
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative molecular dynamics approach guides the tailoring of glycosyltransferases to meet synthetic applications†
Peng
Zhang‡
a,
Shuaiqi
Meng‡
a,
Zhongyu
Li‡
a,
Dennis
Hirtz
b,
Lothar
Elling
 b,
Leilei
Zhu
d,
Yu
Ji
*ac and
Ulrich
Schwaneberg
*a
b,
Leilei
Zhu
d,
Yu
Ji
*ac and
Ulrich
Schwaneberg
*a
aInstitute of Biotechnology, RWTH Aachen University, Worringerweg 3, 52074 Aachen, Germany. E-mail: u.schwaneberg@biotec.rwth-aachen.de; yu.ji@biotec.rwth-aachen.de
bLaboratory for Biomaterials, Institute of Biotechnology and Helmholtz-Institute for Biomedical Engineering, RWTH Aachen University, Pauwelsstraße. 20, 52074 Aachen, Germany
cCollege of Life Science and Technology, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, 100029 Beijing, China
dTianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, 300308 Tianjin, China
First published on 15th July 2024
Abstract
The properties of natural products can be significantly influenced by glycosylation, emphasizing the key role of glycosyltransferases (GTs) in this process. The pursuit of tailored GT catalysts to meet the demands of the glycosylation industry aligns with the principles of green chemistry. However, steering GT engineering towards the desired direction often requires substantial effort. Herein, we employ a comparative molecular dynamics approach to guide the engineering of GTs to alter their catalytic performances. Through comparing the structural flexibility and site-saturation mutagenesis of two GTs BarGT-1 with narrow substrate scope and BarGT-3 with wide substrate scope, the identified substitution K321P in C-loop 5 of BarGT-1 greatly expanded the substrate scope towards diverse pharmaceutically valuable substrates, thereby the catalytic efficiencies were remarkably improved (e.g., 52- and 244-fold for 4′- and 6-hydroxyflavone, kcat/KM). Further, phylogenetic analysis demonstrated that lysine was a highly conserved residue in the GTs within BarGT-1 branch, and its key roles in regulating the substrate scope were validated through site-saturation mutagenesis in two novel GTs, BsyGT (K321F) and BgoGT (K322W). Moreover, the corresponding substitutions G325R/D in C-loop 5 of BarGT-3 also served as switches to regulate the substrate profile of BarGT-3. Finally, through the utilization of BarGT-1 and K321P, we successfully regulated the synthesis of valuable liquiritigenin glycosides with minor effort. The comparative molecular dynamics approach provides insights into the structural dynamics of GTs, optimizes enzymatic processes, and facilitates precise enzyme engineering, offering a directed synthetic paradigm to produce valuable glycosides.
Introduction
Glycosylation of natural products expands the diversity of glycosides, and fine-tunes the properties (e.g., water solubility,1 stability, and bioactivity2) of many valuable compounds.3 Glycosylated products, including phenols (e.g., acetaminophen),4 phenylpropanoids (e.g., coumarins),5 and flavonoids (e.g., hydroxyflavone),6 hold significant value in various fields, such as therapy, nutrition, and industry. Chemical synthesis has long been praised for its remarkable achievements in expanding the repertoire of available compounds, including glycosides.7,8 However, harsh reaction conditions, such as high temperatures, pressure, and the use of toxic reagents, can be challenging, resulting in mixtures of undesired by-products or requiring multiple purification steps for the desired glycosides.9 Remarkably, enzymes serve as biocatalysts aligned with the principles of green chemistry, offering a more environmentally friendly approach for glycoside synthesis by minimizing the use of toxic reagents and waste generation.10Glycosyltransferases (GTs) are the main performers of enzymatic glycosylation, transferring sugar moieties from activated sugar donors to target acceptors.11 Despite the vast diversity of GTs cataloged in the Carbohydrate-Active enZYmes (CAZy) database,12 GT1 family enzymes with GT-B fold and inverting catalytic pattern are well-known for their crucial role in a variety of glycosylation reactions for natural products, and exhibit catalytic versatility to meet the demands of glycosylation industry.13 The GT-B fold confers enzymes more flexible substrates binding pattern, showcasing greater topological plasticity.14 The inverting catalytic pattern (e.g., NDP-α-sugar to β-glycoside) results in the formation of a glycosidic bond between the sugar donor and the acceptor, accompanied by the inversion of the configuration at C1 sugar donor.15 The inherent advantages for natural product glycosylation enable GT1 family enzymes to be applied to various synthetic needs. For instance, a naturally regioselective silibinin GT UGT706F8 from Zea mays could efficiently catalyze the synthesis of silibinin 7-O-β-D-glucoside with quantitative yields.16 A permissive steroid GT UGT74AN1 from Asclepias curassavica showed promiscuity towards 53 structurally diverse compounds to form O-, N-, and S-glycosides.17 Although enzymatic catalysis has gained popularity due to its environmentally friendly manner, the mining of natural enzymes suitable for synthetic applications requires significant effort.18,19 Some of the natural enzymes may need one or several rounds of directed evolution campaign to efficiently catalyze target substrates.20,21 With the elucidation of numerous protein structures, there has been increasingly understanding of the catalytic mechanisms of enzymes.22 Consequently, opportunities have emerged for structure-guided protein strategies, including the engineering of catalytic pockets23 and substrate tunnels.24 In many cases, the advantageous mutations obtained from protein engineering influence the flexibility of the mutation site regions.25,26 This prompts us to employ comparative molecular dynamics to directly correlate the catalytic properties of enzymes with the flexibility of critical regions, and this correlation is also likely to occur among natural enzymes.27
Comparative molecular dynamics involves simulating the dynamics of protein structures to analyze their similarities and differences in behaviors.28 Whereas comparative molecular dynamics currently leans more towards being an explanatory tool,29 it holds promise as a predictive tool for identifying key residues and understanding how variations influence enzyme catalytic preferences or activities. Shende et al. employed molecular dynamics (MD) simulations to reveal the subtle regional conformational differences between two highly homologous (identity >97%) diketopiperazine dimerases NzeB and AspB, and a single interchange variant Q68I of NzeB had the equivalent catalytic function of AspB in C–N bond formation.30 This demonstrates the correlation between catalytic properties and the flexibility of pivotal regions in highly homologous enzymes. However, validating this correlation in distantly related enzymes is more crucial, as they often exhibit greater differences in catalytic properties, and the protein engineering via local dynamic changes can alter the performances of enzyme with minor efforts. Therefore, the enzyme engineering based on dynamic conformational changes holds greater promise, especially considering their potential applications to boost the discovery of tailored efficient catalysts.31
Our previous study has identified two GTs BarGT-1 and BarGT-3 with low homology (33.7%), and exhibited distinct catalytic performances.32 BarGT-1 exhibited typical substrate selectivity, while BarGT-3 possessed substrate promiscuity with broader substrate scope. The catalytic disparities of these two GTs led us to explore the potent avenue of molecular dynamics, identifying key residues regulating their catalytic performances. In this study, we employed MD simulations to analyze the molecular dynamics of the GTs BarGT-1 and BarGT-3, and identified three loop regions N-loop 1 (residues 61–67 in BarGT-1 and residues 56–73 in BarGT-3), C-loop 1 (residues 232–238 in BarGT-1 and residues 236–243 in BarGT-3) and C-loop 5 (residues 317–321 in BarGT-1 and residues 321–325 in BarGT-3, Fig. 1). The corresponding loop regions exhibited different dynamics in flexibility and conformation. The comprehensive site-saturation mutagenesis (SSM) screening identified the substitution K321P, which significantly broadened the substrate scope of BarGT-1. Phylogenetic analysis demonstrated that lysine (K) was a highly conserved residue in the BarGT-1 branch, and its key roles in regulating substrate scope of two novel GTs, BsyGT and BgoGT were validated. Moreover, the corresponding substitutions G325R/D also acted as pivotal switches to regulate the substrate profile of BarGT-3. Finally, the GTs and their beneficial variants were utilized to efficiently synthesize valuable liquiritigenin glycosides, such as neoliquiritin (liquiritigenin 7-O-β-D-glucoside, with anti-proliferative activities33), liquiritin (liquiritigenin 4′-O-β-D-glucoside, with neuroprotective,34 anticancer, and anti-inflammatory activities35), and liquiritigenin 4′,7-O-diglucoside (an excellent anti-oxidant possessing mono-glycosides activities). We successfully employed a comparative molecular dynamics approach to identify crucial regions, providing guidance for altering the catalytic properties of GTs. Our study highlights the close correlation between the regional flexibility and catalytic properties of distantly related enzymes, holding significant importance in advancing the discovery of green and efficient biocatalysts.
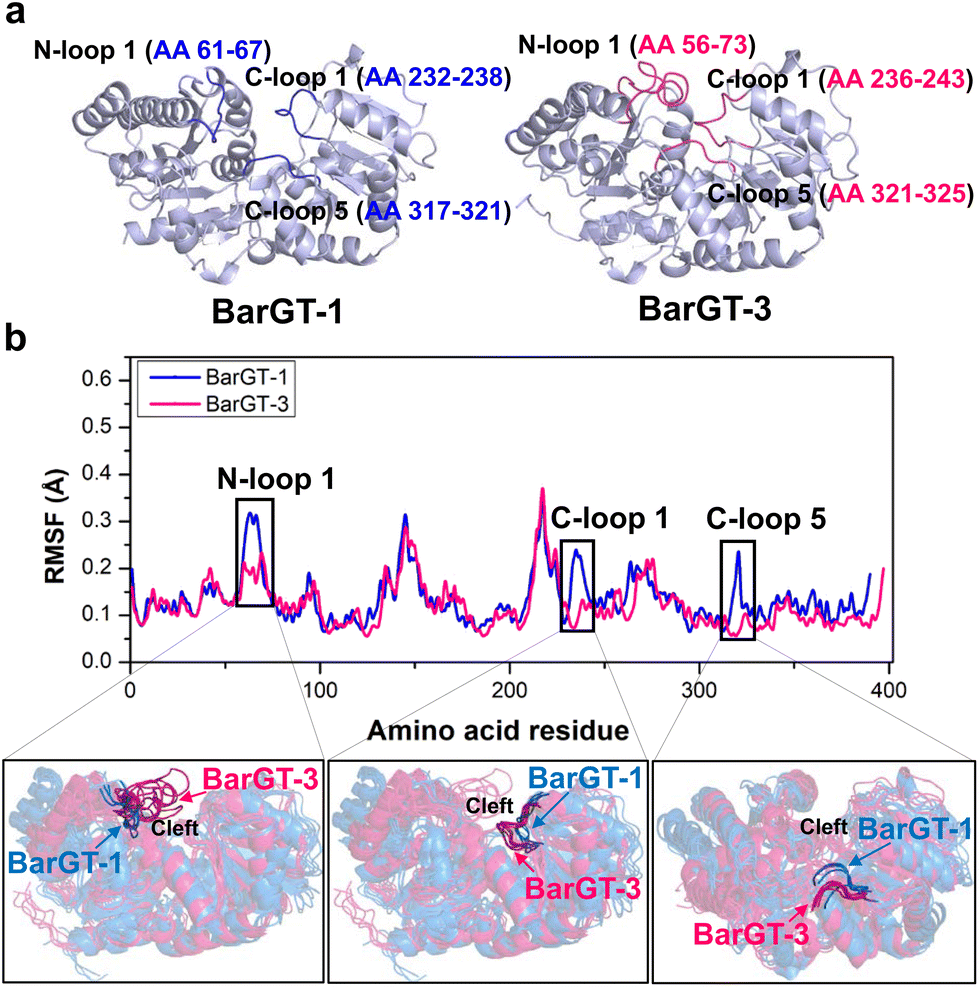 | ||
| Fig. 1 The analysis of MD simulations and conformational dynamics for GTs BarGT-1 and BarGT-3. (a) The structures of BarGT-1 and BarGT-3 are constructed by AlphaFold2. The three loop regions (N-loop 1, C-loop 1 and C-loop 5) in BarGT-1 and BarGT-3 are marked and colored in blue and pink, respectively. (b) The RMSF values for the α-carbon of each residue in BarGT-1 and BarGT-3 after three parallel 1-microsecond MD simulations. The multiple conformational comparisons (every 200 ns over the 1 μs MD simulations) of N-loop 1, C-loop 1, and C-loop 5 of BarGT-1 (in blue) and BarGT-3 (in pink) are highlighted. | ||
Results and discussion
Conformational dynamics analysis of two GTs with distinct catalytic characteristics
The pursuit of versatile catalysts as well as selective (chemo-, regio-, and stereoselectivity) catalysts for synthesizing valuable compounds has always been the goals of green chemistry.36–38 Previous studies showed that two GTs BarGT-1 and -3 exhibited distinct catalytic performances towards diverse natural products.32 GT BarGT-1 showed a typical substrate selectivity, efficiently catalyzing 1-naphthol and 7-hydroxyflavone while exhibiting low activities towards 2-naphthol and 6-hydroxyflavone. However, BarGT-3 is a versatile GT that can catalyze the 4 substrates with high activities.32 Although the GTs BarGT-1 and BarGT-3 showed only 33.7% sequence identity,32 they exhibited structural similarity with a well-aligned arrangement of secondary components (Fig. 1a and Fig. S1†). This motivated us to identify the structural elements that influence the catalytic performances of these two GTs by using molecular dynamics (MD) simulations. Hence, we conducted 1-microsecond simulations for both GTs to identify regions with different flexibility. To begin with, we calculate the RMSF for the α-carbon of each residue (three parallel MD simulations, Fig. 1b and Fig. S2†). Comparing the α-carbon RMSFs of three parallel MD simulations for BarGT-1 and BarGT-3 implied three regions that showed significant difference in flexibility (Fig. 1b): N-loop 1 (NL1, loop 1 in N-domain, residues 61–67 in BarGT-1), C-loop 1 (CL1, loop 1 in C-domain, residues 232–238 in BarGT-1) and C-loop 5 (CL5, loop 5 in C-domain, residues 317–321 in BarGT-1). These three loop regions of BarGT-1 exhibit increased flexibility compared to the three corresponding loops in BarGT-3. In addition, the overlays of representative snapshots (every 200 ns over the 1 μs MD simulations) of the N-loop 1, C-loop 1, and C-loop 5 of BarGT-1 (in blue) and BarGT-3 (in pink) showed greater conformational differences (Fig. 1b). Compared to BarGT-1, BarGT-3 possessed a longer N-loop 1, and both the N-loop 1 and C-loop 1 extended into the catalytic cleft. Conversely, compared to BarGT-3, the C-loop 5 of BarGT-1 was inclined to get closer to the catalytic cleft (Fig. 1b). Due to the differences in flexibilities and conformations of these three loop regions, we hypothesized that these three loop regions in BarGT-1 and -3 are responsible for the distinct catalytic performances.Site-saturation mutagenesis campaign and identification of critical residues
To explore the functional roles of these three loop regions in controlling the catalytic performance of GTs, site-saturation mutagenesis campaign was conducted in selected loop regions (N-loop 1, residues 61–67; C-loop 1, residues 232–238; C-loop 5, residues 317–321) of BarGT-1 (180 colonies every SSM library of each position). The UDP-glucose-recycling high-throughput screening (HTS) system was employed for screening using 2-naphthol (2) and 6-hydroxyflavone (3) as substrates.32,39 Improved variants (>1.2-fold, absorbance at 405 nm) towards substrates 2 or 3 were selected for rescreening. Two key positions (A65 and K321) were identified in N-loop 1 and C-loop 5. Variants A65L and A65P displayed higher activities (15.2-fold for A65L and 5.3-fold for A65P) towards 2 compared to WT (Fig. S3†). Remarkably, substitution K321P exhibited significant improvements towards both substrates 2 and 3 compared to the WT (19.2-fold improvement for 2 and 33.8-fold improvement for 3, Fig. S3†). Therefore, variants