
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable polymers: from synthesis methods to applications of lignin-graft-polyester
Sundol
Kim
 and
Hoyong
Chung
*
and
Hoyong
Chung
*
Department of Chemical and Biomedical Engineering, FAMU-FSU College of Engineering, Tallahassee, FL 32310, USA. E-mail: hchung@eng.famu.fsu.edu
First published on 18th September 2024
Abstract
The issue of non-degradable petroleum-based plastic waste is a global challenge that requires urgent attention due to its harmful impact on humans and the environment. Biomass-based materials have garnered significant attention to address this challenge in recent times. Lignin, with its abundance, low price, and rich aromatic groups, is not only essential biomass but also a crucial material that can be utilized to produce biodegradable polymers. This review paper provides comprehensive knowledge of the synthesis and applications related to lignin, polyester, and lignin–polyester copolymers with a specific focus on organic polymer synthesis techniques and diverse applications. Beginning with lignin, the review explores various extraction methods from raw biomass resources aimed at enhancing its compatibility with polyester matrices. Next, we discuss lignin chemical modification methods that alter its chemical structure and properties, ultimately enhancing its integration with polyesters. Subsequently, the synthesis of polyester is examined, encompassing condensation polymerizations and ring-opening polymerizations. These methods are evaluated for their scalability and capability in producing tailored polymer chains suitable for copolymerization with lignin. The copolymerization strategies involving lignin and polyester are explored in detail, including graft-onto and graft-from approaches. Each method is discussed for its ability to control copolymer composition and properties, which are crucial for achieving desired material characteristics. In terms of applications, the review highlights the wide-ranging utility of lignin–polyester copolymers across industries such as packaging, construction, and separation. These polymers offer improved biodegradability, thermal stability, and mechanical strength compared to conventional polyesters, making them perfect candidates for novel sustainable materials. Overall, this review provides valuable insights into the synthesis methods and applications of lignin, polyester, and lignin–polyester copolymers offering a comprehensive overview of their potential for addressing environmental concerns and expanding the scope of lignin-derived materials in various industrial applications.
1. Introduction
Environmental pollution with nondegradable petroleum-based plastic has become a serious global and national-scale problem.1,2 Discarded plastic contaminates all major ecosystems on Earth, and concerns about its negative impact on wildlife and human health are also increasing.3 Because of this environmental issue, biomass-based or biodegradable polymers have been reported to solve this environmental problem as well as to decrease the dependence on fossil feedstock.It is crucial to remember that biomass-based does not mean biodegradable.4Fig. 1 shows the material coordinate system of bioplastic.5,6 The x-axis means the presence/absence of biodegradation and the y-axis means the origin of raw material, whether it is biomass-based or petroleum-based. Biobased polymers (Fig. 1 top) are a renewable biomass resource leading to sustainable material production, reducing carbon footprint and greenhouse gas emissions.6,7 Biodegradable polymers (Fig. 1 right) are environmentally friendly materials that decompose without leaving behind fragments or harmful products.4
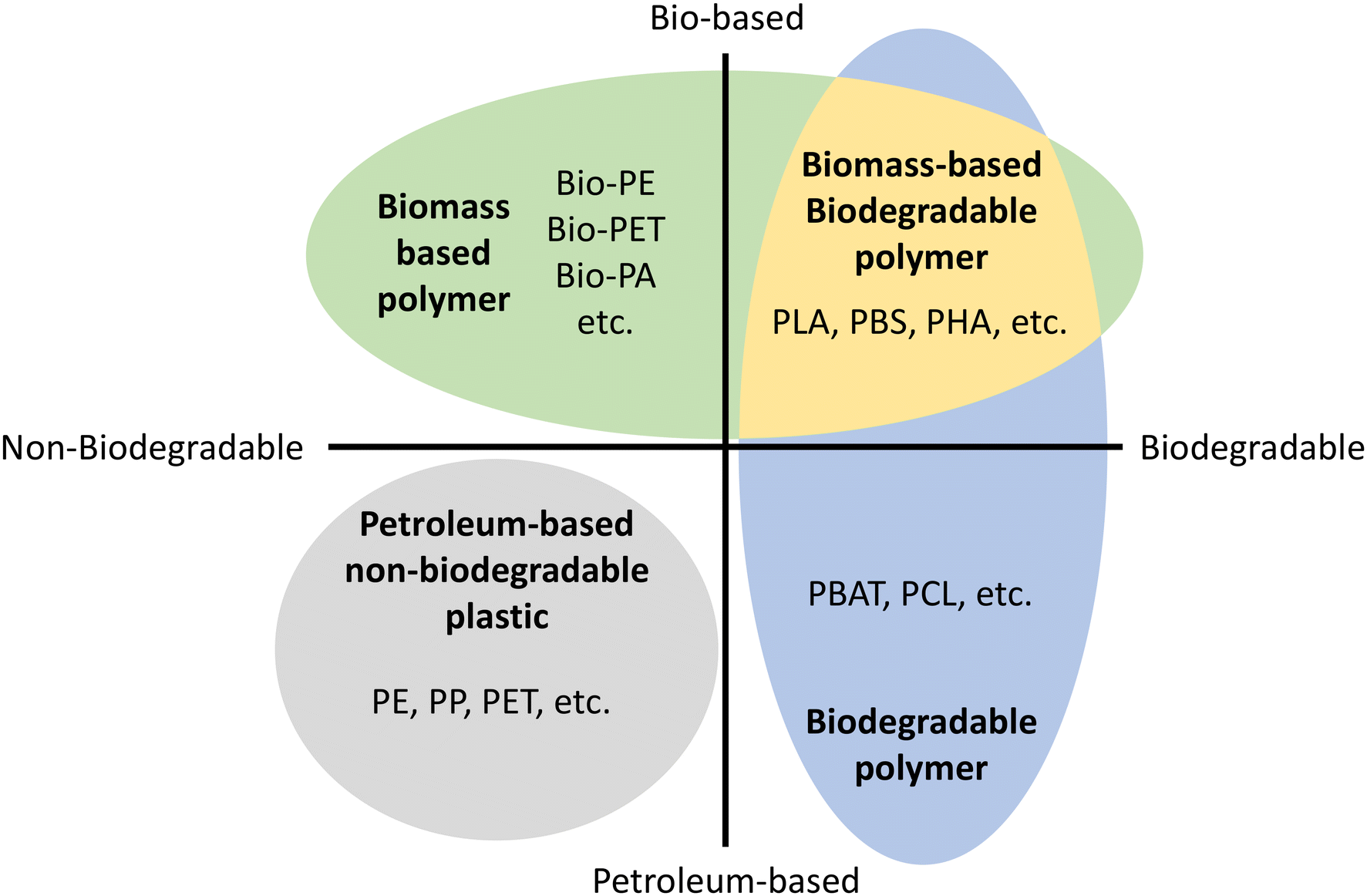 | ||
| Fig. 1 Material coordinate system of bioplastics.5,6 Bio-PE: bio polyethylene, Bio-PET: bio poly(ethylene terephthalate), Bio-PA: bio polyamides, PLA: poly(lactic acid). PBS: poly(butylene succinate), PHA: poly(hydroxyalkanoates), PE: polyethylene, PP: polypropylene, PET: poly(ethylene terephthalate), PBAT: poly(butylene adipate terephthalate), PCL: poly(caprolactone). | ||
Biodegradability is related to a material's chemical and physical structure, not its raw material origin. Therefore, chemical modifications or additives can affect biodegradability, leading to concerns about the biodegradability of chemically modified natural materials. Thus, biomass-based materials can be non-biodegradable such as bio polyethylene (Bio-PE), bio polyethylene terephthalate (Bio-PET), bio trimethylene terephthalate (Bio-PTT), bio polypropylene (Bio-PP), and bio polyamides (Bio-PA).8 On the other hand, petroleum-based polymers can be biodegradable polymers such as poly(caprolactone). As a result, the ultimate and desirable solution to the overall plastic waste problem is biomass-based and biodegradable polymers (Fig. 1 top right).
Biodegradability refers to a material's ability to be broken down by microorganisms, such as bacteria and fungi, into natural substances like water, carbon dioxide, and biomass.9,10 This process can occur in various environments, including soil, water, and anaerobic conditions, with the rate of biodegradation varying significantly depending on factors such as temperature, humidity, and oxygen availability. Materials that decompose into non-toxic components within a specified period under controlled conditions are especially considered compostable, and the resulting compost can be used to enhance soil health.9,10
However, not all biodegradable materials behave the same way in natural environments. This means some plastics are engineered to degrade only under specific conditions, such as elevated temperatures or in the presence of particular microorganisms, which may not be found in nature. For example, polylactic acid (PLA)10 is a widely used biodegradable plastic, commonly found in packaging and disposable tableware. While PLA is compostable, it typically requires temperatures above 55–60 °C to decompose effectively.11,12 However, in natural environments such as the ocean or soil, PLA may not decompose as intended. Even when PLA eventually decomposes, it often fragments into microplastics before it fully decomposes. These tiny plastic particles persist in the environment, accumulating in water bodies and ingested by marine life, which can pose a persistent environmental risk.
2. Lignin
Lignin is one of the biopolymers found in the cells of plant species and some algae.13 It is mainly in the secondary cell wall, cell membrane, and middle lamella. Lignin is firmly combined with cellulose and hemicellulose. It fills the space between cellulose, hemicellulose, and pectin present in the cell wall, especially in the xylem tracheids, vascular structure, and sclereid cells. The primary role of lignin is to provide rigidity to strengthen the structure of cell walls, especially in stems and roots. It crosslinks polysaccharides to improve the mechanical strength of the cell walls and harden the whole plant.14,15 It also blocks pathogen growth and protects plants by lignification of infections and wounds.16In addition, lignin in the vasculature, a pathway for water transport, enables the vascular tissues of plants to transport water efficiently.15 The polysaccharide component of the plant cell wall is hydrophilic and can enable water to permeate, whereas lignin is hydrophobic and impermeable to water. Therefore, hydrophobic lignin crosslinking with polysaccharides prevents moisture from passing through the cell wall.
2.1 Lignin extraction processes
Lignin is separated from lignocellulose and cellulose in the plant during chemical pulping processes. The different pulping processes result in various technical forms of lignin.17 The four main types of technical lignin are classified according to sulfur or sulfur-free processes (Table 1). Sulfur processes include kraft processes and sulfite pulping. The kraft process breaks lignin down with a strong alkali solution, an aqueous solution of sodium hydroxide and sodium sulfide. Kraft-produced lignin is highly hydrophobic and has a high sulfur content at around 1 to 2 wt%.18 Because of its relatively narrow dispersion, abundance, and low price, kraft lignin is commonly used in the development of lignin-based polymers.The second process is sulfite pulping, which is the second most popular process. The sulfite process reacts wood or biomass with calcium or magnesium sulfite at 125–150 °C for 3–7 h, which then undergoes an acidic treatment instead of treatment with an alkali solution in kraft pulping. The product of sulfite pulping is lignosulfonate, which contains a considerable amount of sulfur (approximately 3.5–8 wt%) in the form of sulfonate groups on the aliphatic side chains. Due to the sulfonate polar group, lignosulfonate is soluble in water.26,27
Sulfur-free processes include soda processes and solvent pulping (organosolv). The soda process is the oldest pulping method and is mainly used to treat non-wood materials. The soda process is carried out under strongly basic conditions (pH = 11–13) with NaOH solution at a high temperature (150–170 °C). The difference from the kraft process is that it does not use a sulfur additive, so it is more similar to natural lignin than lignin from other processes. Therefore, it has more economical applications.
Organosolv lignin extraction employs organic solvents to extract lignin from biomass in the presence of acid or base catalysts. Ethanol, methanol, acetic acid, and formic acid are primarily used in this process and water is occasionally used alongside the organic solvent, serving as a solvent but not involved in extraction. During extraction, the α-aryl ether bonds predominantly break, while the β-aryl ether bonds are cleaved to a lesser extent. As a result of ether bond cleavage, new phenolic groups are formed, producing highly phenolic organosolv lignins. Despite their highly phenolic nature, organosolv lignins are relatively hydrophobic, have a low molecular weight, and are free of sulfur and impurities.28–30 Organosolv lignin is hydrophobic, sulfur-free, and has a low molecular weight of around 5 kg mol−1. On the other hand, it is recovered from the solvent by precipitation, which usually requires the adjustment of various parameters such as concentration, pH, and temperature. In this process, an organic solvent is used, which increases the process cost and decreases the economic efficiency.31Fig. 2 briefly describes each process and its major product.
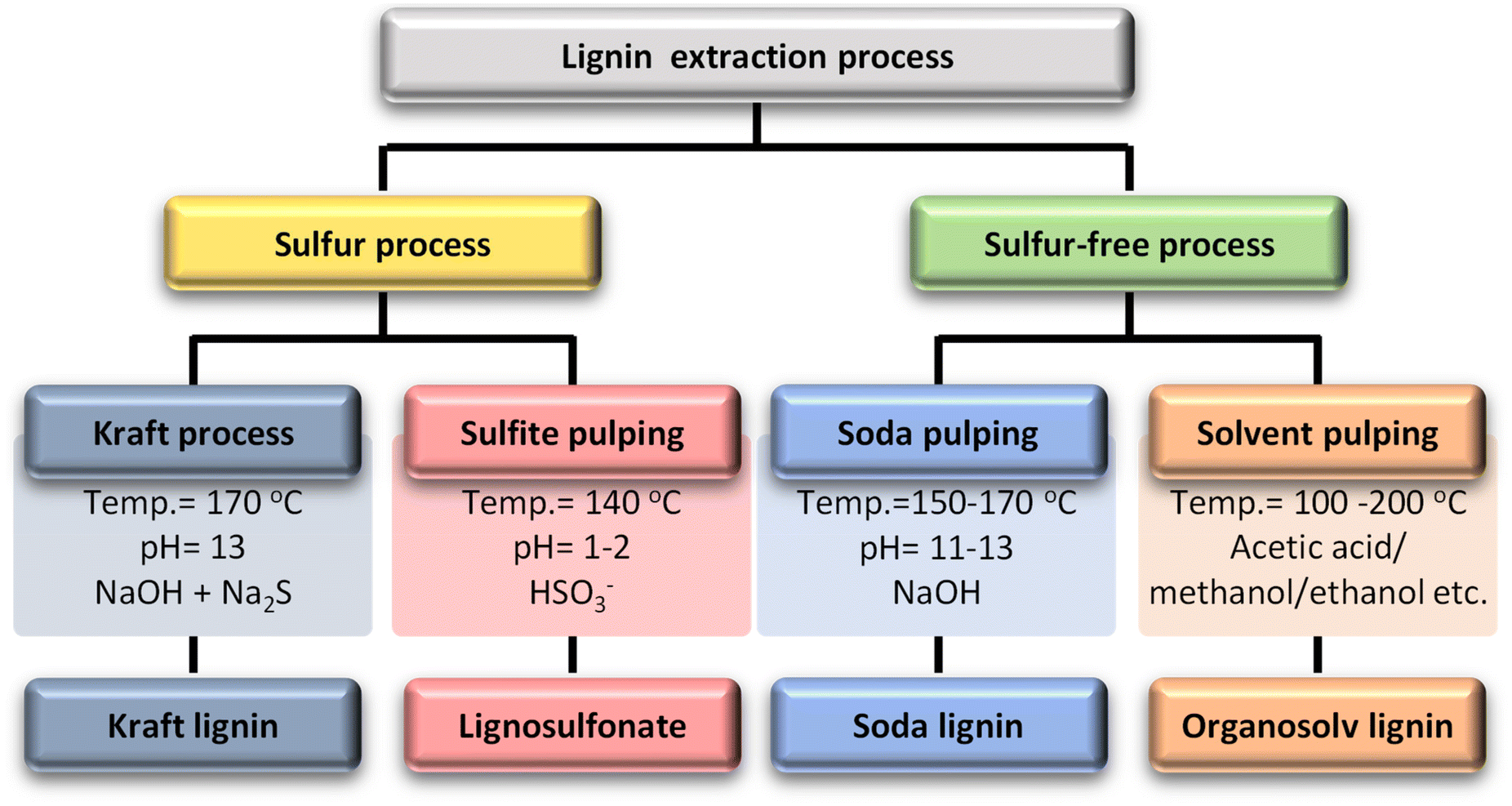 | ||
| Fig. 2 Lignin extraction processes, process conditions, and their dominant products.27 | ||
2.2 Chemical structure of lignin
The chemical structure of lignin is a phenyl propane carbon skeleton with three carbons attached to the benzene ring. These have a three-dimensional network structure in which benzene rings and side chains are combined with each other (Fig. 3). The monomers that participate in the biosynthesis of lignin are collectively called monolignol, and there are three types: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol. Each monolignol is involved in the formation of p-hydroxyphenyl (H), guaiacyl, (G), syringyl (S), which are phenylpropanoid forms (Fig. 4a). The three types of monolignols are distributed in different combinations depending on the type of plant.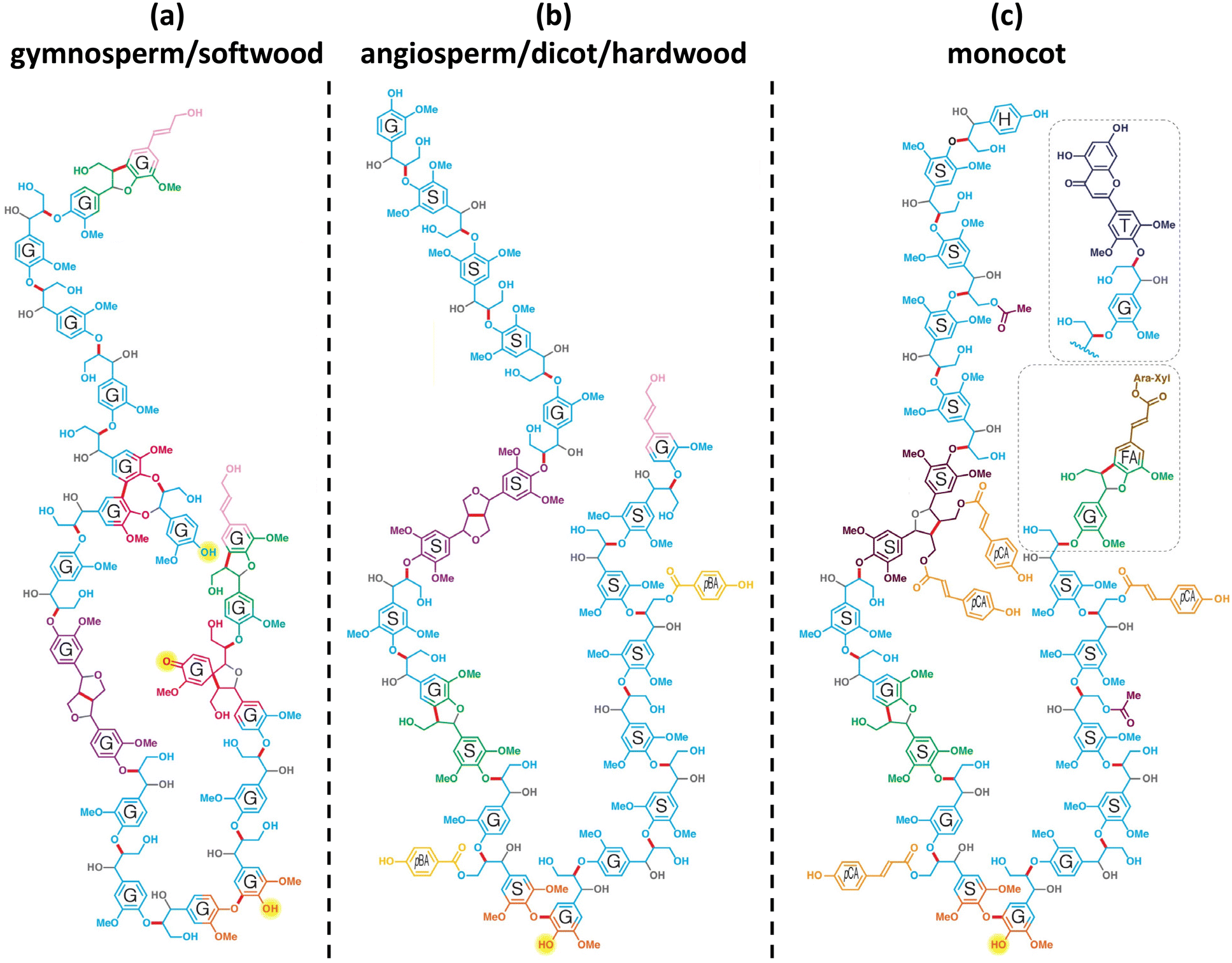 | ||
| Fig. 3 Chemical structures of model lignin 20-mers: (a) gymnosperm/soft wood lignin, (b) angiosperm/dicot/hardwood lignin, and (c) monocot lignin. It is a model structure of lignin since the exact structure of lignin is not known. H: p-hydroxyphenyl unit, G: guaiacyl unit, S: syringyl unit, FA: ferulate, pCA: p-coumarate, pBA: p-hydroxylbenzoate, T: tricin, gray: oxygen or hydroxy groups that derive from water during quinone methide rearomatization, and the bonds to them. Reproduced from ref. 9 with permission from Elsevier Ltd., copyright 2019. | ||
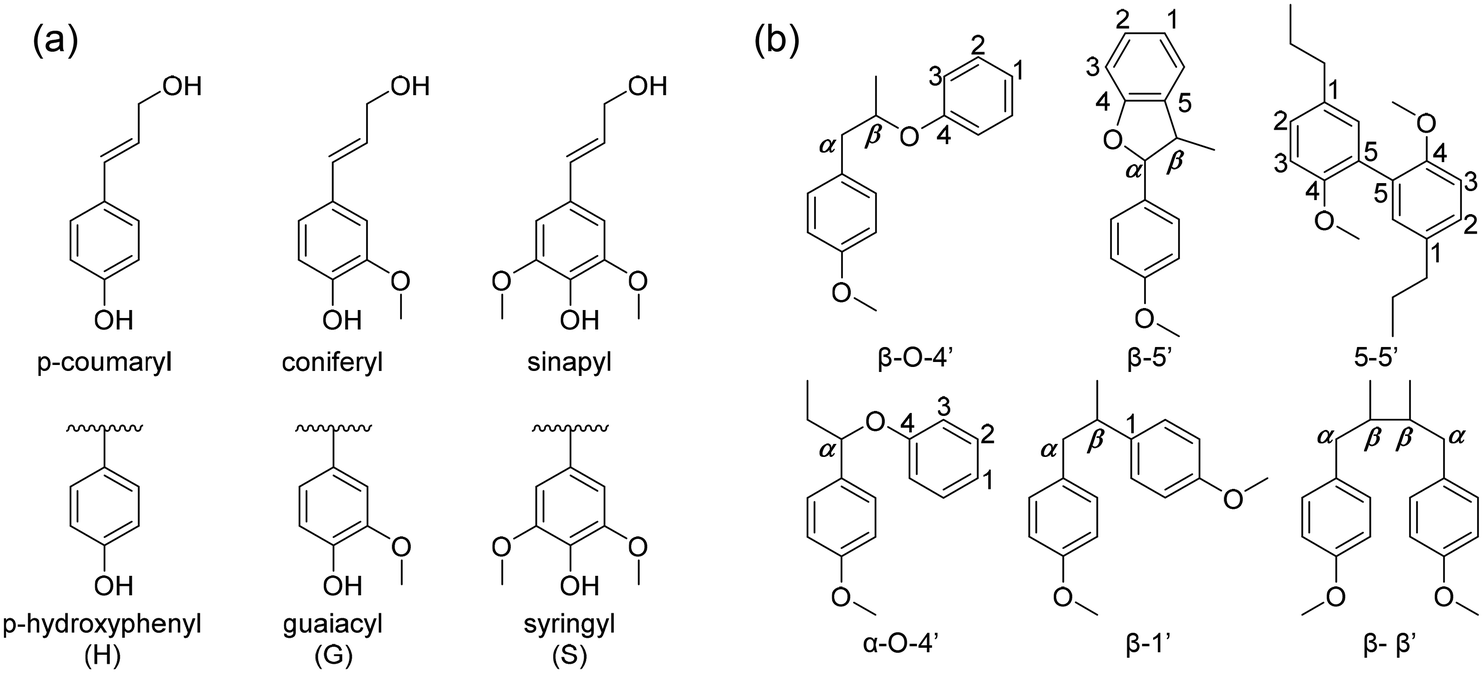 | ||
| Fig. 4 (a) The precursors of lignin (H, G, S from p-coumaryl, coniferyl and synapyl respectively), (b) common linkages between phenyl propane units in lignin. | ||
In biosynthesis, the phenylpropanoid monomers undergo an in situ radical coupling, making interunit linkages, such as aryl ether (α-O-4′ and β-O-4′), resinol (β-β′), phenyl coumaran (β-5′), biphenyl (5-5′), and 1,2-diaryl propane (β-1′) (Fig. 4b).32 Among these interunit linkages, β-O-4′ is the most abundant linkage, almost 50% of the total interunit linkage in lignin. Also, the β-O-4′ linkage is the most important chemical linkage from the aspect of lignin depolymerization and fractionation. While the other linkages (β-5′, β-β′, 5-5′, β-1′) are more resistant to chemical degradation, β-O-4′ is one of the most commonly cleaved chemically, providing a basis for industrial processes such as chemical pulping.16,33–35 Although ethers are usually relatively inert, they can be liable to cleavage because of the presence of lone-pair electrons on oxygen, which is a source of reactivity. The lone-pair electrons increase the polarity of the –C(β)–O– bond and make the neighboring β-carbon atom sensitive to nucleophilic attack. Additionally, the susceptibility of the β-O-4 linkage to undergo a cleavage reaction is dependent on whether the structure is phenolic or non-phenolic due to their reactivity differences.34
Although the chemical structure of lignin cannot be defined exactly in an easy way, it is already known that lignin has various functional groups, including methoxy, phenolic hydroxyl, aliphatic hydroxyl, and other carbonyl containing groups. Various commercial lignins and their hydroxyl group contents are summarized in Table 2. These functional groups provide reactive sites for the modification of lignin.
| Lignin description | Vendor | Hydroxyl group content (mmol g−1) | Reference |
|---|---|---|---|
| Kraft lignin | Jilin Paper Co., Ltd | Phenolic: 2.73 | Lin et al.19 |
| Kraft lignin, alkali | Sigma Aldrich | Phenolic: 3.0 | Liu et al.20 |
| Aliphatic: 4.41 | |||
| Alkali lignin | Sigma Aldrich | 11.24 | Yu et al.21 |
| Hardwood lignin | Sigma Aldrich | Phenolic: 3.2 | Kim and Kadla22 |
| Aliphatic: 2.9 | |||
| Softwood lignin | TCI | 4.48 | Liu and Chung23 |
| Softwood Kraft lignin | Mead Westvaco | Phenolic: 3.8 | Argyropoulos et al.24 |
| Aliphatic: 2.4 | |||
| Organosolv lignin | Lignin Corporation | 1.6 | Korich et al.25 |
2.3 Modification of lignin
One of the most efficient modification methods of lignin is graft copolymerization. Graft copolymerization can functionalize lignin by integrating monomer or polymer chains to improve its properties. Especially, graft copolymerization can produce lignin-based polymers that possess precisely controllable architectures and specialized functional groups. A graft copolymer generally consists of a backbone polymer as the main chain and one or more branch polymers connected to the backbone via covalent bonds.32 The properties of the copolymers produced are controlled by the functional groups on the grafted polymers, length of the graft, and graft density. There are three primary grafting methods: graft-from, graft-onto, and graft-through.The “graft-from” method is a method in which a monomer reacts at an active site on a backbone polymer to grow a grafted polymer (Fig. 5a). In this method, lignin generally acts as a backbone polymer to provide hydroxyl groups as initiation sites. The other way is to modify the hydroxyl group of lignin to make a macroinitiator with functionalized initiation sites. The grafted polymer grows at the beginning of lignin by various polymerization methods, including ATRP (atom transfer radical polymerization), RAFT (reversible addition–fragmentation chain transfer polymerization), ring-opening polymerization, and free radical polymerization. As a result, the graft density of the copolymer synthesized by the graft-from method is generally higher than those from graft-onto method.
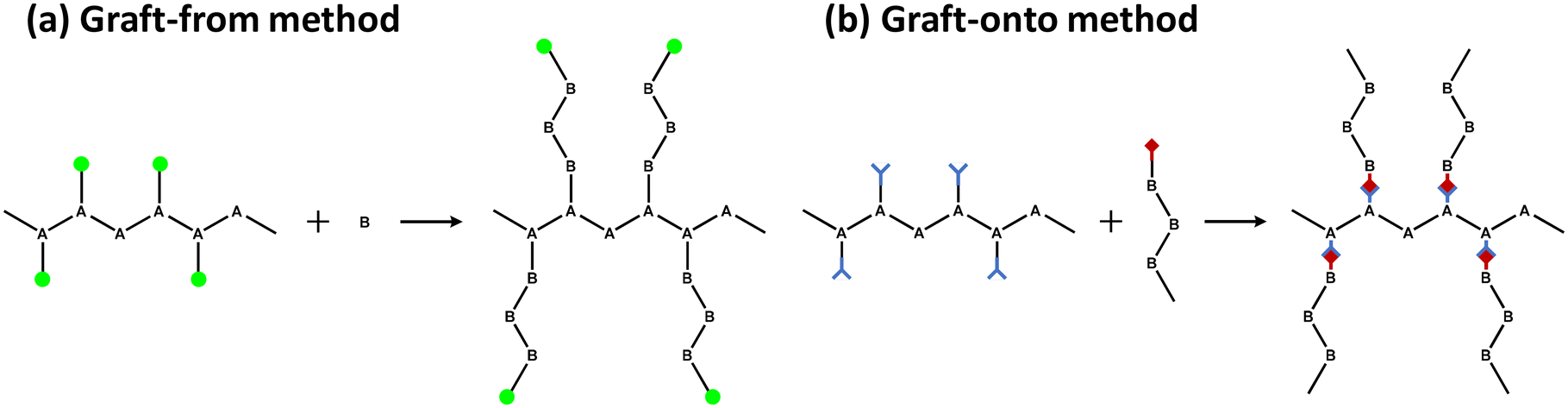 | ||
| Fig. 5 (a) Graft-from and (b) graft-onto methods for the synthesis of graft copolymers. | ||
“Graft-onto” means that polymer chains, which are separately synthesized, covalently link to the surfaces of backbone polymer, lignin (Fig. 5b). The “graft-onto” method requires an efficient covalent bond formation reaction between the lignin backbone and the end groups of the graft polymer. “Graft-onto” can incorporate a variety of integrated reactions and polymers with convenient reaction conditions as well as a wider range of solvent selection and purification methods.36 Click chemistry is the most commonly used synthesis method for grafting polymers onto lignin due to its high efficiency and ease of experimentation. Other reactivity including photoredox catalyzed thiol–ene reactions, base-catalyzed ring-opening reactions of epoxide, in situ free radical reactions, substitution, condensation, boronate ester formation, and crosslinking were successful methods for the preparation of lignin graft copolymers.37
In the “graft-through” method, ordinary monomers are polymerized in the presence of macromonomers so that the main chain is “sewn” through the ends of the side chains during the copolymerization process.38 Due to the lack of a well-defined chemical structure and the irregular three-dimensional network structure of lignin, the number of studies on lignin-based copolymers prepared by this method are limited.
To summarize, lignin is a complex aromatic polymer with unique structural and chemical properties, making it a promising candidate for various applications, including as a feedstock for producing bio-based materials. Among these potential applications, graft copolymerization with aliphatic polyesters has garnered significant attention. Aliphatic polyesters, known for their biodegradability and flexibility, offer complementary properties that can be enhanced when combined with lignin. The copolymerization between lignin and aliphatic polyesters not only provides an opportunity to develop novel composite materials with improved mechanical and thermal properties but also contributes to the advancement of sustainable materials science. This transition from the aromatic, rigid nature of lignin to the aliphatic, flexible characteristics of polyesters sets the stage for exploring the potential benefits and challenges of their integration, which are discussed in the following section.
3. Aliphatic polyester
Because of environmental concerns and increased interest in biomedical applications, aliphatic polyesters have been put in the spotlight as an alternative biodegradable polymer material in recent decades. All polyesters are theoretically considered biodegradable because their main production reaction, esterification, is chemically reversible by hydrolase action and hydrolysis.39 Although aliphatic polyesters are easy to biodegrade, they have low mechanical properties due to their linear chain structure compared to aromatic polyesters. Fig. 6 shows the chemical structures of a few representative biodegradable polymers (aliphatic polyesters). The polyester can be synthesized by a polycondensation reaction between (1) monomers containing a dihydroxyl group (diol) and monomers containing a dicarboxylic acid group,40 (2) a diol and an anhydride,41 (3) a diol and an acid chloride,42 (4) a diol and a nitrile,43 (5) a diol and carbon suboxide,44 (6) a diol and an ester (transesterification),45 and (7) a difunctional monomer with a hydroxyl group and a carboxylic acid,46 and ring opening polymerization. The synthesized aliphatic polyesters showed good potential for applications in multiple fields including drug delivery systems,47 packaging,48 medical implants,48 and films.49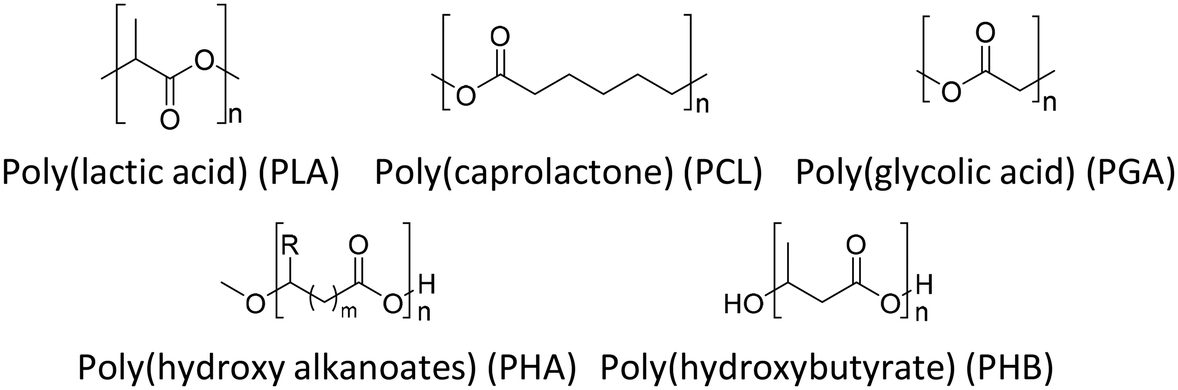 | ||
| Fig. 6 Chemical structures of a few representative biodegradable polymers, aliphatic polyesters. | ||
3.1 Poly(lactic acid) (PLA)
One of the most studied aliphatic polyesters is poly(lactic acid) (PLA). PLA can be synthesized by polycondensation of lactic acid or ring opening polymerization of lactide. Lactic acid and lactide, which are monomers of PLA, are chiral molecules with two forms, L-form (L-lactic acid and L-lactide) and D-form (D-lactic acid and D-lactide). The tacticity of a polymer is determined by which isomers of monomers are bound. The homopolymers of the L-form (PLLA) and D-form (PDLA) are isotactic polymers and the copolymer of the L-form and D-form (PDLLA) is an atactic polymer (Scheme 1). The crystallinity, mechanical properties, and biodegradability rate of polymers determine the tacticity. Isotactic polymers (PLLA and PDLA) are semi-crystalline polymers with relatively high mechanical strength and low degradation rates, whereas an atactic polymer (PDLLA) results in amorphous and low mechanical properties and high degradation rates. Because of these properties, PLLA and PDLA were used in orthopedic and fixation devices.42 On the other hand, PDLLA is used in biomedicine for tissue engineering50 and drug delivery devices.51 | ||
| Scheme 1 Synthesis of (a) poly(L-lactic acid) (PLLA), (b) poly(D-lactic acid) (PDLA), (c) poly(DL-lactic acid) (PDLLA). | ||
3.2 Poly(glycolic acid) (PGA)
Poly(glycolic acid) (PGA) is another aliphatic polyester that can be synthesized by polycondensation and ring opening polymerization (Scheme 2a). Glycolic acid and glycolide, monomers of PGA, are simpler than lactic acid and lactide and have no chirality, thus its polymer, PGA, has no tacticity. It results in high crystallinity (45–55%) with a high melting temperature (Tm, 220–225 °C).52 Also, PGA has a faster degradation rate and better mechanical properties than PLA.53 Based on these properties, glycolic acid is used to synthesize a copolymer, poly(lactic acid-co-glycolic acid) (PLGA), with lactic acid by polycondensation (Scheme 2b). The properties of PLGA are controlled by the molar ratio between lactic acid and glycolic acid. Due to its biodegradable nature, excellent biocompatibility, low toxicity, high miscibility with other polymers, and the ability to form films and capsules, PLGA finds extensive use in drug delivery systems.54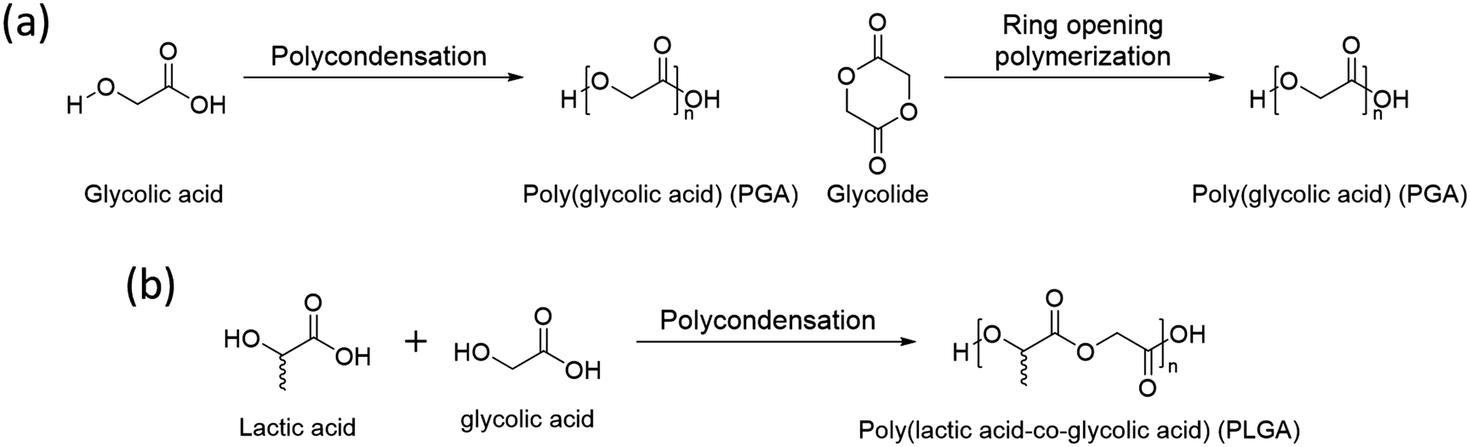 | ||
| Scheme 2 Synthesis of (a) poly(glycolic acid)(PGA) and (b) poly(lactic acid-co-glycolic acid) (PLGA). | ||
3.3 Poly(caprolactone) (PCL)
Recently, aliphatic polyesters obtained from macro lactones have been investigated for their more favorable ductility and strength properties over poly(lactic acid), which are intrinsically brittle.55,56 One of them is poly(caprolactone) (PCL). It is not only an FDA approved material but also a traditional material for applications in biomedicine57 and food packaging.58 PCL was first synthesized by the Carothers group in the early 1930s.59 PCL is mainly synthesized by ring-opening polymerization of ε-caprolactone (Scheme 3). The homopolymer of PCL requires up to four years for complete degradation.60 Because of the biodegradability and biocompatibility of PCL, PCL is employed in various applications, including drug delivery, dentistry, tissue engineering, and several medical devices. Many drug delivery devices made from PCL have been approved by the FDA. Also, diverse medical devices are assembled with PCL copolymers. PCL has good solubility in organic solvents and a low Tm of 60 °C.59 These features enable convenient copolymerization with other synthetic polymers. For example, poly(caprolactone)-block-poly(glycolic acid) is commercialized as a monofilament suture,61 and tissue-reinforcing patches composed of poly(caprolactone)-based polyurethane have been developed.62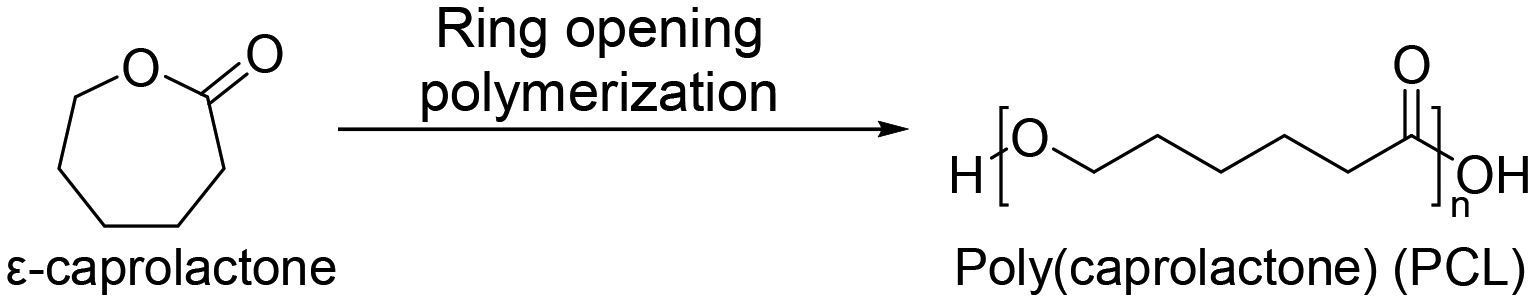 | ||
| Scheme 3 Synthesis of poly(caprolactone) (PCL). | ||
3.4 Poly(ethylene brassylate) (PEB)
An example of an aliphatic polyester obtained from a macro lactone is PEB, a relatively unexplored polymer compared to PCL. Research on PEB began in 2014, and as of August 2024, fewer than 100 references to it are found on Web of Science. The most common method used to obtain PEB is the ring-opening polymerization of ethylene brassylate. The first method reported by Kobayashi et al. in 1999 used lipase enzymes as a catalyst.63 However, this method yielded low molecular weight polymers. In 2014, Pascual et al. reported organic base-catalyzed PEB polymerization.64 The superbase 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) showed the highest polymerization efficiency, the fastest reaction rate, the highest molecular weight, and the narrowest PDI value among all compared conditions. The same research group showed pioneering works in their study of PEB, including utilizing TBD as both the initiator and catalyst of the ring opening polymerization of ethylene brassylate (Scheme 4).64–66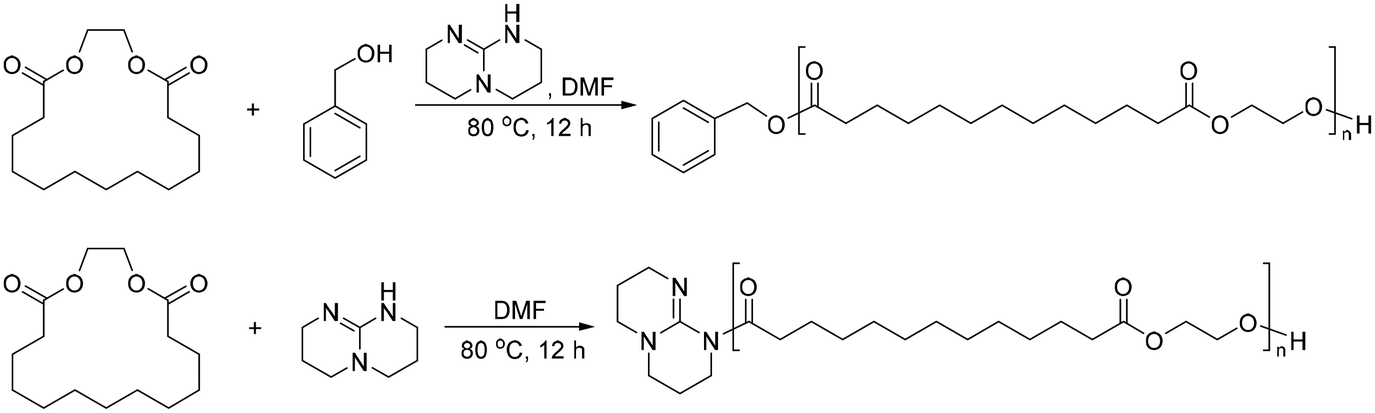 | ||
| Scheme 4 Synthesis of poly(ethylene brassylate) (PEB) via ring-opening polymerization. | ||
PEB has high potential for being a commercially useful, sustainable raw material for polymers. This is because the monomer, ethylene brassylate, is derived from non-human food vegetable oil (castor oil), which is cheap and highly abundant. Ethylene brassylate is a commercially available 17-membered ring lactone and is much cheaper than lactide and other macro lactones, such as ε-caprolactone. A large production infrastructure of ethylene brassylate has already been established in the fragrance industry.67 Briefly, ethylene brassylate can be easily obtained from tridecanoic acid synthesized from 10-undecanoic acid,68 which is an unsaturated fatty acid extracted from castor plants.
The thermal properties of PEB are similar to those of polycaprolactone, but PEB exhibits a slightly higher Tm of 68.1 °C (Mn = 18 kDa, DP = 66) to 73.3 °C (Mn = 137 kDa, DP = 508)83 compared to PCL, which has a Tm of 56.0 °C (Mn = 10 kDa, DP = 87) to 62.7 °C (Mn = 80 kDa, DP = 701).84 The glass transition temperature (Tg) follows the same trend, with a Tg of −30 °C for PEB and a Tg of −60 °C for polycaprolactone.85–87 The renewable and biobased natures of PEB are desirable features (Table 3). However, its mechanical properties need to be improved for advanced applications. Enhanced mechanical properties, such as the tensile strength and modulus, will enable it to replace commercial polyolefins.
| No. | Polymer | Microbial strain | Source | Reference |
|---|---|---|---|---|
| 1 | PHA | Recombinant E. coli | Cheese whey | Pais et al. (2014)69 |
| 2 | Halomonas hydrothermalis | Seaweed-derived crude levulinic acid, containing formic acid, residual sugars, and dissolved minerals | Bera et al. (2015)70 | |
| 3 | T. thermophilus HB8 | Lactose from whey-based media | Pantazaki et al. (2009)71 | |
| 4 | P3HB | Recombinant E. coli BL21 | Glucose | Lin et al. (2017)72 |
| 5 | Cupriavidus necator DSM 545 | Glucose, acetic acid | Marudkla et al. (2018)73 | |
| 6 | Ralstonia eutropha | Sucrose | Park et al. (2015)74 | |
| 7 | Cupriavidus necator NSDG-GG | Glucose, urea | Biglari et al. (2018)75 | |
| 8 | Marine Bacillus megaterium UMTKB-1 strain | Glycerol | Yatim et al. (2017)76 | |
| 9 | Endophytic Bacillus cereus RCL 02 | Valeric acid | Das et al. (2018)77 | |
| 10 | Paracoccus denitrificans DSMZ 413 | Glycerol | Kalaiyezhini et al. (2015)78 | |
| 11 | Acinetobacter junii BP 25 | Parboiled rice mill effluent | Sabapathy et al. (2018)79 | |
| 12 | P4HB | Recombinant Escherichia coli, like JM109 | Propionic acid, glycerol | Malacara et al. (2015)80 |
| 13 | PHO | Recombinant Cupriavidus necator strains | Canola oil | Valdés et al. (2018)81 |
| 14 |
Pseudomonas putida and Bacillus subtilis, at a rate of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
Sodium octanoate | Eremia et al. (2016)82 |
3.5 Poly(hydroxy alkanoate) (PHA)
PHAs are a family of biological polyesters that contain hydroxy alkanoate monomer units.88,89 PHA can usually be obtained from bacteria and archaea90 and synthesized with a polymerizing enzyme. During biosynthesis, all monomer units are in the R configuration due to the enzyme's stereospecificity, which provides the optical activity of the synthesized polymer.89 Examples of PHA families are poly(4-hydroxybutyrate) (P4HB), poly(3-hydroxybutyrate) (P3HB), poly(3-hydroxyoctanoate) (PHO), poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV), and poly(3-hydroxybutyrate-co-3-hydroxyoctanoate) (PHBHHx).90The biosynthetic pathways for the majority of PHA monomers are derived from various carbon sources such as glucose and fatty acids, which are primarily related to essential carbon metabolic pathways, including glycolysis, β-oxidation, and the synthesis of new fatty acids (Fig. 7). Moreover, structurally similar carbon sources such as γ-butyrolactone (GBL)/4HB of 1,4-butanediol (BDO), 3-hydroxyvalerate (3HV) of propionate, and 3-hydroxypropionate (3HP) of 1,3-propiondiol (PDO) can serve as precursors. Additionally, medium and long-chain lengths of other fatty acids, like 3-hydroxyalkanoate (3HA), have a significant impact on the material's properties (Table 4).89,91,92 Consequently, by supplying tailored feedstocks (Fig. 7), various PHA homopolymers and copolymers can be obtained with engineered microorganisms.93
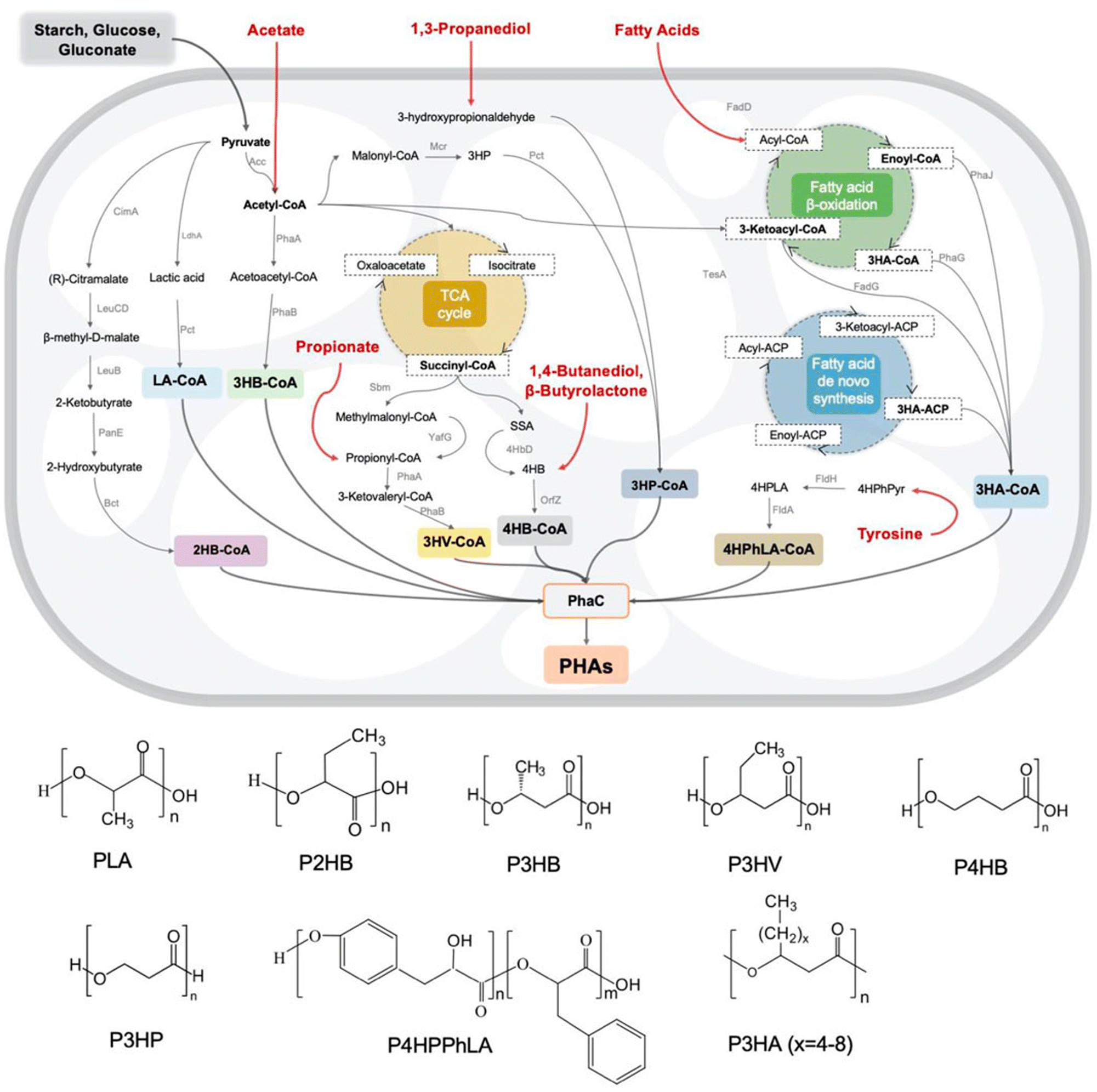 | ||
| Fig. 7 Metabolic pathways and monomer structures of different microbially synthesized PHAs. Reproduced from ref. 93 with permission from Frontiers Media S.A., copyright 2022. | ||
| Polymer | Melting point (°C) | Glass transition temperature (°C) | Crystallinity | Mechanical strength |
|---|---|---|---|---|
| Poly(lactic acid) (PLA) | 170–180 (ref. 84) | 60–65 (ref. 84) | Semi-crystalline (PLLA and PDLA), amorphous (PDLLA) | High strength in isotactic forms (PLLA/PDLA), low in atactic (PDLLA) |
| Poly(glycolic acid) (PGA) | 220–225 (ref. 52) | 35–40 (ref. 52) | High (45–55%) | Higher mechanical strength and faster degradation compared to PLA |
| Poly(caprolactone) (PCL) | 56–63 (ref. 84) | −60 (ref. 84) | Semi-crystalline | Good ductility and strength |
| Poly(ethylene brassylate) (PEB) | 68–73 (ref. 83) | −30 (ref. 83) | Semi-crystalline | Needs to be improved for advanced applications |
| Poly(hydroxy alkanoate) (PHA) | Variable | Variable | Variable | Variable |
3.6 Degradation of aliphatic polyester via hydrolysis
The degradation of polyesters occurs through hydrolysis in the presence of acid or base catalysts.94 Under acidic conditions, the degradation of polyesters begins with protonation of the carbonyl oxygen of the ester group by a hydronium ion, which makes the carbonyl carbon more electrophilic due to the positive charge. The next step is that water molecules attack the carbonyl carbon, which generates a tetrahedral intermediate. The tetrahedral intermediate can decompose into a carboxylic acid and a hydroxyl group, or back the polyester structure (Scheme 5).94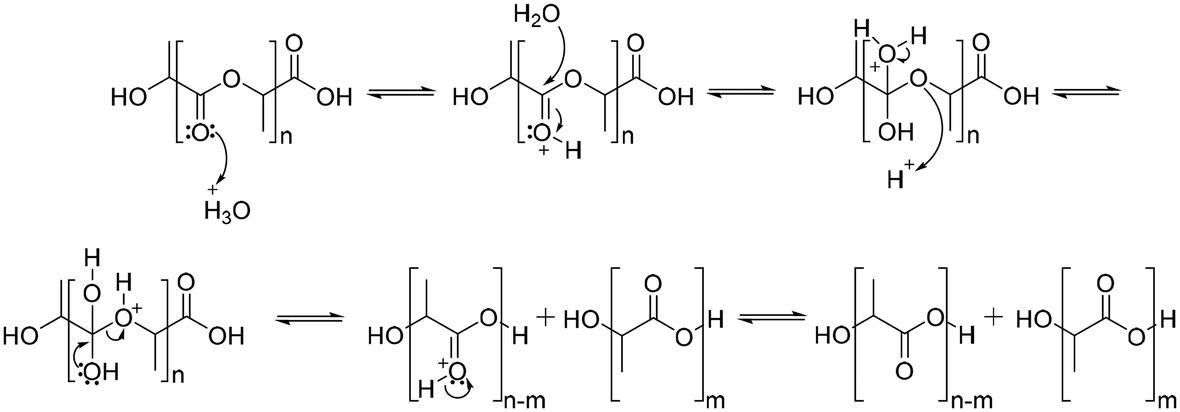 | ||
| Scheme 5 Hydrolysis mechanism of poly(lactic acid) under acidic conditions. | ||
Under basic conditions, degradation begins with attack of the hydroxide anion on the carbonyl carbon of the ester group, generating a tetrahedral intermediate. The tetrahedral intermediate can decompose into a carboxylic acid and a hydroxyl group. The preference of the tetrahedral intermediate toward hydrolysis or ester regeneration is determined by the leaving ability of the hydroxyl group and stabilizing a negative charge (Scheme 6).94,95 The enzyme-mediated biodegradation of aliphatic polyesters is described elsewhere with detailed information.96–100
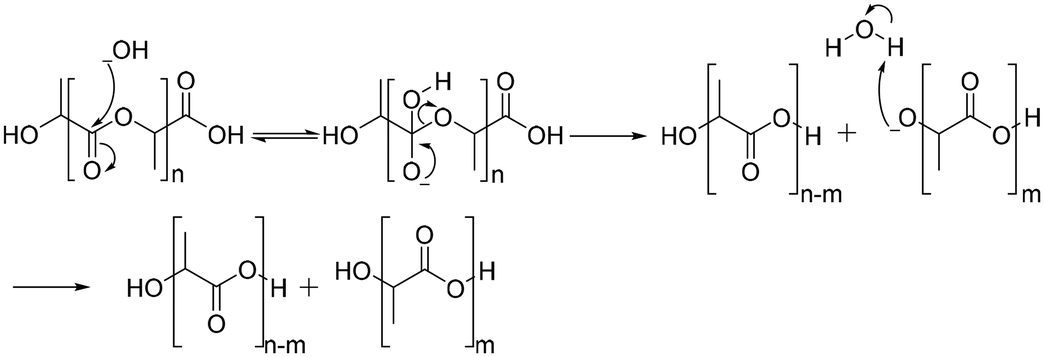 | ||
| Scheme 6 Hydrolysis mechanism of poly(lactic acid) under basic conditions. | ||
4. Lignin-graft-polyester
In this section, we discuss lignin-based copolymers that have covalent linkages between lignin and its counterpart polymers. Especially, we focus on lignin-graft-polyester, not derivatives or depolymerized lignin-based polymer. Lignin-graft-polyester exhibits several characteristics that make it particularly suitable for green chemistry.Firstly, as discussed in section 2, lignin is a natural polymer extracted from wood and other plants, constituting a significant component of biomass. Utilizing lignin leverages renewable resources, reduces reliance on fossil fuel-based materials, and contributes to a lower environmental impact.
Secondly, the direct use of lignin for polymer synthesis circumvents the environmentally harmful depolymerization processes of lignin, thereby conserving energy and minimizing pollutant emissions. Depolymerizing lignin into smaller molecules often requires substantial energy and can produce various environmental pollutants. For instance, the chemical or thermal treatments used to break down lignin into monomers and other fine chemicals involve significant amounts of organic solvents and energy, which can have detrimental environmental effects. The organic solvents typically used in lignin depolymerization do not align with the principles of green chemistry. These solvents are often toxic or highly volatile, posing risks to both the environment and human health. Therefore, the lignin-graft-polyester research discussed supports the principles of a circular economy, decreasing the environmental burden associated with waste management.
Thirdly, lignin-graft-polyester can retain the functional properties of polymers and the biodegradability of natural materials. This ensures that end-of-life lignin-graft-polyesters can decompose naturally in the environment, minimizing their ecological footprint compared to non-degradable conventional petroleum-based polymers. In addition, recent studies have proved that lignin-graft-polyesters are excellent materials that address the shortcomings of previously developed aliphatic polyesters and provide additional functions based on the high aromaticity of lignin.
In summary, lignin-graft-polyesters are well-suited for green chemistry due to the renewable nature of lignin, the avoidance of energy-intensive and polluting depolymerization processes, the minimization of harmful organic solvents, waste reduction, and inherent functionality and biodegradability of the resulting materials. These advantages position lignin–polyester copolymers as promising candidates for sustainable materials.
