
Perspectives on facilitating natural gas and hydrogen storage in clathrate hydrates under a static system
Wonhyeong
Lee†
a,
Kwangbum
Kim†
 a,
Jeongwoo
Lee
a,
Yun-Ho
Ahn
b and
Jae W.
Lee
*a
a,
Jeongwoo
Lee
a,
Yun-Ho
Ahn
b and
Jae W.
Lee
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon, 34141, Republic of Korea. E-mail: jaewlee@kaist.ac.kr
bDepartment of Chemical Engineering, Soongsil University, 369 Sangdo-ro, Dongjak-gu, Seoul 06978, Republic of Korea
First published on 5th June 2024
Abstract
The rising demand for natural gas (NG) and hydrogen, due to their lower carbon footprint and role in storing surplus renewable energy, has highlighted the focus on developing advanced storage technologies. Traditional methods like liquefaction and compression face high energy and safety challenges, prompting the exploration of new solutions. Among these, hydrate-based gas storage stands out for its environmental benefits, using clathrate hydrates to store gas with low energy consumption and carbon emissions. Furthermore, the composition of hydrates, predominantly water (∼85%), and their lack of by-products during repetitive storage–release cycles firmly establish them as environmentally friendly gas storage media. However, kinetic challenges such as stochastic nucleation, limitations in mass and heat transfer, and thermodynamic barriers arising from harsh hydrate formation conditions have hindered the practical application of hydrates. While mechanical methods to improve hydrate formation exist, their use significantly increases the demand for electrical energy. Therefore, developing methods for gas hydrate formation under static conditions is crucial for utilizing this material as a safe and green gas storage medium. This review examines theoretical studies and experimental efforts to enhance hydrate formation kinetics in static systems without additional mechanical methods. Thermodynamic hydrate promoters to increase the driving forces for hydrate formation under mild conditions, surface-modified materials to increase nucleation probabilities for shorter induction times, and porous materials to provide pathways for mass and heat transfer have been widely investigated. The discussion addresses the direction and necessary efforts for utilizing hydrate-based gas storage as a next-generation green technology.
1. Introduction
Global warming is becoming more severe due to human activities in various industrial fields, causing climate change and the destruction of ecosystems.1 Among the contributors, carbon dioxide emissions from energy sectors have the most significant impact.2,3 As part of ongoing efforts to mitigate carbon emissions, the energy sector is transitioning toward the use of renewable energy sources such as wind,4,5 solar,6,7 and waves,8,9 which do not emit carbon sources during electricity generation. However, power generation from renewable sources is still in its infancy, with efficiency varying significantly depending on the operating environment, and it has yet to fully meet global energy demands.Natural gas (NG), primarily composed of methane (approximately 90%), is the cleanest burning fossil fuel, emitting less carbon dioxide during combustion than gasoline and coal.10,11 With its environment-friendly characteristics and abundant reserves, NG is positioned as a cornerstone in the transition to a carbon-neutral energy society.12 However, NG, being a fossil fuel, still emits carbon and poses methane leakage risks during transportation. Despite these issues, the use of NG remains advantageous due to its ability to complement renewable energy sources, in addition to the aforementioned benefits, thereby contributing to a more sustainable energy future. In line with this, in 2018, there was a 4.6% increase in NG consumption, with a projected average annual growth of 0.9% over the next decade.13
On the other side, hydrogen is regarded as the “fuel of the future” and a “clean energy vector”.14–16 It carries nearly three times the energy density of diesel or gasoline and completely eliminates carbon emissions during combustion.17 Moreover, hydrogen can be produced from both renewable sources16,18 and non-renewable sources.17,19 While there are some critiques surrounding production and storage expenses of hydrogen, the aforementioned powerful advantages of hydrogen position it as the ultimate energy source for the future. Due to such increasing demand for NG and hydrogen, the significance of effective storage systems for these gases is being emphasized.
Stable NG and hydrogen storage system is essential for the adequate energy supply and sustainable delivery process. However, the storage of gaseous energy typically demands significant amounts of energy, leading to the use of large amounts of electrical energy and the associated carbon dioxide emissions in its production. Conventional and well-established storage methods for NG and hydrogen are liquefaction20 and compression.20,21 While these technologies can achieve high volumetric storage capacity, they inevitably involve the above pointed substantial energy requirements. For instance, liquefying methane and hydrogen in atmospheric pressure requires a considerable amount of energy due to their extremely low boiling temperatures of at −162 °C and −253 °C, respectively. Compression technology also necessitates high electrical energy to compress the gas to extremely high-pressure conditions (5–70 MPa). These methods also face challenges such as boil-off and high initial costs in liquefaction,22,23 as well as safety concerns in compression.24 Consequently, various technologies based on adsorption25–27 or absorption28,29 have been proposed; these alternatives, however, often involve expensive materials such as metal hydrides30 and inorganic hydrides,31,32 which can be less cost-effective for large-scale operations.
Clathrate hydrates are ice-like crystalline compounds in which small gas or organic molecules are trapped in a hydrogen-bonded water lattice under low-temperature and high-pressure conditions.33–38 Depending on the sizes and properties of guest molecules, hydrates of different crystal structures are formed, and the well-known structures include structure I (sI), structure II (sII), and structure H (sH).39,40 While the initial studies of hydrates were related to harvesting NG hydrates from nature41–44 or inhibiting hydrate formation in the delivery systems,45–52 their unique physicochemical properties have led to the artificial synthesis of hydrates for various industrial applications, including gas separation,53–58 desalination,59–61 cold energy storage,62–64 carbon sequestration,65–67 and gas storage.68–72 In particular, hydrate-based gas storage systems have garnered significant attention as potential replacements for conventional technologies due to several desirable characteristics.
Hydrates can solidify small gas molecules, such as methane and hydrogen, offering high volumetric storage capacity under milder thermodynamic conditions compared to other methods. Table 1 presents the economic and environmental aspects of various hydrogen storage techniques, highlighting the potential of hydrate-based processes. The calculated spent energy-to-stored energy ratio for hydrate-based storage process (0.09) is lower than that of compression (0.12), liquefaction (0.36), metal hydride (0.15), and liquid organic hydrogen carrier (0.23) technologies.73,74 This lower energy requirement for storing hydrogen in hydrates ultimately leads to reduced carbon dioxide emissions. Although the maximum volumetric storage capacity of hydrogen through hydrate-based technology (477.78 v/v) is lower than that of liquefaction (793.52 v/v), metal hydride (1355.14 v/v),75 or liquid organic hydrogen carrier (507.37 v/v),76 the superior energy efficiency and excellent cyclability of the hydrate-based process highlight its significant advantages as a green technology for hydrogen storage. Additionally, the primary component of hydrates, water, is abundantly available on earth, making hydrate technology more suitable for the upcoming hydrogen energy society than metal hydrides or liquid organic hydrogen carrier.
| Compression | Liquefaction | Metal hydridea | Liquid organic hydrogen carrierb | Hydrate | Ref. | |
|---|---|---|---|---|---|---|
| a MgH2 was compared as a representative of metal hydride. b Methylcyclohexane was compared as a representative of liquid organic hydrogen carrier. c CO2 emission were calculated assuming that all utilities required for the process were electric power. d Volumetric capacity is defined as volume of hydrogen at STP/volume of system (for liquefaction: volume of liquid hydrogen, for hydride: volume of Mg alloy, for hydrate: volume of hydrate). e Volumetric capacity value of pure sII H2 hydrate with no thermodynamic promoter. | ||||||
| Spent energy/stored energy | 0.12 | 0.36 | 0.15 | 0.23 | 0.09 | 73 and 74 |
| CO2 emissionc (kg) | 1.34 | 3.83 | 1.58 | 6.61 | 1.01 | 73 and 76 |
| Maximum volumetric capacityd | 268.80 | 793.52 | 1355.14 | 507.37 | 477.78e | 30, 75 and 106 |
| Cyclability | — | — | Poor | Poor | Excellent | 30 and 107 |
| Safety | Explosive | Non-explosive | Non-explosive | Non-explosive | Non-explosive | 30, 75 and 107 |
Furthermore, hydrates serve as an environment-friendly gas storage medium, primarily composed of water (about 85%), with no by-products generated during the process. These water frameworks also ensure the safe, long-term storage of gaseous energy due to their self-extinguishing properties. The hydrate-based gas storage process is also easily handled and offers excellent cyclability due to its straightforward synthesis pathway. Given these advantages, substantial research has focused on storing NG or hydrogen in hydrates.77–82 However, practical applications are still challenged by insufficient formation kinetics83 and the stochastic nature of hydrate nucleation,84 which complicates process design.
For the commercialization of hydrate-based green gas storage systems, current efforts focus on enhancing hydrate formation kinetics.85–87 Traditionally, mechanical stirrers are used to mix water and gas, significantly accelerating hydrate formation compared to unstirred systems.88–90 However, the necessity for continuous mixing of the internal components leads to a significant increase in operating power as hydrate formation drastically elevates the viscosity of the fluid.90 This energy demand undermines the economic and environmental benefits of hydrate-based storage. Therefore, efficient hydrate synthesis with lower energy inputs is essential, necessitating the development of alternative systems that achieve efficient formation without mechanical methods.
For real-life applications, numerous studies on hydrate-based gas storage are thus currently focusing on the development of novel and promising strategies to enhance hydrate formation kinetics in a static system.91–95 In situations where mechanical stirring is absent, the contact between water and gas is not facilitated, and the heat generated by hydrate formation can become easily localized. To overcome these challenges, various strategies, such as using kinetic hydrate promoters to improve water–gas contact, have been proposed and tested.96,97 As a result, numerous studies have dramatically increased the formation rate of methane and hydrogen hydrates by many strategies individually or in combination.98 These steps are crucial in realizing a sustainable and viable hydrate-based gas storage process as a green technology.
Prior to this work, numerous studies have reviewed hydrate-based gas storage processes from different perspectives. Literature includes a perspective on hydrate-based gas storage technologies,71,83 and some reviews specifically focus on the role of specific kinetic additives99,100 or porous media101,102 in the formation kinetics of NG and hydrogen hydrates. However, despite the significance of hydrate formation in a static system, no review has yet been conducted that specifically focuses on this aspect. Aside from reviews of experimental approaches, many research studies have also reviewed existing theories about hydrate nucleation and formation models.103–105 It is crucial to provide a comprehensive explanation of the phenomenological interpretation of the factors that impede the hydrate formation and the strategies to address the challenges. In this work, we present a systematic analysis of the key factors that hinder NG and hydrogen hydrate formation under a static system. In addition, the strategies that have been attempted to overcome such thermodynamic and kinetic hurdles in an unstirred system are presented. Finally, future prospects and directions to commercialize a hydrate-based green gas storage process are presented.
2. Fundamental challenges in hydrate-based gas storage
Despite significant efforts, hydrate-based gas storage has not yet been realized owing to insufficient hydrate formation kinetics. The undesirable hydrate formation kinetics typically arise from a thermodynamic hurdle, along with kinetic hurdles such as a stochastic nucleation, and limitations in mass and heat transfer. In this section, how these thermodynamic and kinetic hurdles affect the performance of hydrate-based gas storage systems and what strategies are available to address them are discussed.2.1. Thermodynamic hurdle for hydrate-based gas storage
As clathrate hydrates are phase change materials, it is essential to carefully consider phase equilibrium when designing a gas storage process based on hydrates.40,108,109 Due to the importance of storing NG and hydrogen, numerous studies have investigated the phase equilibrium of NG (or methane)110–112 and hydrogen hydrates.113,114Fig. 1a summarizes the typical equilibrium points for NG, methane, and hydrogen hydrates, as gathered from various research sources.115–117 In the phase diagram in Fig. 1a, the lower temperature and higher-pressure region along each phase equilibrium line indicate the conditions under which these respective hydrates can form and remain stable. It is accordingly necessary to set the thermodynamic conditions for a hydrate-based gas storage process within this region.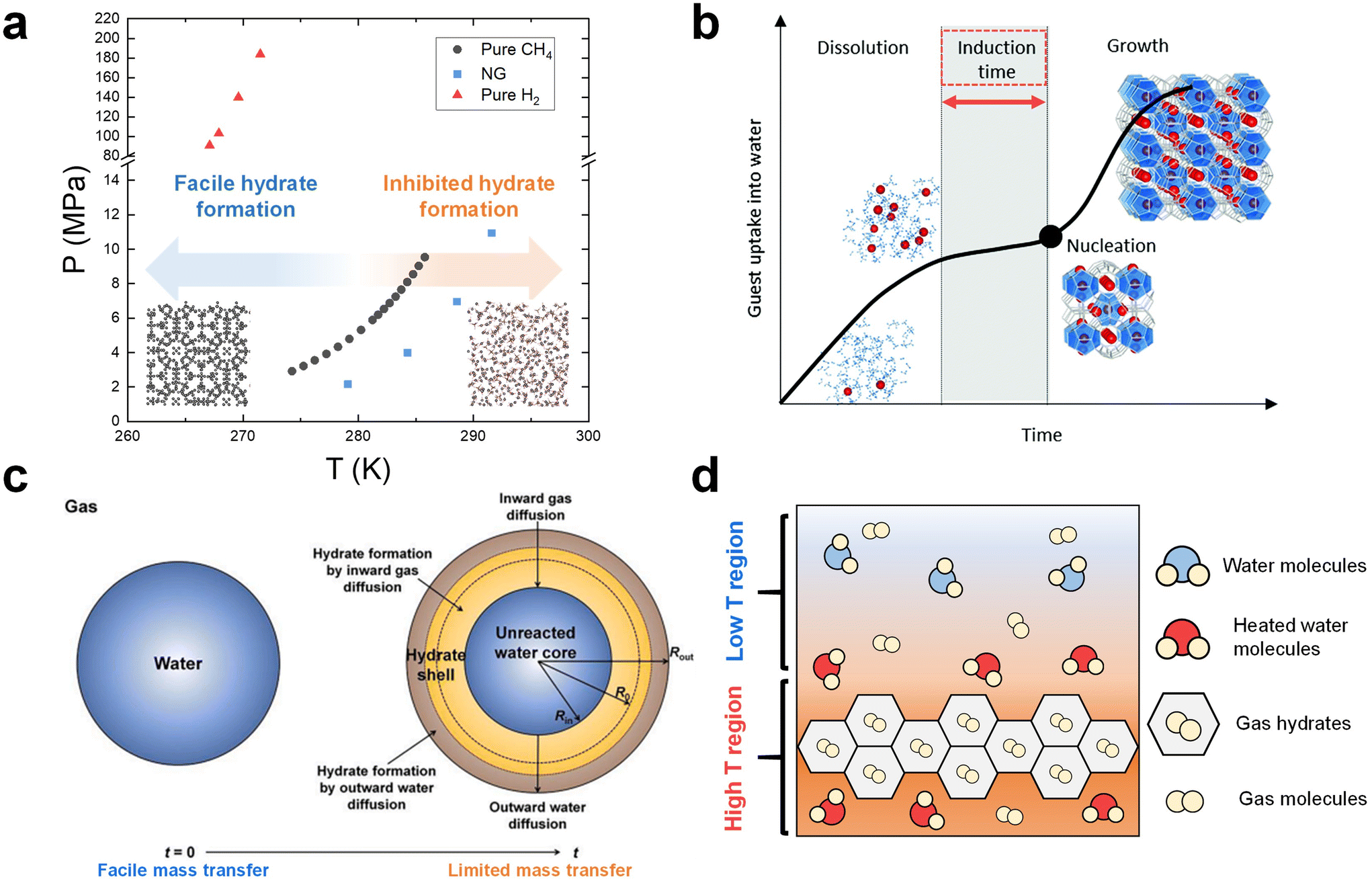 | ||
Fig. 1 (a) Phase equilibrium boundaries of CH4,116 NG,115 and H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 hydrates. (b) General time dependence of the hydrate crystallization process. Reproduced with permission.122 Copyright 2020, Royal Society of Chemistry. (c) Schematic illustration of hydrate formation on a water droplet in the diffusion-controlled regime. Reproduced with permission.143 Copyright 2023 Elsevier. (d) Schematic of heat boundaries in the system where growing hydrates exist. 117 hydrates. (b) General time dependence of the hydrate crystallization process. Reproduced with permission.122 Copyright 2020, Royal Society of Chemistry. (c) Schematic illustration of hydrate formation on a water droplet in the diffusion-controlled regime. Reproduced with permission.143 Copyright 2023 Elsevier. (d) Schematic of heat boundaries in the system where growing hydrates exist. | ||
However, there are more complex factors to consider when determining the process conditions. The hydrate formation kinetics are strongly influenced by the driving force within the system, which implies that harsher conditions are necessary to ensure satisfactory process performance.118 One must also consider the reduction in the system pressure due to gas enclathration into hydrates, highlighting the need for the initial pressure conditions to be significantly higher than the equilibrium point. However, implementing such severe thermodynamic conditions can result in high operational costs. Notably, as depicted in Fig. 1a, the phase equilibria of pure hydrogen hydrates are exceptionally harsh (over than 100 MPa under 270 K), making them unsuitable for practical process conditions.117,119 One potential approach to overcome this challenge is to incorporate thermodynamic hydrate promoters into the system.39 They stabilize the hydrate structure by directly occupying the hydrate cage, significantly alleviating the equilibrium conditions for hydrate formation.120,121 This approach ultimately provides the system with a substantially high driving force for hydrate formation even under much more moderate thermodynamic conditions. More detailed discussions on the utilization of thermodynamic hydrate promoters in NG and hydrogen hydrate formation system are provided in Section 4.
2.2. Kinetic hurdles for hydrate-based gas storage
The hydrate formation process is typically divided into two main phases: nucleation and growth.122 This concept is depicted in Fig. 1b, which provides a schematic representation of the typical time evolution of gas consumption during hydrate formation.122 Initially, the guest species present in the gas phase are dissolved into the liquid phase, resulting in a slight increase of gas uptake. Following the dissolution phase, a state of supersaturation emerges when the pressure and temperature conditions favor the thermodynamic formation of hydrates, even though critical hydrate nuclei have not yet appeared. A critical nucleus of a hydrate phase can be defined as the smallest amount of the new phase that can exist independently. The period between the establishment of supersaturation and the formation of critical nuclei is referred to as the induction time. Once the hydrate nucleation events occur after the induction time, hydrates start to grow further.One of the primary kinetic challenges in the application of hydrates is the inherently stochastic nature of nucleation.104 Multiple experimental studies have shown that hydrate formation does not readily occur within a reasonable time frame due to the stochastic nucleation process.123,124 While increasing the driving force in the system can reduce the induction time, it has been established in numerous studies that harsh formation conditions do not always guarantee hydrate formation.125 To tackle this issue, although understanding hydrate nucleation at a molecular level is difficult due to the small time and length scales involved, many researchers have sought to validate hydrate nucleation pathways through macroscopic experiments126 or molecular dynamics (MD) simulations.127 Consequently, several conceptual theories regarding nucleation pathways have been proposed, including classical nucleation theory,128 the labile cluster hypothesis,129 the local structuring mechanism,130 and the blop hypothesis.131 However, there are still areas in the nucleation process that deviate from these theories and remain unvalidated, necessitating further in-depth investigations. In addition to theoretical studies, experimental strategies to reduce or eliminate the induction time have been rigorously explored.132,133 One prominent approach involves introducing a third-surface substance to induce heterogeneous nucleation.134 In Section 5.1, various strategies for overcoming the stochastic nature of the induction time will be presented.
After the occurrence of hydrate nucleation, a sufficient mass transfer of water and gas molecules to the growing hydrate surface is required for further growth of hydrates.135 This mass transfer process can generally be divided into two distinct steps: the movement of gas molecules from the gas–liquid interface to the bulk liquid phase, followed by the transfer of the gas molecules from the bulk liquid phase to the hydrate solution interface.105,136 Numerous studies have been undertaken considering these mass transfer pathways as potential rate-determining factors of the overall hydrate formation process. Consequently, various hydrate growth models such as the two-film theory have been suggested based on the concentration or fugacity gradients of gas molecules.136,137 Nonetheless, it is worth noting that many of these studies are based on experimental results conducted within an agitated reactor, and the proposed model would be semi-empirical and apparatus dependent. In particular, the rate of hydrate formation would be slower than expected by the proposed models in a static system, because far more time is required for the dissolution of gas molecules into the bulk liquid phase compared to the stirring system.