A65L, A65P, and K321P were further purified and used for the glycosylation reactions towards substrates 1-naphthol (1), 2, 3, 7-hydroxyflavone (4) with UDP-glucose (UDP-Glc) as sugar donor (Fig. S4†). Through HPLC-MS analysis (Fig. S5 and S6†), K321P showed 21.9-fold and 15.9-fold higher conversions towards substrates 2 and 3 compared to the WT. Meanwhile, A65L and A65P only demonstrated improvements (21.6-fold and 6.8-fold) in conversion towards 2 (Fig. 2). Nevertheless, all the variants still retained comparable conversions of WT towards substrates 1 and 4 (Fig. 2). These results indicated that the variants A65L and K321P could expand the substrate scope of BarGT-1.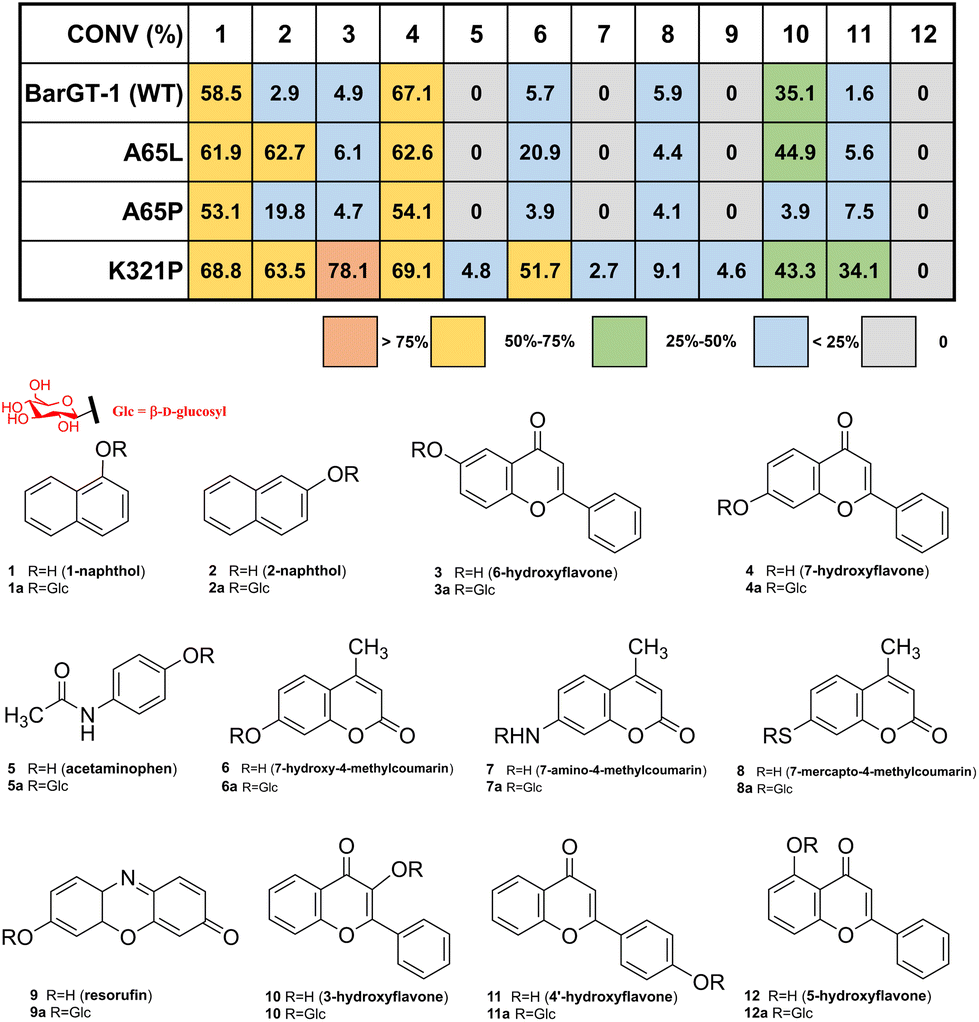 | ||
| Fig. 2 The glycosylation reactions with the purified proteins of BarGT-1 (WT) and its variants. The conversions of BarGT-1, A65L, A65P, and K321P towards a panel of 12 substrates 1–12: 1-naphthol 1, 2-naphthol 2, 6-hydroxyflavone 3, 7-hydroxyflavone 4, acetaminophen 5, 7-hydroxy-4-methylcoumarin 6, 7-amino-4-methylcoumarin 7, 7-mercapto-4-methylcoumarin 8, resorufin 9, 3-hydroxyflavone 10, 4′-hydroxyflavone 11, and 5-hydroxyflavone 12 are shown in a heatmap. A 100 μL reaction mixture contains final concentrations of 50 mM Tris–HCl buffer, 10 mM MgCl2, 2 mM UDP-Glc, 1 mM substrate and 200 μg mL−1 each protein. The corresponding glycosylated products are 1a–11a: 1-naphthol O-β-D-glucoside 1a, 2-naphthol O-β-D-glucoside 2a, 6-hydroxyflavone O-β-D-glucoside 3a, 7-hydroxyflavone O-β-D-glucoside 4a acetaminophen O-β-D-glucoside 5a, 7-hydroxy-4-methylcoumarin O-β-D-glucoside 6a, 7-amino-4-methylcoumarin N-β-D-glucoside 7a, 7-mercapto-4-methylcoumarin S-β-D-glucoside 8a, resorufin O-β-D-glucoside 9a, 3-hydroxyflavone O-β-D-glucoside 10a, 4′-hydroxyflavone O-β-D-glucoside 11a. The structures of a panel of 12 substrates and their corresponding glycosylated products are shown below. | ||
To further explore the substrate scope of BarGT-1 (WT) and variants A65L/P and K321P, a previous spectrum with other 8 substrates was also used to evaluate the catalytic performance of WT, A65L, A65P, and K321P (Fig. 2 and Fig. S6†), and the structures of glycosylated products 1a–9a were already elucidated.32 A65L only showed improved conversion (3.7-fold) towards substrate 7-hydroxy-4-methylcoumarin (6). Notably, K321P showed significantly improved conversions towards a wide range of substrates. Especially, K321P could catalyze the types of substrates that the wild type (BarGT-1) could not (Fig. 2), such as acetaminophen (5), 7-amino-4-methylcoumarin (7), and resorufin (9). Furthermore, double-site variants A65L/K321P and A65P/K321P were generated, but the activities of double-site variants were severely damaged (Fig. S7†). Therefore, we focused on K321P and the kinetic parameters towards a panel of typical 7 substrates with high conversions were evaluated (Table 1 and Fig. S8 and S9†). WT and K321P displayed distinct catalytic efficiencies towards several substrates. In detail, compared to WT, K321P exhibited 43-, 244-, and 52-fold improvements in catalytic efficiencies (kcat/KM) for substrates 2, 3, and 4′-hydroxyflavone (11). Additionally, K321P also showed comparable catalytic efficiencies towards WT in case of 4 and 10 with mild improvements in the catalytic efficiencies (up to 1.4-fold). Especially, K321P exhibited excellent kcat values towards the same scaffold of flavonoids with different hydroxyl positions, such as 3 (6.4 × 103 min−1), 4 (4.7 × 103 min−1), 10 (1.6 × 103 min−1) and 11 (1.8 × 103 min−1). In addition, 1H and 13C NMR spectra were conducted to confirm the glycosylated products 10a and 11a, and the glycosylation at hydroxy group of C-1 was further determined by the key HMBC correlations from H-1′ to C-1. It was indicated that K321P possessed promising performance for the industrial production of glycoside compounds. Although neither K321P nor WT displayed any activity towards 5-hydroxyflavone (12, Fig. 2), K321P could significantly expand the substrate scope with diverse skeletons of substrates, which was of great significance for the expansion of biological activity of natural glycosylated products with an environmentally friendly manner.
