAS.
- M. Wu, H. Ma, Z. Ma, Y. Jin, C. Chen, X. Guo, Y. Qiao, C. M. Pedersen, X. Hou and Y. Wang, ACS Sustainable Chem. Eng., 2018, 6, 9434–9441 CrossRef CAS.
- A. K. L. Yuen, A. F. Masters and T. Maschmeyer, Catal. Today, 2013, 200, 9–16 CrossRef CAS.
- H. Baniamerian, M. Høj, M. J. Beier and A. D. Jensen, Appl. Catal., B, 2023, 330, 122650 CrossRef CAS.
- J. R. Kadam, T. S. Khan and P. L. Dhepe, New J. Chem., 2023, 47, 7548–7555 RSC.
- X. Pan and D. C. Webster, ChemSusChem, 2012, 5, 419–429 CrossRef CAS PubMed.
- S. N. Gunasekara, R. Pan, J. N. Chiu and V. Martin, Appl. Energy, 2016, 162, 1439–1452 CrossRef CAS.
- K. Li, P. H. M. Feron, T. W. Jones, K. Jiang, R. D. Bennett and A. F. Hollenkamp, Fuel, 2020, 263, 116661 CrossRef CAS.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- H. Kobayashi, K. Techikawara and A. Fukuoka, Green Chem., 2017, 19, 3350–3356 RSC.
- K. Techikawara, H. Kobayashi and A. Fukuoka, ACS Sustainable Chem. Eng., 2018, 6, 12411–12418 CrossRef CAS.
- Y. Zheng, L. Lu, W. Chen, A. Zheng, A. Lei and A. D. Chowdhury, Catalysts, 2022, 12, 460 CrossRef CAS.
- S. Xie, C. Jia, S. S. Go Ong, Z. Wang, M. jun Zhu, Q. Wang, Y. Yang and H. Lin, iScience, 2020, 23, 101096 CrossRef CAS PubMed.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi and A. Fukuoka, ACS Sustainable Chem. Eng., 2019, 7, 14883–14888 CrossRef CAS.
- C. Yang, T. Sagawa, A. Fukuoka and H. Kobayashi, Green Chem., 2021, 23, 7228–7234 RSC.
- T. Sagawa, H. Kobayashi, C. Murata, Y. Shichibu, K. Konishi, M. Hashizume and A. Fukuoka, Bull. Chem. Soc. Jpn., 2022, 95, 1054–1059 CrossRef CAS.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS PubMed.
- K. W. Omari, J. E. Besaw and F. M. Kerton, Green Chem., 2012, 14, 1480–1487 RSC.
- S. B. Lee and G. T. Jeong, Appl. Biochem. Biotechnol., 2015, 176, 1151–1161 CrossRef CAS PubMed.
- M. R. Park, H. S. Kim, S. K. Kim and G. T. Jeong, Fuel Process. Technol., 2018, 172, 115–124 CrossRef CAS.
- H. S. Kim, M. R. Park, S. K. Kim and G. T. Jeong, Korean J. Chem. Eng., 2018, 35, 1290–1296 CrossRef CAS.
- Y. Wang, C. M. Pedersen, T. Deng, Y. Qiao and X. Hou, Bioresour. Technol., 2013, 143, 384–390 CrossRef CAS PubMed.
- S. Yu, H. Zang, S. Chen, Y. Jiang, B. Yan and B. Cheng, Polym. Degrad. Stab., 2016, 134, 105–114 CrossRef CAS.
- H. S. Kim, S. K. Kim and G. T. Jeong, RSC Adv., 2018, 8, 3198–3205 RSC.
- H. S. Kim, M. R. Park, Y. J. Jeon, S. K. Kim, Y. K. Hong and G. T. Jeong, Energy Technol., 2018, 6, 1747–1754 CrossRef CAS.
- M. S. Islam, M. Nakamura, N. N. Rabin, M. A. Rahman, M. Fukuda, Y. Sekine, J. N. Beltramini, Y. Kim and S. Hayami, RSC Adv., 2021, 12, 406–412 RSC.
- N. D. Kalane, R. A. Krishnan, V. D. Yadav, R. Jain and P. Dandekar, Cellulose, 2019, 26, 2805–2819 CrossRef CAS.
- T.-W. Tzeng, P. Bhaumik and P.-W. Chung, Mol. Catal., 2019, 479, 110627 CrossRef.
- J. Wang, A. Zhu and L. Li, in Sustainable Catalytic Processes, ed. B. Saha, M. Fan and J. Wang, Elsevier, 2015, pp. 61–98 Search PubMed.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS PubMed.
- L. Zhou, R. Liang, Z. Ma, T. Wu and Y. Wu, Bioresour. Technol., 2013, 129, 450–455 CrossRef CAS PubMed.
- S. K. Singh and P. L. Dhepe, Green Chem., 2016, 18, 4098–4108 RSC.
- B. M. Matsagar and P. L. Dhepe, Catal. Sci. Technol., 2015, 5, 531–539 RSC.
- J. X. Feng, H. J. Zang, Q. Yan, M. G. Li and B. W. Cheng, Adv. Mater. Res., 2015, 1095, 411–414 Search PubMed.
- M. Li, H. Zang, J. Feng, Q. Yan, N. Yu, X. Shi and B. Cheng, Polym. Degrad. Stab., 2015, 121, 331–339 CrossRef CAS.
- Y. Jiang, H. Zang, S. Han, B. Yan, S. Yu and B. Cheng, RSC Adv., 2016, 6, 103774–103781 RSC.
- H. Zang, S. Yu, P. Yu, H. Ding, Y. Du, Y. Yang and Y. Zhang, Carbohydr. Res., 2017, 442, 1–8 CrossRef CAS PubMed.
- M. Zhang, H. Zang, B. Ma, X. Zhang, R. Xie and B. Cheng, ChemistrySelect, 2017, 2, 10323–10328 CrossRef CAS.
- B. Girisuta and H. J. Heeres, Production of Platform Chemicals from Sustainable Resources, Springer Singapore, Singapore, 2017 Search PubMed.
- Á. Szabolcs, M. Molnár, G. Dibó and L. T. Mika, Green Chem., 2013, 15, 439–445 RSC.
- G.-T. Jeong, Ind. Crops Prod., 2014, 62, 77–83 CrossRef CAS.
- M. R. Park, S. K. Kim and G. T. Jeong, J. Ind. Eng. Chem., 2018, 61, 119–123 CrossRef CAS.
- M. R. Park, H. S. Kim, S. K. Kim and G. T. Jeong, Korean Chem. Eng. Res., 2018, 56, 61–65 CAS.
- W. Hou, L. Liu and H. Shen, Carbohydr. Polym., 2018, 195, 267–274 CrossRef CAS PubMed.
- W. Hou, Q. Zhao and L. Liu, Green Chem., 2020, 22, 62–70 RSC.
- Q. Zhao and L. Liu, ACS Sustainable Chem. Eng., 2021, 9, 1762–1771 CrossRef CAS.
- X. Y. Tian, Y. Z. Zheng and Y. C. Zhang, J. Mol. Liq., 2022, 368, 120735 CrossRef CAS.
- R. Dalirfardouei, G. Karimi and K. Jamialahmadi, Life Sci., 2016, 152, 21–29 CrossRef CAS PubMed.
- J. Zhang, W. Xia, P. Liu, Q. Cheng, T. Tahi, W. Gu and B. Li, Mar. Drugs, 2010, 8, 1962–1987 CrossRef CAS PubMed.
- G. Wen and X. Wen, J. Carbohydr. Chem., 2006, 25, 297–301 CrossRef.
- M. Tominaga, M. Nagashima and I. Taniguchi, Electrochem. Commun., 2007, 9, 911–914 CrossRef CAS.
- Y. Ohmi, S. Nishimura and K. Ebitani, ChemSusChem, 2013, 6, 2259–2262 CrossRef CAS PubMed.
- J. Dai, G. Gözaydln, C. Hu and N. Yan, ACS Sustainable Chem. Eng., 2019, 7, 12399–12407 CAS.
- J. Wu, M. Qi, G. Gozaydln, N. Yan, Y. Gao and X. Chen, Ind. Eng. Chem. Res., 2021, 60, 3239–3248 CrossRef CAS.
- K. Kun-asa, P. Reubroycharoen, K. Yamazaki, N. Mimura, O. Sato and A. Yamaguchi, ChemistryOpen, 2021, 10, 308–315 CrossRef CAS PubMed.
- B. M. Matsagar, M. K. Munshi, A. A. Kelkar and P. L. Dhepe, Catal. Sci. Technol., 2015, 5, 5086–5090 RSC.
- A. Pandit, C. Deshpande, S. Patil, R. Jain and P. Dandekar, Carbohydr. Polym., 2020, 230, 115600 CrossRef CAS PubMed.
- C. Erb and F. W. Zerban, Ind. Eng. Chem., Anal. Ed., 1938, 10, 246–250 CrossRef CAS.
- M. Garriga, M. Almaraz and A. Marchiaro, Actas Ing., 2017, 3, 173–179 Search PubMed.
- N. Ghosh and P. L. Dhepe, Carbohydr. Polym. Technol. Appl., 2021, 2, 100139 CAS.
- D. Corradini, Handbook of HPLC, CRC Press, Boca Raton, 2nd edn, 2016 Search PubMed.
| This journal is © The Royal Society of Chemistry 2024 |