Apart from the previously mentioned mass transfer based kinetic models, the shrinking-core model has gained substantial attention within this academic community over the past two decades. Originally proposed to elucidate hydrate formation from ice particles,138 this model has now also been used to explain the hydrate formation in emulsion systems,139–141 and a system using various porous media such as silica particles.142Fig. 1c illustrates a schematic representation of this model, particularly focusing on the formation of a hydrate shell around a water droplet in the diffusion-limited growth stage.143,144 In this model, hydrate growth takes place in both inward and outward directions. The progression of hydrate formation is controlled by the inward diffusion of gas molecules to the hydrate–water interface and the outward diffusion of water molecules to the hydrate–gas interface. As the reaction time progresses, the transfer of water and gas molecules becomes less efficient owing to the increasing thickness of the solid hydrate film, leading to a reduction in the rate of hydrate formation. Ultimately, an unreacted water core remains within the hydrate shell. This model is also relevant in static systems where a solid hydrate film forms at the gas–liquid interface, leaving unreacted bulk water below by physically blocking the transport of gas species into the liquid phase. A number of studies have observed the above-mentioned phenomena in a static system,145–148 suggesting the necessity of employing more effective approaches to ensure efficient mass transfer in an unstirred system.
The shared insight across the various mass transfer models described above is that uninterrupted contact between gas and water is essential for continuous hydrate formation. Consequently, various strategies have been proposed to facilitate mass transfer in static systems. Some of the proposed strategies include utilizing surfactant-type kinetic promoters to facilitate the dissolution of gas into the bulk water phase,149 inducing the growth of porous hydrates with gas channels,150 and forming hydrates within porous media containing internal gas channels.151 Each of these methods, along with their respective advantages and illustrative examples, will be discussed in Section 5.2.
During the process of hydrate formation, a specific quantity of energy is released; this is known as the heat of hydrate formation. The released heat is substantial, for instance, approximately 50 kJ mol−1 of gas for methane hydrates.152 Failure to adequately dissipate this exothermic heat from the growing hydrate surface can result in a local temperature increase, diminishing the driving force for further growth and promoting the decomposition of hydrates.153,154 Due to this substantial impact of heat on hydrate formation, several researchers have suggested kinetic models that assume mass transfer is of minimal significance, with heat transfer predominantly governing the crystallization and growth processes.155–158
In systems with mechanical stirring, the removal of the heat released during hydrate formation would be more efficient compared to static systems due to the physical rearrangements of components in the fluid. In static systems, however, the exothermic heat of hydrate formation can easily become localized, resulting in an elevated temperature region near the growing hydrates (Fig. 1d). This ultimately impedes further hydrate growth, underscoring the critical importance of effective heat removal, particularly in static setups.153,159,160 To address this challenge, various strategies have been experimentally explored. These include the introduction of nanoparticles (NPs) with high thermal conductivity as heat carriers161 and the formation of hydrates within metallic foam structures,162 which serve as pathways for heat transfer. A detailed discussion of these experimental studies can be found in Section 5.3.
3. Kinetic promotion with mechanical techniques
In order to overcome the kinetic challenges in hydrate-based gas storage applications, mechanical techniques have been extensively utilized.88–90,163,164 These mechanical approaches include stirring, bubbling, and spraying, all of which serve to physically increase the gas–liquid interface within the hydrate formation system. This, in turn, increases the nucleation probability and mass transfer of each component in the system. Consequently, mechanical techniques can facilitate hydrate formation even under milder thermodynamic conditions, which means a low driving force. While the adoption of these techniques can significantly accelerate hydrate formation, they do come with drawbacks such as high energy consumption and operational complexities. In this section, we present a brief overview of the principles and examples of these mechanical techniques. At the same time, the relevant issues that can arise when implementing them in large-scale hydrate-based gas storage processes are discussed. Mechanical agitation has been commonly used to promote hydrate formation. This technique achieves its efficacy by continuously regenerating the gas–liquid interface. Additionally, it demonstrates remarkable effectiveness with hydrophobic hydrate-forming gases, as stirring efficiently facilitates the contact between gas and water molecules,165 heightening the probability of nucleation (Fig. 2a).166 In the presence of stirring, nucleated hydrate crystals can also be uniformly dispersed throughout the aqueous phase, facilitating the rapid progression of hydrate formation. Furthermore, stirring enables gas molecules to reach the growth front of hydrates, and it also effectively removes the locally generated exothermic heat resulting from hydrate formation.90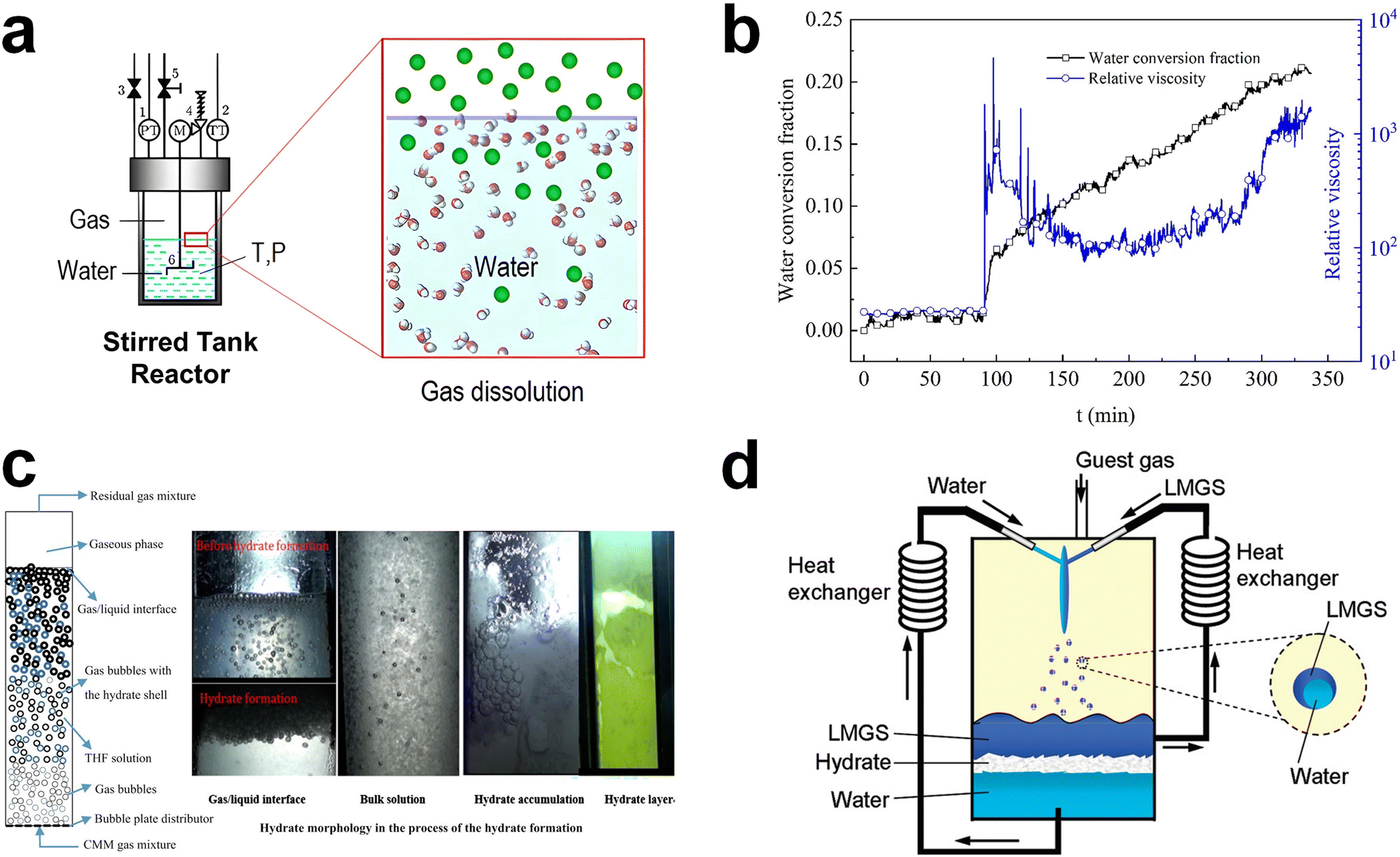 | ||
| Fig. 2 (a) Schematic illustration of a typical stirring system. Gas components in the gas phases are dissolved into the bulk liquid phase by external mechanical mixing. Reproduced with permission.166 Copyright 2022 American Chemical Society. (b) Time-resolved water conversion and relative viscosity during the hydrate formation process. Reproduced with permission.170 Copyright 2020 Elsevier. (c) Schematic representation of a general bubbling reactor for hydrate formation and real images at various regions in the bubbling reactor during the process of the hydrate formation. Reproduced with permission.172 Copyright 2017 Elsevier. (d) Conceptual illustration of a hydrate forming system using a spray nozzle and fluid circulator equipped with heat exchangers. Reproduced with permission.174 Copyright 2009 American Chemical Society. | ||
Numerous studies have investigated the effect of stirring on the efficiency of hydrate-based gas storage systems. Hao et al. compared the methane hydrate formation kinetics with and without stirring, and their results revealed that the use of a stirrer reduced the induction time by a factor of four compared to the static system.90 Furthermore, by employing 320 rpm for a duration of 30 minutes, the time required to attain a comparable methane uptake was also significantly reduced by approximately four-fold relative to that of the unstirred system.90 Numerous additional investigations have explored the influence of stirring rate167 or the shape of the stirrer,168 and all these earlier research efforts suggest that hydrate formation can be promoted through mechanical mixing. Recently, hybrid combinatorial reactor (HCR) approaches, involving both stirred tank reactor and unstirred tank reactor configurations, also have been tested on a lab-scale.169 This HCR approach involves initially using the experimental setup as a stirred tank reactor until hydrate crystal nucleation occurs. Following nucleation, the hydrate formation proceeds without stirring, resulting in reduced energy consumption.169
Despite the advantages associated with mechanical agitation mentioned above, its application in a large-scale hydrate-based gas storage process seems to be inappropriate because of the substantial energy requirements to mix the components within the system. During the hydrate formation, liquid water in the reactor transforms into solid hydrates, leading to an exponential increase in the viscosity of the slurry containing the hydrate particles. Evolution of the relative viscosity during the methane hydrate formation in Fig. 2b provides a good demonstration.170 When the water to hydrate conversion rate reached 2.2%, there was an abrupt rise in the relative viscosity of the slurry, jumping from 27 to 1808. The viscosity of the solution then fluctuated around 600, indicating that the energy consumption for stirring was notably high due to hydrate formation.170 Furthermore, a recent simulation study using Aspen Plus highlighted that the energy cost associated with stirring could account for a substantial portion, specifically 39.0%, of the total energy cost in the hydrate-based gas separation process.171 This result highlights the significant operational expenses stemming from the mechanical mixing in the hydrate-based process.
Generating tiny gas bubbles in a bulk water system has frequently been applied to hydrate formation systems, as the bubble tower is one of the most suitable reactor types for a gas–liquid reactant system.122 Gas bubbles with small size are usually generated at the bottom of the bubbling tower, resulting in significantly increased specific gas–liquid interfacial area in the system (Fig. 2c).172 Hydrates are formed along with the surface of the gas bubbles and then tend to fully cover the gas bubbles. Another crucial aspect of this system is that the generated bubbles naturally rise towards the bulk gas–liquid interface within the reactor due to differences in physical density. During this ascent, bubbles may also aggregate or collide with one another. Once they rise and accumulate below the bulk gas–liquid interface, as depicted in Fig. 2c, the hydrate films on the gas bubbles become thicker over time. The key advantage of this bubbling method is that it allows for easy and continuous hydrate formation. Moreover, by employing a bubbling tower, a multi-stage hydrate formation system can be readily constructed.
Although the bubbling method is a facile way to continuously form hydrates, complete gas bubble conversion to hydrates could not be achieved because hydrates are formed in the form of a solid film on the surface of bubbles. Experimental results revealed that these hydrate shells are difficult to remove from the surface of the bubbles.173 Moreover, bubbles with hydrate shells tend to cluster together instead of merging into larger bubbles, and these bubbles with hydrate shells can accumulate near the bulk gas–liquid interface. Formed hydrate films finally act as the mass transfer barrier between water and gas molecules, leading to low conversion of hydrates and insufficient process efficiency.173
The spraying technique involves the atomization of water or a solution into a gas-filled reaction vessel through a nozzle, as depicted in Fig. 2d.174 In principle, this technology shares similarities with the bubbling method, but it differs in that it atomizes water instead of gas. This approach leads to a significantly increased gas–liquid interfacial area, enabling rapid conversion of the sprayed water or solution into hydrates. Consequently, there has been extensive research into applying water spraying in a guest-gas phase.174–177 After an initial exploration of the water spraying system was reported,178 the significance of effective removal of heat from the hydrate formation sites was highlighted. Several studies have examined the spraying of water onto a solid plate that is consistently cooled and placed in a guest-gas phase to induce the formation of hydrates.154,179,180 A method involving the spraying of water into water pools at the bottom of the reactor also has been investigated.174,176,177 In this approach, water is continuously drained from the water pools, cooled through an external heat exchanger, and then reintroduced into the reactor for further hydrate formation. In particular, water spraying systems using water pools have been explored in various ways for methane storage. One such approach involves storing methane in sH hydrates by incorporating a large-molecular substance as a secondary guest, as illustrated in Fig. 2d.174 During the experiment, both water and an organic liquid were sprayed, and this resulted in the facile enclathration of methane into sH hydrates, suggesting broad applicability of this approach.174
While continuous hydrate formation would be available with water spraying methods, there are some technical hindrances also. One significant issue is the exothermic heat from the hydrate formation, as previously noted. While a cooling plate can be utilized, continuous hydrate formation on the plate would result in the diminished cooling efficiency, leading to decreased process efficiency. Moreover, there is a risk of accidental breaks in the continuous hydrate-forming operations because of the spray nozzle being blocked by hydrate formation.174 In the water pool system, blockages in the water circulating loop can also occur due to the accumulation of tiny hydrate particles.174
Table 2 summarizes the advantages and drawbacks of the aforementioned mechanical techniques. All methods surely offer benefits in accelerating hydrate formation kinetic by increasing the gas–liquid interfacial area. However, these techniques involve high operational energy during the process, and each also has inherent limitations that render them unsuitable for large-scale processes. Although there are alternative mechanical approaches, such as introducing electromagnetic, electric, or acoustic wave fields into the hydrate formation area, these methods also face challenges when it comes to large-scale hydrate formation due to the substantial energy requirements to apply artificial force fields to large volumes of bulk water. Ultimately, the key to commercializing hydrate-based gas storage technology lies in achieving facile hydrate formation within a static system, without any mechanical techniques. The developed hydrate formation strategies in a static system and future aspects for this technology will be covered in the subsequent sections.
| Mechanical techniques | Stirring | Bubbling | Spraying |
|---|---|---|---|
| Advantages | Regenerating the gas–liquid interface | Increasing gas–liquid interfacial area by generating tiny gas bubbles | Enlarging the gas–liquid interface area by dispersing water through a spray nozzle |
| Dispersing hydrate crystals uniformly | Appropriate for a continuous hydrate formation system | ||
| Facilitating heat transfer | |||
| Drawbacks | High energy requirements for mixing components | Energy requirements for generating gas bubbles continuously | Energy requirements for spraying water droplets continuously |
| Dramatically increasing stirring energy with increasing hydrate conversion | Low water-to-hydrate conversion due to the formation of a solid film on the surface of bubbles | Difficulty in heat dispersion | |
| Limited mass transfer between water and gas molecules due to the agglomeration of bubbles | Risk of operational failure due to the blocked spray nozzle |
4. Strategies for overcoming thermodynamic hurdle
In nature, gas hydrates can take on three primary structures, sI, sII, and sH, as mentioned earlier. Among the various types of gas hydrates, sI type is the most commonly observed in NG hydrates. The unit cell of the cubic sI hydrate (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) n) consists of two small cages (512) and six large cages (51262) with a total of 46 water molecules (6(51262)·2(512)·46H2O). Jeffrey (1984) introduced a nomenclature system (nm) for the polyhedral cages, where ‘n’ represents the number of edges on a face type and ‘m’ denotes the number of faces with ‘n’ edges.181 For example, a pentagonal dodecahedron with 12 identical faces and edges of equal length can be described as 512.181 The sII hydrate is meanwhile commonly found in oil production pipelines since it involves high pressure and low temperature conditions with water impurity: propane or iso-butane existing in the oil production pipeline forms sII hydrate. The unit cell of cubic sII hydrate (Fdm) consists of sixteen small cages (512) and eight large cages (51264) with a total of 136 water molecules (8(51264)·16(512)·136H2O). The sH hydrate has the largest cage among all hydrate structures, making it capable of accommodating large guest molecules such as neohexane or cycloheptane.182,183 The unit cell of hexagonal sH hydrate (P6/mmm) consists of three small cages (512), two medium cages (435663), and one large cage (51268) with a total of 34 water molecules ((51268)·2(435663)·3(512)·34H2O).
n) consists of two small cages (512) and six large cages (51262) with a total of 46 water molecules (6(51262)·2(512)·46H2O). Jeffrey (1984) introduced a nomenclature system (nm) for the polyhedral cages, where ‘n’ represents the number of edges on a face type and ‘m’ denotes the number of faces with ‘n’ edges.181 For example, a pentagonal dodecahedron with 12 identical faces and edges of equal length can be described as 512.181 The sII hydrate is meanwhile commonly found in oil production pipelines since it involves high pressure and low temperature conditions with water impurity: propane or iso-butane existing in the oil production pipeline forms sII hydrate. The unit cell of cubic sII hydrate (Fdm) consists of sixteen small cages (512) and eight large cages (51264) with a total of 136 water molecules (8(51264)·16(512)·136H2O). The sH hydrate has the largest cage among all hydrate structures, making it capable of accommodating large guest molecules such as neohexane or cycloheptane.182,183 The unit cell of hexagonal sH hydrate (P6/mmm) consists of three small cages (512), two medium cages (435663), and one large cage (51268) with a total of 34 water molecules ((51268)·2(435663)·3(512)·34H2O).
Small gas molecules, including hydrogen, oxygen, and nitrogen, are known to form sII hydrates. Slightly larger gas molecules, such as methane, carbon dioxide, or hydrogen sulfide, tend to form sI hydrates. The specific hydrate structure depends on the size of the guest molecules. Interestingly, when methane and ethane are combined, sI and sII hydrates can be formed. Gas mixtures containing ethane compositions ranging from 1 to 25 mol% tend to result in sII hydrate.184,185 In general, these gas hydrates can be constructed under low temperature and high pressure conditions, and these conditions can be varied as illustrated in Fig. 3a.186–188 To reduce the required pressure and increase the required temperature, thermodynamic promoters are employed. These promoters are crucial for energy-efficient gas storage in hydrate media and are typically larger in size compared to small gas molecules. They occupy the 51264 cages in sII hydrates or the 51268 cages in sH hydrates, stabilizing the overall hydrate structure. Consequently, small gas molecules can be trapped in the remaining small cages under relatively lower pressure and higher temperature conditions compared to pure gas hydrates. Many chemicals have been identified as effective thermodynamic promoters, providing sufficient driving force to form hydrate structures, and they often feature larger molecular sizes. In some cases, the end-to-end distance of these promoter molecules is slightly larger than the average cavity diameter of hydrates. For example, certain large molecules such as 3-methyl-1-butanol, with an end-to-end distance of 9.04 Å, have been observed to fit within the large cages of sII hydrates, even though their size is slightly larger than the average cavity diameter (51264 cage: 8.66 Å; 51268 cage: 9.46 Å).189 However, since the water framework of hydrates is constructed through hydrogen bonds, the size of hydrate cages can be adjusted to accommodate these larger guest molecules.