| Substrates | Kinetic parameters | |||||
|---|---|---|---|---|---|---|
| BarGT-1_WT | BarGT-1_K321P | |||||
| K M (μM) | k cat (min−1) | k cat/KM (min−1 μM−1) | K M (μM) | k cat (min−1) | k cat/KM (min−1 μM−1) | |
| 1-Naphthol (1) | 66.5 ± 8.1 | 2.2 × 103 ± 122.1 | 33.6 | 84.4 ± 3.7 | 2.8 × 103 ± 109.2 | 33.1 |
| 2-Naphthol (2) | 4.5 × 102 ± 50.7 | 3.1 × 102 ± 12.1 | 0.7 | 1.2 × 102 ± 15.1 | 3.6 × 103 ± 125.8 | 30 |
| 6-Hydroxyflavone (3) | 5.5 × 102 ± 55.6 | 1.5 × 102 ± 5.6 | 0.3 | 87.6 ± 10.6 | 6.4 × 103 ± 230.8 | 73.1 |
| 7-Hydroxyflavone (4) | 65.1 ± 11.7 | 4.1 × 103 ± 279.5 | 63 | 58.3 ± 4.7 | 4.7 × 103 ± 134.8 | 80.6 |
| 7-Hydroxy-4-methylcoumarin (6) | 7.6 × 102 ± 67.2 | 87.9 ± 3.4 | 0.1 | 5.5 × 102 ± 65 | 8.1 × 102 ± 36.6 | 1.5 |
| 3-Hydroxyflavone (10) | 41.3 ± 5.4 | 1.4 × 103 ± 69.6 | 33.9 | 33.8 ± 0.6 | 1.6 × 103 ± 5.2 | 47.3 |
| 4′-Hydroxyflavone (11) | 3.9 × 102 ± 38.3 | 2.1 × 102 ± 5.8 | 0.5 | 69.8 ± 8.3 | 1.8 × 103 ± 60.7 | 25.8 |
Molecular dynamic understanding of BarGT-1 and K321 for the glycosylation pattern
Molecular dynamics (MD) simulations were conducted to gain a molecular understanding on the substrate-switch role of the substitution K321P. By comparing the α-carbon RMSFs of BarGT-1 and K321P (Fig. 3a and Fig. S10†), we observed the reduced flexibility of C-loop 5 in K321P, which is consistent with the low flexibility state of the corresponding region in BarGT-3 (Fig. 1b and 3a). Furthermore, by performing clustering analysis of K321P and WT, the dominant conformations (DC) of K321P and WT (84.5% for WT and 88.4% for K321P) were obtained (Fig. 3b and c). The structural inspection showed that K321 is in the center of the cleft entrance formed by two Rossmann-like binding domains (N- and C-domain), whereas the P321 is relatively distant from the cleft entrance (Fig. 3b and c). The distinct catalytic performances of WT and K321P towards substrates 2 and 3 motivated us to analyze the binding free energies of these two substrates towards WT and K321P. K321P significantly improves the binding affinity of substrate 2 and 3 towards BarGT-1, evidenced by the decreased binding free energies (2, 1.2 kcal mol−1 of WT and −1.8 kcal mol−1 of K321P; 3, 2.7 kcal mol−1 of WT and −0.01 kcal mol−1 of K321P, Fig. 3d and e), and also by the binding free energies of the whole protein BarGT-1 and K321P towards substrates 2 and 3 (2, −65.7 kcal mol−1 of WT and −70.6 kcal mol−1 of K321P; 3, −99.8 kcal mol−1 of WT and −109.9 kcal mol−1 of K321P, Fig. 3d and e). The observed reduction in flexibility around mutation sites, along with the corresponding decrease in binding free energy implied a stable enzyme–substrate complex, thereby facilitating a more favorable microenvironment for catalysis.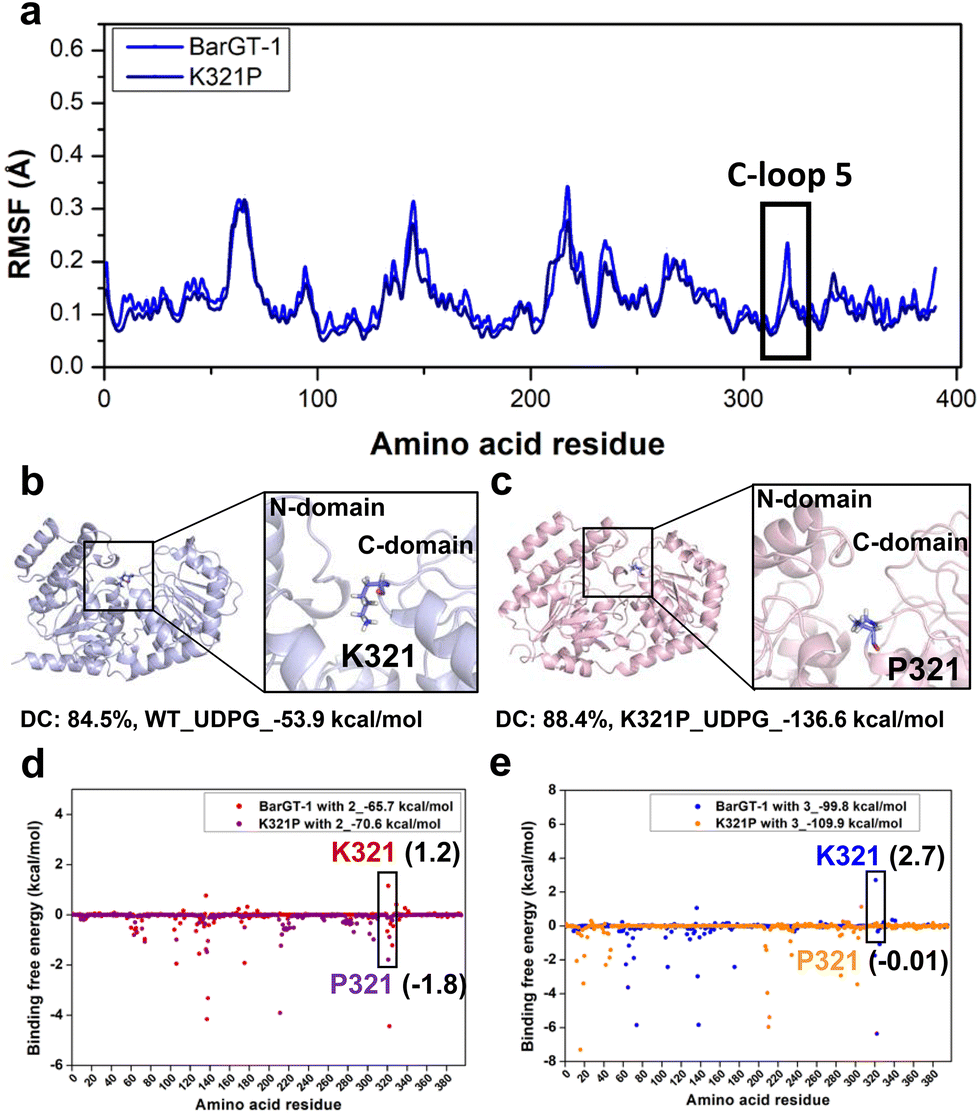 | ||
| Fig. 3 The molecular understanding of the BarGT-1 (WT) and its variant K321P for 2-naphthol (2) and 6-hydroxyflavone (3) glycosylation. (a) The RMSF values for the α-carbon of each residue in BarGT-1 and variant K321P after three parallel 1-microsecond MD simulations. (b and c) Comparison of the locations of 321th site in the dominant conformations (DC) of WT and K321P. (d and e) The binding free energies (MM/PBSA) of 321th site and whole proteins of WT and K321P towards 2 and 3. | ||
To explore the effect of variant K321P on the binding between sugar donor (UDP-Glc) and BarGT-1, we calculated the binding free energies of WT and K321P towards UDP-Glc. The binding free energy of K321P was significantly decreased compared to the WT (−53.9 kcal mol−1 of WT and −136.6 kcal mol−1 of K321P, Fig. 3b and c). Additionally, the kinetic parameters of WT and K321P towards UDP-Glc were determined, and the catalytic efficiencies of K321P increases by 23-fold, and the KM of K321P also decreased by 3.6-fold compared to that of WT using 3 as acceptor (Table 2 and Fig. S11†). These results highlighted the decrease in local flexibility caused by K321P, coupled with the reduction in binding free energy, facilitated the catalysis of both acceptor and sugar donor.
