
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monoliths enabling biocatalysis in flow chemistry
Aleksandra
Lambarska
ab,
Katarzyna
Szymańska
 *a and
Ulf
Hanefeld
*b
*a and
Ulf
Hanefeld
*b
aDepartment of Chemical Engineering and Process Design, Silesian University of Technology, Ks. M. Strzody 7, 44-100 Gliwice, Poland. E-mail: Katarzyna.Szymanska@polsl.pl
bBiokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: U.Hanefeld@tudelft.nl
First published on 27th September 2024
Abstract
This is a review on the feasibility of monolithic porous supports in biocatalysis carried out in a continuous flow system. It discusses factors affecting the efficiency and stability of enzyme immobilisation, kinetic parameters of enzyme processes carried out inside a monolith, biocatalysis in single and two-phase systems, and cascade reactions including cofactor regeneration. It also covers materials engineering (monolith types) and issues related to the flow of reactants through the monolith (chemical engineering). Emphasis is placed on the fact that the application of (bio)catalysis improves selectivity and atom economy, thus lowering the E factor. However, biocatalysts need to be employed in a reactor, which can aid further improvement towards green chemistry goals. The application of enzymes in flow chemistry has been shown to lead to higher space time yields (STYs) compared to batch reactions. In particular, with monolithic reactors a drastic decrease in volume and thus solvent can be achieved. By immobilising very high densities of enzymes directly on the monolith, reaction times dwindle, improving STYs. The small reaction volumes enable excellent heat transfer, helping to save energy. The underlying principles of monolithic flow reactors and their application in mono- and bi-phasic biocatalytic systems will be examined.
 From left to right: Katarzyna Szymańska, Aleksandra Lambarska and Ulf Hanefeld. | Katarzyna Szymańska received her Ph.D. in 2009 from the Faculty of Chemistry, Silesian University of Technology, Gliwice, Poland. Her research interests include engineering of (bio)heterogeneous catalysts, nanostructured materials, and chemical and enzymatic microreactors. She is currently a professor at the Department of Chemical Engineering and Process Design, SUT, Gliwice, Poland, and collaborates with Prof. U. Hanefeld, Prof. P.L. Hagedoorn (TU Delft), E. Magner (University of Limerick), Prof. D. Tischler (RUB), and P. Walde (ETH). Aleksandra Lambarska received her M.Sc. and B.Sc. degrees in biotechnology from the Silesian University of Technology (Poland) in 2022 and 2021 under the supervision of Assoc. Prof. Katarzyna Szymańska. She is currently a Ph.D. student at the same university. She started her Ph.D. journey with an internship at Technische Universiteit Delft (the Netherlands) under the supervision of Prof. Ulf Hanefeld. Her research interests focus on heterogeneous biocatalysis, enzyme immobilisation and their application in continuous flow processes. Ulf Hanefeld received his PhD from the Georg-August-Universität zu Göttingen, having performed the research both with Prof. H. Laatsch (Göttingen) and Prof. H. G. Floss (Seattle). After postdoctoral years with Prof. C. W. Rees (Imperial College London), Prof. J. Staunton (Cambridge) and Prof. J. J. Heijnen and Dr A. J. J. Straathof (TU Delft), he stayed at the Technische Universiteit Delft and his research in Delft focuses on enzymes and their immobilisation and application in organic synthesis. |
1. Introduction
Enzymes provide excellent selectivity in chemical reactions. The industrial application of biocatalysis has therefore increased and it enables processes from bulk to fine chemicals and pharmaceuticals.1–4 These new enzyme-based approaches reduce the number of synthesis steps and decrease the demand for stoichiometric amounts of chemicals while also shortening the time of synthesis,2–4 with the synthesis of molnupiravir being a perfect example of this (Scheme 1).5 Due to this fact, the processes using enzymes as catalysts are often referred to as “white biotechnology”, underlining their positive impact on the environment.1 With this increase in demand, new technologies for the application of enzymes need to be developed.1 In addition to the classical fed-batch reactor many new approaches, such as parallel reactors and flow chemistry, have entered the market for experimental purposes.6–11 | ||
| Scheme 1 Enzymatic strategy for obtaining molnupiravir in just 3 steps. | ||
The industrial use of enzymes on a truly large, multi-ton scale, in particular in a continuous process, requires their immobilisation on a suitable support, enabling the use of a dedicated reactor system.7,12 Most of the carriers used in immobilisation are powders, or more coarsely granulated products, typically applied in batch reactors with intensive mechanical stirring. However, their drawback is gradual size reduction during the process, which negatively affects their properties and hinders their separation. Moreover, due to the necessity to ensure intensive mass transfer in the suspended matter, the amount of the heterogeneous catalyst is limited, which directly affects the efficiency of the process. Additionally, in batch applications, scaling up causes problems related to heat and mass transfer and mixing. Ideally, initial laboratory experiments with immobilised enzymes that ensure recycling could be straightforwardly transferred to industrial settings.
Low-volume flow reactors (microreactors) are a solution that allows amelioration of some of these difficulties.13–17 The problems related to scaling up in the case of using microreactors are resolved by numbering up a single reactor. This eliminates the necessity to design and build a pilot system. However, it does require the correct design of the reactor setup in a parallel or sequential system. In the applied microreactors, the diameters of the channels are usually 0.1–0.5 mm. Such small channel diameters force the use of a laminar flow regime and therefore the mixing (diffusion) time of molecules (t) is proportional to the square of the channel width/diameter (L):
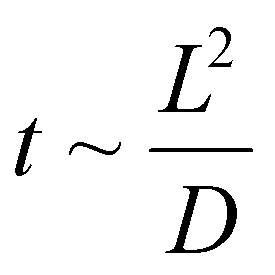
The microreactors (Fig. 1) that attract the most attention are:
• “lab-on-the-chip” type microreactors – usually they are either a single platelet or a stack thereof, with a network of channels with micrometric diameters. These reactors are usually intended for homogeneous reactions and applied in chemical and medical analytics.21,22 The key disadvantage is that no immobilised enzymes can be applied, ruling them out for large-scale applications.
• capillary microreactors – they are a single capillary or a bunch thereof, usually with a very thin catalytic film placed on the internal surface of the walls.23 Here enzymes can be immobilised. The disadvantage is however the straight shape of the channel. This does not intensify the mixing due to laminar flow and therefore the mass and heat exchange can be modest.
• packed bed microreactors – in these reactors, the fine-grained bed of the catalyst fills the capillary/tube and the reactants flow in the annular space between the catalyst grains. These reactors are easy to build but the drawback is the relatively high flow resistance, which can further increase as a result of bed compression.24
• structural microreactors or monoliths – the core of the reactor is constituted by a porous structure/monolith with open pores/channels connected with each other – the catalyst is then immobilised either on the surface of the monolith structure or (optimally) in its pores if it exhibits a two/three-modal porous structure, and reactants flow through the meandering pores.25,26 The continuous monolith structure ensures flow stability and virtually eliminates bed compression (especially with inorganic monoliths).
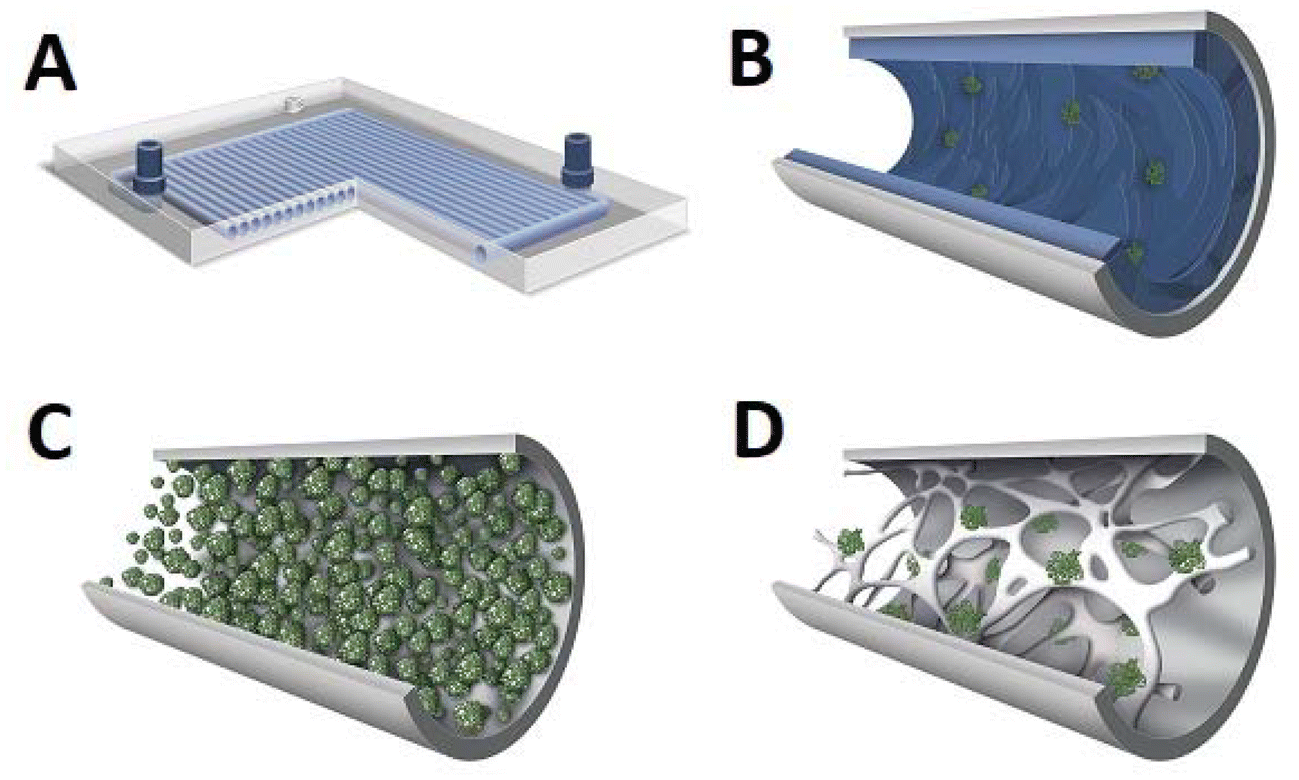 | ||
| Fig. 1 Types of microreactors: lab-on-the-chip (A), capillary (B), packed-bed (C), and structural/monolithic (D). | ||
Overall the monolithic reactors combine all the advantages of continuous flow microreactors. At the same time they do not suffer from back pressure problems and bed compression and create turbulence during flow for ideal mixing, and enzymes can be immobilised on the reactor walls. We have recently summarized our own contribution to that field.27
Microreactor technology is mainly applicable to low-scale processes where large quantities of the final product are not required. The small geometric size of the microreactors makes it possible to create a suitable installation in a short time and in virtually any location, reducing the carbon footprint associated with transport. Adamo et al.28 have recently described a demonstration system that can carry out continuous synthesis and formulation of active pharmaceutical ingredients (APIs) in a refrigerator-sized unit.
In this review the kinetic argument for the advantages of flow chemistry and in particular the application of monolithic microreactors and their application in continuous-flow biosynthesis (Fig. 2) is made. This leads to the possibility of using small reaction volumes and better surface area to volume ratios, which reduces energy consumption. At the same time, better reaction control leads to higher selectivity, thus less waste and straightforward downstream processing (DSP). Improved DSP and higher STY values both contribute to achieving lower environmental (E) factor values. For both single reactions and reaction cascades, in single-phase or biphasic systems and under consideration of cofactors, this will be discussed and the advantages of monolithic microreactors will be demonstrated. In particular the perspective of green chemistry will be considered. The E factor as well as Green Chemistry Principles will be critically applied.
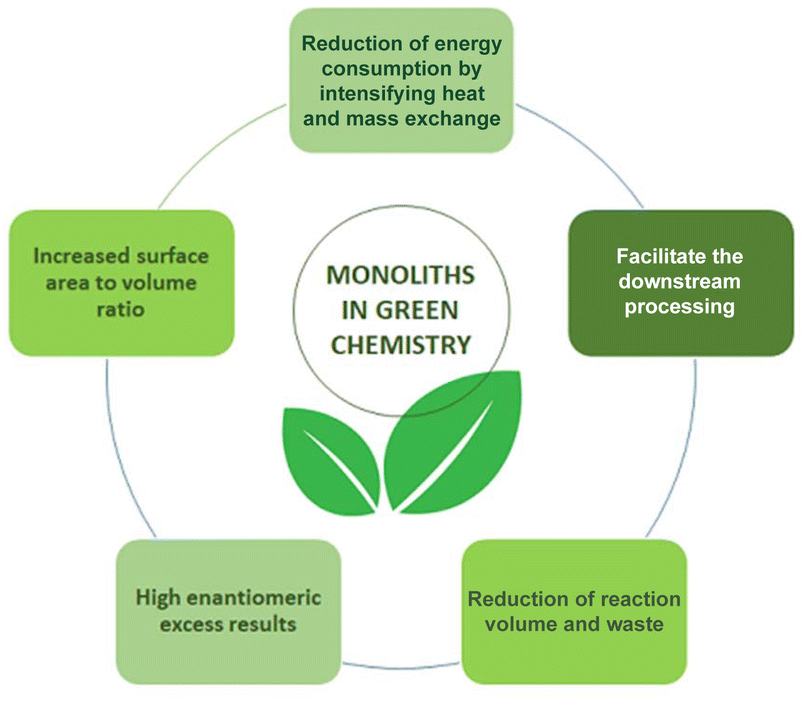 | ||
| Fig. 2 Development of biocatalysis and microreactor technology in the field of green chemistry. | ||
2. Monoliths
2.1 Types of monoliths
The word monolith comes from the Greek word ‘mono lithos’, meaning a single stone. In materials engineering it means a material in ‘one piece’. The automotive industry was one of the first industries to use monoliths. They are produced by extrusion and the most common material obtained is cordierite, which is used as a carrier for catalytic converters in the exhaust of cars.29,30 Monoliths are also made from metals, mullite (a mixture of silicon and aluminum oxides in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio) and silicon carbides. Their disadvantage is a small specific surface area (0.7 m2 g−1 for cordierite) and they are therefore less widely used in other industries.30 Modern monolithic supports overcome these problems. Generally, monolithic carriers are divided into organic/polymer and inorganic (mostly silica and carbon) groups.31–38 The former are characterized by good biocompatibility, pH stability and the presence of various functional groups; however, their limited resistance to organic solvents may lead to changes in the porous structure and thus decrease their mechanical stability.39 Inorganic structures on the other hand are very stable and these kinds of monoliths additionally offer very good permeability, both for organic and inorganic solvents.34–37 Oxide-based inorganic monoliths (e.g. silicon, aluminum and titanium oxides) can be obtained in several ways. Firstly, there are methods using templates such as colloidal particles (e.g. latex or polystyrene beads), ionic liquids, starch, cellulose and many others.40–44 The process involves introducing an inorganic precursor solution into the free spaces created by packing the template. In the next step, the system undergoes condensation and crystallization followed by the removal of the template, usually by calcination or extraction. This leads to the formation of ordered macro/mesoporous materials in which both types of pores are through-going and interpenetrating.
:3 ratio) and silicon carbides. Their disadvantage is a small specific surface area (0.7 m2 g−1 for cordierite) and they are therefore less widely used in other industries.30 Modern monolithic supports overcome these problems. Generally, monolithic carriers are divided into organic/polymer and inorganic (mostly silica and carbon) groups.31–38 The former are characterized by good biocompatibility, pH stability and the presence of various functional groups; however, their limited resistance to organic solvents may lead to changes in the porous structure and thus decrease their mechanical stability.39 Inorganic structures on the other hand are very stable and these kinds of monoliths additionally offer very good permeability, both for organic and inorganic solvents.34–37 Oxide-based inorganic monoliths (e.g. silicon, aluminum and titanium oxides) can be obtained in several ways. Firstly, there are methods using templates such as colloidal particles (e.g. latex or polystyrene beads), ionic liquids, starch, cellulose and many others.40–44 The process involves introducing an inorganic precursor solution into the free spaces created by packing the template. In the next step, the system undergoes condensation and crystallization followed by the removal of the template, usually by calcination or extraction. This leads to the formation of ordered macro/mesoporous materials in which both types of pores are through-going and interpenetrating.