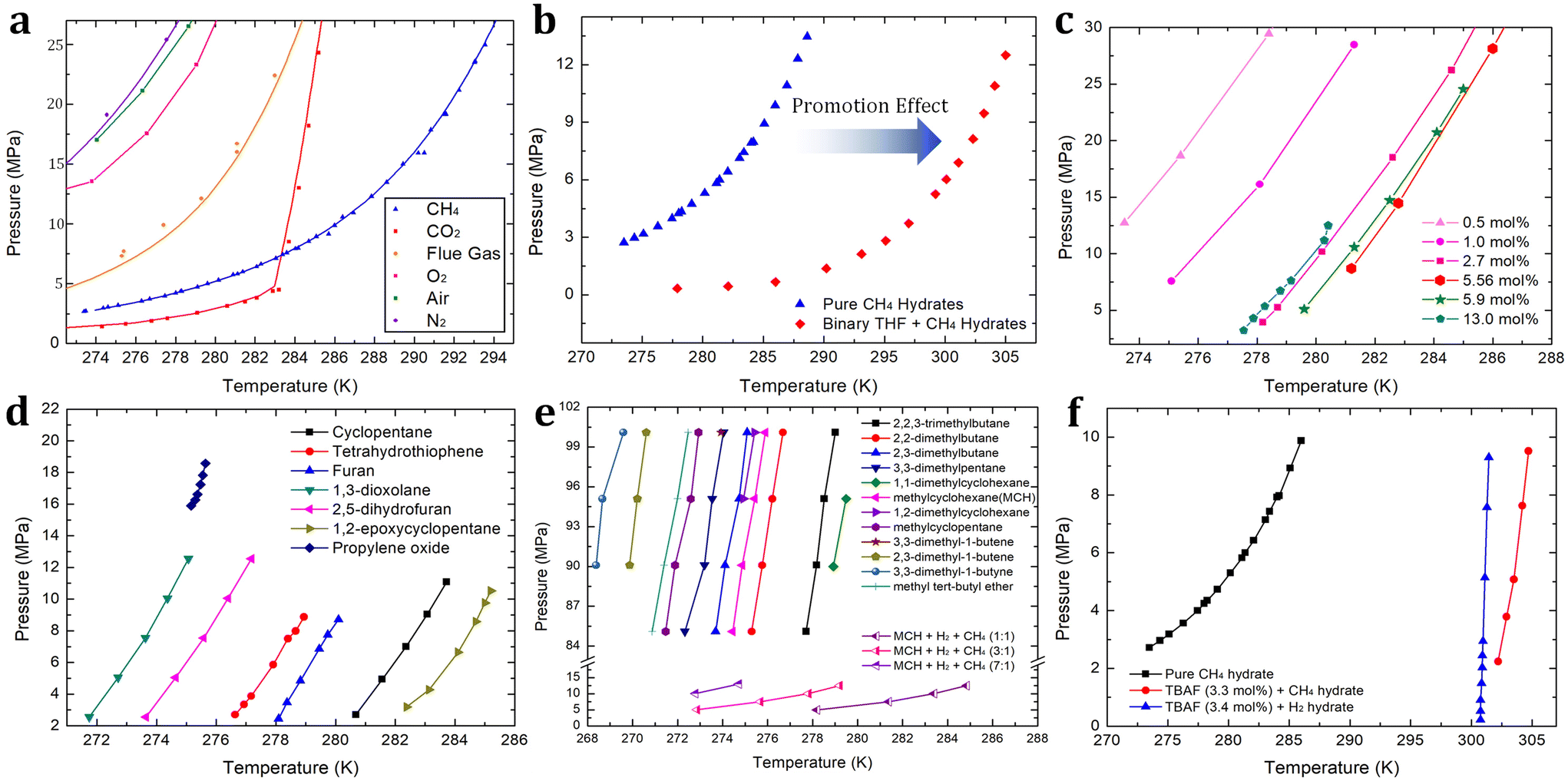 | ||
| Fig. 3 (a) Phase equilibrium boundaries of various gas hydrates containing CH4,115 CO2,186 flue gas (CO2 + N2),187 O2,188 air (O2 + N2),188 and N2.188 (b) Phase equilibrium boundaries of binary THF + CH4 hydrates193 in comparison with pure CH4 hydrate.115 (c) Phase equilibrium boundaries of binary THF + H2 hydrates with various concentrations of THF.194,195 (d) Phase equilibrium boundaries of binary H2 + various sII promoters.198–201 (e) Phase equilibrium boundaries of binary H2 + various sH promoters.202,203 (f) Phase equilibrium boundary of binary TBAF (3.3 mol%) + CH4211 hydrates and that of TBAF (3.4 mol%) + H2210 hydrates in comparison with pure CH4 hydrate.115 | ||
In the following, first, various kinds of sII-forming thermodynamic promoters will be investigated. Vos et al. first reported the enclathration of hydrogen in the ice II phase within a pressure range of 0.75 to 3.1 GPa at a temperature of 295 K.190 As the formation of pure hydrogen hydrate requires extremely high-pressure conditions, many studies have explored various thermodynamic promoters to alleviate this pressure requirement. Florusse et al. introduced an innovative strategy aimed at achieving hydrogen enclathration under more moderate temperature and pressure conditions.191 They successfully incorporated hydrogen into sII hydrate at 5 MPa and 279.6 K, provided that tetrahydrofuran (THF) coexisted within the hydrate medium.191 This breakthrough led to the discovery of an important research avenue within the hydrate field: the search for a suitable thermodynamic promoter to facilitate hydrogen storage via hydrate-based systems. Lee et al. demonstrated that by reducing the THF concentration from the stoichiometric concentration of sII hydrates (5.56 mol%) to 0.2 mol%, the hydrogen storage capacity increased to approximately 4.0 wt%.192 This achievement was made possible through a tuning phenomenon, where the empty large cages of sII hydrates could host between two to four hydrogen molecules. The specifics of this tuning phenomenon are discussed at the end of this section.
Due to the extremely high-pressure conditions required for the existence of pure hydrogen hydrate, comparing the phase equilibrium boundaries of pure sII hydrogen hydrate and binary sII THF + hydrogen hydrate within an identical pressure range is challenging. However, in the case of pure methane hydrate, which can be formed under relatively higher temperatures and lower pressures, we can assess the thermodynamic promotion effect of THF. As illustrated in Fig. 3b, the presence of THF (5.56 mol% to water) in the methane hydrate medium causes a shift in the phase equilibrium boundary.115,193 This shift becomes more pronounced as pressure increases, reaching approximately 17 K at 12 MPa. The extent of this shift is also influenced by the concentration of THF, and this trend can be similarly observed in hydrogen hydrate media. As the concentration of THF increases and reaches the stoichiometric concentration of 5.56 mol%, the promotion effect becomes more significant. However, when the THF concentration exceeds the stoichiometric concentration, as reported by Anderson et al.194 and Hashimoto et al.195 (Fig. 3c), the promotion effect becomes less significant. Motivated by THF offering a sufficient driving force for storing hydrogen or methane in hydrate media, various attempts to apply it to hydrate-based hydrogen or methane storage have been studied. Veluswamy and colleagues reported rapid storage of methane196 or hydrogen197 in hydrates formed from THF solutions. Numerous studies have examined the thermodynamics of hydrogen hydrate promoters, including THF,197 cyclopentane (CP),198 tetrahydrothiophene,199 furan,199 1,3-dioxolane,200 2,5-dihydrofuran,200 1,2-epoxycyclopentane (ECP),201 and propylene oxide.201 The phase equilibrium boundaries of these promoters are summarized in Fig. 3d. Among these thermodynamic promoters forming sII hydrate, ECP stands out as the most potent, with the phase equilibrium boundary of binary ECP + hydrogen hydrate located in the rightmost region (indicating the highest dissociation temperature at any given pressure).
When comparing sII and sH hydrates, sH has higher gas storing capability in terms of the number of gas-storing cages per water molecule. In a unit cell, sH hydrates offer a higher ratio of gas storage cages to water molecules, ((3 small cages + 2 medium cages)/34 water molecules ≈ 0.147) in comparison to sII (16 small cages/136 water molecules ≈ 0.118) hydrates, assuming full occupancy of small or medium cages. While sH hydrates seem promising for energy gas storage due to their excellent gas storage capacity, it is important to note that their formation typically requires much higher pressure than that required for sII hydrates. In 2009, Duarte et al. summarized the phase equilibrium boundaries of sH hydrogen hydrates with various compounds, including alkanes, alkenes, alkynes, cycloalkanes, and ethers.202 They pointed out that the geometry of the promoter can affect the stabilization conditions of sH hydrates containing hydrogen.202 Generally, alkanes and cycloalkanes exhibit better hydrate structure stabilization compared to alkenes, alkynes, and ethers. These phase equilibrium boundaries are summarized in Fig. 3e. Interestingly, in the case of methylcyclohexane (MCH), the phase equilibrium boundary of sH binary MCH + hydrogen hydrate was found in a pressure range of 83 to 100 MPa and a temperature range of 274 to 276 K. However, when methane molecules are introduced alongside MCH and hydrogen in sH hydrate media, the thermodynamic stability is significantly enhanced. For sH ternary MCH + hydrogen + methane hydrates, the phase equilibrium boundaries shift to a lower pressure range of 5 to 15 MPa and a higher temperature range of 273 to 285 K, as shown in Fig. 3e.203 As the hydrogen/methane ratio in the vapor phase increases, the fractional cage occupancy of methane in the medium cage (435663) of sH hydrate decreases since more hydrogen molecules compete with methane molecules to occupy the hydrate lattice. As there should be a trade-off relationship between the moderate hydrogen enclathration conditions (i.e. higher dissociation temperature at any given pressure) and the amount of stored hydrogen (i.e. higher hydrogen/methane ratio, resulting in higher medium cage occupancy of hydrogen), careful consideration is needed to determine the composition of hydrogen + methane blends for storage in sH hydrate media.
Compared to the pressure required for forming pure hydrogen hydrates, thermodynamic promoters that create sH hydrates can reduce the required pressure to some extent. Nevertheless, even with these promoters, the required pressures for storing hydrogen in sH binary hydrogen + promoter hydrates remain relatively high. Therefore, considering both the thermodynamic conditions and the hydrogen storage capacity per water molecule, sII hydrate is a more feasible option. On the other hand, for methane enclathration, the dissociation temperatures of sH binary methane + 2-methylbutane, MCH, and 2,2-dimethylbutane hydrates increase by approximately 1.5, 5.4, and 6.5 K, respectively, compared to pure sI methane hydrate.204 Given that the formation conditions of sH binary methane + promoter hydrates are generally more moderate, sH hydrate offers an advantage as a medium for methane storage.
Finally, ionic clathrate hydrates, which consist of cations or anions enclathrated in cages with counterions incorporated into the host water framework, will be discussed as thermodynamic promoters for hydrate-based gas storage. They are known to remain stable at moderate temperatures without the need for secondary “help” gas molecules. In some cases, small ionic species such as tetramethylammonium cations (Me4N+)205 or hexafluorophosphate anions (PF6−).206,207 Fluoride-incorporated ionic clathrate hydrates can be accommodated within a single cage. However, in the case of larger ionic species such as tetra-n-butyl ammonium ((nBu)4N+) or tetra-iso-amyl ammonium ((i-amyl)4N+), their molecular size exceeds the capacity or size of a single hydrate cage.208 To encage these larger ions, some cages should be partially broken and interconnected, resulting in “semi-clathrate” hydrates. These ionic clathrate hydrates can exhibit various crystal structures, influenced by the hydration number and temperature. For example, Me4NOH hydrates have been observed to have eight different crystal structures.209 A structural transition from β forms of 5.0- or 7.5-hydrates to α forms of 5.0- or 7.5-hydrates occurs as the temperature changes from 42 °C to 6 °C. Additionally, with hydration numbers of 5, 7.5, and 10, Me4N+ cation species are enclathrated in 4668, 51263, and 4151066 cages, respectively.209 Although these Me4NOH hydrates contain identical ions within the hydrate medium, their diverse structures result in variations in thermodynamic stability, gas storage capacity, and conductivity. Therefore, the choice of hydration number and operating temperature range should be carefully considered depending on the intended applications.
Ionic species serve as effective thermodynamic promoters for gas enclathration due to their remarkable hydrate structure stabilization capabilities, which surpass those of conventional sII or sH hydrate promoters. Compared to the sH hydrate former discussed above, hydrogen molecules can stably exist at significantly lower pressures and higher temperatures. For example, tetramethylammonium fluoride (TBAF) semi-clathrate hydrates (at a concentration of 3.4 mol%) can store hydrogen at temperatures above 300 K and pressures below 10 MPa, representing milder conditions compared to the sH hydrates (see Fig. 3f).210 Notably, the TBAF system also offers a stable environment for methane enclathration. At a pressure of 10 MPa, the dissociation point of TBAF (3.3 mol%) + methane hydrate is increased by approximately 19 K (as depicted in Fig. 3f),211 with this degree of structural stabilization slightly surpassing that of the binary sII THF + hydrogen hydrate. Despite their superior thermodynamically promoting effects, sII hydrates remain more favorable for gas storage applications due to their higher gas storage capacity. This difference can be attributed to the increased occupancy and population of small cages in sII hydrates.212
Numerous studies on thermodynamic hydrate promoters demonstrate that the production of NG or hydrogen hydrates can occur in significantly milder conditions when these promoters are utilized. Their use guarantees the economic feasibility of the process and also provides sufficient driving force for hydrate growth. However, it is important to note that as promoter molecules occupy the cages, the maximum gas storage capacity per unit of water decreases. Thus, an optimization step, such as concentration control, should be implemented for practical application.
By employing the aforementioned thermodynamic promoters, it becomes possible to substantially reduce the required pressure for encaging hydrogen or methane due to their ability to stabilize the hydrate structure. Nevertheless, when utilizing thermodynamic promoters there exists a trade-off relationship with their impact on reduced gas storage capacity. These promoter molecules must occupy the large cages of hydrates where hydrogen or methane can be accommodated. As a breakthrough, the “tuning” phenomenon was first introduced by Lee et al.: it involves designating specific large hydrate cages for hydrogen storage when the concentration of the thermodynamic promoter (in this case, THF) is significantly lower than the stoichiometric concentration (below 5.56 mol%).192 As shown in Fig. 3c, the degree of the promoting effect becomes less significant as the concentration of THF decreases. However, the substantial increase in the amount of stored hydrogen (around 4 wt% with 0.15 mol% of THF) compensates for the loss in thermodynamic promotion.
Numerous efforts have been dedicated to inducing the tuning phenomenon within sII hydrate, particularly with the aim of enhancing hydrogen storage in their large cages. Park et al. introduced hydrogen to a pre-synthesized binary THF + N2 hydrate, with the intention of loading multiple hydrogen molecules into the slightly expanded large cages through a guest-exchange reaction.70 Under 35 MPa of pressure, up to four hydrogen molecules were stored in the large cages with this approach.70 Koh et al. utilized a sH hydrate forming-agent, 1-methylpiperidine, at diluted concentrations ranging from 0.5 to 2.0 mol%.213 These diluted promoters induced a structural transformation from sH to sII hydrates, leading to the enclathration of two to four hydrogen molecules in the large cages of sII hydrate at 58 MPa.213 Additionally, certain hydrocarbons, such as propane214 or blends of methane and ethane,215,216 have been reported to induce multiple hydrogen occupancies in both small and large cages of sII hydrates. Furthermore, various intriguing strategies have been introduced to enhance hydrogen storage capacity within hydrate media. These approaches aim to store two to four hydrogen molecules in the large cages or up to two hydrogen molecules in the small cages of sII hydrate through a guest-exchange reaction217 or proton irradiation.218 The overarching goal is to enhance the overall hydrogen storage capacity within hydrates.
5. Strategies for overcoming kinetic hurdles
When sufficient driving force is ensured within the system, hydrate formation depends on effectively overcoming kinetic hurdles, such as stochastic nucleation and limitations in mass and heat transfer. As discussed in Section 3, kinetic promotion strategies using mechanical methods such as stirring, bubbling, and spraying are highly effective for overcoming these obstacles. By applying mechanical techniques, the gas–liquid interfacial areas become enlarged and the heat generated during hydrate formation is easily distributed within the system. On the other hand, various experimental cases introduced in this section are efforts to address kinetic challenges within a static system, utilizing principles similar to those of mechanical techniques. For each kinetic hurdle, strategies to achieve promotion effects within the static system, comparable to those of mechanical approaches, were introduced and their significance was discussed.5.1. Strategies to overcome stochastic nucleation
As mentioned, the stochastic nature of hydrate nucleation is a critical hindrance for utilizing hydrates in various industrial fields. Therefore, numerous strategies have been developed to facilitate hydrate nucleation and, at the same time, fundamental investigations of the nucleation process are ongoing. Similar to the typical nucleation systems, the nucleation of hydrates can be classified into two types: homogeneous and heterogeneous nucleation. While homogeneous nucleation rarely occurs, heterogeneous nucleation mostly occurs in hydrate-forming systems.219 There are two possible nucleation sites for a typical hydrate-forming system: (i) a gas–liquid interface and (ii) a liquid–substrate interface. Most of the methods for the kinetic promotion of gas hydrates under a static system add an additional substrate to the system for efficient heterogeneous nucleation. This is done because the surface properties of the additional substrate can highly increase the nucleation possibilities and lower energy hurdles for the nucleation process.Classical nucleation theory describes the energy barriers for the heterogeneous nucleation process in terms of the energy barrier for homogeneous nucleation and the contact angle between the hydrate crystal-liquid–solid surface in the system (Fig. 4a).220 Total excess energy for homogeneous nucleation (ΔGtotal) is equal to the sum of the surface excess energy (ΔGs) and volume excess energy (ΔGv), and therefore their relationship can be written as eqn (1):
| ΔGtotal = ΔGs + ΔGv = 4πr2γ + ¾πr3Δgv | (1) |
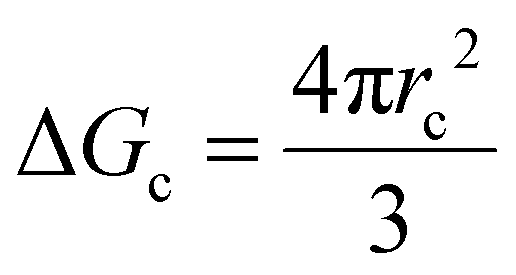 | (2) |
| ΔG′c = ϕΔGc | (3) |
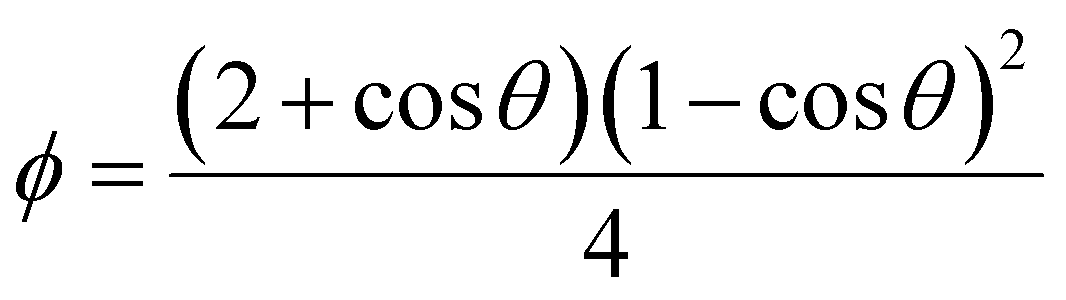 | (4) |
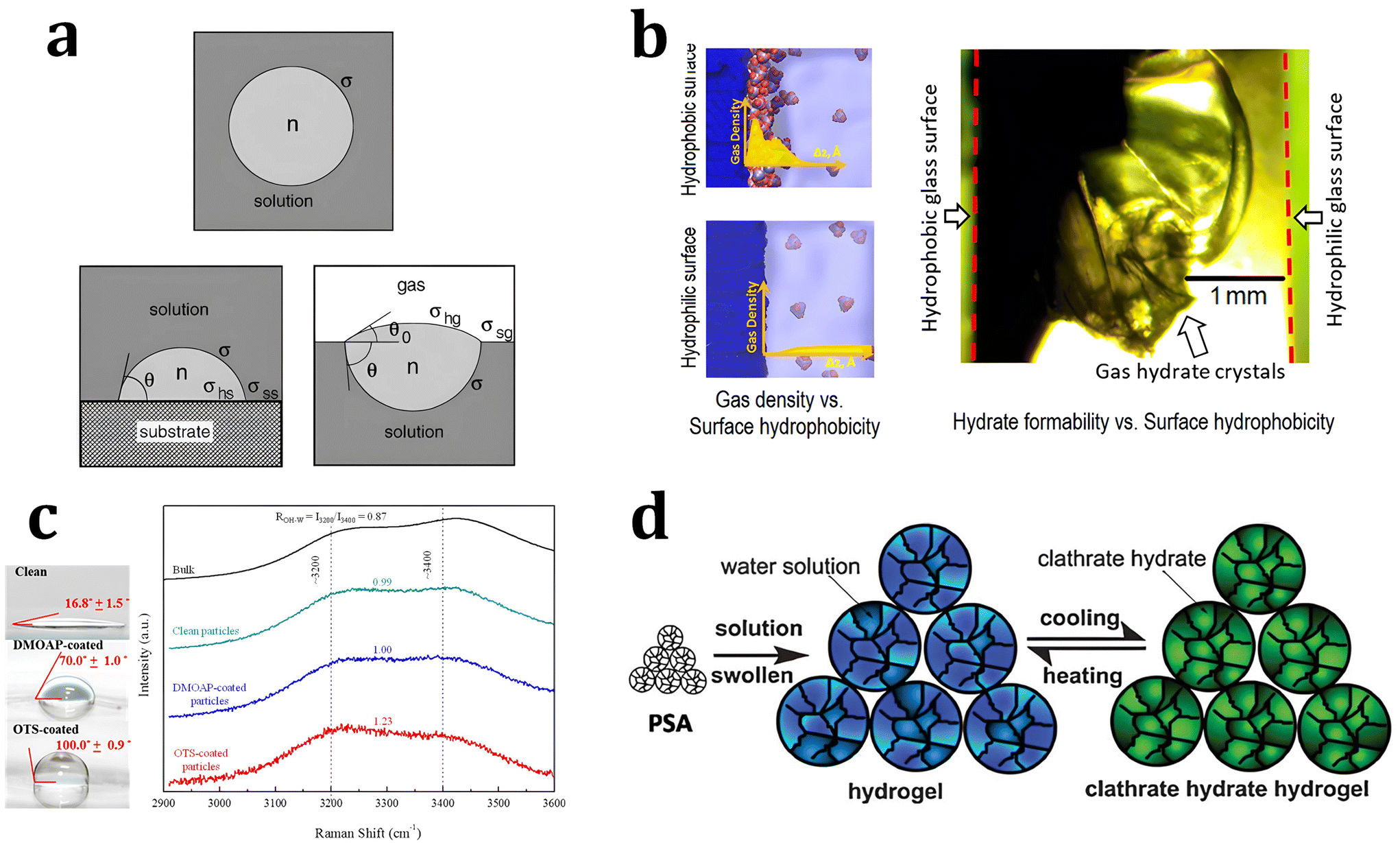 | ||
| Fig. 4 (a) Schematic illustration of hydrate clusters in homogeneous nucleation and heterogeneous nucleation. Reproduced with permission.220 Copyright 2002 Elsevier. (b) Gas density profile at the interfacial region with hydrophobic and hydrophilic surfaces. Reproduced with permission.221 Copyright 2017 American Chemical Society. (c) Raman spectra for water measured in the bulk and near various particles. Reproduced with permission.223 Copyright 2016 American Chemical Society. (d) Schematic illustration of hydrogen hydrate formation with hydrogel. Reproduced with permission.234 Copyright 2009 John Wiley and Sons. | ||
Some theoretical and MD studies have reported that hydrophobic substrates have an advantage in promoting the heterogeneous nucleation of hydrates under static conditions. Nguyen et al. investigated the local gas density near the surfaces of substrates with varying degrees of hydrophobicity using MD to understand the promotion effect of hydrophobic surfaces on gas hydrate formation.221 Their simulation results showed that hydrate nucleation processes were promoted with hydrophobic substrates owing to the enrichment of hydrophobic gas molecules near the substrates. In contrast, hydrophilic surfaces decrease the local density of hydrophobic gas molecules near the surface (Fig. 4b). This indicates that the hydrate nucleation process could be highly dependent on the properties of substrates, where the heterogeneous nucleation processes occur. Subsequent experimental results also support their MD studies, showing that CO2 gas hydrate preferentially forms near the hydrophobic glass. With hydrophobic glass having contact angles of 130 degrees, CO2 hydrate formation occurred under static conditions within 12 hours of reaction time, while CO2 hydrate did not form on hydrophilic glass with a contact angle of 5 degrees.
Apart from the effect of hydrophobic/hydrophilic substrates on increasing/decreasing the local gas density at the hydrate nucleation sites, the behavior of water molecules near those substrates has also been regarded as a key factor affecting the nucleation probability of hydrates. Most studies consistently show that hydrophobic substrates are more effective in promoting hydrate nucleation than hydrophilic substrates by altering the behavior water molecules near the substrates. However, the mechanism underlying the nucleation promotion effect of hydrophobic substrates remains controversial. Bai et al. conducted MD studies to investigate the effect of water molecule ordering with substrates of varying hydrophilicity on CO2 hydrate nucleation.222 They found that substrates with strong hydrophilicity exhibited a local ice-like water structure near the surfaces, along with a three-step nucleation mechanism: (i) an ice-like layer, (ii) an intermediate thin layer, and (iii) CO2 hydrate formation. In the case of substrates with weak hydrophobicity, however, the amorphous ice-like layer did not form near the liquid–solid interfaces, decreasing the number of nucleation steps from three to two steps, which promotes the formation of gas CO2 gas hydrates.
However, from the results of other MD and spectroscopic studies, it has been argued that the local water structure near hydrophobic hydrate surfaces promotes hydrate nucleation. Nguyen et al. observed the local water structure near hydrophobic substrates using MD studies, and suggested a synergistic effect with a high local density of gas molecules for the promotion of hydrate nucleation.221 Li et al. investigated the local water structure at the surface of substrates using Raman spectroscopic analysis.223 In Raman spectra, the O–H stretching bonds of water molecules are indicated in the range of 3000 to 3600 cm−1, and the ratio of intensity maxima of two major bands at 3200 cm−1 (ordered water molecules) and 3400 cm−1 can indicate the ordered state of the water molecules (Fig. 4c). Their experimental studies showed that the ordering parameters of the water molecules have an inverse relationship with the mean induction time for hydrate formation, indicating a promotion effect of hydrophobic substrates on hydrate nucleation.
Numerous types of materials have been used to enhance hydrate nucleation under static conditions, providing preferred nucleation sites for gas hydrates. Silica gel, which comprises amorphous silicon dioxide (SiO2) particles, is a well-known porous material that offers a high surface area for rapid and immediate nucleation in static systems. Many studies on methane hydrate formation under static conditions have shown that silica gel can effectively eliminate the induction time due to its nano-pore structure and high surface area, increasing the probability of nucleation by providing ample sites for heterogeneous nucleation.224,225 However, due to the interaction between water molecules and silicon dioxide, the nano-pore structure of silica gel slightly shifts the equilibrium conditions for methane hydrate towards harsher hydrate formation conditions, as confirmed by several studies.226–228 Therefore, silica gel can be used as a kinetic promoter under experimental conditions with sufficiently high driving forces (e.g., low temperature and high pressure) where its equilibrium-shifting effect can be minimized.
Fixed-bed media containing silica sand can also promote the nucleation of gas hydrates under static conditions by increasing the number of nucleation sites for hydrate formation. Pan et al. reported that the induction time for NG hydrate formation was eliminated by a combination of silica sand and a sodium dodecyl sulfate (SDS) solution, which can decrease the interfacial energy barrier for nucleation.229 It was established that the water saturation ratio in the fixed bed highly affects hydrate nucleation, where no induction time is exhibited at 50% saturation under static conditions, while 100% saturation results in several hours of induction time. This superior nucleation promotion effect with a low water saturation level is attributed to more abundant gas–liquid–solid interfaces than in the case of a high level of water saturation, where a three-phase interface exists only at the top of the bed. Moreover, Linga et al. reported that the induction time for fixed-bed silica sand systems under static conditions is highly affected by the volume and the dimension size of the fixed bed.230 When the reactor volume was reduced by using a copper cylinder to minimize heat transfer resistance, the induction time for methane hydrate formation was significantly reduced. This reduction occurred because heat was easily removed from the reactor, as indicated by the small exothermic temperature peak during the hydrate formation process.
Activated carbon, one of the most common adsorbents for hydrocarbons and pollutants, can also be used to promote the nucleation of methane hydrates due to its high specific surface area and porosity. Zhou et al. first investigated the usage of water-adsorbed activated carbon for enhancing the methane storage capacity by forming hydrates at sufficiently high pressure.231 Many studies have shown that the use of activated carbon in the system significantly reduces the induction time for the methane hydrate formation process. Govindaraj et al. found that the induction time for the methane hydrate formation process was significantly shorter with a suspension of activated carbon particles compared to a system with pure water or silica suspensions.232 They argued that the decrease in induction time with activated carbon suspension is due to the presence of multiple nucleation sites for hydrate formation and the increased solubility of methane gas, which is a result of enhanced methane gas absorption.
Hard supporting matrices such as silica gel and silica sands, however, have their own mass and volume, and this will decrease the overall volumetric energy density and increase the storage and shipping costs. Moreover, to raise the storage capacity for an energy-efficient transportation process, a pelletizing process for gas hydrates would be necessary,233 which would be challenging when using hard supporting matrices. Therefore, soft and lightweight materials with low density are now widely used to trigger an immediate nucleation event. Superabsorbent polymers, also known as hydrogels, can be used to reduce or completely eliminate the induction time for NG and hydrogen hydrate formation in a static system. Su et al. first suggested the use of superabsorbent polymers (polyacrylic acid sodium salt) as kinetic promoters for THF–hydrogen hydrate formation in a static system (Fig. 4d).234 Furthermore, Kang et al. studied the repetitive formation of methane hydrates with superabsorbent polymers and THF as thermodynamic promoters to develop a sustainable methane hydrate formation and storage system.235 They found that the structure of the superabsorbent polymers remained intact during the repetitive formation–dissociation process of methane hydrate, and under all conditions in the range of 275 to 285 K and 50 to 70 bar, the formation of methane hydrate occurred immediately without any induction time.
Aside from utilizing additional kinetic additives or supporting matrices, which decrease the volumetric and mass storage capacity for energy-efficient gas storage, the use of tiny amounts of pre-constructed hydrate particles has garnered attention owing to their powerful ability to trigger nucleation events even under static conditions. Baek et al. revealed that pre-constructed hydrate crystals can provide powerful nucleation sites for the immediate growth of methane hydrate in a static system.236 The metastable zone width, which represents the required extent of subcooling for hydrate formation, was significantly reduced with the addition of pre-constructed CP hydrate crystals.237 These crystals provide nucleation sites for the heterogeneous nucleation of methane hydrate that are much stronger than the nucleation sites on a stainless-steel reactor wall. The promotion effect of pre-constructed hydrate seed crystals was also confirmed in a hydrogen hydrate formation system. Lee et al. utilized them to induce immediate nucleation of hydrogen hydrate with a combination of hydrocarbon gases or liquid-phase promoters such as CP or THF, confirming facile hydrogen storage in hydrates.238,239
Although hydrate nucleation is a stochastic and complex phenomenon that is challenging to fully understand, experimental and simulation studies have shown that rapid or immediate nucleation can be induced using appropriate strategies. Table 3 summarizes the representative strategies demonstrated in this section for enhancing hydrate nucleation probability. Introducing a hydrophobic substrate into the hydrate formation system could increase the local density of gas molecules and rearrange water molecules to more favorable positions for hydrate nucleation. Additionally, employing porous materials, which provide numerous heterogeneous nucleation sites with a high specific surface area, appears promising for triggering hydrate nucleation. Injecting pre-constructed hydrate seeds, which serve as powerful nucleation sites, can also be considered a viable strategy to eliminate the induction time. The individual or combined implementation of these strategies should be considered when designing a hydrate-based NG/hydrogen storage process operating under static conditions.
| Strategies | Materials used | Mechanisms promoting hydrate nucleation |
|---|---|---|
| Utilizing hydrophobic substrate | Hydrophobic glass | Increasing local gas density near substrate |
| Activated carbon | Ordering water molecules | |
| Graphite | ||
| Utilizing porous material | Silica gel | Enlarging water–gas interfacial area |
| Silica sand | Providing numerous heterogeneous nucleation sites | |
| Activated carbon | ||
| Hydrogel | ||
| Utilizing pre-constructed hydrate seed | CP hydrate seeds | Providing powerful nucleation sites |
| THF hydrate seeds |
5.2. Strategies to overcome mass transfer limitations
When the facile nucleation of hydrates is assured, the overall performance of the gas storage process using hydrates would be most significantly influenced by mass transfer within the system. Specifically, insufficient mass transfer in a static system usually results in premature termination of hydrate formation, as mentioned earlier. To overcome the mass transfer limitation in hydrate formation, two strategies are widely adopted in a static system: one is increasing the interfacial area of the gas–liquid or liquid–substrate system, and the other is providing mass transfer pathways to the bottom of the system. The growth kinetics of gas hydrates can be interpreted by considering the mass balance equation of hydrate-forming systems. The hydrate kinetic growth model suggested by Englezos et al. considers the mass balance equation between a gas–liquid interface and liquid–hydrate surface system.136 The gas consumption rate per hydrate particles can be expressed using eqn (5) and (6):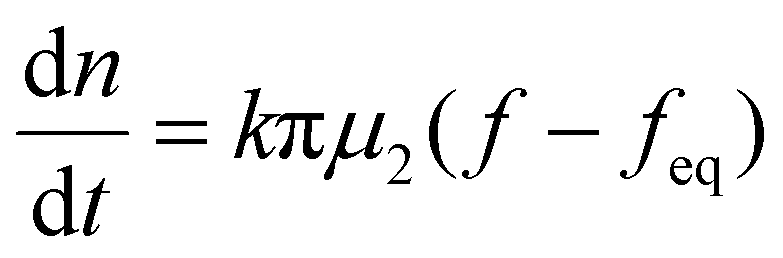 | (5) |
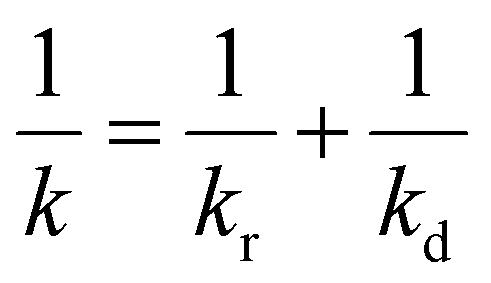 | (6) |
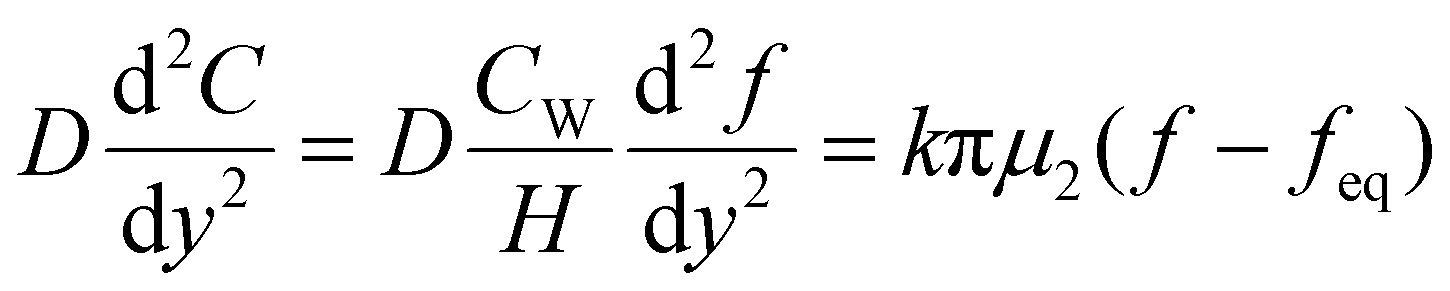 | (7) |
| f = fg at y = 0 | (8) |
| f = feq at y = δ | (9) |
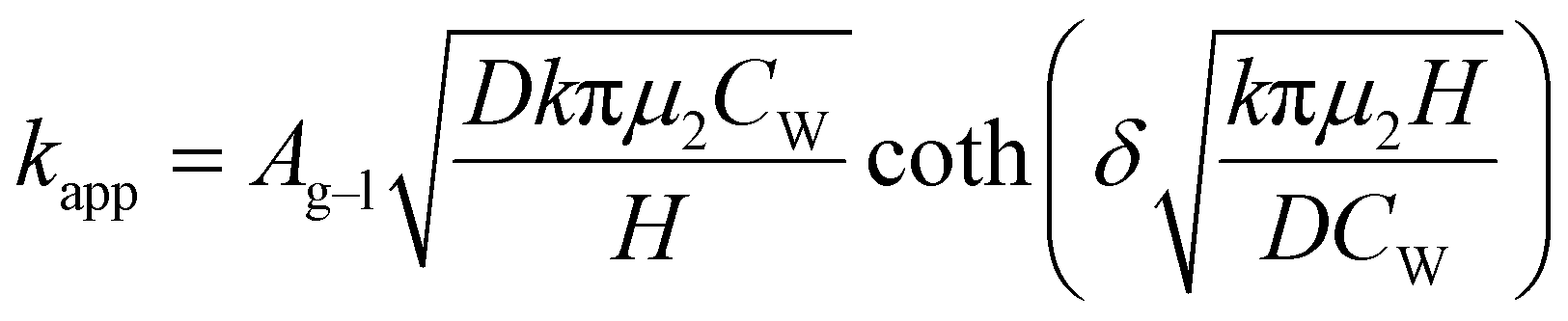 | (10) |
Surfactants or surfactant-like molecules, such as SDS or hydrophobic amino acids, are used to increase the formation rate of gas hydrates by reducing the interfacial tension between the gas–liquid interface, increasing the interfacial area of the hydrate surfaces, or raising the local gas density of hydrophobic gas molecules with the incorporation of hydrophobic substrates (Fig. 5a).240–242 On the other hand, the adsorption of surfactants on the hydrate surface prevents the agglomeration of hydrate particles, which is why surfactants are used as anti-agglomerants to prevent plugging issues in submarine oil or gas transportation pipelines caused by hydrate plugs. Based on this role, some surfactants, such as Span 20 or Tween 20, are used as hydrate inhibitors that inhibit agglomeration and further growth of gas hydrates.243 Nonetheless, an appropriate amount of surfactants can be utilized as kinetic promoters under static conditions.49,243,244
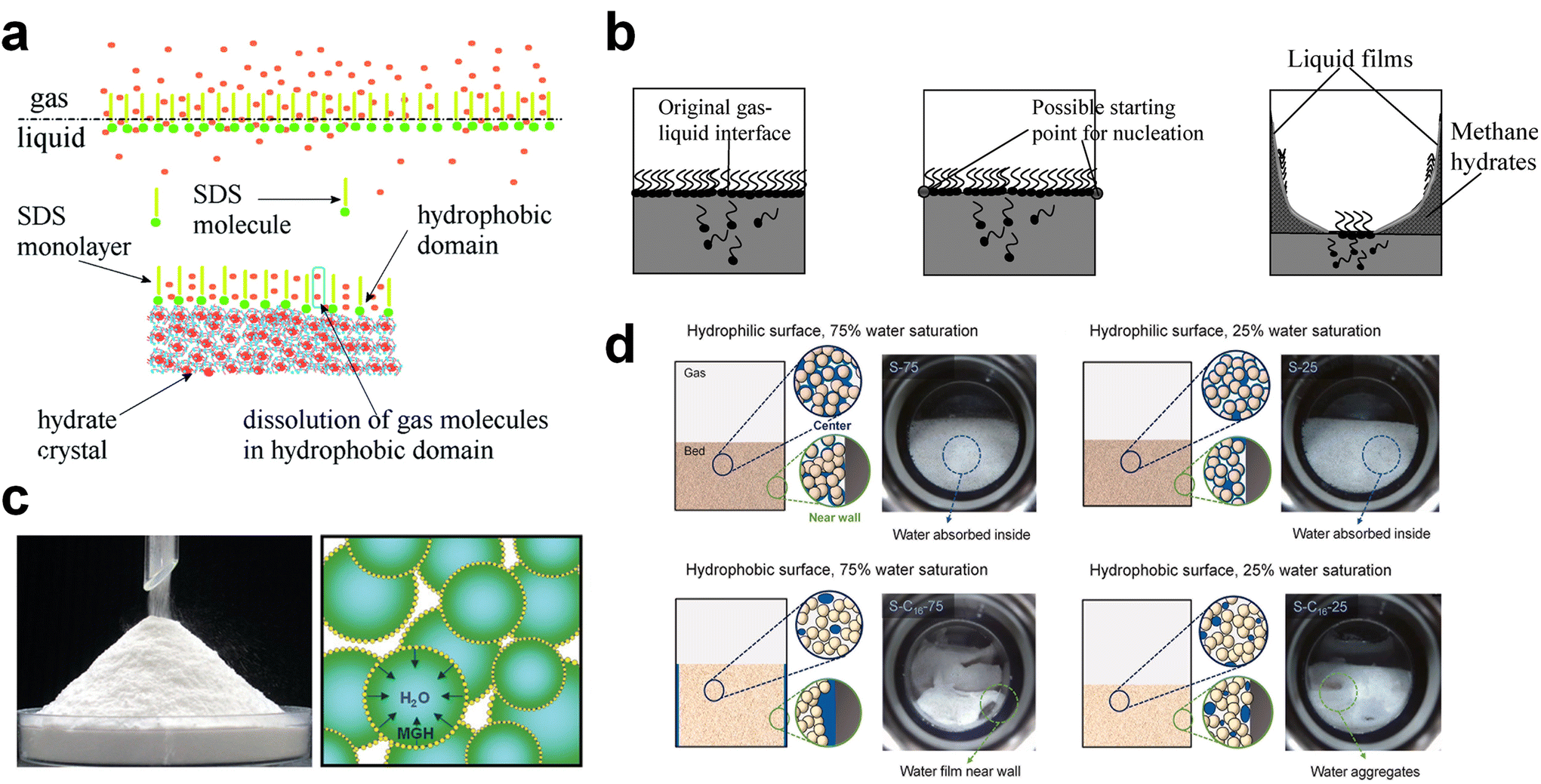 | ||
| Fig. 5 (a) Schematic illustration of surfactant adsorption on the hydrate surface. Reproduced with permission.242 Copyright 2019 Royal Society of Chemistry. (b) Schematic illustration of possible mechanism for methane hydrate formation under SDS. Reproduced with permission.124 Copyright 2007 American Chemical Society. (c) Picture of dry water and schematic of methane hydrate formation with dry water. Reproduced with permission.252 Copyright 2008 American Chemical Society. (d) Schematic representations describing the influence of interfacial interactions between liquid water, gas, and silica sand particles on hydrate formation kinetics. Reproduced with permission.253 Copyright 2023 Elsevier. | ||
Zhong and Rogers first reported that a small amount (284 ppm) of SDS could enhance the growth of ethane hydrates by increasing the hydrate formation rate by 700 times.245 Gayet et al. suggested that only a small amount (10–3 wt%) of SDS was sufficient to promote hydrate formation by preventing the agglomeration of hydrate particles and inhibiting the formation of a rigid hydrate film at the gas–liquid interface.246 Among previous studies on the SDS promotion effect, Zhang et al. calculated the growth kinetics parameters of methane hydrate with the addition of SDS in the reaction system and observed the growth morphologies of the methane hydrate.124 By increasing the SDS concentration, the initial apparent rate constant of hydrate growth also increased due to the increased value of μ2 (in eqn (10)), which represents the total surface area of hydrate particles. This is because SDS prevents the agglomeration of hydrate particles, which increases the number of particles around the crystallization zone. Additionally, Zhang et al. suggested that SDS can promote mass transfer and the apparent rate constant over time by increasing Ag–l, the gas–liquid interfacial area (in eqn (10)). This increase is achieved by inducing vertical growth of methane hydrates as the aqueous solution is drawn up through the generated porous structure of hydrates by capillary force (Fig. 5b). Interestingly, the formation of the porous structure of hydrates is due to the adsorption of SDS molecules on the hydrate surface, which is also related to the inhibition mechanism of some surfactant molecules. Therefore, it can be concluded that an appropriate amount of surfactant molecules such as SDS can significantly promote the hydrate growth by increasing the total particle area and gas–liquid interfacial area without any mechanical technique.
Some amino acids, which are protein-building molecules, have recently been widely investigated as bio- and eco-friendly hydrate kinetic promoters. Amino acids can be classified into hydrophilic or hydrophobic amino acids depending on the hydrophilicity/hydrophobicity of their side chain. Hydrophobic amino acids share a similar molecular structure with surfactants, featuring hydrophilic heads (carboxyl and amino groups) and hydrophobic chains in their side chains. Many studies have revealed that hydrophobic amino acids have a promotion effect on the formation of NG or hydrogen hydrates, while hydrophilic amino acids do not.96,247–249 Veluswamy et al. investigated several amino acids as kinetic promoters for methane hydrate formation under both stirred and unstirred reactor systems.250L-Tryptophan, a hydrophobic amino acid with an aromatic side chain, showed a significant kinetic promotion effect for methane hydrate formation in both static and stirred reaction systems. However, for polar amino acids, the hydrate promotion effect was not as pronounced as that of nonpolar hydrophobic amino acids.
Jeenmuang et al. suggested a possible hypothesis explaining how the hydrophobicity of amino acids affects the growth kinetics of methane–THF hydrate.89 They proposed that the hydrophobicity of amino acids creates a hydrophobic area around the amino acid molecules, which enhances the diffusivity of methane molecules and, consequently, promotes hydrate growth. Furthermore, water molecules, pushed away from methane clusters by the hydrophobic domain, were localized through hydrogen-bonding interactions, further promoting the growth of methane hydrate. Therefore, they concluded that the hydrophobic portion of the amino acids plays a significant role in localizing water molecules and increasing the diffusivity of methane or hydrogen molecules around the ‘hydrate embryo’.
The porous or high-surface area materials mentioned in Section 5.1, such as porous media or NPs, can also be used to enhance the growth kinetics and the formation rate of hydrates by increasing the interfacial area or providing pathways for mass transfer. Many studies have shown that silica gel with a high surface area can increase the growth rate and reduce the required reaction time for methane or hydrogen hydrate formation by expanding the surface area in static systems.225,251 Zhang et al. suggested kinetic models for hydrate formation with silica gel pores of different diameters based on the shrinking core model.224 Applying these kinetic models, it was found that the dominant rate-determining process for hydrate formation varied depending on the pore size of the hydrogel. For silica gels with inner pores of 129.5 nm and 179.6 nm, the dominant process was the gas diffusion process, whereas for silica gels with a pore size of 332 nm, the reaction process was dominant. When hydrate formation was reaction-controlled (i.e., with a large pore size), the formation rate was mainly influenced by the hydrate formation conditions (temperature and pressure). When hydrate formation was diffusion-controlled, the formation rate was affected by hydrate saturation within the silica gels.
Silica particles can also be used to enhance mass transfer in various forms. One such application of silica NPs is in the creation of “dry water,” a free-flowing powder. Dry water consists of water droplets surrounded by a network of hydrophobic fumed silica, forming a water-in-air inverse foam (Fig. 5c). It was initially investigated as a kinetic additive for methane hydrate formation by Wang et al.252 They proposed that gas hydrates based on dry water could be a practical platform for methane storage within a reasonable time frame under static conditions. By utilizing dry water prepared by mixing hydrophobic silica NPs and water (95 wt% water), significantly rapid methane uptake can be obtained, achieving 90% saturation within 160 minutes of reaction time in a static system. Moreover, constructing a fixed-bed system consisting of micro-sized silica sand can result in rapid and high gas uptake under static conditions due to the pore structure in the bed system.249 Engineering functional groups on the surface of silica particles would also offer the opportunity to significantly facilitate the transport of gas molecules in the system. Kim et al. investigated the use of surface-modified silica sand in a fixed bed reactor for promoting methane hydrate formation under static conditions (Fig. 5d).253 By modifying the surface of the silica sand to render it hydrophobic, the interfacial interactions between the silica sand, water, and gas are significantly altered. This surface modification prevents the silica sand particles from becoming wet, providing stable gas diffusion pathways during the hydrate formation process and, consequently, leading to faster hydrate formation kinetics compared to the system using typical hydrophilic silica sand.
Table 4 summarizes the strategies discussed in this section that enhance mass transfer and facilitate hydrate formation. The addition of surfactants or surfactant-like molecules in the hydrate-forming system holds promise for improving the mass transfer of water and gas molecules. Their amphiphilic nature enables the dissolution of gas molecules into the solution and facilitates the transport of water molecules by forming a porous hydrate structure. However, the use of surfactant molecules often leads to foam generation during the dissociation process of hydrates, necessitating careful handling of these materials. Utilizing dry water or porous materials also presents promising strategies, as their use results in a significant increase in gas–liquid interfaces within the system. Nonetheless, as mentioned in Section 5.1, their volume and mass negatively affect the achievement of high volumetric gas storage capacity. They would be a suitable option for stationary hydrate-based NG/hydrogen storage systems positioned near large gas fields or hydrogen production facilities.
| Strategies | Materials used | Mechanisms promoting hydrate formation |
|---|---|---|
| Injecting amphiphilic molecules | Surfactant molecules (e.g. SDS) | Enabling the dissolution of gas molecules into water |
| Amino acids (e.g.L-tryptophan) | Making hydrate growth patterns porous | |
| Encapsulating water droplet by forming dry water | Hydrophobic fumed silica nanoparticle | Enlarging water–gas interfacial area |
| Utilizing porous materials | Silica gel | Enlarging water–gas interfacial area |
| Surface-modified silica sand | Providing a mass transfer pathway for gas molecules |
5.3. Strategies to overcome heat transfer limitations
Aside from the kinetic hurdles discussed in the previous sections, the exothermic heat generated from the hydrate formation could hinder the growth of hydrates by elevating the system temperature to the low driving force region. Therefore, extensive models regarding heat transfer as the significant factor of hydrate growth have been studied, as mentioned in Section 2.2. The empirically driven models can be classified according to the mechanism of heat transfer (by convective or conductive) and the shape of the film (straight or curved). The model proposed by Uchida et al., which is based on conductive heat transfer, assumed that the film is hemispherical. In this model, the propagation rate of the film is proportional to the difference between the film temperature and the equilibrium hydrate formation temperature.155 The model presented by Freer et al. suggests complete wetting of the one-dimensional straight film, assuming that conductive heat transfer occurs from the hydrate formation film to the bulk water.157 On the other hand, Mochizuki and Mori introduced an advanced model based on the model proposed by Freer et al., which assigned additional physical significance to the film's shape.158 They advocate a semi-circular shaped film with two-dimensional conductive heat transfer. The Mori model, proposed as an alternative to the conductive heat transfer model, accounts for convective heat transfer in films growing at a uniform velocity and presents a model where the growth rate of the film is proportional to the 1.5 power of the driving force.156 Based on the model suggested by Mori et al., the Peng model considers that the thickness of the film can vary with the driving force, and designs a model where the growth rate of the film is proportional to the 2.5 power of the temperature difference.254 A common feature among these models is that the temperature driving force has a significant effect on the hydrate film growth, indicating that heat transfer is crucial in the hydrate growth system.Typically, NG consists of light paraffins such as methane (C1 alkane), ethane (C2 alkane), and propane (C3 alkane). The heat of formation for C1/C2/C3 alkane hydrate is −54.49 kJ molgas−1,255 −71.80 kJ molgas−1,256 and −129.2 kJ molgas−1,256 respectively. Since the formation of NG hydrates generates a significant amount of heat, it can interfere with the further growth of hydrates. Therefore, heat must be transferred from the growing hydrate surface to the surrounding solution or gas phases to guarantee a high driving force in the hydrate formation system. To accomplish this, various strategies to increase the thermal conductivity in the system have been developed and tested.
For transferring heat generated during hydrate formation, two primary approaches are employed. The first method emphasizes enhancing the thermal conductivity of the liquid phase itself. This is achieved through the incorporation of metal and non-metallic NPs characterized by high thermal conductivity, effectively serving as heat carriers. The use of small-sized particles results in a substantial specific surface area, thereby augmenting the thermal conductivity of the NPs.257 Consequently, NPs with elevated thermal conductivity effectively enhance the thermal conductivity of the solution,99 enabling efficient distribution of heat within static systems. Based on this kinetic promotion effect of nanofluids, their utilization often leads to improved gas uptake and enhanced conversion rates.
In a recent study, a solution introducing both SDS and graphite NPs and a typical SDS solution were compared as hydrate-forming fluids.258 Graphite NPs, possessing a high effective surface area and thermal conductivity, offer abundant nucleation sites and facilitate heat transfer in the hydrate-forming system when used at an appropriate concentration that avoids NP aggregation. For methane hydrate formation, a solution containing SDS and 0.4 wt% NPs exhibited a maximum 17% increase and 56% decrease in gas uptake and induction time, respectively, compared to the SDS solution (Fig. 6a). This promotional effect of NPs was also evident in a fixed-bed system. In a porous media system, the incorporation of SiO2 NPs in glass bead media resulted in a 67.7% increase in gas consumption compared to pure water.259 Surface treatment of NPs by doping with other substances was also studied. Some studies implemented surface-coated Fe3O4 NPs to improve the heat transfer in the hydrate forming system, and one study in particular examined the impact of Fe3O4 NPs doped with SDS and sodium oleate (SO) on methane hydrate formation.260 The structure scheme of the engineered NPs is shown in Fig. 6b. While surface-doped NPs could increase the total thermal conductivity in the system, the grafted functional group on the surface facilitated the dissolution of methane into the solution, resulting in escalated hydrate formation kinetics. Therefore, in the presence of NPs compared to the solution with the same weight fraction of SDS, the induction time was decreased about 77.9% and the storage capacity was elevated about 19.5%. Another study proposed SDS-doped Fe3O4 NPs and demonstrated higher methane consumption when compared to an SDS solution due to the high thermal conductivity of the NP solution.261
 | ||
| Fig. 6 Representative methodologies to overcome heat transfer limit; (a) effect of concentration of graphite NPs on the formation kinetics of CH4 hydrates. Reproduced with permission.258 Copyright 2020 Elsevier. (b) Schematic illustration of hydrate formation aspects at the surface of SDS–SO doped Fe3O4 NPs. Reproduced with permission.260 Copyright 2021 Elsevier. (c) MD simulations of CH4 + THF hydrate formation system with and without metal rod (red: THF molecule, blue: water molecule, grey: methane, black: metal). Reproduced with permission.266 Copyright 2023 Elsevier. (d) Schematic representation of heat propagation through an Al honeycomb framework in a CH4 hydrate growth system. Reproduced with permission.267 Copyright 2019 Elsevier. (e) Schematic interpretation of heat transfer mechanism through floating EG NPs in a CH4 hydrate formation system. Reproduced with permission.270 Copyright 2021 Elsevier. | ||
The second approach involves the introduction of additional heat pathways for heat transfer. This strategy employs auxiliary frameworks with high thermal conductivity to induce the outward flow of heat from the growing hydrate surface. The thermal conductivity of DI water and methane hydrate is 0.61 W m−1 K−1 and 0.57 W m−1 K−1 respetively,262 which are considerably lower values than those of metals (e.g., aluminium, 237 W m−1 K−1263). Therefore, the utilization of metal frameworks as heat transfer pathways results in more facile heat transfer, thereby accelerating hydrate formation. Experimental investigations on hydrate formation in the presence of metal rods have demonstrated that hydrates tend to grow vertically along the metal rods, allowing for the facile release of formation heat.264 Similarly, other experimental studies have indicated accelerated hydrate growth in systems featuring metal plates, attributable to heat transfer toward the plates. Various geometries of metal plates have been explored, revealing rapid hydrate growth in the presence of metal plates with large surface areas.265 This improvement in hydrate formation kinetics in the presence of a metallic framework was also validated in an MD simulation study investigating the growth of THF + methane hydrates in the presence of metal rods. This study showed that the presence of the metal rods increased the curvature of the gas–liquid interface due to the rapid heat transfer through the rods, as illustrated in Fig. 6c.266 This phenomenon leads to an increase of the effective Laplace pressure near the metal rods (according to the Young–Laplace equation), which increases the concentration of aqueous methane. Consequently, the induction time decreased by 76.3% and the methane permeation energy barrier decreased by 15% compared to the bulk system.
Rather than simple metal rods or plates, the incorporation of auxiliary open-cell structured frameworks can ensure significantly enhanced heat transfer within the system. In a previous study, an Al honeycomb-shaped framework was introduced into an SDS solution, resulting in a 14.3% increase in gas uptake compared to a methane hydrate formation system without a framework. As the Al framework provides nucleation sites and dissipates the heat generated during hydrate formation due to its high thermal conductivity, the methane consumption rate was also elevated. The illustration in Fig. 6d depicts the heat transfer-based promotion mechanism, where the hydrate formation heat propagates outside of growing hydrate surface through the well-structured metallic frameworks composed of Al with a high thermal conductivity.267 Another study utilized open-cell copper foam for heat transfer and conducted a sensitivity analysis with respect to porosity.268 With enough high porosity of copper foam (15 pores per inch), the hydrate conversion rate was exceeded by 8.72% compared to the system without a supplementary framework. To induce facile heat conduction, SiC foam was utilized in another experimental study on methane hydrates, resulting in a substantial enhancement in gas uptake of 122.5% compared to a typical SDS solution.269 Research in which NPs play a role in providing a heat pathway has also been carried out. In this study, extended graphite (EG) NPs with a high specific surface area and high thermal conductivity were introduced into an SDS solution.270 The EG NPs, floating on the SDS solution, provided a linked pathway for heat transfer at the gas–liquid interface (Fig. 6e). As a result, when EG was introduced, it was observed that gas uptake enhanced by up to 26.18%, and the maximum reaction rate improved by up to 51.67% compared to a typical SDS solution. These abovementioned strategies also have been readily combined in some research. Yang et al. utilized both stainless steel fibers and Cu NPs with a SDS solution to improve the formation kinetics of NG. The presence of both stainless-steel fiber and Cu NPs resulted in an increase in storage capacity of up to 24.3% and 10.6%, when compared to systems without both additives and just with NPs, respectively.271
Strategies to enhance heat transfer during hydrate formation are summarized in Table 5. One promising approach is the introduction of NPs, which possess high thermal conductivity. Their introduction significantly increases the thermal conductivity of the solution, facilitating the effective dissipation of heat throughout the system. Additionally, incorporating an auxiliary framework composed of materials with high thermal conductivity provides an alternate pathway for heat transfer, allowing for the external emission of heat from the system. It is evident that the utilization of such strategies can maintain the dynamic driving force for hydrate formation to a desire extent. Therefore, strategies that can ensure facile heat transfer in the system should be considered and combined with other strategies introduced in the previous sections for improved NG and hydrogen hydrate formation kinetics in static systems.
| Strategies | Materials used | Mechanisms promoting hydrate nucleation |
|---|---|---|
| Nano particles | Silicon NPs | Enhancing the averaged thermal conductivity of the solution |
| Doped Fe3O4 NPs | ||
| Cu NPs | ||
| Auxiliary framework | Metal rod/plate/fibers | Introducing the additional heat transfer pathways |
| Alumina honeycomb | ||
| SiC foam | ||
| Extended graphite |
6. Future aspects
To utilize clathrate hydrates as a green gas storage medium, a key challenge lies in the development of energy-efficient gas hydrate formation techniques. Achieving cost-effective and large-scale gas hydrate formation necessitates rapid processes without resorting to mechanical techniques. However, facilitating hydrate formation under static conditions is challenging due to thermodynamic and kinetic hurdles, such as limited gas–liquid interfacial area. This review discusses strategies for overcoming these challenges in a quiescent system. As mentioned throughout the manuscript, various studies have investigated methods to overcome thermodynamic and kinetic hurdles in static systems. While all the introduced strategies have yielded significant kinetic improvements compared to a pure water system, these strategies should now be combined for the development of hydrate formation techniques at a commercial level. For instance, hydrate formation systems using porous materials could be tested with additional materials such as NPs to enhance heat transfer. Engineering the surface properties of porous materials to be hydrophobic is also recommended to increase nucleation probability. Other combinations of strategies are also viable, such as utilizing both pre-constructed hydrate seeds and engineered NPs. While some studies have employed multiple strategies to achieve superior hydrate formation kinetics,249,254,267 such efforts should be carried out more systematically in future research. Additionally, a comprehensive consideration of other factors is imperative for real-life hydrate applications, as detailed below.First and foremost, attention must be directed towards ensuring the long-term stability and sustainability of hydrate-based gas storage technology. This technology is employed for the secure storage of energy carriers, such as NG or hydrogen, which are flammable and explosive. The process involves dissociating hydrates to utilize the stored energy gases and reforming hydrates to store them again. Consequently, careful consideration is needed for the system changes arising from the repeated formation and dissociation of gas hydrates. For instance, the use of SDS as a kinetic promoter can significantly induce foam formation during the hydrate dissociation step, impeding the repetitive gas hydrate formation process. To address such issues, a pinpoint strategy, such as incorporating anti-foaming agents, are required for practical application. Additionally, for the use of volatile thermodynamic promoters such as THF, compensatory replenishment of the promoters is needed to offset THF loss during repetitive process cycles.272 Moreover, for systems using porous support materials, a systematic analysis is essential to understand how the physical and morphological properties of the porous media are affected by the repeated formation and dissociation of hydrates. While these issues can be effectively addressed at the laboratory scale, scaling up hydrate-based gas storage processes necessitates the consideration of additional parameters, particularly regarding induction time. Repetitive cycles of hydrate formation and dissociation reduce induction time due to memory effects, offering a degree of predictability in process operation. However, there is still uncertainty due to the inherently random nature of hydrate nuclei formation. Providing a sufficiently large driving force considering the phase equilibrium boundary or injecting small amounts of pre-synthesized hydrate seeds can be employed to pass through or circumvent the metastable region, enabling hydrate-based gas storage process on a large scale.
For the utilization of gas hydrates as transportation media,273 other considerations are necessary. Ultimately, gas hydrates should be prepared in the form of pellets: pelletizing gas hydrates enhances the energy density per unit volume, making them suitable for gas transportation. In large-scale gas hydrate formation through batch reactions, as previously discussed, the utilization of porous support materials becomes imperative. However, the incorporation of these materials may result in a decrease in volumetric energy storage density. Therefore, the development and utilization of porous support materials with low density and large surface area should be prioritized. A comprehensive study on the pelletization process of these materials, their property changes after pelletization, and their reusability also should be conducted. Finally, analysing the long-term gas storage performance of pelletized gas hydrates with self-preservation effects is crucial, as this contributes to ensuring the long-term utilization and sustainability of hydrate-based gas transportation technology. The latest research trends related to the long-term stability and pelletization of gas hydrates are summarized in Table 6, and these studies suggest the future directions for real-life applications of gas hydrates.
| Year | Works | Ref. |
|---|---|---|
| Repeated formation/dissociation process for gas hydrates | ||
| 2020 | Utilized antifoam A concentrate (AAC) to eliminate the foam generated by the addition of SDS | Ko et al.274 |
| 2021 | Investigated long-term stability of SAP and SAP-supported gas hydrates with 20 cycles of formation–dissociation process | Kang et al.235 |
| 2021 | Investigated hydrate formation kinetics using various pore-size media through repeated formation–dissociation processes with magnetic resonance imaging | Farahani et al.275 |
| 2023 | Utilized amino acids to prevent foam generation during the dissociation process and investigated the effect of hydrophobicity on hydrate formation | Jeenmuang et al.89 |
| 2024 | Investigated long-term stability of melamine sponge-supported gas hydrates to increase the mass storage density of gas hydrates | Kang et al.272 |
| Pelletizing process for hydrate storage | ||
| 2021 | Investigated long-term (2 years) stability of pelletized methane–THF hydrate at atmospheric pressure | Bhattacharjee et al.276 |
| 2022 | Utilized dioxane as thermodynamic promoters for methane hydrate and investigated long-term (120 days) stability of pelletized hydrate at moderate (268.3 K) temperature | Zhang et al.277 |
| 2024 | Investigated the stability (30 days) of hythane 15 (H2:CH4 – 15:85) hydrate pellets without any additional thermodynamic promoters to analyze the feasibility of transporting hythane gas hydrate pellets |
Mahant et al.278 |
7. Conclusions
This review discusses the challenges in achieving energy-efficient and safe storage of NG and hydrogen through hydrate-based greener gas storage processes. We emphasize the importance of discussing the phase equilibrium of the gas hydrates and the stochastic nature of nucleation. Moreover, the limitation in mass or heat transfer is regarded as a key obstacle to the rapid formation of gas hydrates. Although various mechanical techniques, including stirring, bubbling, or spraying, can improve the formation kinetics of NG or hydrogen hydrate, it is essential to establish methods for gas hydrate formation under static conditions due to their high cost-effectiveness and energy efficiency. In light of this, several strategies to overcome the kinetic hurdles for gas hydrate formation under a static system have been widely investigated.Thermodynamic hydrate promoters, which occupy several cages of the hydrate to stabilize the gas hydrates under mild conditions, can be used to enhance the formation kinetics of gas hydrates by increasing the driving forces for the target gas hydrate formation. Aside from thermodynamic promoters, kinetic hydrate promoters also can accelerate hydrate formation kinetics by increasing the nucleation probability or facilitating mass and heat transfer. Because surface properties of the substrate strongly affect the nucleation probability for gas hydrates by altering local gas density, and the ordering of water molecules, the addition of a porous substrate and pre-constructed hydrate crystals strongly decreases the induction time for hydrogen and NG hydrates without any mechanical agitation. Moreover, as the hydrate formation rate is proportional to the amount of surface area for hydrate growth, surfactants such as SDS can enhance the hydrate formation kinetics by preventing the agglomeration of hydrate particles and forming a porous hydrate structure. Porous materials such as silica gel or silica NPs also can enhance the formation kinetics of hydrate by providing a large efficient surface area for hydrate growth. As exothermic heat released from the hydrate formation process is unfavorable for further hydrate formation, which is facilitated at low temperature, the hydrate formation kinetics should be significantly enhanced by the addition of NPs or an auxiliary framework with high thermal conductivity, which can release the exothermic heat sufficiently. This review paper emphasizes the importance of the hydrate formation process under static conditions and provides a fundamental understanding of static hydrate formation processes by investigating theoretical and experimental studies on challenging hurdles for rapid formation kinetics of gas hydrates.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support by the ERC Center funded by the National Research Foundation of Korea (NRF-2022R1A5A1033719), Republic of Korea.References
- B. Duchemin, Green Chem., 2022, 24, 2653–2679, 10.1039/D1GC03262C.
- J. Lee, M. Lee, D. Kim, Y. Shin and J. W. Lee, Korean J. Chem. Eng., 2023, 40, 2815–2825, DOI:10.1007/s11814-023-1574-0.
- M. Lee, H. Lee, C. Seo, J. Lee and J. W. Lee, Sep. Purif. Technol., 2022, 287, 120598, DOI:10.1016/j.seppur.2022.120598.
- M. I. Blanco, Renewable Sustainable Energy Rev., 2009, 13, 1372–1382, DOI:10.1016/j.rser.2008.09.004.
- S. A. Vargas, G. Roberta, T. Esteves, P. M. Maçaira, B. Q. Bastos, F. Luiz, C. Oliveira and R. C. Souza, J. Cleaner Prod., 2019, 218, 850–870, DOI:10.1016/j.jclepro.2019.02.015.
- N. Pirrone and F. Bella, Green Chem., 2022, 24, 5379–5402, 10.1039/D2GC00292B.
- X. Peng, T. W. Root and C. T. Maravelias, Green Chem., 2017, 19, 2427–2438, 10.1039/C7GC00023E.
- S. Astariz and G. Iglesias, Renewable Sustainable Energy Rev., 2015, 45, 397–408, DOI:10.1016/j.rser.2015.01.061.
- J. Falnes and J. Løvseth, Energy Policy, 1991, 19, 768–775, DOI:10.1016/0301-4215(91)90046-Q.
- K. Huang, J. B. Miller, G. W. Huber, J. A. Dumesic and C. T. Maravelias, Joule, 2018, 2, 349–365, DOI:10.1016/j.joule.2018.01.001.
- Y. Kim, H. S. Lim, M. Lee, M. Kim, D. Kang and J. W. Lee, ACS Sustainable Chem. Eng., 2021, 9, 14800–14810, DOI:10.1021/acssuschemeng.1c04619.
- C. M. Simon, J. Kim, D. A. Gomez-Gualdron, J. S. Camp, Y. G. Chung, R. L. Martin, R. Mercado, M. W. Deem, D. Gunter, M. Haranczyk, D. S. Sholl, R. Q. Snurr and B. Smit, Energy Environ. Sci., 2015, 8, 1190–1199, 10.1039/C4EE03515A.
- G. Bhattacharjee, M. N. Goh, S. E. K. Arumuganainar, Y. Zhang and P. Linga, Energy Environ. Sci., 2020, 13, 4946–4961, 10.1039/D0EE02315A.
- S. Aslam, S. Rani, K. Lal, M. Fatima and T. Hardwick, Green Chem., 2023, 25, 9543–9573, 10.1039/D3GC02849F.
- Y. Kim, H. Seok, D. Kang, M. Kim and J. W. Lee, Chem. Eng. J., 2023, 468, 143662, DOI:10.1016/j.cej.2023.143662.
- O. O. James, S. Maity, M. A. Mesubi, K. O. Ogunniran and T. O. Siyanbola, Green Chem., 2011, 13, 2272–2284, 10.1039/C0GC00924E.
- T. Abbasi and S. A. Abbasi, Renewable Sustainable Energy Rev., 2011, 15, 3034–3040, DOI:10.1016/j.rser.2011.02.026.
- M. Bowker, Green Chem., 2011, 13, 2235–2246, 10.1039/C1GC00022E.
- H. S. Lim, M. Kim, Y. Kim, H. S. Kim, D. Kang, M. Lee, A. Jo and J. W. Lee, ACS Appl. Energy Mater., 2022, 5, 8437–8442, DOI:10.1021/acsaem.2c00662.
- M. I. Khan, T. Yasmin and A. Shakoor, Renewable Sustainable Energy Rev., 2015, 51, 785–797, DOI:10.1016/j.rser.2015.06.053.
- G. Sdanghi, G. Maranzana, A. Celzard and V. Fierro, Renewable Sustainable Energy Rev., 2019, 102, 150–170, DOI:10.1016/j.rser.2018.11.028.
- D. Kwak, J. H. Heo, S. Park, S. Seo and J. Kim, Energy, 2018, 148, 915–929, DOI:10.1016/j.energy.2018.01.154.
- S. Z. Al Ghafri, S. Munro, U. Cardella, T. Funke, W. Notardonato, J. P. M. Trusler, J. Leachman, R. Span, S. Kamiya, G. Pearce, A. Swanger, E. D. Rodriguez, P. Bajada, F. Jiao, K. Peng, A. Siahvashi, M. L. Johns and E. F. May, Energy Environ. Sci., 2022, 15, 2690–2731, 10.1039/D2EE00099G.
- R. Moradi and K. M. Groth, Int. J. Hydrogen Energy, 2019, 44, 12254–12269, DOI:10.1016/j.ijhydene.2019.03.041.
- P. A. Julien, C. Mottillo and T. Friščić, Green Chem., 2017, 19, 2729–2747, 10.1039/C7GC01078H.
- W. Zhou, H. Wu, M. R. Hartman and T. Yildirim, J. Phys. Chem. C, 2007, 111, 16131–16137, DOI:10.1021/jp074889i.
- V. Rozyyev, D. Thirion, R. Ullah, J. Lee, M. Jung, H. Oh, M. Atilhan and C. T. Yavuz, Nat. Energy, 2019, 4, 604–611, DOI:10.1038/s41560-019-0427-x.
- Y. Liu, J. Cui, H. Wang, K. Wang and Y. Tian, Green Chem., 2023, 25, 4981–4994, 10.1039/D3GC01003A.
- R. Dickson, M. S. Akhtar, A. Abbas, E. D. Park and J. Liu, Green Chem., 2022, 24, 8484–8493, 10.1039/D2GC02079C.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140, DOI:10.1016/j.ijhydene.2006.11.022.
- J. Zhang, Y. Zhao, D. L. Akins and J. W. Lee, J. Phys. Chem. C, 2011, 115, 8386–8392, DOI:10.1021/jp200049y.
- J. Zhang, Y. Zhao, D. L. Akins and J. W. Lee, J. Phys. Chem. C, 2010, 114, 19529–19534, DOI:10.1021/jp105014t.
- O. Salako, C. Lo, A. Couzis, P. Somasundaran and J. W. Lee, J. Colloid Interface Sci., 2013, 412, 1–6, DOI:10.1016/j.jcis.2013.09.007.
- S. Baek, J. Min and J. W. Lee, RSC Adv., 2015, 5, 58813–58820, 10.1039/C5RA08335D.
- W. Lee, J. Min, Y. H. Ahn, S. Baek, C. A. Koh and J. W. Lee, Ind. Eng. Chem. Res., 2019, 58, 5064–5070, DOI:10.1021/acs.iecr.8b05869.
- Y. Ahn, S. Baek, J. Min, M. Cha and J. W. Lee, ChemPhysChem, 2019, 20, 429–435, DOI:10.1002/cphc.201800943.
- S. Potdar, J. W. Lee and S. Lee, Korean J. Chem. Eng., 2016, 33, 3216–3221, DOI:10.1007/s11814-016-0172-9.
- A. Ahuja, A. Iqbal, M. Iqbal, J. W. Lee and J. F. Morris, Energy Fuels, 2018, 32, 5877–5884, DOI:10.1021/acs.energyfuels.8b00795.
- E. D. Sloan and C. A. Koh, Clathrate hydrates of natural gases, CRC Press, New York, 3rd edn, 2007 Search PubMed.
- H. Kang, Y. H. Ahn, D. Y. Koh, S. Baek, J. W. Lee and H. Lee, Ind. Eng. Chem. Res., 2016, 55, 6079–6084, DOI:10.1021/acs.iecr.6b01286.
- J. Mok, W. Choi, J. Lee and Y. Seo, Energy, 2022, 239, 122153, DOI:10.1016/j.energy.2021.122153.
- C.-G. Xu, J. Cai, F.-h. Lin, Z.-Y. Chen and X.-S. Li, Energy, 2015, 79, 111–116, DOI:10.1016/j.energy.2014.10.068.
- C. Yan, Y. Chen, W. Tian, Y. Cheng and Y. Li, J. Cleaner Prod., 2022, 354, 131703, DOI:10.1016/j.jclepro.2022.131703.
- Q. Yuan, C. Y. Sun, B. Liu, X. Wang, Z. W. Ma, Q. L. Ma, L. Y. Yang, G. J. Chen, Q. P. Li, S. Li and K. Zhang, Energy Convers. Manage., 2013, 67, 257–264, DOI:10.1016/j.enconman.2012.11.018.
- C. Lo, J. S. Zhang, A. Couzis, P. Somasundaran and J. W. Lee, J. Phys. Chem. C, 2010, 114, 13385–13389, DOI:10.1021/jp102846d.
- J. Peixinho, P. U. Karanjkar, J. W. Lee and J. F. Morris, Langmuir, 2010, 26, 11699–11704, DOI:10.1021/la101141j.
- J. S. Zhang, C. Lo, A. Couzis, P. Somasundaran, J. Wu and J. W. Lee, J. Phys. Chem. C, 2009, 113, 17418–17420, DOI:10.1021/jp907796d.
- J. H. Song, A. Couzis and J. W. Lee, Langmuir, 2010, 26, 9187–9190, DOI:10.1021/la101309j.
- J. Min, D. W. Kang, Y. H. Ahn, W. Lee, M. Cha and J. W. Lee, Chem. Eng. J., 2020, 389, 124461, DOI:10.1016/j.cej.2020.124461.
- J. Zhang and J. W. Lee, Ind. Eng. Chem. Res., 2009, 48, 4703–4709, DOI:10.1021/ie8019328.
- S. Baek, J. Min, Y. H. Ahn, M. Cha and J. W. Lee, Energy Fuels, 2019, 33, 523–530, DOI:10.1021/acs.energyfuels.8b03210.
- W. Lee, S. Baek, J. Kim and J. W. Lee, Energy Fuels, 2015, 29, 4245–4254, DOI:10.1021/acs.energyfuels.5b00768.
- J. Zhang, P. Yedlapalli and J. W. Lee, Chem. Eng. Sci., 2009, 64, 4732–4736, DOI:10.1016/j.ces.2009.04.041.
- K. Kim and J. Sa, Sep. Purif. Technol., 2023, 310, 123135, DOI:10.1016/j.seppur.2023.123135.
- H. Dashti, L. Z. Yew and X. Lou, J. Nat. Gas Sci. Eng., 2015, 23, 195–207, DOI:10.1016/j.jngse.2015.01.033.
- P. Linga, R. Kumar and P. Englezos, J. Hazard. Mater., 2007, 149, 625–629, DOI:10.1016/j.jhazmat.2007.06.086.
- J. H. Yoon, Korean J. Chem. Eng., 2012, 29, 1670–1673, DOI:10.1007/s11814-012-0137-6.
- G. Ko, E. Kim, D. Lee and Y. Seo, Korean J. Chem. Eng., 2023, 40, 1725–1730, DOI:10.1007/s11814-023-1385-3.
- K. Chan, P. Linga, K. Park, S. Choi and J. Dong, Desalination, 2014, 353, 84–90, DOI:10.1016/j.desal.2014.09.007.
- P. Babu, A. Nambiar, T. He, I. A. Karimi, J. D. Lee, P. Englezos and P. Linga, ACS Sustainable Chem. Eng., 2018, 6, 8093–8107, DOI:10.1021/acssuschemeng.8b01616.
- J.-n. Zheng and M. Yang, Desalination, 2020, 478, 114284, DOI:10.1016/j.desal.2019.114284.
- Z. Yin, J. Zheng, H. Kim, Y. Seo and P. Linga, Adv. Appl. Energy, 2021, 2, 100022, DOI:10.1016/j.adapen.2021.100022.
- H. Kim, J. Zheng, Z. Yin, S. Kumar, J. Tee, Y. Seo and P. Linga, Appl. Energy, 2022, 308, 118397, DOI:10.1016/j.apenergy.2021.118397.
- Q. Sun and Y. T. Kang, Renewable Sustainable Energy Rev., 2016, 62, 478–494, DOI:10.1016/j.rser.2016.04.062.
- J. Zheng, Z. R. Chong, M. F. Qureshi and P. Linga, Energy Fuels, 2020, 34, 10529–10546, DOI:10.1021/acs.energyfuels.0c02309.
- Y. Kuang, L. Zhang and Y. Zheng, Energy, 2022, 252, 124082, DOI:10.1016/j.energy.2022.124082.
- D. Riestenberg and O. R. West, Environ. Sci. Technol., 2003, 37, 3701–3708, DOI:10.1021/es026301l.
- D. W. Kang, W. Lee, Y. Ahn and J. W. Lee, J. Mol. Liq., 2022, 349, 118490, DOI:10.1016/j.molliq.2022.118490.
- P. Yedlapalli, S. Lee and J. W. Lee, J. Thermodyn., 2010, 2010, 1–6, DOI:10.1155/2010/651819.
- S. Park, D. Y. Koh, H. Kang, J. W. Lee and H. Lee, J. Phys. Chem. C, 2014, 118, 20203–20208, DOI:10.1021/jp5061254.
- H. P. Veluswamy, A. Kumar, Y. Seo, J. D. Lee and P. Linga, Appl. Energy, 2018, 216, 262–285, DOI:10.1016/j.apenergy.2018.02.059.
- H. P. Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2014, 122, 112–132, DOI:10.1016/j.apenergy.2014.01.063.
- P. Di Profio, S. Arca, F. Rossi and M. Filipponi, Int. J. Hydrogen Energy, 2009, 34, 9173–9180, DOI:10.1016/j.ijhydene.2009.09.056.
- F. B. Juangsa, L. A. Prananto, Z. Mufrodi, A. Budiman, T. Oda and M. Aziz, Appl. Energy, 2018, 226, 31–38, DOI:10.1016/j.apenergy.2018.05.110.
- Y. Zhang, G. Bhattacharjee, R. Kumar and P. Linga, Chem. Eng. J., 2022, 431, 133702, DOI:10.1016/j.cej.2021.133702.
- A. S. Mehr, A. D. Phillips, M. P. Brandon, M. T. Pryce and J. G. Carton, Int. J. Hydrogen Energy, 2024, 70, 786–815, DOI:10.1016/j.ijhydene.2024.05.182.
- M. Cha, S. Baek, H. Lee and J. W. Lee, RSC Adv., 2014, 4, 26176–26180, 10.1039/C4RA03680H.
- J. Min, Y. H. Ahn, S. Baek, K. Shin, M. Cha and J. W. Lee, J. Phys. Chem. C, 2019, 123, 20705–20714, DOI:10.1021/acs.jpcc.9b04125.
- X. Lang, S. Fan and Y. Wang, J. Nat. Gas Chem., 2010, 19, 203–209, DOI:10.1016/S1003-9953(09)60079-7.
- N. J. Kim, J. Hwan Lee, Y. S. Cho and W. Chun, Energy, 2010, 35, 2717–2722, DOI:10.1016/j.energy.2009.07.020.
- M. Cha, S. Baek, W. Lee, K. Shin and J. W. Lee, J. Chem. Eng. Data, 2015, 60, 278–283, DOI:10.1021/je500568f.
- J. Min, D. W. Kang, W. Lee and J. W. Lee, J. Phys. Chem. C, 2020, 124, 4162–4171, DOI:10.1021/acs.jpcc.9b11459.
- Y. Lee, H. Kim, W. Lee, D. W. Kang, J. W. Lee and Y. H. Ahn, J. Environ. Chem. Eng., 2023, 11, 110933, DOI:10.1016/j.jece.2023.110933.
- P. J. Metaxas, V. W. S. Lim, C. Booth, J. Zhen, P. L. Stanwix, M. L. Johns, Z. M. Aman, G. Haandrikman, D. Crosby and E. F. May, Fuel, 2019, 252, 448–457, DOI:10.1016/j.fuel.2019.04.131.83.
- G. Zhang, X. Shi and F. Wang, AIChE J., 2021, 68, e17423, DOI:10.1002/aic.17423.
- G. Zhang, R. Zhang, Y. Kong and F. Wang, Chem. Eng. J., 2023, 467, 143471, DOI:10.1016/j.cej.2023.143471.
- Y. Qin, Z. Pan, L. Shang, X. Sun, J. He, Y. Yang and B. Yuan, Fuel, 2023, 336, 127129, DOI:10.1016/j.fuel.2022.127129.
- L. Mu, Q. Tan, X. Li, Q. Zhang and Q. Cui, Energy, 2023, 264, 126310, DOI:10.1016/j.energy.2022.126310.
- K. Jeenmuang, P. Pornaroontham, K. Inkong, G. Bhattacharjee, S. Kulprathipanja, P. Linga and P. Rangsunvigit, Chem. Eng. J., 2023, 454, 140326, DOI:10.1016/j.cej.2022.140326.
- W. Hao, J. Wang, S. Fan and W. Hao, Energy Convers. Manage., 2007, 48, 954–960, DOI:10.1016/j.enconman.2006.08.007.
- J. Du, H. Li and L. Wang, Adv. Powder Technol., 2014, 25, 1227–1233, DOI:10.1016/j.apt.2014.06.002.
- A. Farhadian, U. Z. Mirzakimov, M. E. Semenov, M. Maddah, Y. F. Chirkova, R. S. Pavelyev, A. Heydari, S. A. Nazarychev, A. M. Aimaletdinov and M. A. Varfolomeev, ACS Appl. Energy Mater., 2023, 6, 4119–4132, DOI:10.1021/acsaem.2c03240.
- X. Niu, J. Zhong, D. Lei, H. Zhang and W. Wang, Ind. Eng. Chem. Res., 2022, 61, 3775–3780, DOI:10.1021/acs.iecr.1c04642.
- Z. Cheng, S. Wang, N. Xu, W. Liu, Y. Zhao, J. Zhao, L. Jiang and J. Zheng, Fuel, 2021, 301, 121021, DOI:10.1016/j.fuel.2021.121021.
- M. K. Kim, G. Han, H. Kim, J. Yu, Y. Lee, T. Song, J. Park, Y. H. Kim and Y. H. Ahn, Korean J. Chem. Eng., 2023, 39, 23–26, DOI:10.1007/s11814-022-1301-2.
- G. Bhattacharjee and P. Linga, Energy Fuels, 2021, 35, 7553–7571, DOI:10.1021/acs.energyfuels.1c00502.
- Q. Nasir, H. Suleman and Y. A. Elsheikh, J. Nat. Gas Sci. Eng., 2020, 76, 103211, DOI:10.1016/j.jngse.2020.103211.
- J. S. Zhang, J. A. Salera and J. W. Lee, Ind. Eng. Chem. Res., 2010, 49, 8267–8270, DOI:10.1021/ie100759p.
- O. Nashed, B. Partoon, B. Lal, K. M. Sabil and A. Mohd, J. Nat. Gas Sci. Eng., 2018, 55, 452–465, DOI:10.1016/j.jngse.2018.05.022.
- C. B. Bavoh, B. Lal, H. Osei, K. M. Sabil and H. Mukhtar, J. Nat. Gas Sci. Eng., 2019, 64, 52–71, DOI:10.1016/j.jngse.2019.01.020.
- L. Wang, M. Dou, Y. Wang, Y. Xu, Y. Li, Y. Chen and L. Li, ACS Omega, 2022, 7, 33666–33679, DOI:10.1021/acsomega.2c03048.
- Y. Qin, L. Shang, Z. Lv, J. He, X. Yang and Z. Zhang, J. Energy Chem., 2022, 74, 454–480, DOI:10.1016/j.jechem.2022.07.019.
- W. Ke, T. M. Svartaas and D. Chen, J. Nat. Gas Sci. Eng., 2019, 61, 169–196 CrossRef CAS.
- M. Khurana, Z. Yin and P. Linga, ACS Sustainable Chem. Eng., 2017, 5, 11176–11203, DOI:10.1021/acssuschemeng.7b03238.
- C. P. Ribeiro Jr and P. L. C. Lage, Chem. Eng. Sci., 2008, 63, 2007–2034, DOI:10.1016/j.ces.2008.01.014.
- J. S. Lee, A. Cherif, H. J. Yoon, S. K. Seo, J. E. Bae, H. J. Shin, C. Lee, H. Kwon and C. J. Lee, Renewable Sustainable Energy Rev., 2022, 165, 112556, DOI:10.1016/j.rser.2022.112556.
- M. C. Massaro, R. Biga, A. Kolisnichenko, P. Marocco, A. H. A. Monteverde and M. Santarelli, J. Power Sources, 2023, 555, 232397, DOI:10.1016/j.jpowsour.2022.232397.
- G. Zylyftari, J. W. Lee and J. F. Morris, Chem. Eng. Sci., 2013, 95, 148–160, DOI:10.1016/j.ces.2013.02.056.
- M. Cha, H. Lee and J. W. Lee, J. Phys. Chem. C, 2013, 117, 23515–23521, DOI:10.1021/jp4076564.
- T. Y. Makogon and E. D. Sloan, J. Chem. Eng. Data, 1994, 39, 351–353, DOI:10.1021/je00014a035.
- S. O. Yang, S. H. Cho, H. Lee and C. S. Lee, Fluid Phase Equilib., 2001, 185, 53–63, DOI:10.1016/S0378-3812(01)00456-3.
- S. Peddireddy, S. Y. Lee and J. W. Lee, AIChE J., 2006, 52, 1228–1234, DOI:10.1002/aic.10702109.
- J. W. Du, D. Q. Liang, X. X. Dai, D. L. Li and X. J. Li, J. Chem. Thermodyn., 2011, 43, 617–621, DOI:10.1016/j.jct.2010.11.018.
- H. Komatsu, H. Yoshioka, M. Ota, Y. Sato, M. Watanabe, R. L. Smith and C. J. Peters, J. Chem. Eng. Data, 2010, 55, 2214–2218, DOI:10.1021/je900767h.
- J. Nixdorf and L. R. Oellrich, Fluid Phase Equilib., 1997, 139, 325–333, DOI:10.1016/S0378-3812(97)00141-6.
- T. Nakamura, T. Makino, T. Sugahara and K. Ohgaki, Chem. Eng. Sci., 2003, 58, 269–273, DOI:10.1016/S0009-2509(02)00518-3.
- Y. A. Dyadin, E. G. Larionov, A. Y. Manakov, F. V. Zhurko, E. Y. Aladko, T. V. Mikina and V. Y. Komarov, Mendeleev Commun., 1999, 9, 209–210, DOI:10.1070/MC1999v009n05ABEH001104.
- D. Kashchiev and A. Firoozabadi, J. Cryst. Growth, 2002, 241, 220–230, DOI:10.1016/S0022-0248(02)01134-X.
- J. W. Lee, P. Yedlapalli and S. Lee, J. Phys. Chem. B, 2006, 110, 2332–2337, DOI:10.1021/jp0531311.
- A. A. A. Majid, J. Worley and C. A. Koh, Energy Fuels, 2021, 35, 19288–19301, DOI:10.1021/acs.energyfuels.1c02786.
- D. Mech, P. Gupta and J. S. Sangwai, J. Nat. Gas Sci. Eng., 2016, 35, 1519–1534, DOI:10.1016/j.jngse.2016.06.013.
- A. Hassanpouryouzband, E. Joonaki, V. M. Farahani, S. Takeya, C. Ruppel, J. Yang, N. J. English, J. M. Schicks, K. Edlmann, H. Mehrabian, Z. M. Aman and B. Tohidi, Chem. Soc. Rev., 2020, 49, 5225–5309, 10.1039/C8CS00989A.
- S. Wang, M. Yang, W. Liu, J. Zhao and Y. Song, J. Pet. Sci. Eng., 2016, 145, 565–572, DOI:10.1016/j.petrol.2016.06.003.
- J. S. Zhang, S. Lee and J. W. Lee, Ind. Eng. Chem. Res., 2007, 46, 6353–6359, DOI:10.1021/ie070627r.
- M. Arjmandi, B. Tohidi, A. Danesh and A. C. Todd, Chem. Eng. Sci., 2005, 60, 1313–1321, DOI:10.1016/j.ces.2004.10.005.
- R. Ohmura, M. Ogawa, K. Yasuoka and Y. H. Mori, J. Phys. Chem. B, 2003, 107, 5289–5293, DOI:10.1021/jp027094e.
- M. R. Walsh, C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Science, 2009, 326, 1095–1098, DOI:10.1126/science.117401.
- E. D. Sloan, Jr., Nature, 2003, 426, 353–359, DOI:10.1038/nature02135.
- A. M. Gambelli and F. Rossi, Chem. Eng. J., 2021, 425, 130706, DOI:10.1016/j.cej.2021.130706.
- R. Radhakrishnan and B. L. Trout, J. Chem. Phys., 2002, 117, 1786–1796, DOI:10.1063/1.1485962.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Am. Chem. Soc., 2010, 132, 11806–11811, DOI:10.1021/ja1051445.
- D. Riestenberg, O. West, S. Lee, S. Mccallum and T. J. Phelps, Mar. Geol., 2003, 198, 181–190, DOI:10.1016/S0025-3227(03)00100-2.
- K. U. Heeschen, J. M. Schicks and G. Oeltzschner, Fuel, 2016, 181, 139–147, DOI:10.1016/j.fuel.2016.04.017.
- R. S. Defever and S. Sarupria, J. Chem. Thermodyn., 2018, 117, 205–213, DOI:10.1016/j.jct.2017.08.021.
- Z. Yin, M. Khurana, H. K. Tan and P. Linga, Chem. Eng. J., 2018, 342, 9–29, DOI:10.1016/j.cej.2018.01.120.
- P. Englezos, N. Kalogerakis, P. D. Dholabhai and P. R. Bishnoi, Chem. Eng. Sci., 1987, 42, 2647–2658, DOI:10.1016/0009-2509(87)87015-X.
- J. M. Herri, J. S. Pic, F. Gury and M. Cournil, AIChE J., 1999, 45, 590–602, DOI:10.1002/aic.690450316.
- E. D. Sloan and F. Fleyfel, AIChE J., 1991, 37, 1281–1292, DOI:10.1002/aic.690370902.
- B. H. Shi, J. Gong, C. Y. Sun, J. K. Zhao, Y. Ding and G. J. Chen, Chem. Eng. J., 2011, 171, 1308–1316, DOI:10.1016/j.cej.2011.05.029.
- D. J. Turner, K. T. Miller and E. Dendy Sloan, Chem. Eng. Sci., 2009, 64, 3996–4004, DOI:10.1016/j.ces.2009.05.051.
- P. U. Karanjkar, J. W. Lee and J. F. Morris, Chem. Eng. Sci., 2012, 68, 481–491, DOI:10.1016/j.ces.2011.10.014.
- F. Zhang, X. Wang, B. Wang, X. Lou and W. Lipiński, Chem. Eng. J., 2022, 446, 137247, DOI:10.1016/j.cej.2022.137247.
- H. Liang, D. Guan, K. Shi, L. Yang, L. Zhang and J. Zhao, Chem. Eng. J., 2022, 428, 132626, DOI:10.1016/j.cej.2021.132626.
- J. Zhang and J. W. Lee, Ind. Eng. Chem. Res., 2009, 48, 5934–5942, DOI:10.1021/ie801170u.
- S. R. Davies, E. D. Sloan, A. K. Sum and C. A. Koh, J. Phys. Chem. C, 2010, 114, 1173–1180, DOI:10.1021/jp909416y.
- A. N. Nesterov and A. M. Reshetnikov, J. Nat. Gas Sci. Eng., 2022, 99, 104424, DOI:10.1016/j.jngse.2022.104424.
- R. Ohmura, S. Matsuda, T. Uchida and T. Ebinuma, Cryst. Growth Des., 2005, 5, 953–957, DOI:10.1021/cg049675u.
- H. P. Veluswamy, G. Bhattacharjee, J. Liao and P. Linga, Energy Fuels, 2020, 34, 15257–15269, DOI:10.1021/acs.energyfuels.0c01862.
- H. P. Veluswamy, J. Y. Chen and P. Linga, Chem. Eng. Sci., 2015, 126, 488–499, DOI:10.1016/j.ces.2014.12.052.
- G. Bhattacharjee, H. P. Veluswamy, R. Kumar and P. Linga, Appl. Energy, 2020, 269, 115142, DOI:10.1016/j.apenergy.2020.115142.
- J. Liu and D. Liang, RSC Adv., 2019, 9, 15022–15032, 10.1039/C9RA01973A.
- I. N. Tsimpanogiannis, V. K. Michalis and I. G. Economou, Fluid Phase Equilib., 2019, 489, 30–40, DOI:10.1016/j.fluid.2019.01.024.
- R. E. T. Meindinyo, T. M. Svartaas, T. N. Nordbø and R. Bøe, Energy Fuels, 2015, 29, 587–594, DOI:10.1021/ef502366u.
- S. Matsuda, H. Tsuda and Y. H. Mori, AIChE J., 2006, 52, 2978–2987, DOI:10.1002/aic.10890.
- T. Uchida, T. Ebinuma, J. Kawabata and H. Narita, J. Cryst. Growth, 1999, 204, 348–356, DOI:10.1016/S0022-0248(99)00178-5.
- Y. H. Mori, J. Cryst. Growth, 2001, 223, 206–212, DOI:10.1016/S0022-0248(01)00614-5.
- E. M. Freer, M. S. Selim and E. D. Sloan Jr, Fluid Phase Equilib., 2001, 185, 65–75, DOI:10.1016/S0378-3812(01)00457-5.
- T. Mochizuki and Y. H. Mori, J. Cryst. Growth, 2006, 290, 642–652, DOI:10.1016/j.jcrysgro.2006.01.036.
- M. Yang, Y. Song, L. Jiang, W. Jing, W. Liu and B. Dou, J. Chem. Thermodyn., 2014, 78, 167–174, DOI:10.1016/j.jct.2014.06.025.
- M. Tian, Y. Song, J. Zheng, G. Gong and M. Yang, Energy, 2022, 261, 125220, DOI:10.1016/j.energy.2022.125220.
- W. Zhang, H. Li, C. Xu, X. Li and Z. Huang, RSC Adv., 2022, 12, 20227–20238, 10.1039/D2RA03376C.
- L. Yang, S. Fan, Y. Wang, X. Lang and D. Xie, Ind. Eng. Chem. Res., 2011, 50, 11563–11569, DOI:10.1021/ie200825e.
- X. Li, C. Chen, Y. Chen, Y. Li and H. Li, Energy Fuels, 2015, 29, 2277–2288, DOI:10.1021/ef5028923.
- I. Ahmed, O. Bamaga, M. H. Albeirutty, H. Abulkhair, A. Alsaiari, H. Organji and P. Linga, Energy Fuels, 2022, 36, 7676–7686, DOI:10.1021/acs.energyfuels.2c00395.
- T. Uchida, R. Sugibuchi, K. Yamazaki and K. Gohara, Energy Fuels, 2022, 36, 10444–10457, DOI:10.1021/acs.energyfuels.2c01403.
- N. N. Nguyen and A. V. Nguyen, ACS Nano, 2022, 16, 11504–11515, DOI:10.1021/acsnano.2c04640.
- W. Wijayanti, AIP Conf. Proc., 2018, 1983, 020012, DOI:10.1063/1.5046208.
- S. Hashimoto, H. Ito, K. Ohgaki and Y. Inoue, Int. J. Chem. Eng., 2012, 2012, 1–6, DOI:10.1155/2012/856120.
- H. P. Veluswamy, A. Kumar, R. Kumar and P. Linga, Appl. Energy, 2017, 188, 190–199, DOI:10.1016/j.apenergy.2016.12.002.
- Z. Liu, Y. Song, W. Liu, R. Liu, C. Lang and Y. Li, Fuel, 2020, 275, 117961, DOI:10.1016/j.fuel.2020.117961.
- Q. Li, S. Fan, Q. Chen, G. Yang, Y. Chen, L. Li and G. Li, J. Nat. Gas Sci. Eng., 2019, 72, 103008, DOI:10.1016/j.jngse.2019.103008.
- J. Cai, C. G. Xu, Z. M. Xia, Z. Y. Chen and X. S. Li, Appl. Energy, 2017, 204, 1526–1534, DOI:10.1016/j.apenergy.2017.05.010.
- Y. T. Luo, J. H. Zhu, S. S. Fan and G. J. Chen, Chem. Eng. Sci., 2007, 62, 1000–1009, DOI:10.1016/j.ces.2006.11.004.
- T. Murakami, H. Kuritsuka, H. Fujii and Y. H. Mori, Energy Fuels, 2009, 23, 1619–1625, DOI:10.1021/ef800880f.
- R. Ohmura, S. Kashiwazaki, S. Shiota, H. Tsuji and Y. H. Mori, Energy Fuels, 2002, 16, 1141–1147, DOI:10.1021/ef0200727.
- H. Tsuji, R. Ohmura and Y. H. Mori, Energy Fuels, 2004, 18, 418–424, DOI:10.1021/ef034054g.
- H. Tsuji, T. Kobayashi, R. Ohmura and Y. H. Mori, Energy Fuels, 2005, 19, 869–876, DOI:10.1021/ef049785a.
- R. E. Rogers, G. Y. Yevi and M. Swalm, in Proceedings of the 2nd International Conference on Natural Gas Hydrates, 1996, pp. 423–429.
- S. Fujita, K. Watanabe and Y. H. Mori, AIChE J., 2009, 55, 1056–1064, DOI:10.1002/aic.11744.
- K. Fukumoto, T. Jun-ichhiro, R. Ohmura and Y. H. Mori, AIChE J., 2001, 47, 1899–1904, DOI:10.1002/aic.690470821.
- G. A. Jeffrey, J. L. Atwood, J. E. D. Davies and D. D. MacNichol, Inclusion Compounds, Academic Press, London, 1984, vol. 1 Search PubMed.
- R. Susilo, S. Alavi, S. Lang, J. Ripmeester and P. Englezos, J. Phys. Chem. C, 2008, 112, 9106–9113, DOI:10.1021/jp8006848.
- J. A. Ripmeester, J. Tse, C. I. Ratcliffe and B. M. Powellt, Nature, 1987, 325, 135–136, DOI:10.1038/325135a0.
- S. Subramanian, R. A. Kini, S. F. Dec and E. D. Sloan Jr, Chem. Eng. Sci., 2000, 55, 1981–1999, DOI:10.1016/S0009-2509(99)00389-9.
- M. Kwon, J. Lee and H. Lee, J. Phys. Chem. C, 2014, 118, 28906–28913, DOI:10.1021/jp5102219.
- S. Adisasmro, R. J. Frank and E. D. Sloan, J. Chem. Eng. Data, 1991, 36, 68–71, DOI:10.1021/je00001a020.
- H. Bruusgaard, J. G. Beltrán and P. Servio, J. Chem. Eng. Data, 2008, 53, 2594–2597, DOI:10.1021/je800445x.
- A. H. Mohammadi, B. Tohidi and R. W. Burgass, J. Chem. Eng. Data, 2003, 48, 612–616, DOI:10.1021/je025608x.
- M. Cha, K. Shin and H. Lee, J. Phys. Chem. B, 2009, 113, 10562–10565, DOI:10.1021/jp9056412.
- W. L. Vos, L. W. Finger, R. Hemley and H. Mao, Phys. Rev. Lett., 1993, 71, 3150–3153, DOI:10.1103/PhysRevLett.71.3150.
- L. J. Florusse, C. J. Peters, J. Schoonman, K. C. Hester, C. A. Koh, S. F. Dec, K. N. Marsh and E. D. Sloan, Science, 2004, 306, 469–472, DOI:10.1126/science.1102076.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. T. Seo, H. Zeng, I. L. Moudrakovskr, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743–746, DOI:10.1038/nature03457.
- Y. J. Lee, T. Kawamura, Y. Yamamoto and J. H. Yoon, J. Chem. Eng. Data, 2012, 57, 3543–3548, DOI:10.1021/je300850q.
- R. Anderson, A. Chapoy and B. Tohidi, Langmuir, 2007, 23, 3440–3444, DOI:10.1021/la063189m.
- S. Hashimoto, T. Sugahara, H. Sato and K. Ohgaki, J. Chem. Eng. Data, 2007, 52, 517–520, DOI:10.1021/je060436f.
- H. P. Veluswamy, A. J. H. Wong, P. Babu, R. Kumar, S. Kulprathipanja, P. Rangsunvigit and P. Linga, Chem. Eng. J., 2016, 290, 161–173, DOI:10.1016/j.ijhydene.2013.01.123.
- H. P. Veluswamy and P. Linga, Int. J. Hydrogen Energy, 2013, 38, 4587–4596, DOI:10.1016/j.ijhydene.2013.01.123.
- J. S. Zhang and J. W. Lee, J. Chem. Eng. Data, 2009, 54, 659–661, DOI:10.1021/je800219k.
- T. Tsuda, K. Ogata, S. Hashimoto, T. Sugahara, M. Moritoki and K. Ohgaki, Chem. Eng. Sci., 2009, 64, 4150–4154, DOI:10.1016/j.ces.2009.06.018.
- A. T. Trueba, L. J. Rovetto, L. J. Florusse, M. C. Kroon and C. J. Peters, Fluid Phase Equilib., 2011, 307, 6–10, DOI:10.1016/j.fluid.2011.04.025.
- S. Chen, Y. Wang, X. Lang, S. Fan and G. Li, J. Chem. Eng. Data, 2023, 68, 1184–1190, DOI:10.1021/acs.jced.2c00701.
- A. R. C. Duarte, A. Shariati and C. J. Peters, J. Chem. Eng. Data, 2010, 54, 1628–1632, DOI:10.1021/je800993w.
- M. N. Khan, L. J. Rovetto, C. J. Peters, E. D. Sloan, A. K. Sum and C. A. Koh, J. Chem. Eng. Data, 2015, 60, 418–423, DOI:10.1021/je500675q.
- A. P. Mehta and E. D. Sloan, J. Chem. Eng. Data, 1993, 38, 580–582, DOI:10.1021/je00012a027.
- Y. H. Ahn, D. Lim, J. Min, J. Kim, B. Lee, J. W. Lee and K. Shin, Chem. Eng. J., 2019, 359, 1629–1634, DOI:10.1016/j.cej.2018.10.238.
- H. Bode and G. Teufer, Acta Crystallogr., 1955, 8, 611, DOI:10.1107/s0365110x5500193x.
- B. Lee and K. Shin, Korean J. Chem. Eng., 2023, 40, 2520–2528, DOI:10.1007/s11814-023-1462-7.
- L. S. Aladko, Y. A. Dyadin, T. V. Rodionova and I. S. Terekhova, J. Struct. Chem., 2002, 43, 990–994, DOI:10.1023/A:1023698728342.
- D. Mootz and R. Seidel, J. Inclusion Phenom. Macrocyclic Chem., 1990, 8, 139–157, DOI:10.1007/BF01131293.
- J. Sakamoto, S. Hashimoto, T. Tsuda, T. Sugahara, Y. Inoue and K. Ohgaki, Chem. Eng. Sci., 2008, 63, 5789–5794, DOI:10.1016/j.ces.2008.08.026.
- S. Lee, Y. Lee, S. Park, Y. Kim, J. D. Lee and Y. Seo, J. Phys. Chem. B, 2012, 116, 9075–9081, DOI:10.1021/jp302647c.
- S. Hashimoto, S. Murayama, T. Sugahara, H. Sato and K. Ohgaki, Chem. Eng. Sci., 2006, 61, 7884–7888, DOI:10.1016/j.ces.2006.09.039.
- D. Y. Koh, H. Kang and H. Lee, Chem. Commun., 2013, 49, 6782–6784, 10.1039/C3CC43376E.
- D. Y. Koh, H. Kang, J. Jeon, Y. H. Ahn, Y. Park, H. Kim and H. Lee, J. Phys. Chem. C, 2014, 118, 3324–3330, DOI:10.1021/jp410632q.
- S. Moon, Y. Lee, D. Seo, S. Lee, S. Hong, Y. H. Ahn and Y. Park, Renewable Sustainable Energy Rev., 2021, 141, 110789, DOI:10.1016/j.rser.2021.110789.
- Y. H. Ahn, S. Moon, D. Y. Koh, S. Hong, H. Lee, J. W. Lee and Y. Park, Energy Storage Mater., 2020, 24, 655–661, DOI:10.1016/j.ensm.2019.06.007.
- H. Lu, J. Wang, C. Liu, C. I. Ratcliffe, U. Becker, R. Kumar and J. Ripmeester, J. Am. Chem. Soc., 2012, 134, 9160–9162, DOI:10.1021/ja303222u.
- S. Moon, S. Hong, Y. Lee, J. S. Lee, Y. H. Ahn and Y. Park, J. Phys. Chem. C, 2021, 125, 1767–1773, DOI:10.1021/acs.jpcc.0c10508.
- R. P. Sear, CrystEngComm, 2014, 16, 6506–6522, 10.1039/C4CE00344F.
- D. Kashchiev and A. Firoozabadi, J. Cryst. Growth, 2002, 243, 476–489, DOI:10.1016/S0022-0248(02)01576-2.
- N. N. Nguyen, A. V. Nguyen, K. M. Steel, L. X. Dang and M. Galib, J. Phys. Chem. C, 2017, 121, 3830–3840, DOI:10.1021/acs.jpcc.6b07136.
- D. Bai, G. Chen, X. Zhang, A. K. Sum and W. Wang, Sci. Rep., 2015, 5, 12747, DOI:10.1038/srep12747.
- H. Li, P. Stanwix, Z. Aman, M. Johns, E. May and L. Wang, J. Phys. Chem. A, 2016, 120, 417–424, DOI:10.1021/acs.jpca.5b11247.
- Y. Zhang, X. Li, Z. Chen, Z. Xia, Y. Wang and G. Li, Appl. Energy, 2018, 225, 827–834, DOI:10.1016/j.apenergy.2018.05.059.
- Y. Zhang, X. S. Li, Z. Y. Chen, G. Li and Y. Wang, J. Nat. Gas Sci. Eng., 2016, 35, 1463–1471, DOI:10.1016/j.jngse.2016.04.026.
- M. Zarifi, J. Javanmardi, H. Hashemi, A. Eslamimanesh and A. H. Mohammadi, Fluid Phase Equilib., 2016, 423, 17–24, DOI:10.1016/j.fluid.2016.03.018.
- D. H. Smith, J. W. Wilder and K. Seshadri, AIChE J., 2002, 48, 393–400, DOI:10.1002/aic.690480222.
- Y. Seo, H. Lee and T. Uchida, Langmuir, 2002, 18, 9164–9170, DOI:10.1021/la0257844.
- Z. Pan, Z. Liu, Z. Zhang, L. Shang and S. Ma, J. Nat. Gas Sci. Eng., 2018, 56, 266–280, DOI:10.1016/j.jngse.2018.06.018.
- P. Linga, C. Haligva, S. C. Nam, J. A. Ripmeester and P. Englezos, Energy Fuels, 2009, 23, 5496–5507, DOI:10.1021/ef900542m.
- L. Zhou, Y. Sun and Y. Zhou, AIChE J., 2002, 48, 2412–2416, DOI:10.1002/aic.690481030.
- V. Govindaraj, D. Mech, G. Pandey, R. Nagarajan and J. S. Sangwai, J. Nat. Gas Sci. Eng., 2015, 26, 810–818, DOI:10.1016/j.jngse.2015.07.011.
- S. Ota, H. Kawano, K. Hirai and M. Kamei, Oceans, 2004, 2, 1115–1120, DOI:10.1109/OCEANS.2004.1405666.
- F. Su, C. L. Bray, B. O. Carter, G. Overend, C. Cropper, J. A. Iggo, Y. Z. Khimyak, A. M. Fogg and A. I. Cooper, Adv. Mater., 2009, 21, 2382–2386, DOI:10.1002/adma.200803402.
- D. W. Kang, W. Lee, Y. H. Ahn and J. W. Lee, Chem. Eng. J., 2021, 411, 128512, DOI:10.1016/j.cej.2021.128512.
- S. Baek, Y. H. Ahn, J. Zhang, J. Min, H. Lee and J. W. Lee, Appl. Energy, 2017, 202, 32–41, DOI:10.1016/j.apenergy.2017.05.108.
- S. Baek, W. Lee, J. Min, Y. H. Ahn, D. W. Kang and J. W. Lee, Korean J. Chem. Eng., 2020, 37, 341–349, DOI:10.1007/s11814-019-0451-3.
- W. Lee, D. W. Kang, Y. H. Ahn and J. W. Lee, ACS Sustainable Chem. Eng., 2021, 9, 8414–8424, DOI:10.1021/acssuschemeng.1c00807.
- W. Lee, D. W. Kang, Y. H. Ahn and J. W. Lee, Renewable Sustainable Energy Rev., 2023, 177, 113217, DOI:10.1016/j.rser.2023.113217.
- C. Lo, J. Zhang, P. Somasundaran and J. W. Lee, J. Colloid Interface Sci., 2012, 376, 173–176, DOI:10.1016/j.jcis.2012.03.012.
- J. S. Zhang, C. Lo, P. Somasundaran, S. Lu, A. Couzis and J. W. Lee, J. Phys. Chem. C, 2008, 112, 12381–12385, DOI:10.1021/jp801963c.
- Y. He, M. Sun, C. Chen, G. Zhang, K. Chao, Y. Lin and F. Wang, J. Mater. Chem. A, 2019, 7, 21634–21661, 10.1039/C9TA07071K.
- J. Min, S. Baek, P. Somasundaran and J. W. Lee, Langmuir, 2016, 32, 9513–9522, DOI:10.1021/acs.langmuir.6b02729.
- A. Phan, M. Stamatakis, C. A. Koh and A. Striolo, ACS Appl. Mater. Interfaces, 2021, 13, 40002–40012, DOI:10.1021/acsami.1c06309.
- Y. Zhong and R. E. Rogers, Chem. Eng. Sci., 2000, 55, 4175–4187, DOI:10.1016/S0009-2509(00)00072-5.
- P. Gayet, C. Dicharry, G. Marion, A. Graciaa, J. Lachaise and A. Nesterov, Chem. Eng. Sci., 2005, 60, 5751–5758, DOI:10.1016/j.ces.2005.04.069.
- P. S. R. Prasad and B. S. Kiran, J. Nat. Gas Sci. Eng., 2018, 52, 461–466, DOI:10.1016/j.jngse.2018.02.001.
- R. Li, Z. Sun and J. Song, J. Mol. Liq., 2021, 344, 117880, DOI:10.1016/j.molliq.2021.117880.
- S. Srivastava, A. M. Kollemparembil, V. Zettel, T. Claßen, B. Gatternig, A. Delgado and B. Hitzmann, Sci. Rep., 2022, 12, 8359, DOI:10.1038/s41598-022-12538-1.
- H. P. Veluswamy, P. Y. Lee, K. Premasinghe and P. Linga, Ind. Eng. Chem. Res., 2017, 56, 6145–6154, DOI:10.1021/acs.iecr.7b00427.
- S. P. Kang, Y. Seo and W. Jang, Energy Fuels, 2009, 23, 3711–3715, DOI:10.1021/ef900256f.
- W. Wang, C. L. Bray, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 11608–11609, DOI:10.1021/ja8048173.
- K. Kim, H. S. Truong-Lam, J. D. Lee and J. H. Sa, Energy, 2023, 270, 126902, DOI:10.1016/j.energy.2023.126902.
- B. Z. Peng, A. Dandekar, C. Y. Sun, H. Luo, Q. L. Ma, W. X. Pang and G. J. Chen, J. Phys. Chem. B, 2007, 111, 12485–12493, DOI:10.1021/jp074606m.
- N. Goel, J. Pet. Sci. Eng., 2006, 51, 169–184, DOI:10.1016/j.petrol.2006.01.005.
- Y. Seo, S. Lee, I. Cha, J. D. Lee and H. Lee, J. Phys. Chem. B, 2009, 113, 5487–5492, DOI:10.1021/jp810453t.
- T. P. Teng, Y. H. Hung, T. C. Teng, H. E. Mo and H. G. Hsu, Appl. Therm. Eng., 2010, 30, 2213–2218, DOI:10.1016/j.applthermaleng.2010.05.036.
- L. Yang, X. Wang, D. Liu, G. Cui, B. Dou, J. Wang and S. Hao, Chin. J. Chem. Eng., 2020, 28, 1112–1119, DOI:10.1016/j.cjche.2019.12.009.
- Z. Cheng, Y. Zhao, W. Liu, Y. Liu, L. Jiang and Y. Song, J. Nat. Gas Sci. Eng., 2020, 79, 103375, DOI:10.1016/j.jngse.2020.103375.
- Y. Wu, T. Tang, L. Shi and Y. He, Fuel, 2021, 303, 121248, DOI:10.1016/j.fuel.2021.121248.
- G. Q. Liu, F. Wang, S. J. Luo, D. Y. Xu and R. B. Guo, J. Mol. Liq., 2017, 230, 315–321, DOI:10.1016/j.molliq.2016.12.050.
- R. Wei, Y. Xia, A. Qu, X. Lv, Q. Fan, L. Zhang, Y. Zhang, J. Zhao and L. Yang, Fuel, 2022, 317, 123565, DOI:10.1016/j.fuel.2022.123565.
- J. Wang, J. J. Li, G. J. Weng and Y. Su, Acta Mater., 2020, 185, 461–473, DOI:10.1016/j.actamat.2019.12.032.
- Y. Xie, K. Guo, D. Liang, S. Fan and J. Gu, Chem. Eng. Sci., 2005, 60, 777–786, DOI:10.1016/j.ces.2004.09.003.
- J. Lee, C. Shin and Y. Lee, Energy Fuels, 2010, 24, 1129–1134, DOI:10.1021/ef901020g.
- P. Hu, G. Wu, M. Zi, L. Li and D. Chen, Fuel, 2019, 255, 115720, DOI:10.1016/j.fuel.2019.115720.
- R. Li, D. Liu, L. Yang, G. Cui, J. Wang, X. Wang and Z. Liu, Fuel, 2019, 252, 574–580, DOI:10.1016/j.fuel.2019.04.160.
- L. Yang, C. Li, J. Pei, X. Wang, N. Liu, Y. Xie, G. Cui and D. Liu, Chem. Eng. J., 2022, 440, 135912, DOI:10.1016/j.cej.2022.135912.
- L. Tian and G. Wu, Fuel, 2020, 260, 116307, DOI:10.1016/j.fuel.2019.116307.
- Z. Deng, Y. Wang, C. Yu, G. Li, X. Lang, S. Wang and S. Fan, Fuel, 2021, 299, 120867, DOI:10.1016/j.fuel.2021.120867.
- L. Yang, Z. Liu, D. Liu, G. Cui, B. Dou, J. Wang and S. Hao, J. Mol. Liq., 2020, 301, 112410, DOI:10.1016/j.molliq.2019.112410.
- D. W. Kang, W. Lee, Y.-H. Ahn, K. Kim and J. W. Lee, Energy, 2024, 292, 130631, DOI:10.1016/j.energy.2024.130631.
- G. Han, W. Lee, M.-K. Kim, J. W. Lee and Y.-H. Ahn, Chem. Eng. J., 2024, 483, 149409, DOI:10.1016/j.cej.2024.149409.
- G. Ko and Y. Seo, Chem. Eng. J., 2020, 400, 125973, DOI:10.1016/j.cej.2020.125973.
- M. V. Farahani, X. Guo, L. Zhang, M. Yang, A. Hassanpouryouzband, J. Zhao, J. Yang, Y. Song and B. Tohidi, Sustainable Energy Fuels, 2021, 5, 1567–1583, 10.1039/D0SE01705A.
- G. Bhattacharjee, H. P. Veluswamy, A. Kumar and P. Linga, Chem. Eng. J., 2021, 415, 128927, DOI:10.1016/j.cej.2021.128927.
- Y. Zhang, J. Zhao, G. Bhattacharjee, H. Xu, M. Yang, R. Kumar and P. Linga, Energy Environ. Sci., 2022, 15, 5362–5378, 10.1039/D2EE01968J.
- B. Mahant, O. S. Kushwaha and R. Kumar, Fluid Phase Equilib., 2024, 582, 114089, DOI:10.1016/j.fluid.2024.114089.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |