
Mechanochemical aerobic oxidative Heck coupling by polymer-assisted grinding: cyclodextrin additive facilitating regioselectivity control†
Keyu
Xiang
 a,
Haowen
Shou
ab,
Chenhui
Hu
a,
Weike
Su
a and
Jingbo
Yu
*a
a,
Haowen
Shou
ab,
Chenhui
Hu
a,
Weike
Su
a and
Jingbo
Yu
*a
aLaboratory of Pharmaceutical Engineering of Zhejiang Province, Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: yjb@zjut.edu.cn
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, P.R. China
First published on 9th April 2024
Abstract
The oxidative Heck reaction is a well-known organic transformation. However, previous reactions have been limited by the need for sophisticated directing or auxiliary groups, reliance on bulk solvents, lack of catalyst recycling, and the formation of a mixture of isomeric products, thereby hindering its broader application. Herein, we present a novel approach utilizing the polymer-assisted grinding (POLAG) technique to facilitate the regioselective oxidative Heck coupling of aryl boronic acids with electronically unbiased olefins under solvent-free conditions. This catalytic reaction is facilitated by a ligand-free Pd(TFA)2 catalyst in conjunction with cyclodextrin (CD) serving as a POLAG additive and molecular oxygen as the green oxidant. Mechanistic studies have indicated that cyclodextrin not only stabilizes the palladium catalyst but also influences the reaction selectivity by imposing steric hindrance on the substrates. Recovery experiments and electron microscopy images demonstrate an effective recovery capability of Pd/CD, with the catalyst morphology largely preserved after five cycles. XPS experiments confirm the formation of a new palladium catalytic species during the ball-milling process. Remarkably, this reaction takes place at ambient temperature, obviating the need for additional stoichiometric amounts of hazardous oxidants and toxic solvents, aligning with environmentally friendly practices upon green metrics evaluation.
Introduction
The emergence of mechanochemical synthesis as an efficient and sustainable technology has been witnessed in recent decades.1,2 This technology has been recognized by the International Union of Pure and Applied Chemistry (IUPAC) in 2019 as one of the top ten emerging technologies in the field of chemistry for its potential to advance society and improve our quality of life.3 In comparison with traditional solution-based methods, the mechanochemical protocol provides numerous advantages such as minimal solvent usage, milder conditions, shorter reaction time and the capability to alter chemical reactivity and selectivity, thereby leading to a reduced environmental footprint and favourable results in green metric assessment.4,5Some mechanochemical reactions exhibit unique chemical behaviours and different reaction pathways when a small amount of additive is introduced.1h,6 These reactions can be classified into two distinct groups. First, a specialized approach to mechanochemical reactions that involves the use of catalytic liquid additives, known as liquid-assisted grinding (LAG),7,8 which was introduced nearly two decades ago and has been shown to significantly enhance reaction rates and reactivity (Fig. 1a). For instance, Browne and co-workers9 reported an elegant finding when using LAG to selectively favor mono-fluorination over di-fluorination in the presence of a catalytic amount of acetonitrile. Our recent work10 on LAG reactions demonstrated that trace amounts of tert-butanol could accelerate the asymmetric Csp3–H/Csp3–H coupling of glycine esters and β-indanone esters, wherein tert-butanol not only provides a sub-stoichiometric solvent environment, but also contributes to the formation of active transition species, thereby boosting catalytic activity and enantiocontrol.
 | ||
| Fig. 1 (a) Rate enhancement and selectivity alteration by LAG ball-milling. (b) Rate enhancement and catalyst stabilization by POLAG ball-milling. (c) General Heck reaction and chain-walking process. (d) Directing/auxiliary group assisted oxidative Heck reaction. (e) Regioselective oxidative Heck coupling by CD assisted grinding and its recycling process (this work). | ||
The other type of mechanochemical technique, which utilizes polymer additives to improve chemical transformations by stabilizing catalysts or altering intermolecular interactions, is referred to as polymer-assisted grinding (POLAG)11 (Fig. 1b). A representative example was showcased by Lamaty and co-workers12 using polyethylene glycol (PEG) additives to accelerate the palladium-catalyzed Mizoroki–Heck cross-coupling reaction for the first time, leading to a significant improvement in reaction efficiency. More recently, the Ito group2i reported that the use of the PEGylated phosphine ligand in POLAG could effectively suppress the deactivation of palladium in a Suzuki–Miyaura coupling reaction. Despite advances, these methods are still facing sustainability challenges, primarily due to the lack of emphasis on the recycling of additives/catalysts, leading to unsatisfactory results in green metric assessments. In these scenarios, some polymer-assisted grindings using natural source additives, such as saccharides, have attracted our attention.13 Considering that saccharides can act as reducing agents under ball-milling conditions,13a we envision that such POLAG could also be used to facilitate catalytic redox processes to enable selectivity control through topology matching between the oligosaccharide additives and the substrates, or by forming a specific metal–additive complex.