| Donor | Enzyme | K M (μM) | k cat (min−1) | k cat/KM (min−1 μM−1) |
|---|---|---|---|---|
| UDP-Glc | BarGT-1 | 4.7 × 102 ± 54.6 | 1 × 102 ± 4.5 | 0.2 |
| K321P | 1.3 × 102 ± 23.8 | 6 × 102 ± 37.3 | 4.6 |
Furthermore, the potential interactions between BarGT-1, UDP-Glc and substrate (3) were investigated by docking studies. 3 was docked to BarGT-1 and K321P to generate a binary complex and the dominant conformation (clustering with a cut-off value of 0.2 nm) was obtained after 200 ns MD simulation. The dominant complexes were selected for further molecular docking with UDP-Glc, and MD simulations (1 μs) for ternary complexes of BarGT-1 and K321P were performed. BarGT-1 is a representative of inverting GTs with a GT-B fold, which are known to employ a direct displacement SN2-like mechanism.15 During the glycosylation process, the catalytic base abstracts a proton from hydroxyl group of an acceptor (deprotonation), facilitating nucleophilic attack at the anomeric carbon (C1) of the sugar donor. Structural studies have revealed that the catalytic bases are typically situated near the nucleophilic oxygen of the acceptor.15 By multiple sequence alignment (MSA) analysis with typical GT-B fold GTs, H14 was identified as the key catalytic residue of BarGT-1 (Fig. S12†). In the dominant ternary complex of K321P, a closer distance between nucleophilic oxygen of 3 and H14 was observed compared to that of WT (2.9 Å vs. 5.5 Å, Fig. 4a and b). In addition, the distance between C1 of UDP-Glc and hydroxyl group of 3 was also closer in the dominant ternary complex of K321P (3.5 Å vs. 6.5 Å, Fig. 4a and b). To further investigate the dynamic interactions between the catalytic residue, acceptor, and sugar donor, distance changes between the nucleophilic oxygen of 3 and catalytic residue H14, as well as between the nucleophilic oxygen of 3 and the C1 of UDP-Glc during the last 200 ns of MD simulation were also analyzed. Fig. 4c showed that, in the complex of K321P, the nucleophilic oxygen of 3 exhibited a significant tendency to approach the catalytic residue H14 compared to WT. It is reported that a histidine (His) residue is crucial for initiating GT-mediated glycosylation,15 and His would act as an effective general base to deprotonate the OH-group of the acceptor within a permissible distance (typically within 3.5 Å).40 Therefore, frequency for distances within 3.5 Å between the hydroxyl group of 3 and H14 was calculated, and the results revealed that the hydroxyl group of 3 in the K321P complex exhibited a significantly higher probability of being positioned near the catalytic residue for deprotonation compared to the hydroxyl group of 3 in the WT complex (444/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in K321P vs. 5/10000 for WT, Fig. 4c). Further, the distance between C1 of UDP-Glc and hydroxyl group (–OH) of 3 was also calculated to evaluate the probability of nucleophilic attack at the anomeric carbon (C1) of the sugar donor to form the glycosidic bond.41 Throughout the last 200 ns of MD simulation, it was consistently observed that the C1-OH distance between UDP-Glc and 3 in the K321P complex (around 3.5 Å) maintained closer proximity in comparison to the C1-OH distance in the WT complex (around 6 Å) (Fig. 4d), which indicates that the K321P complex encountered higher frequency of nucleophilic attacks and faster rate of glycosidic bond formation.
000 in K321P vs. 5/10000 for WT, Fig. 4c). Further, the distance between C1 of UDP-Glc and hydroxyl group (–OH) of 3 was also calculated to evaluate the probability of nucleophilic attack at the anomeric carbon (C1) of the sugar donor to form the glycosidic bond.41 Throughout the last 200 ns of MD simulation, it was consistently observed that the C1-OH distance between UDP-Glc and 3 in the K321P complex (around 3.5 Å) maintained closer proximity in comparison to the C1-OH distance in the WT complex (around 6 Å) (Fig. 4d), which indicates that the K321P complex encountered higher frequency of nucleophilic attacks and faster rate of glycosidic bond formation.
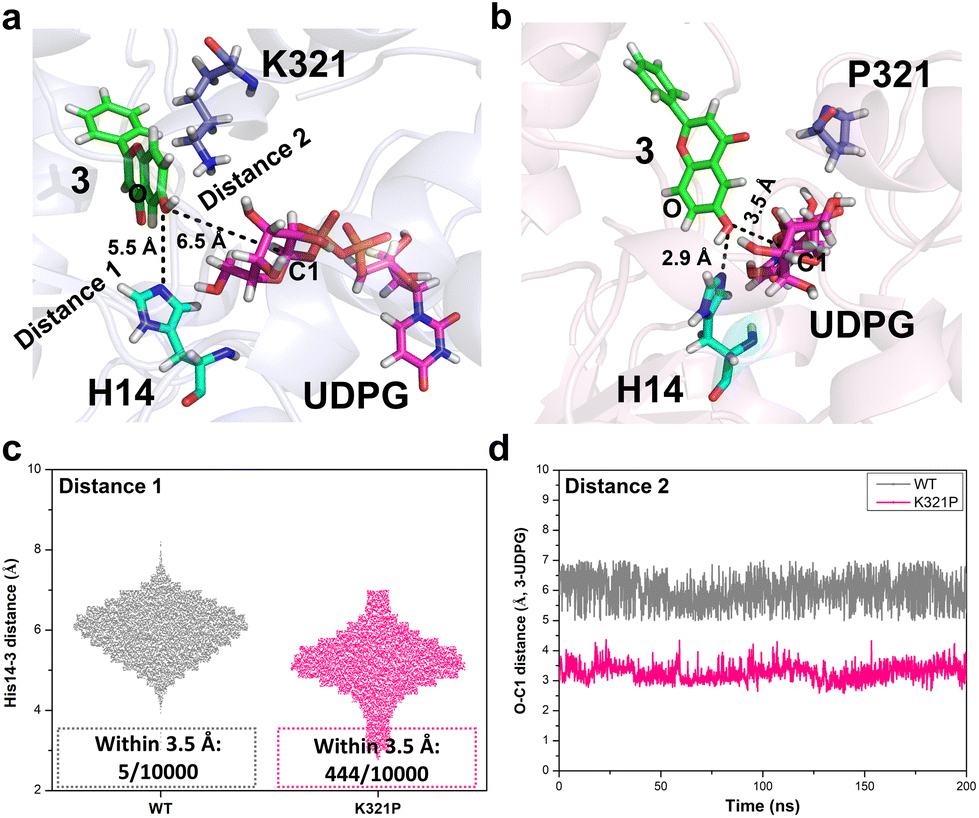 | ||
| Fig. 4 Mechanism analysis of the BarGT-1 (WT) and its variant K321P for 6-hydroxyflavone (3) glycosylation. (a and b) The distances of catalytic residue (H14), acceptor (3) and sugar donor (UDP-Glc) in the dominant conformations of WT and K321P, distance 1, from the catalytic residue H14 to the hydroxyl group of 3; distance 2, from C1 of UDP-Glc to hydroxyl group (–OH) of 3. (c and d) The analysis of the distances for catalytic residue (H14), 3 and UDP-Glc during the last 200 ns MD simulations, distance 1, from the catalytic residue H14 to the hydroxyl group of 3; distance 2, from C1 of UDP-Glc to hydroxyl group of 3. | ||
Phylogenetic insights into the general functions of lysine (K) in BarGT-1 branch