A macro/mesoporous structure can also be created by chemical reaction-induced phase separation. This phenomenon was first used by Nakanishi et al.45–49 and the method was later modified by, among others, Smått.50 A polymer (e.g. polyethylene oxide) is used to induce phase separation. Phase separation results in the formation of a solvent-rich phase and a polymer-rich phase. The polymer phase solidifies by gelation of the silica precursors within it, forming a micrometric skeleton, while the solvent-rich phase forms macropores after evaporation. The ratio of reactants and the process conditions allow the control of the structural parameters, i.e. the macro/mesopore sizes, the specific surface area and the total volume of the pores, covering a wide range.26,36–38 Oxide-based inorganic monoliths can be used in catalysis51–57 as liquid chromatography columns58,59 or controlled drug release systems.60,61
The most common route to synthesize self-supported carbon monoliths with multilevel porous structures relies on the carbonization of porous polymers62–66 and biomass.67–69 Another route makes use of an inorganic porous material with a uniform channel system as a hard template. The template is filled with carbon precursors which are internally carbonized and then the inorganic template is removed.70–72 Carbon monoliths have been used as supercapacitors,62,69 microextraction fibers,66 microreactors,63,73 electrochemical sensors,67,68,70 and sorbents.74
Organic polymer monoliths have been considered good supports for enzyme immobilisation owing to their good biocompatibility, excellent pH stability and ease of modification with various functional groups.75–79 The known procedures for making polymer monoliths are simple ‘one-pot’ synthesis, where the monomers, initiators, cross-linkers and pyrogenic solvents are simply pre-mixed before being filled into a capillary and then exposed to factors such as UV light or heat to initiate polymerization.31,80,81 Polymer monoliths are widely used primarily in different kinds of chromatography methods,82,83 solid-phase extraction,84,85 for the preparation of flow-through immobilised enzyme reactors (IMERs) suitable for proteomics,86–90 microfluidic analysis,91,92 pharmaceutics,93,94 biodiesel95 and oligosaccharide96,97 production, etc.
Monoliths can also be produced using additive manufacturing, commonly referred to as 3D printing, which is an emerging bottom-up manufacturing technology with the potential to achieve rapid prototyping of complex geometries and structures, even with sophisticated internal structures (pores or complex channels).98,99 A wide range of materials can be utilized in 3D fabrication, which can be classified into three main categories: polymers, ceramics, and metals.100 Furthermore, the chemical modification of printed materials is also attracting increasing attention: by modifying the printed carriers, enzymes can be immobilised on them.101–106
2.2 Flow behaviour in monoliths
While considering the potential of microreactors for continuous processes, one should note that the flow in small-diameter straight channels is laminar in the entire range of the applied conditions. For that reason, mixing and transportation to catalytic centres located on the surface is slow compared to the mixing observed in turbulent flow.107,108 Mixing is diffusion limited in the absence of turbulent flow. Although it is possible to elongate the channel system to allow diffusion to complete as the fluid is transported through the microsystem, it also requires a longer channel and thus reaction time. Several strategies have been devised to speed up mixing processes actively in microfluidic systems. Bessoth et al.109 proposed to split a liquid flow into even smaller channels, where the diffusion distances become so small that the corresponding mixing times by diffusion were in the order of milliseconds. Later, all divided streams were combined again downstream.109 To intensify the mixing, the introduction of baffles was proposed110 as well as using ultrasound111 or introducing fragments of porous structures.112,113 For these reasons, monoliths characterized by a very high number of interconnected tortuous channels and micrometric diameters seem to be an interesting solution. The tortuous character of the channels causes sudden changes in the direction of the flow of the liquid, which fosters the formation of swirls, the intensification of mixing and the contact between the liquid and the wall of the microchannel (Fig. 3). Their small diameter additionally intensifies the exchange of mass and heat. However, this could increase the back pressure. The flow of liquid through monolith structures is complex from a microscale perspective and tends to obey Darcy's law.26,114 The relationships between the pressure drop gradient (ΔP/L) and the volumetric flow rate of liquid (![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) ), its viscosity (η) and the cross-sectional area of the monolith (A) are given by the Darcy–Weisbach equation:
), its viscosity (η) and the cross-sectional area of the monolith (A) are given by the Darcy–Weisbach equation:where the permeability coefficient (K) is proportional to the diameter of the macropores (K ∼ d2). As was shown by Szymańska et al.26 the monoliths with macropore diameters in the range 20–40 μm allowed for applying flow ranges of 200–700 cm3 (cm2− min)−1 at pressure gradients falling in the range of 15–65 kPa cm−1. A decrease of macropore diameter to 10–12 μm caused comparable decreases in pressure, however, at much smaller flow rates (50–200 cm3 (cm2− min)−1). A low pressure decrease allows for simple and thus cost-efficient metering pumps while permitting them to operate at increased linear speeds of flow (intensification of the mass transport) and at flow rates of reacting substances measured in tens and hundreds of cm3 min−1. This opens the possibility of multi-kilogram syntheses in cm3 reactors. These amounts are in stark contrast with the values characterizing typical microfluidic reactors, where the applied flow rates are limited to tens and hundreds of μm3 min−1.86,115

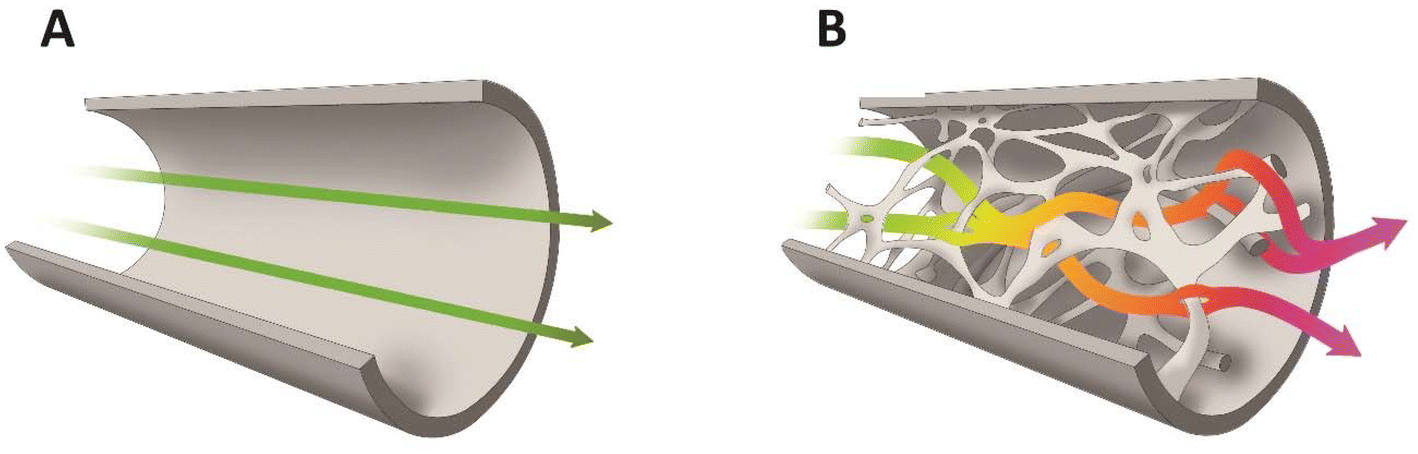 | ||
| Fig. 3 Scheme of liquid flow in a straight channel (A) and in structured monoliths (B). | ||
In processes where the reactants are in two phases (biphasic processes), mass transfer in microchannels becomes even more important and depends on the flow patterns. A common pattern is segmented or plug flow, where one phase forms drops, the sizes of which are as large as the channel diameter (plugs) separating the continuous phase into segments (slugs) (Fig. 4A).116 The overall mass transfer rates depend on the plug geometry. In addition, the wall wetting properties might result in the formation of a thin film between the plugs and the wall, which increases the specific interfacial area available for mass transfer. The latter is also intensified through internal circulation within the slugs or the plugs caused by the shear between the continuous phase/wall surface and the slug or plug axis, which enhances diffusive penetration.117–121 In certain situations, especially in straight channels, the advantage provided by the increased interfacial area and mixing effects may be insufficient to balance the short residence time. Many methods for increasing the mixing effects in a straight channel microreactor were developed such as ultrasound122–125 and magnetic nanoparticle addition.126–129 A straightforward and elegant strategy was demonstrated by Abraham et al.,130 who changed the dimension of the circular microchannel (via a sudden expansion and then a sudden contraction), increasing the slug and droplet diameter and reducing the length (and vice versa). This type of transformation alters the concentration field and positively interferes with the internal mixing in the slugs and droplets. As the shear stress plays an important role in triggering the internal circulations inside the fluid elements, the flow through a non-uniform channel improves the extraction efficiency. A similar effect will be obtained using the structural/monolithic microreactors.26,131–133 The use of monolithic microreactors ensures a favourable surface-to-volume ratio and the presence of meandering channels further improves mixing and thereby increases the transport of mass and heat between phases (Fig. 4B). The meandering structure of the silica monolith will induce changes in the flow of a single droplet and thus not only enhance the interface between the continuous and dispersed phases but also intensify the internal circulation within the slugs or the plugs, which in turn has a positive effect on mass transport.
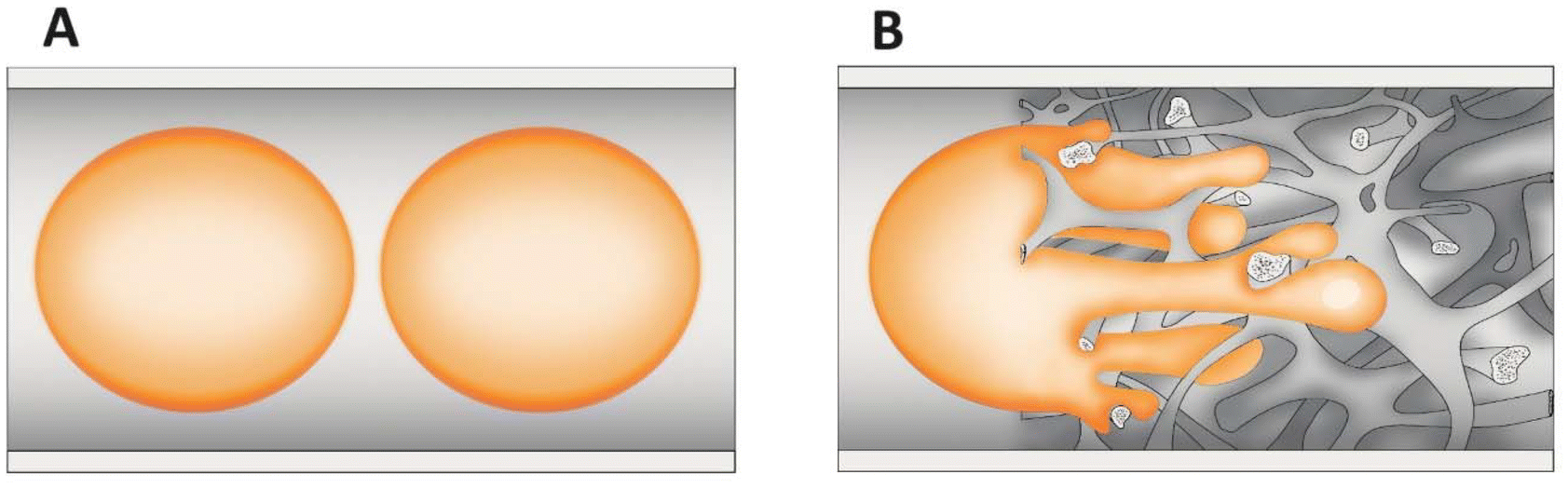 | ||
| Fig. 4 Scheme of biphasic flow in a straight channel (A) and in structured monoliths (B). | ||
3. Biocatalysis in monoliths
3.1 Attaching the enzyme to the monolith
The application of catalysts in general and specifically enzymes always requires their removal at the end of the reaction. Immobilisation of the enzymes greatly eases this downstream processing (DSP) and at the same time often improves the stability of the biocatalysts. This is achieved by single or multi-point interaction with the carrier's surface and it generates a favourable micro-environment and protection against intermolecular interactions.134–137 As an additional advantage it enables reuse and reduction of process costs.134,138 The application of immobilised enzymes furthermore allows for a significant simplification of the reactor's structure and for controlling the process, e.g. stopping it by separating the catalyst from the reaction mixture. Due to the above, the immobilisation of enzymes is often performed in large-scale industrial applications.136,137 The first reports on immobilised enzymes date back to 1916,139 while their commercial use first took place in the 1960s.140 Today immobilised Penicillin G acylase is reused141 up to 1000 times and immobilised glucose isomerase7,142 is at the core of the high fructose corn syrup process.There is no universal immobilisation technique to acquire an active and stable preparation. While designing the immobilisation method, many factors must be considered, including:134,136,143
• interactions: enzyme–substrate/product, enzyme–carrier, substrate/product–carrier;
• resistance of the carrier to reaction conditions (solvent, temperature, pH, etc.);
• the reactor type used: tank reactor with mixing, membrane reactor, microreactor.
However, the most important factor influencing the effective attachment of enzymes is the presence of appropriate functional groups on the carrier surface. Functional groups must provide a suitable microenvironment for the expression of enzyme activity and ensure the stability of the biocatalyst.
By immobilising enzymes on the surface/inside the pore of a monolith, it is possible to use all known techniques for binding the enzyme to a solid support (covalent binding, physical and ionic adsorption, transition metal complexation). These techniques have already been described in detail in numerous reviews137,144–146 and will therefore not be discussed here. Due to the number of factors affecting the immobilisation process, it is very difficult to determine a priori which of the mentioned techniques is superior. The dominant opinion is that it is necessary to determine the parameters individually for each selected enzyme, especially considering that the forces acting on the immobilised enzyme in a flow system are significantly different from those in a batch reactor.15,69–74
Enzyme immobilisation is critical when considering continuous processes. To highlight its importance, a few representative examples are given. One of the most important performance parameters of flow microreactors is their process stability. The use of different methods of enzyme immobilisation results in different strengths of enzyme–carrier interactions. The continuous flow of reactants through the monolith can favour leaching of the enzyme from its surface. In a study by van den Biggelaar et al.,147 transaminases were immobilised on macrocellular silica monoliths functionalised with amino groups. Two immobilisation techniques were used: adsorption via ionic interactions and a covalent method using an amino-functionalised monolith and glutaraldehyde as a coupling agent, but only the latter method gave stable bioreactors. Decreases in activity were explained by the leaching of the enzyme from the support. Similar results were obtained in the work of Szymańska et al.26 where trypsin was adsorbed on cyano-functionalised silica and covalently immobilised on amino-functionalised silica using glutaraldehyde as a linker. The adsorbed trypsin showed a higher initial activity, but there was a significant decrease in the activity over time. In the case of covalent binding, the enzyme retained its initial activity. As in previous work, the observed effects (decrease in activity) were explained by the leaching of the adsorbed enzyme from the support surface. Adsorption was also used for the deposition of lipase on a hydrophobised silica monolith,133 and the obtained catalyst/reactor was used in the kinetic resolution of the non-equimolar mixture (S:R = 85:15) of the secondary allylic alcohol (+)-1-[(1S,5R)-6,6-dimethylbicyclo[3.1.0]hex-2-en-2-yl]ethanol. Interestingly, the adsorption method used in this case allowed the reactor to operate stably for about 120 h. The main difference between the cases described is that in the first two processes, the reactions were carried out using an aqueous solvent, whereas the kinetic resolution was carried out in an organic solvent. Enzymes have a high affinity for water, so the use of this solvent favours the leaching of the enzyme from the support. By using an organic solvent, the leaching can be drastically reduced.