On the other hand, the oxidative Heck-type reaction is one of the most attractive transformations that converts vinylic C–H bonds into C–C bonds, finding wide applications in synthetic chemistry.14 However, a serious limitation is that the use of inactive alkenes often leads to an inseparable mixture of styrene/allylic isomers. This issue primarily results from a chain-walking process,15 wherein reinsertion of the metal-hydride species is followed by β-hydride elimination. Moreover, imperfect selectivity also arises from the inability of metal-hydride species to distinguish between HS and HA during β-hydride elimination14c,16 (Fig. 1c). Over the past few years, a wealth of methods for the regulation of the oxidative Heck-type reactions involving electronically unbiased olefins have been developed by the groups of White,17 Liu,18 Xu,19 Yu,20 Maiti,21 Jeganmohan,22 Sigman23 and others.24 The selectivity aspect of these transformations can be affected by the use of a specially designed ligand, which has proved to be an attractive tool for achieving catalyst recycling in some cases.24c,d Nevertheless, there often remains a necessity for stoichiometric hazardous oxidants, bulk solvent, and preinstalled auxiliary/directing groups (Fig. 1d). Such requirements greatly limit the utility of the oxidative Heck reaction. The development of an operationally simple protocol for oxidative Heck coupling without the need for solvent, ligands, toxic oxidant and auxiliary/directing groups would therefore be highly beneficial.
We herein report a practically useful mechanochemical strategy for solvent-free oxidative coupling of phenylboronic acid with electronically unbiased olefins using polysaccharides as POLAG additives (POLAGs) under an oxygen atmosphere (Fig. 1e). We found that commercial cyclodextrins (CDs) were capable of regulating the regioselectivity of the oxidative Heck reaction via interactions between the substrates and the CD cavity, as well as through the formation of a new metal/CD complex in situ. The inclusion of CD not only led to high dispersion of the substrate within the solid mixture but also facilitated the stabilization of the metal catalysts. To the best of our knowledge, there have been no reports thus far regarding the selective oxidative Heck reaction with unbiased olefins under solvent-free mechanochemical conditions. Furthermore, the use of oxygen as a green oxidant, CD as a renewable natural additive for POLAG, which can be recycled and reused alongside the metal catalyst, aligns this protocol with the principles of green chemistry.
Results and discussion
The initial studies of the oxidative Heck reaction involved the utilization of phenylboronic acid (1a) and 6-bromo-1-hexene (2a) as coupling substrates in the presence of Pd(TFA)2 (10 mol%), and α-CD (1.0 equiv.) under flowing oxygen. The reaction was conducted in a custom-made ventilated stainless-steel jar (see the ESI† for details) under optimized mechanical parameters (20 Hz, 30 min, see Fig. S1†). After screening several metal catalysts, we found that Pd(TFA)2 gives the desired product in 30% yield (Table 1, entries 1–3). Further additive screening indicated that only CDs and soluble starch led to product formation, whereas trace amounts of the product were observed when methyl α-D-glucopyranoside and α-lactose were used as POLAGs (Table 1, entries 4–8). We also surveyed the molar amount of α-CD (Table 1, entries 9–14), finding that while excess α-CD inhibited the formation of the product, lowering its equivalence to 0.2 and 0.1 also led to significant decreases of yield. Moreover, reducing the amount of Pd(TFA)2 in the presence of α-CD lowered the yield of 3a (Table 1, entries 15 and 16). It is worth noting that both the oxygen flow rate and the size of milling balls significantly impact the reaction outcomes. In Table 1, entry 17 demonstrates that augmenting the oxygen flow rate from 4 to 8 mL min−1 correspondingly increases the reaction yield, achieving peak conversion under an oxygen flow of 6 mL min−1 (for more details, see ESI section 3.2†). Conversely, reducing the ball size to ∅ 1.0 cm notably hampers the reaction efficiency, especially in a larger 50 mL milling jar, likely due to a decrease in the mechanical force that results in inferior dispersion of reagents and increased aggregation (for more details, see ESI section 3.3†). However, using even larger balls with ∅ 1.4 cm did not lead to any additional enhancement (Table 1, entry 18). Our control experiments are depicted in entries 19 and 20, which were a subset of reactions that were conducted in a round-bottomed flask. It is noteworthy that stirring the reaction mixture in a DMF solution resulted in a heterogeneous state and led to poor conversion to the alkenylation product 3a. To understand the role of agitation during the ball-milling process, gaseous oxygen was flushed over the mixture of 1a, 2a, α-CD and Pd(TFA)2 for 4h without any agitation, followed by storing the mixture under an O2 atmosphere at ambient pressure for 7 days. No transformation was observed after the “aging”25 process (Table 1, entry 20), confirming that the milling process is necessary for the conversion.|
|
||
|---|---|---|
| Entry | Variation | Yieldf |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), Pd(TFA)2 (10 mol%) and an additive were placed in a 25 mL stainless-steel jar with two stainless-steel balls (∅ = 1.2 cm), milling at 20 Hz for 30 min under an oxygen atmosphere. b A gas flowmeter was utilized to precisely monitor and regulate the oxygen flow rate. c 50 mL stainless-steel jar was used. d Comparative experiment: 1a (0.5 mmol), 2a (0.5 mmol), Pd(TFA)2 (10 mol%), α-CD (0.4 equiv.) and DMF (5 mL) were added in a round-bottomed flask, stirring at 50 °C for 24 h under an oxygen atmosphere. e Comparative experiment: 1a (0.5 mmol), 2a (0.5 mmol), Pd(TFA)2 (10 mol%) and α-CD (0.4 equiv.) were placed in a stainless-steel jar without agitation for 4 h under an oxygen atmosphere, then aged in an flask for 7 days under an oxygen atmosphere. f Isolated yields. | ||