BarGT-1 is situated within the previous reported nBGT-1 branch.32 The key roles of K321 in regulating the selective and promiscuous catalysis of BarGT-1 sparked our interest in this nBGT-1 branch. The phylogenetic analysis of the nBGT-1 branch revealed significant divergent evolution of the enzymes of this branch (Fig. 5a). Generally, GTs with higher homology (higher sequence identity) are more likely to exhibit similar catalytic activities.14 Hence, we analyzed the sequence identity of all GTs in the nBGT-1 branch and identified two novel enzymes, BsyGT and BgoGT, which are the top two GTs with the lowest sequence identities (56.2% and 59.8%, respectively) compared to BarGT-1. Similarly, the sequence identity between BsyGT and BgoGT was also only 62.8% (Fig. 5a). It was indicated that these three enzymes could represent the general characteristics of GTs in the nBGT-1 branch. Interestingly, Multiple sequence alignments (MSA) also showed that the residue of lysine exhibited 100% conservation among all GTs in the nBGT-1 branch (Fig. 5b). We synthesized and characterized the GTs BsyGT and BgoGT. The wild-type BsyGT and BgoGT only showed moderate activities toward the substrate profile 1–12 (Fig. 5c). To further explore the general functions of lysine in BarGT-1 branch, site-saturation mutagenesis libraries (SSM) of lysine of BsyGT and BgoGT were generated and screened (180 colonies for each SSM library) with 3 as substrate. The lysine variants with tryptophan, tyrosine and phenylalanine exhibited significantly enhanced catalytic activities, and the K321F and K322W were the most advantageous variants in BsyGT and BgoGT, respectively (Fig. S13†). The purified variants K321F and K322W also exhibited significantly increased conversion towards the whole substrate profile compared to the WTs (Fig. 5c and Fig. S4†). Especially, the variants K321F and K322W were effective in enabling the wild types to exhibit catalytic activity that they previously lacked, particularly in catalyzing substrates 7 and 9 with significantly increased activity (Fig. 5c).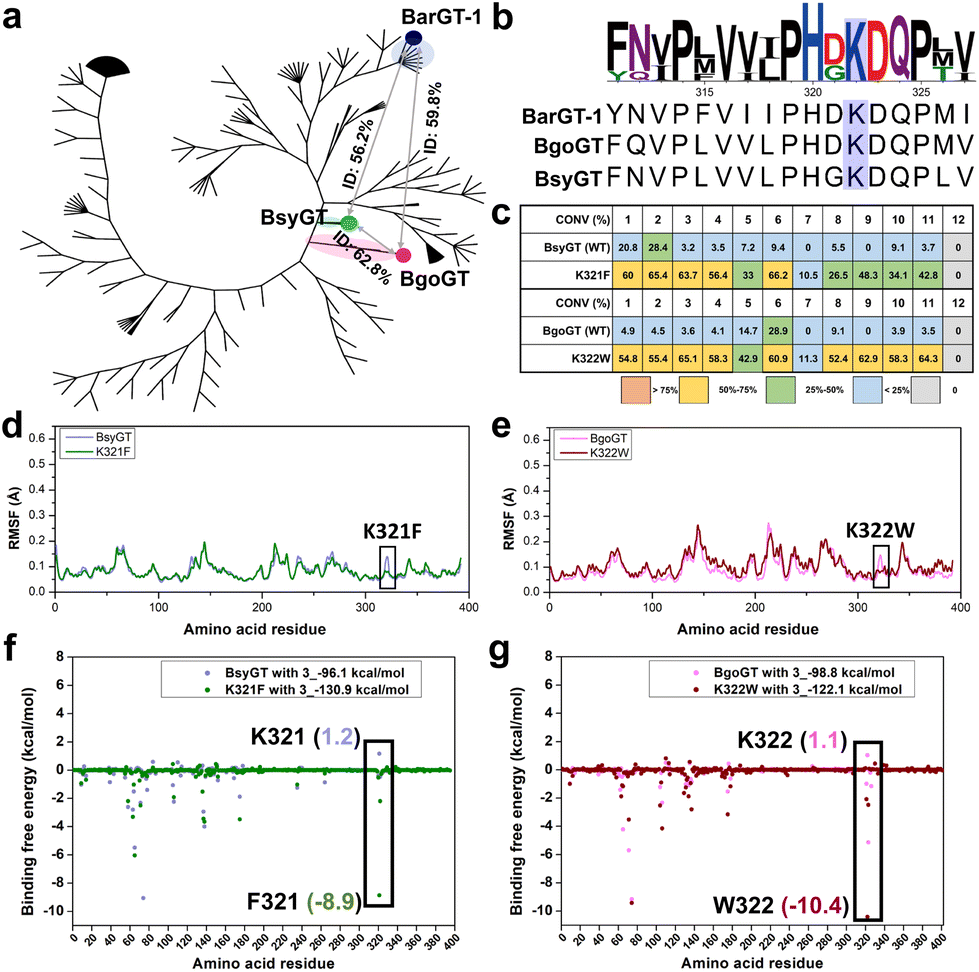 | ||
| Fig. 5 Phylogenetic tree, substrate profile and molecular dynamics (MD) simulations analysis of BsyGT, BgoGT and their lysine (K) variants. (a) Phylogenetic analysis of GTs in the nBGT1 branch. The positions of BarGT-1, BsyGT and BgoGT in the phylogenetic tree and sequence identities of these three enzymes are labeled. (b) Multiple sequence alignment for BarGT-1, BsyGT and BgoGT and the conservation analysis of lysine site in GTs of nBGT1 branch. (c) The substrate profile of BsyGT, BgoGT and their corresponding most advantageous lysine (K) variants K321F and K322W. (d and e) The RMSF values for the α-carbon of each residue in BsyGT, BgoGT and their corresponding most advantageous lysine (K) variants K321F and K322W after three parallel 400 ns MD simulations. (f and g) The binding free energy of each residue in BsyGT and BgoGT with their most advantageous lysine variant (K321F of BsyGT, K322W of BgoGT) toward 6-hydroxyflavone (3), the values of the binding free energies of K321/F321 of BsyGT and K322/W322 of BgoGT are marked. | ||
MD simulations were also conducted for BsyGT and BgoGT with the corresponding advantageous variants K321F and K322W. The RMSF values indicated the increases in flexibility of loop regions in variants K321F and K322W compared to WTs (Fig. 5d, e and Fig. S14†). Correspondingly, the binding free energy analysis showed that both K321 in BsyGT and K322 in BgoGT exhibited highest binding free energies among all the residues towards substrate 3, whereas the binding free energies of both K321F in BsyGT and K322W in BgoGT were significantly decreased (1.2 kcal mol−1 of BsyGT and −8.9 kcal mol−1 of K321F; 1.1 kcal mol−1 of BgoGT and −10.4 kcal mol−1 of K322W, Fig. 5f and g). This indicates that the decreases in loop flexibility and binding free energy are conductive to augmenting the promiscuity of GTs in the nBGT1 branch.
G325R/D switch the substrate profile of BarGT-3
To further characterize the role of the corresponding position of BarGT-3 in regulating substrate profile, the position G325 was identified, because both K321 and G325 are close to the corresponding typical and highly conserved Asp/Glu-Gln motif (D322-Q323 in BarGT-1 and D326-Q327 in BarGT-3, Fig. S12†).1,14 To verify the key roles of G325 in the promiscuous BarGT-3, a SSM library was constructed for G325 and the UDP-Glc-recycling HTS system was employed for screening for four substrates 1, 2, 3, and 4. Most BarGT-3 variants retained activities comparable to the WT of BarGT-3. However, the substitutions G325D/R showed different substrate preferences (Fig. S15†). Further glycosylation reactions with purified BarGT-3 and variants G325D/R revealed that variant G325R still maintained catalytic activity towards substrates 1 and 2 but the activity towards 3 and 4 was reduced, while variant G325D showed catalytic preferences towards 3 and 4 (Fig. 6a and Fig. S4†). Notably, WT and its variants G325D/R also exhibited different catalytic performances towards more scaffolds of substrates (additional 8 substrates). For example, the variant G325R exhibited preferences towards 1, 2, and 5 with high activities, whereas the substitution G325D showed preferences towards 3, 4, and 7. Moreover, both G325R and G325D displayed high activity towards 8 compared to WT. In general, the variants G325R/D narrowed the substrate profile and enhanced the specificity of BarGT-3. To further explore the relationship between the flexibility of C-loop 5 and promiscuity of BarGT-3, 1-microsecond MD simulations were conducted for BarGT-3 and its variants G325R/D. The RMSF values indicated the increases in flexibility of C-loop 5 in variants G325R/D compared to WT (Fig. 6b and Fig. S16†).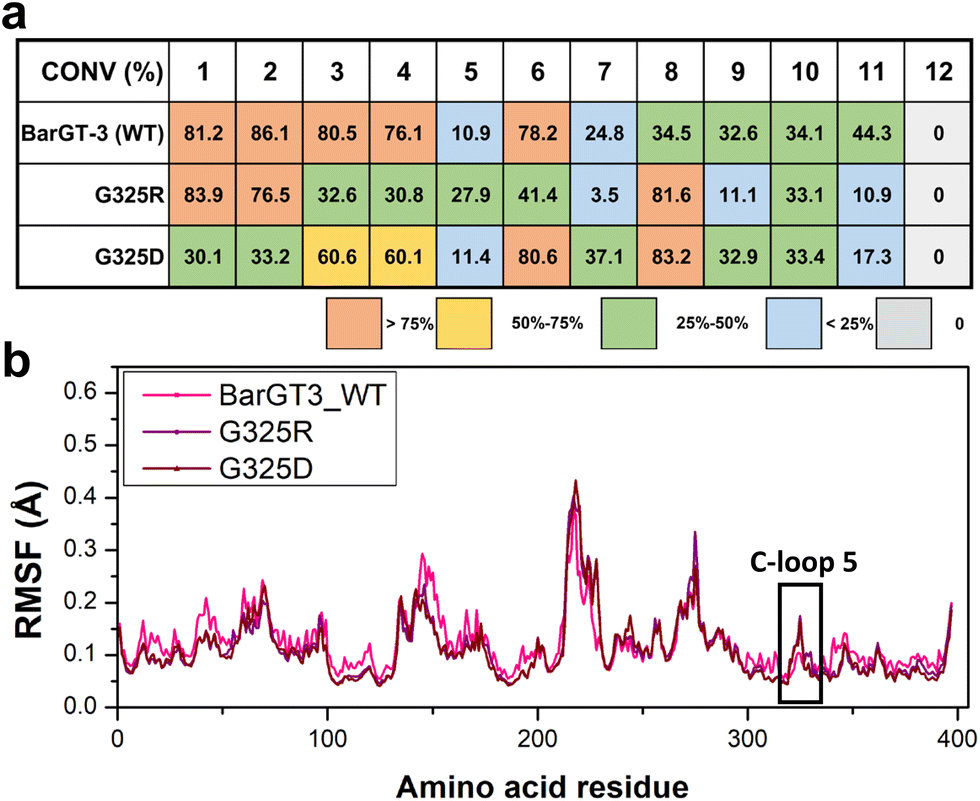 | ||
| Fig. 6 The catalytic characteristics and molecular understanding of BarGT-3 (WT) and its variants G325R/D. (a) The glycosylation conversions of BarGT-3 (WT) and its variants G325R/D towards a panel of 12 substrates. (b) The comparison of RMSF values for the α-carbon of each residue in BarGT-3 and its variants G325R/D. | ||
The charged residues (R/D) restrict the expansion of the substrate scope of BarGT-3 by introducing electrostatic interactions that increase the perturbations of C-loop 5. These interactions result in greater loop flexibility, thereby enhancing the substrate selectivity of BarGT-3.
K321P expanded the regioselectivity of BarGT-1 in liquiritigenin glycosylation
Efforts towards achieving both non-regioselective and regioselective catalysis with minor efforts are highly sought after.6 While the substitution K321P significantly expanded the substrate scope of BarGT-1, to further explore the key roles of K321P in regioselectivity of BarGT-1, the liquiritigenin (13) was selected as candidate substrate due to its dihydroxyl groups and high synthetic values of its glycosides (Fig. 7a). Neoliquiritin (13a), liquiritin (13b), and liquiritigenin 4′,7-O-β-D-diglucoside (13c) are important pharmaceutical compounds with significant therapeutic efficacies.33,34 We plotted the conversion curves of WT and K321P and observed that both WT and K321P could produce three glycoside products, 13a, 13b, and 13c, albeit in different proportions. The WT predominantly catalyzed the formation of 13a, with a conversion of 83.1% after 2 h (Fig. 6b), while the conversions of 13b and 13c were both below 15% (Fig. 6c and d). Clearly, the WT demonstrated significant regioselectivity towards the 7-OH of 13. In contrast, the K321P exhibited a non-regioselective catalytic pattern for glycosylation of 13. The highest conversions of 13a and 13b by K321P reached 54.6% and 32.3%, respectively (Fig. 6b and c). Subsequently, 13a and 13b were further converted to produce the diglycoside 13c, with a conversion of 60.8% after 10 h (Fig. 6d). Additionally, the conversions from monoglycosides (13a and 13b) to diglycoside (13c) were also analyzed. K321P showed superior activity over WT in producing 13c from monoglycosides 13a and 13b, with conversions of 49.5% and 80.1%, respectively (Fig. 6e and f). 1H and 13C NMR spectra were also conducted to confirm the glycosylated products 13a, 13b and 13c, and the glycosylation at hydroxy group of C-1 of 13a and 13b was further determined by the key HMBC correlations from H-1′ to C-1, respectively. The catalytic preference of BarGT-1 and its variant K321P towards 13 are quite similar to that of GTs GuGT10 and GuGT11 in Glycyrrhiza uralensis,18 while the control of regioselectivity was achieved by a single mutation (K321P) with minor effort in this study. Overall, it was indicated that K321P significantly weakened the regioselectivity of WT towards the 7-OH of 13 and enhanced the non-regioselectivity towards both the 7-OH and 4′-OH of 13. The utilization of regioselectivity and non-regioselectivity of GTs is crucial for enzymatic synthesis to regulate the yield and proportions of glycosides, and BarGT-1 and K321P can serve as efficient biocatalysts for the synthesis of liquiritigenin glycosides and other related valuable flavonoid glycosides.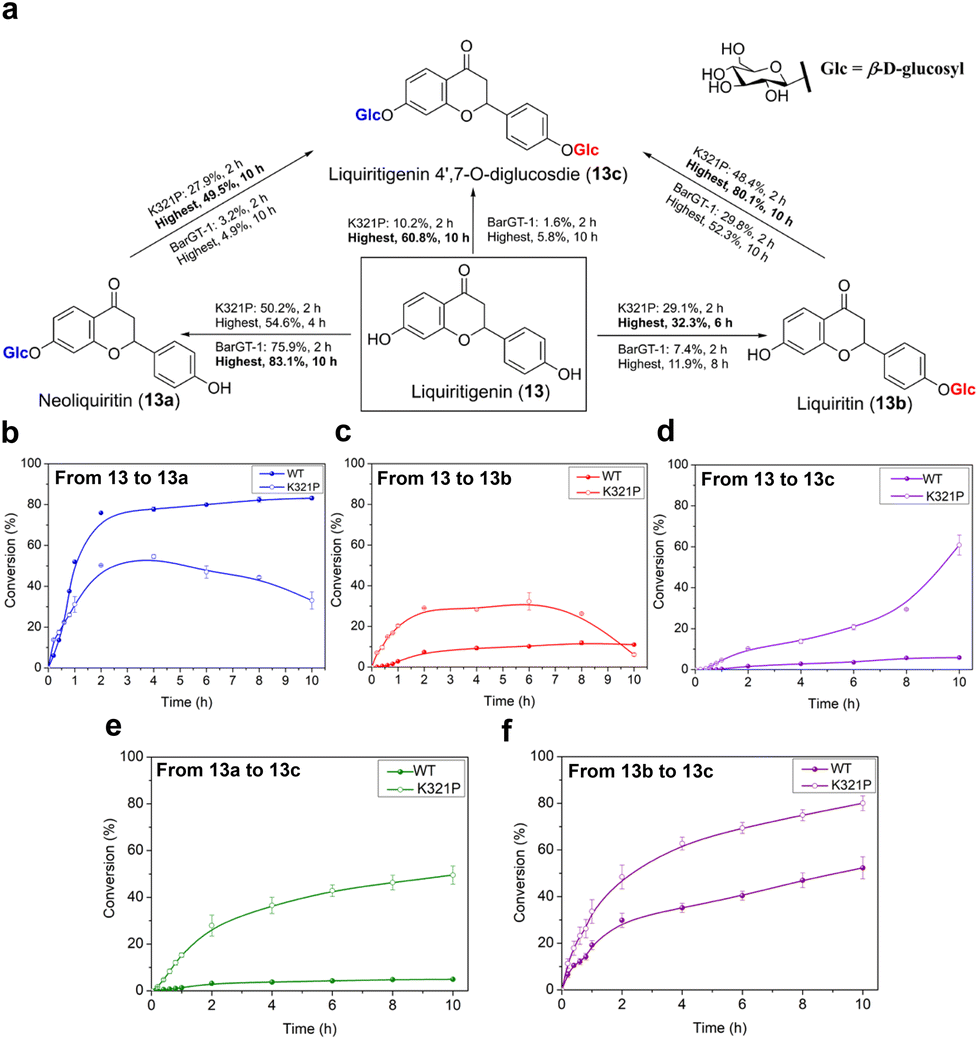 | ||
| Fig. 7 Catalytic characteristics of BarGT-1 (WT) and K321 variant towards liquiritigenin (13) and its glycosides neoliquiritin (13a), liquiritin (13b), and liquiritigenin 4′,7-O-β-D-diglucoside (13c). (a) The scheme of BarGT-1 and K321P convert 13 to 13a, 13b and 13c, and convert 13a and 13b to 13c. (b) The conversion curves of WT and K321P towards 13 to produce 13a. (c) The conversion curves of WT and K321P towards 13 to produce 13b. (d) The conversion curves of WT and K321P towards 13 to produce 13c. (e) The conversion curves of WT and K321P towards 13a to produce 13c. (f) The conversion curves of WT and K321P towards 13b to produce 13c. | ||
Conclusions
In this study, a comparative molecular dynamics approach was used to identify a key residue (lysine) in regulating selective and promiscuous catalysis of two GTs BarGT-1 and -3, and revealed its universal roles in a branch of GTs. The substitution K321P of BarGT-1, together with K321F of BsyGT and K322W of BgoGT significantly expanded the substrate scope of wild types towards diverse substrates (naphthols, coumarins, and flavonoids, etc.), and the kcat value could be improved towards a variety of pharmaceutically important synthons (e.g., flavonoids, up to 6.4 × 103 min−1). Similarly, the substitution G325R/D switched BarGT-3 from a broad to a narrow substrate profile thereby also enhancing catalytic efficiencies for specific scaffolds. The shift of enzyme performance enables more controlled reactions, minimizes by-product generation, and promotes sustainable industrial development. Moreover, comparative molecular dynamics simulations allow for the efficient screening of key residues with minor effort to alter the catalytic characteristics of GTs, which is of great significance for the selective catalysis and promiscuous catalysis in glycosylation applications. Overall, the integration molecular dynamics simulations and site-saturation mutagenesis of identified key positions in regulating the substrate profile of GTs proved to be an efficient protein engineering strategy to empower GTs and other enzymes for enzymatic synthesis of valuable compounds.Experimental
Please see the Experimental section of ESI.†Author contributions
Peng Zhang: conceptualisation, data curation, formal analysis, investigation, methodology, validation, visualisation, funding acquisition, and writing of the original draft. Shuaiqi Meng: data curation, formal analysis, resources, and software. Zhongyu Li: data curation, resources, and software. Dennis Hirtz: methodology, discussion, resources. Lothar Elling: methodology, discussion, resources. Leilei Zhu: discussion, resources. Yu Ji: conceptualisation, data curation, validation, discussion, methodology, supervision, and writing and review of the original draft. Ulrich Schwaneberg: conceptualisation, data curation, validation, discussion, methodology, supervision, and writing and review of the original draft.Data availability
The data supporting this article have been included as part of the ESI.† This study was carried out using publicly available data from [Carbohydrate-Active enZYmes (CAZy) database] at [https://www.cazy.org/].Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Sino-German (China Scholarship Council, No. 202006220278 and Deutscher Akademischer Austauschdienst, No. 91810743) Postdoc Scholarship Program (57575640) for Dr P. Zhang.References
- P. Zhang, L. Zhang, X. Jiang, X. T. Diao, S. Li, D. D. Li, Z. Zhang, J. Fang, Y. J. Tang, D. L. Wu, C. Wu and Y. Z. Li, Commun. Biol., 2022, 5, 100 CrossRef PubMed.
- P. Zhang, L. Zhang, X. J. Yue, Y. J. Tang, C. Wu and Y. Z. Li, Appl. Microbiol. Biotechnol., 2020, 104, 9125–9134 CrossRef PubMed.
- N. C. Veitch and R. J. Grayer, Nat. Prod. Rep., 2011, 28, 1626–1695 RSC.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef.
- K. Robe, E. Izquierdo, F. Vignols, H. Rouached and C. Dubos, Trends Plant Sci., 2021, 26, 248–259 CrossRef PubMed.
- J. Xiao, T. S. Muzashvili and M. I. Georgiev, Biotechnol. Adv., 2014, 32, 1145–1156 CrossRef PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef PubMed.
- Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331–1355 RSC.
- H. M. Christensen, S. Oscarson and H. H. Jensen, Carbohydr. Res., 2015, 408, 51–95 CrossRef.
- M. M. Palcic, Curr. Opin. Chem. Biol., 2011, 15, 226–233 CrossRef PubMed.
- L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, in Annu Rev Biochem, Annual Reviews, Palo Alto, 2008, vol. 77, pp. 521–555 Search PubMed.
- V. Lombard, H. G. Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef.
- B. He, X. Bai, Y. Tan, W. Xie, Y. Feng and G.-Y. Yang, Synth. Syst. Biotechnol., 2022, 7, 602–620 CrossRef PubMed.
- P. Zhang, Z. Zhang, L. Zhang, J. Wang and C. Wu, Comput. Struct. Biotechnol. J., 2020, 18, 1383–1390 CrossRef.
- D. M. Liang, J. H. Liu, H. Wu, B. B. Wang, H. J. Zhu and J. J. Qiao, Chem. Soc. Rev., 2015, 44, 8350–8374 RSC.
- G. N. Bidart, N. Putkaradze, F. Fredslund, C. Kjeldsen, A. G. Ruiz, J. Ø. Duus, D. Teze and D. H. Welner, ACS Sustainable Chem. Eng., 2022, 10, 5078–5083 CrossRef.
- C. Wen, W. Huang, X.-L. Zhu, X.-S. Li, F. Zhang and R.-W. Jiang, Org. Lett., 2018, 20, 534–537 CrossRef PubMed.
- K. Chen, Z.-m. Hu, W. Song, Z.-l. Wang, J.-b. He, X.-m. Shi, Q.-h. Cui, X. Qiao and M. Ye, ACS Synth. Biol., 2019, 8, 1858–1866 CrossRef.
- L.-J. Zhang, D.-G. Wang, P. Zhang, C. Wu and Y.-Z. Li, ACS Synth. Biol., 2022, 11, 812–819 CrossRef.
- Y. Ji, S. Islam, A. M. Mertens, D. F. Sauer, G. V. Dhoke, F. Jakob and U. Schwaneberg, Appl. Microbiol. Biotechnol., 2019, 103, 3761–3771 CrossRef.
- J. Li, J. Yang, S. Mu, N. Shang, C. Liu, Y. Zhu, Y. Cai, P. Liu, J. Lin and W. Liu, ACS Catal., 2020, 10, 3629–3639 CrossRef.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- S. Meng, Y. Ji, L. Zhu, G. V. Dhoke, M. D. Davari and U. Schwaneberg, Biotechnol. Adv., 2022, 61, 108051 CrossRef CAS PubMed.
- K. Prakinee, A. Phintha, S. Visitsatthawong, N. Lawan, J. Sucharitakul, C. Kantiwiriyawanitch, J. Damborsky, P. Chitnumsub, K.-H. Van Pee and P. Chaiyen, Nat. Catal., 2022, 5, 534–544 CrossRef CAS.
- B. M. Nestl and B. Hauer, Journal, 2014, 4, 3201–3211 CAS.
- J. P. Richard, J. Am. Chem. Soc., 2019, 141, 3320–3331 CrossRef CAS.
- C. S. Karamitros, K. Murray, B. Winemiller, C. Lamb, E. M. Stone, S. D'Arcy, K. A. Johnson and G. Georgiou, Proc. Natl. Acad. Sci., 2022, 119, e2118979119 CrossRef CAS.
- Y.-w. Dong, M.-l. Liao, X.-l. Meng and G. N. Somero, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1274–1279 CrossRef CAS PubMed.
- H. Yu and P. A. Dalby, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E12192–E12200 CAS.
- V. V. Shende, N. R. Harris, J. N. Sanders, S. A. Newmister, Y. Khatri, M. Movassaghi, K. N. Houk and D. H. Sherman, Angew. Chem., Int. Ed., 2023, 62, e202210254 CrossRef CAS.
- R. M. Crean, J. M. Gardner and S. C. Kamerlin, J. Am. Chem. Soc., 2020, 142, 11324–11342 CrossRef CAS PubMed.
- P. Zhang, Y. Ji, S. Meng, Z. Li, D. Hirtz, L. Elling and U. Schwaneberg, Green Chem., 2023, 25, 8108–8116 RSC.
- H. Ohno, D. Araho, Y. Uesawa, H. Kagaya, M. Ishihara, H. Sakagami and M. Yamamoto, Anticancer Res., 2013, 33, 3061–3068 CAS.
- Y. Nakatani, A. Kobe, M. Kuriya, Y. Hiroki, T. Yahagi, I. Sakakibara, K. Matsuzaki and T. Amano, Eur. J. Pharmacol., 2017, 815, 381–390 CrossRef CAS PubMed.
- J. Qin, J. Chen, F. Peng, C. Sun, Y. Lei, G. Chen, G. Li, Y. Yin, Z. Lin and L. Wu, J. Ethnopharmacol., 2022, 293, 115257 CrossRef.
- E. O. Romero, A. T. Saucedo, J. R. Hernández-Meléndez, D. Yang, S. Chakrabarty and A. R. Narayan, JACS Au, 2023, 3, 2073–2085 CrossRef CAS PubMed.
- R. Li, H. J. Wijma, L. Song, Y. Cui, M. Otzen, Y. E. Tian, J. Du, T. Li, D. Niu and Y. Chen, Nat. Chem. Biol., 2018, 14, 664–670 CrossRef CAS.
- D. Ma, Z. Cheng, L. Peplowski, L. Han, Y. Xia, X. Hou, J. Guo, D. Yin, Y. Rao and Z. Zhou, Chem. Sci., 2022, 13, 8417–8428 RSC.
- R. W. Gantt, P. Peltier-Pain, W. J. Cournoyer and J. S. Thorson, Nat. Chem. Biol., 2011, 7, 685–691 CrossRef CAS.
- H. Shao, X. He, L. Achnine, J. W. Blount, R. A. Dixon and X. Wang, Plant Cell, 2005, 17, 3141–3154 CrossRef CAS.
- Z.-L. Wang, W. Wei, H.-D. Wang, J.-J. Zhou, H.-T. Wang, K. Chen, R.-S. Wang, F.-D. Li, X. Qiao and H. Zhou, Chem. Sci., 2023, 14, 4418–4425 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc01508h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |