
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D structure-functional design of a biomass-derived photocatalyst for antimicrobial efficacy and chemical degradation under ambient conditions†
Wan
Zhang
 ab,
Yuanhao
Liang
ab,
Cheng
Hu
ab,
Weiwei
Li
c,
Jingru
Lai
ab,
Kainan
Chen
ab,
Sisi
Xiang
d,
Dariusz
Niedzwiedzki
f,
Jing
Wu
d,
Andrew
Li
e and
Susie Y.
Dai
*abg
ab,
Yuanhao
Liang
ab,
Cheng
Hu
ab,
Weiwei
Li
c,
Jingru
Lai
ab,
Kainan
Chen
ab,
Sisi
Xiang
d,
Dariusz
Niedzwiedzki
f,
Jing
Wu
d,
Andrew
Li
e and
Susie Y.
Dai
*abg
aDepartment of Plant Pathology and Microbiology, Texas A&M University, College Station, TX 77843, USA. E-mail: sydai@tamu.edu
bSystems and Synthetic Biology Innovation Hub, Texas A&M University, College Station, TX 77843, USA
cDepartment of Energy, Environmental, and Chemical Engineering, Washington University in St Louis, St Louis, MO 63130, USA
dMaterials Characterization Facility, Texas A&M University, College Station, TX 77843, USA
eDepartment of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA
fCenter for Solar Energy and Energy Storage and Department of Energy, Environmental and Chemical Engineering, Washington University in St Louis, St Louis, MO 63130, USA
gDepartment of Civil and Environmental Engineering, Texas A&M University, College Station, TX, USA
First published on 4th September 2024
Abstract
Surface sterilization and hazardous chemical degradation under ambient conditions can provide significant benefits for public and environmental health. Materials with sterilization and chemical degradation capacity under sunlight can efficiently reduce infectious disease incidence rates and toxic chemical exposure. Utilizing renewable energy for sustainable sterilization and degradation is more desirable as it reduces the potential secondary contamination. Herein, we report functional structure design using lignin, a renewable carbon heterogeneous polymer, to synthesize a highly efficient and stable photocatalyst that degrades environmentally hazardous organic compounds rapidly. Through a hydrolysis reaction between Ti–OH and the hydroxyl groups of lignin, Ti–O–C and Ti–O–Ti bonds were established and a lignin based photocatalyst with a hollow sphere structure (Clignin@H-TiO2) was formed. The presence of a homozygous carbon modified TiO2 structure contributes to the enhanced photodegradation activity with solar light. The close hetero-interfacial contact between carbonized lignin and TiO2 further improves the photocatalytic efficiency by facilitating effective charge carrier separation. After synthesis optimization, the resulting Clignin@H-TiO2 photocatalyst exhibits excellent performance in the degradation of atenolol under solar light irradiation with 100% degradation within five minutes. Additionally, it efficiently removes approximately 50% of PFOA and kills about 90% of bacteria within three hours. The uniform distribution of lignin within the crosslinking structures ensures a durable carbon modified TiO2 framework, which remains stable after 10 cycles of usage. The robustness of the lignin-based photocatalyst enables incorporating the catalyst into diversified material formats and various usages. Coating of the photocatalyst onto device surfaces shows bacterial killing efficacy under sunlight. The photocatalysts based on lignin valorization present a green chemistry approach for environmental remediation and surface sterilization, which has long-term environmental protection benefits, with broad applications in toxin treatment and health protection against pathogen infection.
1. Introduction
Our modern society with highly dense populations calls for convenient and rapid viral aerosol deactivation, surface bacterial sterilization and chemical decontamination methods to reduce the public and environmental health risks in routine life. Most of the current sterilization techniques and decontamination processes require harsh reaction conditions, use non-renewable resources, or are not environmentally friendly.1 Using solar energy as a sustainable energy source, photocatalysis is a promising technology for environmental remediation and bacterial sterilization, gaining significant attention in recent years due to its mild reaction conditions, high efficiency, strong oxidation capacity, less harmful byproducts, and rendering of the least environmental impact of treated effluents.2–4 However, the existing photocatalytic system faces challenges in widespread industrial implementation due to its dependency on UV radiation, inefficient utilization of visible light, rapid recombination of charges, and low efficiency in transferring photo-generated electrons and holes.5 As such, visible light range photocatalysis offers supreme advantages in routine lifestyle as contaminant removal can be achieved under ambient conditions.6 Among various visible light range catalysts, metal-based visible-light photocatalysts show promising catalytic activity, but some are hindered by the presence of toxic elements,7,8 limiting their practical applications and potentially causing secondary pollution. Additionally, precious metal based photocatalyst design creates challenges for commercialization due to the high cost.9 Among various photocatalytic materials, titanium dioxide (TiO2) is a cost-effective, photostable, avirulent, and highly efficient oxide semiconductor catalyst used extensively for gas purification, sterilization, and the degradation of refractory organic pollutants.10 However, TiO2 has several disadvantages in photocatalysis. First, the structure limitations result in rapid quenching of photoactive species.11 Second, the activation energy is primarily under the ultraviolet region,11,12 which limits the broad applications in various environmental settings.As one of the most abundant biopolymers on earth, lignin presents a sustainable means to synthesize functional materials.13–18 Traditionally, the inert heterogeneous polymeric structure of lignin challenges the conversion and degradation of lignin for the manufacture of high-value products. However, the polymeric structure and rich functional groups of lignin provide opportunities for chemical bond modification to catalyze multifunctional material synthesis. Particularly, the unique phenolic structure and high carbon content enable lignin to serve as the precursor for carbon-based materials with broad applications in engineering for our society. Lignin is composed of numerous crosslinked phenylpropanoid monomers such as coniferyl alcohol, sinapyl alcohol, and p-coumaryl alcohol, which provide the 3D structural framework and the hydroxyl groups for chemical modification.19,20 Lignin thus serves as an ideal sustainable material for structural functional design to overcome the weakness using TiO2 for photocatalysis mentioned above.
Based on the lignin polymeric crosslinking structure and the photocatalytic properties of TiO2, we thus functionally designed a 3D photocatalyst, which achieved highly efficient catalytic properties in the visible light wavelength range. Using lignin as a sustainable carbon source and doping agent, we developed a hollow nanoparticle structure photocatalyst that could detoxify organic contaminants and sterilize surface areas under solar irradiation by crafting lignin with TiO2 to form lattice 3D particles. The thus-formed spherical material absorbed visible light and inhibited charge recombination, which enabled efficient catalytic properties. The hollow photocatalyst (Clignin@H-TiO2) resulted from crosslinking the lignin–OH surface functional groups with hydrolyzed Ti(OH)4 to form the stable Ti–O–C bonds. The bonding between Clignin@H-TiO2 lattices guaranteed a uniform and stable carbon modification, which improved the resistance to photocorrosion and extended the catalyst lifespan. Furthermore, the shell structure of Clignin@H-TiO2 strengthened the light scattering in the hollow cavity and provided adequate active sites on a large surface area, thus significantly improving the photo-utilization and stabilizing the photogenerated charge. The material could degrade highly persistent environmental contaminants such as PFAS into shorter-chain products, completely destroy pharmaceutical compounds within minutes, deconstruct biologically relevant macromolecules, and kill bacteria under visible light. Incorporation of the material onto surfaces shows a sterilization effect on routine devices, suggesting broad applications in our daily life.
2. Materials and methods
2.1 Materials
99% tetrabutyl titanate was obtained from Beantown chemical. Atenolol was purchased from EMA Millipore Corp. Alkali lignin, acetone and 95% perfluorooctanoic acid (PFOA) were purchased from Sigma Aldrich. The isotope-labeled PFOA (perfluoro-n-[1,2-13C2] octanoic acid) was purchased from Wellington Laboratory (Guelph, Ontario). The isotope-labeled 99.8% (±) – atenolol-d7 (iso-propyl-d7) was purchased from CDN isotopes, Canada. All the chemicals were analytical grade and used as received.2.2 Preparation of Clignin@H-TiO2
Colloidal lignin particles were first initiated by dissolving 2 g of kraft lignin (dry basis) in 200 mL of an acetone/water 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture and stirred for 3 h, followed by filtration using a glass microfiber filter (Titan3™ Glass Microfiber Syringe Filters, Thermo Fisher Scientific, pore size 0.7 μm) to remove the undissolved lignin. The obtained solution was rapidly poured into 400 mL of Milli-Q water under vigorous stirring for 3 h. Acetone was further removed by reduced pressure distillation at 40 °C to obtain the lignin nanoparticle dispersion. The dispersed lignin nanoparticles were freeze-vacuum dried and then stored in a desiccator for further characterization and use. To prepare lignin@H-TiO2, 340 mg titanium(IV) n-butoxide was added to 50 mL lignin nanoparticle ethanol dispersion with magnetic stirring for 3 h until a clear brown uniform solution was obtained. Subsequently, 10 mL Milli-Q water was added dropwise to the above solution, which gradually turned into an opaque brown-yellow dispersion due to the hydrolysis reaction of titanium(IV) n-butoxide. The opaque brown-yellow solution was left to react for 12 h, ensuring complete hydrolysis of the titanium(IV) n-butoxide and its successful copolymerization with lignin. After the reaction, the dispersion was centrifuged with ethanol 2 times and Milli-Q water 5 times to remove any unreacted lignin and residual ethanol. The resulting precipitate was subsequently dried at 60 °C for 3 h, yielding brown powder identified as lignin@H-TiO2. To obtain lignin@H-TiO2 with different grafted lignin ratios, 0.4 mg, 0.8 mg, 4 mg, 8 mg, 12 mg and 50 mg lignin nanoparticles were used to prepare lignin@H-TiO2 samples with lignin contents of 0.5%, 1.0%, 5%, 10%, 15% and 50%, respectively. The lignin@H-TiO2 samples were calcined into Clignin@H-TiO2 in a split tube furnace with a vacuum system (GSL 1600X, MTI Corporation, Richmond, CA). The thermostabilization process was carried out with heating from room temperature to 200 °C at a heating rate of 5 °C min−1 and then holding at 200 °C for 20 min. Afterwards, the thermostabilized lignin@H-TiO2 was carbonized under a nitrogen atmosphere (240 cm3 min−1). The temperature was increased from 200 °C to the calcination temperature with a heating rate of 5 °C min−1. The holding time at calcination temperature was 1.5 h. After the calcination process, a black powder was obtained as Clignin@H-TiO2. For the investigation of the impact of the calcination temperature, various calcination temperatures within the range of 550 °C to 800 °C were employed. Calcined TiO2 (H-TiO2) is prepared with the same synthesis process, without adding lignin. TiO2 without calcination was synthesized by adding 340 mg titanium(IV) n-butoxide dropwise into 50 mL ethanol and dispersion with magnetic stirring for 3 h. After baking at 80 °C for 24 h, a white powder was obtained.