| 1 | None | 30 |
| 2 | Pd(OAc)2 instead of Pd(TFA)2 | 22 |
| 3 | Pd[O2C(CH3)3]2 instead of Pd(TFA)2 | Trace |
| Variation of additive | ||
| 4 | β-CD instead of α-CD | 20 |
| 5 | γ-CD instead of α-CD | 17 |
| 6 | Methyl α-D-glucopyranoside instead of α-CD | Trace |
| 7 | α-Lactose instead of α-CD | Trace |
| 8 | Soluble starch instead of α-CD | 21% |
| 9 | 2.0 equiv. α-CD | Trace |
| 10 | 0.8 equiv. α-CD | 51 |
| 11 | 0.6 equiv. α-CD | 57 |
| 12 | 0.4 equiv. α-CD | 63 |
| 13 | 0.2 equiv. α-CD | 50 |
| 14 | Without CD | 10 |
| Reaction in the presence of 0.4 equiv. α-CD | ||
| 15 | 5 mol% Pd(TFA)2 | 22 |
| 16 | 7 mol% Pd(TFA)2 | 46 |
| 17b | O2 flow rate (4/6/8 mL min−1) | 51/63/63 |
| 18 | Milling ball sizes (∅ = 1.0/1.4 cm) | 59 (57c)/63 |
| 19d | 10 mol% Pd(TFA)2 | 9% |
| 20e | 10 mol% Pd(TFA)2 | Trace |

With the optimal reaction conditions in hand, we set out to explore the scope of functionalized aliphatic olefins 2 in the oxidative Heck reaction, using 1a as a model substrate (Scheme 1). The mild reaction conditions allowed for the tolerance of a wide range of functional groups, including alkyl bromine (3a), alcohol (3b), ether (3c), acid (3d), ester (3e), amide (3f) and aryl groups (3h–3j). All reactions delivered the styrenyl products with exclusive E-stereoselectivity. Owing to the lack of chelation/directing effects, performing a ligand-free oxidative Heck reaction with electronically unbiased olefins is generally challenging. Such reactions have been rarely reported in the literature, typically achieving unsatisfactory yield and selectivity.26 Unfortunately, this synthesis was found to be barely suitable for substituted arylboronic acids. While some arylboronic acids with different substituents exhibited excellent selectivity and satisfactory yields when coupled with methyl 3,3-dimethylpent-4-enoate (3k–3p), undesired isomers (allylic and branch) were observed when using other olefins (for details see ESI Scheme S1†).
 | ||
| Scheme 1 Substrate scope for the functionalized aliphatic olefins. Reaction conditions: 1 (0.5 mmol), 2 (0.5 mmol), Pd(TFA)2 (10 mol%), and α-CD (40 mol%) were placed in a stainless-steel vessel with two stainless-steel balls (∅ = 1.2 cm) milling at 20 Hz for 30 min under an oxygen atmosphere. Isolated yields. | ||
We further elaborated the scope of this reaction with several aliphatic olefins derived from natural products and active pharmaceutical ingredients. Specifically, the substituted olefins obtained from menthol (3q), cholesterol (3r), furfuryl alcohol (3s), and the embedded olefins in amantadine (3t), ibuprofen (3u), and gemfibrozil (3v) produced the expected products in 35–58% yields, highlighting the excellent functional group compatibility of this strategy.
A wide range of unfunctionalized aliphatic olefins were then examined (Scheme 2), including long linear olefins (5a–5c), branched olefins (5d–5h, 5j), camphene (5i), exocyclic olefin (5k) and cyclic olefins (5l–5n). They all demonstrated excellent reactivity and yielded the E-styrenyl products with high selectivity. The longer-chain olefins gave the target product in a slightly higher yield than the shorter ones (5c > 5b > 5a). The use of phenylboronic acid with different substituents also yielded the desired products 5d–5h in good yield. Initially, relatively low yields were obtained, possibly due to the emission of neohexene (bp. 41 °C) during the milling process under flowing oxygen. As anticipated, increasing the amount of neohexene to 2 equiv. led to the desired products in high yields (up to 85%), and the products could be easily isolated by filtration and distillation (for details, see ESI section 3.6†). Interestingly, under the standard conditions, cyclopentene (4l) hardly participated in the reaction, yielding no product. However, when employing β-CD instead of α-CD, an allylic-type product 5l was obtained in 70% yield. Similarly, the reactions with cyclohexene and cyclooctene produced the allylic products 5m and 5n with yields of 68% and 74%, respectively.
 | ||
Scheme 2 Substrate scope for the unfunctionalized aliphatic olefins.a a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1 (0.5 mmol), 4 (0.5 mmol), Pd(TFA)2 and α-CD (40 mol%) were placed in a stainless-steel vessel with two stainless-steel balls (∅ = 1.2 cm) milling at 20 Hz for 30 min under an oxygen atmosphere. Isolated yields. bTwo equiv. of olefins were added. cβ-CD was used instead of α-CD. Reaction conditions: 1 (0.5 mmol), 4 (0.5 mmol), Pd(TFA)2 and α-CD (40 mol%) were placed in a stainless-steel vessel with two stainless-steel balls (∅ = 1.2 cm) milling at 20 Hz for 30 min under an oxygen atmosphere. Isolated yields. bTwo equiv. of olefins were added. cβ-CD was used instead of α-CD. | ||
In the previous report, it was suggested that the size of the CD cavity significantly influences the reaction selectivity.13d,e To validate this hypothesis, we expanded the study to include several substrates with varying sizes of CDs. The results of the reaction between 6-bromine-1-hexene (2a) and phenylboronic acid (1a) are shown in Fig. S6.† When α-CD was used as the POLAG additive, only the styrenyl product 3a was formed, whereas when β-CD was used it led to a mixture of styrenyl and allylic products. No products were detected when γ-CD was utilized. In the absence of POLAGs, the reaction afforded a mixture of styrenyl, allylic, branched, and homocoupled products, highlighting the significant role of CD in regulating the selectivity for this oxidative Heck reaction. Additionally, the reactions between phenylboronic acid (1a) and camphene (4i), cyclopentane (4l) or cyclooctene (4n) were also carried out with or without the addition of α-CD/β-CD/γ-CD. The reactivity and selectivity of the POLAG reaction were found to be highly dependent on the matching of the cavity size of the CD and the substrate (for more details, see ESI sections 3.3 and 4†).