This was also noted for hydroxynitrile lyases (HNLs). When applied for cyanohydrin synthesis, the reactions were performed in buffer saturated organic solvents and no enzyme leaching was observed.148–151 In contrast when the non-natural Henry reaction was catalysed, water was necessary and leaching occurred.152 The strength of the enzyme–carrier interaction is not the only factor affecting stable reactor performance. A critical issue may be the immobilisation method itself. In the work of Shortall et al.153 lactate dehydrogenase (LDH) was immobilised via two different methods: electrochemical entrapment in poly(3,4-ethylenedioxypyrrole) and covalent attachment on glyoxyl agarose. Both reactors allowed for up to 100% conversion of NADH; however, LDH@agarose proved superior in terms of reuse and storage. In this case, no leaching of the enzyme was observed and the loss of activity was explained by the stability of the enzyme itself.
The covalent bond between the enzyme and the support can be direct, i.e. the functional groups of the support bind covalently to the side group of the amino acid, or indirect through the introduction of a linker (e.g. glutaraldehyde). In principle, this does not affect the leaching of the enzyme, but it can affect the activity and stability of the enzyme. Yao et al.31 compared two methods for the covalent immobilisation of trypsin on polymer monoliths. The first, termed ‘one-step’, involved the direct formation of a covalent bond between the epoxy group of the support and the enzyme. The second, termed ‘multi-step’, involved the modification of the epoxide group with a diamine followed by activation with glutaraldehyde. The researchers showed that the ‘multi-step’ method produced catalysts/reactors with higher activity. Similar studies can be found in ref. 154 where ribonuclease A was immobilised by the direct reaction between the amino groups of the enzyme and the epoxy groups of the sorbent and the reaction of the enzyme with the aldehyde-containing polymer spacer preliminarily bound to the surface of the monolith. In this case, the introduction of the linker did not result in any change in enzyme activity. This shows that the introduction of the linker, and therefore the distance of the enzyme from the surface of the monolith, does not have a unidirectional effect on the activity of the enzyme and that each case must be considered individually.
Another important parameter affecting the immobilisation of enzymes within the monolith structure is the uniform distribution of functional groups. This is particularly important when functional groups are introduced after the monolith preparation/synthesis step. The work of van den Biggelaar et al.155 compared methods for introducing an amino group onto the surface of a silica monolith and found that the essential parameters that allow reaching high catalytic performance were the functionalisation mode (dry versus wet) and water availability during the functionalisation process. In practice, briefly dipping the monolith in the functionalisation solution followed by aging under a solvent-saturated atmosphere allowed obtaining a homogeneous functional group dispersion throughout the entire monolith. Total water control during the process (by using dried monoliths and water-saturated solvent) allowed minimizing the batch-to-batch variability which affects enzyme immobilisation and biocatalyst efficiency. Equally important, the immobilisation of biocatalysts will enable the reuse of enzymes and support the DSP process.
3.2 Microbial infection in the monolith
Optimal aqueous reaction conditions for most enzymes (neutral pH values, room temperature) are also optimal conditions for the growth of various microorganisms. The risk increases even more when the substrates are an easy source of carbon or nitrogen as a growth medium. Moreover, there is a whole range of microorganisms, primarily fungi, that are able to not only survive but also thrive under slightly more extreme conditions, such as high temperature (above 40 °C) and low pH values (below pH 5). A solution of standard antibiotics such as penicillin can be utilised to eliminate the microbiological infection while preserving the initial catalytic activity of the immobilised enzyme protein. However, carrying out such a procedure only allows curing the symptoms but not elimination of the cause. Therefore, microbial contamination inside monolithic industrial microreactors can be a serious problem due to difficulties in dismantling for cleaning purposes. To overcome this obstacle, a practical solution is to filter all solutions before use and to maintain sterile conditions throughout the process to prevent bacterial/fungal infections. Also low concentrations of microbial growth inhibitors, such as for example sodium azide (NaN3),156,157 can be employed. However, the most important factor is the hygiene of the process.Interestingly, monolithic microreactors can be used to detect bacteria in biological samples. One solution is to use an immunoreactive filter in a microporous polyacrylamide monolith. Due to size exclusion, bacteria interact with the surface of the filter and become stained. This solution can provide an alternative for infection diagnosis.158,159
3.3 Kinetics in monoliths
As established by Michaelis and Menten, the rate of an enzymatic reaction depends on the substrate concentration. The initial increase in the rate is linear and proportional to the increase in substrate concentration. The maximum reaction rate (Vmax) is reached when the enzyme is completely saturated with the substrate. Further increases in substrate concentration will not increase the reaction rate. For enzymes, this typically occurs at lower substrate concentrations than for other catalysts. The reaction rate can be increased by the amount of enzyme available, provided of course that the amount of substrate is significantly higher than the amount of enzyme. The above considerations apply to enzymatic reactions catalysed by a non-immobilised enzyme.The case is slightly different when an immobilised enzyme is considered. In general the increase of the amount of enzyme on the support is desirable to enhance the enzyme/substrate ratio and thus the reaction rate. On the other hand, the kinetics of the reaction is significantly influenced by the accessibility of the active site of the immobilised enzyme. Consequently, the effect of enzyme density on the surface of the solid support must be considered as a critical parameter affecting the availability of the active site of the enzyme.157 This means that diffusion is also now a key parameter.
To study the influence of substrate size on diffusion, Volokitina et al.160 immobilised ribonuclease A on polymeric monoliths for the hydrolysis of a small and a large substrate (cytidine-2′,3′-cyclic monophosphate monosodium salt (CCP) and RNA). Columns with pore sizes of 450, 830, 1220 and 2020 nm were prepared and the enzyme was embedded in them. Both the small molecule substrate (CCP) and the large molecule substrate (RNA) were hydrolysed. As expected for the small-molecule substrate, they showed that both the molecular recognition process (convergence of KM values) and the biocatalytic activity did not depend on the average pore size of the monolithic stationary phase. The results obtained indirectly prove that the mass transfer mechanism is the same in all the columns tested and, consequently, that the process of enzyme–substrate complex formation takes place in the same way.
Completely different results were obtained for the macromolecular substrate.160 In this case, a clear effect of pore size on RNA hydrolysis was observed, as it was found that the degree of substrate hydrolysis increased with increasing pore size. With macromolecular substrates, the enzyme must sequentially hydrolyse all possible bonds in the macromolecule to convert it into a series of resulting products. Unlike the reaction in solution, this situation may be complicated for immobilised enzymes due to steric limitations. They reduce the accessibility of the enzyme's active site for the large substrate molecule. Furthermore, the use of macromolecular substrates leads to a reduction in mass transfer, i.e. a reduction in the contact time between the enzyme and the substrate, which in turn can lead to a reduction in reactor efficiency. In this regard, pore characteristics161 can play a significant role in the degradation of high molecular weight substrates.160
Another parameter that influences the kinetics of the reaction taking place inside the monolith is the chemical nature of the monolith surface and, related to this, the way in which the enzyme is immobilised. Luangon et al.63 adsorbed Candida rugosa lipase on a hierarchical porous carbon monolith with micropores on an interconnected macroporous skeleton synthesized through the sol–gel polycondensation of resorcinol and formaldehyde. The effects of the oxygenated surface of micro-/macroporous carbon monoliths on lipase immobilisation were investigated. The estimated values of Vmax and KM of immobilised lipase on a crude carbon monolithic column are approximately 2 times lower than the values of Vmax and KM obtained from an oxygenated carbon monolithic column. Although the protein binding efficiencies of crude and oxygenated columns are similar, the carbon monolith with the oxygenated surface exhibits clearly higher efficiency compared with the untreated surface carbon monolith. Thus, the oxygenated surface of the micro-/macroporous carbon monolith exhibits a better transesterification rate. An improvement in the transesterification rate can be explained by an induction of a slightly polar substrate p-nitrophenyl palmitate (p-NPP) by the oxygenated surface of the micro-/macroporous carbon monolith or by better stabilization of lipase by the oxygenated carbon surface.
The influence of the chemical nature of the monolith surface and the associated immobilisation method is of importance162 but not relevant in all cases. The work described by Volokitina et al.154 compared two methods of immobilising ribonuclease A (RNase A) on a methacrylate-based polymer monolith. In the first case, the biocatalyst molecule was attached to the solid surface by direct covalent bonding, while in the second case, a flexible-chain synthetic polymer was used as an intermediate spacer. The values of KM determined for the hydrolysis of polynucleotide by bound RNase were 0.76 mM for direct immobilisation and 0.56 mM for immobilisation with a polymer spacer. The similar KM values confirm their practically identical enzyme–substrate binding strength. The specific activity values were found to be quite similar for both immobilisation methods, demonstrating the maximum activity of the biocatalyst bound in different ways.
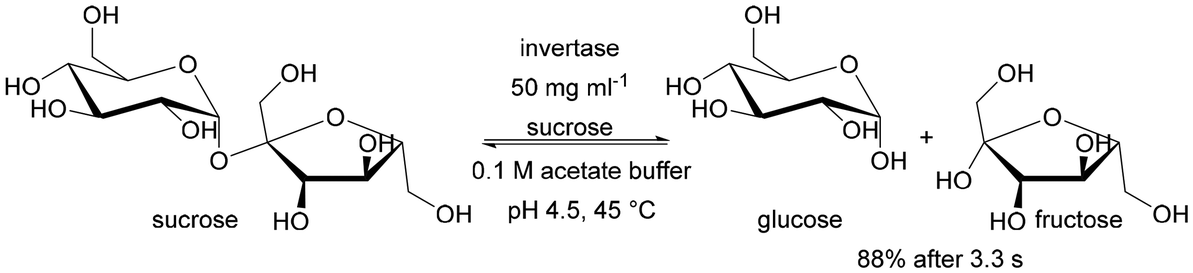
From the point of view of flow processes, it is also interesting to compare the kinetic parameters of enzymes immobilised inside the monolith with those immobilised on silica powder and applied in a batch system. Does flow efficiently suppress diffusion limitations caused by immobilisation? When immobilising invertase in a silica monolith,56 the KM value was similar for those immobilised on particles, but the Vmax value was approximately 1500 times higher for invertase immobilised in the monolith. While at first glance unlikely, it is justified by the amount of enzyme present in the reaction zone. For the powder immobilised enzyme this was 0.037 mg of enzyme per 24 mL of reaction volume, while for the enzyme immobilised in the monolith, it was 0.106 mg per 0.03 mL of reaction volume. Immobilising such a large amount of enzyme per unit reaction volume is virtually impossible with conventional batch reactor solutions. Only the use of flow-through microreactors, particularly of the packed bed and structural (monolithic) type, allows such a favourable ratio of surface area (on which the enzyme is deposited) to volume of the reaction mixture. Therefore this type of reactor is the focus of our review.
Even though large amounts of enzyme have a positive influence on the reaction, as shown above, it is important to investigate the limits thereof. The effect of the amount of enzyme and thus the suppression of diffusion limitations was the focus of Temporini et al.157 and Zhang et al.75 They immobilised different amounts of trypsin on the surface of silica monolithic columns157 or pepsin on polymer monoliths.75 It was found that when the amount of enzyme immobilised increased, there was an initial increase in activity, as expected. But further increases caused a drastic decrease in the enzyme activity. Those results indicate that either the high enzyme density on the solid support hinders access to the active site or the higher enzyme loading induces an aggregation and/or denaturation of the enzyme before or after immobilisation.
In the light of the above considerations, several factors can be identified that influence the kinetic parameters of enzymes embedded in the structure of monoliths. The first and foremost is the amount of enzyme immobilised. Initially, an increase in the amount of enzyme immobilised resulted in an increase in activity. This increase is however not unlimited. Once oversaturation occurs, a drastic decrease in activity was observed. Another factor is the correlation between the pore size of the monolith and the size of the substrate molecules. Fortunately, the pore size of the monolith has no significant effect on the kinetic parameters of processes using small-molecule substrates. For processes using macromolecular substrates, the pore size has a significant effect. Attention should also be paid to the chemical nature of the monolith surface and the associated method of enzyme immobilisation. The influence of these parameters is ambiguous and must be determined on a case-by-case basis for individual enzyme–monolith pairs. From a chemical engineering point of view, the mode of operation of the monolith itself is also important. It allows a very favorable ratio of enzyme to reactant volume, which is reflected in the kinetic parameters.
