
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free aerobic photooxidation of sensitive benzylic alcohols with chemoselectivity controlled using DMSO as the solvent†
Ivana
Weisheitelová
 a,
Naisargi
Varma
b,
Josef
Chudoba
c,
Gotard
Burdziński
d,
Marek
Sikorski
*b and
Radek
Cibulka
*a
a,
Naisargi
Varma
b,
Josef
Chudoba
c,
Gotard
Burdziński
d,
Marek
Sikorski
*b and
Radek
Cibulka
*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, 16628 Prague, Czech Republic. E-mail: cibulkar@vscht.cz
bFaculty of Chemistry, Adam Mickiewicz University, 61-614 Poznań, Poland. E-mail: sikorski@amu.edu.pl
cCentral Laboratories, University of Chemistry and Technology, Prague, 16628 Prague, Czech Republic
dFaculty of Physics, Adam Mickiewicz University, 61-614 Poznań, Poland
First published on 12th March 2024
Abstract
The drawbacks commonly observed in synthetic methods for alcohol oxidation often stem from the utilization of complex, toxic, hazardous, or waste-producing oxidants. When sensitive or complex substrates bearing several functional groups are to be transformed, the selectivity of oxidation becomes another significant challenge. Herein, a chemoselective and operationally simple catalyst-free and additive-free method is presented for the aerial oxidation of 1-phenylpropargyl and 1-phenylallyl alcohols to their corresponding ketones, requiring only a solvent and visible light irradiation. The crucial role of dimethylsulfoxide (DMSO) as the solvent lies in achieving high chemoselectivity. Singlet oxygen, whose formation is photosensitized by the substrate and the product, is captured by DMSO, thereby preventing the undesired over-oxidation that occurs in other solvents. The application of DMSO to protect the substrate against singlet oxygen represents a novel approach that is potentially applicable to other aerobic photocatalytic processes.
Introduction
Most syntheses of valuable organic compounds, such as pharmaceuticals, dyes and agrochemicals, involve an alcohol oxidation reaction to form their corresponding aldehyde or ketone derivative.1 This transformation has been routinely achieved using stoichiometric agents based on high-valent metals, for example, chromium(VI) compounds or MnO2;2 alternatively hypervalent iodine reagents,3 activated dimethyl sulfoxide (DMSO)4 and organic peroxides have been used.5,6 Unfortunately, these stoichiometric procedures always produce a lot of waste and many of the reagents involved in these reactions are highly toxic.Molecular oxygen is considered a green oxidant producing only water as the by-product.7 However, oxidation with oxygen requires either the substrate or oxygen to be activated (Scheme 1A), which is traditionally achieved using transition metal-containing homogeneous or heterogeneous catalysts,1b,8 although some metal-free or biocatalytic procedures have also been developed.6a,9,10 Recent progress in oxidation reactions with oxygen has been made within photoredox catalysis using light and a photoexcited catalyst.11 In particular, methods using organic dyes12,13 seem to be promising from an environmental point of view, although they still suffer from the low stability of the photocatalyst and/or, with a few exceptions, the need for an additive. Thus, an oxidation reaction with oxygen, which does not require both a catalyst and an additive, could be considered as an optimal process. However, such methods for alcohol oxidation are rare and require high temperature and pressure conditions or a strong light source.14 Harsh conditions similar to those for excitation with light cause another problem not mentioned yet – low chemoselectivity, which is manifested by the production of undesired by-products. Thus, the discovery of a green method without the need for additives utilizing mild conditions, which would also be suitable for sensitive substrates, remains an unsolved problem.
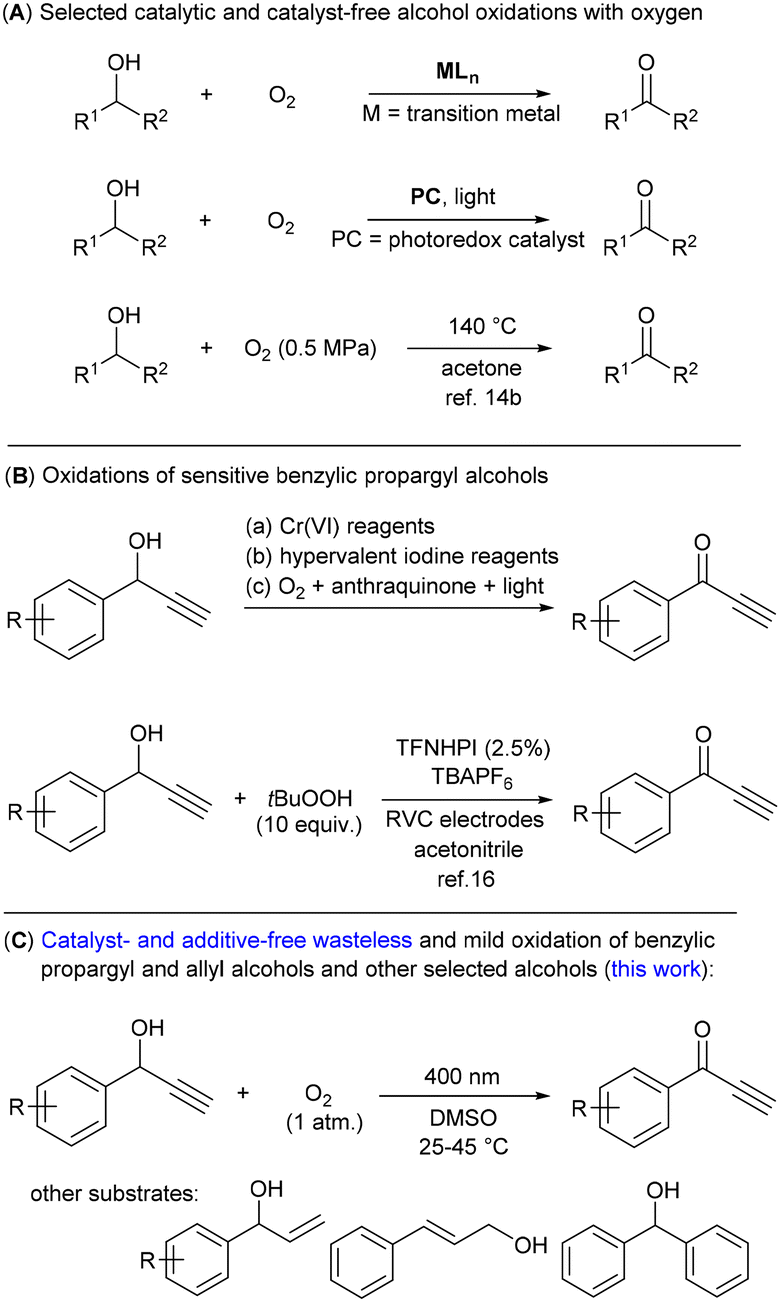 | ||
| Scheme 1 Oxidation of alcohols to ketones with molecular oxygen and overview of oxidations of sensitive 1-phenylpropargyl alcohols. | ||
1-Phenyl propargyl alcohols 1 contain a triple bond and an activated benzylic position, which are prone to undesired side- or over-oxidation reactions.15 On the other hand, they are considered as difficult to oxidize substrates.16 In addition to their importance in organic synthesis,15 alcohols 1 are suitable representatives of sensitive substrates. Some procedures have been reported in the literature that can transform benzylic alcohols bearing multiple bonds to their corresponding ketone derivatives (Scheme 1B). However, most of them use stoichiometric amounts of Cr(VI) or other metal-based reagents.17 Alternatively, oxidation reactions using hypervalent iodine reagents18 or photocatalytic procedures with 2-bromoanthraquinone19 have been shown in a limited number of examples. Very recently, the most effective and general procedure based on N-hydroxy-tetrafluorophtalimide (TFNHPI) as an electrochemical mediator and tert-butylhydroperoxide as a stoichiometric oxidizing agent has been developed.16 Herein, we report the catalyst- and additive-free chemoselective photooxidation of alcohols 1 to ketones 2 using oxygen performed at room temperature (Scheme 1C). The operational simplicity and lack of waste generation characterize this method, which requires only irradiation by visible light and the use of DMSO as the solvent. The choice of solvent was crucial to achieve the desired chemoselectivity; DMSO exhibits an important protective role against singlet oxygen processes, described in this paper for the first time.
Results and discussion
Development and optimization of the reaction
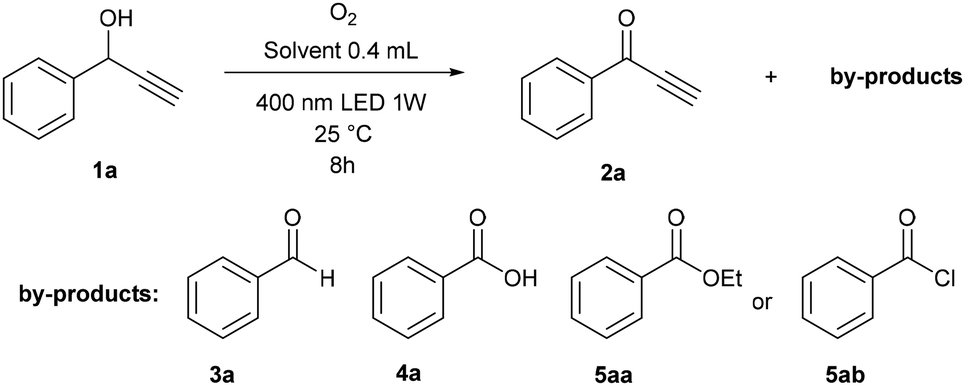
With the aim of developing the selective oxidation of sensitive 1-phenylpropargyl alcohols 1 to give the corresponding ketones 2 using photoredox catalysis (see ESI S4† for preliminary experiments), we observed that the remarkable oxidation of model substrate 1a also occurred in the absence of a catalyst when irradiated with 400 nm light (Table 1). Interestingly, the reaction in dimethyl sulfoxide (DMSO) took place and gave ketone 2 in an excellent yield with high chemoselectivity (entry 1), while those performed in other solvents formed several undesired by-products (entries 2–8). Benzoic acid (4a) was the main product formed in N,N-dimethylformamide (DMF), ethanol, dioxane and toluene, and also appeared in high amounts in acetonitrile. In dioxane, ethanol and chloroform, significant amounts of benzaldehyde (3a), ethyl benzoate (5aa) or benzoylchloride (5ab) were also observed, respectively. For the reaction in DMSO, we investigated the effect of temperature (entries 9 and 10) and wavelength (entries 11 and 12), but the original conditions, i.e. 25 °C and 400 nm appeared to be the best. We successfully demonstrated the use of sunlight by performing the reaction at a window, which achieved a good yield, albeit a longer reaction time was necessary (entry 13). Under air instead of oxygen, the system was less efficient (entry 14). Control experiments showed that oxidation does not occur in the absence of oxygen or light (entries 15 and 16).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Condition alternation | Yieldc [%] | |||
| 1a | 2a | 3a | 4a | |||
| a Selected data; for further experiments, see ESI S5.† b Conditions: 1a (0.1 mmol), solvent (0.4 mL), 400 nm LED 1 W, oxygen (balloon), 25 °C, 8 h. c Determined by GC-MS. d 20 hours. e 22% of 5aa. f 52% of 5ab. | ||||||
| 1 | DMSO | No | 4 | 90 | 2 | 4 |
| 2 | DMF | No | 42 | 0 | 0 | 58 |
| 3 | EtOH | No | 7 | 6 | 2 | 40e |
| 4 | Acetonitrile | No | 0 | 62 | 0 | 38 |
| 5 | Dioxane | No | 2 | 3 | 47 | 48 |
| 6 | CHCl3 | No | 0 | 0 | 0 | 47f |
| 7 | Acetone | No | 0 | 7 | 1 | 90 |
| 8 | Toluene | No | 8 | 16 | 17 | 43 |
| 9 | DMSO | 45 °C | 2 | 91 | 3 | 4 |
| 10 | DMSO | 0 °C | 38 | 55 | 3 | 4 |
| 11 | DMSO | 450 nm | 64 | 34 | 1 | 0 |
| 12 | DMSO | 385 nm | 45 | 48 | 3 | 4 |
| 13 | DMSO | Sunlightd | 25 | 65 | 10 | 0 |
| 14 | DMSO | Air | 90 | 9 | 1 | 0 |
| 15 | DMSO | Ar | 100 | 0 | 0 | 0 |
| 16 | DMSO | Under darkness | 100 | 0 | 0 | 0 |
Substrate scope investigation
Having in hand such a photooxidation method that requires only a solvent (DMSO), visible light (400 nm) and oxygen, we were interested in its limitations regarding the substrate structure (Table 2). We found that in addition to the unsubstituted derivative 1a (entries 1 and 2), the method can be used for para-substituted 1-phenylpropargyl alcohols containing electron-withdrawing (1b) or electron-donating (1c and 1d) groups (entries 3–8). Substrates 1e and 1f containing strongly electron-withdrawing nitro- and trifluoromethyl groups gave their corresponding ketone products in a lower yield, which were not enhanced by an elevated temperature (45 °C) or prolonging the reaction time (entries 9–14). A very low yield of the ketone product was observed with the ortho-substituted derivative 1g (entries 15 and 16), which corresponds to its low ability to absorb 400 nm light (see ESI S3† and discussion below). In contrast, the meta-substituted derivative 1h with an absorbing well in the 400 nm region (similar to para-derivative 1b) gave a significant amount of ketone after irradiation (entries 17 and 18).| Entry | Substrate | Temperature [°C] | Time [h] | Yieldc [%] |
|---|---|---|---|---|
| a Selected data; for further experiments, see the ESI.† b Conditions: 1a (0.1 mmol), DMSO (0.4 mL), 400 nm LED 1 W, oxygen (balloon), 8 h. c Determined by GC-MS. d Determined by 1H NMR. | ||||
| 1 |
|
25 | 8 | 90 |
| 2 | 45 | 8 | 91 | |
| 3 |
|
25 | 8 | 89 |
| 4 | 45 | 8 | 39 | |
| 5 |
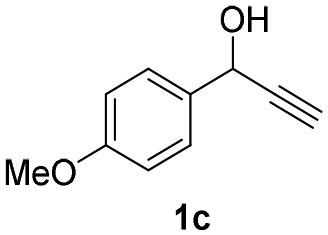
|
25 | 8 | 93 |
| 6 | 45 | 8 | 79 | |
| 7 |
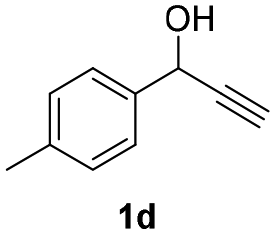
|
25 | 8 | 85 |
| 8 | 45 | 8 | 86 | |
| 9 |
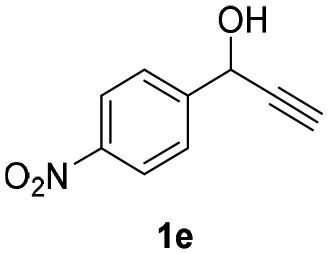
|
25 | 8 | 14 |
| 10 | 25 | 16 | 4 | |
| 11 | 45 | 8 | 12 | |
| 12 |
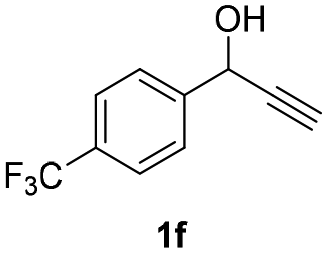
|
25 | 8 | 27 |
| 13 | 25 | 16 | 28 | |
| 14 | 45 | 8 | 32 | |
| 15 |
|
25 | 8 | 2 |
| 16 | 45 | 8 | 5 | |
| 17 |
|
25 | 8 | 49 |
| 18 | 45 | 8 | 19 | |
| 19 |
|
25 | 8 | 47 |
| 20 | 45 | 8 | 42 | |
| 21 |
|
25 | 8 | 44 |
| 22 | 45 | 8 | 26 | |
| 23 |
|
25 | 8 | 70 |
| 24 | 45 | 8 | 16 | |
| 25 |
|
25 | 8 | 75 |
| 26 | 45 | 8 | 64 | |
| 27 |
|
25 | 8 | 45d |
| 28 | 45 | 8 | 72d | |
| 29 |
|
25 | 8 | 17d |
| 30 | 45 | 8 | 41d | |
Our method can also be applied to 1-phenylallyl alcohols 6 (entries 19–24), although they are less reactive in oxidation to ketones 7, which are, on the other hand, more susceptible to over-oxidation to their benzoic acid derivatives, as especially evident at 45 °C. This tendency was demonstrated by the decreased yield of ketone 7 (see ESI S5† for reactions performed under different conditions). On the other hand, cinnamyl alcohol 8 (entries 25 and 26) was sufficiently oxidized to aldehyde 9 with high yield and selectivity. Diphenylmethanol (10) and 1-(4-methoxyphenyl)ethanol (12), which are representatives of 1,1-diaryl- and 1-arylalkanols, were also effectively oxidized at higher temperatures (entries 27–30).
In selected cases, we demonstrated the effectiveness of our method on a preparative scale giving the corresponding ketones in good to high isolated yields (Fig. 1). Preparative experiments occurred mainly at 40 °C because the reaction mixture was heated upon irradiation by the LED. For compounds 1b, 1c and 8, the mixtures were cooled to 25 °C to maintain the chemoselectivity of the reaction. The reaction times were optimized to achieve the highest conversions to carbonyl compounds (see data in Fig. 1). Thus, with the exception of 1c, some alcohol remained unreacted in the mixture. Moreover, a minor amount of the corresponding benzaldehyde or benzoic acid was present in the crude reaction mixtures after preparative experiments with 1a–d and 8 (see ESI S3†). Decreased isolated yields of ketones as compared with conversions were caused by purification with column chromatography.
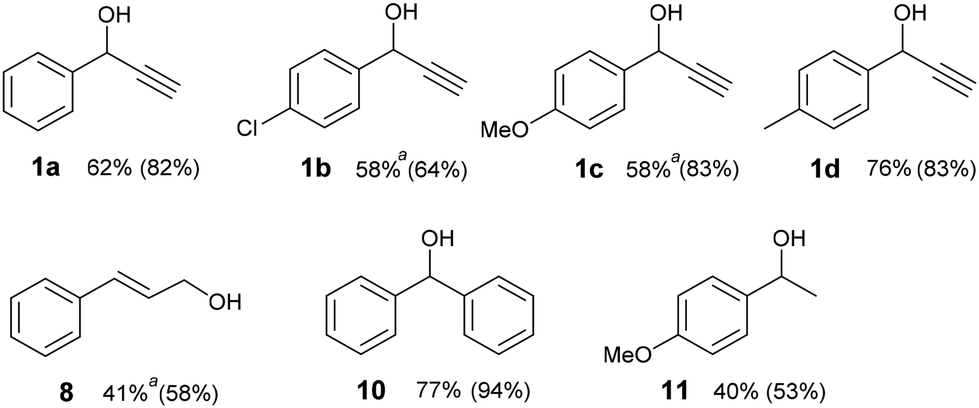 | ||
| Fig. 1 Isolated yields of catalyst-free photooxidation on a 1 mmol scale. Conversions to ketones analysed by 1H NMR are given in parentheses. Conditions: alcohol (1 mmol), DMSO (4 mL), 400 nm LED 1 W, oxygen (balloon), 40 °C, 8–10 hours, see ESI S5† for details. aCooling to 25 °C. | ||
The essence of the ease of oxidization upon LED irradiation under oxygen was based on the ability of the respective substrates to absorb visible light at the concentration used in the photooxidation reactions. 1-Phenyl propargyl alcohol 1a exhibits a broad absorption band at around 260 nm and does not absorb 400 nm light at “analytical” concentrations. However, a highly concentrated 0.25 M solution of 1a in DMSO (the concentration of 1a in the reaction mixture) and acetonitrile display some absorption at around 400 nm, which is probably caused by the aggregation of 1a (see Fig. 2A and ESI S3†). A similar situation was observed with other substrates, giving a remarkable yield of the ketone product. On the other hand, an ortho-substituted derivative 1g exhibited a relatively small absorption around 400 nm even at high concentrations (see ESI S3†), which is probably the reason why only a small amount of ketone 2g (together with starting material) was found in the reaction mixture after the oxidation reaction. Interestingly, alcohol 1a exhibits fluorescence in the visible light region (see ESI S8†). Thus, we measured the excitation spectra of 1a in DMSO showing a clear band at around 400 nm (see Fig. 2B).
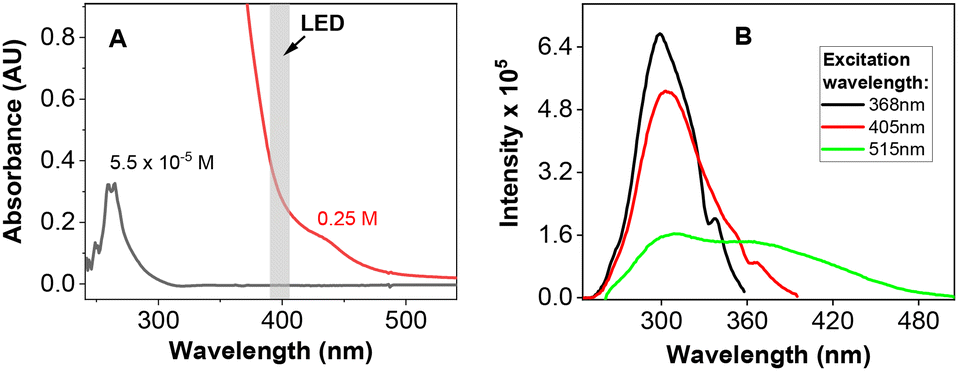 | ||
| Fig. 2 (A) Absorption spectra of 1a at a low concentration (5 × 10–5 mol L−1, black) and at the concentration of photocatalytic experiments (0.25 mol L−1, red) in DMSO. Dominant LED emission is highlighted in grey. (B) Excitation spectra of 1a in DMSO. | ||
Mechanistic investigations
In our mechanistic investigations, we focused on explaining the overall reaction scheme and origin of the high chemoselectivity observed in the oxidation reactions performed in DMSO. For detailed studies, we selected acetonitrile as a solvent representative, allowing the undesired over-oxidation reaction.First, we monitored the concentrations of the starting alcohol 1a, ketone 2a and over-oxidation products 3 and 4 under photooxidation conditions over 24 h (Fig. 3).
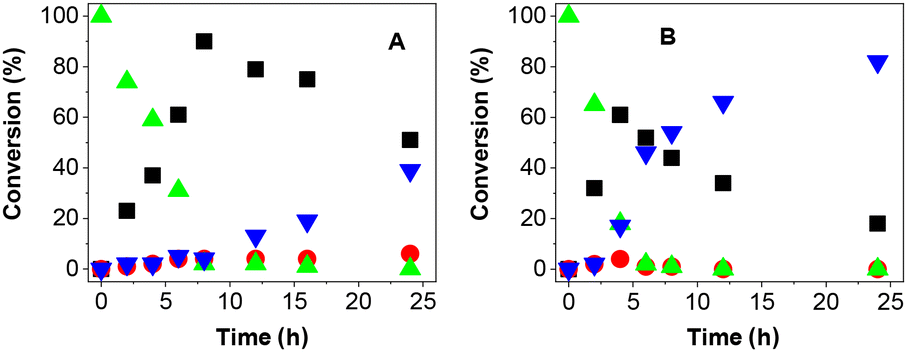 | ||
| Fig. 3 Reaction course of 1a photooxidation under standard conditions in (A) DMSO and (B) acetonitrile. Symbols mean conversion of 1a (▲), 2a (■), 3a (●) and 4a (▼). | ||
The reaction courses in both DMSO and acetonitrile correspond to a sequence of consecutive reactions: alcohol 1a was oxidized to ketone 2a, which was further oxidized to carboxylic acid. In both solvents, ketone 2a oxidation likely occurred via aldehyde 3a, which was detected in small concentrations most of the time. Overall, the oxidation is slower in DMSO. However, the most significant difference can be found in the rate of the transformation from 2a to 4a, which was much faster in acetonitrile. Moreover, carboxylic acid 4a was formed immediately in acetonitrile after 2a appeared, even in small concentrations. Thus, the direct oxidation of alcohol 1a to form 4a in acetonitrile cannot be excluded.
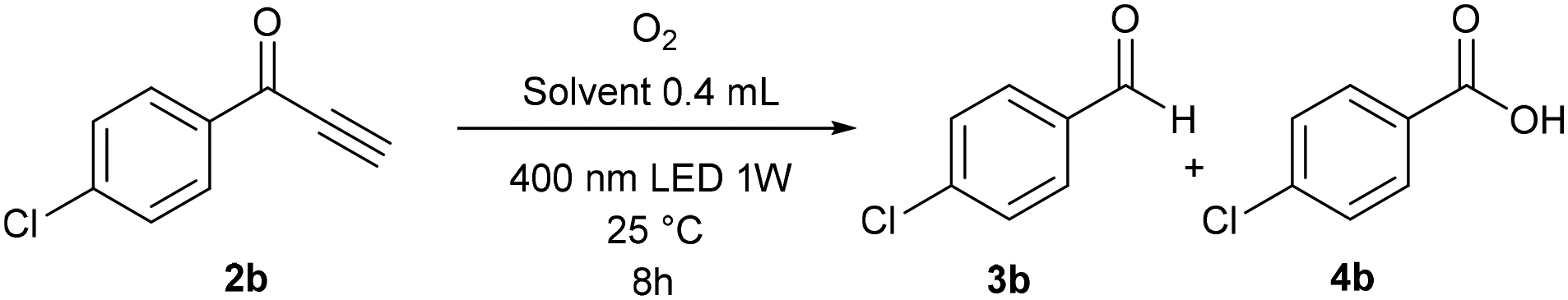
Analogous conclusions can be drawn from independent experiments on the oxidation of ketone 2b (Table 3). In DMSO, only small amounts of the over-oxidation products were observed after 8 h of irradiation (entries 1–3). On the other hand, all of ketone 2b was consumed in acetonitrile and quantitatively converted into benzoic acid 4b, even after 3 h (entries 4 and 5). Interestingly, a faster reaction was observed in deuterated acetonitrile when compared to non-deuterated acetonitrile (entries 6 and 7), which indicates the participation of singlet oxygen (see the mechanistic studies below).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Condition alternation | Conversionb [%] | ||
| 2b | 3b | 4b | |||
| a Conditions: 2a (0.1 mmol), solvent (0.4 mL), 400 nm LED 1 W, oxygen (balloon), 25 °C, 8 h. b Determined by GC-MS. c Determined by 1H NMR. | |||||
| 1 | DMSO | — | 69 | 6 | 25 |
| 2 | DMSO | Under darkness | 92 | 4 | 0 |
| 3 | DMSO | Ar | 100c | 0 | 0 |
| 4 | Acetonitrile | — | 0 | 0 | 100 |
| 5 | Acetonitrile | 3 h | 2 | 0 | 98 |
| 6 | Acetonitrile | 1 h | 77 | 2 | 21 |
| 7 | Acetonitrile-d3 | 1 h | 67 | 1 | 32 |
The photophysical and spectral properties of 1a and 2a were studied using steady state and transient absorption spectroscopy in acetonitrile (Fig. 4). After irradiation, both 1a and 2a enter short-lived singlet excited states, followed by fast intersystem crossing (ISC) to the triplet T1 state (Fig. 4A and B), which was found to be quenched by oxygen: the lifetimes of T1 for 1a and 2a were found to be 1.4 μs and 19.6 μs under argon and 115 ns and 0.4 μs under an air atmosphere, respectively, measured by transient absorption spectroscopy (Fig. 4C and D). Both compounds show phosphorescence spectra (Fig. 4E and F), which are measurable even at room temperature. Taking into account the concentration of oxygen in acetonitrile under normal pressure (2.42 × 10−3 mol L−1, 1 atm. air),20 the rate constants of 1a (kT1q = 3.15 × 109 L mol−1 s−1) and 2a (kT1q = 1.0 × 109 L mol−1 s−1) T1 state quenching by oxygen were determined. The energy transfer mechanism was confirmed as kT1q/kdiff, which was close to 1/9.21 We also found that the quantum yield of singlet oxygen formation sensitized by 1a or 2a was 0.17 and 0.31, respectively (see ESI S8†). Thus, singlet oxygen seems to be, in addition to molecular oxygen, the stoichiometric oxidation agent in oxidative processes in acetonitrile. In DMSO, similar data were found for the excited 1a and 2a states as in acetonitrile (see ESI S8†). On the other hand, a significant difference was found in the production of singlet oxygen, which was barely detected after 1a or 2a was irradiated in DMSO (cf.Fig. 4G and H for 1a as an example).
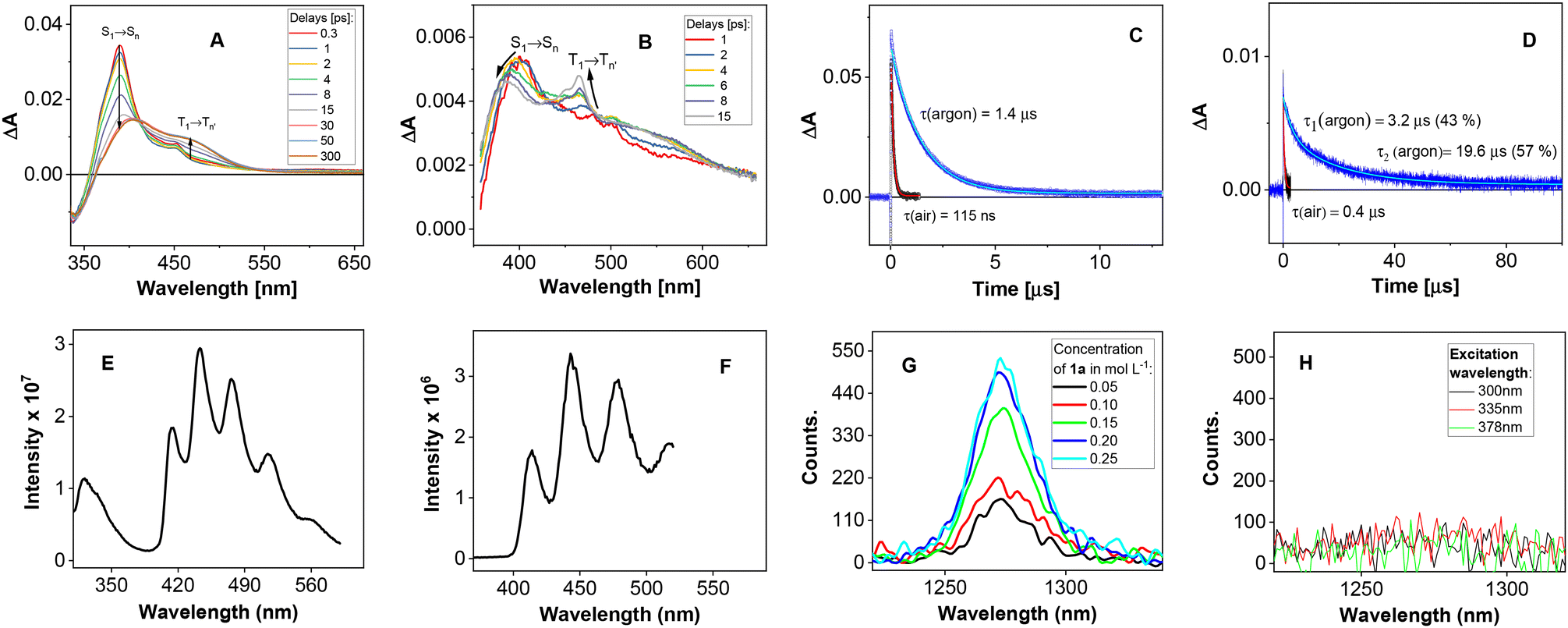 | ||
| Fig. 4 Time resolved absorption data for (A) 1a and (B) 2a in acetonitrile demonstrating formation of a triplet state. T1 kinetic traces for (C) 1a and (D) 2a in acetonitrile under argon (blue) and air (black). Phosphorescence spectra of (E) 1a (λext = 300 nm) and (F) 2a (λext = 263 nm) at 77 K in acetonitrile. The emission spectra of singlet oxygen in the presence of 1a (G) in acetonitrile (different concentrations) and (H) in DMSO (different excitation wavelengths). | ||
The phosphorescence spectra obtained for both 1a and 2a have allowed us to estimate the triplet state energies and, in combination with the ground state oxidation potentials, the excited state oxidation potentials characterizing the ease of oxidation of both compounds in their excited T1 states (see Scheme 2). When comparing the data obtained for ET1ox(1a) = −0.52 V vs. SCE and ET1ox(2a) = −0.20 V vs. SCE with the oxygen species reduction potentials [E(3O2/O2˙−) = −0.43 V vs. SCE; E(1O2/O2˙−) = +0.56 V vs. SCE],22 it can be concluded that while oxidation of 1a can occur with both molecular oxygen and singlet oxygen, 2a was probably oxidized by only singlet oxygen.
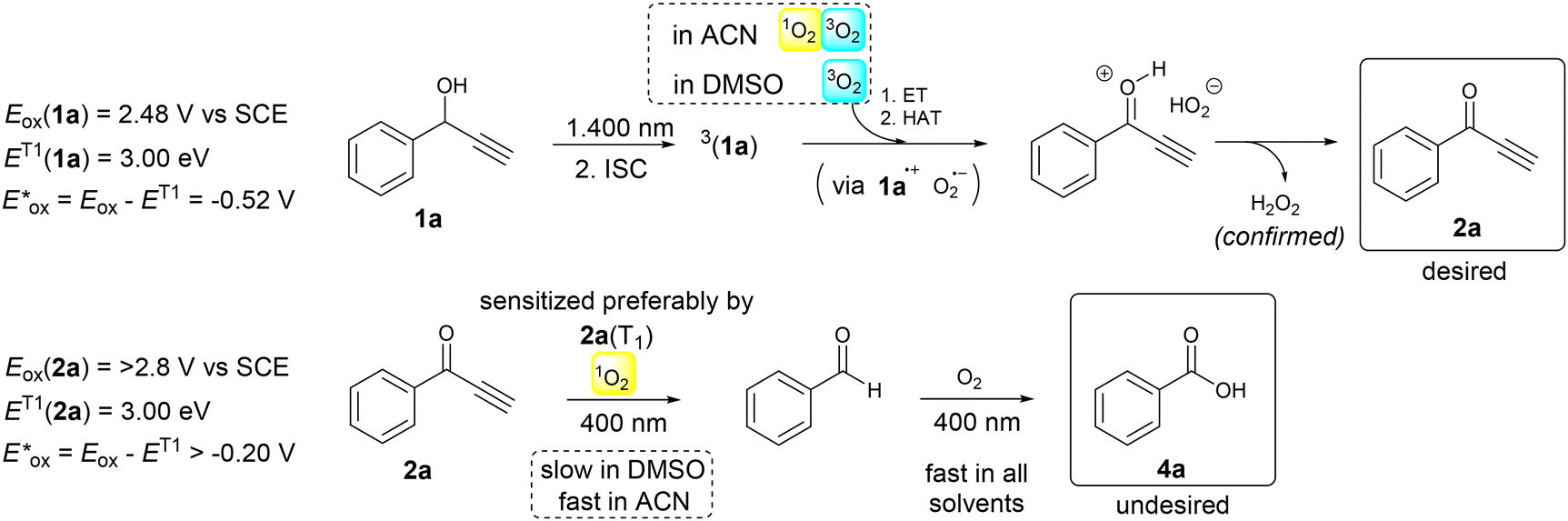 | ||
| Scheme 2 Proposed mechanism of 1a photooxidation with oxygen under catalyst- and additive-free conditions. In DMSO, ground state oxygen (3O2) is involved in photooxidation of alcohol 1a to ketone 2a, while in acetonitrile, singlet oxygen (1O2) can be involved. In acetonitrile, the first step oxidation is followed by fast oxidation of 2a with singlet oxygen. The second process is inhibited in DMSO. Thermodynamic parameters based on cyclic voltammetry and phosphorescence measurements are given. For oxidation potential measurements, see ESI S7.† | ||
Looking for a difference between DMSO and acetonitrile to elucidate the potential origin of the DMSO-based chemoselectivity of the photooxidation reactions, one should keep in mind the lower oxygen solubility in DMSO (2.2 × 10−3 mol L−1, 1 atm. O2) compared to acetonitrile (9.1 × 10−3 mol L−1, 1 atm. O2).23 We were also interested in the singlet oxygen lifetime in both solvents. While singlet oxygen in acetonitrile has been well studied, the data for DMSO have rarely been reported. The singlet oxygen lifetime is also significantly influenced by solvent deuteration, as demonstrated by the values observed for acetonitrile (81 μs) and acetonitrile-d3 (1613 μs).24 We were surprised to observe only a slight effect of deuteration on the singlet oxygen lifetimes in DMSO (8.4 μs) and DMSO-d6 (9.7 μs) (see ESI S9†). This can be attributed to the fact that DMSO and DMSO-d6 readily react with singlet oxygen to form the corresponding sulfone.25 Thus, the singlet oxygen lifetime was controlled by consumption due to sulfoxide oxidation rather than physical quenching in DMSO.
The findings described above enabled us to propose the overall reaction mechanism as well as explain the unique chemoselectivity observed during the photooxidation of 1a in DMSO (Scheme 2). Initially, alcohol 1a in a triplet excited state undergoes a photoinduced electron transfer primary with molecular oxygen in DMSO. In solvents like acetonitrile, characterized by longer singlet oxygen lifetimes, electron exchange with singlet oxygen is also expected. After hydrogen atom and proton transfer, ketone 2a is formed along with hydrogen peroxide, whose amount corresponds to that calculated theoretically and confirmed using independent experiments (see ESI S6†). The oxidation of ketone 2a needs singlet oxygen as photoinduced electron transfer with molecular oxygen is not thermodynamically feasible. Alternatively, [2 + 2] cycloaddition of singlet oxygen to a multiple bond can occur to form a dioxetane species,26 which undergoes C–C bond fragmentation to benzaldehyde or benzoic acid. This photooxidative cleavage is favoured in acetonitrile and represents an interesting reaction which usually needs transition metal catalysis or peroxides.27
In DMSO, singlet oxygen was rapidly consumed by solvent oxidation to dimethylsulfone, which was found in significant amounts in all the reaction mixtures upon photooxidation using 1H NMR spectroscopy and mass spectrometry (see ESI S6†). Therefore, the undesired oxidation of ketone 2a was very slow in DMSO, allowing its isolation after an appropriate time in high yield. In contrast, singlet oxygen in acetonitrile is relatively long-lived, which supports the over-oxidation reactions. Thus, ketone 2a was, in fact, “protected” against oxidation by DMSO. Additionally, the overall low concentration of oxygen in DMSO can also contribute to chemoselectivity, slowing down the oxidative processes.
Conclusions
A novel, operationally simple chemoselective oxidation methodology for sensitive benzylic propargyl alcohols and allyl alcohols has been presented. This method has been shown to be also useful for other sensitive substrates, such as diphenylmethanol and 1-phenylethanol. The method is catalyst-free and additive-free requiring only solvent and visible light irradiation (400 nm), thus significantly distinguishing itself from all other methodologies published to date for similar sensitive substrates. In general, it is also unique among the few catalyst- and additive-free procedures reported for alcohol oxidation, functioning at room temperature or a slightly elevated temperature with a relatively weak light source (1 W LED). Previous methodologies have typically required high temperature and pressure along with a strong light source (45 W LED).14An original and unique source of chemoselectivity in oxidation reactions using molecular oxygen has been presented in this work, based on the use of DMSO as the solvent. Due to its ability to react with singlet oxygen, DMSO in fact “protects” the substrate from undesired over-oxidation. This novel concept might find application in other oxidative processes suffering from poor selectivity due to singlet oxygen. Regarding green chemistry, our procedure fulfills several criteria: (i) atom economy as it uses oxygen as the sole oxidant, (ii) selectivity, leading to the production of a single product, and (iii) low energy consumption, using a weak light source (1 W LED) at room temperature and under atmospheric pressure conditions. The relatively low amount of a non-toxic solvent (400 μL for 1 mmol) used in the reaction represents another advantage.
Author contributions
R. C., I. W. and M. S. wrote the paper with input from all of the authors. I. W. performed synthesis of all the used compounds and conducted all photochemical reactions. J. C. performed all mass spectroscopy analyses. N. V. together with M. S. collected the stationary spectroscopy and singlet oxygen data. G. B. performed all other time-resolved spectroscopic experiments. Experimental spectroscopy data were analysed by M. S. together with G. B. The project was conceived by R. C., I. W., and M. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (Grant No. 21-14200K) and by the research grant CEUS-UNISONO 2020/02/Y/ST4/00042 from The National Science Centre of Poland (NCN). The authors thank Ludmila Šimková for the cyclic voltammetry measurements.References
- (a) F. Cavani, Catal. Today, 2010, 157, 8–15 CrossRef CAS; (b) G. D. Yadav, R. K. Mewada, D. P. Wagh and H. G. Manyar, Catal. Sci. Technol., 2022, 12, 7245–7269 RSC; (c) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed; (d) J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, in Ullmann's Encyclopedia of Industrial Chemistry, 2015, pp. 1–103 Search PubMed.
- (a) Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Eds. G. Tojo and M. Fernández, Springer US, Boston, MA, 2006, pp. 1–95 Search PubMed; (b) G. Cahiez, M. Alami, R. J. K. Taylor, M. Reid, J. S. Foot, L. Fader, V. Sikervar and J. Pabba, in Encyclopedia of Reagents for Organic Synthesis (EROS), 2017, pp. 1–16 Search PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS.
- L. De Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2001, 66, 7907–7909 CrossRef CAS PubMed.
- (a) E. T. Poursaitidis, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem. Sci., 2024, 15, 1177–1203 RSC; (b) I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS; (c) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (d) R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS; (e) G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- (a) J.-E. Bäckvall, Modern Oxidation Methods, 2nd Completely Revised, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed; (b) G. Tojo and M. I. Fernández, Oxidation of Alcohols to Aldehydes and Ketones: A Guide to Current Common Practice, Springer, 2006 Search PubMed.
- (a) R. A. Sheldon, in Biomimetic Oxidations Catalyzed by. Transition Metal Complexes, ed. B. Meunier, Imperial College Press, 2000, pp. 613–662 Search PubMed; (b) L. Vanoye, M. Abdelaal, K. Grundhauser, B. Guicheret, P. Fongarland, C. De Bellefon and A. Favre-Réguillon, Org. Lett., 2019, 21, 10134–10138 CrossRef CAS; (c) C. S. Batsika, C. Koutsilieris, G. S. Koutoulogenis, M. G. Kokotou, C. G. Kokotos and G. Kokotos, Green Chem., 2022, 24, 6224–6231 RSC; (d) L. Vanoye and A. Favre-Réguillon, Org. Process Res. Dev., 2022, 26, 335–346 CrossRef CAS.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS.
- (a) J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS; (b) D. Romano, R. Villa and F. Molinari, ChemCatChem, 2012, 4, 739–749 CrossRef CAS.
- (a) N. Gunasekaran, Adv. Synth. Catal., 2015, 357, 1990–2010 CrossRef CAS; (b) X. Wang, R. Liu, Y. Jin and X. Liang, Chem. – Eur. J., 2008, 14, 2679–2685 CrossRef CAS PubMed.
- (a) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (b) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (c) V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS; (d) D. Tang, G. Lu, Z. Shen, Y. Hu, L. Yao, B. Li, G. Zhao, B. Peng and X. Huang, J. Energy Chem., 2023, 77, 80–118 CrossRef CAS.
- (a) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596–4614 RSC; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) R. Obertík, J. Chudoba, J. Šturala, J. Tarábek, L. Ludvíková, T. Slanina, B. König and R. Cibulka, Chem. – Eur. J., 2022, 28, e202202487 CrossRef PubMed; (b) A. Pokluda, Z. Anwar, V. Boguschová, I. Anusiewicz, P. Skurski, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2021, 363, 4371–4379 CrossRef CAS; (c) A. H. Tolba, F. Vávra, J. Chudoba and R. Cibulka, Eur. J. Org. Chem., 2020, 1579–1585 CrossRef CAS; (d) E. Skolia, P. L. Gkizis, N. F. Nikitas and C. G. Kokotos, Green Chem., 2022, 24, 4108–4118 RSC; (e) N. F. Nikitas, D. I. Tzaras, I. Triandafillidi and C. G. Kokotos, Green Chem., 2020, 22, 471–477 RSC; (f) Y.-H. Wang, Q. Yang, P. J. Walsh and E. J. Schelter, Org. Chem. Front., 2022, 9, 2612–2620 RSC; (g) C. Liu, H. Liu, X. Zheng, S. Chen, Q. Lai, C. Zheng, M. Huang, K. Cai, Z. Cai and S. Cai, ACS Catal., 2022, 12, 1375–1381 CrossRef; (h) H. Zhang, T. Guo, M. Wu, X. Huo, S. Tang, X. Wang and J. Liu, Tetrahedron Lett., 2021, 67, 152878 CrossRef CAS; (i) W. Schilling, D. Riemer, Y. Zhang, N. Hatami and S. Das, ACS Catal., 2018, 8, 5425–5430 CrossRef CAS.
- (a) M. Xu, J. Ou, K. Luo, R. Liang, J. Liu, N. Li, B. Hu and K. Liu, Molecules, 2023, 28, 3031 CrossRef CAS PubMed; (b) X. Tian, X. Cheng, X. Yang, Y.-L. Ren, K. Yao, H. Wang and J. Wang, Org. Chem. Front., 2019, 6, 952–958 RSC.
- H. Qian, D. Huang, Y. Bi and G. Yan, Adv. Synth. Catal., 2019, 361, 3240–3280 CrossRef CAS.
- C. E. Hatch, M. I. Martin, P. H. Gilmartin, L. Xiong, D. J. Beam, G. P. A. Yap, M. J. Von Bargen, J. Rosenthal and W. J. Chain, Org. Lett., 2022, 24, 1423–1428 CrossRef CAS.
- (a) K. C. Weerasiri and A. E. V. Gorden, Tetrahedron, 2014, 70, 7962–7968 CrossRef CAS; (b) G. Ieronimo, G. Palmisano, A. Maspero, A. Marzorati, L. Scapinello, N. Masciocchi, G. Cravotto, A. Barge, M. Simonetti, K. L. Ameta, K. M. Nicholas and A. Penoni, Org. Biomol. Chem., 2018, 16, 6853–6859 RSC; (c) J. Muzart and A. N. Ajjou, Synthesis, 1993, 785–787 CrossRef CAS; (d) M. Hunsen, Tetrahedron Lett., 2005, 46, 1651–1653 CrossRef CAS; (e) F. Shi, S.-W. Luo, Z.-L. Tao, L. He, J. Yu, S.-J. Tu and L.-Z. Gong, Org. Lett., 2011, 13, 4680–4683 CrossRef CAS PubMed; (f) K. Bowden, I. M. Heilbron, E. R. H. Jones and B. C. L. Weedon, J. Chem. Soc., 1946, 39–45 RSC.
- (a) S. H. Stickley and J. C. Martin, Tetrahedron Lett., 1995, 36, 9117–9120 CrossRef CAS; (b) G.-C. Ge, D.-L. Mo, C.-H. Ding, L.-X. Dai and X.-L. Hou, Org. Lett., 2012, 14, 5756–5759 CrossRef CAS PubMed; (c) M. C. Bagley, C. Brace, J. W. Dale, M. Ohnesorge, N. G. Phillips, X. Xiong and J. Bower, J. Chem. Soc., Perkin Trans. 1, 2002, 1663–1671 RSC; (d) S. Chassaing, M. Kueny-Stotz, G. Isorez and R. Brouillard, Eur. J. Org. Chem., 2007, 2438–2448 CrossRef CAS.
- S. Liao, J. Liu, L. Yan, Q. Liu, G. Chen and L. Ma, RSC Adv., 2020, 10, 37014–37022 RSC.
- M. Quaranta, M. Murkovic and I. Klimant, Analyst, 2013, 138, 6243–6245 RSC.
- A. A. Abdel-Shafi and D. R. Worrall, J. Photochem. Photobiol., A, 2005, 172, 170–179 CrossRef CAS.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radical Biol. Med., 2010, 49, 317–322 CrossRef CAS PubMed.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Taylor & Francis, 2nd edn, 1993 Search PubMed.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- L. V. Lutkus, S. S. Rickenbach and T. M. McCormick, J. Photochem. Photobiol., A, 2019, 378, 131–135 CrossRef CAS.
- M. Orfanopoulos, Photochem. Photobiol., 2021, 97, 1182–1218 CrossRef CAS.
- (a) G. Urgoitia, R. SanMartin, M. T. Herrero and E. Domínguez, Chem. Commun., 2015, 51, 4799–4802 RSC; (b) B. P. Taduri, S. M. A. Sohel, H.-M. Cheng, G.-Y. Lin and R.-S. Liu, Chem. Commun., 2007, 2530–2532 RSC; (c) S. R. Hart, D. C. Whitehead, B. R. Travis and B. Borhan, Org. Biomol. Chem., 2011, 9, 4741–4744 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, copies of NMR, UV-vis spectra and details of the mechanistic investigation. See DOI: https://doi.org/10.1039/d4gc00087k |
| This journal is © The Royal Society of Chemistry 2024 |