Next, the recyclability of the Pd/α-CD catalyst was examined in the reaction of phenylboronic acid (1a) and olefins 2j/4c/4d/4i (see ESI Fig. S11†). The results showed that Pd/α-CD can be reused in five runs without significant loss of efficiency. After each run, Pd/α-CD was recovered by washing with cyclohexane, dried under vacuum, and directly used for the next reaction. However, a slight loss of yield was observed after five cycles, potentially due to the loss of Pd content during the recycling process (for ICP-MS results, see ESI Table S3†).
Moreover, in order to rule out the potential catalytic effects stemming from leached metals such as Fe, Co, and Ni present in stainless-steel materials during the mechanochemically activated reaction, control experiments were carried out using alternative zirconium oxide/Teflon jars and balls. These tests revealed that there was virtually no discernible difference in the reaction outcome between jars and balls made of stainless-steel versus those made of zirconium (for details, see ESI section 3.11†). It is noteworthy that the observed decrease in yield with the Teflon jar could be attributed to the lower mechanical force exerted due to its lighter weight material.
To gain mechanistic insight into the observed catalytic effect upon the addition of CD, we used scanning electron microscopy (SEM) to confirm the changes in the microstructures of the Pd/CD mixture. In Fig. 2a, a high dispersion of palladium with CD is evident after the first run of the Pd/CD mixture, whereas Fig. 2b depicts a decrease in the dispersion density of Pd after 5 runs, indicating the possibility of the loss of Pd content during the recycling process, which is consistent with the ICP-MS results. However, the Pd catalyst remained attached to α-CD even after 5 runs, which was tentatively confirmed by energy-dispersive spectroscopy (EDS) mapping experiments, as shown in Fig. 2c–f. These EDS mapping results showed a uniform distribution of Pd, C and O in the Pd/CD mixture, suggesting that Pd(TFA)2 and α-CD may form relatively stable Pd/CD dispersions/complexes after mechanochemical ball-milling, with α-CD potentially serving as a dispersant for the Pd catalyst.
 | ||
| Fig. 2 Scanning electron microscopy images of the Pd/α-CD (a) after the first run and (b) after the fifth run. (c–f) EDS-SEM images of the Pd/α-CD after the fifth run. | ||
Transmission electron microscopy (TEM) was further utilized to validate the in situ generation of Pd/CD dispersions/complexes during the ball-milling process (Fig. 3). The observed images clearly show the formation of evenly distributed Pd nanoparticles on CD in the reaction mixture (Fig. 3a). The morphology of Pd/CD remained unchanged over 5 consecutive catalytic runs, with only a slight aggregation of the Pd particles observed (Fig. 3c), and no significant alteration in particle size (approximate size: 2–4 nm).
 | ||
| Fig. 3 Transmission electron micrographs of the Pd/α-CD and size distributions. (a) After the first run; (b) after the third run; (c) after the fifth run; (d) size distributions for a; (e) size distributions for b; (f) size distributions for c. | ||
On the other hand, the image obtained for the palladium species derived from the reaction mixture in the presence of α-D-glucopyranoside, α-lactose monohydrate and soluble starch (Fig. 4) indicates significant aggregation of the Pd species into dense particles (Fig. 4). These findings suggest that α-CD might act as a dispersant for the Pd-based catalyst to inhibit excessive aggregation of the nanoparticles.
 | ||
| Fig. 4 Transmission electron micrographs of the Pd catalyst with saccharide additives after the first run. (a) Pd/methyl α-D-glucopyranoside, (b) Pd/α-lactose monohydrate, (c) Pd/soluble starch. | ||
For further investigation of the interaction between Pd catalyst and α-CD, X-ray photoelectron spectroscopy (XPS) was carried out for the commercial Pd(TFA)2 (Fig. 5a), Pd(TFA)2/α-CD (Sample A, Fig. 5b), reaction mixture with CD (Sample B, Fig. 5c), and reaction mixture without CD (Sample C, Fig. 5d). As determined by XPS, the binding energy of Pd 3d 5/2 in Sample A (for Pd II, 336.64 eV) was lower than that in Pd(TFA)2 (for Pd II, 338.17 eV), which is attributed to PdIIO species and comparable to the literature binding energy value in the NIST X-ray photoelectron spectroscopy database.27 The presence of the PdIIO peak indicates that the formation of the new catalyst takes place after ball-milling with α-CD (Sample A, Fig. 5b). Besides, PdIIO species were also observed in the reaction mixture with CD (Sample B), with appropriate amounts of metallic Pd0 formed (Fig. 5c). For the reaction mixture without CD (Sample C), only Pd(TFA)2 and Pd(0) species were confirmed by XPS, and no signal characteristic of PdIIO was detected (Fig. 5d).
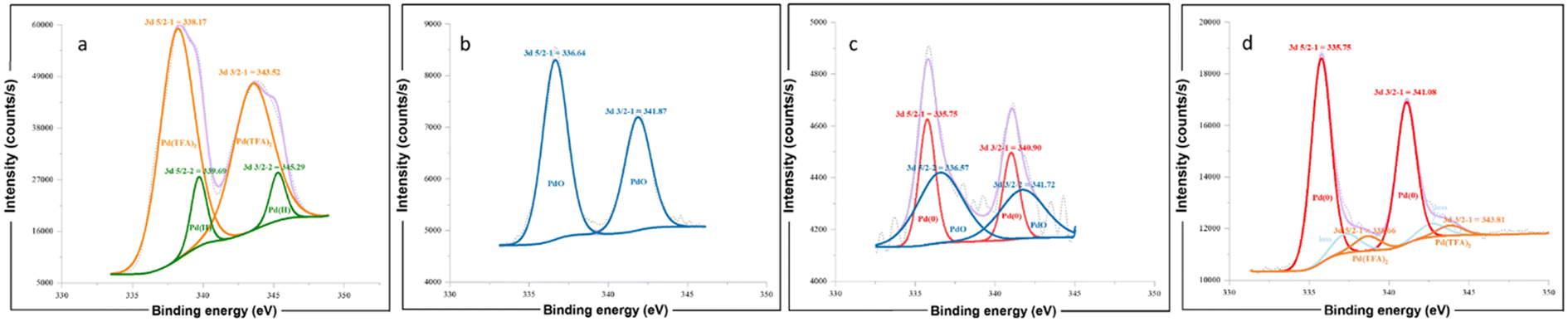 | ||
| Fig. 5 X-ray photoelectron spectroscopy of the mixtures after the reaction. (a) Commercial Pd(TFA)2; (b) the mixture of Pd(TFA)2 and α-CD after 30 min ball-milling at 20 Hz; (c) the mixture of Pd(TFA)2, olefin, phenylboronic acid and α-CD after 30 min ball-milling at 20 Hz, followed by rinsing with cyclohexane and drying; (d) the mixture of Pd(TFA)2, olefin and phenylboronic acid after 30 min ball-milling at 20 Hz, followed by rinsing with cyclohexane and drying. | ||
From the above observations, we can conclude that a new Pd–O bond was formed between Pd(TFA)2 and CD induced by mechanical force, leading to the formation of stable Pd/CD complexes. These complexes can stabilize the Pd(0) originating from the reductive elimination process and facilitate its re-oxidation to Pd(II), thereby improving the catalytic efficiency of the reaction. Conversely, in the absence of CD, the catalytic efficiency of Pd(TFA)2 was suppressed, and the re-oxidation of the deactivated Pd(0) was challenging due to aggregation, which significantly inhibited the catalytic cycle (see also Table 1, entry 14).
Finally, a direct comparison of this work with previous literature studies28 is shown in Fig. 6. Our POLAG reaction demonstrated time economy and avoids the use of bulky solvents and purpose-made catalysts. The formation of (E)-5-phenylpent-4-enoic acid (3d), (E)-prop-1-ene-1,3-diyldibenzene (3g), (E)-prop-1-ene-1,2-diyldibenzene (3h), and (E)-(3,3-dimethylbut-1-en-1-yl)benzene (5d) highlights the avoidance of severe conditions to facilitate the transformations (80–140 °C vs. rt). In the case of solvent-free oxidative Heck coupling, our previous conditions utilized DDQ as the oxidant.28d Mechanochemical olefinations with methyl 3,3-dimethylpent-4-enoate, 3,3-dimethylbut-1-ene and neohexene under an oxygen atmosphere are all illustrative of the greenness of this methodology as compared to our previous approach.
 | ||
| Fig. 6 Direct comparison with literature processes. | ||
Moreover, various green metrics, including effective mass yield (EMY), atom economy (AE), atom efficiency (AEF), reaction mass efficiency (RME), optimum efficiency (OE), process mass intensity (PMI), mass intensity (MI), solvent intensity (SI), and E-factor parameters, were employed to simultaneously evaluate the efficiency and environmental impact of the process in the synthesis of (E)-dodec-1-en-1-ylbenzene (5c) compared with a reported solution-based method,29 as per the literature.4a,30 As seen in Fig. 7a, a value of 1 for each pentagon radius is considered as the ideal condition for a sustainable process, and the closer the value of the different metrics to unity, the greener the process. In contrast, lower PMI, MI, E-factor, and SI scores signify a more environmentally friendly process (Fig. 7b).
 | ||
| Fig. 7 Summary of green chemistry metrics. The parameters in (a): the higher the better; in (b): the lower the better. | ||
It is noteworthy that the atom efficiency (AE) value is the highest for this work, probably due to the negligible contribution of lower molecular weight of the leaving group. Besides, the mechanochemical POLAG approach exhibits significantly superior parameters in terms of EMY, PMI, MI, SI and E-factor compared to those of previous solution-based methods. These findings underscore the importance of advancing solvent-free reactions and incorporating material recovery.
Conclusions
In this study, a mild and highly regioselective oxidative Heck cross-coupling of aryl boronic acids and electronically unbiased olefins was developed using the polymer-assisted grinding (POLAG) methodology. The method integrates eco-friendly components such as employing oxygen as an oxidant and naturally sourced cyclodextrin (CD) as a recyclable POLAG additive, establishing a sustainable and green synthesis pathway. This approach exhibits compatibility with a range of olefins that were previously ineffective in direct oxidative Heck coupling, while delivering products with excellent regioselectivity. Mechanistic study showed that cyclodextrin was efficient for the dispersion of Pd catalyst and also prevented the formation of undesired isomers. Analyses by transmission electron microscopy, scanning electron microscopy and X-ray photoelectron spectroscopy suggest the formation of a new form of PdIIO catalyst during the ball-milling process, which prevented catalyst deactivation caused by palladium species aggregation, despite a slight loss of palladium observed after five runs. Furthermore, the regioselectivity of the Pd/CD-catalyzed oxidative Heck coupling strongly relies on the CD/substrate interaction, suggesting topology matching between the CD and the substrate. Notably, a quantitative assessment of green chemistry metrics further demonstrates the environmental friendliness of this methodology compared to that of traditional solution-based approaches.This study presents a sustainable approach for regioselective oxidative Heck reactions, demonstrating significant potential for the efficient and selective synthesis of organic products by a polymer-assisted grinding method.
Author contributions
JB suggested the idea. KY, HW and CH performed the experiments and the greenness evaluation. HW carried out the electron microscopy experiments and processed the data. WK and JB supervised the project. Writing – review and editing were conducted by KY and JB. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of Zhejiang Province (LY23B060005), the National Key R&D Program of China (2021YFC2101000) and the National Natural Science Foundation of China (21978270) for financial support.References
- For selected reviews discussing mechanochemical studies see: (a) V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS; (b) J. Reynes, V. Isoni and F. García, Angew. Chem., Int. Ed., 2023, 62, e2023008 Search PubMed; (c) A. Jones, J. Leitch, S. Raby-Buck and D. Browne, Nat. Synth., 2022, 1, 763–775 CrossRef; (d) I. Egorov, A. Mukherjee, S. Santra, D. Kopchuk, I. Kovalev, Y. Liu, G. Zyryanov, A. Majee, O. Chupakhin and B. Ranu, Adv. Synth. Catal., 2022, 364, 2462–2478 CrossRef CAS; (e) M. Williams, L. Morrill and D. Browne, ChemSusChem, 2022, 15, e2021021 CrossRef; (f) E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS; (g) P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS; (h) T. Friščić, C. Mottillo and H. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef; (i) W. Pickhardt, S. Grätz and L. Borchardt, Chem. – Eur. J., 2020, 26, 12903–12911 CrossRef CAS PubMed; (j) A. Porcheddu, E. Colacino, L. Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS; (k) M. Perez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef CAS; (l) N. Egorov, S. Santra, D. Kopchuk, I. Kovalev, G. Zyryanov, A. Majee, B. Ranu, V. Rusinov and O. Chupakhin, Green Chem., 2020, 22, 302–315 RSC; (m) D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC; (n) J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC; (o) C. Bolm and J. Hernández, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed; (p) D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 18–33 CrossRef CAS; (q) J. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed; (r) J. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed; (s) G. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC; (t) S. James and T. Friščić, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC; (u) K. Ralphs, C. Hardacre and S. James, Chem. Soc. Rev., 2013, 42, 7701–7718 RSC; (v) S. James, C. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. Orpen, I. Parkin, W. Shearouse, J. Steed and D. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- For selected examples of mechanochemical studies recently see: (a) K. Kubota, J. Jiang, Y. Kamakura, R. Hisazumi, T. Endo, D. Miura, S. Kubo, S. Maeda and H. Ito, J. Am. Chem. Soc., 2024, 146, 1062–1070 CrossRef CAS PubMed; (b) K. Kubota, T. Endo and H. Ito, Chem. Sci., 2024, 15, 3365–3371 RSC; (c) F. Cuccu, F. Basoccu, C. Fattuoni and A. Porcheddu, Green Chem., 2024, 26, 1927–1934 RSC; (d) F. Cuccu and A. Porcheddu, Green Chem., 2024, 26, 2684–2691 RSC; (e) S. Pan, F. Mulks, P. Wu, K. Rissanen and C. Bolm, Angew. Chem., Int. Ed., 2024, 63, e2023167 Search PubMed; (f) J. Templ and M. Schnürch, Angew. Chem., Int. Ed., 2024, 63, e2023146 CrossRef PubMed; (g) G. Shao, P. Li, Z. Yin, J. Chen, X. Xia and G. Wang, RSC Mechanochem., 2024 10.1039/D3MR00010A; (h) T. Čarný, P. Kisszékelyi, M. Markovič, T. Gracza, P. Koóš and R. Šebesta, Org. Lett., 2023, 25, 8617–8621 CrossRef PubMed; (i) T. Seo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2023, 145, 6823–6837 CrossRef CAS PubMed; (j) X. Gu, T. Wang and K. Yan, Org. Lett., 2023, 25, 7287–7292 CrossRef CAS PubMed; (k) H. Luo, F. Liu, Y. Liu, Z. Chu and K. Yan, J. Am. Chem. Soc., 2023, 145, 15118–15127 CrossRef CAS PubMed; (l) C. Lennox, T. Borchers, L. Gonnet, C. Barrett, S. Koenig, K. Nagapudi and T. Friščić, Chem. Sci., 2023, 14, 7475–7481 RSC; (m) M. Passia, M. Amer, J. Demaerel and C. Bolm, ACS Sustainable Chem. Eng., 2023, 11, 6838–6843 CrossRef CAS; (n) C. Chen, J. Chen and C. To, Green Chem., 2023, 25, 2559–2562 RSC; (o) C. Patel, E. André-Joyaux, J. Leitch, X. Irujo-Labalde, F. Ibba, J. Struijs, M. Ellwanger, R. Paton, D. Browne, G. Pupo, S. Aldridge, M. Hayward and V. Gouverneur, Science, 2023, 381, 302–306 CrossRef CAS PubMed; (p) P. Bonn, J. Ke, C. Weike, J. Ward, K. Rissanen and C. Bolm, CCS Chem., 2023, 5, 1737–1744 CrossRef.
- (a) F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 CrossRef; (b) F. Gomollón-Bel and J. García-Martínez, Nat. Chem., 2022, 14, 113–114 CrossRef PubMed.
- For selected reviews discussing green metric assessment in mechanochemistry see: (a) N. Fantozzi, J. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC; (b) K. Ardila-Fierro and J. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- For selected examples discussing green metric assessment in mechanochemistry see: (a) Z. Zhao, S. Ikawa, S. Mori, Y. Sumii, H. Adachi, T. Kagawa and N. Shibata, ACS Sustainable Chem. Eng., 2024, 12, 3565–3574 CrossRef CAS; (b) V. Canale, M. Kamiński, W. Trybała, M. Abram, K. Marciniec, X. Bantreil, F. Lamaty, J. Parkitna and P. Zajdel, ACS Sustainable Chem. Eng., 2023, 11, 16156–16164 CrossRef CAS; (c) L. Pontini, J. Leitch and D. Browne, Green Chem., 2023, 25, 4319–4325 RSC; (d) S. Niu, W. Yuan, X. Gong, B. Bao, Z. Wu, B. Xu, R. Zeng, Q. Yang and Q. Ouyang, ACS Sustainable Chem. Eng., 2023, 11, 17816–17825 CrossRef CAS; (e) X. Yang, H. Wang, Y. Zhang, W. Su and J. Yu, Green Chem., 2022, 24, 4557–4565 RSC.
- D. Hasa, G. S. Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS PubMed.
- (a) G. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC; (b) T. Friščić, S. Childs, S. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- For selected examples discussing liquid-assisted grinding see: (a) J. Jia, Q. Wang, J. Li, Z. Xu, H. Li, D. Wei and B. Yuan, ACS Sustainable Chem. Eng., 2024, 12, 111–119 CrossRef CAS; (b) M. Williams, L. Morrill and D. Browne, Adv. Synth. Catal., 2023, 365, 1477–1484 CrossRef CAS; (c) Y. Hou, H. Wang, J. Xi, R. Jiang, L. Zhang, X. Li, F. Sun, Q. Liu, Z. Zhao and H. Liu, Green Chem., 2023, 25, 2279–2286 RSC; (d) T. Peňaška, V. Modrocká, K. Stankovianska, M. Mečiarová, E. Rakovský and R. Šebesta, ChemSusChem, 2022, 15, e2022000 CrossRef PubMed; (e) T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
- (a) Q. Cao, J. Howard, D. Crawford, S. James and D. Browne, Green Chem., 2018, 20, 4443–4447 RSC; (b) J. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. Browne, Green Chem., 2017, 19, 2798–2802 RSC.
- P. Ying, T. Ying, H. Chen, K. Xiang, W. Su, H. Xie and J. Yu, Org. Chem. Front., 2024, 11, 127–134 RSC.
- (a) K. Kubota, T. Seo and H. Ito, Faraday Discuss., 2023, 241, 104–113 RSC; (b) L. Germann, S. Emmerling, M. Wilke, R. Dinnebier, M. Moneghini and D. Hasa, Chem. Commun., 2020, 56, 8743–8746 RSC; (c) D. Scaramuzza, G. Rauber, D. Voinovich and D. Hasa, Cryst. Growth Des., 2018, 18, 5245–5253 CrossRef CAS; (d) A. Mascitti, M. Lupacchini, R. Guerra, I. Taydakov, L. Tonucci, N. d'Alessandro, F. Lamaty, J. Martinez and E. Colacino, Beilstein J. Org. Chem., 2017, 13, 19–25 CrossRef CAS PubMed.
- V. Declerck, E. Colacino, X. Bantreil, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11778–11780 RSC.
- (a) S. Mkrtchyan, V. Purohit, J. Zapletal, O. Shalimov and V. Iaroshenko, ACS Sustainable Chem. Eng., 2024, 12, 1–9 CrossRef CAS; (b) U. Kanchana, E. J. Diana, T. Mathew and G. Anilkumar, Carbohydr. Res., 2020, 489, 107954 CrossRef CAS PubMed; (c) Ö. Laçin, J. Kwiczak-Yiğitbaşı, M. Erkan, Ş. Cevher and B. Baytekin, Polym. Degrad. Stab., 2019, 168, 108945 CrossRef; (d) K. Cousin, S. Menuel, E. Monflier and F. Hapiot, Angew. Chem., Int. Ed., 2017, 56, 10564–10568 CrossRef CAS PubMed; (e) S. Menuel, B. Léger, A. Addad, E. Monfliera and F. Hapiot, Green Chem., 2016, 18, 5500–5509 RSC.
- (a) R. Heck, Acc. Chem. Res., 1979, 12, 146–151 CrossRef CAS; (b) R. Heck, Org. React., 1982, 27, 345–390 CAS; (c) I. Beletskaya and A. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed; (d) M. Oestreich, The Mizoroki–Heck Reaction, Wiley, New York, 2009 CrossRef.
- (a) Y. Li and G. Yin, Acc. Chem. Res., 2023, 56, 3246–3259 CrossRef CAS PubMed; (b) T. Kochi, S. Kanno and F. Kakiuchi, Tetrahedron Lett., 2019, 60, 150938 CrossRef CAS; (c) H. Sommer, F. Juliá-Hernández, R. Martin and I. Marek, ACS Cent. Sci., 2018, 4, 153–165 CrossRef CAS PubMed; (d) E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816–9826 RSC; (e) A. Dounay and L. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed.
- (a) M. Hilton, L. Xu, P. Norrby, Y. Wu, O. Wiest and M. Sigman, J. Org. Chem., 2014, 79, 11841–11850 CrossRef CAS PubMed; (b) J. Knowles and A. Whiting, Org. Biomol. Chem., 2007, 5, 31–44 RSC.
- J. Delcamp, A. Brucks and M. White, J. Am. Chem. Soc., 2008, 130, 11270–11271 CrossRef CAS PubMed.
- Z. Li, Y. Zhang and Z. Liu, Org. Lett., 2012, 14, 74–77 CrossRef CAS PubMed.
- Z. Jiang, L. Zhang, C. Dong, Z. Cai, W. Tang, H. Li, L. Xu and J. Xiao, Adv. Synth. Catal., 2012, 354, 3225–3230 CrossRef CAS.
- M. Lu, X. Chen, H. Xu, H. Dai and J. Yu, Chem. Sci., 2018, 9, 1311–1316 RSC.
- S. Maity, P. Dolui, R. Kancherla and D. Maiti, Chem. Sci., 2017, 8, 5181–5185 RSC.
- S. Jambu, C. Shambhavi and M. Jeganmohan, Org. Lett., 2021, 23, 2964–2970 CrossRef CAS PubMed.
- E. Werner and M. Sigman, J. Am. Chem. Soc., 2010, 132, 13981–13983 CrossRef CAS PubMed.
- (a) V. Bhoyare, E. Carrizo, C. Chintawar, V. Gandon and N. Patil, J. Am. Chem. Soc., 2023, 145, 8810–8816 CrossRef CAS PubMed; (b) C. K. Giri, S. Mondal and M. Baidya, Chem. – Asian J., 2023, 18, e202300243 CrossRef CAS PubMed; (c) J. Han, X. Sun, X. Wang, Q. Wang, S. Hou, X. Song, Y. Wei, R. Wang and W. Ji, Org. Lett., 2020, 22, 1480–1484 CrossRef CAS PubMed; (d) W. Li, C. Li, H. Xiong, Y. Liu, W. Huang, G. Ji, Z. Jiang, H. Tang, Y. Pan and Y. Ding, Angew. Chem., Int. Ed., 2019, 58, 2448–2453 CrossRef CAS PubMed; (e) Y. Zhou, Y. Wang, L. Ning, Z. Ding, W. Wang, C. Ding, R. Li, J. Chen, X. Lu, Y. Ding and Z. Zhan, J. Am. Chem. Soc., 2017, 139, 3966–3969 CrossRef CAS PubMed; (f) R. Li, Z. Ding, C. Li, J. Chen, Y. Zhou, X. An, Y. Ding and Z. Zhan, Org. Lett., 2017, 19, 4432–4435 CrossRef CAS PubMed; (g) R. Sharma, R. Kumar, I. Kumar and U. Sharma, Eur. J. Org. Chem., 2015, 7519–7528 CrossRef CAS; (h) C. Hu, K. Xiang, T. Ying and J. Yu, Adv. Synth. Catal., 2024 DOI:10.1002/adsc.202400014.
- K. Xiang, P. Ying, T. Ying, W. Su and J. Yu, Green Chem., 2023, 25, 2853–2862 RSC.
- Previously, there was almost no use of oxidative Heck reaction to synthesis 3 with selectivity challenge, we only found some examples of synthesizing 3h. (a) Z. Zhou, Z. Hou, F. Yang and B. Yao, Tetrahedron, 2018, 74, 7228–7236 CrossRef CAS; (b) X. Mi, M. Huang, H. Guo and Y. Wu, Tetrahedron, 2013, 69, 5123–5128 CrossRef CAS; (c) P. Sun, Y. Zhu, H. Yang, H. Yan, L. Lu, X. Zhang and J. Mao, Org. Biomol. Chem., 2012, 10, 4512–4515 RSC; (d) Y. Jung, R. Mishra, C. Yoon and K. Jung, Org. Lett., 2003, 5, 2231–2234 CrossRef CAS PubMed.
- The NIST X-ray Photoelectron Spectroscopy Database Online: https://srdata.nist.gov/xps/SpectralByElm/Pd, (accessed February 2024).
- (a) F. Singh and T. Wirth, Org. Lett., 2011, 13, 6504–6507 CrossRef CAS PubMed; (b) T. Furukawa, T. Yanagi, A. Kaga, H. Saito and H. Yorimitsu, Helv. Chim. Acta, 2021, 104, e202100195 CrossRef; (c) D. Meena, Deepa, M. Aalam, P. Chaudhary, G. Yadav and S. Singh, Polyhedron, 2022, 222, 115931 CrossRef; (d) J. Yu, H. Shou, W. Yu, H. Chen and W. Su, Adv. Synth. Catal., 2019, 361, 5133–5139 CrossRef CAS; (e) P. Liu, Y. Pan, K. Hu, X. Huang, Y. Liang and H. Wang, Tetrahedron, 2013, 69, 7925–7930 CrossRef CAS.
- (a) E. Werner and M. Sigman, J. Am. Chem. Soc., 2011, 133, 9692–9695 CrossRef CAS PubMed; (b) Y. Zou and J. Zhou, Chem. Commun., 2014, 50, 3725–3728 RSC.
- L. Wei, J. Zhang and L. Xu, ACS Sustainable Chem. Eng., 2020, 8, 13894–13899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization data, mechanism study, ICP-MS and XPS analyses, green metrics calculation, characterization data and NMR spectra. See DOI: https://doi.org/10.1039/d4gc01006j |
| This journal is © The Royal Society of Chemistry 2024 |