:1 (v/v) mixture and stirred for 3 h, followed by filtration using a glass microfiber filter (Titan3™ Glass Microfiber Syringe Filters, Thermo Fisher Scientific, pore size 0.7 μm) to remove the undissolved lignin. The obtained solution was rapidly poured into 400 mL of Milli-Q water under vigorous stirring for 3 h. Acetone was further removed by reduced pressure distillation at 40 °C to obtain the lignin nanoparticle dispersion. The dispersed lignin nanoparticles were freeze-vacuum dried and then stored in a desiccator for further characterization and use. To prepare lignin@H-TiO2, 340 mg titanium(IV) n-butoxide was added to 50 mL lignin nanoparticle ethanol dispersion with magnetic stirring for 3 h until a clear brown uniform solution was obtained. Subsequently, 10 mL Milli-Q water was added dropwise to the above solution, which gradually turned into an opaque brown-yellow dispersion due to the hydrolysis reaction of titanium(IV) n-butoxide. The opaque brown-yellow solution was left to react for 12 h, ensuring complete hydrolysis of the titanium(IV) n-butoxide and its successful copolymerization with lignin. After the reaction, the dispersion was centrifuged with ethanol 2 times and Milli-Q water 5 times to remove any unreacted lignin and residual ethanol. The resulting precipitate was subsequently dried at 60 °C for 3 h, yielding brown powder identified as lignin@H-TiO2. To obtain lignin@H-TiO2 with different grafted lignin ratios, 0.4 mg, 0.8 mg, 4 mg, 8 mg, 12 mg and 50 mg lignin nanoparticles were used to prepare lignin@H-TiO2 samples with lignin contents of 0.5%, 1.0%, 5%, 10%, 15% and 50%, respectively. The lignin@H-TiO2 samples were calcined into Clignin@H-TiO2 in a split tube furnace with a vacuum system (GSL 1600X, MTI Corporation, Richmond, CA). The thermostabilization process was carried out with heating from room temperature to 200 °C at a heating rate of 5 °C min−1 and then holding at 200 °C for 20 min. Afterwards, the thermostabilized lignin@H-TiO2 was carbonized under a nitrogen atmosphere (240 cm3 min−1). The temperature was increased from 200 °C to the calcination temperature with a heating rate of 5 °C min−1. The holding time at calcination temperature was 1.5 h. After the calcination process, a black powder was obtained as Clignin@H-TiO2. For the investigation of the impact of the calcination temperature, various calcination temperatures within the range of 550 °C to 800 °C were employed. Calcined TiO2 (H-TiO2) is prepared with the same synthesis process, without adding lignin. TiO2 without calcination was synthesized by adding 340 mg titanium(IV) n-butoxide dropwise into 50 mL ethanol and dispersion with magnetic stirring for 3 h. After baking at 80 °C for 24 h, a white powder was obtained.
2.3 Strain construction and culture
The plasmid roGFP2-Orp1 that expresses fused green fluorescent protein (roGFP2) and hydrogen peroxide (H2O2)-sensitive proteins (glutaredoxin 1 or Orp1) was used to investigate intracellular redox states of microorganisms.21 To construct an engineered strain, the roGFP2-Orp1 plasmid was transformed into Pseudomonas putida A514 competent cells via electroporation using a Bio-Rad Gene Pulser Electroporation system with parameters of 2.0 kV, 200 Omega, and 25 μF. The successful transformants were selected on an LB agar plate with 100 mg L−1 ampicillin using fluorescence as a marker. Then Pseudomonas putida A514 with oxygen reduction potential (ORP) capabilities was cultured in Luria–Bertani (LB) medium overnight at 30 °C under agitation at 200 rpm. Following the culture period, the bacterial cells were harvested through centrifugation and subsequently re-suspended in phosphate-buffered saline (PBS) to achieve an optical density at 600 nm (OD600) of 0.2.2.4 Contaminant photodegradation
10 mg Clignin@H-TiO2 was added in 5 mL porcelain crucibles with 2 mL PFAS solution or atenolol solution (2.5 ppm concentration). After 5 min adsorption equilibrium, the porcelain crucibles were placed under a solar simulator equipped with a xenon lamp (CME-SL500, Microenerg Beijing Technology Co., Ltd) with an intensity of 1800 W m−2 and a working distance of 15 mm to test the photodegradation performance. After a certain time of light treatment, a 40 μL sample solution was collected from the porcelain crucibles, and then 460 μL HPLC grade water was added into the above collected PFAS or atenolol solution to dilute the sample concentration to lower than 200 ppb for fitting the calibration curve. HPLC-MS was used to detect the PFOA and PFOS concentrations. Control samples were prepared in the same manner but without light treatment or without Clignin@H-TiO2 modification.2.5 Plasmid photodegradation
0.5 mg of Clignin@H-TiO2 was mixed with 20 μL of pLM231 at a concentration of 50 ng μL−1 (equivalent to 1 μg of DNA). Then the mixture was exposed to solar light for 5 to 30 min. The control groups included samples of anatase TiO2 (purchased from Sigma) + pLM231, TiO2 without calcination + pLM231, active carbon + pLM231, and Clignin@H-TiO2 + pLM231 (subjected to dark treatment). The quantities of the materials and the concentration of the plasmid remained consistent across all groups. For the experimental group, 1 μL of the sample was taken out every 5 min during the light treatment period. Following the treatment, gel electrophoresis was performed to assess the concentration of the DNA plasmid.2.6 Antibacterial performance under coating conditions
A binary mixture containing Clignin@H-TiO2 (100 mg) and waterborne polyurethane (5 g) was introduced into a 15 mL beaker. The mixture was subjected to magnetic stirring at 500 rpm for 5 min. Subsequently, this mixture was carefully transferred into a printing frame positioned atop a phone case. The printed pattern was then squeegeed by a scraper for three times, and cured under 80 °C for 20 min. Pseudomonas putida A514 ORP was inoculated in LB media overnight at 30 °C with agitation at 200 rpm. The bacterial culture was collected via centrifugation, and subsequently resuspended in PBS to achieve an OD 600 of 0.2. The printed phone case was placed under a solar simulator and 20 μL of the above Pseudomonas putida A514 ORP solution was dropped on the printed and un-printed parts, respectively, and then the setup was treated under light for 2 min. After light stimulation, the bacterial solutions from the printed and un-printed parts were collected and diluted to a concentration of 1 × 10−5. The experiments were repeated three times. Subsequently, 200 μL of the cultured solutions were evenly spread onto Luria–Bertani agar plates supplemented with ampicillin (LBAmp). The agar plates were incubated at 30 °C overnight to facilitate colony enumeration.2.7 Preparation of a Clignin@H-TiO2 membrane
0.5 g of a binary mixture of Clignin@H-TiO2 and water-borne polyurethane was carefully transferred onto a glass slide. The coated glass slide was then cured under 80 °C for 10 minutes. After the mixture was fully cured, the coated glass slide was put into methanol and finally the Clignin@H-TiO2 membrane was obtained by peeling it off from the glass slide.3. Results and discussion
3.1 Clignin@H-TiO2 preparation and characterization
The lignin constructed Clignin@H-TiO2 was prepared via an environmentally friendly sol–gel method (Fig. 1a). Specifically, lignin was dispersed freely in ethanol due to the –OH functional groups outside the particle surface. Ti(OH)4 was formed when tetrabutyl titanate slowly hydrolyzed with ethanol. Consequently, the TiO2 growth was positively influenced by the reticular molecular structure of lignin, as the hydrolysis reaction happened between the –OH groups of lignin and Ti(OH)4, forming the Ti–O–C and Ti–O–Ti crosslinking structures.22 The shell chemical structures of the 3D hollow particles were verified by Fourier-transform infrared spectroscopy (FTIR) and X-ray photoelectron spectroscopy (XPS), as shown in Fig. 1b and c–e. As a result, lignin was uniformly distributed among the Ti–O–C and Ti–O–Ti crosslinking structures after the polymerization reaction, which ensured a durable carbon-doped TiO2 structure. The peaks (Fig. 1d) at the binding energies of 458 and 464 eV can be attributed to Ti 2p3/2 and Ti 2p1/2, respectively, suggesting that the valence state of Ti is +4.23 The O 1s XPS spectrum is shown in Fig. 1e and can be divided into four peaks. The peaks at 531.6 eV and 532.9 eV are ascribed to C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively. The peaks at 529.15 eV are commensurate with the oxygen anions in the lattice (Ti–O–Ti), and the peak at 529.7 eV is attributed to C–O–Ti bonds.24,25 The dispersion structure obtained through grafting thus increased the 3D structure stability compared to other carbon modification structures such as composition.26 After calcination, the crosslinked lignin transformed into a carbon framework for the photocatalyst.
O bonds, respectively. The peaks at 529.15 eV are commensurate with the oxygen anions in the lattice (Ti–O–Ti), and the peak at 529.7 eV is attributed to C–O–Ti bonds.24,25 The dispersion structure obtained through grafting thus increased the 3D structure stability compared to other carbon modification structures such as composition.26 After calcination, the crosslinked lignin transformed into a carbon framework for the photocatalyst.
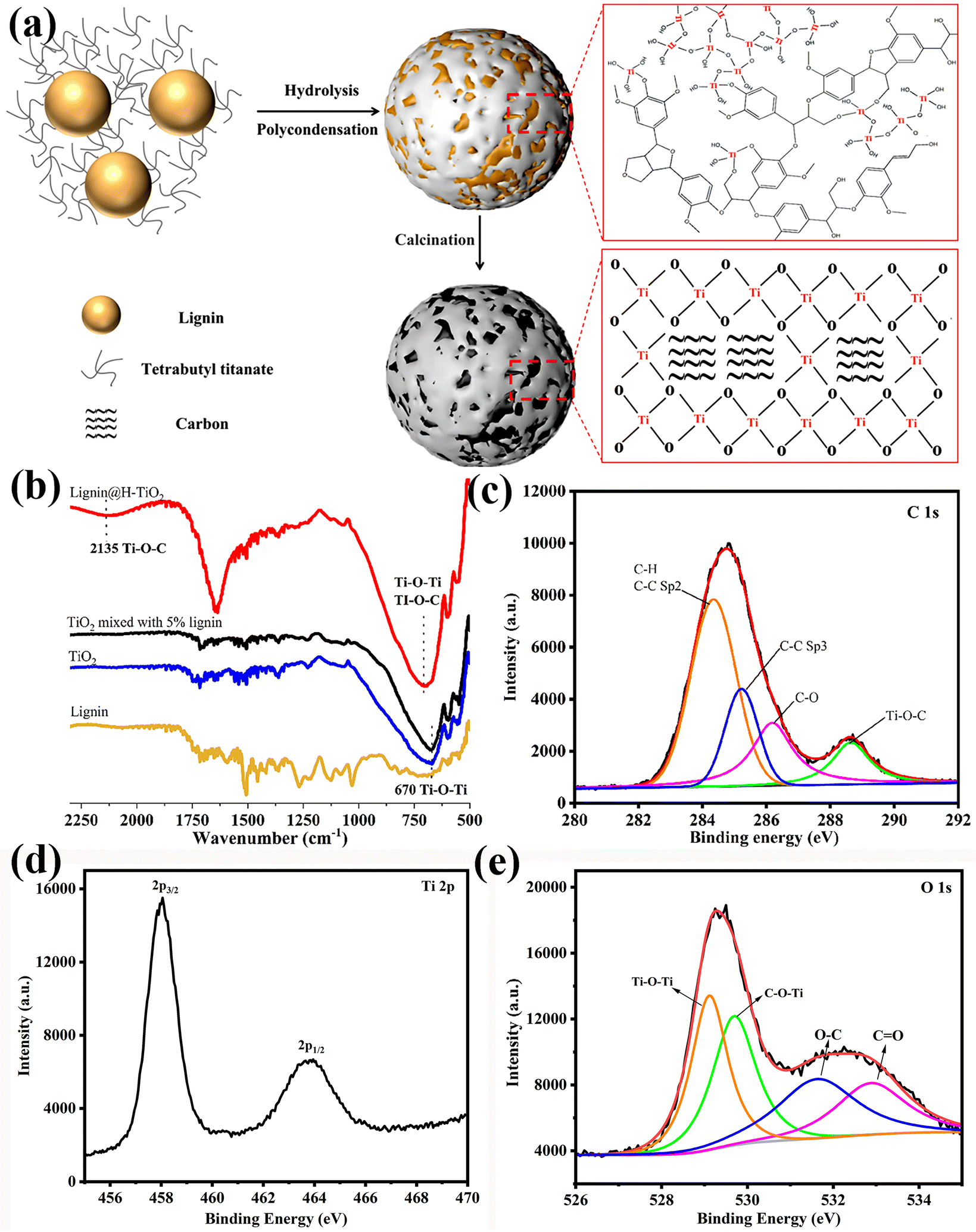 | ||
| Fig. 1 (a) Schematic illustration of the Clignin@H-TiO2 synthetic procedure. (b) FTIR of lignin@H-TiO2, TiO2 mixed with 5% lignin, TiO2 and lignin. XPS spectra of (c) C 1s, (d) Ti 2p and (e) O 1s for lignin@H-TiO2. | ||
The morphology and microstructure of Clignin@H-TiO2 with 5% lignin were revealed through scanning electron microscopy (SEM) and transmission electron microscopy (TEM) analyses. Fig. 2a shows the field emission scanning electron microscopy (SEM) images of the prepared Clignin@H-TiO2. The particle diameters ranged from 250 to 600 nm with a mean value of about 350 nm from the SEM measurements and the DLS test (Fig. 2a and S1†). The TEM images of Clignin@H-TiO2 are shown in Fig. 2b, suggesting that the spherical structure was rather uniform. The transparent appearance could be due to the hollow structure. As for a photocatalyst, the relative uniform size and rough structure could be beneficial for light harvesting. The lattice fringes and the crystal pattern were clearly visible (Fig. S2†), and the inter-planar distance was about 0.347 nm on average, which could be attributed to the (101) plane of anatase TiO2.27 In addition to the covalent Ti–O–C, the (101) surface of TiO2 also directly attached to the (101) surface of graphite carbon via weak van der Waals force to form a stable contact interface.28 Likewise, on the Clignin@H-TiO2 interface, the lattice fringes for the TiO2 surface became disordered, possibly due to the presence of a certain orbital hybridization between the π and O 2p orbitals,29 indicating that the TiO2 and the carbon were in tight contact, which was conducive to charge carriers between the two substances. Moreover, the X-ray (EDX) mapping image of the corresponding spherical particle of Clignin@H-TiO2 showed that Ti, O and C were homogeneously distributed in the shell (Fig. 2c, d and e), further suggesting a close hetero-interfacial contact between the carbonized lignin and TiO2, and a uniform binary carbon hybrid nanoarchitecture due to the polymerization reaction.
 | ||
| Fig. 2 (a) SEM morphology, (b) TEM morphology, and (c–e) EDS morphology of Clignin@H-TiO2. Red: carbon; green: Ti; and yellow: oxygen. | ||
TG analysis was conducted to investigate the Clignin content of Clignin@H-TiO2 synthesized with varying amounts of lignin (Fig. 3a). The initial weight loss observed from approximately 400 to 580 °C was due to the oxidation of carbon materials. A slight weight loss was observed for anatase TiO2 from 520 to 550 °C, which could be attributed to the decomposition of titanium-bonded groups such as –OH.18 As the weight loss due to the decomposition of titanium-bonded groups was negligible,18,30 the Clignin content of Clignin@H-TiO2 synthesis with 5% lignin was estimated to be 2.67%.
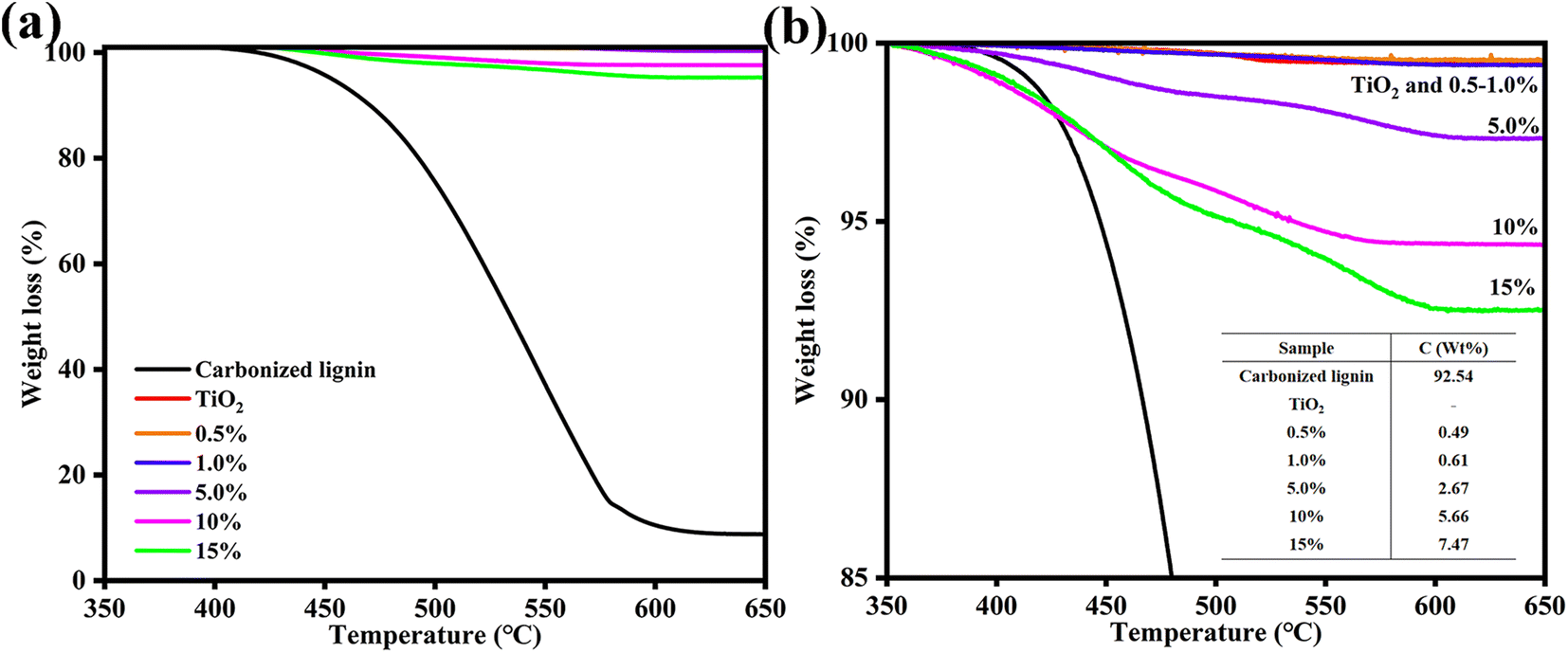 | ||
| Fig. 3 (a) Tg and (b) the calculated Clignin content of Clignin@H-TiO2 synthesized with different lignin weight percentages. | ||
3.2 Degradation performance of Clignin@H-TiO2
As highlighted, enhanced usage of visible lights can broaden the photocatalytic application in routine life. Using solar light irradiation, we thus evaluated the photocatalytic performance of Clignin@H-TiO2 by monitoring the photodegradation of atenolol, a model pharmaceutical compound. To show the effectiveness of our structural functional design, three other material designs were compared, which included H-TiO2 mixed with carbonated lignin (H-TiO2/Clignin), carbon@TiO2 synthesized by adding boric acids, according to a previous report,31 and anatase TiO2. All these materials showed an anatase phase crystalline structure upon calcination at 600 °C for 90 minutes (Fig. S3a†). Before the degradation performance testing, the atenolol solution and atenolol solution mixed with Clignin@H-TiO2 were kept in the dark. The atenolol solution with solar light irradiation was compared as the control (Fig. 4a). In the ten-minute testing period, the atenolol control was stable without degradation under solar light irradiation. Compared with other materials, Clignin@H-TiO2 showed the highest and fastest degradation rate, demonstrating the effective structural functional design with homozygous carbon-doped TiO2 hollow particle structure. For example, the carbon-based carbon@TiO2 only reached 75% removal efficiency compared to that of Clignin@H-TiO2, which almost reached 100% with five minutes of solar irradiation. The lignin and TiO2 crosslinking structure of Clignin@H-TiO2 assembled Clignin-doped mesoporous TiO2 while transforming lignin to carbon during calcination treatment.32 Consequently, the solar lights likely transmitted through the holes and form multiple reflectances inside the hollow structure, enhancing the photocatalytic activity.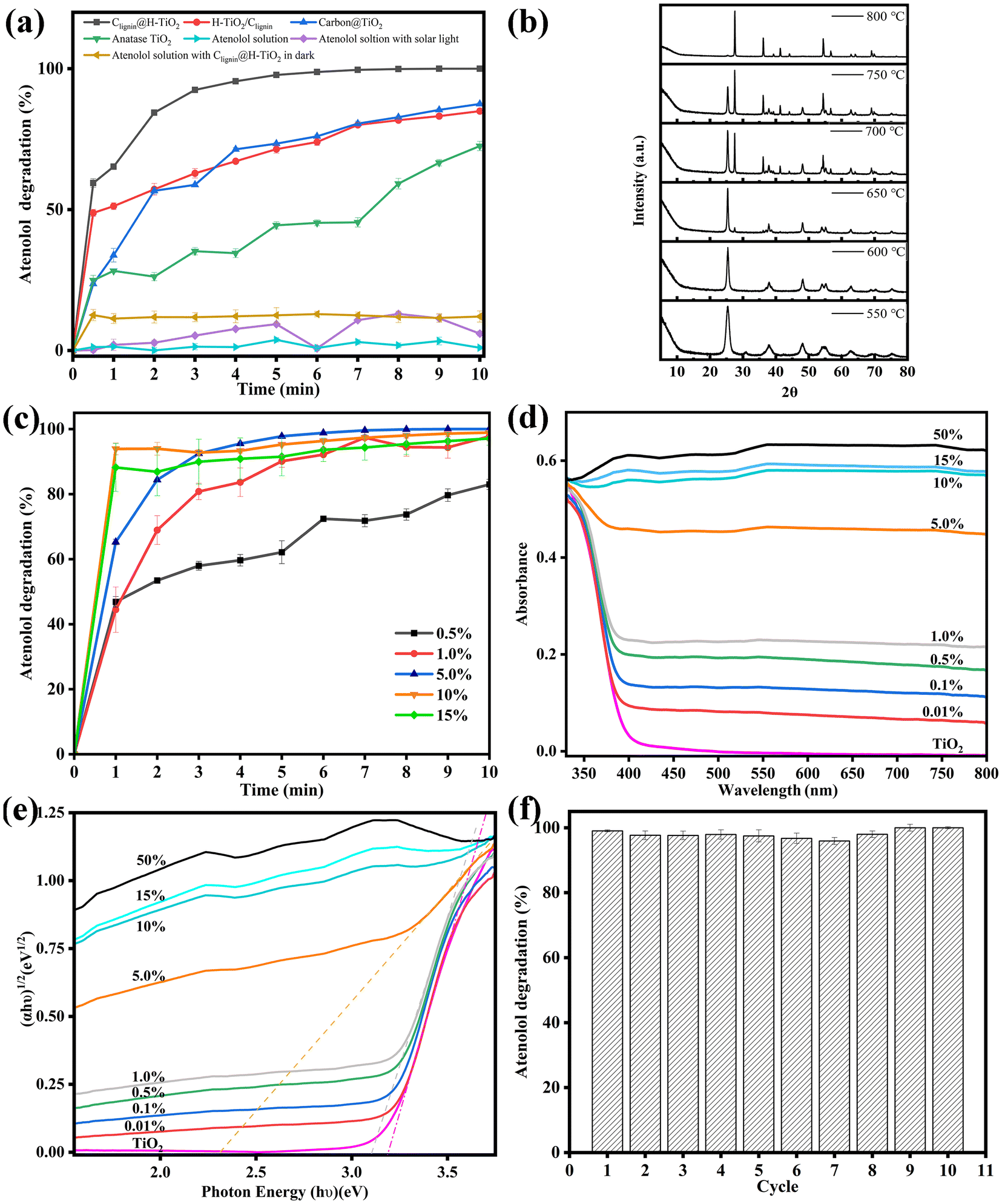 | ||
| Fig. 4 (a) Atenolol degradation performance of TiO2 based samples (each point represents triplicate measurements). (b) XRD of Clignin@H-TiO2 materials synthesized at varying temperatures. (c) Atenolol degradation with different lignin content (each point represents triplicate measurements). (d) UV-Vis absorption spectra. (e) An (αhυ)1/2versus (hυ) plot. (f) Performance of the Clignin@H-TiO2 photocatalyst upon successive 10 cycles. | ||
As shown in Table S1,† previous reports have applied various light sources using the TiO2 based photocatalysts. Our design of Clignin@H-TiO2 showed the fastest efficiency even with solar light. Furthermore, Clignin@H-TiO2 removed all atenolol from the solution phase within the first five minutes, in which the rapid kinetics could be attributed to both the enhanced photocatalysis due to the hollow structure and increased surface area for adsorption. As such, the BET surface area was measured for the synthesized materials. The surface areas were found to be 30.2, 41.9, 51.8, 50.1, 132.4 and 147.5 m2 g−1 for the samples of pure TiO2 and Clignin@H-TiO2 with 0.5 wt%, 1.0 wt%, 5.0 wt%, 10 wt%, and 15 wt% of lignin, respectively. The increased surface area could contribute to improved chemical adsorption and photocatalytic degradation. Pore volumes were calculated to be 91.9, 102.6, 95.0, 110.8, 84.4 and 79.4 cm3 g−1 Å−1 for the samples of pure TiO2 and Clignin@H-TiO2 with 0.5 wt%, 1.0 wt%, 5.0 wt%, 10 wt%, and 15 wt% of lignin, respectively. Nevertheless, H-TiO2/Clignin reached about 75% degradation for five minutes of solar irradiation and 80% degradation for ten minutes of solar irradiation, with much better performance than the individual anatase TiO2.
To investigate the effects of calcination on the as-designed sample performance, XRD measurements were conducted to analyze their crystalline structures. Fig. 4b illustrates the formation of crystalline structures upon calcination for 90 minutes. When synthesized with 550 °C calcination, the sample showed much more anatase phase than the brookite phase. With the calcination temperature increased, the crystalline structure gradually changed to the anatase phase and finally reached the rutile phase. The rutile TiO2 exhibited its most pronounced orientation preference along the (110) plane, which appeared at 2θ = 27.44°, as well as the second and third strongest orientation preferences along the (101) and (211) planes, which appeared at 2θ = 36.09° and 2θ = 54.33°, respectively.33 The observed diffraction peaks at 2θ = 25.3, 36.9, 37.9, 38.6, 48.0, 53.9, 55.1, and 62.1° corresponded to the anatase (101), (103), (004), (112), (200), (105), (221), and (213) planes, respectively.28,34 The diffraction peak observed at 2θ = 31° could be attributed to brookite (121).35 Rutile has the smallest band gap of approximately 3.0 eV among the polymorphs, but it typically exhibits photocatalytic activity that is about an order of magnitude lower than that of anatase.36 The optimal calcination condition was selected as 600 °C, which resulted in excellent atenolol degradation with a pure anatase crystalline structure (Fig. S3b†).
We further discovered that the lignin content had a significant impact on the photocatalytic capacity. Lignin composition could influence the spheroidal morphology, reduce the TiO2 band gap, and help match with the visible-light spectrum for light absorption.37 As shown in Fig. S4,† Clignin@H-TiO2 synthesized with 5.0% lignin showed a spherical shape with more and uniform holes. Additionally, as shown in Fig. S5,† the incorporation of lignin into the TiO2 increased the material conductivity, which could promote transferring the charges from the bulky TiO2 structure to the oxidation reaction sites and facilitate photocatalysis. Fig. 4c showed the photocatalytic degradation of Clignin@H-TiO2 with varying amounts of lignin. In the ten-minute testing period, the degradation percentage increased as the amount of lignin increased from 0.5% to 5%. However, further increasing lignin content (i.e., 10% and 15%) resulted in little improvement of the degradation efficiency. Consequently, Clignin@H-TiO2 with 5% lignin content was selected as the material composition for this study. The XRD measurements in Fig. S6† showed that all samples prepared formed the anatase phase TiO2 after calcination at 600 °C for 90 minutes. Furthermore, it has no influence on the crystalline structures of Clignin@H-TiO2 when the lignin content is lower than 15%.
To investigate the effects of the Clignin content on the degradation performance, UV–Vis and UV–Vis DRS were conducted to analyze their visible light absorption and band gap. The UV–Vis absorption spectra of the synthesized photocatalysts are shown in Fig. 4d. The anatase type TiO2 sample exhibited an absorption edge near 400 nm, similar to that of the other samples with varying Clignin content. The addition of Clignin led to red shifts in the light absorption in the visible region (>400 nm), which was also responsible for the gray/black color of the samples.38 With the increasing Clignin content, the absorption shifted to longer wavelengths, particularly in the visible range, correlating to the darker color of the materials.39,40 This effect was more evident with higher Clignin content, which led to much higher light absorption. The samples containing higher than 5% lignin showed black colors due to the carbon composite, which was responsible for the light absorption in the visible wavelength region. The samples were further characterized by UV–Vis DRS with the reflectance mode to estimate the band gap values, as shown in Fig. 4e. With the increasing content of Clignin, the Clignin@H-TiO2 photocatalysts exhibited lower band gap energies, except for the samples with more than 5% Clignin, where the deep black color did not allow estimation of the band gap value of the titania phase.38 The doped carbon is able to substitute the oxygen from the TiO2 lattice and bring down the band gap by contributing to its 2p orbitals.41 The band gap energy of TiO2 was found to be about 3.2 eV and the band gap energy of Clignin@H-TiO2 with 5% Clignin was found to be about 2.33 eV (Fig. 4e). A smaller band gap typically means that it is more capable of absorbing visible light and more effective in utilizing visible light for photocatalytic reactions. The results elucidate the reasons behind the influence of Clignin on the degradation performance and indicate that Clignin@H-TiO2 with 5% Clignin exhibits superior photocatalytic performance under visible light conditions.
The photocatalysts stability is crucial for broad applications. Fig. 4f displays the evolution of the atenolol degradation percentage vs. irradiation time upon ten consecutive cycles with Clignin@H-TiO2. The results suggested that the catalyst degradation capacity remained about the same after 10 usage cycles, degrading 100% of the atenolol. The remarkable stability could be attributed to the uniform distribution of lignin within the crosslinking structures, ensuring a durable carbon-modified TiO2 framework, which efficiently separated photogenerated electrons and holes to sustain excited electrons and holes, introduced by the engineered Clignin lattice structure.
3.3 Photocatalytic degradation mechanism
To understand the highly efficient photocatalytic capacity, we then evaluated the mechanism of the designed photocatalyst. Selected scavengers were used to identify the primary reactive oxygen species. Benzoquinone (BQ), AgNO3, isopropanol and potassium iodide (KI) were chosen as the capture agents of superoxide radicals (˙O2−), electron (e−), hydroxyl radicals (˙OH) and hole (h+), respectively.42,43 The efficiency of photocatalytic degradation of atenolol by Clignin@H-TiO2 was reduced significantly after adding 0.5 mM of BQ, isopropanol and KI, individually (Fig. 5a). Fig. 5a indicates that ˙O2−, ˙OH and h+ played key roles in the photocatalytic degradation of atenolol. However, addition of 0.5 mM AgNO3 did not significantly reduce the photocatalytic performance as about 100% degradation was achieved after 10 min, comparable to the catalysts’ performance by itself (Fig. 5a). Since the AgNO3 molecules scavenge e−, the observation suggested that e− might be present in the Clignin@H-TiO2 photocatalytic degradation system at trace amounts, but not as the main photocatalytic active species. Apparently, the primary reactive oxygen species involved in acetaminophen photodegradation include ˙O2−, ˙OH radicals and h+. The ESR spin-trapping technique with DMPO is used to further verify the ˙OH and ˙O2− radicals.44 As shown in Fig. S7,† when irradiated with solar light, the ESR spectra of Clignin@H-TiO2 show a stronger signal than those of the dark control, which suggests the detection of ˙OH and ˙O2− radicals. Since the ˙OH radicals are generally generated by holes,45 the results are also strong evidence for the existence of h+.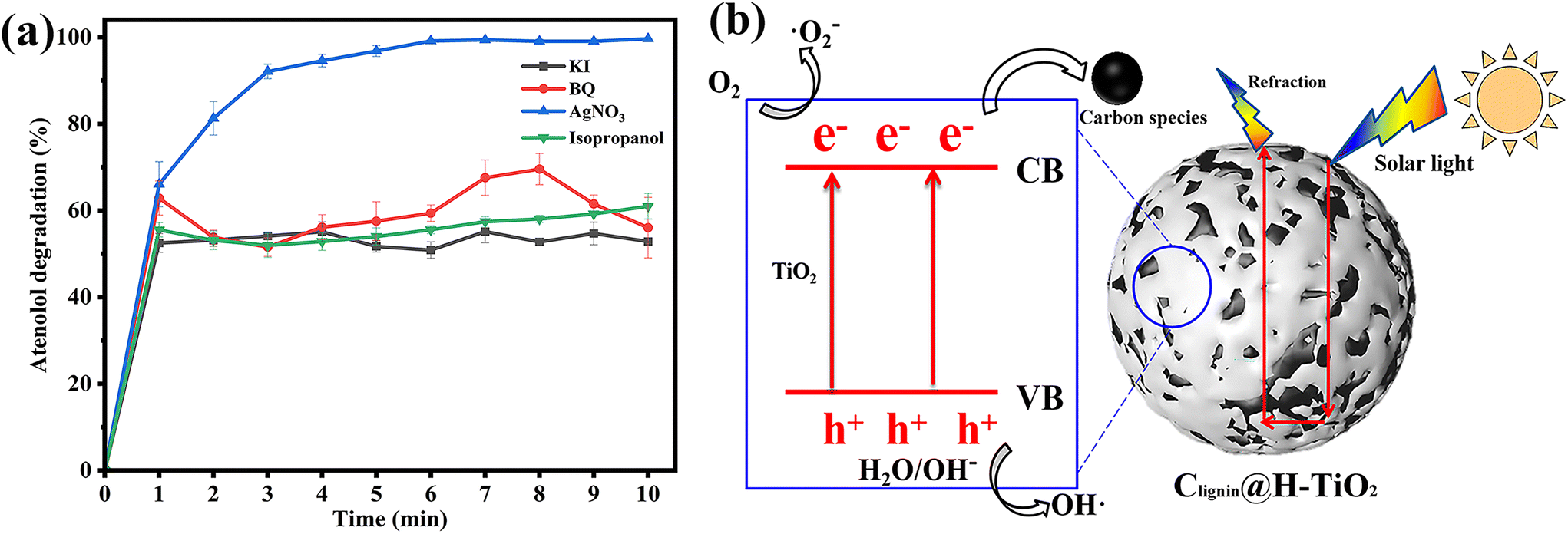 | ||
| Fig. 5 (a) Effect of quenchers on the atenolol degradation performance (each point represents triplicate measurements). (b) Schematic diagram of degradation mechanism (VB: valence band; CB: conduction band). | ||
Accordingly, we proposed the potential mechanisms of the high photodegradation activity of Clignin@H-TiO2 nanocomposites as shown in Fig. 5b. The photocatalytic process included the photogenerated electron–hole separation and the free radical formation. The photocatalytic TiO2 is well-known for the fast and easy recombination of photo-generated electron–hole pairs.46,47 Doping of Clignin would have introduced lattice defects into TiO2, which resulted in a narrower bandgap for visible light absorption. Under visible light, photons generated electrons and holes in Clignin modified TiO2. Furthermore, the available charge carriers could form more reactive species and facilitate photocatalysis. Besides, the separated electrons could react with the adsorbed O2 to form ˙O2− and catalyze the photo-oxidation reaction. The remaining holes in TiO2 could also facilitate the redox reactions by forming ˙OH radicals.48 The hollow structure with potential sphere interior may give rise to surface reflectivity, which could further expand the light path within the Clignin@H-TiO2 and facilitate the light/matter interaction. As such, the photo-generated electrons could react with O2 to form active oxidative species such as ˙O2− and the holes react with H2O to generate ˙OH.45
To further characterize the mechanism leading to the improved photocatalytic capacity, we have measured the photoluminescence emission, electrochemical impedance, and photocurrent response of Clignin@H-TiO2 and compared them with those of TiO2 (Fig. S8†). Electrochemical impedance spectroscopy (EIS) is applied to characterize the charge transfer properties of the samples, where Nyquist plots are presented in Fig. S8a.† The Clignin@H-TiO2 exhibits a smaller arc radius than anatase TiO2, indicating that Clignin@H-TiO2 is more efficient in transferring and separating photogenerated carriers.49 The photocurrent is measured for Clignin@H-TiO2 and anatase TiO2 (Fig. S8b†). The photocurrent density of Clignin@H-TiO2 is higher than that of anatase TiO2, demonstrating that the separation rate of the photogenerated electrons and holes was enhanced.50 From Fig. S8c,† the decreased PL intensity of that Clignin@H-TiO2 compared to that of the anatase TiO2 at 350 nm suggested a potential better charge separation and a complicated overall recombination process for the Clignin@H-TiO2 material.51
3.4 Degradation of biological molecules and implication for sterilization applications
With the capacity of producing superoxide radicals (˙O2−) and hydroxyl radicals (˙OH), we further evaluated the sterilization mechanism and examined cellular oxidative stress upon the photocatalyst treatment. The model bacterium used in this study was an environmental bacterial strain, Pseudomonas putida, which is a robust strain that could survive under extreme environmental conditions. The engineered strain has a fluorescence tag that produces florescence signal when the cells are under oxidative stress.52 We observed that the engineered Pseudomonas putida strain had a strong fluorescence signal when incubating with 0.5 mg of the photocatalyst (Fig. 6a). Furthermore, the cellular oxidative stress was clearly dependent on the light intensity. During the three-hour irradiation treatment, the cells experienced much stronger stress under the 50%, 75% and 100% light intensity. Interestingly, the cell population presented higher fluorescence signal intensity under the 50% light intensity than under the 100% light intensity, which could be attributed to a percentage of cells could have died from the strong oxidative stress, which prevented the gene expression and fluorescence signal production.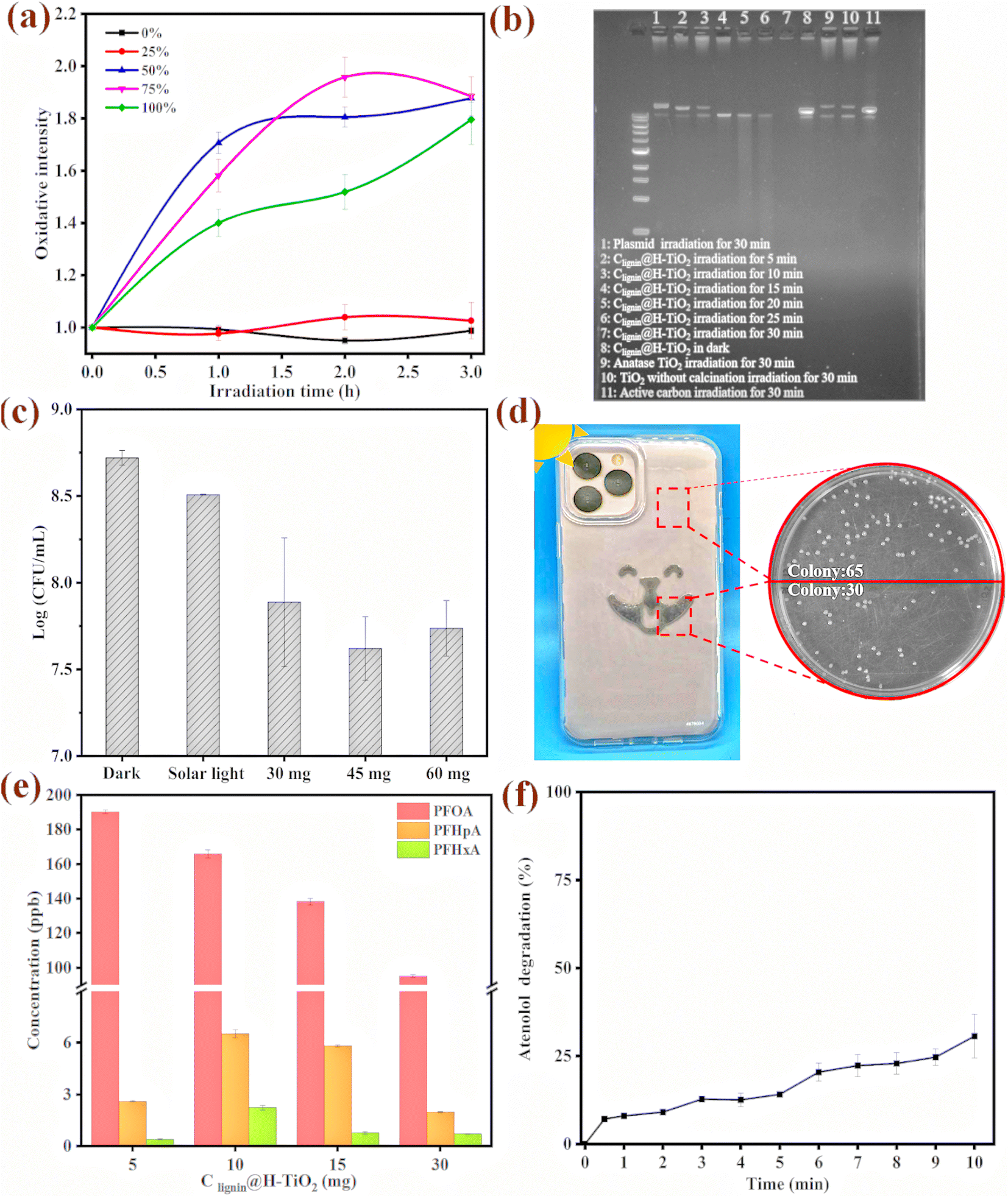 | ||
| Fig. 6 (a) Fluorescence signal intensity, suggesting bacterial oxidative stress (the stress is probed by transforming a redox-sensitive green fluorescence protein fusion (i.e., ORP-GFP) into the cell) under solar light in the presence of Clignin@H-TiO2 (percentage represents light intensity). (b) Plasmid degradation at different radiation time. (c) Antibacterial performance of Clignin@H-TiO2 with different catalyst dosages. (d) Antibacterial performance of Clignin@H-TiO2 applied on a protection mobile case. (e) PFOA degradation by Clignin@H-TiO2 at different catalyst quantities. (f) Atenolol degradation of the Clignin@H-TiO2 membrane. | ||
We further reviewed possible mechanisms for cell damage and used biological molecules as a surrogate to evaluate the molecular level oxidative damage. A plasmid of colibacillus origin with 11k base pairs was used to test the photocatalytic performance on large biological molecules. The plasmid molecule was also an ideal surrogate for DNA/RNA viruses in vivo. The plasmid was transferred and expressed in the E. coli HB101 competent cells to obtain 50 ng μL−1 for the degradation experiment. The purified plasmid was subjected to Clignin@H-TiO2 treatment for five to 30 minutes and electro gel analysis. As shown in Fig. 6b, the plasmid was exposed to irradiation for different time periods. Lane one shows the intact plasmid that has not been treated with the photocatalysts. When the plasmid was mixed with the photocatalyst and gradually exposed to irradiation for five minutes, the plasmid began to degrade and produce small DNA fragments, as shown in lane three. No more visible fragments were presented after 30 minutes of irradiation treatment (Fig. 6b lane 7). The Clignin@H-TiO2 catalysts performed much better than the anatase TiO2, as shown in lane 9, where DNA fragments were visible after the 30-minute irradiation. The plasmid/DNA degradation mechanism highlighted the potential application toward deactivating DNA/RNA viruses from the routine environment under ambient conditions.