4. Single- and two-phase reactions inside monoliths
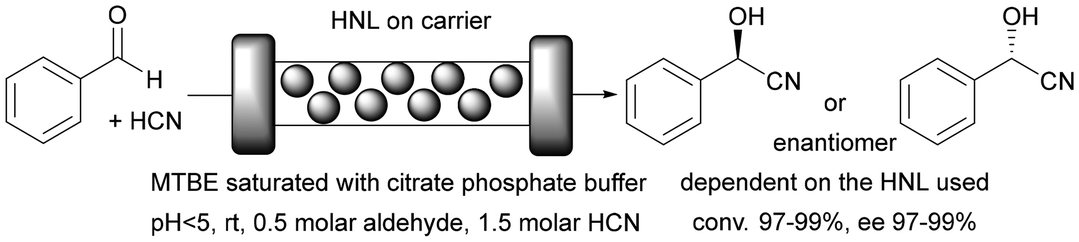
The use of a biocatalyst immobilised on monolithic microreactors results in at least two phase processes: a liquid solution of substrates and an enzyme immobilised on a solid carrier. There are also situations where the reactants are in two different liquid phases, in which case the whole process should be considered as a three-phase process. In order to quantitatively describe these processes and their environmental impact, we will use factors such as space–time yield (STY) and environmental factor (EF). The STY is an important engineering parameter that determines the amount of product generated during a certain residence time in the reactor used, as well as the productivity of the reactor.163 In turn, a significant parameter in the context of green chemistry assumptions is primarily the EF. This parameter makes it possible to determine the amount of waste generated in relation to the product produced.164 Here we examine single- and two-phase liquid phase systems in the monolith reactor and give full STY and EF details, always where it was available.Enzymes are mostly used in aqueous media, their natural environment. However, most biocatalysts also work well in organic solvents. Typically buffer saturated solvents are used as it has been shown that most enzymes require a minimal amount of water molecules to maintain their activity in a non-aqueous environment,165 lipases being the exception.166 This is important especially for the pharmaceutical industry, as most active compounds dissolve much better in organic solvents than in the aqueous phase. In some cases, the use of a monophasic organic medium effectively suppresses unwanted side reactions.149,150 This suppression of unwanted side reactions can be achieved by using a suitable medium or, as in the case of microreactors, by reducing the process time, or more precisely, the contact time between the reactants and the catalyst. Achieving high substrate conversions in batch reactors typically requires several to tens of hours, whereas in a microreactor, the same effect can be achieved in a matter of seconds to tens of minutes, depending on the enzyme used and the type of reaction. This is due to the favourable ratio of enzyme covered surface area to reaction volume. This effect can be critical if an unwanted competing/side reaction is observed in the system. An example is the use of (S)-hydroxynitrile lyases from Hevea brasiliensis (HbHNL) immobilised on a silica support in the (S)-mandelonitrile synthesis reaction carried out in a batch and continuous system.149 In the batch system, 90% conversion was achieved after about 30 h, but the enantiomeric excess (ee) was only 40%. This low enantiomeric excess is due to the long contact time of the reactants with the support on which the enzyme is deposited, that is, the background reaction. By using a silica-structured microreactor, the contact time could be reduced to only about 20 min, resulting in high substrate conversion (∼100%) and enantiomeric excess (ee ∼ 100%). This effect has been confirmed in other work by the same team.150 Furthermore, the use of monolithic flow microreactors significantly increases the STY compared to a batch system. For the monolithic microreactor with immobilised HbHNL,149 it was 613 g L−1 h−1 or after taking into account the amount of protein, it was 71 g L−1 h−1 mgprotein−1, whereas in batch, it was ∼2 g L−1 g−1 or 1 g L−1 h−1 mgprotein−1. To date, many types of enzymes have been successfully immobilised on monolithic microreactors, conducting reactions in monophasic water or organic systems (Table 1). The main applications included enantioselective synthesis,149,150,167,168 protein digestion26 or demonstration of microreactor operation.169
| Monolith type | Enzyme | Immobilisation technique | Biocatalysis result | Application | EF [gwaste gproduct−1] | STY [gproduct L−1 h−1] | Ref. |
|---|---|---|---|---|---|---|---|
| HRP, horse radish peroxidase; pNPG, 4-nitrophenyl-β-D-glucopyranoside. | |||||||
| Silica porous monolith obtained by the sol–gel method | HNLs (AtHNL, MeHNL, HbHNL) | Covalent binding via amino or epoxide groups | Higher STY yield and enantioselectivity compared to the batch system | Highly enantioselective chiral cyanohydrin synthesis | 22.95 | At: 1290; Me: 1235; Hb: 613 | 149 and 150 |
|
|||||||
| Trypsin | Covalent binding via amino, cyano or epoxide groups | High practical potential in protein proteolysis without any earlier pretreatment | Protein digestion | n.a. | n.a. | 26, 39 and 157 | |
| Invertase | Covalent binding via amino groups | 1000 times faster sucrose hydrolysis than in the batch system | Proof of concept of applying a monolith microreactor | 0.29 | 6009 | 56 | |
|
|||||||
| Alginate–silica hybrid | β-Glucosidase | Cross-linking | High pNPG hydrolysis yield, 94% after 24 h | Obtaining saponins from plant materials and scaffolds for tissues | 0.59 | 1.34 | 167 |
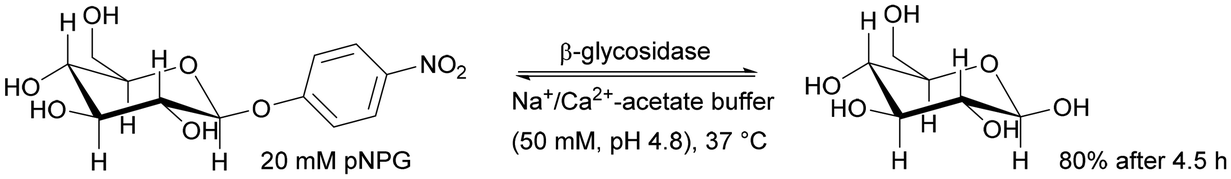
|
|||||||
| Monolith coated with polyaniline | Penicillin G acylase (PenGA) | Cross-linking | 95% of original activity maintained after 48 h of continuous operation | Semisynthetic penicillin production | 0.23 | 17.39 | 170 |

|
|||||||
| Poly(vinyl alcohol) monolith functionalised via reacting glutaraldehyde and polyethylenimine | Lipase, HRP | Covalent binding via aldehyde groups | Higher activity and stability of immobilised enzymes compared to their free form | Biotechnology and biomedical fields’ potential applications | n.a. | n.a. | 168 |
The single phase processes are to date the most thoroughly investigated and optimised approach. Consequently, the EF here is typically particularly low. With EF below 1 for aqueous processes and 23 for synthesis in organic solvents, it shows the power of monolithic microreactors to achieve the green chemistry targets.
In the 1990s, non-aqueous solvents were extensively explored. With our current understanding of their influence on activity and stability, it is possible to achieve similar activities in organic solvents as in aqueous media. However, this often requires a higher degree of control.171 Although the use of the biphasic reaction system (BRS) has been known since the last century (Pickering interfacial catalysis),172,173 the trend of using BRS in enzymatic reactions carried out in a continuous system has only gained momentum in the last decade. BRS enables high substrate concentrations in the organic phase while saturating the aqueous layer.174 Solutions in which the aqueous phase is made up of ionic liquids (ILs) are also successful.165 One of the most commonly used groups of enzymes in this case are lipases, due to their interfacial activation, opening their hydrophobic cap that shields the active center.166,175 In two-phase reactions, it is necessary to ensure intensive mixing165,169 of the two phases, which is provided in monolithic microreactors due to their large reaction area.176 The use of monolithic microreactors in a biphasic system allows for increased enantioselectivity and STY (up to 28 gproduct L−1 h−1 in flow compared to 0.7 gproduct L−1 h−1 in the batch system) in kinetic resolution processes.177 Enzymatic chemical processes in a multiphase system are primarily successful in biorefinery178,179 and pharmaceutical drug synthesis.180 They represent a green way to synthesize pharmaceuticals and chemical compounds according to the sustainable chemical industry principles.180
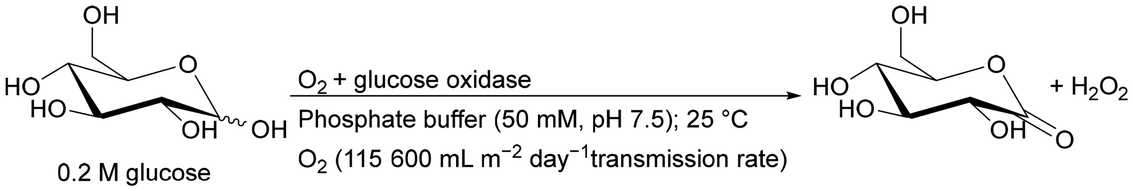
The two-phase reactions are not limited to aqueous–organic systems but include also liquid–gas systems. Efficient gas dosing into monolithic microreactors is an extremely difficult issue due to their wall structure. Recently, an innovative solution has emerged for the use of a monolithic microreactor enclosed by a semi-permeable wall, capable of continuously and efficiently dosing oxygen. This allows the system to achieve operational stability for a minimum of 40 h.181 A summary of the application of a biphasic reaction system in monolithic reactors is presented in Table 2 and Fig. 5.
 | ||
| Fig. 5 Opportunities and limitations in biphasic reaction system applications. | ||
| Monolith type | Immobilisation technique | Biocatalysis result | Application | EF [gwaste gproduct−1] | STY [gproduct L−1 h−1] | Ref. |
|---|---|---|---|---|---|---|
| CALB, lipase B from Candida antarctica; MsAcT, acyltransferase; PDMS, polydimethylsiloxane. | ||||||
| Silica porous monolith obtained by the sol–gel method | Covalent binding via amino groups and transition metals | Full and rapid (in a minute) transesterification | Transesterification of neopentylglycol | 7.62 | 820 | 174 |
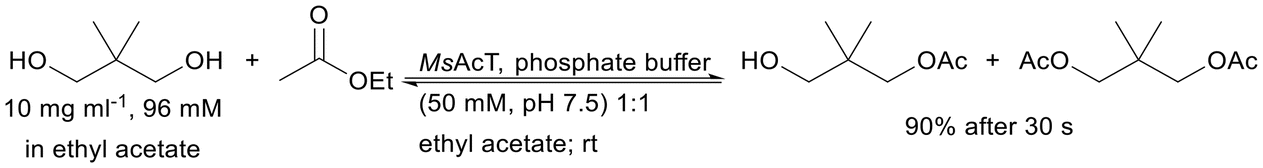
|
||||||
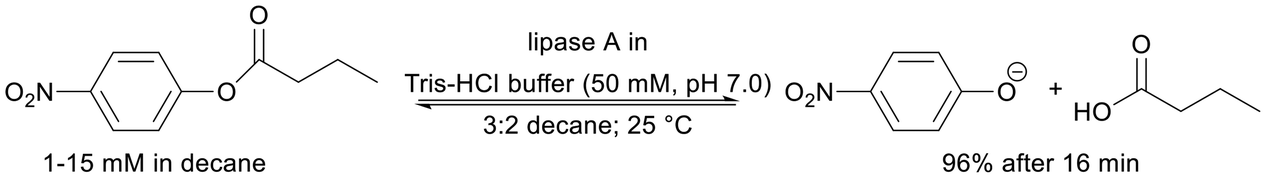
| Physical adsorption via multipoint interactions between the enzyme and the carrier surface | High productivities in the hydrolysis of 4-NPB in media: Tris-HCl buffer and decane solvent | Proof of higher activity in immobilised enzymes compared to free enzymes | 4294 | 1042 | 175 | |
|
||||||
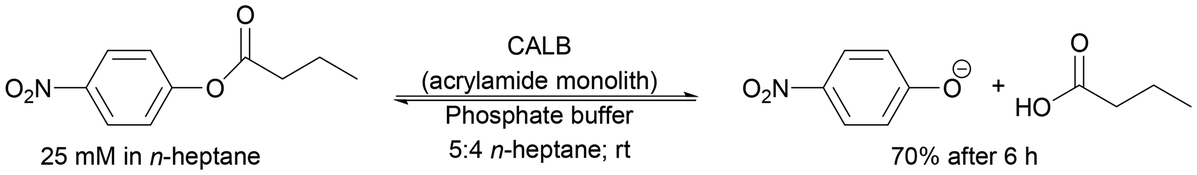
| Acrylamide porous monolith | Encapsulation and cross-linking | Specific activity above 90% in 15 cycles | Development of a system to efficiently immobilise enzymes and perform reactions | 282 | 19241 |
176 |
|
||||||
| Hybrid polyurethane monolith with cellulase microbeads | Encapsulation and cross-linking | Higher productivities compared to batch-type reactions | Creation of an efficient system for chiral ester and amide synthesis | n.a. | 5 | 177 |
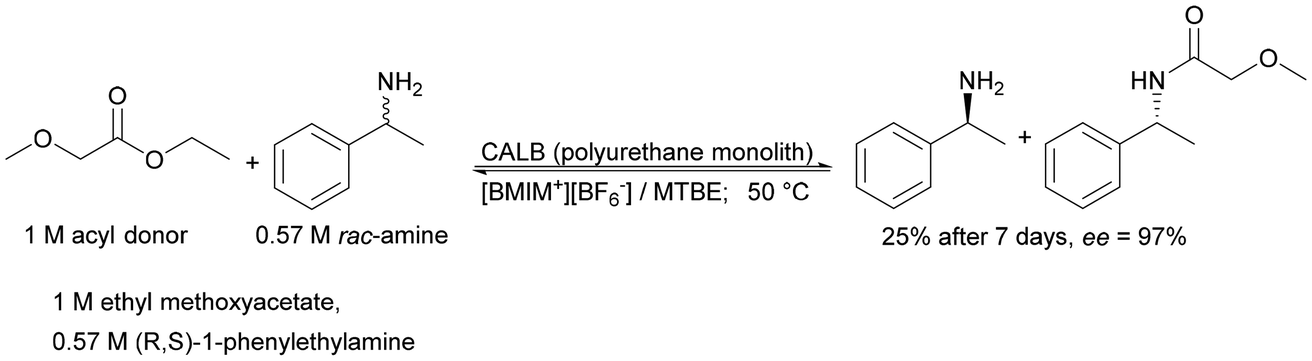
|
||||||
| Hybrid polymeric monoliths: from styrene or acrylate, supported with ILs | Covalently supported IL phase with an absorbed enzyme solution | The best results were obtained with the most hydrophobic monolith | Synthesis of citronellal propionate in supercritical carbon dioxide by transesterification | n.a. | 20 | 182 |
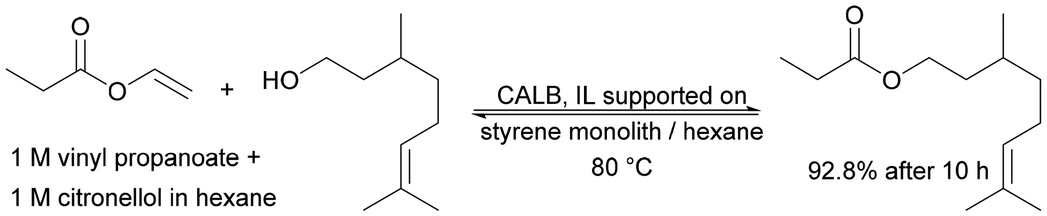
|
||||||
| Thiolene monolith with PDMS wall | Adsorption via thiol groups | 40 h operational stability with diffused oxygen supply | Alternative approach for efficient enzymatic synthesis with continuous gas delivery | n.a. | n.a. | 181 |
|
||||||
The two-phase processes are in many cases still in a development stage. Therefore, reliable calculations of EF and STY are often not possible. The example of MsAcT in an aqueous organic system does however also indicate the potential to achieve the green chemistry principles.
5. Reaction cascades in monoliths
Multi-enzymatic cascade reactions have become very popular in recent years and represent a promising route to the production of pharmaceuticals, cosmetics, biofuels or fine chemicals.183,184 In contrast to classical multi-step synthesis, multi-enzymatic cascade reactions eliminate the need to isolate and purify intermediates after each step, thus reducing the number of steps and DSP. This saves time, reduces the amount of solvent used and therefore generates less waste, reducing the environmental and economic costs while at the same time helping to implement the principles of green chemistry. In many cases, higher yields are also achieved because, in cascade systems, the product formed in one step is rapidly consumed in the next, so the balance of the enzymatic reaction can be shifted towards the product, avoiding the need for excess reactants.156 In addition, the immobilisation of multiple enzymes enables the use of cascade processes in industry. Multiple enzyme immobilisation can be achieved by immobilising individual enzymes on a separate support or by co-immobilising all enzymes on a single support (Fig. 6). The advantage of the former is undoubtedly the flexibility of unit operation and the ability to determine the activity and stability of each immobilised enzyme, which is unfortunately very difficult for co-immobilised enzymes. However, co-immobilisation of enzymes reduces or eliminates the lag time, which can contribute to increased reaction rates and catalytic efficiency. This lag occurs when several enzymes are immobilised on separate supports and is due to the fact that the concentration of intermediates is initially low, preventing subsequent enzymes in the reaction chain from showing activity at the beginning of the reaction. Co-immobilisation of enzymes also removes the diffusion limitations in the transfer of intermediates from the active site of the first enzyme to the second, which is the case when immobilised on separate supports. However, when immobilising several enzymes on a single support, it is not only the enzyme/support interactions that are significant, but also the properties of the different enzymes. It is extremely important to find optimal conditions (temperature, pH and reaction medium) where all the enzymes involved in the cascade show high activity and stability. In the case of independently immobilised enzymes, a different, specifically optimised immobilisation protocol can be used for each enzyme, the most appropriate support and its modification (e.g., a functional group on the support).185,186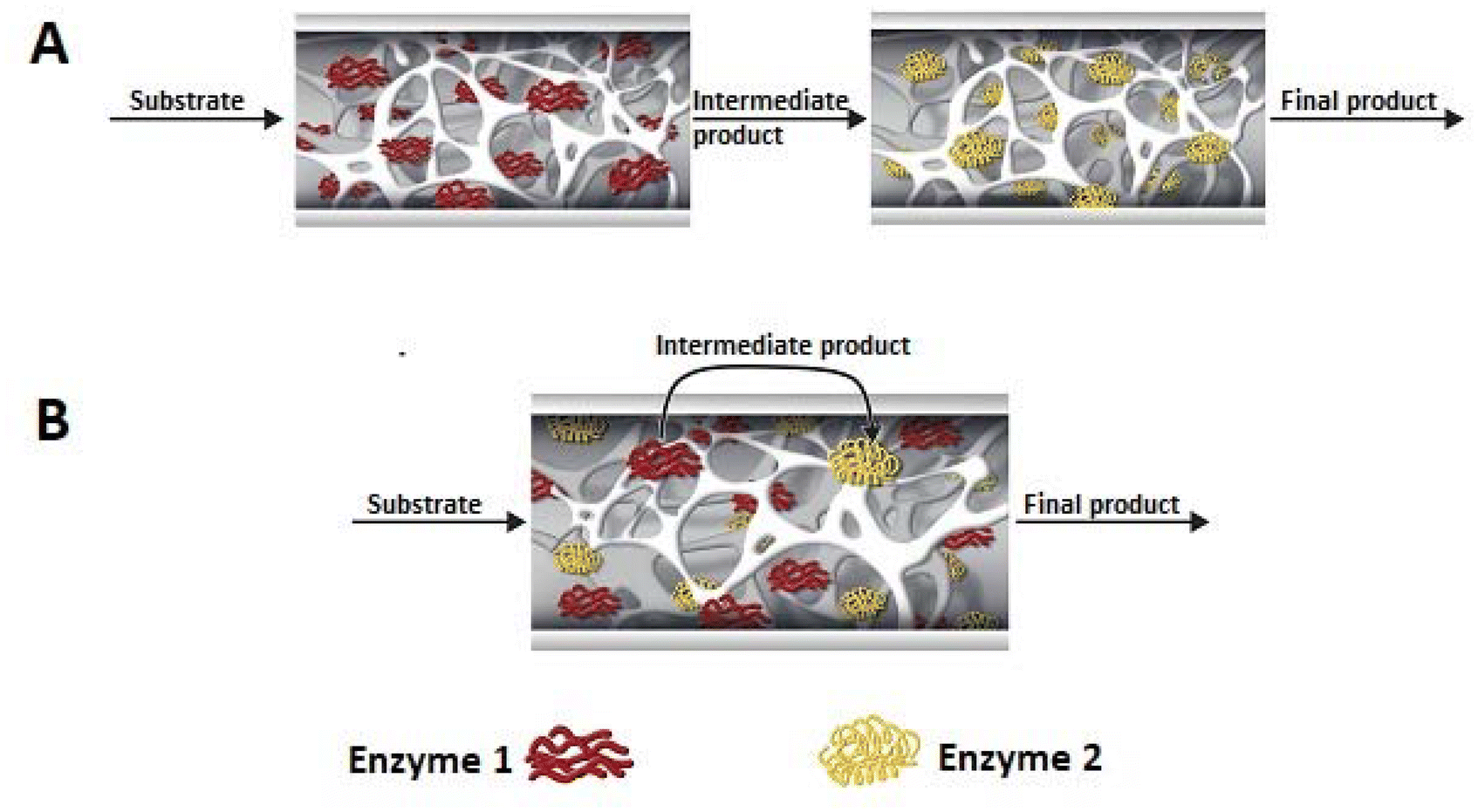 | ||
| Fig. 6 Classification of multienzyme cascade reactions in monoliths in flow: catalysed by a sequential immobilised enzyme system (stepwise) (A) and catalysed by a co-immobilised one-monolith enzyme system (B). | ||
5.1 Cascades in one monolith
One of the variants of the multienzyme cascade processes is a single reactor system, where all enzymes are immobilised on the same monolith. Co-immobilising enzymes is greatly facilitated by the use of the same immobilisation conditions for each biocatalyst as well as the same range of temperature and pH optimum of enzymes subjected to the co-immobilisation process. Meeting these conditions is necessary to obtain the most efficient co-immobilised multi-enzymatic system possible. Recent reports on the use of co-immobilised multienzyme systems on monolithic carriers mainly refer to the co-immobilisation of oxidoreductases and lyases. A summary of the application of co-immobilised multienzymatic monolithic reactors is presented in Table 3.| Immobilised enzymes | Monolith type | Application | Ref. |
|---|---|---|---|
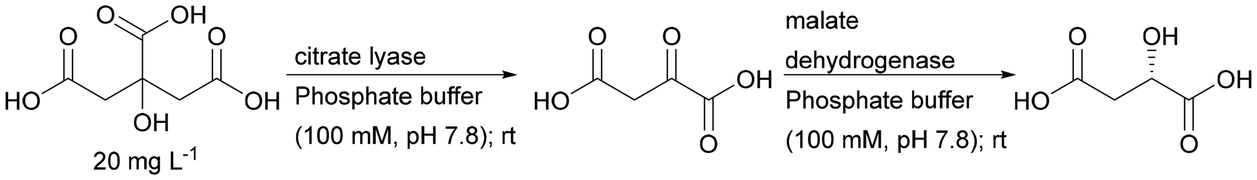
| Citrate lyase + malate dehydrogenase | CIM disk – commercial polymethacrylate monolithic column functionalised with epoxide groups | Indirect analysis of citrate | 187 |
|
|||
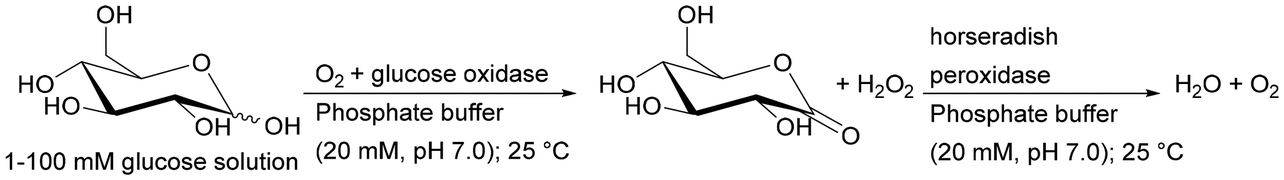
| Horseradish peroxidase + glucose oxidase | pH-responsive polymethacrylate monolithic capillary grafted with poly(4-vinylphenylboronic acid) | Glucose detection, obtaining reusable biosensors | 188 |
|
|||
| Trypsin + chymotrypsin | Chromolith column – commercial monolithic silica column functionalised with epoxide groups | Protein digestion | 189 |
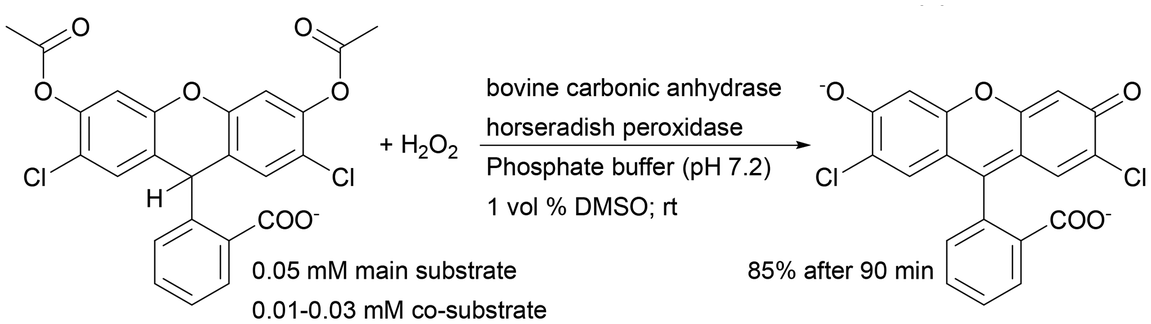
| Horseradish peroxidase + bovine carbonic anhydrase | Silica monolith with a dendronized polymer–enzyme conjugate | Controllable immobilisation method application | 55 |
|
|||
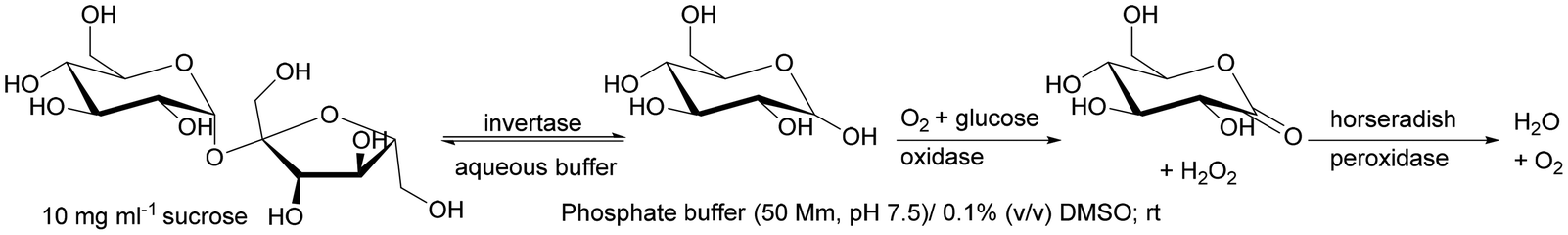
| Invertase + glucose oxidase + horseradish peroxidase | Lab-made polymethacrylate monolithic capillary column grafted with vinyl azlactone | Directional multienzymatic reactions | 190 |
|
|||
An interesting example demonstrating the superiority of using a two-enzyme system in a single monolith is the cascade created by citrate lyase (CL) and malate dehydrogenase (MDH). Initially, the enzymes were immobilised separately and worked in a sequential arrangement of CL and the following MDH. For its catalytic activity, CL requires Mg2+ ions. This can however also form a complex with the product of the reaction that inhibits the enzyme CL. The use of sequential immobilisation promoted the formation of this complex and inhibited the process. Only the embedding of both enzymes in one monolith allowed immediate transformation of the product, significantly improving the stability of CL.187 Monoliths based on the silica skeleton have found applications in the construction of heterogeneous enzyme systems using dendronised polymer–enzyme conjugates. This solution allowed for the reproducible immobilisation of predetermined amounts of catalytically active enzyme molecules.
Recently, such a system has been used for immobilisation of horseradish peroxidase isoenzyme C (HRP) and bovine erythrocyte carbonic anhydrase (BCA) and has been tested as a heterogeneous biocatalyst in a one-monolith cascade reaction with co-immobilised enzymes and two bioreactors in a sequential system in 2′,7′-dichlorofluorescein synthesis. For both reactor systems, the final product formation was the same with 30 μM hydrogen peroxide (co-substrate). However, the application of 10 μM H2O2 shows about 20% higher product formation with co-immobilised enzymes.55 This clearly indicated a higher reaction efficiency when the co-immobilisation system is used, which additionally shortens the delay time even if lower substrate concentrations are used.
An interesting example is the co-immobilisation of invertase (INV), glucose oxidase (GOX) and HRP inside a polymeric monolith structure within microfluidic devices by photo-patterning. The enzymes were co-immobilised in different orders of occurrence in the monolith, which was made possible by the use of photo-patterned vinyl azlactone grafting, through which immobilisation occurred. The first step of the reaction was the hydrolysis of sucrose (first substrate) to glucose and fructose catalysed by INV, then GOX oxidises glucose to gluconolactone and hydrogen peroxide, which was further used by HRP to oxidise Amplex Red (second substrate) to fluorescent resorufin. The authors established that only the sequence of co-immobilised enzymes INV-GOX-HRB gave high product yield.190 This demonstrates how important not only the close contact of enzymes involved in the cascade but also placing the co-immobilised biocatalysts in the correct order on the monolith (if possible) is to control the reaction sequence.
All of these examples are proof of principle to solve a given problem (product inhibition, order of immobilisation) that were not directed towards high productivity and therefore no meaningful EF or STY could be calculated.
5.2 Monoliths with separate reactions in a row/parallel
The second possibility for the multienzyme cascade processes is to link multiple microreactor (monolithic) systems, where each enzyme is immobilised on a separate monolith under optimal conditions and can be applied at different reaction parameters (Fig. 6A). The advantages of separate immobilisation of enzymes on individual monoliths are the availability of a single reactant that can be used either independently or in various combinations in sequence as well as separate replacements once the individual enzymes lose activity. A summary of the application of sequential multienzyme monolithic reactors is presented in Table 4.| Monolith type | Application | EF [gwaste gproduct−1] | STY [gproduct L−1 h−1] | Ref. |
|---|---|---|---|---|
| AhPNP, purine nucleoside phosphorylase; CpUP, uridine phosphorylase; mCherry-TuTreT, trehalose transferase; TaGalU, UDP-glucose pyrophosphorylase | ||||
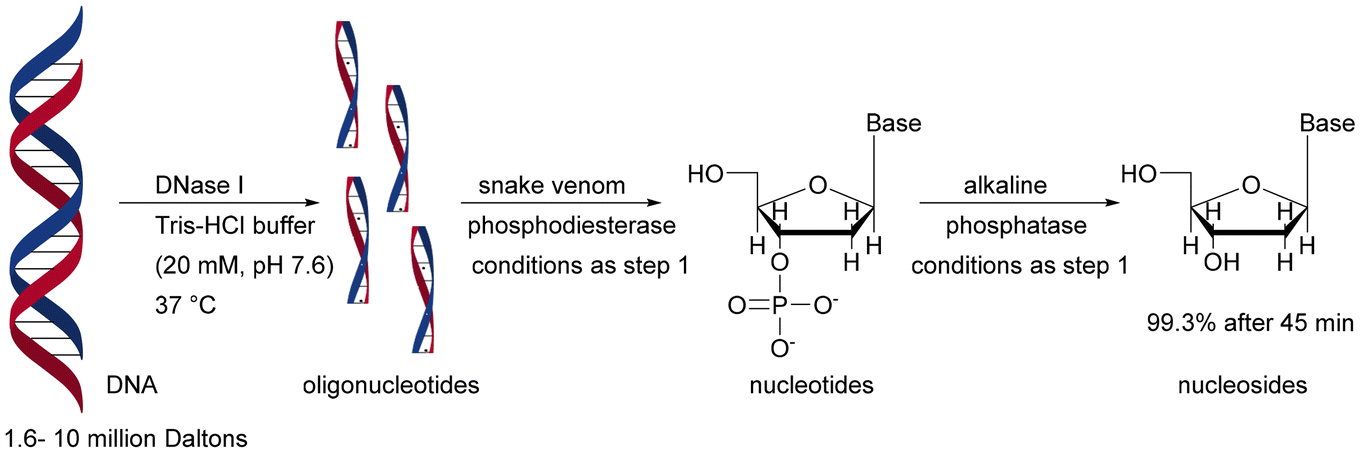
| Silica based capillary monolith functionalised via amino groups using glutaraldehyde as a cross-linking agent | Digestion of genomic DNA, analysis and identification of structural DNA modifications | n.a. | n.a. | 191 and 192 |
|
||||
| Chromolith flash silica support | Synthesis of arabinosyladenine, an antiviral nucleoside | 1.42 | 4.32 | 193 |
|
||||
| Lab-made silica monoliths functionalised via amino groups | Trehalose synthesis, transformation of sugar derivatives | 5.18 | I step: 12–91 | 156 |
| II step: 12–70 | ||||
|
||||
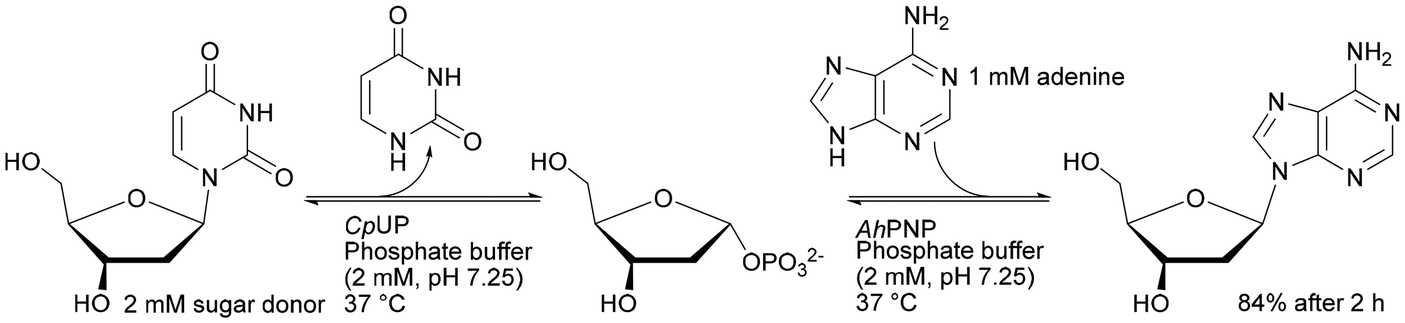
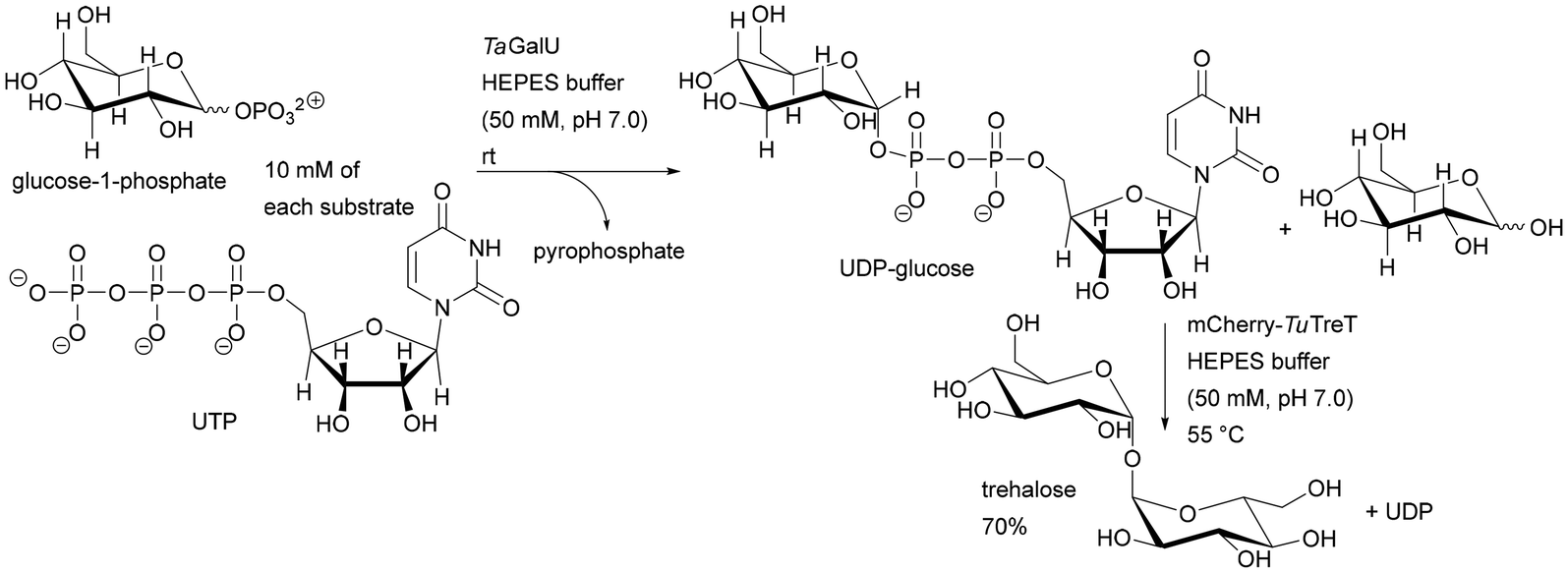
The importance of the order of using a single-enzyme microreactor in a multienzyme sequential cascade was demonstrated in studies on the enzymatic digestion of genomic DNA. Three enzymes – deoxyribonuclease I (DNase I), snake venom phosphodiesterase (SVP), and alkaline phosphatase (ALPase) – were constructed in cascade bioreactors assembled by tandem connection (DNase I – SVP – ALPase) on silica-based capillary monoliths. The developed solution is promising in highly sensitive detection and rapid identification of adducts in genomic DNA samples. The correct connection of the bioreactor sequence has an influence on the digestion performance. Removing the SVP bioreactor or placing it before the cascade (instead of the DNase I bioreactor) reduced the digestion efficiency from 99.3% to 59.8–83.7%.191 Both DNase I and SVP are capable of generating mononucleotides, but DNase I tends to digest double-stranded DNA to generate small DNA fragments. Thus, combining with DNase I at the outset provided a high yield of genomic DNA digestion. The authors of this study succeeded in developing a system that contained benzonase endonuclease instead of DNase I, resulting in a reduction in genomic DNA digestion time from 45 to 10 minutes.192 Stability tests showed less than 20% variation of enzyme activity when the immobilised bioreactor was stored in Tris-HCl buffer (10 mM, pH 7.4) at 4 °C for 30 days. As expected from earlier data,134–138,144–146 the free enzyme solution mixture, stored under the same conditions, lost its activity within 1 day. Therefore, the cascade monolithic bioreactors have an advantage in terms of stability and reusability. A direct comparison of immobilised enzymes for batch and in a monolith is given above (Table 1), displaying advanced reactivity and stability for the monolith.
Monolith microreactors were also employed for the synthesis of APIs or pharmaceutical intermediates. Purine nucleoside phosphorylase from Aeromonas hydrophila (AhPNP) was earlier covalently immobilised on a silica particle based support, which caused a high backpressure resulting in a loss of enzyme activity.194 Two years later, a study was published that used a cascade reaction with immobilised uridine phosphorylase from Clostridium perfringens (CpUP) on a commercial Chromolith Flash monolithic support. This allowed reducing the back-pressure and obtaining the desired nucleoside products.195 The co-immobilised CpUP and AhPNP on one monolith support in a one-reactor system provided 84% conversion over 120 min in flow (0.5 ml min−1) reaction for the synthesis of 2′-deoxyadenosine.193 However, the step-by-step two bioreactor system approached 95% conversion yield after 90 min reaction with the same flow.195 Co-immobilised systems mimic the “one-pot” transglycosylation system involving two enzymes, but independent bioreactors turned out to be better for controlling the multienzyme reaction.
Heterogeneous multienzyme systems have applications, especially in sugar synthesis and transformation of sugar derivatives. Traditional chemical pathways to sugars usually suffer from poor stereoselectivity and the downstream processes and purification of intermediate products are difficult. Several multienzyme systems suitable for sugar coupling systems have been found. An excellent example is the enzymatic synthesis of trehalose catalysed by UDP-glucose pyrophosphorylase from Thermocrispum agreste (TaGalU) and trehalose transferase from Thermoproteus uzoniensis (mCherry-TuTreT) immobilised on monolithic silica supports in two variants: co-immobilisation and step-by-step synthesis. Significant differences in temperature requirements affected both the activity and stability of each immobilised enzyme, preventing both reactions from occurring simultaneously in one microreactor. Thus, the application of a sequential cascade allowed the reaction to be carried out in separate monoliths, maintaining ideal temperature conditions for each enzyme, allowing the highest activity for each of the immobilised biocatalysts. Under optimised conditions, the productivity values varied, depending on the applied flow rate, from 1.9 to 14.4 g UDP-glucose per L per h per mgprotein obtained by immobilised TaGalU and 8.3 to 49.6 g trehalose per L per h per mgprotein obtained by immobilised mCherry-TuTreT.57
The latter examples have excellent EF (1.42 and 5.18) and STY, again demonstrating the power of flow chemistry to achieve the green chemistry principles.
5.3 Cofactors and considerations concerning them
The selective syntheses catalysed by enzymes are very versatile and straightforward to perform. However, a wide range of enzymes requires a cofactor. This can be a metal or an organic cofactor such as thiamine or Fe-containing porphyrins. These cofactors are tightly bound to the enzyme and need to be replenished only occasionally. Therefore they do not need to be taken into consideration during enzyme immobilisation. For redox enzymes, the cofactor, especially nicotinamide adenine dinucleotide (NADH) and its phosphorylated form (NADPH), needs to be regenerated. This is particularly important when considering industrial scale processes and has to date been solved for batch processes. There are a number of methods for regenerating cofactors4,196,197i.e. enzymatic, chemical, electrochemical or photocatalytic. Of the methods listed here, those using enzymes are the most popular. The use of isolated enzymes rather than whole cells allows much better process control and we will focus on this approach. Two main strategies for cofactor regeneration are known (Fig. 7). In the easiest case, found mainly for alcohol dehydrogenases (ADH), a ‘coupled substrate’ allows the application of a single enzyme that catalyses both substrate conversion and cofactor regeneration. The cofactor does not leave the active site and the same considerations as for the cofactors like thiamine apply. In the case of ADHs, high concentrations of the sacrificial substrate are necessary to drive the equilibrium toward the desired product. While straightforward, this in turn leads to a loss of activity in the main target reaction as a result of competition among substrates and cosubstrates for the same active site on the ADH.198 The ‘coupled enzyme’ involves the use of two enzymes, the first to produce the main product and the second to regenerate a cofactor with the production of a by-product. The by-product is typically chosen to ensure an irreversible reaction and thus maximal driving force and optimal yields in the desired reaction. Clearly here the cofactor has to be mobile and its immobilisation has to be considered separately.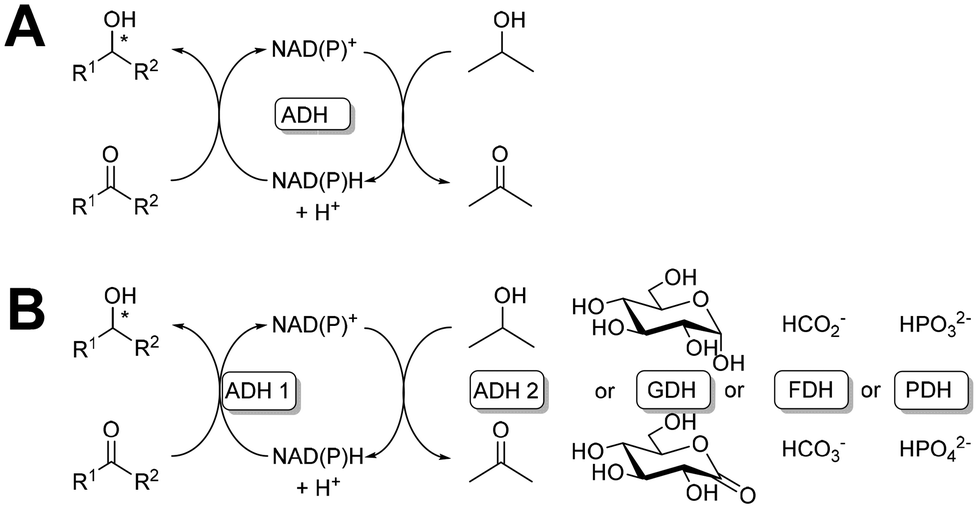 | ||
| Fig. 7 Schematic representation of cofactor enzymatic regeneration: a coupled substrate system (A) and a coupled enzyme system (B). | ||
Combining oxidation and reduction in a cascade leads to economical and efficient transformation. This is why co-immobilisation is so eagerly used to design multienzyme cascade systems for cofactor recycling. It allows us to obtain self-sufficient heterogeneous biocatalysts that do not require exogenous cofactor delivery.199,200
A novel micromonolith bioreactor with co-immobilised ADH from Thermoanaerobacter brockii (TbSADH) and NADP(H) has been recently developed to provide a cost-effective basis for cofactor-dependent biocatalysis.201 The cell extract with TbSADH was cross-linked by a hydrogel (cEAG) and then the cofactor was cross-linked to form a NADP@cEAG monolithic microreactor. There is no need to leave the active site of the enzyme to fully regenerate the cofactor during the catalytic cycle (Fig. 7A). The microreactor with a heterogeneous biocatalyst and cofactor was characterized by high enantioselectivity (<99%) and a high STY of 46.3 g L−1 h−1 with 94.8% conversion in the flow synthesis of (R)-1-phenylethanol at 0.01 ml min−1 flow rate, which indicates the unique advantage of co-immobilisation on monolith microreactors in recycling cofactors in flow. The additional co-substrate was isopropanol, which did not generate a significant increase in the process costs.
Application of a coupled enzyme system in cofactor regeneration was tested in an enzymatic reaction for the synthesis of formate from carbon dioxide gas in a tandem reaction. The production enzyme was a high-performance formate dehydrogenase from Methylobacterium extorquens AM1 (MeFDH1), while the regeneration of NADH was performed by phosphite dehydrogenase from Pseudomonas stutzeri (PsPDH). Both enzymes and cofactors were immobilised by cross-linking with poly(ethylene glycol) diglycidyl ether (PEGDGE) on hierarchical porous inorganic monoliths, from silica or carbon. This allowed the fabrication of a system of two heterogeneous biocatalysts with a flexibly bound cofactor capable of moving between active centers for regeneration6 (Fig. 7B). After testing, this carbon monolithic reactor proved superior to achieve the highest productivity reported to date for NAD-dependent biocatalytic systems for producing formate from carbon dioxide gas without the need for additional hydrogenation and under atmospheric conditions.202
The immobilisation of cofactors for the coupled enzyme approach and their effective recovery through their reuse203,204 is a prerequisite for the economic application of these enzymes. However, immobilised cofactors may have lower availability than free cofactors dissolved in the reaction mixture. This is as a result of slower mass transfer, which must be compensated by the cofactor recycling and reuse.198 Both ionic and covalent immobilisation have been studied. At low buffer strengths, ionic immobilisation on ionic carriers has been successful. Permanent binding of cofactors with long-lasting activity is still a challenge. The use of encapsulation leads to rapid leaching of the cofactor in the reaction solution.205 One way to improve stability is to link the cofactor to another macromolecule (e.g. dextran,206 chitosan207) via a permanent covalent bond. Noncovalent attachment of NAD+ on carbon nanotubes208 or covalent binding of NAD+via epoxy groups to the surface of a silica matrix obtained by the sol–gel method has also been proposed.205 However, permanent binding of cofactors on monoliths is still a relatively new topic, with challenging research questions209,210 and solutions.
6 Conclusions
To summarise the above discussion from a green chemistry perspective, biocatalysis in a continuous flow system carried out in monolithic microreactors fulfils most of the principles of green chemistry. The high selectivity of enzymes ensures that virtually no side-products are formed and thus no waste is generated. The combination of biocatalysis and flow chemistry additionally minimises energy consumption by intensifying mass and heat exchange. In addition, flow chemistry in monolithic microreactors allows for a significant reduction in reaction volume while maintaining a high efficiency of the process carried out. As exemplified in Table 1 for the conversion of benzaldehyde with hydrogen cyanide149 and in Table 2 for transesterifications,174 the residence time in a monolithic microreactor at which near 100% conversion and enantioselectivity is achieved is often less than a few minutes. While biocatalysis in flow is not yet fully applicable on an industrial scale, its potential for accelerating the use of enzymes towards green chemistry principles is obvious.Author contributions
AL, KS and UH wrote the review. Together they edited and designed the structure of the review.Data availability
This is a review; therefore, we do not have any data except for the references in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for generous financial support from the European Union's European Innovation Council programme under grant agreement 101136310 (cassaFLOW for U. H.) and from the Ministry of Education and Science (Poland) programme under grant agreement PN/01/0267/2022 (The Pearls of Science for A. L.).References
- A. R. Alcántara, P. Domínguez de María, J. A. Littlechild, M. Schürmann, R. A. Sheldon and R. Wohlgemuth, ChemSusChem, 2022, 15, e202102709 CrossRef PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55–71 CrossRef PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef PubMed.
- U. Hanefeld, F. Hollmann and C. E. Paul, Chem. Soc. Rev., 2022, 51, 594–627 RSC.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H. I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef PubMed.
- J. Coloma, Y. Guiavarc'h, P. L. Hagedoorn and U. Hanefeld, Chem. Commun., 2021, 57, 11416–11428 RSC.
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437–6474 RSC.
- S. C. Cosgrove and A. P. Mattey, Chem. – Eur. J., 2022, 28, e202103607 CrossRef CAS PubMed.
- A. Naramittanakul, S. Buttranon, A. Petchsuk, P. Chaiyen and N. Weeranoppanant, React. Chem. Eng., 2021, 6, 1771–1790 RSC.
- T. Sokač Cvetnić, A. Šalić, M. Benković, T. Jurina, D. Valinger, J. Gajdoš Kljusurić, B. Zelić and A. Jurinjak Tušek, Catalysts, 2023, 13, 708 CrossRef.
- Y. J. Hu, J. Chen, Y. Q. Wang, N. Zhu, Z. Fang, J. H. Xu and K. Guo, Chem. Eng. J., 2022, 437, 135400 CrossRef CAS.
- M. B. Ansorge-Schumacher and O. Thum, Chem. Soc. Rev., 2013, 42, 6475–6490 RSC.
- Y. Asano, S. Togashi, H. Tsudome and S. Murakami, Pharm. Eng., 2010, 30, 1–9 Search PubMed.
- J. M. Woodley and N. J. Titchener-Hooker, Bioprocess Eng., 1996, 14, 263–268 CrossRef.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef PubMed.
- Z. Tang, Y. Oku and T. Matsuda, Org. Process Res. Dev., 2024, 28, 1308–1326 CrossRef.
- P. Žnidaršič-Plazl, Biotechnol. J., 2019, 14, 1800580 CrossRef.
- D. Pirozzi, M. Abagnale, L. Minieri, P. Pernice and A. Aronne, Chem. Eng. J., 2016, 306, 1010–1016 CrossRef.
- K. S. Elvira, X. C. I. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef PubMed.
- A. deMello and R. Wootton, Lab Chip, 2002, 2, 7–13 RSC.
- J. Lawrence, B. O'Sullivan, G. J. Lye, R. Wohlgemuth and N. Szita, J. Mol. Catal. B: Enzym., 2013, 95, 111–117 CrossRef.
- H. Kim, A. Nagaki and J. I. Yoshida, Nat. Commun., 2011, 2, 264 CrossRef.
- J. González-Álvarez, P. Arias-Abrodo, M. Puerto, M. E. Viguri, J. Perez and M. D. Gutiérrez-Álvarez, New J. Chem., 2015, 39, 8560–8568 RSC.
- A. Abdul Halim, N. Szita and F. Baganz, J. Biotechnol., 2013, 168, 567–575 CrossRef PubMed.
- M. Oelschlägel, A. Riedel, A. Zniszczoł, K. Szymańska, A. B. Jarzębski, M. Schlömann and D. Tischler, J. Biotechnol., 2014, 174, 7–13 CrossRef PubMed.
- K. Szymańska, M. Pietrowska, J. Kocurek, K. Maresz, A. Koreniuk, J. Mrowiec-Białoń, P. Widłak, E. Magner and A. Jarzębski, Chem. Eng. J., 2016, 287, 148–154 CrossRef.
- K. Szymańska, A. Ciemięga, K. Maresz, W. Pudło, J. Malinowski, J. Mrowiec-Białoń and A. B. Jarzębski, Front. Chem. Eng., 2021, 3, 1–14 Search PubMed.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 352, 61–67 CrossRef PubMed.
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev. - Sci. Eng., 2001, 43, 345–380 CrossRef.
- J. L. Williams, Catal. Today, 2001, 69, 3–9 CrossRef.
- C. Yao, L. Qi, W. Hu, F. Wang and G. Yang, Anal. Chim. Acta, 2011, 692, 131–137 CrossRef PubMed.
- Y. Liang, D. Tao, J. Ma, L. Sun, Z. Liang, L. Zhang and Y. Zhang, J. Chromatogr. A, 2011, 1218, 2898–2905 CrossRef PubMed.
- S. Wu, L. Sun, J. Ma, K. Yang, Z. Liang, L. Zhang and Y. Zhang, Talanta, 2011, 83, 1748–1753 CrossRef.
- A. Sachse, A. Galarneau, F. Di Renzo, F. Fajula and B. Coq, Chem. Mater., 2010, 22, 4123–4125 CrossRef.
- A. Koreniuk, K. Maresz and J. Mrowiec-Białoń, Catal. Commun., 2015, 64, 48–51 CrossRef.
- A. Sachse, A. Galarneau, F. Fajula, F. Di Renzo, P. Creux and B. Coq, Microporous Mesoporous Mater., 2011, 140, 58–68 CrossRef.
- A. Koreniuk, K. Maresz, K. Odrozek and J. Mrowiec-Białoń, Microporous Mesoporous Mater., 2016, 229, 98–105 CrossRef.
- A. Galarneau, A. Sachse, B. Said, C. H. Pelisson, P. Boscaro, N. Brun, L. Courtheoux, N. Olivi-Tran, B. Coasne and F. Fajula, C. R. Chim., 2016, 19, 231–247 CrossRef.
- J. Ma, Z. Liang, X. Qiao, Q. Deng, D. Tao, L. Zhang and Y. Zhang, Anal. Chem., 2008, 80, 2949–2956 CrossRef PubMed.
- P. Yang, T. Deng, D. Zhao, P. Feng, D. Pine, B. F. Chmelka, G. M. Whitesides and G. D. Stucky, Science, 1998, 282, 2244–2246 CrossRef.
- C. G. Oh, Y. K. Baek and S. K. Ihm, Adv. Mater., 2005, 17, 270–273 CrossRef.
- M. Antonietti, B. Berton, C. Göltner and H. P. Hentze, Adv. Mater., 1998, 10, 154–159 CrossRef.
- Q. Luo, L. Li, B. Yang and D. Zhao, Chem. Lett., 2000, 29, 378–379 CrossRef.
- Y. Shin, L. Wang, J. H. Chang, W. D. Samuels and G. J. Exarhos, Stud. Surf. Sci. Catal., 2003, 146, 447–451 CrossRef.
- K. Nakanishi, J. Porous Mater., 1997, 4, 67–112 CrossRef.
- K. Nakanishi, H. Minakuchi, N. Soga and N. Tanaka, J. Sol-Gel Sci. Technol., 1997, 8, 547–552 CAS.
- R. Takahashi, K. Nakanishi and N. Soga, J. Non-Cryst. Solids, 1995, 189, 66–76 CrossRef CAS.
- K. Nakanishi, J. Sol-Gel Sci. Technol., 2000, 19, 65–70 CrossRef CAS.
- K. Nakanishi, Y. Sato, Y. Ruyat and K. Hirao, J. Sol-Gel Sci. Technol., 2003, 26, 567–570 CrossRef CAS.
- J. H. Smått, S. Schunk and M. Lindén, Chem. Mater., 2003, 15, 2354–2361 CrossRef.
- A. Ciemięga, K. Maresz and J. Mrowiec-Białoń, Appl. Catal., A, 2018, 560, 111–118 CrossRef.
- A. Ciemięga, K. Maresz and J. Mrowiec-Białoń, Microporous Mesoporous Mater., 2017, 252, 140–145 CrossRef.
- K. Maresz, A. Ciemięga and J. Mrowiec-Białoń, Chem. Eng. J., 2020, 379, 122281 CrossRef.
- N. Ghéczy, S. Tao, S. Pour-Esmaeil, K. Szymańska, A. B. Jarzębski and P. Walde, Macromol. Biosci., 2023, 23, 1–20 CrossRef PubMed.
- N. Ghéczy, W. Xu, K. Szymańska, A. B. Jarzębski and P. Walde, ACS Omega, 2022, 7, 26610–26631 CrossRef PubMed.
- K. Szymańska, W. Pudło, J. Mrowiec-Białoń, A. Czardybon, J. Kocurek and A. B. Jarzębski, Microporous Mesoporous Mater., 2013, 170, 75–82 CrossRef.
- D. Kowalczykiewicz, M. Przypis, L. Mestrom, A. Kumpf, D. Tischler, P. L. Hagedoorn, U. Hanefeld, A. Jarzębski and K. Szymańska, Chem. Eng. J., 2022, 427, 1–8 CrossRef.
- H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka and N. Tanaka, Anal. Chem., 1996, 68, 3498–3501 CrossRef CAS PubMed.
- N. Ishizuka, H. Minakuchi, K. Nakanishi, N. Soga and N. Tanaka, J. Chromatogr. A, 1998, 797, 133–137 CrossRef CAS.
- M. Kozakiewicz-Latała, D. Marciniak, K. Krajewska, A. Złocińska, K. Prusik, B. Karolewicz, K. P. Nartowski and W. Pudło, Mol. Pharm., 2023, 20, 641–649 CrossRef PubMed.
- W. Pudło, P. Borys, G. Huszcza, A. Staniak and R. Zakrzewska, Eur. J. Pharm. Biopharm., 2019, 141, 12–20 CrossRef.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, T. Abe and K. Nakanishi, Chem. Mater., 2016, 28, 3944–3950 CrossRef CAS.
- B. Luangon, A. Siyasukh, P. Winayanuwattikun, W. Tanthapanichakoon and N. Tonanon, J. Mol. Catal. B: Enzym., 2012, 75, 80–85 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155–166 CrossRef CAS.
- W. Kiciński, M. Norek and M. Bystrzejewski, J. Phys. Chem. Solids, 2013, 74, 101–109 CrossRef.
- Z. G. Shi, F. Chen, J. Xing and Y. Q. Feng, J. Chromatogr. A, 2009, 1216, 5333–5339 CrossRef CAS.
- L. Wang, Q. Zhang, S. Chen, F. Xu, S. Chen, J. Jia, H. Tan, H. Hou and Y. Song, Anal. Chem., 2014, 86, 1414–1421 CrossRef CAS.
- Y. Song, X. Lu, Y. Li, Q. Guo, S. Chen, L. Mao, H. Hou and L. Wang, Anal. Chem., 2016, 88, 1371–1377 CrossRef CAS.
- P. A. Goodman, H. Li, Y. Gao, Y. F. Lu, J. D. Stenger-Smith and J. Redepenning, Carbon, 2013, 55, 291–298 CrossRef CAS.
- C. C. Kung, P. Y. Lin, F. J. Buse, Y. Xue, X. Yu, L. Dai and C. C. Liu, Biosens. Bioelectron., 2014, 52, 1–7 CrossRef CAS PubMed.
- L. Y. Xu, Z. G. Shi and Y. Q. Feng, Microporous Mesoporous Mater., 2008, 115, 618–623 CrossRef CAS.
- G. Hasegawa, K. Kanamori and K. Nakanishi, Microporous Mesoporous Mater., 2012, 155, 265–273 CrossRef CAS.
- C. Bernal, S. Escobar, L. Wilson, A. Illanes and M. Mesa, Carbon, 2014, 74, 96–103 CrossRef CAS.
- W. Sebai, S. Ahmad, N. Brun, T. Cacciaguerra, D. Cot, A. Boccheciampe, P. Chaurand, C. Levard, M. P. Belleville, J. Sanchez-Marcano and A. Galarneau, Chem. Mater., 2023, 35, 8464–8482 CrossRef.
- Y. Zhang, Y. Wang, Y. Tang, R. Li and Y. Ji, Anal. Methods, 2019, 11, 2465–2472 RSC.
- L. Wang, Y. Zhao, Y. Zhang, T. Zhang, J. Kool, G. W. Somsen, Q. Wang and Z. Jiang, J. Chromatogr. A, 2018, 1563, 135–143 CrossRef PubMed.
- J. Ma, L. Zhang, Z. Liang, W. Zhang and Y. Zhang, J. Sep. Sci., 2007, 30, 3050–3059 CrossRef PubMed.
- N. Li, W. Zheng, Y. Shen, L. Qi, Y. Li, J. Qiao, F. Wang and Y. Chen, J. Sep. Sci., 2014, 37, 3411–3417 CrossRef.
- T. Hong, X. Yang, Y. Xu and Y. Ji, Anal. Chim. Acta, 2016, 931, 1–24 CrossRef.
- Ç. Kip, C. Demir and A. Tuncel, J. Chromatogr. A, 2017, 1502, 14–23 CrossRef PubMed.
- Q. C. Wang, F. Svec and J. M. J. Fréchet, Anal. Chem., 1993, 65, 2243–2248 CrossRef CAS PubMed.
- A. Kurganov, Anal. Chim. Acta, 2013, 775, 25–40 CrossRef PubMed.
- D. Moravcová, A. H. Rantamäki, F. Duša and S. K. Wiedmer, Electrophoresis, 2016, 37, 880–912 CrossRef PubMed.
- E. Candish, A. Khodabandeh, M. Gaborieau, T. Rodemann, R. A. Shellie, A. A. Gooley and E. F. Hilder, Anal. Bioanal. Chem., 2017, 409, 2189–2199 CrossRef PubMed.
- E. G. Vlakh, M. A. Stepanova, Y. M. Korneeva and T. B. Tennikova, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1029–1030, 198–204 CrossRef.
- E. Calleri, C. Temporini, F. Gasparrini, P. Simone, C. Villani, A. Ciogli and G. Massolini, J. Chromatogr. A, 2011, 1218, 8937–8945 CrossRef.
- D. Josic and J. G. Clifton, J. Chromatogr. A, 2007, 1144, 2–13 CrossRef PubMed.
- S. Jiang, Z. Zhang and L. Li, J. Chromatogr. A, 2015, 1412, 75–81 CrossRef CAS PubMed.
- M. Naldi, U. Černigoj, A. Štrancar and M. Bartolini, Talanta, 2017, 167, 143–157 CrossRef CAS.
- K. Meller, P. Pomastowski, M. Szumski and B. Buszewski, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1043, 128–137 CrossRef CAS.
- J. Křenková and F. Foret, Electrophoresis, 2004, 25, 3550–3563 CrossRef.
- K. W. Ro, R. Nayak and D. R. Knapp, Electrophoresis, 2006, 27, 3547–3558 CrossRef CAS.
- M. Bartolini, N. H. Greig, Q. Yu and V. Andrisano, J. Chromatogr. A, 2009, 1216, 2730–2738 CrossRef CAS.
- J. Sproß and A. Sinz, Anal. Bioanal. Chem., 2009, 395, 1583–1588 CrossRef.
- J. Urban, F. Svec and J. M. J. Fréchet, Biotechnol. Bioeng., 2012, 109, 371–380 CrossRef CAS.
- C. Delattre, P. Michaud and M. A. Vijayalakshmi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 861, 203–208 CrossRef CAS.
- M. V. Volokitina, K. S. Bobrov, K. Piens, E. V. Eneyskaya, T. B. Tennikova, E. G. Vlakh and A. A. Kulminskaya, Biotechnol. J., 2015, 10, 210–221 CrossRef CAS PubMed.
- F. Coro, L. Di Pietro, S. Micalizzi, A. Bertei, G. Gallone, A. M. Raspolli Galletti, A. Ahluwalia and C. De Maria, Chem. Eng. Sci., 2024, 285, 119590 CrossRef CAS.
- J. F. A. Valente, T. S. Carreira, J. R. Dias, F. Sousa and N. Alves, Pharmaceutics, 2022, 14, 2266 CrossRef CAS.
- S. Fernandez-Velayos, G. Vergara, J. M. Olmos, J. Sanchez-Marcos, N. Menendez, P. Herrasti and E. Mazarío, Sci. Total Environ., 2024, 906, 167376 CrossRef CAS.
- J. Ye, T. Chu, J. Chu, B. Gao and B. He, ACS Sustainable Chem. Eng., 2019, 7, 18048–18054 CrossRef CAS.
- J. E. Song, W. S. Song, S. Y. Yeo, H. R. Kim and S. H. Lee, Text. Res. J., 2017, 87, 1177–1191 CrossRef.
- L. Yang, Y. Guo, W. Zhan, Y. Guo, Y. Wang and G. Lu, Microporous Mesoporous Mater., 2014, 197, 1–7 CrossRef.
- M. Maier, C. P. Radtke, J. Hubbuch, C. M. Niemeyer and K. S. Rabe, Angew. Chem., Int. Ed., 2018, 57, 5539–5543 CrossRef.
- F. Kazenwadel, E. Biegert, J. Wohlgemuth, H. Wagner and M. Franzreb, Eng. Life Sci., 2016, 16, 560–567 CrossRef.
- M. S. Sarwar, U. Simon and S. Dimartino, J. Chromatogr. A, 2021, 1646, 462125 CrossRef PubMed.
- G. M. Whitesides and A. D. Stroock, Phys. Today, 2001, 54, 42–48 CrossRef.
- W. Reschetilowski, Microreactors in Preparative Chemistry Practical Aspects in Bioprocessing, Natanotechnology, Catalysis and more, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- F. G. Bessoth, A. J. DeMello and A. Manz, Anal. Commun., 1999, 36, 213–215 RSC.
- A. D. Stroock, S. K. W. Dertinger, A. Ajdari, I. Mezić, H. A. Stone and G. M. Whitesides, Science, 2002, 295, 647–651 CrossRef CAS.
- Z. Yang, S. Matsumoto, H. Goto, M. Matsumoto and R. Maeda, Sens. Actuators, A, 2001, 93, 266–272 CrossRef CAS.
- T. Rohr, C. Yu, M. H. Davey, F. Svec and J. M. J. Fréchet, Electrophoresis, 2001, 22, 3959–3967 CrossRef CAS.
- M. Heule, K. Rezwan, L. Cavalli and L. J. Gauckler, Adv. Mater., 2003, 15, 1191–1194 CrossRef CAS.
- S. Whitaker, Ind. Eng. Chem. Res., 1969, 61, 15–28 Search PubMed.
- J. Duan, Z. Liang, C. Yang, J. Zhang, L. Zhang, W. Zhang and Y. Zhang, Proteomics, 2006, 6, 412–419 CrossRef CAS PubMed.
- V. Dore, D. Tsaoulidis and P. Angeli, Chem. Eng. Sci., 2012, 80, 334–341 CrossRef CAS.
- M. N. Kashid and D. W. Agar, Chem. Eng. J., 2007, 131, 1–13 CrossRef.
- P. Garstecki, M. J. Fuerstman, H. A. Stone and G. M. Whitesides, Lab Chip, 2006, 6, 437–446 RSC.
- A. L. Dessimoz, L. Cavin, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2008, 63, 4035–4044 CrossRef.
- M. N. Kashid, I. Gerlach, S. Goetz, J. Franzke, J. F. Acker, F. Platte, D. W. Agar and S. Turek, Ind. Eng. Chem. Res., 2005, 44, 5003–5010 CrossRef.
- N. D. M. Raimondi, L. Prat, C. Gourdon and P. Cognet, Chem. Eng. Sci., 2008, 63, 5522–5530 CrossRef.
- H. Katou, R. Miyake and T. Terayama, JSME Int. J., 2005, 48, 350–355 CrossRef.
- M. Riccaboni, E. La Porta, A. Martorana and R. Attanasio, Tetrahedron, 2010, 66, 4032–4039 CrossRef.
- D. F. Rivas, P. Cintas and H. J. G. E. Gardeniers, Chem. Commun., 2012, 48, 10935–10947 RSC.
- J. J. John, S. Kuhn, L. Braeken and T. Van Gerven, Chem. Eng. Process., 2016, 102, 37–46 CrossRef.
- P. Hajiani and F. Larachi, Chem. Eng. J., 2012, 203, 492–498 CrossRef.
- M. Rahimi, O. Jafari and A. Mohammdifar, Chem. Eng. Process., 2017, 111, 79–88 CrossRef.
- Y. Wang, J. Zhe, B. T. F. Chung and P. Dutta, Microfluid. Nanofluid., 2008, 4, 375–389 CrossRef.
- N. Azimi, M. Rahimi and N. Abdollahi, Chem. Eng. Process., 2015, 97, 12–22 CrossRef.
- E. Abraham, G. N. Mukunthan Sulochana, B. Soundarajan and S. Narayanasamy, Ind. Eng. Chem. Res., 2017, 56, 10845–10855 CrossRef.
- K. Szymańska, K. Odrozek, A. Zniszczoł, W. Pudło and A. B. Jarzębski, Chem. Eng. J., 2017, 315, 18–24 CrossRef.
- K. Zielińska, K. Szymańska, R. Mazurkiewicz and A. Jarzębski, Tetrahedron: Asymmetry, 2017, 28, 146–152 CrossRef.
- D. J. Strub, K. Szymańska, Z. Hrydziuszko, J. Bryjak and A. B. Jarzębski, React. Chem. Eng., 2019, 4, 587–594 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- Y. Zhang, J. Ge and Z. Liu, ACS Catal., 2015, 5, 4503–4513 CrossRef.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- E. G. Griffin and J. M. Nelson, J. Am. Chem. Soc., 1916, 38, 1109–1115 CrossRef.
- T. Tosa, T. Mori, N. Fuse and I. Chibata, Biotechnol. Bioeng., 1967, 9, 603–615 CrossRef.
- A. I. Kallenberg, F. Van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2005, 347, 905–926 CrossRef.
- E. M. M. Abdelraheem, H. Busch, U. Hanefeld and F. Tonin, React. Chem. Eng., 2019, 4, 1878–1894 RSC.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- R. C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef CAS PubMed.
- N. A. Mohidem, M. Mohamad, M. U. Rashid, M. N. Norizan, F. Hamzah and H. b. Mat, J. Compos. Sci., 2023, 7, 488 CrossRef CAS.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS.
- L. Van Den Biggelaar, P. Soumillion and D. P. Debecker, Catalysts, 2017, 7, 54 CrossRef.
- J. Coloma, Y. Guiavarc'h, P. L. Hagedoorn and U. Hanefeld, Catal. Sci. Technol., 2020, 10, 3613–3621 RSC.
- M. P. Van Der Helm, P. Bracco, H. Busch, K. Szymańska, A. B. Jarzębski and U. Hanefeld, Catal. Sci. Technol., 2019, 9, 1189–1200 RSC.
- D. Stradomska, J. Coloma, U. Hanefeld and K. Szymańska, Catal. Sci. Technol., 2022, 12, 3356–3362 RSC.
- U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6321 RSC.
- J. Coloma, L. Teeuwisse, M. Afendi, P. L. Hagedoorn and U. Hanefeld, Catalysts, 2022, 12, 161 CrossRef CAS.
- K. Shortall, S. Arshi, S. Bendl, X. Xiao, S. Belochapkine, D. Demurtas, T. Soulimane and E. Magner, Green Chem., 2023, 25, 4553–4564 RSC.
- M. V. Volokitina, E. G. Vlakh, G. A. Platonova, D. O. Vinokhodov and T. B. Tennikova, J. Sep. Sci., 2013, 36, 2793–2805 CrossRef CAS.
- L. Van Den Biggelaar, P. Soumillion and D. P. Debecker, RSC Adv., 2019, 9, 18538–18546 RSC.
- D. Kowalczykiewicz, M. Przypis, L. Mestrom, A. Kumpf, D. Tischler, P. L. Hagedoorn, U. Hanefeld, A. Jarzębski and K. Szymańska, Chem. Eng. J., 2022, 427, 1–8 CrossRef.
- C. Temporini, E. Perani, F. Mancini, M. Bartolini, E. Calleri, D. Lubda, G. Felix, V. Andrisano and G. Massolini, J. Chromatogr. A, 2006, 1120, 121–131 CrossRef CAS PubMed.
- J. Mai, V. V. Abhyankar, M. E. Piccini, J. P. Olano, R. Willson and A. V. Hatch, Biosens. Bioelectron., 2014, 54, 435–441 CrossRef CAS.
- O. Behrmann, M. Hügle, F. Eckardt, I. Bachmann, C. Heller, M. Schramm, C. Turner, F. T. Hufert and G. Dame, Micromachines, 2020, 11, 1–17 CrossRef PubMed.
- M. V. Volokitina, A. V. Nikitina, T. B. Tennikova and E. G. Korzhikova-Vlakh, Electrophoresis, 2017, 38, 2931–2939 CrossRef CAS PubMed.
- L. Bayne, R. V. Ulijn and P. J. Halling, Chem. Soc. Rev., 2013, 42, 9000–9010 RSC.
- A. Basso, P. Braiuca, S. Cantone, C. Ebert, P. Linda, P. Spizzo, P. Caimi, U. Hanefeld, G. Degrassi and L. Gardossi, Adv. Synth. Catal., 2007, 349, 877–886 CrossRef.
- P. De Santis, L. E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- P. Lozano and E. García-Verdugo, Green Chem., 2023, 25, 7041–7057 RSC.
- P. Bracco, N. van Midden, E. Arango, G. Torrelo, V. Ferrario, L. Gardossi and U. Hanefeld, Catalysts, 2020, 10, 15–19 CrossRef.
- R. Onbas and O. Yesil-Celiktas, Eng. Life Sci., 2019, 19, 37–46 CrossRef PubMed.
- X. Sun, Y. Xin, X. Wang and H. Uyama, Colloid Polym. Sci., 2017, 295, 1827–1833 CrossRef CAS.
- K. Szymańska, W. Pudło, J. Mrowiec-Białoń, A. Czardybon, J. Kocurek and A. B. Jarzębski, Microporous Mesoporous Mater., 2013, 170, 75–82 CrossRef.
- Y. D. Ahn and J. H. Lee, Biotechnol. Bioprocess Eng., 2018, 23, 349–354 CrossRef CAS.
- A. M. Klibanov, Nature, 2001, 409, 241–246 CrossRef CAS.
- K. Martinek and A. N. Semenov, Biochim. Biophys. Acta, Enzymol., 1981, 658, 90–101 CrossRef CAS.
- A. Ismail, S. Soultani and M. Ghoul, J. Biotechnol., 1999, 69, 135–143 CrossRef CAS.
- K. Szymańska, K. Odrozek, A. Zniszczoł, G. Torrelo, V. Resch, U. Hanefeld and A. B. Jarzębski, Catal. Sci. Technol., 2016, 6, 4882–4888 RSC.
- P. He, G. Greenway and S. J. Haswell, Process Biochem., 2010, 45, 593–597 CrossRef CAS.
- Z. Yin, Y. Zhou, X. Liu, S. Zhang and B. P. Binks, J. Colloid Interface Sci., 2023, 648, 308–316 CrossRef CAS.
- B. Sandig and M. R. Buchmeiser, ChemSusChem, 2016, 9, 2917–2921 CrossRef CAS PubMed.
- I. Dávila and J. Labidi, Curr. Opin. Green Sustainable Chem., 2021, 28, 100435 CrossRef.
- C. J. Zimmermann, N. V. Bollar and S. G. Wettstein, Biomass Bioenergy, 2018, 118, 163–171 CrossRef CAS.
- P. Lozano, Green Chem., 2010, 12, 555–556 RSC.
- L. Zverina, M. Pinelo, J. M. Woodley and A. E. Daugaard, J. Chem. Technol. Biotechnol., 2021, 96, 2488–2495 CrossRef CAS.
- P. Lozano, E. García-Verdugo, R. Piamtongkam, N. Karbass, T. De Diego, M. I. Burguete, S. V. Luis and J. L. Iborra, Adv. Synth. Catal., 2007, 349, 1077–1084 CrossRef CAS.
- S. F. Mayer, W. Kroutil and K. Faber, Chem. Soc. Rev., 2001, 30, 332–339 RSC.
- R. J. Conrado, J. D. Varner and M. P. DeLisa, Curr. Opin. Biotechnol., 2008, 19, 492–499 CrossRef PubMed.
- Q. Ji, B. Wang, J. Tan, L. Zhu and L. Li, Process Biochem., 2016, 51, 1193–1203 CrossRef.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef.
- M. Berovic and M. Vodopivec, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2003, 795, 105–113 Search PubMed.
- J. Luo, L. Ma, F. Svec, T. Tan and Y. Lv, Biotechnol. J., 2019, 14, 1–10 CrossRef.
- C. Temporini, E. Calleri, K. Cabrera, G. Felix and G. Massolini, J. Sep. Sci., 2009, 32, 1120–1128 CrossRef.
- T. C. Logan, D. S. Clark, T. B. Stachowiak, F. Svec and J. M. J. Fréchet, Anal. Chem., 2007, 79, 6592–6598 Search PubMed.
- J. Yin, T. Xu, N. Zhang and H. Wang, Anal. Chem., 2016, 88, 7730–7737 CrossRef CAS PubMed.
- J. Yin, S. Chen, N. Zhang and H. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 21883–21890 CrossRef CAS PubMed.
- F. Rinaldi, J. Fernández-Lucas, D. de la Fuente, C. Zheng, T. Bavaro, B. Peters, G. Massolini, F. Annunziata, P. Conti, I. de la Mata, M. Terreni and E. Calleri, Bioresour. Technol., 2020, 307, 123258 CrossRef CAS PubMed.
- E. Calleri, G. Cattaneo, M. Rabuffetti, I. Serra, T. Bavaro, G. Massolini, G. Speranza and D. Ubiali, Adv. Synth. Catal., 2015, 357, 2520–2528 CrossRef CAS.
- G. Cattaneo, M. Rabuffetti, G. Speranza, T. Kupfer, B. Peters, G. Massolini, D. Ubiali and E. Calleri, ChemCatChem, 2017, 9, 4614–4620 CrossRef CAS.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- K. Bachosz, J. Zdarta, M. Bilal, A. S. Meyer and T. Jesionowski, Sci. Total Environ., 2023, 868, 161630 CrossRef CAS.
- X. Wang, T. Saba, H. H. P. Yiu, R. F. Howe, J. A. Anderson and J. Shi, Chem, 2017, 2, 621–654 CAS.
- A. I. Benítez-Mateos, B. Nidetzky, J. M. Bolivar and F. López-Gallego, ChemCatChem, 2018, 10, 654–665 CrossRef.
- B. Reus, M. Damian and F. G. Mutti, J. Flow Chem., 2024, 14, 219–238 CrossRef CAS.
- Q. Chen, Y. Wang and G. Luo, ChemSusChem, 2022, 16, e202201654 CrossRef PubMed.
- M. Baccour, A. Lamotte, A. Lamotte, K. Sakai, E. Dubreucq, A. Mehdi, K. Kano, A. Galarneau, J. Drone and N. Brun, Green Chem., 2020, 22, 3727–3733 RSC.
- G. Chen, Z. Wu and Y. Ma, J. Biotechnol., 2015, 196–197, 52–57 CrossRef CAS PubMed.
- Y. Li, H. Liang, L. Sun, J. Wu and Q. Yuan, Biotechnol. Lett., 2013, 35, 915–919 CrossRef CAS PubMed.
- Z. Wang, M. Etienne, F. Quilès, G. W. Kohring and A. Walcarius, Biosens. Bioelectron., 2012, 32, 111–117 CrossRef CAS.
- M. Montagné and J. L. Marty, Anal. Chim. Acta, 1995, 315, 297–302 CrossRef.
- M. Zhang, C. Mullens and W. Gorski, Anal. Chem., 2007, 79, 2446–2450 CrossRef CAS.
- H. Zhou, Z. Zhang, P. Yu, L. Su, T. Ohsaka and L. Mao, Langmuir, 2010, 26, 6028–6032 Search PubMed.
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918 Search PubMed.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |