
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhombohedral ZnIn2S4-catalysed anodic direct electrochemical oxidative cleavage of C–O bond in α-O-4 linkages in ambient conditions†
Qi
Zhu
a,
Bo
Gong
a,
Shuquan
Huang
ac,
Yangxin
Jin
a,
Shengqin
Liu
a,
Shan
Shao
a,
Yuwei
Yang
d,
Taren
Cataldo
 d,
Nicholas M.
Bedford
d and
Jason Chun-Ho
Lam
*ab
d,
Nicholas M.
Bedford
d and
Jason Chun-Ho
Lam
*ab
aSchool of Energy and Environment, City University of Hong Kong, Kowloon Tong, Hong Kong SAR. E-mail: jason.lam@cityu.edu.hk
bState Key Laboratory of Marine Pollution, City University of Hong Kong, Kowloon Tong, Hong Kong SAR
cFaculty of Chemical Engineering, Kunming University of Science and Technology, Kunming, PR China
dSchool of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia
First published on 29th February 2024
Abstract
The electrochemical selective oxidative transformation of lignin feedstocks into valuable oxygenated aromatics is essential to establish a sustainable biorefinery. In this study, we used a rhombohedral ZnIn2S4 (R-ZIS) electrocatalyst to realise the efficient anodic cleavage of C–O bonds in benzyl phenyl ether (BPE), an α-O-4 lignin model compound, to a series of industrially relevant oxygenated mono-aromatics. The reaction occurred at +2 VAg/AgCl, 65 °C, and atmospheric pressure. In optimised conditions, the reaction achieved over 99% conversion rate with a Faraday efficiency of 51.6% at 91.3% conversion of BPE, that is greater than most known electrocatalytic oxidative cleavage examples. The ZnIn2S4 electrocatalyst was deposited on a carbon cloth support, and its structural properties and surface morphology were extensively investigated. Systematic potential controlled electrolysis coupled with 18O isotopic labelling confirmed that C–O scission occurred exclusively between the benzylic carbon and phenolic oxygen. Furthermore, substrate competition studies were conducted to compare the electrocatalytic performance of the R-ZIS catalyst with that of its structural analogue, hexagonal ZnIn2S4, in terms of BPE and mono-aromatics conversion. The experimental results were supported by density functional theory calculations. The substrate scoping study revealed the suitability of the R-ZIS induced electrocatalytic system for a variety of substituted α-O-4/β-O-4 model dimers. Overall, this work demonstrates an efficient anodic process that can enable the atom-efficient valorisation of lignin to produce oxygenated aromatics. The proposed approach can complement the numerous existing reductive lignin cleavage methods.
Introduction
The catalytic cleavage of lignin linkages is essential for establishing sustainable biorefinery processes for producing renewable chemicals and biofuels. Lignin is a complex three-dimensional recalcitrant polymer composed of three phenylpropane monomers (syringyl, guaiacyl, and p-hydroxyphenyl units) bonded through seven types of known linkages: β-O-4, α-O-4, 4-O-5, β-5, β-1, 5-5, and β–β. Many of these linkages contain C–O bonds in various locations. Thus, an efficient and mild strategy for achieving C–O scission is essential to promote lignin valorisation.1,2 Thus far, β-O-4 linkages have received the most attention due to their abundance in lignin.3,4 On the other hand, α-O-4 linkages remain underexplored, even though their cleavages are of comparable importance for generating aromatic products that can be applied in industries such as pharmaceuticals, food additives, fragrances, and fuels.5,6 At present, commercial chemicals and fuels are predominantly manufactured from petroleum feedstocks, and their oxygen-derived nature necessitates additional chemical oxidation processes to functionalise the structure with the desired oxygenated groups. For example, the petroleum synthesis route for guaiacol involves alkylating petroleum benzene with propylene to yield cumene, which is oxidised at the isopropyl chain to yield phenol. This phenol undergoes hydroxylation to yield catechol, and one of the hydroxyl groups is methylated to produce guaiacol.7 In contrast, guaiacol can be directly obtained from lignin through hydrothermal degradation or pyrolytic cleavage.8 However, other oxidative transformations typically necessitate additional stoichiometric oxidants, such as PhNO2,9 KMnO4,10 peracetic acid,11 and hypervalent(III) iodine,12 resulting in poor atom economy and waste generation.13A promising solution to address these challenges is the catalytic valorisation of lignin for the production of small aromatic precursors. Lignin-derived aromatics are naturally oxygenated and thus provide a more atom-efficient pathway to access oxygenated compounds.2,4 Moreover, if this process could be achieved in ambient conditions powered by renewable energy,14 it would be even more attractive from the perspective of green chemistry.15
Electrocatalytic upgrading represents a suitable approach to realise this process, as it can enable chemical redox reactions in mild conditions by applying a potential bias across an electrode pair.16,17 Unlike conventional thermal catalytic degradation, which typically occurs in high-temperature (several hundred degrees) and highly pressurised environments, electrocatalysis can achieve comparable redox outcomes at temperatures below the boiling point of water and atmospheric pressure.18 The redox processes are driven by the applied potential bias without the need for chemical reagents (unless the redox is electrochemically mediated). In some cases, electrochemical reactions can even be designed to simultaneously co-process two different streams to maximise the reaction outputs.19,20
However, while environmentally friendly, the electrocatalytic oxidation of organic substrates in an aqueous electrolyte often experiences competition from the oxygen evolution reaction (OER), resulting in a low faradaic efficiency (FE). The issue is exacerbated when oxidising recalcitrant substrates, such as lignin, as the anodic reaction may prioritise OER or even promote electrode surface corrosion instead of lignin oxidation. To address this problem, researchers have developed non-active electrodes that can generate reactive oxygenated species (ROS), e.g., OH radicals (OH*) to induce C–O cleavage (Fig. 1). For example, Wan's group used OH* generated from H2O2 to cleave α-O-4 lignin models under an O2 atmosphere. However, the electrocatalytic depolymerisation rate of α-O-4 dimers was only 1.87%–65.1% at 20–80 °C. The same group reported another OH*-triggered cleavage strategy using a RuO2–IrO2/Ti mesh electrode, resulting in improved conversion efficiency of 92.5%–96.2% and better FE of 7.3%–29.5% in ionic liquids.21–23 Although this approach is promising, the continuous supply of O2 and use of ionic liquids may incur significant costs when scaled. Moreover, the majority of electrocatalytic oxidation of lignin linkages (e.g. β-O-4 model dimers) are conducted in pure organic solvent (e.g. CH3CN, THF) or strong alkaline electrolytes (e.g. KOH), and is often accompanied by the use of extra mediators (e.g. TEMPO, tBuOOH) for the cleavage (Table S1†). The use of a mediators, which are typically a homogeneous reagent, necessitates post-reaction product separation and poses additional cost that may diminish the feasibility of the biorefinery process.16,24 Therefore, the development of an effective electrocatalyst that can directly induce oxidative cleavage without any stoichiometric oxidants or redox mediator would provide more mild and green pathways to access renewable aromatic products from lignin.
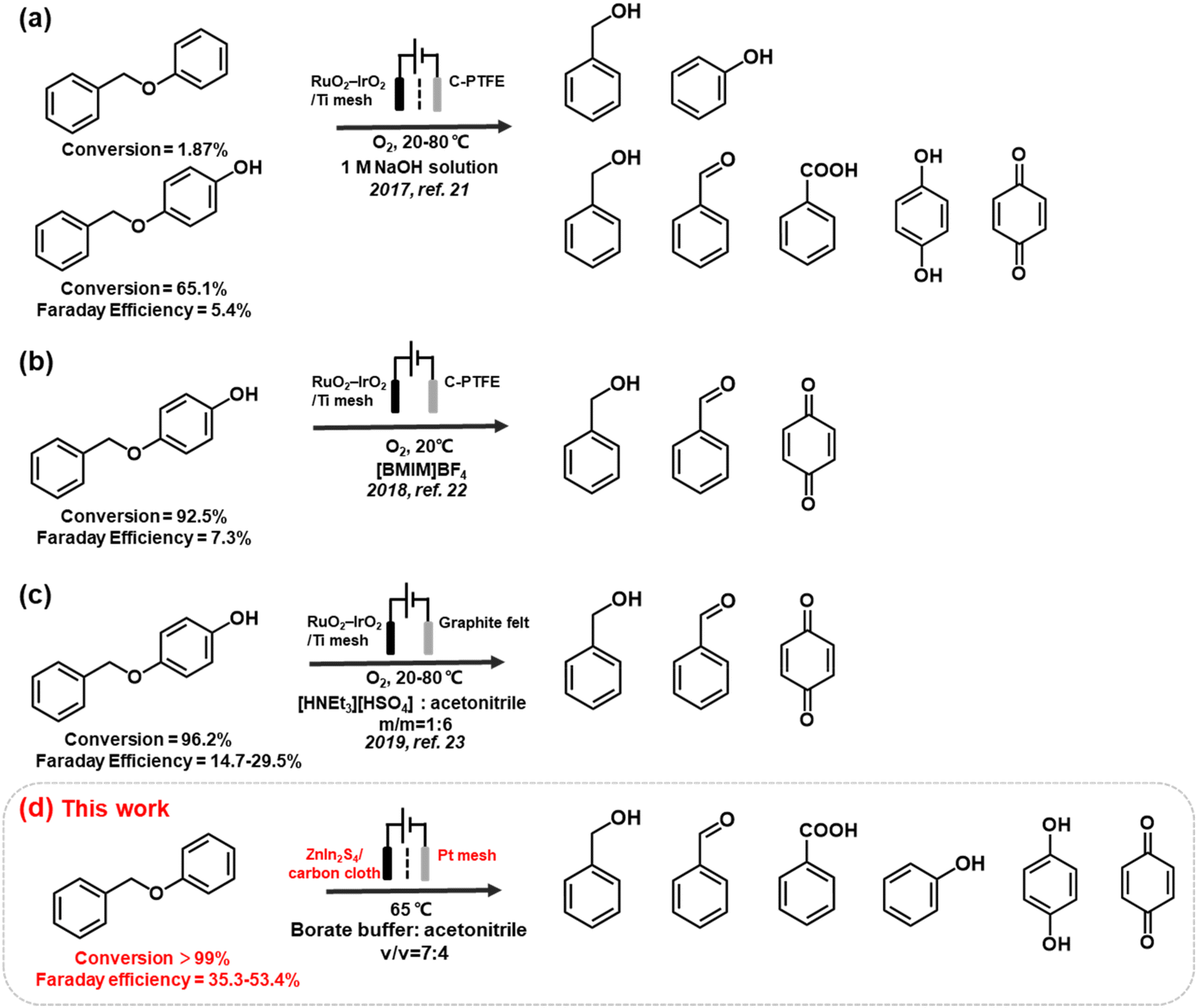 | ||
| Fig. 1 Electrocatalytic oxidative cleavage strategies used for the depolymerisation of α-O-4 lignin dimers. | ||
In this study, we developed an anodic material, ZnIn2S4, which is a ternary chalcogenide material,25,26 to enable the direct oxidative C–O cleavage of α-O-4 linkages at 65 °C in mild pH = 9 borate buffer solution with ∼35% acetonitrile as cosolvent. Recently, ZnInS4 have shown its promising photocatalytic performances in selective upgrading of lignin linkages into high-value aromatic products and have attracted much research attention in different catalysis fields.27 As a chalcogenide, ZnInS4 contains multiple phases, including cubic, rhombohedral, and hexagonal phases, and their transitions can be controlled via surface thermal treatment.28,29 We developed a facile protocol to deposit ZnIn2S4 onto carbon cloth support and modified the distribution of the surface phases through annealing treatment. The electrocatalytic performances of the hexagonal and rhombohedral phases and their mixtures were evaluated and optimized for α-O-4 cleavage. The results indicated that rhombohedral ZnIn2S4 (R-ZIS) achieved a conversion rate of >99% and generated a variety of aromatic products with widespread industrial applications. The cleavage mechanism was investigated through 18O-isotope labelling experiments, substrate competition studies, and density functional theory (DFT) calculations. All organic transformations were electrocatalytically driven under ambient conditions. We then expand the substrate scope a list of β-O-4 compounds to showcase versatility and all showed great conversion. The as-prepared electrodes demonstrated great recyclability, and it contained only 2.5 wt.% of In deposited on a carbon support, a loading comparable to those of commercially available catalysts, such as 5% Pt/C, 5% Pd/C or Pt sheet electrode used in related lignin valorization study.
This novel and mild strategy represent a safer and more environmentally friendly protocol to promote biorefinery by enabling a direct cleavage of lignin-relevant C–O linkage in ambient conditions to generate oxygenated aromatic chemicals. Moreover, electrochemical oxidation bypasses the need for stoichiometric oxidants, which 1) enhance the atom efficiency of the reaction and 2) avoid generating potentially hazardous byproducts. These reasons all align well with the green chemistry principles.
Experimental section
Preparation of ZnIn2S4 electrode on carbon support
The carbon support (carbon cloth, CeTech Co. Ltd, Type: WOS1009) measuring 1 × 1 cm was washed in 98% H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70% HNO3:deionised (DI) water (1:1:1) at 70 °C for 24 h. The acidified cloth was then rinsed with DI water and calcined at 500 °C for 2 h, thereby improving the surface hydrophilicity.30 The layered hexagonal ZnIn2S4 electrode was prepared through a one-step hydrothermal reaction. Specifically, 136 mg zinc chloride (ZnCl2), 442 mg indium chloride (InCl3), and 600 mg thioacetamide (TAA) were dissolved in 30 mL of N,N-dimethylformamide (DMF) with vigorous stirring for 20 min. The resulting mixture was transferred to a 50 mL Teflon-lined stainless-steel autoclave containing the treated carbon cloth, which was then heated in an oven at 170 °C for 24 h. After cooling to room temperature (23 ± 2 °C), the ZnIn2S4-decorated carbon electrode was washed with ethanol and DI water and labelled as H-ZIS, as the ZnIn2S4 exhibited the hexagonal phase in this stage.
:70% HNO3:deionised (DI) water (1:1:1) at 70 °C for 24 h. The acidified cloth was then rinsed with DI water and calcined at 500 °C for 2 h, thereby improving the surface hydrophilicity.30 The layered hexagonal ZnIn2S4 electrode was prepared through a one-step hydrothermal reaction. Specifically, 136 mg zinc chloride (ZnCl2), 442 mg indium chloride (InCl3), and 600 mg thioacetamide (TAA) were dissolved in 30 mL of N,N-dimethylformamide (DMF) with vigorous stirring for 20 min. The resulting mixture was transferred to a 50 mL Teflon-lined stainless-steel autoclave containing the treated carbon cloth, which was then heated in an oven at 170 °C for 24 h. After cooling to room temperature (23 ± 2 °C), the ZnIn2S4-decorated carbon electrode was washed with ethanol and DI water and labelled as H-ZIS, as the ZnIn2S4 exhibited the hexagonal phase in this stage.
To induce the hexagonal-to-rhombohedral phase transition, the H-ZIS electrodes were annealed in Ar at various temperatures for 2.5 h, with a heating rate of 3 °C min−1 in a tube furnace. After the heat treatment, the tube was allowed to cool to room temperature under a supply of Ar gas. The resulting ZnIn2S4 electrodes with mixed hexagonal and rhombohedral phases obtained at 400 °C and 600 °C were labelled as ZIS-400 and ZIS-600, respectively. The pure rhombohedral ZnIn2S4 electrode was obtained at 800 °C and labelled as R-ZIS.
Material characterisation
Powder X-ray diffraction (XRD) measurements were conducted using a PANalytical X'Pert3 powder (Netherlands) X-ray diffractometer using Cu Kα radiation. Scanning electron microscopy (SEM) images were obtained using a Zeiss Sigma 500 scanning electron micro-analyser operating at 15 kV and equipped with an energy-dispersive X-ray (EDS) spectrometer. Elemental analysis was performed using an inductively coupled plasma (ICP) optical emission spectrometer (OPTIMA 8000, PerkinElmer, USA). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were conducted using an FEI Talos F200X G2 transmission electron microscope. Raman spectra were obtained using a dispersive Raman microscope (Renishaw InVia Qontor) with a solid-state 532 nm excitation laser. Attenuated Total Reflectance Fourier-transform infrared (ATR-FTIR) spectra were acquired using the Shimadzu IR Affinity-1 spectrometer. X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA) measurements were performed using monochromatic Al Kα radiation to determine the valence state and phase transition of elements. All collected data were calibrated against the C 1s peak at 284.8 eV. The Zn K edge X-ray absorption spectroscopic (XAS) measurements were obtained at the SAMBA beamline of the Soleil Synchrotron. The samples were measured using transmission mode and probed from ∼150 eV below to ∼700 eV above the edge. X-ray absorption near-edge structure (XANES) measurements of S K edge were acquired at the SXRMB beamline of the Canadian Light Source. The samples were measured under vacuum using an X-ray fluorescence yield detector and probed from ∼20 eV below to ∼100 eV above the edge. The In K edge XAS measurements were obtained at the XAS beamline of the Australian Synchrotron. The samples were measured using fluorescence geometry and probed from ∼150 eV below to ∼900 eV above the edge. Data were processed and modeled using the Demeter XAS software package (Athena and Artemis).31 For the extended X-ray absorption fine structure (EXAFS) modeling, the reported structure for Zn(InS2)2 was used to generate Zn–S and In–S contributions.32S02 values for In (0.84), and Zn (0.79) were obtained by modeling the EXAFS of reference In and Zn foils, respectively.General electrolysis for electrocatalytic oxidation
An H-cell with air-tight covers (purchased from Gaoss Union Technology Co., Ltd) separated by a Nafion 1110 membrane was used for the general electrolysis reactions. The prepared ZnIn2S4 electrode was placed in the anode chamber as the working electrode in 10 mL of an electrolyte composed of 7 mL of 0.2 M pH 9 borate buffer and 3 mL of acetonitrile (MeCN) cosolvent. The model compound, BPE (20.27 mg, >98% (GC), Aladdin Bio-Chem Technology Co., Ltd), was dissolved in 1 mL of MeCN and then added to the anolyte. The working potential was measured against a 3.5 M Ag/AgCl reference electrode. A platinum (Pt) mesh sized 1 × 1 cm was used as the counter electrode, placed inside the cathode compartment loaded with the same electrolyte but without the BPE. The setup was immersed in a 65 °C oil bath, and the anolyte was stirred at 400 rpm. An initial sample (T0) was collected from the anolyte before the potential bias was applied. For the substrate scope expansion work, the synthesis procedures of various lignin model compounds are included in the ESI.†Product analysis
For qualitative analysis, the electrolysis products were extracted, dehydrated, and acidified before being analysed by a gas chromatography (GC, Shimadzu QP2010 Ultra) device equipped with a mass spectrometry (MS) detector and HP-5 column. Helium was used as the carrier gas, and the injected sample volume was 1 μL. A temperature programme was implemented during the measurement: 2-min isothermal hold at 60 °C, ramp of 15 °C min−1 to 250 °C with a 10-min hold, and a ramp of 20 °C min−1 to 220 °C with a 5-min hold. For quantitative analysis, the liquid sample was filtered using a 0.22 μm filter, diluted, and analysed through high-performance liquid chromatography (HPLC, Agilent 1200). Compound separation was achieved using a gradient method, in which the MeCN:H2O ratio was varied from 30:70 (v/v) to 70:30 (v/v) over 40 min at a flow rate of 0.6 mL min−1 on a dC18 column (Waters Atlantis).
The substrate conversion rates (Conv.), carbon balance (C. Bal.), FE, and product yields were calculated using eqn (1)–(4).
 | (1) |
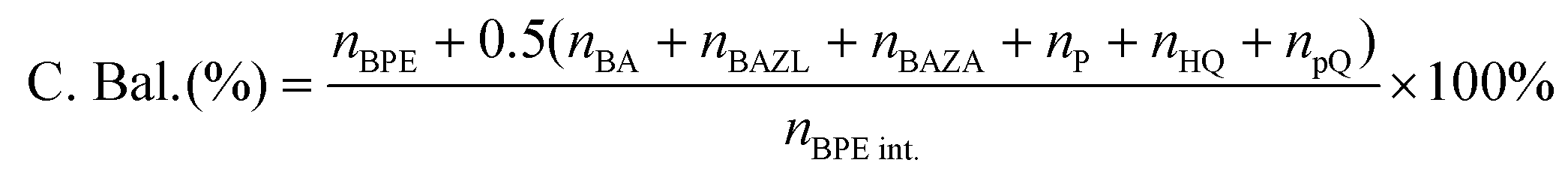 | (2) |
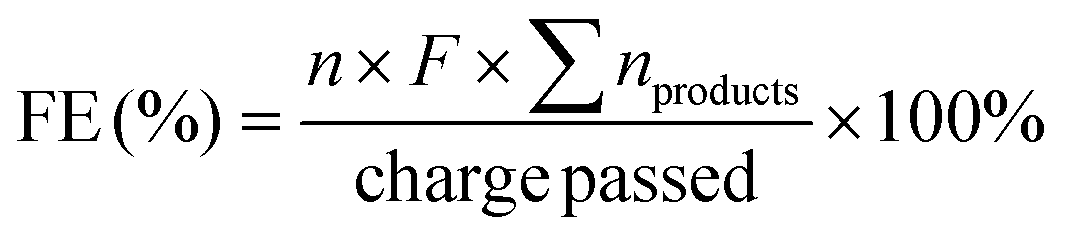 | (3) |
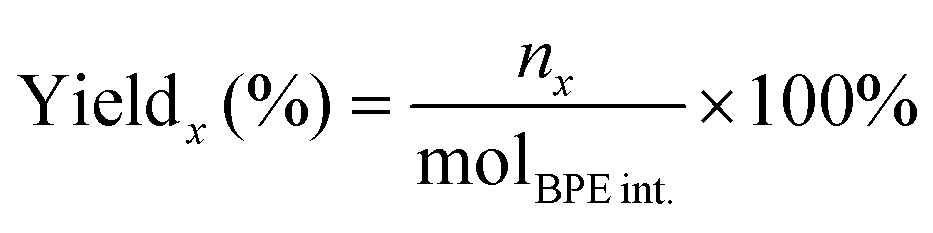 | (4) |
485 C mol−1), respectively.
DFT calculations
All DFT calculations were performed using the PWscf module in Quantum ESPRESSO.33 The generalised gradient approximation in the form of the functional developed by Perdew, Burke, and Ernzerhof (GGA-PBE) and ultrasoft pseudopotential were chosen to describe the exchange–correlation energy and simulate the electron–ion core interactions.34,35 The DFT-D2 method was used to consider dispersion effects, a form of van der Waals interaction.36 The cutoff energies for the wavefunctions and augmented part of the charge density were set as 30 and 240 Ry, respectively. Geometrical relaxations were performed with an energy threshold of 1.0 × 10−4 Ry and force threshold of 1.0 × 10−3 Ry per Bohr. A 2 × 2 × 1 Monkhorst–Pack k-point mesh was used for all calculations. Bulk crystals of hexagonal ZnIn2S4 (a = b = 3.92 Å, c = 25.31 Å, α = β = 90°, γ = 120°) and rhombohedral ZnIn2S4 (a = b = 3.93 Å, c = 37.89 Å, α = β = 90°, γ = 120°) were used. The (110) facet with an initial four-layer model, consisting of eight and twelve ZnIn2S4 units for hexagonal and rhombohedral phases, respectively, was constructed for the calculations.Discussion
Electrode morphology and structural phase analysis
Uniform hexagonal ZnIn2S4 (H-ZIS) nanosheets were fabricated on carbon cloth through solvothermal treatments (Fig. 2a). Compared with the plain carbon cloth that is smooth and black surface (Fig. S1†), the SEM and EDS analyses revealed that H-ZIS formed a cluster of nanoflower arrays perpendicular to the surface of the carbon cloth (Fig. S2a–c†). XRD analysis confirmed that the H-ZIS single crystals (yellow podwer) exhibited a hexagonal pattern (JCPDS card No. 72-0773) with no impurity peaks (Fig. 2b).37 When H-ZIS was annealed at 400–600 °C in Ar, the ZIS elemental composition remained unchanged, but the surface phase transitioned from hexagonal to rhombohedral, resulting in a more aggregated morphology (Fig. S2d–i†). The XRD analyses of ZIS-400, ZIS-600, and R-ZIS indicated that their crystallinity improved during the phase transition. The appearance of three new peaks at ∼28.3°, 33°, and 46.9° in ZIS-600 corresponded to the (104), (018), and (110) crystal planes of rhombohedral ZnIn2S4. At 800 °C, the XRD pattern matched well with the JCPDS (No. 49-1562), which confirmed that the observed phase was pristine rhombohedral with the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.29 The woven structure of carbon cloth maintained after the R-ZIS catalyst coating, and the catalysts were deposited with great uniformity across the woven strands (Fig. 2c and d). The cross-section of the electrode after deliberately scratched off randomly to show the R-ZIS was evenly coated on the carbon cloth surface with a thickness of 100–300 nm (Fig. S3†).
m space group.29 The woven structure of carbon cloth maintained after the R-ZIS catalyst coating, and the catalysts were deposited with great uniformity across the woven strands (Fig. 2c and d). The cross-section of the electrode after deliberately scratched off randomly to show the R-ZIS was evenly coated on the carbon cloth surface with a thickness of 100–300 nm (Fig. S3†).
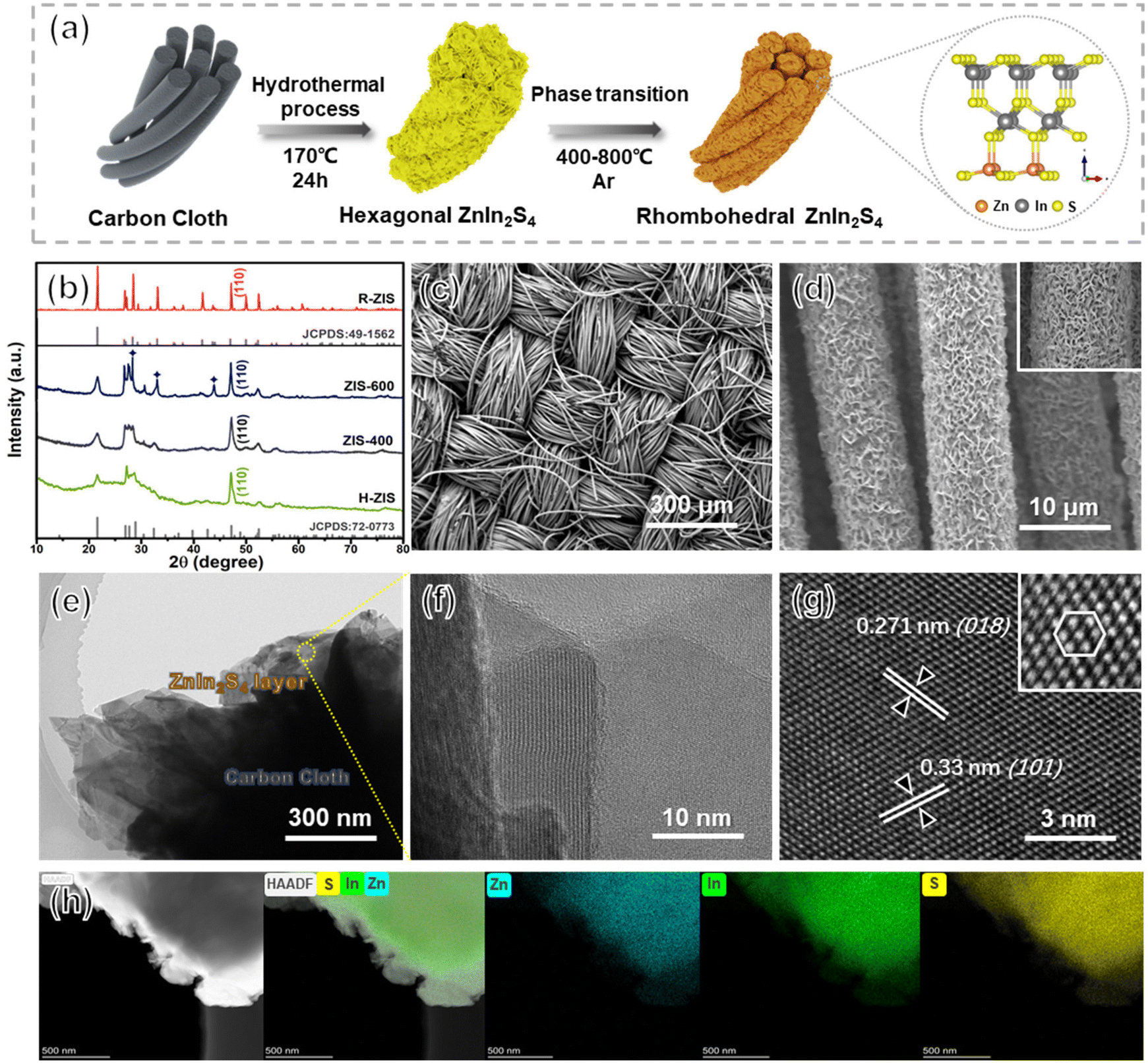 | ||
| Fig. 2 (a) Preparation of the ZnIn2S4 electrodes through hydrothermal and annealing treatment processes. (b) XRD patterns of the as-prepared H-ZIS, ZIS-400, ZIS-600, and R-ZIS samples and their reference cards. (c and d) FESEM images of the R-ZIS electrode. (e and f) TEM images, (g) HRTEM image, and (h) EDS mappings of the R-ZIS electrode. | ||
The interfacial arrangement of R-ZIS was examined using TEM. The well-ordered epitaxial growth of R-ZIS showed a distinguishable layered structure, suggesting the intimate combination between the components (Fig. 2e).38 The crystallinity of the R-ZIS was supported by a clear lattice planes image in the layered R-ZIS at nanometer level (Fig. 2f). According to the HRTEM analysis (Fig. 2g), the d-spacing between the atomic layers was 0.271 nm and 0.33 nm, corresponding to the (018) and (101) lattice planes of the Rm rhombohedral structure, consistent with the XRD pattern. The inset HRTEM image in Fig. 2g indicated that a clear hexagon was formed by the In and Zn atoms, reflecting the rhombohedral crystallographic morphology.39,40 The EDS element mapping disclosed the uniform distribution of Zn, In, and S elements in the R-ZIS electrode with less than 0.5% of the O element was detected, which originated from adsorbed water molecules in atmospheric environment (Fig. 2h). The ICP analysis revealed that the In content of the R-ZIS electrode was 1.27 mg cm−2, corresponding to a loading of 2.5 wt% of In on the carbon cloth (Table S2†). Meanwhile, the element ratio of Zn and In was 1:2, which was consistent with the elemental distribution in ZnIn2S4 material. The collective observations from Fig. 2b–h confirmed that the R-ZIS electrode was successfully deposited onto the carbon support with a high level of crystallinity.
Raman spectroscopy was performed to gain additional insights into the crystal structure and phonon vibrations (Fig. 3a). The H-ZIS sample synthesised through the solvothermal process exhibited the characteristic band of Raman shifts, consistent with a typical ZnIn2S4 sample.41 As the temperature increased from 400 to 800 °C, the 200–400 cm−1 characteristic peak intensity gradually increased, potentially because of the enhanced crystallinity and morphological changes. The vibration peaks at 241 and 301 cm−1 were attributable to the longitudinal optical mode (LO1) and transverse optical mode (TO2), originating from A1g-like vibrations corresponding to the out-of-plane breathing modes of layered ZnIn2S4.42 The 350 cm−1 peak represented Eg-like features associated with the in-plane breathing modes. The blue shift of the A1g-like peaks from H-ZIS to R-ZIS was attributable to the increase in the van der Waals force in the thicker R-ZIS samples.40 This observation was consistent with the surface topography changes observed in the SEM analysis. Additionally, a peak at 335 cm−1 emerged in ZIS-600 and R-ZIS due to the formation of the rhombohedral phase and enhanced crystallinity.28
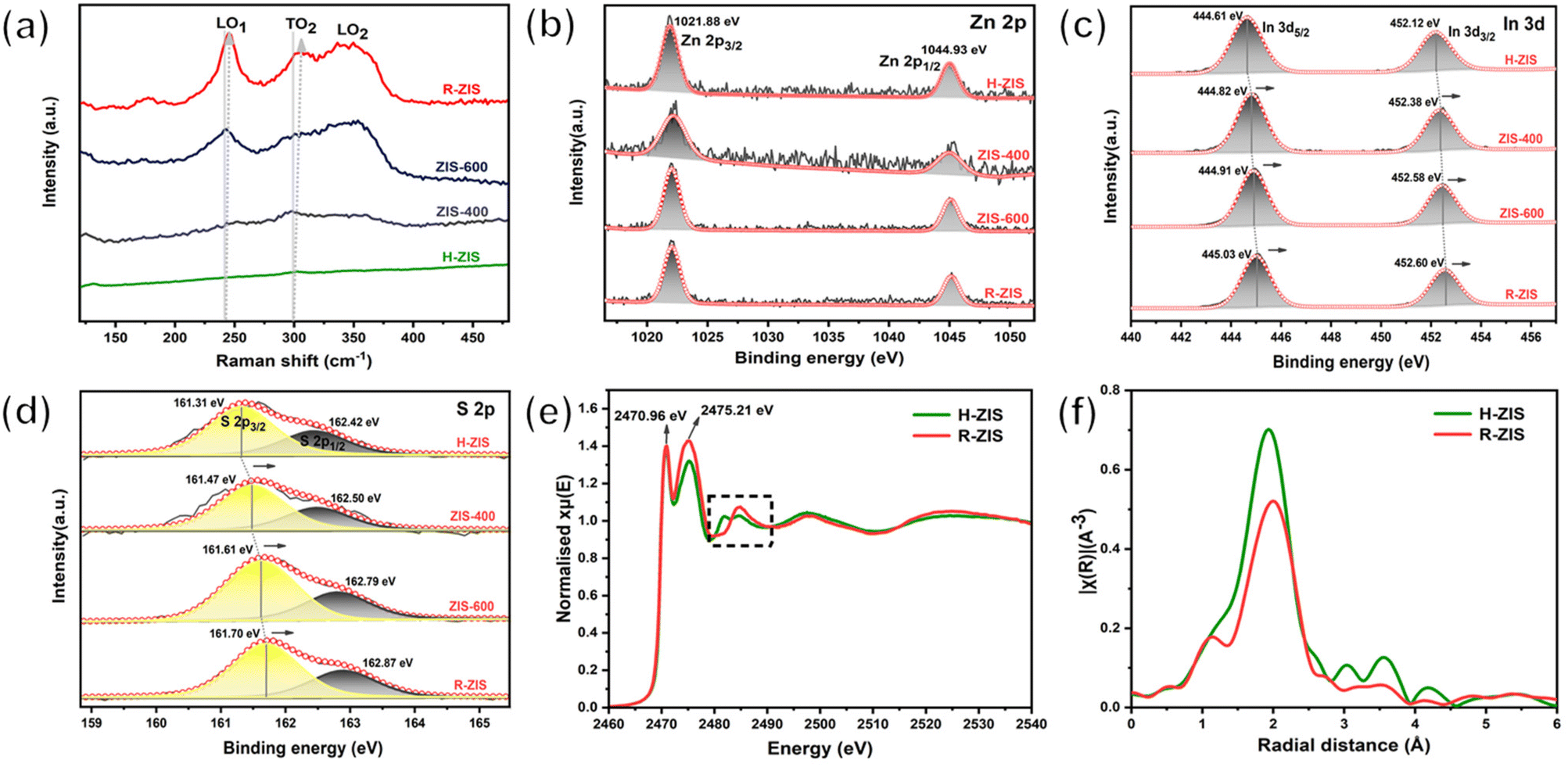 | ||
| Fig. 3 (a) Raman spectra of H-ZIS, ZIS-400, ZIS-600, and R-ZIS electrodes. (b–d) XPS spectra of Zn 2p, In 3d, and S 2p for H-ZIS, ZIS-400, ZIS-600, and R-ZIS electrodes. (e) Normalised S K-edge XANES spectra and (f) Fourier transform of In K-edge EXAFS spectra in the R-space of the H-ZIS and R-ZIS electrodes. | ||
The chemical states of Zn, In, and S in the ZIS electrodes were confirmed by high-resolution XPS. The ZnIn2S4 samples exhibited two stable characteristic peaks at 1021.88 eV and 1044.93 eV in Zn 2p spectra, attributable to Zn 2p3/2 and Zn 2p1/2, respectively, consistent with the +2 valence state of Zn ions (Fig. 3b).40 The fitted In 3d spectra of H-ZIS exhibited two well-defined peaks at 444.61 and 452.12 eV (Fig. 3c), characteristic of the In 3d5/2 and In 3d3/2 branches of In3+ ions, respectively.42 For the S 2p states of H-ZIS, the main peak could be fitted into two peaks at 161.31 and 162.42 eV, attributable to S 2p1/2 and S 2p3/2, respectively (Fig. 3d). Notably, both the In 3d and S 2p characteristic peaks shifted to higher binding energies during the formation of a two-phase state from the pure hexagonal crystal phase, indicating crystal phase transition and interfacial electronic coupling between the hexagonal and rhombohedral ZnIn2S4.39 To investigate the alterations in local structure accompanying the hexagonal-to-rhombohedral phase transition, X-ray absorption spectroscopy (XAS) was conducted, focusing on the Zn K-edge, In K-edge, and S K-edge. Analysis of the XANES spectra at the Zn and In K-edges revealed a decrease in the intensity of the white line peak after heating treatment of ZnIn2S4 to 800 °C (Fig. S4a and b†). Conversely, an increase in the white line intensity was noted in the S K-edge spectrum, indicative of Zn 4s-S 3p and In 5s-S 3p hybridization at 2470.96 eV and 2475.21 eV, respectively (Fig. 3e). Additionally, alternations in peak characteristics were discerned in the black squared features at the S K-edge, typically associated with alterations in the second shell coordination around the S atom. These changes in the electronic structure collectively imply a transformation in the local coordination geometry surrounding the Zn, In, and S atoms, with the most pronounced changes observed near the In atoms. This observation aligns with the expectation that Zn maintains octahedral local coordination in both hexagonal and rhombohedral structures, whereas a significant shift in the octahedron/tetrahedron coordination ratio is anticipated in the In-S local environment due to the hexagonal-to-rhombohedral phase transition. To corroborate these findings, the EXAFS spectra at the In K-edge and Zn K-edge were examined, along with the corresponding fitting results (as depicted in Fig. 3f and Fig. S4c–h†). The EXAFS spectra and wavelet transform (WT) contour for both Zn and In K-edges exhibited a singular first coordination shell, corresponding to Zn–S and In–S local coordination, respectively. Notably, the In–S peak in the R-ZIS phase exhibited a negative shift towards a shorter bond length, potentially attributable to a more distorted structure and an altered octahedron/tetrahedron coordination ratio in the In–S local environment due to the phase transition (Fig. 3f). This extended bond length was further substantiated by the fitting results, which indicated an In–S bond length of 2.56 ± 0.01 Å in the R-ZIS, elongated by 0.04 Å compared to the H-ZIS (Fig. S4h†). Additionally, the fitting results for both Zn and In K-edges suggested that the phase transition leads to a reduction in coordination numbers, with Zn–S decreasing from 4.16 ± 0.39 to 3.49 ± 0.68, and In–S decreasing from 6.27 ± 0.53 to 5.22 ± 0.56 (Fig. S4h†). These findings further support the notion of an altered local coordination environment and the potential formation of S defects following annealing treatment.
Electrocatalytic performance evaluation via potential controlled electrolysis
The electrolytic performance of the ZIS electrodes for the cleavage of α-O-4 bonds in BPE was evaluated through potential controlled electrolysis in an H-shaped divided cell separated by a Nafion 1110 membrane at 65 °C. Each ZIS electrode (H-ZIS, ZIS-400, ZIS-600, and R-ZIS) was individually used as an anode, operating at +2.0 VAg/AgCl (Fig. 4a–d) to deliver a total charge of 140 C for the electrolysis. Samples were collected periodically based on the passed charge. After 80 C, samples from the cathode analyte were also collected and combined with the anode analyte to account for any product loss due to membrane crossover. In all trials, benzyl alcohol, benzaldehyde, benzoic acid, phenol, hydroquinone, and para-benzoquinone were detected and quantified by GC-MS and HPLC using external references. The product distribution was consistent with our expectation: C–O bond cleavage in BPE yielded benzyl alcohol and phenol, followed by their secondary and tertiary oxidations.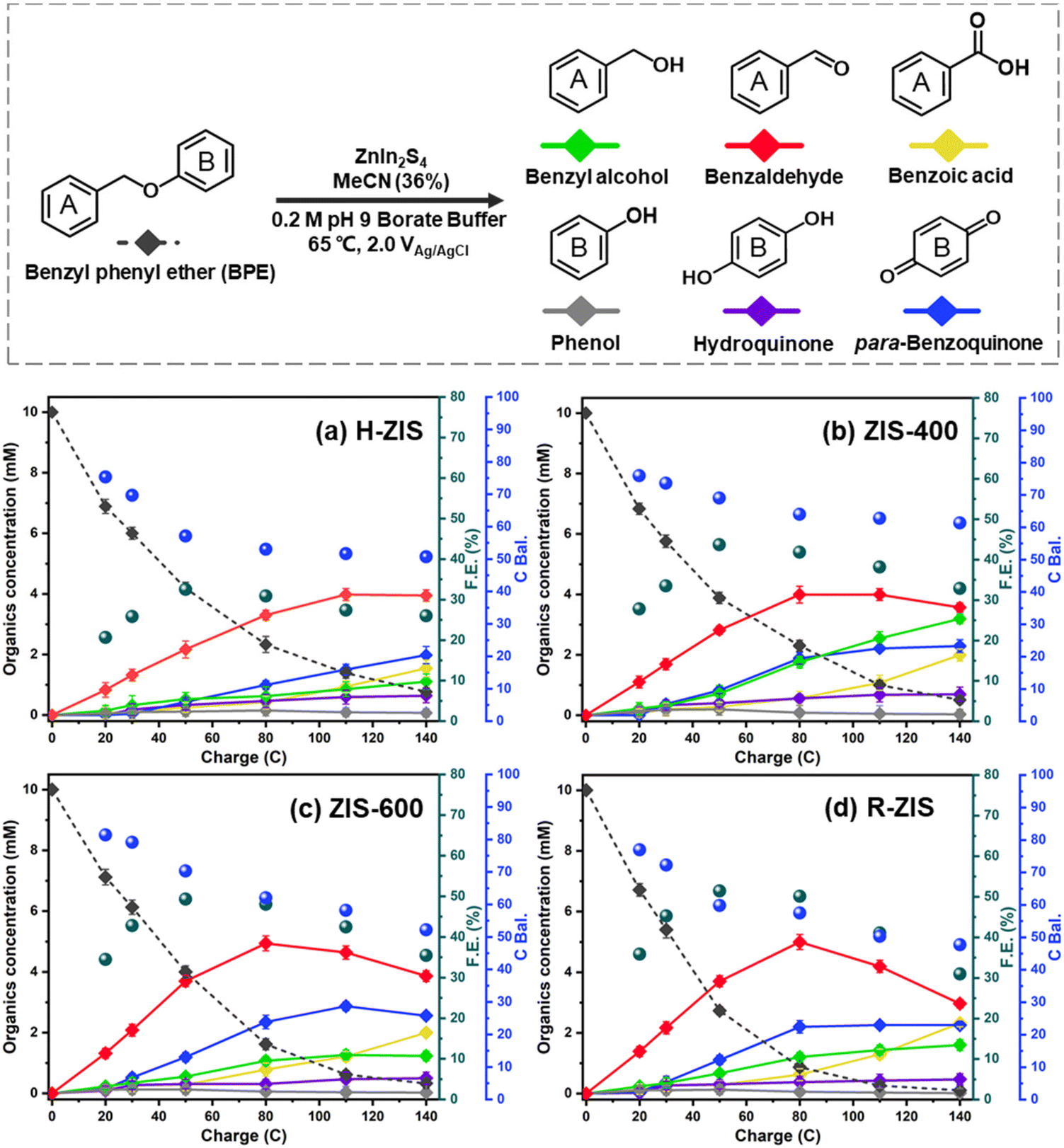 | ||
| Fig. 4 Kinetic profiles of product distribution during electrolysis on different electrodes: (a) H-ZIS, (b) ZIS-400, (c) ZIS-600, and (d) R-ZIS. | ||
As shown in Fig. 4, the BPE consumption increased from ∼93% for H-ZIS to >99% for R-ZIS, indicating that the C–O cleavage was promoted by the emergence of the rhombohedral phase. The improvement in C–O bond cleavage led to increased yields of benzyl alcohol, benzaldehyde, and benzoic acid. Comparing the amount of benzaldehyde at 80 C across the electrodes, a progressive increase of benzaldehyde from 3.30 mM with the H-ZIS to 5.05 mM with the R-ZIS was observed. Similar trends were observed for benzoic acid, which is expected, given that it is produced from benzaldehyde. The superior catalytic performance of R-ZIS suggested BPE is adsorbed and degraded more effectively on the R-ZIS surface than the H-ZIS, ZIS-400, and ZIS-600 electrodes, resulting in improved oxidative cleavage activity. To confirm the mechanism, a linear sweep voltammetry (LSV) analysis was performed on the ZIS electrodes to analyse the adsorption of BPE in the potential range of 0 to +2.7 VAg/AgCl at a scan rate of 50 mV s−1 (Fig. 5a).
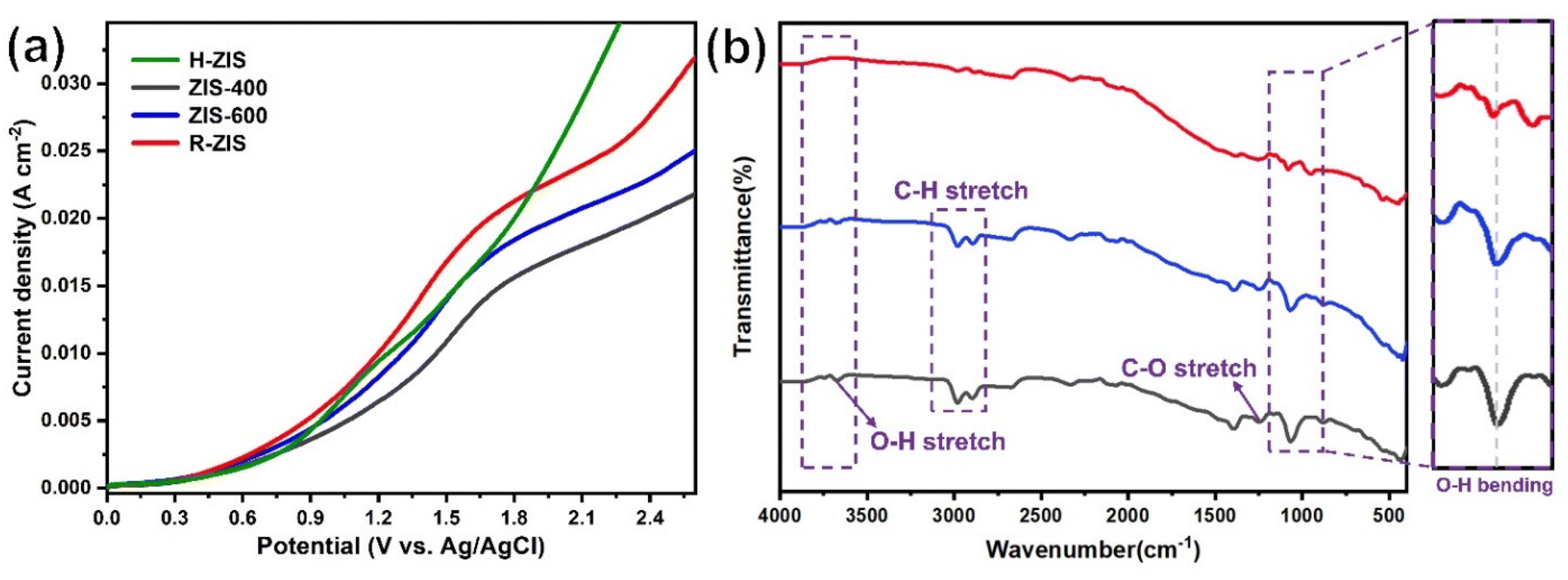 | ||
| Fig. 5 (a) Linear sweep voltammetry (LSV) curves of BPE on different ZIS electrodes at a scan rate of 50 mV s−1 in standard reaction conditions. (b) ATR-FTIR spectra of post-reaction tainted carbon cloths after adsorption and oxidation–catalysis reaction of phenol in the R-ZIS electrode. (Red) pristine R-ZIS electrode. (Blue) Adsorption of phenol in open-circuit conditions for 8 h on R-ZIS. (Black) Electrolysis of phenol for 8 h on R-ZIS. | ||
The LSV result showed that a current suppression occured between +1.6–2.0 VAg/AgCl for all the ZIS electrodes with the ZIS-400, ZIS-600, and R-ZIS showed a more apparent suppression (Fig. 5a). The suppression only appeared in the presence of BPE, which confirmed the adsorptive behavior of BPE on the electrodes’ surface (Fig. S5†). In the absence of BPE, the current continued to rise with the working potential due to OER, as water was the only oxidizable component in the electrolyte. The current inhibition is comparable to what is observed during the oxidative electrolysis of carboxylic acid, aka Kolbe electrolysis, where the carboxylate anion forms a layer of inhibitive film on the anode catalyst surface to inhibit the OER current.43 Another worth noting trend is that the H-ZIS only showed a weak adsorptive inhibition compared to the other ZIS variants, suggesting it was mainly performing OER, and the result is consistent with the lower overall FE observed with H-ZIS. The observation is also consistent with the DFT calculation that revealed BPE was better adsorbed on the R-ZIS than the H-ZIS surface.
In terms of the phenolic compounds, the amounts of phenol and hydroquinone remained low on all the ZIS electrodes, and their oxidised terminal product (based on the detectable amount), i.e., para-benzoquinone, experienced a small growth as the electrode transitioned from H- to R-ZIS. We hypothesised that the limited amounts of phenolics observed in the electrolyte were attributable to their adsorptive retention on the ZIS surface and subsequent oxidative degradation. Post-electrolysis analysis of the R-ZIS electrodes with surface ATR-FTIR (Fig. 5b) indicated the presence of four notable peaks at 3610–3670 cm−1 (O–H stretching), 2895–2982 cm−1 (C–H stretching), 1248 cm−1 (C–O stretching), and 1070 cm−1 (O–H bending), originating from phenol.44 These peaks were observed in both the post-electrolysis R-ZIS electrodes and open-circuit adsorption control, which confirmed that the R-ZIS could accommodate phenol adsorption and oxidation. The amount of the phenolic end-product, para-hydroquinone, increased from H-ZIS to ZIS-600 and then decreased at R-ZIS. These changes were attributable to further oxidation, as confirmed by the C. Bal. analysis: Overall trend of C. Bal. peaked at the ZIS-600 and then declined. Besides, the overall C. Bal. trend was decreasing as the reaction proceeds, indicating the gradual oxidation and formation of low-chain carboxylic acids as BPE depletes.45 The limited production of phenolic compounds (phenol, hydroquinone, and para-benzoquinone) compared with the substantial yield of the benzylic alcohol products (benzyl alcohol, benzaldehyde, and benzoic acid) provided indicative evidence that the phenolic products were more vulnerable to degradation.
The emergence of the rhombohedral phase also influenced the FE. Comparing the FE at 50 C across the ZIS, where the FE peaked, the FE was noted to increase from 32.5% (H-ZIS) to 43.6% (ZIS-400), 49.3% (ZIS-600), and 51.4% (R-ZIS). The lower FE observed for H-ZIS can be attributed to weak interaction between the BPE and H-ZIS phase, as indicated by the LSV analysis (Fig. 5a). This phenomenon resulted in less efficient C–O cleavage, lack of oxidisable organics in the electrolyte, and shifted reaction selectivity towards the OER. The progressive improvement in FE was well correlated with the increasing coverage of the rhombohedral phase, providing strong evidence that the rhombohedral surface led to enhanced adsorption and organic oxidation. This conclusion was confirmed by DFT calculations, as discussed in a later section. Meanwhile, all ZnIn2S4 electrodes exhibited a typical FE trend, where it declined gradually as the BPE depleted, shifting the reaction selectivity from BPE cleavage to OER. Because R-ZIS was the most active to deplete the BPE, its FE trend exhibited a more apparent decline compared to others. Nonetheless, it is worth noting that the FE reached 51.6% at 91.3% conversion of BPE, which is significantly greater than most known electrocatalytic oxidative cleavage of α-O-4 lignin dimers (5.4%–29.5%).21–23
Effect of the applied potential value in bulk controlled potential electrolysis on optimised ZIS crystalline phase
Having established R-ZIS as the most active electrocatalyst for BPE C–O cleavage, we further investigated the influence of working potentials on the reaction. Although the operation at 2.0 VAg/AgCl was effective, it appeared to favour the electrochemical oxidation of the mono-aromatic products over the cleavage of BPE. Thus, bulk controlled potential electrolysis at different potential values was conducted at +1.6 to +1.9 VAg/AgCl, selected based on the faradaic peak observed in the LSV analysis, to investigate the catalytic outcomes at different potentials.As shown in Fig. 6a–d, the BPE consumption rate improved as the working potential decreased. The BPE consumption rate, determined as the linear slope of the first four sample points, increased from 0.108 mM C−1 to 0.151 mM C−1 as the working potential decreased from +1.9 to +1.6 VAg/AgCl. This improvement could be attributable to either the OER suppression at a low working potential or a shift in the reaction pathway selectivity from the small organics to BPE. Upon examining the FE values across the ZIS electrodes, we observed that the BPE cleavage enhancement was attributable to the shift in the reaction pathway selectivity. In particular, the FE was not significantly affected by the working potential, indicating that the proportion of electrons used for oxidising the organic compounds remained nearly constant. If the decrease in the working potential led to OER suppression, a more dramatic shift in the FE would be expected. Moreover, the lower working potentials of +1.6 and +1.7 VAg/AgCl resulted in the retention of larger amounts of benzyl alcohol and benzaldehyde in the electrolyte as the reaction selectivity shifted to BPE oxidation. The benzyl alcohol yield at the end of electrolysis reached 3.99 mM and 3.67 mM at +1.6 and +1.7 VAg/AgCl, respectively, nearly two times the amount observed in the +1.8 and +1.9 VAg/AgCl trials. However, the change in the working potential did not improve the phenol yield, and it remained low in all conditions, owing to its adsorption and oxidation on the ZIS surface, as observed in the surface ATR-FTIR analysis (Fig. 5b). Nevertheless, the amount of para-benzoquinone increased with the working potential, suggesting that the adsorbed phenol was oxidised and released back into the electrolyte.
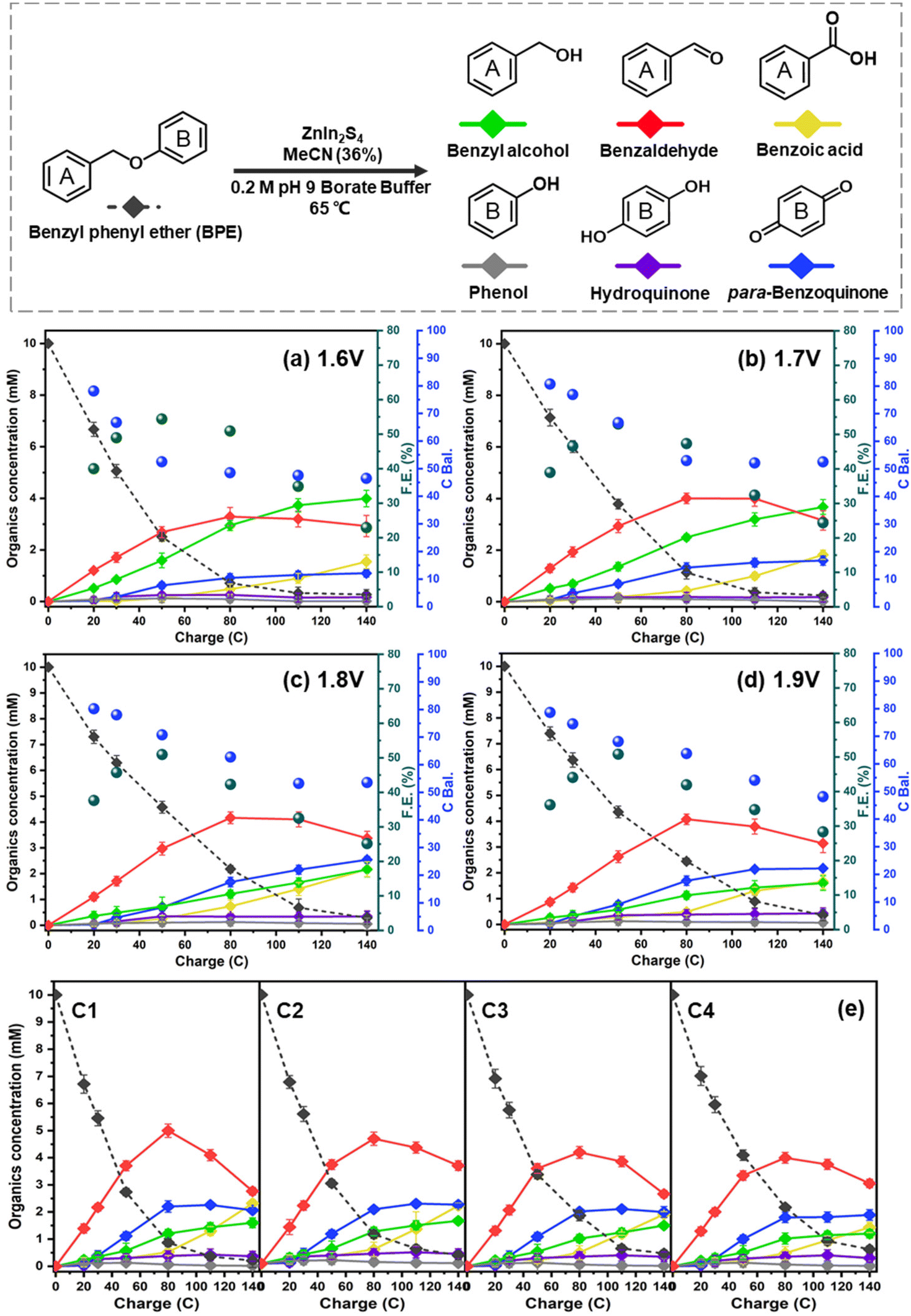 | ||
| Fig. 6 Electrolysis of BPE using the R-ZIS electrode in pH 9 buffer solutions at (a) 1.6 V, (b) 1.7 V, (c) 1.8 V, and (d) 1.9 VAg/AgCl in 65 °C. (e) Formation of products in four stability cycle tests in pH 9 buffer solutions at 2 VAg/AgCl in 65 °C. | ||
The stability of the R-ZIS electrode was evaluated by performing bulk electrolysis for four consecutive cycles at +2 VAg/AgCl and 65 °C. Even at the maximum working potential (2 V), the product-stream results exhibited consistency across the cycling tests (Fig. 6e). XRD, SEM and ICP analyses performed on the post-electrolysis samples resulted in reproducible patterns and elemental components consistent with those of the freshly prepared electrode (Fig. S6 and Table S2†).
Individual and competitive electrolysis of BPE and key intermediates
The nearly stable FE values, despite the different product distributions, suggested that BPE and its cleaved products likely competed for the same catalytic oxidation sites. To verify this hypothesis, a series of competition reactions were examined to clarify the influence of different substrates on the oxidative transformation of other substrates. Equal amounts of BPE, phenol, and benzyl alcohol were introduced into the anode compartment of an H-cell at +2.0 VAg/AgCl to investigate how they compete with BPE for electrocatalytic oxidation.In the case of H-ZIS, the phenol consumption rate (0.150 mM C−1) was nearly double that of benzyl alcohol (0.082 mM C−1). However, in the case of R-ZIS, the consumption rates of the two chemicals were nearly identical: benzyl alcohol (0.124 mM C−1) and phenol (0.149 mM C−1). When phenol and benzyl alcohol were subjected to the same electrolysis conditions on the H-ZIS surface, the conversion rate of benzyl alcohol decreased by 23.2% from 0.082 mM C−1 to 0.063 mM C−1 in the presence of an equal amount of phenol, whereas the phenol consumption rate was almost unaffected by benzyl alcohol: the corresponding values with and without benzyl alcohol were 0.153 mM C−1 and 0.150 mM C−1, respectively (Fig. 7a). On the R-ZIS electrode, the competition between benzyl alcohol and phenol improved as the consumption rate of benzyl alcohol increased by 28.6% from 0.063 mM C−1 (H-ZIS) to 0.081 mM C−1 (R-ZIS). However, the presence of phenol decelerated the oxidation of benzyl alcohol by 34.7% from 0.124 mM C−1 to 0.081 mM C−1 on the R-ZIS surface, whereas the effect of benzyl alcohol on the consumption rate of phenol was negligible (Fig. 7b). These observations confirmed that owing to its superior oxidation power, R-ZIS had a catalytic impact on benzyl alcohol but not phenol. Phenol degradation remained unchanged, regardless of the electrode surface. For the product distribution of phenol and benzaldehyde competition experiment, benzaldehyde was produced steadily, and some of which was oxidized to benzoic acid (Fig. S7†). Similar to the bulk controlled potential electrolysis, phenol was oxidized gradually to the hydroquinone, and later to para-benzoquinone.
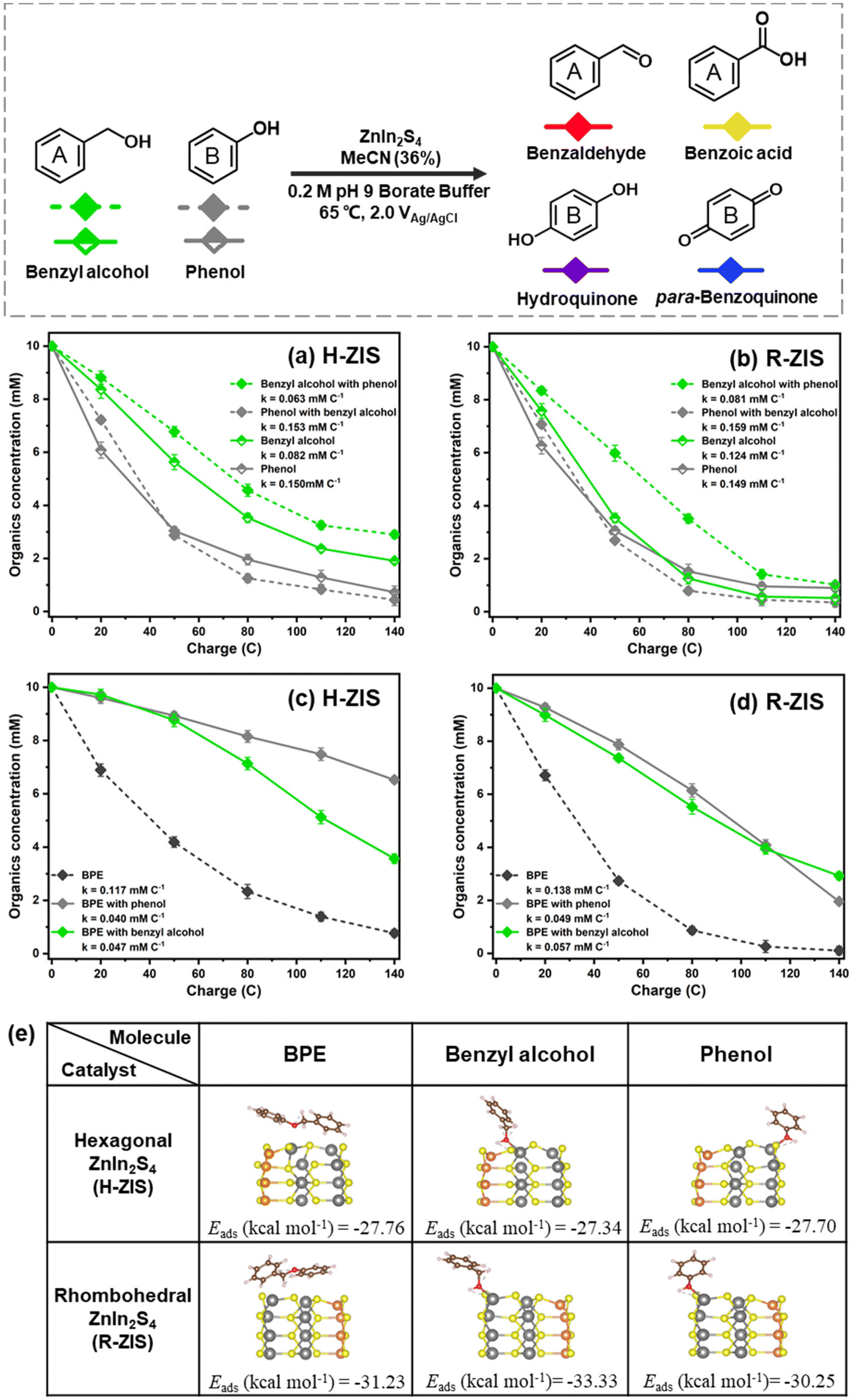 | ||
| Fig. 7 Kinetic profiles of control experiment with the addition of phenol and benzyl alcohol intermediates on (a) H-ZIS and (b) R-ZIS electrodes. (c and d) Kinetic profiles of competition experiment with the addition of phenol and benzyl alcohol intermediates on the H-ZIS and R-ZIS electrodes. All reactions were performed in a pH 9 buffer at 2 VAg/AgCl in 65 °C. Only the reactant consumption rates are shown for clarity. Product distributions are shown in Fig. S7 and S8.† (e) Geometrical structures (side view) of BPE, benzyl alcohol, and phenol adsorption on the In sites of hexagonal and rhombohedral ZnIn2S4 samples. The colour coding is orange (Zn), grey (In), yellow (S), brown (C), red (O), and white (H). | ||
The effects of benzyl alcohol and phenol on BPE consumption were investigated to examine whether they impeded the BPE C–O scission. On the H-ZIS surface, the BPE consumption was impeded by phenol, resulting in a consumption rate of 0.040 mM C−1. The degree of competition with benzyl alcohol was similar, as the BPE consumption rate reduced to 0.047 mM C−1 (Fig. 7c). As a reference, the BPE consumption rate was 0.117 mM C−1 when no competing substrates were added. However, the cleavage of BPE still generated benzyl alcohol and phenol, which could compete with BPE. Despite this, it was not possible to isolate the sole consumption rate for BPE. These results demonstrated that both benzyl alcohol and phenol could compete with BPE for the oxidation site, with phenol having a more notable effect. A similar trend was observed on the R-ZIS surface, but the impact of phenol was less severe than that in the case of H-ZIS. As shown in Fig. 7d, both benzyl alcohol and phenol hindered the BPE consumption, but to a lesser degree compared to the H-ZIS shown in Fig. 7c. Towards the end of the electrolysis, the BPE consumption rate in the presence of phenol declined more rapidly, attributable to the depletion of phenol. The competitive products from phenol and BPE substrates was similar to that of the BPE bulk controlled potential electrolysis trials on both H-ZIS and R-ZIS electrodes (Fig. S8†). When an equal amount of benzyl alcohol was added, smaller amounts of phenolic compounds (phenol, hydroquinone and para-benzoquinone) was yieled on both H-ZIS and R-ZIS electrodes. The benzylic ring products, such as benzyl alcohol and benazlahyde, showed a more apparent accumulating trend compared to the phenolic products.
Several key messages can be derived from the competitive study. First, the reactivity trend is phenol > benzyl alcohol > BPE. During the reaction, phenol reacted preferentially over the other two. Once the competing substrate was depleted, the reaction continued to oxidise the next most readily oxidisable substrates. Second, the R-ZIS surface was more active than the H-ZIS in oxidising BPE and benzyl alcohol. However, the phenol consumption rate was insensitive to the catalyst surface. Third, both phenol and benzyl alcohol competed with BPE at different rates, indicating that all three compounds were oxidised on similar reaction sites on the catalyst surface.
DFT calculations
DFT calculations were performed to clarify the surface adsorption behaviours of BPE and its intermediates. The adsorption energies of the BPE dimer and primary products on the H-ZIS and R-ZIS crystal plane (110) were calculated. The BPE dimer adsorbed onto the H-ZIS and R-ZIS structures with the two phenyl rings positioned parallel to the surface (Fig. 7e). Regardless of the initial placement of the phenyl rings, the adsorption geometry automatically converged to the most relaxed structure with minimum energy. Compared with the H-ZIS phase, the R-ZIS phase exhibited a higher adsorption energy with the BPE dimer by 3.47 kcal mol−1, consistent with the superior catalytic performance of the R-ZIS phase observed in all the experiments.Similarly, phenol and benzyl alcohol exhibited enhanced adsorption on the R-ZIS phase, with energy differences of 2.55 and 5.99 kcal mol−1, respectively. In the minimum energy structure, the O atoms in phenol and benzyl alcohol formed chemical bonds with the In sites (Fig. 7e, distances of 2.31 and 2.28 Å for phenol on R-ZIS and H-ZIS, respectively; and 2.27 and 2.25 Å for benzyl alcohol on R-ZIS and H-ZIS, respectively). In addition, intermolecular hydrogen bonds between the H atoms in the hydroxyl groups of phenol or benzyl alcohol and the S atoms on the ZIS surface were observed, as indicated by the dashed lines. When benzyl alcohol adsorbed on the ZnIn2S4 surface, the benzene ring was further from the surface sites because of the additional benzylic carbon compared with phenol, resulting in a weak inhibition effect. The DFT calculation results revealed R-ZIS was more accommodating for BPE, phenol, and benzyl alcohol compared to the H-ZIS surface, which is consistent with the experimental observation that the R-ZIS surface was more electrocatalytic active for the organic transformations.
Tentative electrocatalytic oxidation route of BPE with O18 isotopic confirmation
Based on the experimental findings, we propose the following tentative mechanism for the BPE cleavage and formation of the secondary and tertiary oxidation products (Fig. 8). First, BPE underwent electrochemical oxidation at the phenolic oxygen to yield BPE˙+. This electrochemical ionisation process was supported by control experiments. An open circuit did not produce any cleaved product, which ruled out the possibility of −OH-anion-promoted SN2-type nucleophilic cleavage occurring at the benzylic carbon. Moreover, we considered whether aerobic catalysis occurred by O2 on the ZIS catalyst. This hypothesis was dismissed after another open-circuit control experiment using O2 bubbled with the ZIS catalyst for 8 h (Fig. S9†). Less than 1% of the benzyl alcohol and benzaldehyde was observed with the R-ZIS, and only a small amount of benzyl alcohol was converted to benzaldehyde after 4 h. Therefore, oxidation at the phenolic oxygen facilitated C–O cleavage to yield a phenyl anion and benzylic radical. The phenolic anion likely followed one of two pathways: 1) surface polymerisation or 2) further oxidation to yield hydroquinone, which may be oxidised to para-benzoquinone. The benzylic radical could react with H2O from the aqueous electrolyte to yield benzyl alcohol, which could be electrochemically oxidised to benzaldehyde and benzoic acid. The involvement of H2O and the subsequent secondary and tertiary oxidation pathways were confirmed using 18O-isotopic labelled H2O as an electrolyte solvent. Analysis of the 18O-labelled product using GC-MS in the selected ion monitoring mode revealed that the 18O label was incorporated exclusively onto the benzylic ring rather than phenol during the BPE cleavage (Fig. S10†). An open-circuit control experiment confirmed that benzyl alcohol, benzaldehyde, and benzoic acid did not undergo 18O exchanges independently, and thus, the 18O installation was catalysed electrochemically. The 18O isotopic label was also observed on hydroquinone and para-benzoquinone during post-electrolysis analysis.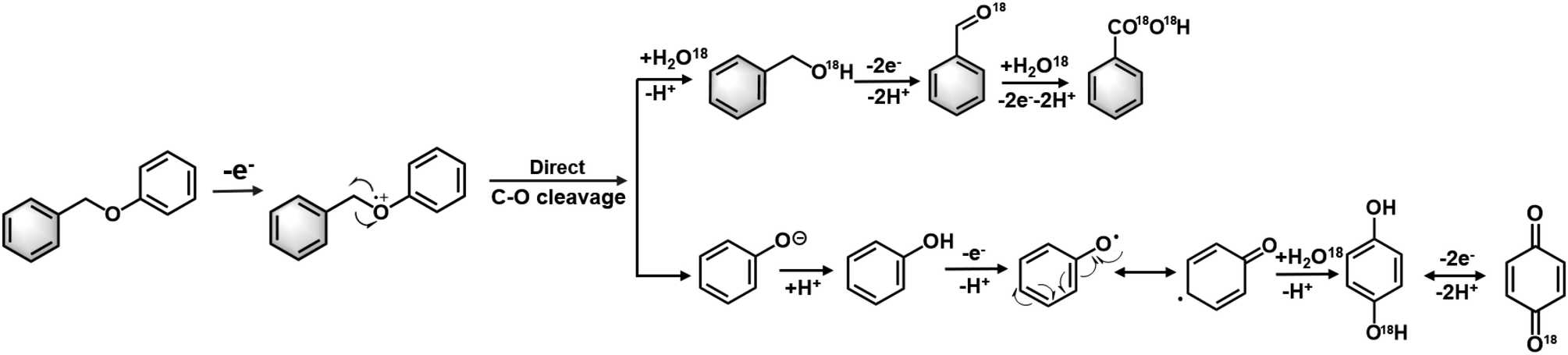 | ||
| Fig. 8 Proposed tentative mechanism of ZnIn2S4-catalysed conversion of BPE dimer. | ||
Electrooxidative cleavage of additional lignin model compounds
The optimized ECO condition for oxidative cleavage was applied toward a series α-O-4 and β-O-4 substrates equipped with different lignin-relevant groups, such as hydroxy (–OH), methoxy (–OCH3), and alkyl (–Me and -tert-butyl), on the aromatic ring (Table 1). All the α-O-4 substrates (a–e) cleaved efficiently with 95–99% conversion. Only substrate c suffered a minor setback conversion presumably due to the bulky tert-butyl group that obstructed its adsorption on the electrode surface. Other substrates, a, b, d, and e showed at least 99% conversion. All α-O-4 substrates cleavaged selectively at the Cbenzyl–O bond, produced the corresponding substituted aldehydes as the main product. Based on the product distribution, the Cbenzyl–O bond cleavage led to the formation of the corresponding benzylic alcohols and phenols; both underwent secondary oxidation to aldehydes/carboxylic acids and hydroquinone/para-benzoquinone, respectively. For meta-methoxy-substituted BPE, d, high selectivity of aromatic aldehydes were obtained under the present conditions without benzoic acid derivatives detected. All phenolics suffered a greater material loss compared to the benzylic products due to surface adsorption, as the ATR-FTIR study showed (Fig. 5b), and its more oxidizable nature, as the competitive electrolysis control study confirmed earlier. Nonetheless, the the R-ZIS electrode demonstrated an extremely selective cleavage at the Cbenzyl–O bond to is crucial to the lignin depolymerization scheme.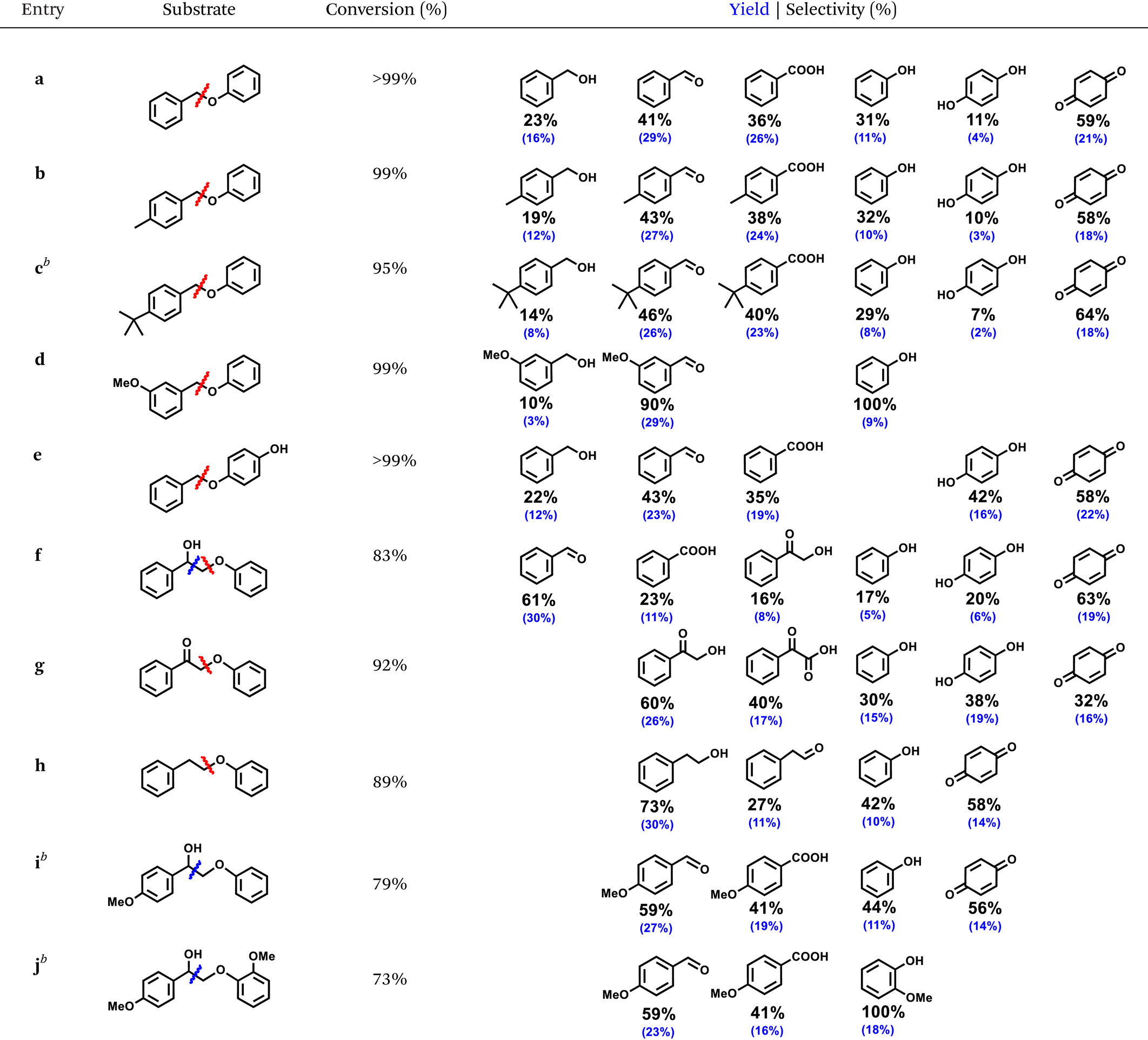
To verify the versatility of the R-ZIS electrode, the β-O-4 linkage was also examined as they are universally abundant in lignin. Under the same reaction condition, β-O-4 linkages were also cleaved efficiently, yielding 73–92% conversion. The cleavage of f, 2-phenoxyphenylethanol, produced benzaldehyde, benzoic acid, 2-hydroxyacetophenone (2-HACP), phenol, hydroquinone, and para-benzoquinone. The Cbenzyl–O bond cleavage should produce 2-HACP and phenolics similar to the α-O-4 dimers. While the Cα–Cβ bond cleavage produced benzaldehyde, benzoic acid, and unstable phenyl formate, which could easily decompose into phenol.46 Interestingly, during the oxidative cleavage of f, no g was detected, which is commonly expected from thermocatalytic,47–50 and occasionally electrocatalytic oxidative cleavage of β-O-4.51 We thus investigated g and observed it was converted at a similar rate with f, and the cleavage occurred exclusively at the CBenzyl–O bond. The result confirmed the Cα–OH remains intact during the cleavage of f because if f and g cleaved at a similar rate, as observed, both of them should be observed during the cleavage of f. Moreover, unlike f, subtrate g cleaved exclusively at the Cbenzyl–O bond, providing another evidence that f did not become g during its oxidative cleavage treatment. When cleaved at Cα–Cβ bond, f produced benzaldehyde instead of benzyl alcohol, which was not observed. The observation is consistent with our mechanistic expectation as the Cα–Cβ bond cleavage should produce a phenylmethanediol, a geminal diol that is readily dehydrated to benzaldehyde.46 The possible formation of anisole from the Cα–Cβ bond cleavage was also considered, though none was detected. Under the same reaction condition, anisole did not react, confirming phenol did not originate from anisole hydrolysis but rather from the Cα–Cβ/Cbenzyl–O bond cleavage. Substrate h produced mainly 2-phenylethanol, indicating the Cbenzyl–O bond occurred similar to the α-O-4 compounds. Extending the study on f, substrate i and j were examined and both cleaved efficiently, producing 79% and 73% conversion, respectively. They cleaved exclusively at the Cα–Cβ bond, producing 4-methoxybenzaldehyde, 4-methoxybenzoic acid, and the corresponding methoxylated phenolics. The slightly lower conversion compared to f can be attributed to the substituents, which induced steric hinderance to the electrode surface similar the case of c.
Conclusion
We developed a novel facile protocol for synthesising ZnIn2S4 electrodes with adjustable distributions of hexagonal and rhombohedral crystal phases, to achieve selective C–O bond oxidative cleavage in BPE, a lignin model compound. The electrocatalytic performance evaluation confirmed that the rhombohedral phase (R-ZIS) exhibited better catalytic properties for C–O bond cleavage and efficiently converted BPE to phenol and benzylic alcohol. In optimised conditions, a conversion rate of over 99% was achieved with a Faraday efficiency of 51.6% at 91.3% conversion of BPE, exceeding the performance of oxidant-driven BPE cleavage.21–23 The catalysts could be easily deposited on a low-cost carbon cloth support, and the deposited catalyst was characterised via XRD, SEM, EDS, ICP, TEM, Raman, XPS, and XAFS techniques to gain insights into the loading, phase, crystallinity, and surface morphology. The reaction mechanism was clarified through the systematic investigation of the working potentials, substrate competition studies, LSV tests, and 18O-isotopic labelling experiments. The DFT calculation results supported the experimental results and confirmed that the rhombohedral crystal phase provided a more active surface for the adsorption of BPE and its subsequent products. Experimental investigation of the underlying mechanism suggested that the reaction proceeds through direct C–O bond cleavage on the ZnIn2S4 surface, followed by subsequent electron loss processes to produce other oxidative products. Substrate scopeing work revealed that the as-prepared R-ZIS anode have universal applicability to a variety of substituted α-O-4 and β-O-4 model dimers. The investigation on substituting the Pt cathode with an earth-abundant electrocatalyst is underway to boost the green chemistry aspect of this work.52–54 If the cathodic reaction is replaced with electrochemical reductive upgrading of biomass-relevant substrates, paired electrolysis can be realised to diversify the product outcome.55–57 This study represents a significant advancement in the development of high-performance electrodes for the direct valorisation of lignin through a green and sustainable electrochemical route, enabling atom-efficient electro-refinery for oxygenated chemicals production via the green chemistry principles.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The author acknowledges the generous financial support from the National Natural Science Foundation of China (22109133) and the State Key Laboratory of Marine Pollution (SKLMP) Seed Collaborative Research Fund (SKLMP/IRF/0028). N. M. B. and Y. Y. acknowledge financial support from the Faculty of Engineering at the University of New South Wales. Also we would like to acknowledge Dr Bernt Johannessen's assistance in In K-edge measurements performed at the XAS beamline of the Australian Synchrotron, part of ANSTO. We acknowledge SOLEIL for provision of synchrotron radiation facilities and we would like to thank Gautier Landrot for assistance in using beamline SAMBA. We acknowledge CLS for provision of synchrotron radiation facilities and we would like to thank Mohsen Shakrouri for assistance in using beamline SXRMB.References
- Z. Luo, Y. Wang, M. He and C. Zhao, Green Chem., 2016, 18, 433–441 RSC.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- K. Ye, Y. Liu, S. Wu and J. Zhuang, Ind. Crops Prod., 2021, 172, 114008 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- T. Wang, M. W. Nolte and B. H. Shanks, Green Chem., 2014, 16, 548–572 RSC.
- P. Feng, H. Wang, H. Lin and Y. Zheng, Carbon Resour. Convers., 2019, 2, 1–12 CrossRef CAS.
- X. Shen, Q. Meng, Q. Mei, H. Liu, J. Yan, J. Song, D. Tan, B. Chen, Z. Zhang and G. Yang, Chem. Sci., 2020, 11, 1347–1352 RSC.
- D. Gao, D. Ouyang and X. Zhao, Green Chem., 2022, 24, 8585–8605 RSC.
- M. V. Doble, A. C. Ward, P. J. Deuss, A. G. Jarvis and P. C. Kamer, Bioorg. Med. Chem., 2014, 22, 5657–5677 CrossRef CAS PubMed.
- J. Kim and C.-H. Huang, ACS ES&T Water, 2020, 1, 15–33 Search PubMed.
- H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- Q. Tu, A. Parvatker, M. Garedew, C. Harris, M. Eckelman, J. B. Zimmerman, P. T. Anastas and C. H. Lam, Environ. Sci. Technol., 2021, 55, 3240–3249 CrossRef CAS PubMed.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC.
- M. Garedew, C. H. Lam, L. Petitjean, S. Huang, B. Song, F. Lin, J. E. Jackson, C. M. Saffron and P. T. Anastas, Green Chem., 2021, 23, 2868–2899 RSC.
- M. Garedew, F. Lin, T. M. DeWinter, B. Song, C. M. Saffron, J. E. Jackson, C. H. Lam and P. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS.
- C. H. Lam, W. Deng, L. Lang, X. Jin, X. Hu and Yi Wang, Energy Fuels, 2020, 34(7), 7915–7928 CrossRef CAS.
- C. A. Martinez-Huitle and S. Ferro, Chem. Soc. Rev., 2006, 35, 1324–1340 RSC.
- B. E. Logan and K. Rabaey, Science, 2012, 337, 686–690 CrossRef CAS PubMed.
- L. Wang, Y. Chen, S. Liu, H. Jiang, L. Wang, Y. Sun and P. Wan, RSC Adv., 2017, 7, 51419–51425 RSC.
- L. Wang, S. Liu, H. Jiang, Y. Chen, L. Wang, G. Duan, Y. Sun, Y. Chen and P. Wan, J. Electrochem. Soc., 2018, 165, H705 CrossRef CAS.
- H. Jiang, L. Wang, L. Qiao, A. Xue, Y. Cheng, Y. Chen, Y. Ren, Y. Chen and P. Wan, Int. J. Electrochem. Sci., 2019, 14, 2645–2654 CrossRef CAS PubMed.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS.
- R. Yang, L. Mei, Y. Fan, Q. Zhang, R. Zhu, R. Amal, Z. Yin and Z. Zeng, Small Methods, 2021, 5, 2100887 CrossRef CAS.
- F. Cai, X. Hu, F. Gou, Y. Chen, Y. Xu, C. Qi and D.-K. Ma, Appl. Surf. Sci., 2023, 611, 155696 CrossRef CAS.
- X. Wu, S. Xie, H. Zhang, Q. Zhang, B. F. Sels and Y. Wang, Adv. Mater., 2021, 33, 2007129 CrossRef CAS.
- S. Shen, P. Guo, L. Zhao, Y. Du and L. Guo, J. Solid State Chem., 2011, 184, 2250–2256 CrossRef CAS.
- H. Jing, G. Xu, B. Yao, J. Ren, Y. Wang, Z. Fang, Q. Liang, R. Wu and S. Wei, ACS Appl. Energy Mater., 2022, 5, 10187–10195 CrossRef CAS.
- K. Kordek, L. Jiang, K. Fan, Z. Zhu, L. Xu, M. Al-Mamun, Y. Dou, S. Chen, P. Liu and H. Yin, Adv. Energy Mater., 2019, 9, 1802936 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, APL Mater., 2013, 1 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- H. Qian, Z. Liu, Z. Guo, M. Ruan and J. Ma, J. Alloys Compd., 2020, 830, 154639 CrossRef CAS.
- S. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- G. Lv, L. Long, X. Wu, Y. Qian, G. Zhou, F. Pan, Z. Li and D. Wang, Appl. Surf. Sci., 2023, 609, 155335 CrossRef CAS.
- W. Zhen, X. Zhou, S. Weng, W. Zhu and C. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 12571–12582 CrossRef CAS.
- C. Du, Q. Zhang, Z. Lin, B. Yan, C. Xia and G. Yang, Appl. Catal., B, 2019, 248, 193–201 CrossRef CAS.
- S. Shen, L. Zhao and L. Guo, J. Phys. Chem. Solids, 2008, 69, 2426–2432 CrossRef CAS.
- A. J. Fry, Synthetic organic electrochemistry, John Wiley & Sons, 1989 Search PubMed.
- I. K. Oikonomopoulos, M. Perraki, N. Tougiannidis, T. Perraki, M. J. Frey, P. Antoniadis and W. Ricken, Int. J. Coal Geol., 2013, 115, 1–12 CrossRef CAS.
- J. L. A. de Queiroz, D. C. de Moura, E. Santos, B. A. Frontana-Uribe and C. A. Martínez-Huitle, Quim. Nova, 2020, 43, 253–260 Search PubMed.
- T. Cui, L. Ma, S. Wang, C. Ye, X. Liang, Z. Zhang, G. Meng, L. Zheng, H.-S. Hu and J. Zhang, J. Am. Chem. Soc., 2021, 143, 9429–9439 CrossRef CAS PubMed.
- M. Liu, Z. Zhang, X. Shen, H. Liu, P. Zhang, B. Chen and B. Han, Chem. Commun., 2019, 55, 925–928 RSC.
- M. Liu, Z. Zhang, J. Song, S. Liu, H. Liu and B. Han, Angew. Chem., 2019, 131, 17554–17559 CrossRef.
- L. Zhao, S. Shi, G. Zhu, M. Liu, J. Gao and J. Xu, Green Chem., 2019, 21, 6707–6716 RSC.
- H.-R. Tian, Y.-W. Liu, Z. Zhang, S.-M. Liu, T.-Y. Dang, X.-H. Li, X.-W. Sun, Y. Lu and S.-X. Liu, Green Chem., 2020, 22, 248–255 RSC.
- L. Ma, H. Zhou, X. Kong, Z. Li and H. Duan, ACS Sustainable Chem. Eng., 2021, 9, 1932–1940 CrossRef CAS.
- P. Nilges and U. Schröder, Energy Environ. Sci., 2013, 6, 2925–2931 RSC.
- S. Huang, Y. Jin, M. Zhang, K. Yan, S.-P. Feng and J. C.-H. Lam, Green Chem., 2022, 24, 7974–7987 RSC.
- S. Huang, B. Gong, Y. Jin, P. H. L. Sit and J. C.-H. Lam, ACS Catal., 2022, 12, 11340–11354 CrossRef CAS.
- S. Liu, Y. Jin, S. Huang, Q. Zhu, S. Shao and J. C.-H. Lam, Nat. Commun., 2024, 15, 1141 CrossRef CAS.
- H. Liu, T.-H. Lee, Y. Chen, E. W. Cochran and W. Li, Green Chem., 2021, 23, 5056–5063 RSC.
- N. Sbei, T. Hardwick and N. Ahmed, ACS Sustainable Chem. Eng., 2021, 9, 6148–6169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: https://doi.org/10.1039/d4gc00338a |
| This journal is © The Royal Society of Chemistry 2024 |