The antibacterial properties of Clignin@H-TiO2 were further evaluated by treating bacterial colonies (Pseudomonas putida A514) with the photocatalyst, as shown in Fig. 6c and Fig. S9.† The initial concentration of the bacterial concentration was approximately 1 × 109 CFU mL−1, and the survival bacterial concentrations in different Clignin@H-TiO2 treatment groups were compared with those under dark conditions, and solar light exposure with one hour irradiation time. The bacterial concentration of the irradiated groups showed varying degrees of reduction. 45 mg of the photocatalyst presented the best sterilization effect, compared with those of the 30 mg and 60 mg materials. A log reduction of more than one suggested that more than 90% of the bacteria had been killed by the photocatalyst upon light irradiation. The cell death and damage mechanism would be more complex than the plasmid degradation.53 However, the strong oxidative species produced by the photocatalyst could potentially lyse the cell, permeate the cell membrane and induce molecular level degradation and intracellular damage.
To further demonstrate the applicability of the as-designed photocatalyst in routine sterilization, Clignin@H-TiO2 was used in combination with other polymers for material surface coating. We incorporated Clignin@H-TiO2 into polyurethane resin and made a viscous solution, which was then applied to phone cases via screen printing. As displayed in Fig. 6d and Table S2,† the Clignin@H-TiO2-coated areas show a significantly lower number of bacterial colonies compared to the uncoated areas. Meanwhile, we used a commercial product (i.e., Clorox disinfecting wet wipes) as the control to demonstrate the photocatalyst's effectiveness. The Clignin@H-TiO2-coated areas showed a significantly lower number of bacterial colonies compared to the uncoated areas and a similar antibacterial effect compared to that of the Clorox disinfecting wet wipe wiped areas, indicating a remarkable antibacterial effect. Per fixed area, the bacterial colonies were reduced by about 50% upon minutes of light irradiation. This highlighted the potential of Clignin@H-TiO2 for routine use for sterilizing material surfaces.
3.5 Chemical degradation and indication of detoxification
After using atenolol as the model compound to evaluate the photocatalytic efficiency, we further evaluated other persistent chemicals of a broader scope. We tested the degradation capacity of Clignin@H-TiO2 on per- and poly-fluoroalkyl substances (PFAS). PFOA (perfluorooctanoic acid), one of the so-called stable “forever chemicals”, was selected to test the degradation efficiency. As such, Clignin@H-TiO2 was mixed with the PFOA solution and tested under different conditions. The degradation behaviors of different amounts of Clignin@H-TiO2 are shown in Fig. 6e. The PFOA removal rate increased with the increasing amount of Clignin@H-TiO2, and, consequently, the degraded products were clearly observed by LCMS measurements. In the three-hour testing time, 30 mg of the Clignin@H-TiO2 removed the most PFOA (∼50%) from the solution phase and produced degraded products of perfluoroheptanoic acid (PFHpA, C7) and perfluorohexanoic acid (PFHxA, C6). Interestingly, the disappearance of the PFOA molecule was positively correlated with the catalyst quantity, which was not observed with the degraded products (C7 and C6). This could be due to the degradation kinetics, that the catalyst continuously degrades into the shorter chain degraded products. The degradation of perfluorooctanoic acid (PFOA) can potentially be initiated through two pathways: the hole oxidation pathway and the ˙OH attack pathway.54–56 Both pathways involve the cleavage of the C–C bond between the carbon chain and the carboxylic group, generating shorter chain perfluorocarboxylic acids (PFCAs) as intermediates.57 The degradation process could begin at the terminal carboxylic end, where the h+ trapped e− triggered formation of the perfluorocarboxylate radicals (C7F15COO˙). These unstable radicals initiated C–C bond cleavage between the carboxyl carbon and its adjacent carbon atoms, generating perfluorinated alkyl radicals that could react with ˙OH to produce perfluorinated alcohol (C7F15OH). The alcohol would be further defluorinated to produce C6F13COF and C6F13COOH.58 The intermediate radicals could then undergo successive loss of the terminal carboxylate group, addition of H2O, elimination of HF, and hydrolysis, ultimately leading to the formation of CO2 and F−.Inspired by the antibacterial property when Clignin@H-TiO2 was mixed with polymers and coated on the device surface, we further evaluated the application of Clignin@H-TiO2 as a membrane material for chemical removal. A membrane composed of Clignin@H-TiO2 and polyurethane was fabricated to test atenolol removal. As shown in Fig. 6f, the membrane with Clignin@H-TiO2 achieved about 30% removal of atenolol after 10 minutes, which was lower than that of Clignin@H-TiO2 itself (100% in 5 min, Fig. 4a). The reduction of photocatalytic degradation efficiency could be attributed to several factors, such as treatment on the membrane surface, rather than in the colloid solution; the mixture of the photocatalyst with polyurethane could reduce the active percentage of the material per unit area. Additionally, the polyurethane coating may also affect the reactivity of ˙O2−, e−, ˙OH and h+, further contributing to the reduction of degradation efficiency. Nevertheless, given the short irradiation time (10 minutes) applied in this study, we expect that for routine applications, the irradiation time would not be the constraint as the constant sunlight can catalyze chemical degradation continuously.
4. Conclusion
In this study, we have successfully designed a sunlight driven photocatalyst Clignin@H-TiO2 with a hollow spherical structure, which can achieve persistent chemical degradation and solar sterilization under ambient conditions. The structural design is inspired by the polymeric carbon framework of lignin, a sustainable carbon source that can be functionalized to achieve desired properties. By crafting lignin and TiO2 to form a 3D structure that effectively separates the electrons and holes (i.e., the active species for photocatalysis), the as-designed photocatalyst achieved highly efficient oxidative capacity (i.e., removing 100% of the atenolol at 200 ppb concentration in the solution phase within 10 minutes) in the visible light wavelength range. Doping lignin carbon into the TiO2 and forming the crystalline structure narrowed the band gap of TiO2, generating more powerful photocatalysts. The potential catalyst mechanism was studied, suggesting the primary oxidative species include h+, ˙OH, and ˙O2−. The broad utility of photocatalytic sterilization and chemical degradation under ambient conditions was demonstrated by coating the catalyst onto device surfaces and manufacturing membranes. Under sunlight, the photocatalyst can kill bacteria on a cell phone plastic cover when coated with a polymer mixture (kills 90% of the bacteria under visible light within one hour). The Clignin@H-TiO2 was further coated onto a membrane surface to show the chemical degradation application under ambient conditions. Using sustainable solar energy and renewable materials, our ambient condition treatment technology presents broad routine application opportunities for lignin doped photocatalysts for protecting environmental and public health.Author contributions
Conceptualization: W. Z. and S. Y. D.; methodology: W. Z. and Y. L.; investigation: W. Z., Y. L., C. H., J. L., K. C. and A. L.; resources: C. H., J. L., S. X., D. N., W. L., and J. W.; writing – original draft: W. Z. and S. Y. D.; writing – review & editing: W. Z. and S. Y. D.; funding acquisition: S. Y. D.; supervision; S. Y. D.; cover page: S. Y. D.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
Use of the TAMU Materials Characterization Facility (RRID:SCR_022202) is acknowledged. We would like to acknowledge the X-ray diffraction laboratory at Texas A&M University for the material characterization instruments. The research was partially supported by National Institute of Environmental Health Sciences R01ES032708 to S. Y. D.References
- M. S. Mauter, I. Zucker, F. Perreault, J. R. Werber, J.-H. Kim and M. Elimelech, Nat. Sustain., 2018, 1, 166–175 CrossRef.
- W. Zhao, M. Adeel, P. Zhang, P. Zhou, L. Huang, Y. Zhao, M. A. Ahmad, N. Shakoor, B. Lou, Y. Jiang, I. Lynch and Y. Rui, Environ. Sci.: Nano, 2022, 9, 61–80 RSC.
- A. M. Nasir, N. Awang, J. Jaafar, A. F. Ismail, M. H. D. Othman, M. A. Rahman, F. Aziz and M. A. M. Yajid, J. Water Process Eng., 2021, 40, 101878 CrossRef.
- S. Mishra and B. Sundaram, Mater. Today: Proc., 2023 Search PubMed.
- W. S. Koe, J. W. Lee, W. C. Chong, Y. L. Pang and L. C. Sim, Environ. Sci. Pollut. Res., 2020, 27, 2522–2565 CrossRef CAS PubMed.
- Y. Sun and D. W. O'Connell, Water Environ. Res., 2022, 94, e10781 CrossRef CAS PubMed.
- Q. Huang, W. Yuan, Q. Ke, Y. Guo, L. Wang and C. Wang, J. Environ. Chem. Eng., 2023, 11, 109696 CrossRef CAS.
- Z. Ji, X. Yang, X. Qi, H. Zhang, Y. Zhang, X. Xia and Y. Pei, Chemosphere, 2022, 308, 136144 CrossRef CAS PubMed.
- X. Ye, Y. Zhang, L. Chen, S. Chen, L. Ma, Y. Wu and Q. Wang, Opt. Mater., 2022, 134, 113201 CrossRef CAS.
- S. Banerjee, S. C. Pillai, P. Falaras, K. E. O′shea, J. A. Byrne and D. D. Dionysiou, J. Phys. Chem. Lett., 2014, 5, 2543–2554 CrossRef CAS PubMed.
- H. Dong, G. Zeng, L. Tang, C. Fan, C. Zhang, X. He and Y. He, Water Res., 2015, 79, 128–146 CrossRef CAS PubMed.
- S. Yin, Q. Zhang, F. Saito and T. Sato, Chem. Lett., 2003, 32, 358–359 CrossRef CAS.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- R. Madhu, A. P. Periasamy, P. Schlee, S. Hérou and M.-M. Titirici, Carbon, 2023, 207, 172–197 CrossRef CAS.
- M. Vasudevan, S. Remesh, V. Perumal, P. B. Raja, M. N. M. Ibrahim, S. C. B. Gopinath, S. Karuppanan and M. Ovinis, Chem. Eng. J., 2023, 468, 143613 CrossRef CAS.
- J. Li, C. Hu, Y.-Y. Wang, X. Meng, S. Xiang, C. Bakker, K. Plaza, A. J. Ragauskas, S. Y. Dai and J. S. Yuan, Matter, 2025, 5, 3513–3529 CrossRef.
- R. Li, D. Huang, L. Lei, S. Chen, Y. Chen, G. Wang, L. Du, W. Zhou, J. Tao and H. Chen, J. Mater. Chem. A, 2023, 11, 2595–2617 RSC.
- M. Chen, L. Wu, S. Zhou and B. You, Adv. Mater., 2006, 18, 801–806 CrossRef CAS.
- M. Bilal, S. A. Qamar, M. Qamar, V. Yadav, M. J. Taherzadeh, S. S. Lam and H. M. N. Iqbal, Biomass Convers. Biorefin., 2022, 14, 4457–4483 CrossRef.
- M. J. Gan, Y. Q. Niu, X. J. Qu and C. H. Zhou, Green Chem., 2022, 24, 7705–7750 RSC.
- W. H. Reuter, T. Masuch, N. Ke, M. Lenon, M. Radzinski, V. V. Loi, G. Ren, P. Riggs, H. Antelmann, D. Reichmann, L. I. Leichert and M. Berkmen, Redox Biol., 2019, 26, 101280 CrossRef CAS PubMed.
- R. Djellabi, B. Yang, K. Xiao, Y. Gong, D. Cao, H. M. A. Sharif, X. Zhao, C. Zhu and J. Zhang, J. Colloid Interface Sci., 2019, 553, 409–417 CrossRef CAS PubMed.
- G. An, W. Ma, Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and K. Ding, Carbon, 2007, 45, 1795–1801 CrossRef CAS.
- W. Wu, T. Liu, X. Deng, Q. Sun, X. Cao, Y. Feng, B. Wang, V. A. L. Roy and R. K. Li, Int. J. Biol. Macromol., 2019, 126, 1030–1036 CrossRef CAS PubMed.
- R. Kumar, B. K. Singh, A. Soam, S. Parida, V. Sahajwalla and P. Bhargava, Nanoscale Adv., 2020, 2, 2376–2386 RSC.
- X. Zheng, M. Gao, C. Liang, S. Wang and X. Wang, Electrochim. Acta, 2022, 428, 140910 CrossRef CAS.
- N. Srisasiwimon, S. Chuangchote, N. Laosiripojana and T. Sagawa, ACS Sustainable Chem. Eng., 2018, 6, 13968–13976 CrossRef CAS.
- Y. Li, Q. Shen, R. Guan, J. Xue, X. Liu, H. Jia, B. Xu and Y. Wu, J. Mater. Chem. C, 2020, 8, 1025–1040 RSC.
- L.-Y. Gan, Q. Zhang, C.-S. Guo, U. Schwingenschlogl and Y. Zhao, J. Phys. Chem. C, 2016, 120, 2119–2125 CrossRef CAS.
- W. Zhang, X. Ji, C. Zeng, K. Chen, Y. Yin and C. Wang, J. Mater. Chem. C, 2017, 5, 8169–8178 RSC.
- Z. Zhang, Z. Xiong, C. Zhao, P. Guo, H. Wang and Y. Gao, Appl. Surf. Sci., 2021, 565, 150554 CrossRef CAS.
- M. Li, J. Qiu, J. Xu and J. Yao, Ind. Eng. Chem. Res., 2020, 59, 13997–14003 CrossRef CAS.
- G. Chen, J. Chen, Z. Song, C. Srinivasakannan and J. Peng, J. Alloys Compd., 2014, 585, 75–77 CrossRef CAS.
- M. Zarattini, C. Dun, L. H. Isherwood, A. Felten, J. Filippi, M. P. Gordon, L. Zhang, O. Kassem, X. Song, W. Zhang, R. Ionescu, J. A. Wittkopf, A. Baidak, H. Holder, C. Santoro, A. Lavacchi, J. J. Urban and C. Casiraghi, J. Mater. Chem. A, 2022, 10, 13884–13894 RSC.
- E. Solati, Z. Aghazadeh and D. Dorranian, J. Cluster Sci., 2022, 31, 961–969 CrossRef.
- G. Odling and N. Robertson, ChemSusChem, 2015, 8, 1838–1840 CrossRef CAS PubMed.
- Z. Shayegan, F. Haghighat and C.-S. Lee, J. Environ. Chem. Eng., 2020, 8, 104162 CrossRef CAS.
- A. Gómez-Avilés, M. Peñas-Garzón, J. Bedia, J. J. Rodriguez and C. Belver, Chem. Eng. J., 2019, 358, 1574–1582 CrossRef.
- K. Chen, X. Zhou, D. Wang, J. Li and D. Qi, Int. J. Biol. Macromol., 2022, 218, 33–43 CrossRef CAS PubMed.
- X. Chen, H. Sun, J. Zhang, O. A. Zelekew, D. Lu, D.-H. Kuo and J. Lin, Appl. Catal., B, 2019, 252, 152–163 CrossRef CAS.
- C. Negi, P. Kandwal, J. Rawat, M. Sharma, H. Sharma, G. Dalapati and C. Dwivedi, Appl. Surf. Sci., 2021, 554, 149553 CrossRef CAS.
- Y. Wang, L. Lin, Y. Dong and X. Liu, New J. Chem., 2022, 46, 16584–16592 RSC.
- Y. Wen, A. Rentería-Gómez, G. S. Day, M. F. Smith, T.-H. Yan, R. O. K. Ozdemir, O. Gutierrez, V. K. Sharma, X. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2022, 144, 11840–11850 CrossRef CAS PubMed.
- Y. Zhu, Y. Wang, Z. Chen, L. Qin, L. Yang, L. Zhu, P. Tang, T. Gao, Y. Huang and Z. Sha, Appl. Catal., A, 2015, 498, 159–166 CrossRef CAS.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., 2005, 44, 8269 CrossRef CAS.
- J. Yu, T. Ma and S. Liu, Phys. Chem. Chem. Phys., 2011, 13, 3491–3501 RSC.
- C. Wang, M. Cao, P. Wang, Y. Ao, J. Hou and J. Qian, Appl. Catal., A, 2014, 473, 83–89 CrossRef CAS.
- Y. Huang, S. Kang, Y. Yang, H. Qin, Z. Ni, S. Yang and X. Li, Appl. Catal., B, 2016, 196, 89–99 CrossRef CAS.
- Z. Liang, X. Bai, P. Hao, Y. Guo, Y. Xue, J. Tian and H. Cui, Appl. Catal., B, 2019, 243, 711–720 CrossRef CAS.
- W. Jiao, L. Zhang, R. Yang, J. Ning, L. Xiao, Y. Liu, J. Ma, N. Mahmood and X. Jian, Appl. Mater. Today, 2022, 27, 101498 CrossRef.
- R. Velmurugan, B. Krishnakumar, B. Subash and M. Swaminathan, Sol. Energy Mater. Sol. Cells, 2013, 108, 205–212 CrossRef CAS.
- L. N. Jayakody, C. W. Johnson, J. M. Whitham, R. J. Giannone, B. A. Black, N. S. Cleveland, D. M. Klingeman, W. E. Michener, J. L. Olstad, D. R. Vardon, R. C. Brown, S. D. Brown, R. L. Hettich, A. M. Guss and G. T. Beckham, Energy Environ. Sci., 2018, 11, 1625–1638 RSC.
- S. Adhikari, A. Banerjee, N. K. Eswar, D. Sarkar and G. Madras, RSC Adv., 2015, 5, 51067–51077 RSC.
- Z. Song, X. Dong, N. Wang, L. Zhu, Z. Luo, J. Fang and C. Xiong, Chem. Eng. J., 2017, 317, 925–934 CrossRef CAS.
- N. Wang, H. Lv, Y. Zhou, L. Zhu, Y. Hu, T. Majima and H. Tang, Environ. Sci. Technol., 2019, 53, 8302–8313 CrossRef CAS PubMed.
- Y. Shen, C. Zhu, S. Song, T. Zeng, L. Li and Z. Cai, Environ. Sci. Technol., 2019, 53, 9091–9101 CrossRef CAS PubMed.
- Z. Song, X. Dong, J. Fang, C. Xiong, N. Wang and X. Tang, J. Hazard. Mater., 2019, 377, 371–380 CrossRef CAS PubMed.
- H. Tang, Q. Xiang, M. Lei, J. Yan, L. Zhu and J. Zou, Chem. Eng. J., 2012, 184, 156–162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01246a |
| This journal is © The Royal Society of Chemistry 2024 |