The representative grafted polymers on lignin are PLA and PCL. The lignin-graft-polyesters provide cost-efficiency, high mechanical properties, adhesion properties, flame retardancy, antioxidant properties, and antimicrobial properties.101
4.1 Lignin-graft-poly(lactic acid) (lignin-graft-PLA)
Lignin-graft-PLA can be synthesized by both “graft-from” and “graft-onto” methods. The graft-from methods for the synthesis of a lignin-graft-PLA copolymer are reported in the literature several times.102–105In 2013, Chung et al. reported the synthesis of lignin-graft-PLA through ring-opening polymerization in the presence of a TBD catalyst by initiating the hydroxyl group of lignin. The authors found that the grafting efficiency in lignin-graft-PLA synthesis from aliphatic hydroxyl groups was higher than for phenolic hydroxyl groups (Scheme 7a).102 Also, Liu et al. and Dai et al. introduced a synthesis method using 4-dimethyl pyridine (DMAP) as a catalyst (Scheme 7b).103,104
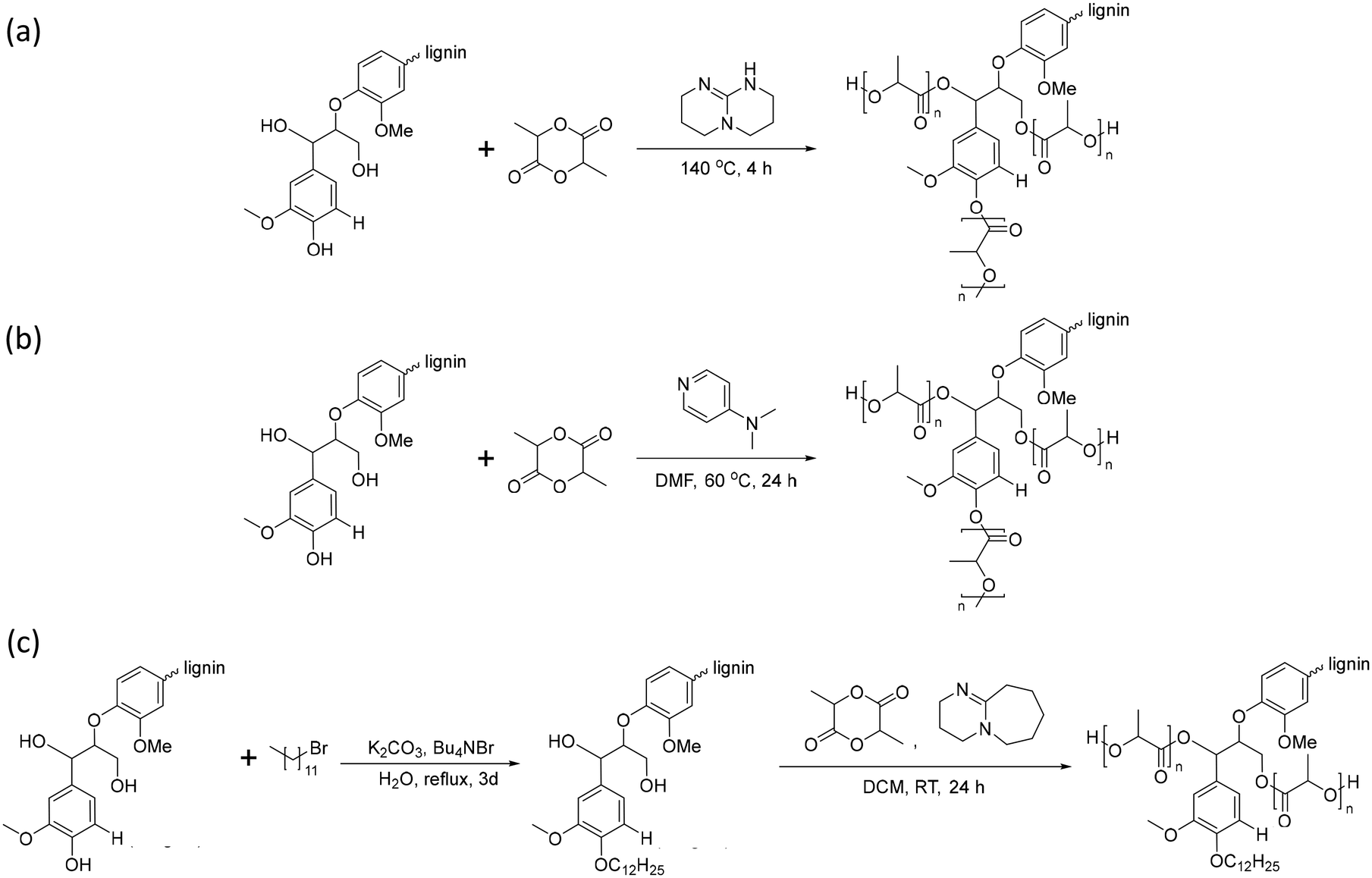 | ||
| Scheme 7 (a) Synthesis of lignin-graft-PLA with TBD as a catalyst through a graft-from method. (b) Synthesis of lignin-graft-PLA with DMAP as a catalyst through a graft-from method. (c) Synthesis of dodecylated lignin-graft-PLA. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
In 2016, Ren et al. presented dodecylated lignin-graft-PLA to grow the high molecular weight of PLA (Scheme 7c).105 Acetylation and butylation of lignin have been applied to reduce the number of hydroxyl groups. Reactions usually occur at the aliphatic hydroxyl groups, which are the preferred position for growing PLA. The authors reported a method for selective dodecylation to the phenolic hydroxyl group without interfering with the growth of PLA.
Lignin-graft-PLA synthesized by a graft-onto method was reported by Chile et al. in 2018.106 PLA was separately synthesized by ring opening polymerization in the presence of [(NNO)InCl]2 (μ-Cl)(μ-OEt), and the end group of PLA was modified as the chloride derivative. The modified PLA-Cl was grafted onto lignin with K2CO3 in DMF. When comparing the ratio of lignin to PLA in the produced polymer, the graft-onto method showed a higher lignin portion than the graft-from method for lignin-graft-PLA (Scheme 8).
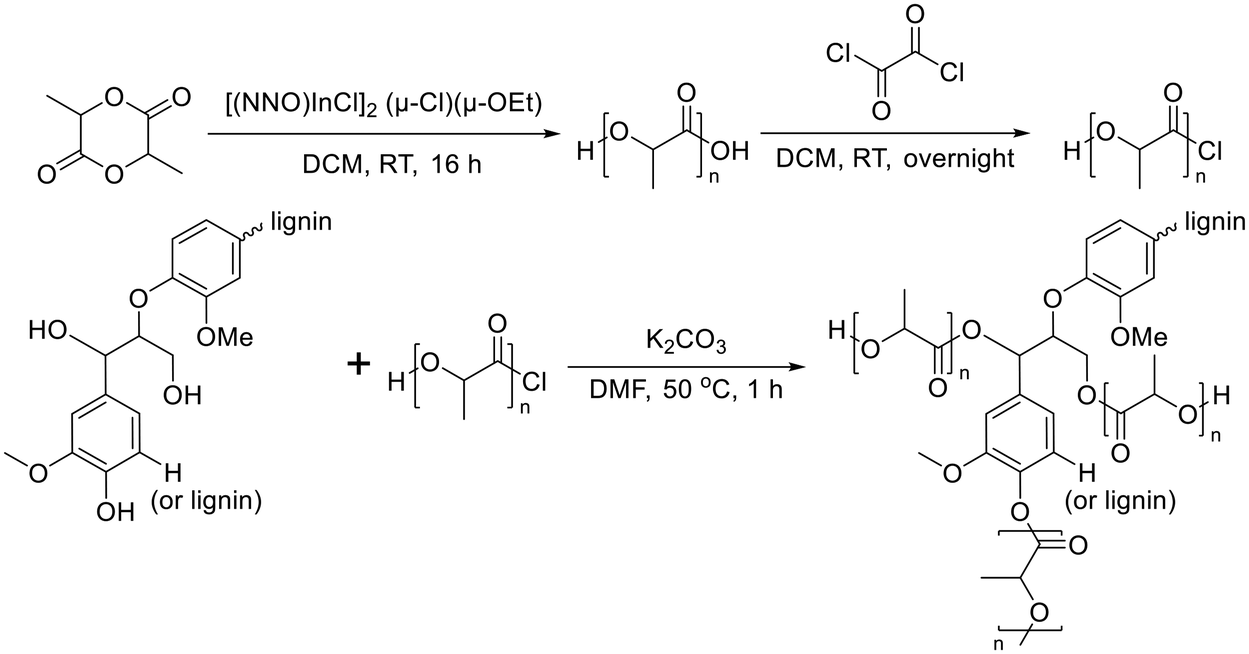 | ||
| Scheme 8 Synthesis of PLA and graft copolymerization of PLA onto lignin. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
Recently, grafting methods, including graft-from and graft-onto, via reactive extrusion were reported.107–109 Reactive extrusion is a process that combines chemical reactions with extrusion, typically used in polymer processing. In this technique, the chemical reaction occurs within the extruder, allowing for the continuous formation and shaping of polymers or composites. This process is particularly useful for polymer modifications such as crosslinking, grafting, or blending, as it offers advantages like controlled reaction conditions, efficient mixing, and reduced processing time. The key benefits of reactive extrusion include the ability to conduct reactions under high shear and temperature, precise control over the reaction environment, and the potential for industrial-scale production.
In 2023, Makri et al. synthesized lignin-graft-PLA through reactive extrusion, employing both graft-from and graft-onto methods.108 For the graft-from approach, the appropriate amount of lignin was premixed with L-lactide and a catalyst system consisting of triphenyl phosphine (TPP) and Sn(Oct)2. This mixture was then fed into a rheometer (Brabender® Plasti-Corder® Lab-Station torque rheometer) set at 180 °C with a screw speed of 50 rpm, under a nitrogen atmosphere. The reaction was carried out for approximately 20 min. For the graft-onto method, PLA and lignin were premixed and fed into the same rheometer using a pneumatic ram, mixed at 180 °C with a screw speed of 50 rpm for 5 min. In this step, the catalyst is not required to form a graft copolymer.
The main application of lignin-graft-PLA is as an additive for a commercial polymer matrix to improve its optical and mechanical properties.102,105,106,110 In 2013, Chung et al. reported on a PLA/lignin-graft-PLA composite.102 The synthesized lignin-graft-PLA showed good dispersity, whereas unmodified lignin formed phase separated particles in the PLA matrix (Fig. 8a–e).
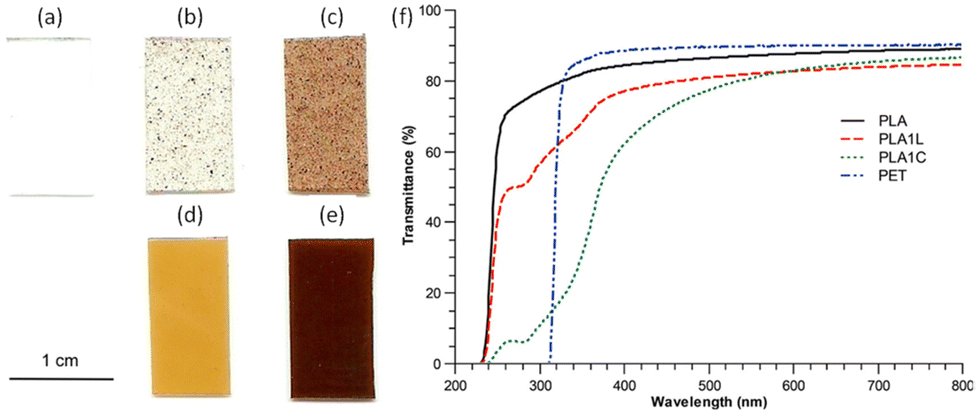 | ||
| Fig. 8 Images of PLA–lignin composite samples suggest increased dispersion of lignin in composites made from lignin-g-PLA copolymer compared to those made with unmodified lignin: (a) PLA, (b) PLA with 1 wt% of lignin, (c) PLA with 5 wt% of lignin, (d) PLA with 1 wt% of lignin and 10 wt% of lignin-g-PLA copolymers, (e) PLA with 5 wt% of lignin and 12.5 wt% of lignin-g-PLA copolymer. (f) UV–vis spectra of PLA, PET, and PLA–lignin composites with unmodified lignin (PLA1L) and lignin-g-PLA copolymers (PLA1C). The amounts of lignin in the PLA films with unmodified lignin (PLA1L) and with lignin-g-PLA copolymers (PLA1C) are 1% and 0.9%, respectively. Reproduced from ref. 102 with permission from American Chemical Society, copyright 2013. | ||
The PLA/lignin-graft-PLA composite with 1% lignin blocked the entire UV-C (100–280 nm) and UV-B (280–315 nm) regions, and it blocked 40–85% of UV-A (315–400 nm) depending on the wavelength. However, the PLA matrix without the lignin component blocked only 25% of UV light above 250 nm (Fig. 8f). Also, PLA/lignin-graft-PLA composite with 1% lignin showed an increase in the tensile strength (+16%) and strain (+9%) compared to PLA (Fig. 9).
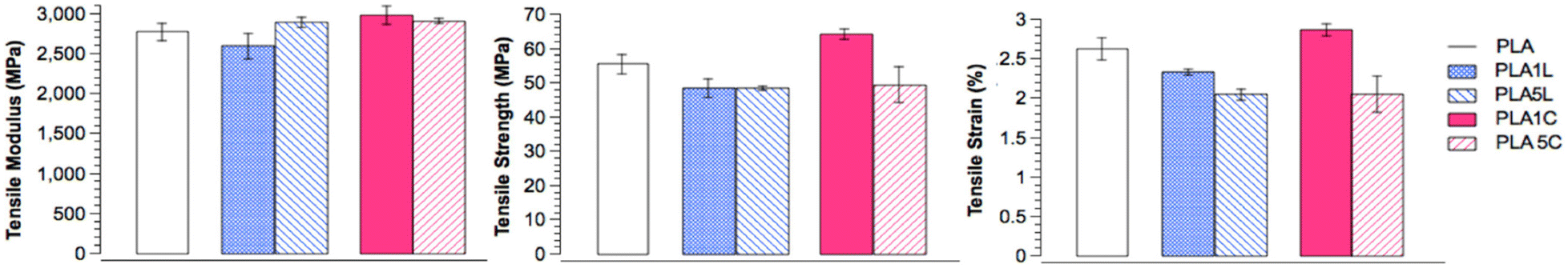 | ||
| Fig. 9 Mechanical properties of PLA–lignin composites. The lignin contents in PLA1L/PLA1C and PLA5L/PLA5C are 0.9–1.0 and 4.4–4.8 wt%, respectively. Reproduced from ref. 102 with permission from American Chemical Society, copyright 2013. | ||
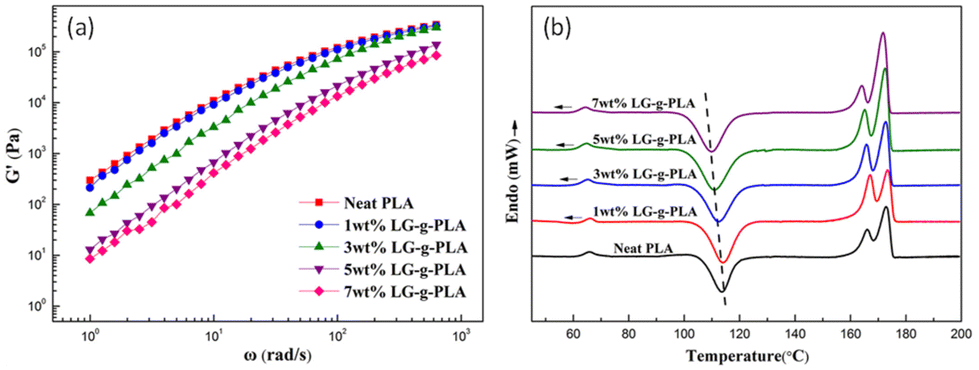
Zong et al. introduced PLA foam with lignin-graft-PLA in 2020.111 First, they blended PLA and lignin-graft-PLA to make PLA/lignin-graft-PLA composite sheets. The sheets were placed in a high-pressure vessel and 15 MPa of CO2 was applied to manufacture PLA/lignin-graft-PLA composite foam. The rheological, mechanical, and thermal properties of the prepared foam are investigated. The storage modulus of the PLA/lignin-graft-PLA samples decrease as the lignin-graft-PLA content increases. This behavior is attributed to the plasticization effect of lignin-graft-PLA on PLA. It increases the polymer chain spacing (free volume) of PLA and reduces entanglement between molecular chains (Fig. 10a).
 | ||
| Fig. 10 (a) Storage modulus of PLA with different LG-g-PLA contents and (b) DSC curves of the PLA/LG-g-PLA and PLA samples. Reproduced from ref. 111 with permission from John Wiley and Sons, copyright 2020. | ||
The Tg and cold crystallization temperature of the PLA/lignin-graft-PLA samples decrease with the addition of lignin-graft-PLA. In addition, the crystallinity was increased to 7.1–12.4% compared to neat PLA. This is due to the good compatibility between lignin-graft-PLA and the PLA matrix, which prevents the uneven distribution of lignin-graft-PLA and ensures superior nucleation of lignin-graft-PLA. As a result, the diffusion of chain segments and the speed of chain arrangement improved the crystallization rate of PLA/lignin-g-PLA (Fig. 10b).
The size distribution of pores in foam samples was confirmed by SEM images of PLA/lignin-graft-PLA foams (Fig. 11a–e). The composite form of PLA with 3% of lignin-graft-PLA showed a smaller average cavity cell size (30.6% smaller), higher cell density (two orders of magnitude higher), and a higher expansion ratio (about 150% higher) than pure PLA foams. This is attributed to the formation of a relatively smaller size and larger number of spherocrystals of PLA, resulting from the heterogeneous nucleation of lignin-graft-PLA.
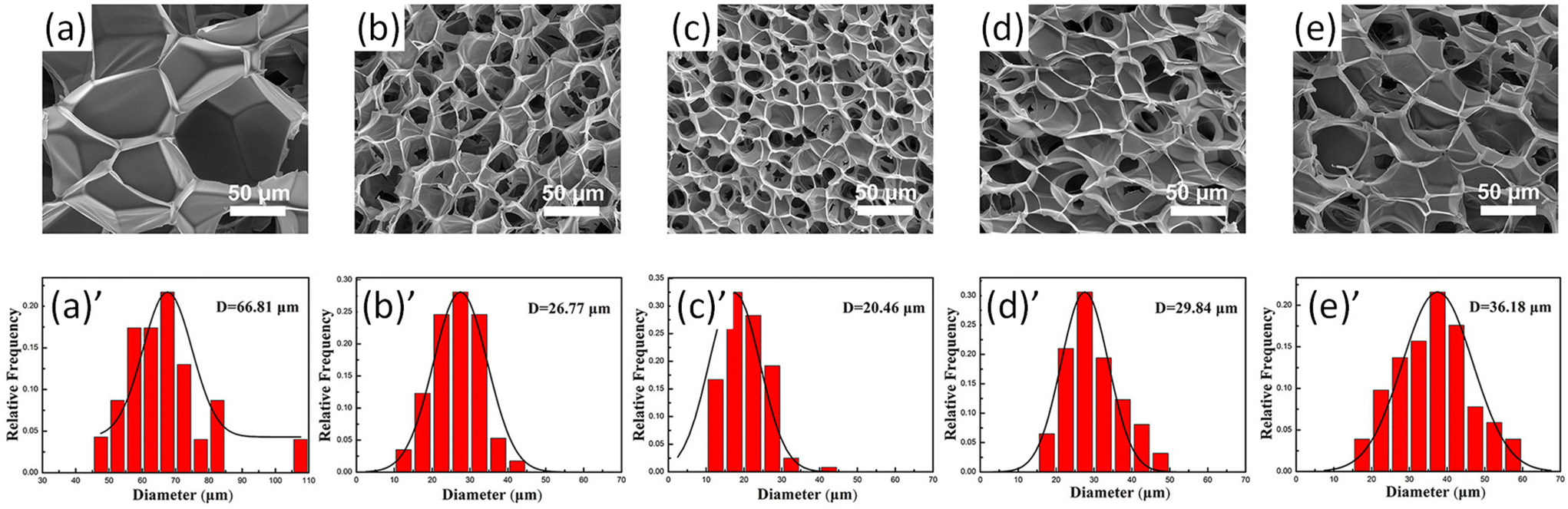 | ||
| Fig. 11 (a)–(e) SEM images of cell morphology; (a)′–(e)′ corresponding cell size distributions of the PLA and PLA/lignin-graft-PLA foams: (a) and (a)′ neat PLA, (b) and (b)′ 1 wt% lignin-graft-PLA, (c) and (c)′ 3 wt% lignin-graft-PLA, (d) and (d)′ 5 wt% lignin-graft-PLA, and (e) and (e)′ 7 wt% lignin-graft-PLA. Reproduced from ref. 111 with permission from John Wiley and Sons, copyright 2020. | ||
Liu et al. investigated the influence of PLA tacticity on the thermal and mechanical properties (Fig. 12).112 Lignin-graft-PDLA, lignin-graft-PLLA, and lignin-PDLLA were prepared by ring opening polymerization of L-lactide, D-lactide, and L-lactide/D-lactide mixture, respectively and these were used for making a blend film with PLLA. The crystallization behavior and thermal stability of lignin-graft-PLAs were investigated. PLLA/lignin-graft-PDLA showed the fastest crystallization half-time, the highest crystallinity, and the highest Tg. Also, PLLA/lignin-graft-PDLA was the most stable at high temperature. On the other hand, PLLA/lignin-graft-PLLA showed the slowest crystallization half-time, the lowest crystallinity, the lowest Tg, and the lowest thermal stability (Fig. 13). The reason is that PDLA and PLLA form a triclinic racemic structure, known as a stereocomplex. Within the stereocomplex, left- and right-handed molecules pack side by side via van der Waals interactions, increasing the likelihood of forming zip-lock hooks. This zip-lock hook structure influences the crystallization behavior of PLLA.113
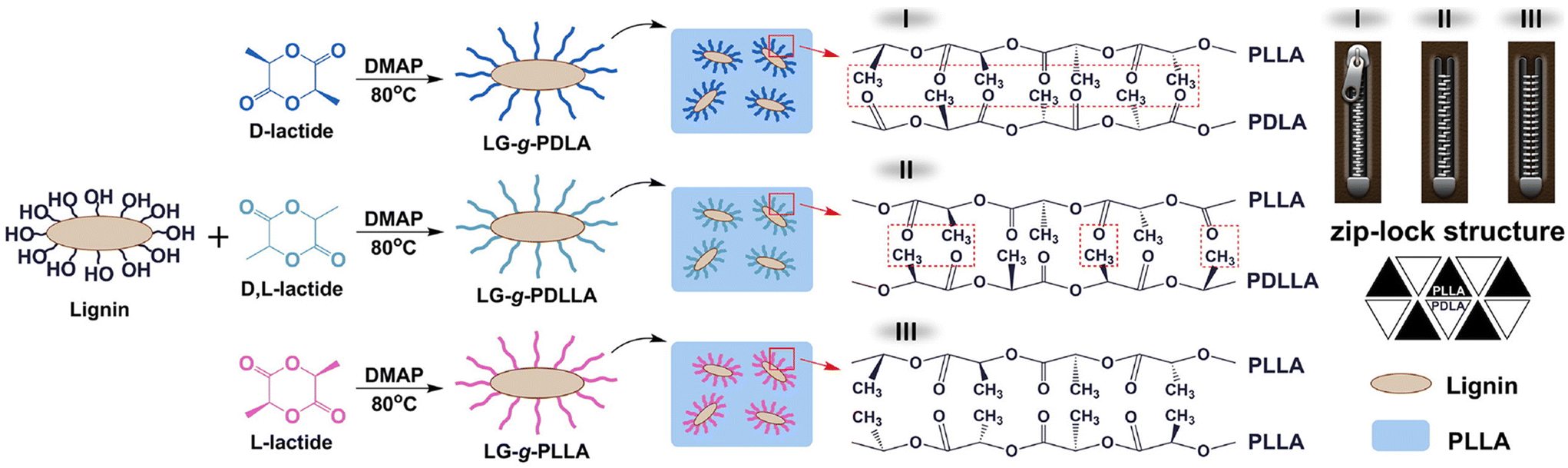 | ||
| Fig. 12 Illustration of the synthesis of LG-g-PDLA (-PDLLA, -PLLA) and structures of the stereocomplex films. Reproduced from ref. 112 with permission from Elsevier, copyright 2017. | ||
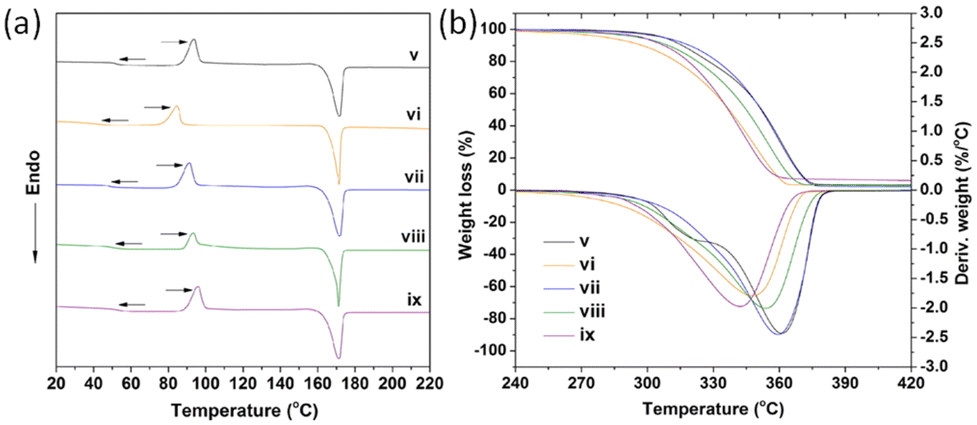 | ||
| Fig. 13 (a) DSC heating curves related to the second heating scan, (b) TGA and DTG curves of PLLA and PLLA complex films. v: PLLA, vi: PLLA/3% lignin, vii: PLLA/3% lignin-g-PDLA, viii: PLLA/3% lignin-g-PDLLA, ix: PLLA/3% lignin-g-PLLA. Reproduced from ref. 112 with permission from Elsevier, copyright 2017. | ||
4.2 Lignin-graft-poly(caprolactone) (lignin-graft-PCL)
For lignin-graft-PCL, the most common synthesis method is graft-from copolymerization.114–119 In 2013, Laurichesse et al. reported the synthesis of lignin-graft-PCL by ring opening polymerization in the presence of stannous octanoate (SnOct2) catalyst and hydroxyl initiation sites on lignin.117 The crystallinity of lignin-graft-PCL increased with a higher molar ratio of caprolactone. Lauricheese et al. employed dry toluene solvent, while Wang et al. and Park et al. utilized graft-from methods for lignin-graft-PCL, which was synthesized under solvent-free conditions using the same catalyst, SnOct2 (Scheme 9).118,119 Another catalyst, ZnCl2, was used by Abdollahi et al. to prepare lignin-graft-PCL.116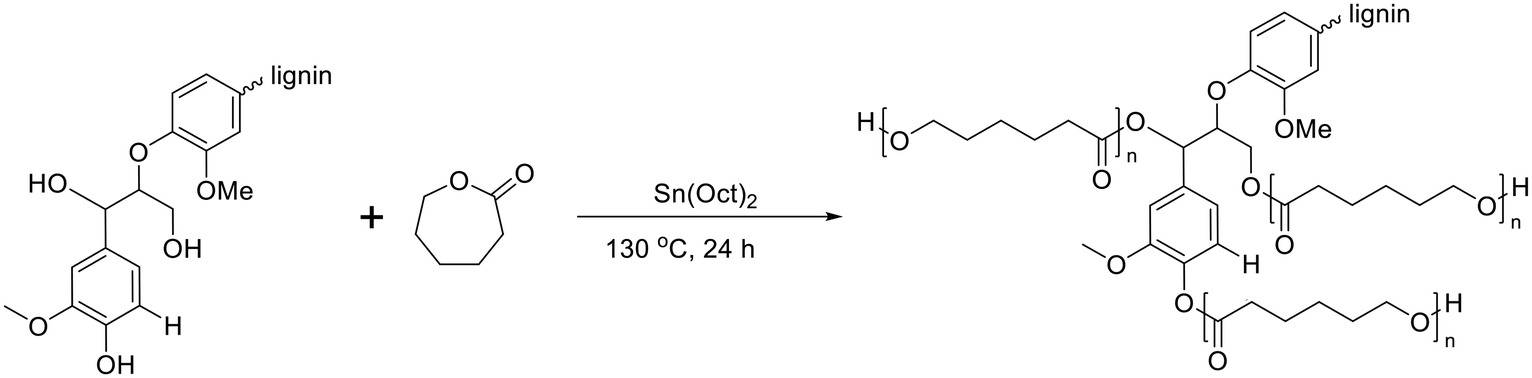 | ||
| Scheme 9 Synthesis of lignin-graft-PCL using the graft-from method. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
The polymerization of ε-CL with Sn(Oct)2/lignin is proposed to occur through a coordination mechanism, as depicted in Scheme 10.120–124 Initially, Sn(II) alkoxide is generated through exchange reactions between Sn(Oct)2 and the hydroxyl group of lignin. Once the authentic initiator is formed, initiation commences with the coordination of the carbonyl oxygen of ε-CL with tin. Subsequently, the nucleophilic oxygen in Sn–O-lignin attacks the carbonyl carbon of ε-CL, leading to cleavage of the acyl–oxygen bond in the ε-CL ring. This process results in the creation of the propagating species (–SnO–), and the propagation step advances by inserting ε-CL into the reactive Sn–O bond found in these active species.
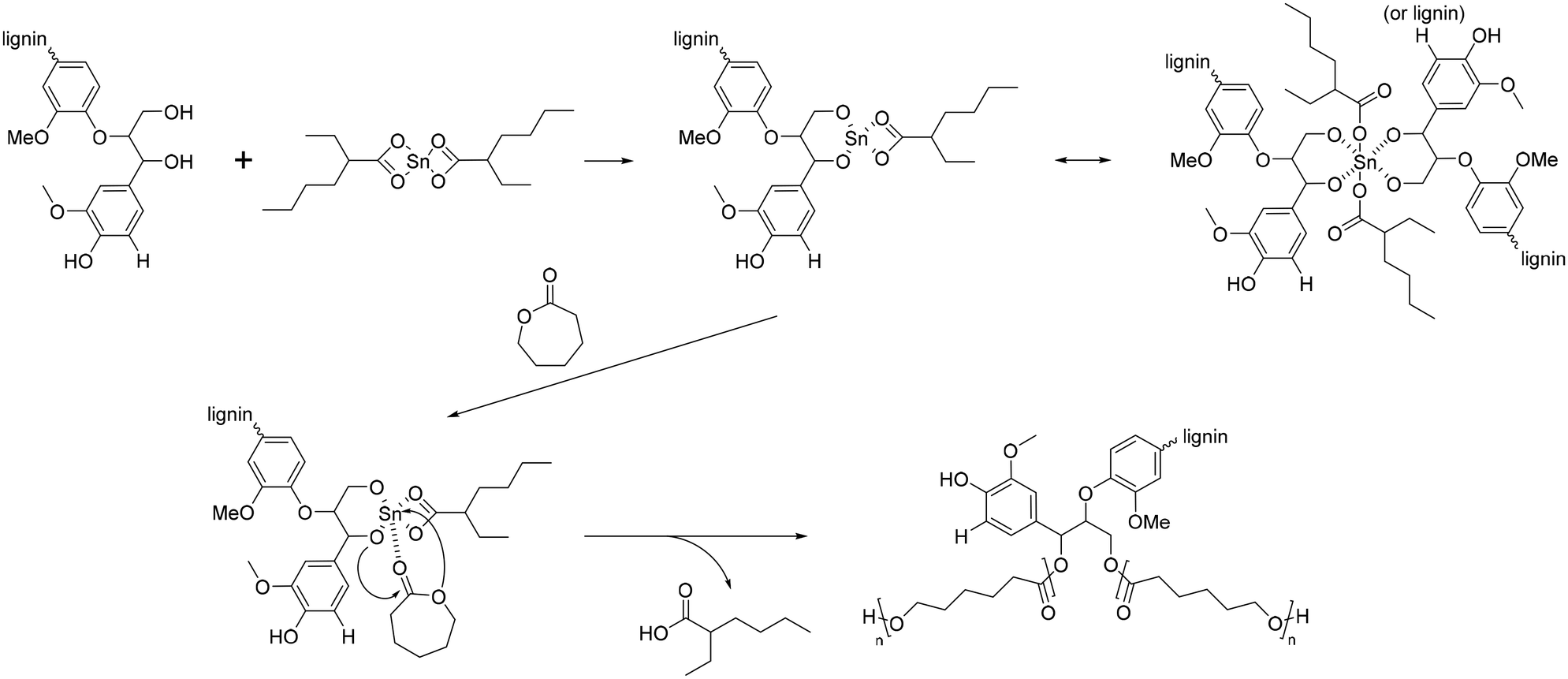 | ||
| Scheme 10 Mechanism of ring-opening polymerization of caprolactone with Sn(Oct)2. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
In 2012, Korich et al. reported a graft-onto method to prepare lignin-graft-PCL.25 In the first step, Bpin-PCL-OH was synthesized by ring opening polymerization using a pinacol boronate ester (Bpin)-containing initiator. Then, the hydroxyl end group of Bpin-PCL-OH was functionalized as an acylate end group and Bpin was deprotected as B(OH)2 on ion exchange resin. As-prepared B(OH)2-PCL-OAc was reacted with the hydroxyl groups of lignin to form aryl boronate ester bonds (Scheme 11).
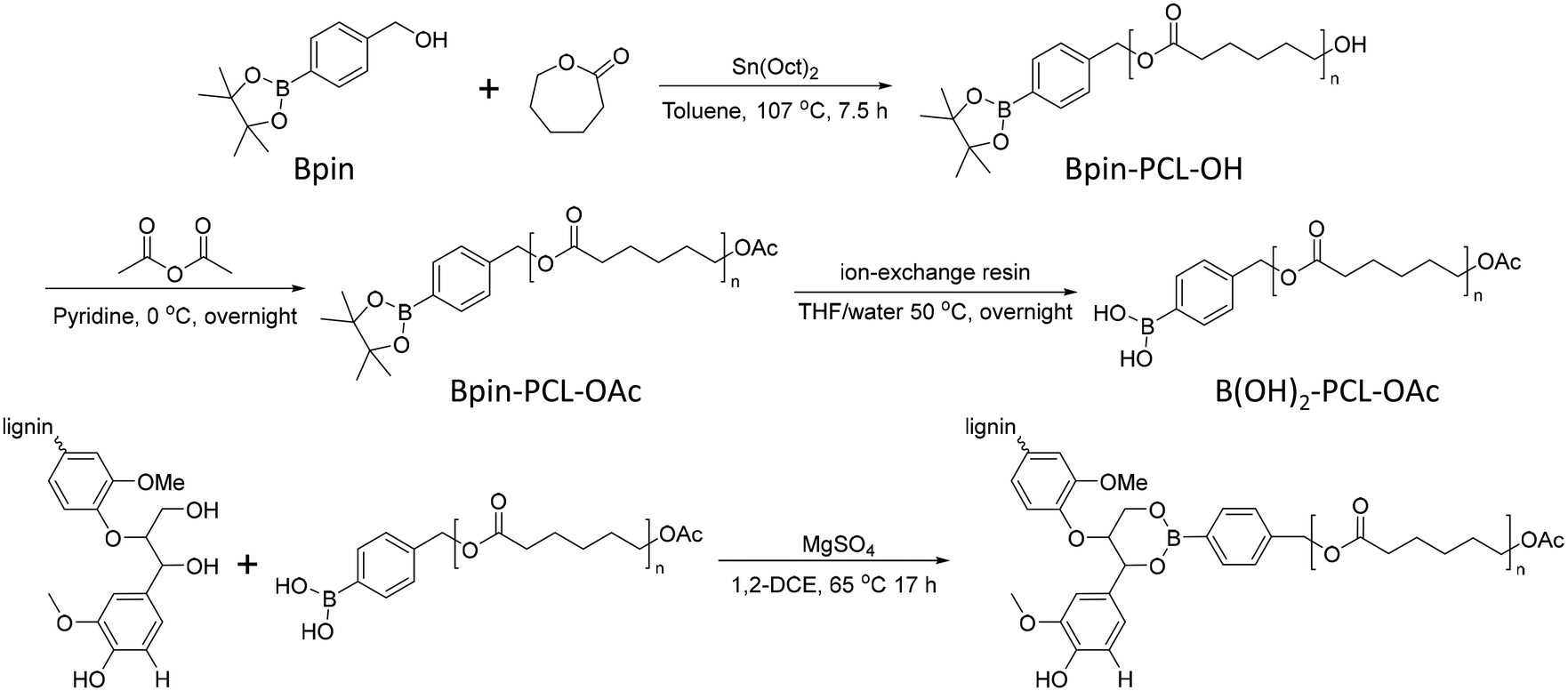 | ||
| Scheme 11 Synthesis of separately prepared PCL and graft copolymerization of PCL onto lignin. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
Another graft-onto method for lignin-graft-PCL, using click chemistry, was studied by Han et al.162 In this work, lignin molecules are linked by PCL to form lignin-graft-PCL (Scheme 12). Lignin served as the initiator for the graft-from polymerization of PCL, followed by the synthesis of lignin-graft-PCL. Then, the hydroxyl terminus of PCL was functionalized as an alkyne group. Simultaneously, the hydroxyl group of the other natural lignin was reacted with bromobutyryl chloride by esterification and the bromo terminus was modified to become an azide terminus. The lignin-azide and lignin-graft-PCL-alkyne were linked by click chemistry (Scheme 12).
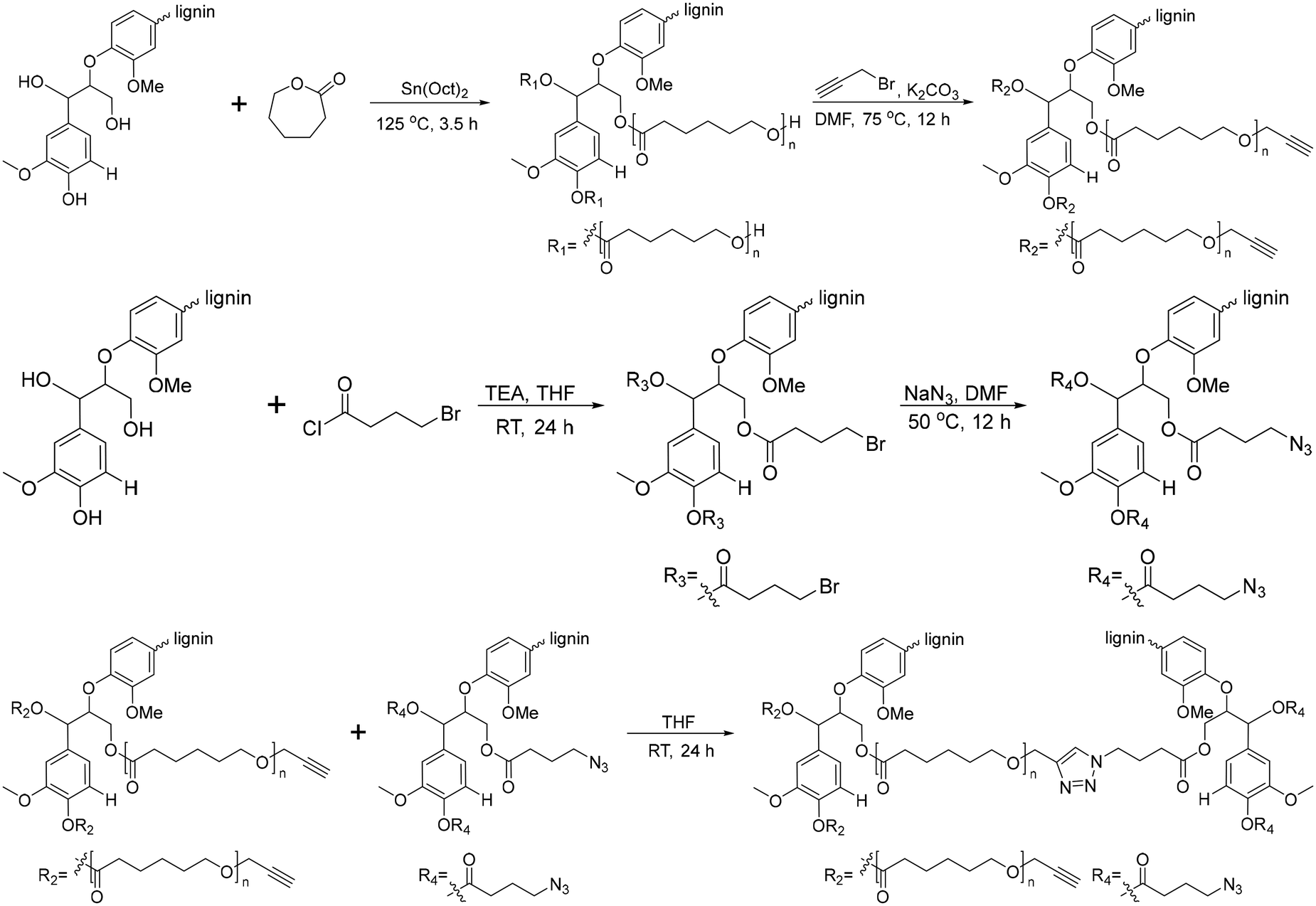 | ||
| Scheme 12 Synthesis of lignin-graft-PCL by the graft-from method and lignin-graft-PCL-graft-lignin by the graft-onto method. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
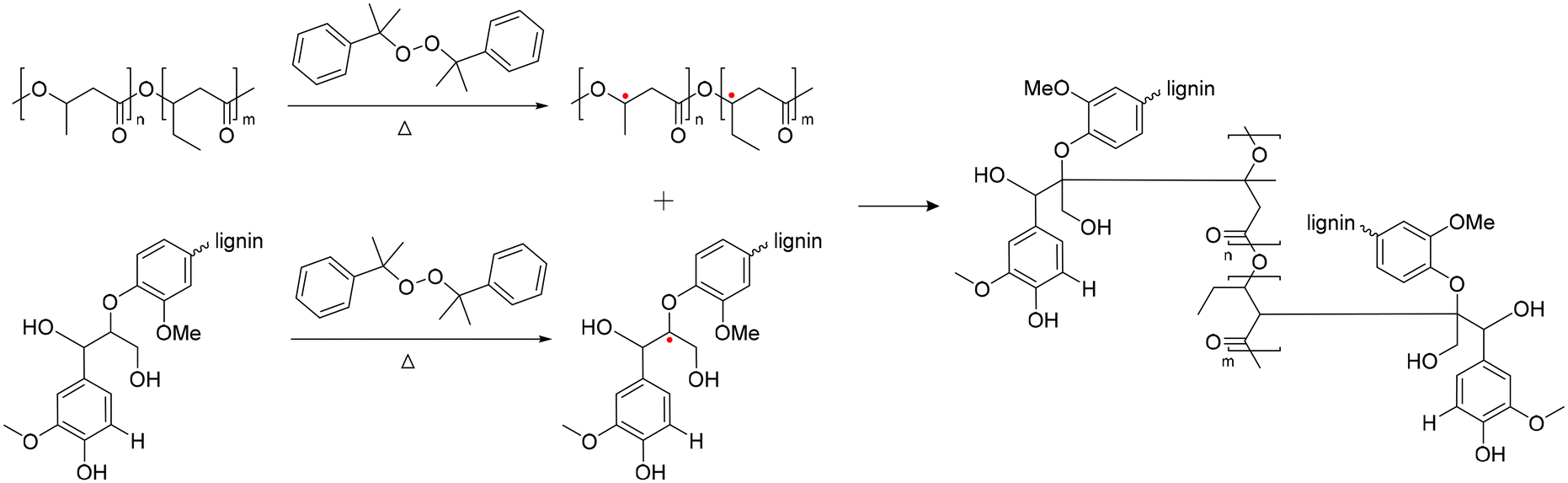
In 2019, Liu et al. reported crosslinked lignin-graft-PCL, which was synthesized by a graft-onto method through a Ru-catalyzed photoredox thiol–ene reaction.56 Lignin was modified to have an alkene terminus by esterification and PCL was prepared with pentaerythritol, a tetraol, as an initiator. The four hydroxyl terminal groups of four-armed PCL were modified to possess thiol terminal groups. The four-armed PCL-SH was reacted with lignin–alkene to make PCL-S-C-C-lignin via a photoredox thiol–ene reaction in the presence of Ru(bpy)3Cl2 catalyst and p-toluidine under blue light (Scheme 13).
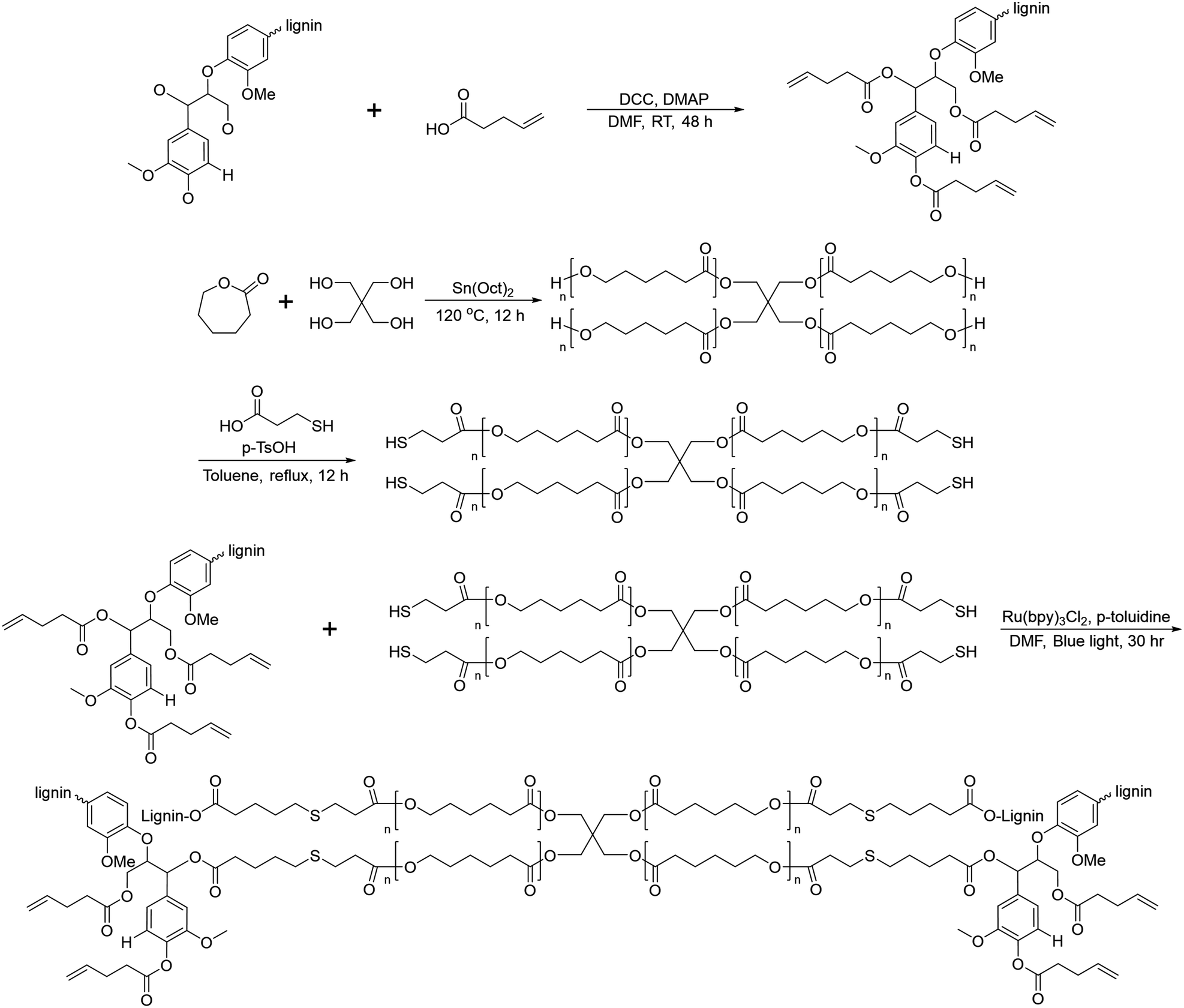 | ||
| Scheme 13 Synthesis of lignin-graft-PCL by the graft-onto method using the thiol–ene reaction. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
Common applications of lignin-graft-PCL include as an additive in the PCL matrix,119 as a replacement for PCL,126 as an antioxidant for biomedical applications,118 as a nucleating agent for PCL,115 as a shape memory material,56 for thermal energy storage,127 separation,128 to coat a metal surface,124 and as a polyol for polyurethane synthesis.129
Park et al.119 and Tian et al.126 introduced lignin-graft-PCL as a component for blending with PCL. Separately synthesized lignin-graft-PCL was blended with the PCL matrix to enhance its mechanical properties. Compared to natural lignin, lignin-graft-PCL showed much better dispersity in PCL (Fig. 14). Also, lignin graft-PCL showed enhanced mechanical properties compared to PLA.
 | ||
| Fig. 14 Photographic images of melt mixing of PCL at 150 °C with (a) lignin and (b) lignin-graft-PCL. Reproduced from ref. 119 with permission from Springer Nature, copyright 2019. | ||
The antioxidant application of lignin-graft-PCL was reported by Wang et al.118 Nanofibers of a mixture of lignin-graft-PCL and PCL were prepared by an electrospinning process (Fig. 15a and b). PCL10LP2 is a composite fiber with 10% lignin-graft-PCL (mass feed ratio of lignin:CL was 2:8) and PCL10LP4 is a composite fiber with 10% lignin-graft-PCL (mass feed ratio of lignin:CL was 4:6) in PCL. The nanofibers of lignin-graft-PCL/PCL mixture (PCL10LP4 and PCL10LP2) showed 95.7% and 43.2% free radical inhibition, respectively, whereas PCL showed only 11.5% (Fig. 15c and d). The addition of lignin copolymers to the composition improved the antioxidant activities of the nanofibers, with a higher amount of lignin copolymers leading to increased free radical inhibition. In addition, the nanofibers showed excellent biocompatibility. Neurofilaments, mainly found in cells of neuronal origin, showed almost double the growth rate on the lignin-graft-PCL/PCL nanofibers than on PCL fibers (Fig. 15e–g).
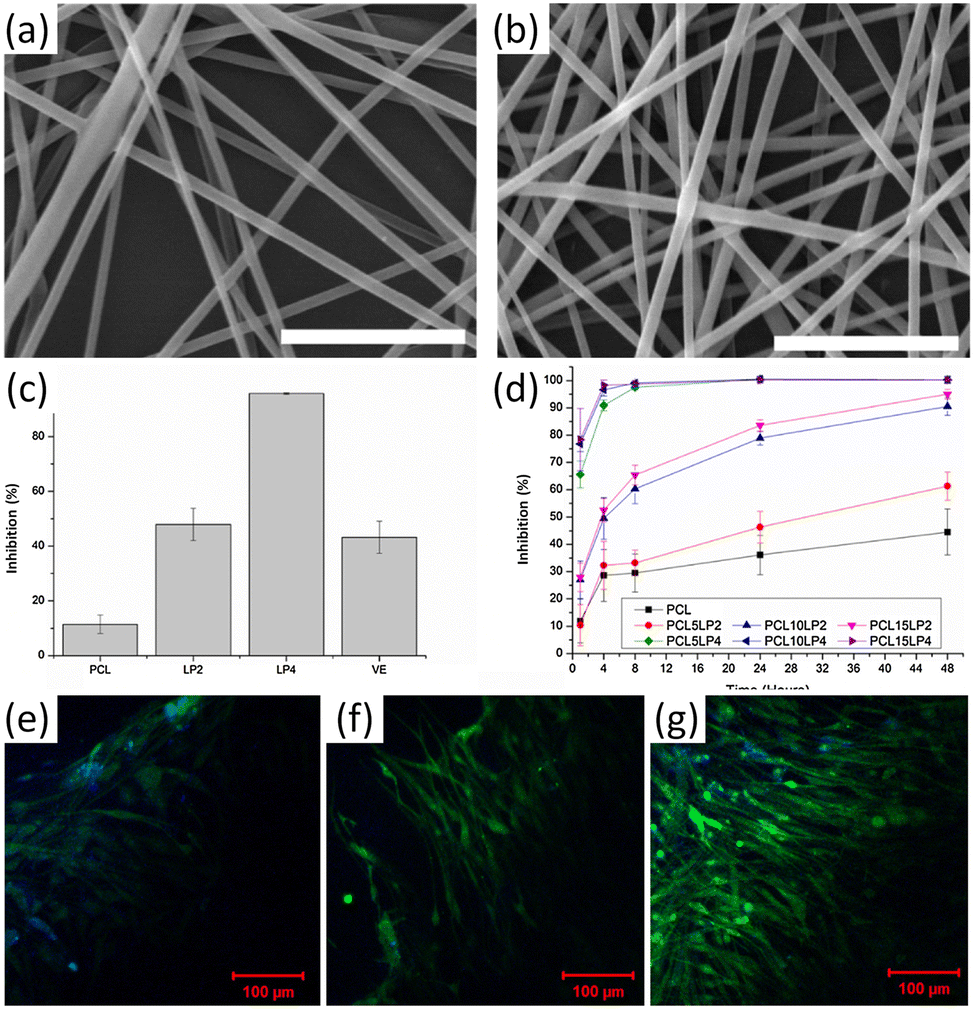 | ||
| Fig. 15 (a and b) SEM images of different lignin-g-PCL nanofibrous scaffolds: (a) PCL10LP2, (b) PCL10LP4; (c) BMSC proliferation on different nanofibers; (d) Schwann cell proliferation on different nanofibers after H2O2 treatment; (e–g) MBP expression of Schwann cells on different nanofibers: (e) PCL, (f) PCL10LP2, (g) PCL10LP4. Green color: MBP; blue color: the nuclei. Reproduced from ref. 118 with permission from Elsevier, copyright 2018. | ||
In 2015, Pérez-Camargo et al. reported the effect of lignin on the morphology, nucleation and crystallization kinetics of PCL using lignin-graft-PCL prepared by a graft-from method.115 Lignin displayed a nucleating reaction in the PCL matrix to yield an intersecting lamellar structure. Especially at low contents (2–5 wt%) of lignin, a high efficiency value (close to 100%) of nucleation was observed. This led to an increase in crystallization and Tm as well as the isothermal crystallization rate of lignin-graft-PCL.
However, when the lignin content exceeds 18 wt%, antinucleation effects become apparent. The antinucleation effects refer to the inhibition of lignin on the crystallization process of PCL. This causes a decrease in both crystallization and Tm, reduces the degree of crystallinity, hinders annealing during thermal fractionation and significantly retards isothermal crystallization. The results can be explained by competition between the nucleating effects of lignin and intermolecular interactions caused by hydrogen bonding between PCL and lignin building blocks (Fig. 16).
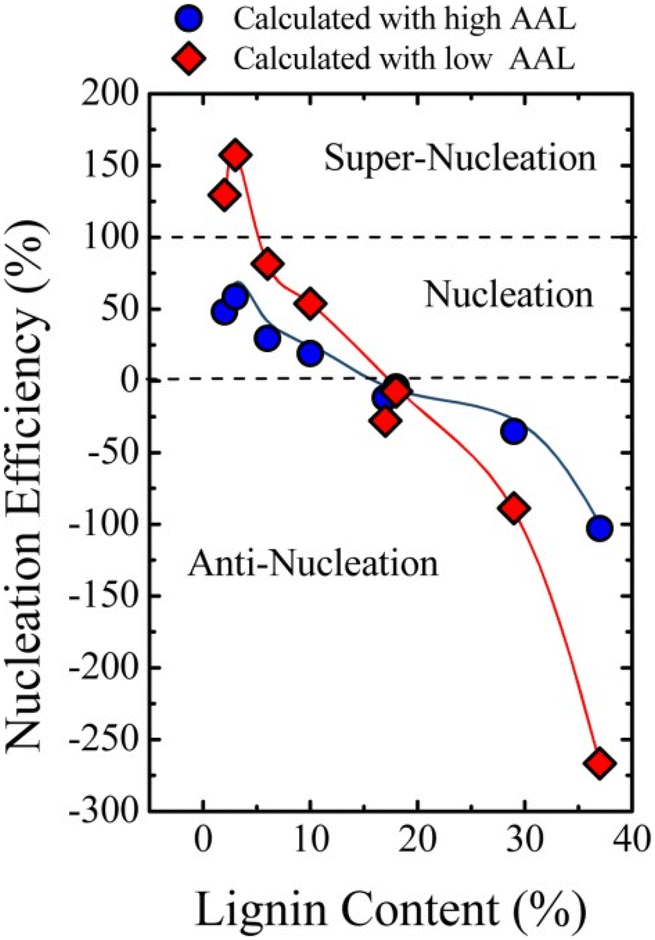 | ||
| Fig. 16 Nucleation efficiency of lignin in PCL-g-lignin samples. The efficiency was calculated using reference crystallization parameters from both low average arm length (AAL) PCL and high AAL PCL. Reproduced from ref. 115 with permission from John Wiley and Sons, copyright 2015. | ||
Lignin-graft-PCL as a shape memory material was reported by Liu et al.56 The shape memory mechanism of crosslinked polymers involves the interaction of net points and switching segments.130–133
Net points, formed through chemical and physical crosslinking, are essential for facilitating permanent shape recovery. On the other hand, switching segments exhibit distinct behaviors depending on the temperature.
The crosslinked lignin-graft-PCL was synthesized from lignin with 4-armed PCL with different weight % of lignin. Increased lignin amounts lead to higher gel contents and lower Tm and Tg. The prepared crosslinked lignin-graft-PCL showed an efficient and prompt thermal responsive shape memory function. Above Ttrans (80 °C), the polymer exhibits enough chain flexibility for it to be deformed into a temporary shape under the application of an external force. Once the material is cooled below Tcool (10 °C), the temporary shape becomes solidified and is maintained at room temperature. When the material is reheated above Ttrans, in the absence of an external force, it recovers its original shape, returning to its initial form (Fig. 17).
 | ||
| Fig. 17 Pictures of the shape memory behavior demonstrated by crosslinked lignin–PCL. The permanent shape (left) takes on the temporary shape (middle, shape change at 80 °C and then fixation at 10 °C) and is recovered again (right, shape recovery at 80 °C). All photos were taken at room temperature. Reproduced from ref. 56 with permission from John Wiley and Sons, copyright 2019. | ||
In 2022, Lee et al. reported the thermal energy storage function of lignin-graft-PCL.127 Lee et al. prepared four different physically mixed samples with varying weight ratios, and prepared one lignin-graft-PCL (26 wt% lignin). The samples were prepared in a rectangular block on filter paper and exposed to a temperature beyond their Tm overnight. Then, the samples were removed from the filter paper, and the difference in weight from the initial state was measured. Fig. 18 shows the initial sample, the sample after heating, and leakage stains from melted block samples. The physically mixed samples (L4P6, L3P7, L2P8 and L1P9) melted and leaked as a gel upon exposure to high temperature, while lignin-graft-PCL retained its shape even at temperatures higher than its Tm. Also, leakage stains from melted block samples were observed after removing the samples from the filter paper. The leakage from lignin-graft-PCL on filter paper had the lowest weight, and a larger amount of lignin showed a lower leakage weight in the physically mixed samples. This is because PCL is grafted to the lignin backbone, preventing leakage of the PCL as separate polymer components, as observed in the physically mixed samples.
 | ||
| Fig. 18 Photographs taken during the leakage test at the initial stage, after high-temperature exposure and the leakage stain left on filter paper. Reproduced from ref. 127 with permission from Elsevier, copyright 2022. | ||
The thermal response was investigated using IR imaging, as shown in Fig. 19b. Lignin-graft-PCL was placed on a heating module and heated to 80 °C, followed by waiting for 200 s for maximum thermal energy storage in the sample. Subsequently, the sample was cooled down to room temperature. The test results indicated a slow cooling process for lignin-graft-PCL, highlighting its potential as a heat storage material (Fig. 19b).
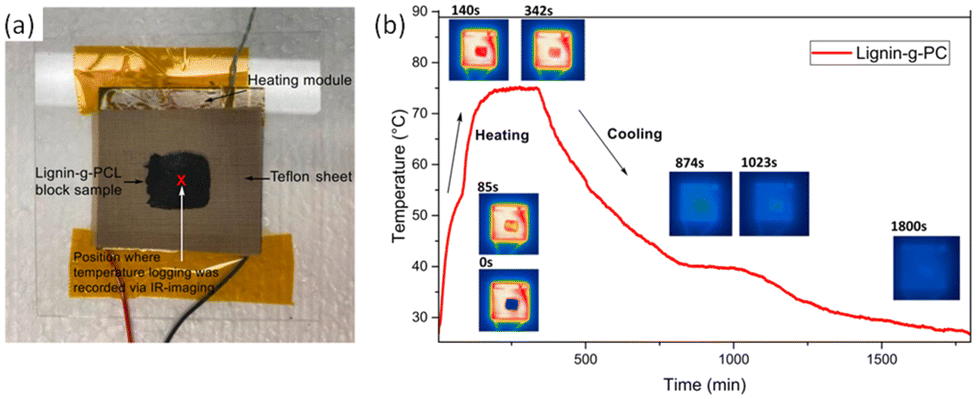 | ||
| Fig. 19 (a) IR-imaging set-up and position of temperature logging, and (b) IR-imaging of lignin-g-PCL and its temperature profile over time for a block of lignin-g-PCL sample being heated beyond its melting temperature (∼80 °C) and subsequently cooled. Reproduced from ref. 127 with permission from Elsevier, copyright 2022. | ||
In 2022, Xie et al. reported the use of lignin-graft-PCL in a separation application.128 In this work, lignin was copolymerized with caprolactone by solvent-free ring opening polymerization. Subsequently, filter paper was immersed in the solution of the synthesized lignin-graft-PCL for 12 h and then thoroughly dried. After that, SiO2 (silicone oil) was dispersed on the sample as a second impregnation step. The organic solvent (chloroform, bottom phase, orange color in Fig. 20) and water (top phase and purple color in Fig. 20) mixture was added to the funnel and then passed through the prepared filter paper for separation. The separation efficiency (%) was calculated as the weight percentage of stopped water in the funnel over the originally added water in the mixture. The CFP-C (filter paper coated with lignin-graft-PCL) effectively filters low surface energy organic solvents into the vial, while retaining high surface energy water in the funnel. Lignin-graft-PCL's separation efficiency was 99.0%, whereas CFP-P (filter paper coated with homoPCL) showed only 12.8% due to the low hydrophobicity of the PCL coating. In the case of FP (uncoated filter paper), it filtered most of the water because of its hydrophilicity (Fig. 20).
 | ||
| Fig. 20 Images of the original water (purple top layer)/chloroform (orange bottom layer) mixture and filtrates of FP, CFP-P, and CFP-C obtained. Reproduced from ref. 128 with permission from American Chemical Society, copyright 2022. | ||
Lignin graft copolymer as a coating agent on metal surfaces was reported in 2020 by Najarro et al.124 The prepared lignin–PCL copolymer, which was synthesized by the graft-from method, was coated on a stainless steel surface. The lignin–PCL copolymer and cross-linker mixture served as the coating solution on the polished and rinsed steel. The author tested the adhesion properties and coating stability on the stainless-steel surface. In a water bath, the coating with neat lignin showed clear differences from the coating with lignin–PCL copolymer. The neat lignin coating disintegrated in less than 10 min, whereas the coating with copolymers remained stable even after 30 days. This is attributed to the ester polymer arm from the copolymer enabling film formation, whereas neat lignin only exhibits rigidity without forming a film.
The crosslinker further enhanced the adhesion properties, which were measured by pull-off and cross-cut tape tests, by forming networks. The copolymer without crosslinker needs 0.6 MPa for pull-off adhesion tests and it showed only 1–2 on the scale for the cross-cut tape test. In this study, the results were evaluated using a 0–5 scale, where 5 indicates perfect adhesion. In contrast, the copolymer with crosslinker showed an adhesion force of 6.2 MPa for pull off and 4–5 on the scale for the cross-cut tape test (Fig. 21).
 | ||
| Fig. 21 (a) The stability test in a water bath at room temperature. (b) Pictures of adhesion tests for the lignin–PCL and lignin–PCL with crosslinker. Reproduced from ref. 124 with permission from American Chemical Society, copyright 2020. | ||
In 2017, Zhang et al. reported lignin-graft-PCL based polyurethane.125 As shown in Scheme 14, PCL served as the soft segment along with hexamethylene diisocyanate (HDI). The lignin-graft-PCL containing polyurethane (LPU) was synthesized by the graft-onto method. In the first step of the synthesis procedure, the hydroxyl groups of lignin were activated by HDI, and then the PCL's hydroxyl groups reacted with isocyanate on activated lignin (top scheme in Scheme 14).
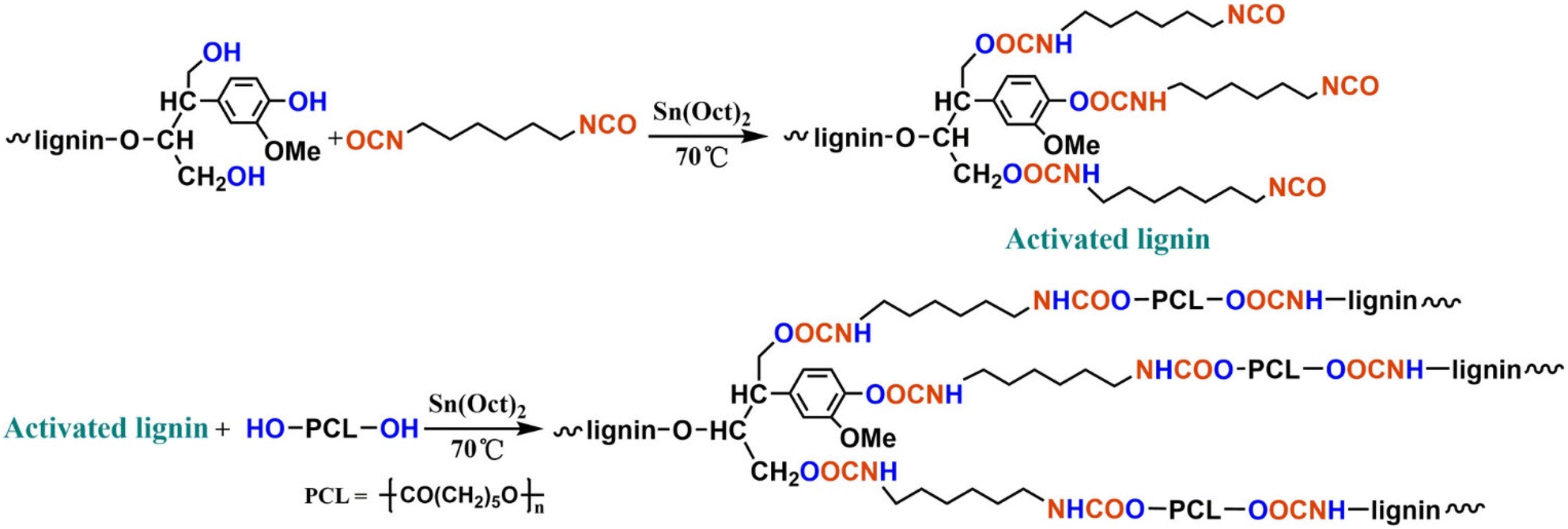 | ||
| Scheme 14 Synthesis route to the lignin–PCL based polyurethane. Reproduced from ref. 125 with permission from American Chemical Society, copyright 2017. | ||
Several samples were prepared with different ratios of –NCO/OH and varying weight percentages of lignin/PCL to compare their mechanical properties. The LPU with a higher ratio of –NCO/OH showed improved mechanical properties, such as higher tensile strength, higher elongation break, and right-angle tear strength, because more isocyanate groups led to more cross-linking with hydroxyl groups. However, ratios of –NHC/OH higher than 1.75 resulted in decreased mechanical properties. The negative effect was caused by restricted movement of polymer chain segments due to increased cross-linking and enhanced intermolecular forces. Also, the polymer length of PCL affected LPU's properties. LPU with a lower molecular weight of PCL showed poorer mechanical properties because of a higher crosslinking density (shorter chain length between crosslinking points). The effect of the overall lignin content in the polymer was also examined. The tensile strength increased as the lignin content rose to 37.30%. However, a subsequent decrease occurred when the lignin content further increased from 37.30% to 43.15% because of excess lignin agglomeration.
4.3 Lignin-graft-poly(ethylene brassylate) (lignin-graft-PEB)
In 2021, Kim and Chung reported lignin-graft-poly(ethylene brassylate) (lignin-graft-PEB).134 Lignin was chemically modified by sebacic acid to introduce a carboxylic acid functionality into the hydroxyl group of lignin. Another precursor of the copolymer, PEB, was prepared by ring-opening polymerization of ethylene brassylate in the presence of the 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) catalyst without another initiator. The condensation copolymerization of the modified lignin and PEB occurred by the reaction between the TBD terminus of PEB and the carboxylic acid of the modified lignin (Scheme 15). The thermal properties of lignin-graft-PEB exhibit high stability, with a Tm of 78 °C and a Tg of 12 °C. Mechanical property tests reveal that lignin-graft-PEB possesses comparable properties to commercial polyolefins such as LDPE, with a tensile strength of 7.34 MPa and a Young's modulus of 442.9 MPa, surpassing that of LDPE. The crucial factors influencing the mechanical properties include the lignin/PEB ratio and the molecular weight of PEB, as identified through comprehensive property tests. Especially, the presence of covalent bond linkages between lignin and PEB is essential for achieving enhanced mechanical properties, emphasizing the significance of their chemical integration.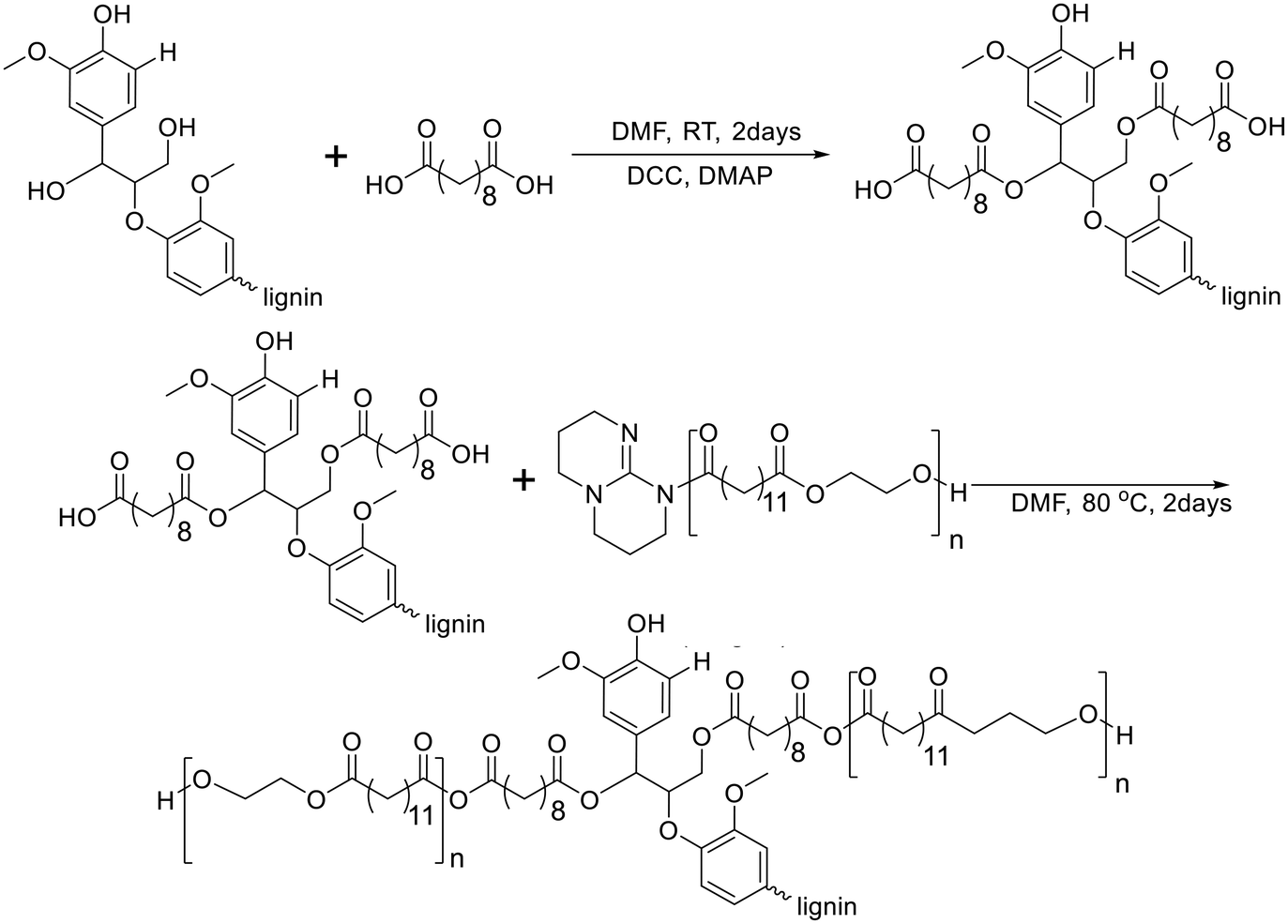 | ||
| Scheme 15 Synthesis of lignin-graft-poly(ethylene brassylate). Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
4.4 Lignin-graft-poly(hydroxybutyrate) (lignin-graft-PHB)
Poly(3-hydroxybutyrate) (P3HB) stands out as the first isolated and characterized polymer within the polyhydroxyalkanoate (PHA) family. P3HB possesses a highly crystalline structure due to its linear chain configuration.135 Three primary methods for producing PHB are available. The first involves the ring opening polymerization of β-butyrolactone.136–138 Another method utilizes natural or transgenic plants, capable of synthesizing PHAs in their cells due to the abundance of acetyl-CoA, a key substrate in PHA biosynthesis. Examples include Linum usitatissimum L., commonly known as flax.139 The third method entails bacterial fermentation, where optimal conditions can yield PHA materials constituting over 90% of the cell's dry weight.140 This bacterial fermentation approach is the one most widely employed for PHB synthesis.Lignin-graft-PHB was reported by Kai et al. in 2019.141 Lignin-graft-PHB was synthesized by a graft-from method with lignin and butyrolactone without solvent. Sn(Oct)2 served as a catalyst; it is commonly employed in ring opening polymerizations (Scheme 16). The prepared lignin-graft-PHB was used to produce nanofibers by electrospinning with PHB. The prepared lignin-graft-PHB/PHB nanofibers were tested for their mechanical properties, antioxidant ability, and biocompatibility. Only 2% of such a copolymer is required to increase the tensile strength by ∼4 times, stiffness by 1.5 times, and elongation by ∼10 times compared to pure PHB. Increasing the lignin content to 5% resulted in improved mechanical properties; however, beyond 5%, enhancements decreased due to the brittle nature of lignin (Fig. 22a). Furthermore, lignin-graft-PHB/PHB nanofibers display tunable antioxidant activity and good biocompatibility both in vitro and in vivo. Especially, higher lignin contents showed a higher antioxidant ability (Fig. 22b–h). These properties indicate that lignin-graft-PHB can have broader applications in the biomedical field.
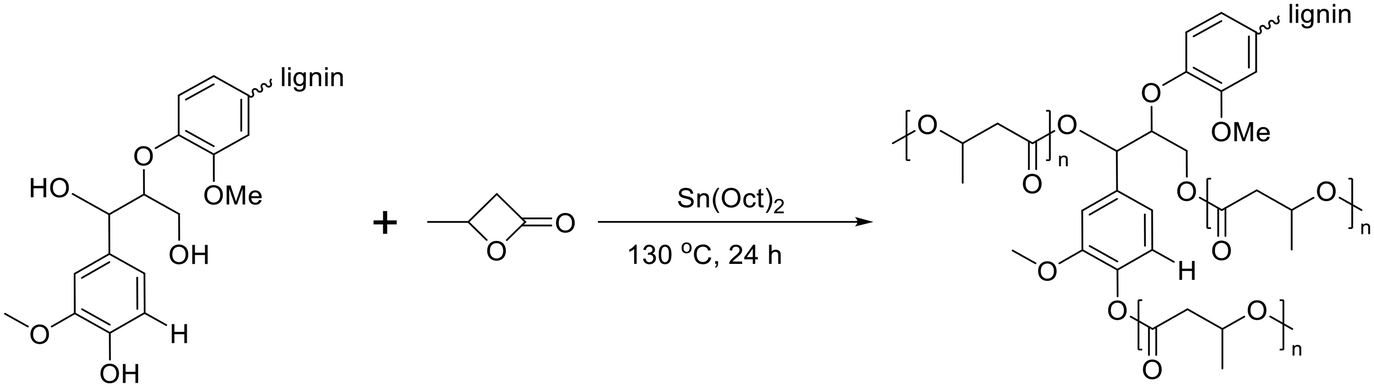 | ||
| Scheme 16 Synthesis of lignin-graft-poly(3-hydroxybutyrate). Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
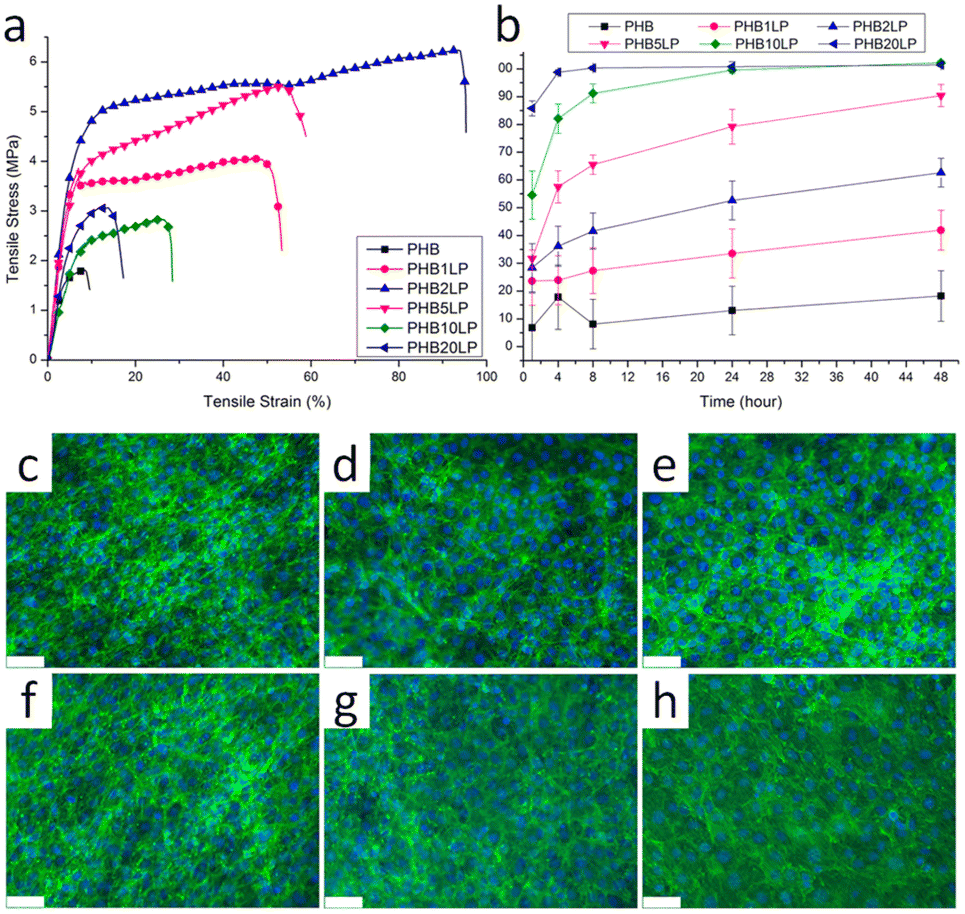 | ||
| Fig. 22 (a) Typical tensile stress–strain curves of PHB and PHB/lignin–PHB nanofibers. (b) Antioxidant activity of PHB/lignin–PHB nanofibers (DPPH assay). (c–h) NIH-3T3 fibroblasts stained with DAPI and phalloidin after 8 days of culture on various nanofibers: (c) PHB, (d) PHB1LP, (e) PHB2LP, (f) PHB5LP, (g) PHB10LP, and (h) PHB20LP nanofibers (scale bar = 75 μm). The tight mesh of cells after 8 days of culture indicates very high confluency and good biocompatibility. Reproduced from ref. 141 with permission from American Chemical Society, copyright 2019. | ||
4.5 Lignin-graft-poly(caprolactone-co-lactic acid) (lignin-graft-PCLLA)
Lignin can undergo graft copolymerization not only with a single polyester but also with copolymers such as poly(caprolactone-co-lactic acid) (PCLLA).142–144Sun et al. reported lignin-graft-PCLLA synthesized by a graft-from method in 2015 (Scheme 17a).142 PCLLA was polymerized by ring opening polymerization, initiated by the hydroxyl group on lignin in toluene solvent. Then, separately synthesized PDLA was polymerized onto the prepared lignin-graft-PCLLA to use it as a compatibilizer. Lignin-graft-PCLLA-PDLA provides a strong filler–matrix interfacial attraction, even improving the dispersity of polymers in the matrix. In addition, lignin-graft-PCLLA-PDLA has a role as a reinforcement agent in PLLA blend, resulting in improved mechanical properties compared to pure PLLA.
 | ||
| Scheme 17 Synthesis of lignin-graft-PCLLA through the graft-from method (a) with toluene as a solvent and (b) without solvent. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
In 2017, Kai et al. introduced the solvent-free synthesis of lignin-graft-PCLLA (Scheme 17b).143 The synthesized lignin-graft-PCLLA acts as an additive for both PCL and PLLA, to prepare uniform nanofibers via electrospinning (Fig. 23). The mechanical properties and antioxidant effects of these fibers were investigated. The blend of lignin-graft-PCLLA in PCL matrix reinforced the PCL nanofibers. In addition, the multipolymer blend of lignin-graft-PCLLA/PCL/PLA blend nanofibers exhibited good antioxidant activity and biocompatibility. This outcome highlights the potential of lignin-graft-PCLLA/PLA and lignin-graft-PCLLA/PCL blends as materials for biomedical applications.
 | ||
| Fig. 23 SEM images of electrospun (a) PCL, (b) PCL/LP1, (c) PLLA, and (d) PLLA/LP1 nanofibers. Scale bars = 1 μm. Reproduced from ref. 143 with permission from American Chemical Society, copyright 2017. | ||
4.6 Lignin-graft-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (lignin-graft-PHBV)
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) is commonly named poly(3-hydroxybutyric acid-co-3-hydroxyvaleric acid) (PHBV). PHBV is copolymerized with PHB and 3-hydroxyvalerate (HV) units in a bacterial fermentation process, aiming to increase flexibility and to decrease Tm for easier processing.145 The Tm of PHBV is lower than that of PHB and diminishes with an increased fraction of HV in the polymer, thereby widening the processing range and results in an improvement in the ductility and flexibility of the polymer.146 PHBV also exhibits resistance to hydrolysis and better resilience to ultraviolet radiation.147–149 Consequently, PHBV has attracted considerable attention from various research groups due to its high flexibility/strength and reduced chain packing/toughness.150–153 Moreover, the increased proportion of HV within the PHB matrix make the overall polymer more amorphous, which is advantageous for drug release. Drugs typically exhibit easier diffusion through the amorphous region of a polymer.154In 2016, Luo et al. reported lignin-graft-PHBV, as shown in Scheme 18.155 Lignin-graft-PHBV was prepared by a graft-onto method to improve the interfacial adhesion of lignin and PHBV. In the first step of polymerization, a radical initiator, dicumyl peroxide (DCP), was added to lignin and PHBV to form radicals during melt extrusion. The generated radicals lead to covalent bond grafting (Scheme 18). Four different types of lignin-graft-PHBV were prepared with different amounts of radical initiator. The mechanical properties of those prepared lignin-graft-PHBV samples were studied. Lignin-graft-PHBV showed a higher modulus but a lower strain at break compared to homo PHBV. This can be attributed to the presence of lignin, a rigid three-dimensional macromolecule, which restricts chain movement during deformation. In particular, lignin-graft-PHBV, which was linked with 2% radical initiator, exhibited significantly enhanced mechanical properties, a higher modulus, and a higher tensile strength and strain at break, compared to the blend of lignin and PHBV. According to the spherulite morphology study, pure PHBV had an average radius of approximately 700 μm of spherulites. Crosslinked PHBV showed similar spherulites, but the size was substantially decreased to 220 μm. On the other hand, lignin-graft-PHBV showed a smaller size and significantly higher number of spherulites compared to both PHBV and crosslinked PHBV. The grafting point between PHBV and lignin likely served as a nucleation site. The increased number of grafting points hindered diffusion, resulting in a higher concentration of radical initiator regionally, leading to a larger number of smaller spherulites (Fig. 24).
 | ||
| Scheme 18 Synthesis of lignin-graft-PHBV. Terminal β-O-4 softwood lignin unit is used for simplicity. | ||
 | ||
| Fig. 24 Micrographs of (a) PHBV, (b) crosslinked PHBV, (c) PHBV–lignin blend, (d) PHBV-1-lignin with 1% DCP, and (e) PHBV-2-lignin with 1% DCP crystallized at 90 °C. Scale bar: 200 μm. Reproduced from ref. 155 with permission from American Chemical Society, copyright 2016. | ||
4.7 Lignin-graft-poly(caprolactone)-poly(hydroxybutyrate) (lignin-graft-PCL-PHB)
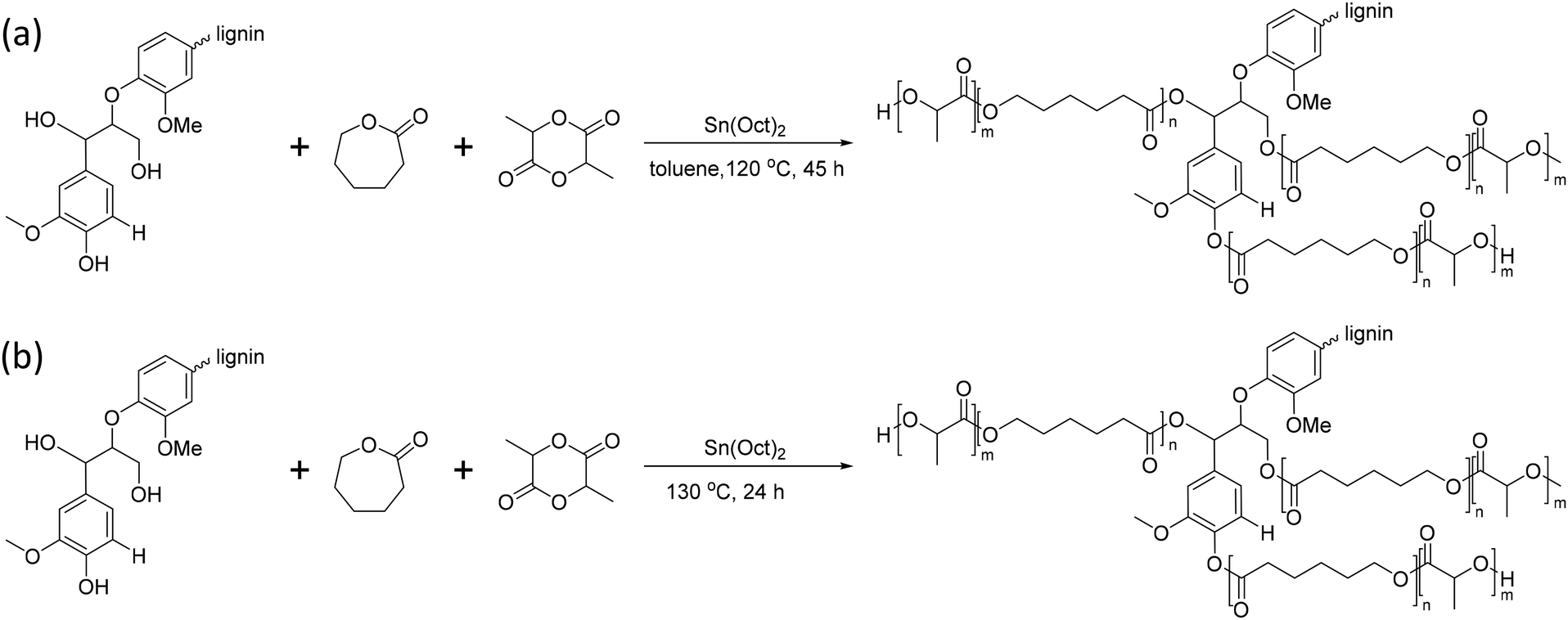
In 2018, Kai et al. reported lignin-graft-PCL-PHBs and investigated their role as a filler of PHB nanofibers.156 The lignin copolymers are synthesized by solvent-free ring-opening polymerization of butyrolactone and caprolactone in the presence of a tin catalyst. They prepared lignin-graft-PHB (LPH), lignin-graft-random copolymer of PHB and PCL (LPHC), lignin-graft-PHV-PCL (LPH + C), and lignin-graft-PCL-PHB (LPC + H) (Scheme 19). Then, these lignin copolymers are mixed with PHB at a mass ratio of 5% and produce nanofibers through electrospinning. The PHB nanofibers showed a tensile strength of 1.81 MPa, a modulus of 80.9 MPa, and an elongation at break of 15%. The addition of LPH decreased the tensile strength and Young's modulus, while the elongation at break was increased. The addition of LPHC also resulted in decreased strength and modulus; additionally, it showed a decreased elongation at break. The nanofibers that consisted of PHB and LPH + C showed an enhanced tensile strength and modulus, while they showed the smallest value of elongation at break of 8%. The addition of LPC + H exhibited the best enhancement of the mechanical properties, the highest tensile strength, Young's modulus, and elongation at break. These results indicated that lignin acted as a rigid core for nanofibers, and PCL segments acted as a rubber phase between the PHB matrix and lignin core. Also, the PHB blocks improve the interaction between the lignin copolymer and PHB matrix. Thus, the addition of LPC + H showed the best performance due to the PHP outer segments of LPC + H mostly enhancing the interaction with the PHP matrix (Fig. 25).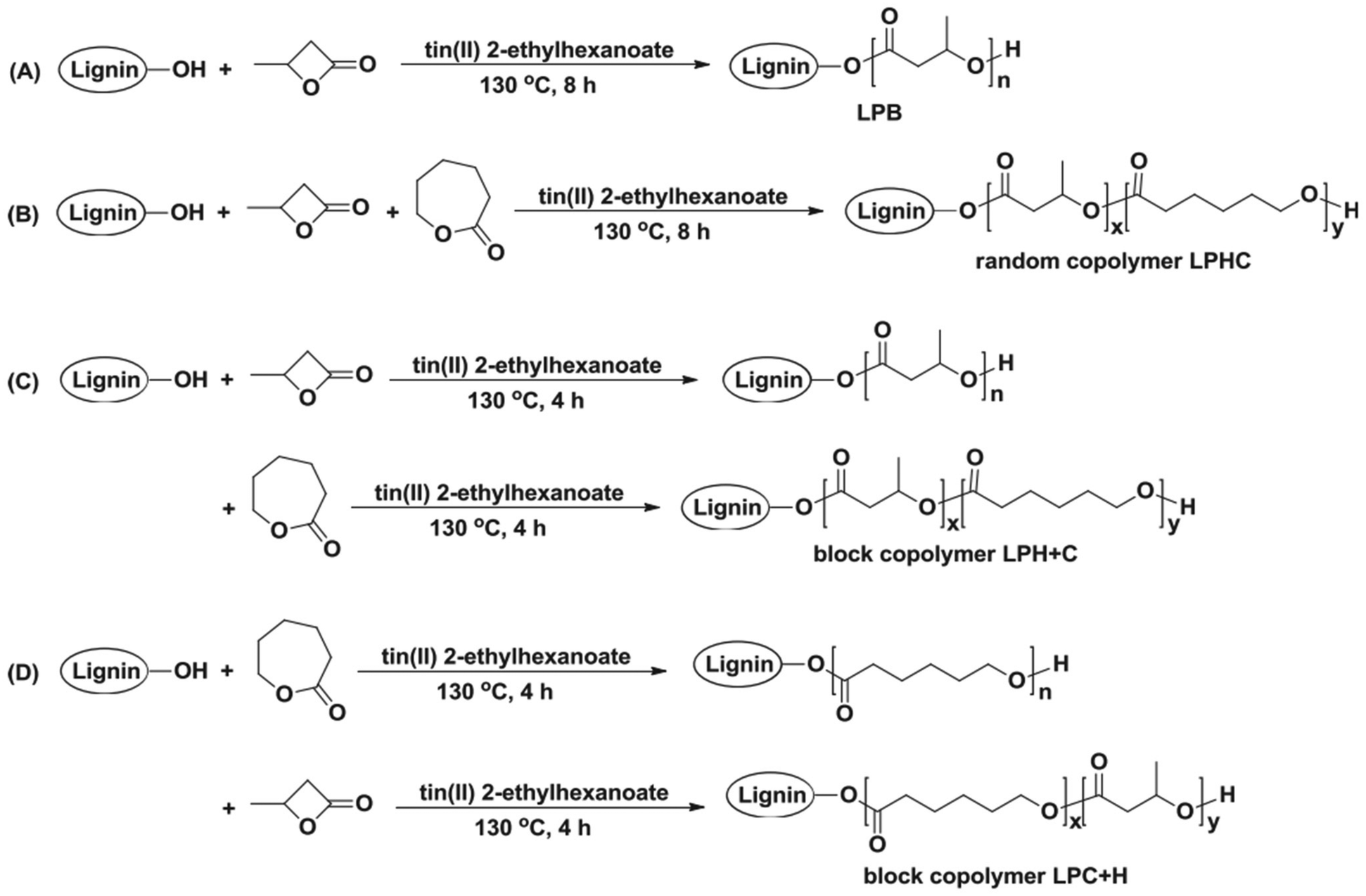 | ||
| Scheme 19 The synthetic routes to lignin PHB copolymers: (A) lignin-graft-PHB (LPB), (B) lignin-graft-random copolymer of PHB and PCL (LPHC), (C) lignin-graft-PHV-PCL (LPH + C) and (D) block copolymer lignin-graft-PCL-PHB (LPC + H). Reproduced from ref. 156 with permission from Elsevier, copyright 2018. | ||
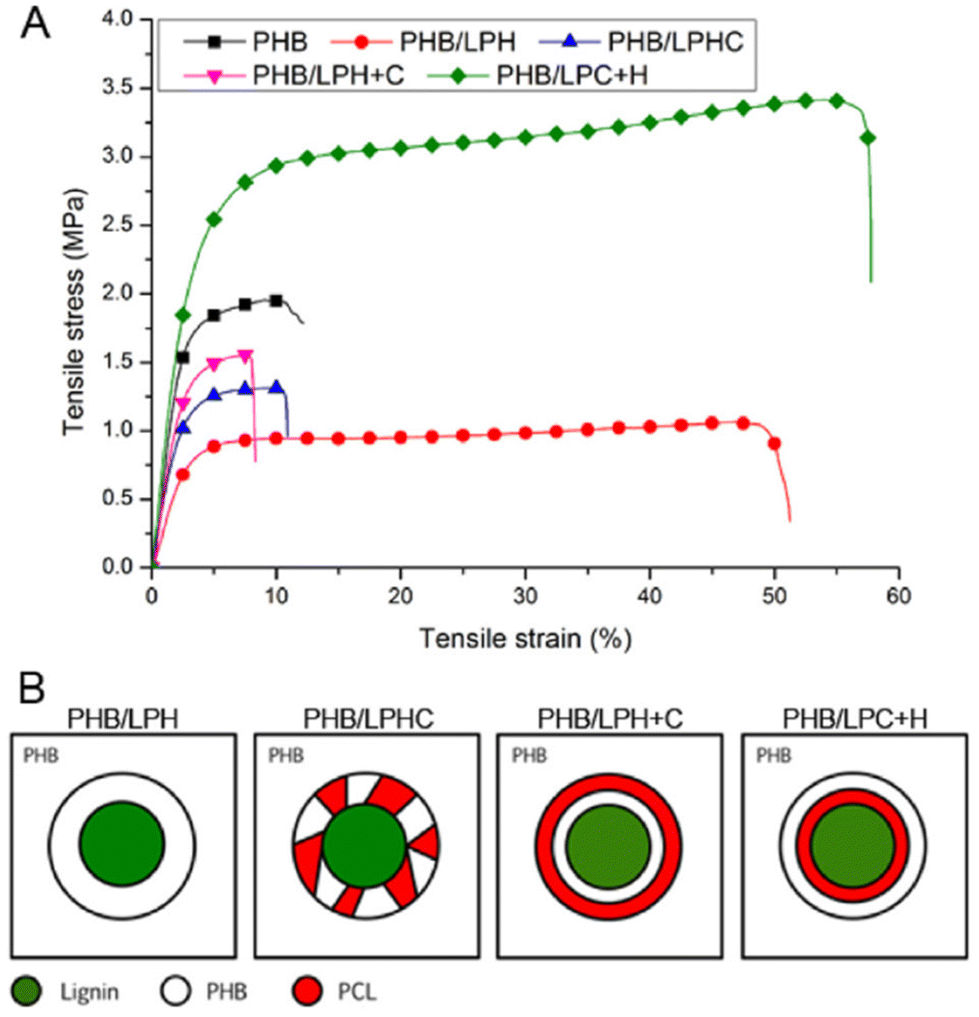 | ||
| Fig. 25 (A) Typical stress–strain curves of electrospun nanofibers obtained from a tensile test. (B) Proposed dispersion models to show how different lignin copolymers disperse in PHB matrix. Reproduced from ref. 156 with permission from Elsevier, copyright 2018. | ||
4.8 Lignin-graft-polyester/amide
In 2021, a graft copolymer with lignin and polyester/amide consisting of a short-chain diacid (succinic acid, adipic acid, and suberic acid), butanediol, and diamino butane was reported by Young et al. as shown in Scheme 20.157 The study aimed to develop fully biobased thermoplastic lignin copolymers using a two-step, solvent-free melt condensation process. The first step involved creating a low-molecular-weight prepolymer from butanediol (BD), diaminobutane (DAB), and one of three diacids: succinic acid (SA), adipic acid (AA), or suberic acid (SuA). These prepolymers were designed to terminate predominantly with carboxylic acid functionalities. In the second step, the prepolymers were reacted with softwood kraft lignin to form lignin–copolyester/amides. The study monitored the progression of polymerization using FTIR spectroscopy and characterized the resulting lignin copolymers’ mechanical, thermal, and structural properties. These copolymers demonstrated thermoplastic behavior and varied tensile properties based on their diacid chain length, lignin content, and DAB content, showing potential for creating sustainable, biobased thermoplastic materials.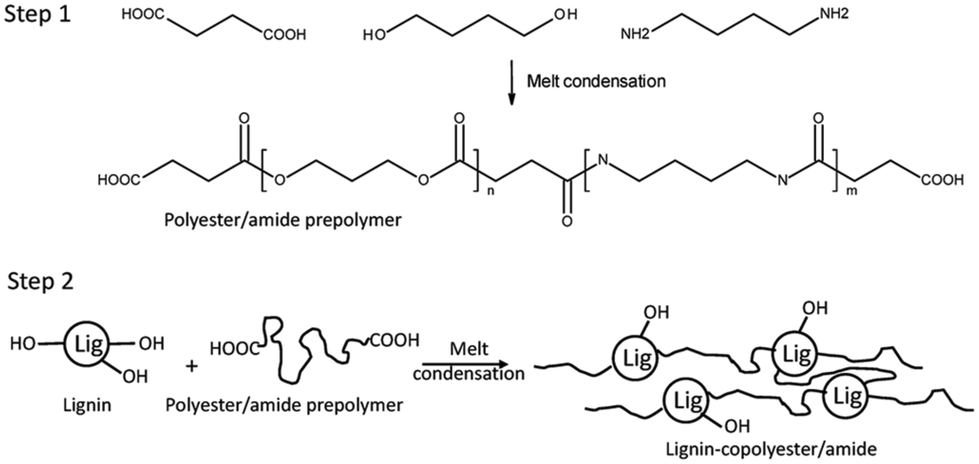 | ||
| Scheme 20 Schematic showing the 2-step process for producing the polybutylene–succinate/succinamide prepolymer and lignin–copolyester/amides (lignin–copolybutylene–succinate/succinamide). Reproduced from ref. 157 with permission from MDPI, copyright 2021. | ||
The synthesized lignin–copolyester/amides were found to have semi-crystalline structures with properties influenced by their composition. The succinic acid-based copolymers were stiff and brittle, while adipic acid-based ones were more flexible, and suberic acid-based copolymers displayed intermediate characteristics. Furthermore, lignin contents above 30% hindered the melt behavior, suggesting a limit for practical applications.
4.9 Summary
In this section, we discussed lignin-graft-polyesters. The copolymers are synthesized by either graft-from or graft-onto methods. Ring-opening polymerization is usually used for the graft-from method. Polycondensation, the thiol–ene reaction, azide–alkyne click chemistry, and radical reactions are used for the graft-onto method. Lignin copolymers have diverse applications across several fields due to their unique properties and the benefits they confer when combined with other polymers. The common applications of lignin-graft-polyesters include as additives to achieve higher mechanical properties, improved thermal stability, greater dispersity, and antioxidant applications. These benefits result from lignin's high mechanical properties (such as high Young's modulus, strain at break, and tensile strength), high glass transition temperature, and antioxidant properties.One prominent example is lignin-graft-polycaprolactone (lignin-graft-PCL). These materials are widely used as additives in PCL matrices to enhance the mechanical properties, providing improvements in flexibility, strength, and durability. They serve as a replacement for PCL, offering a cost-effective and environmentally friendly alternative. Additionally, lignin-graft-PCL copolymers act as antioxidants, making them suitable for biomedical applications where oxidative stress needs to be minimized. They are also used as nucleating agents for PCL, aiding in the crystallization process and improving the material's overall properties. These properties are affected by lignin's free volume due to its bulk polymer size. Moreover, lignin-graft-PCL copolymers have applications in shape memory materials, which can return to their original shape after deformation when exposed to an external stimulus like heat. They are also utilized in thermal energy storage systems due to their capacity to absorb and release heat efficiently. In separation processes, these copolymers are employed for their excellent barrier properties, making them suitable for filtration and purification applications. Additionally, they are used to coat metal surfaces to enhance corrosion resistance and provide a protective layer. In the field of polyurethane synthesis, lignin-graft-PCL copolymers serve as polyols, contributing to the creation of foams, adhesives, and elastomers with improved mechanical and thermal properties.
In the realm of biomaterials, lignin copolymers such as lignin-graft-polyhydroxybutyrate (lignin-graft-PHB) show significant promise. These copolymers can be processed into nanofibers through electrospinning, a technique that produces fibers with enhanced mechanical properties, antioxidant activity, and biocompatibility. When incorporated into materials, even in small amounts, lignin-graft-PHB copolymers can significantly improve tensile strength, stiffness, and elongation, making them ideal for medical devices, tissue engineering, and other biomedical applications. The incorporation of lignin-graft copolymers can increase tensile strength by up to four times, stiffness by 1.5 times, and elongation by ten times, demonstrating their potential to create more durable and versatile biomaterials. These applications highlight the versatility and potential of lignin copolymers for creating sustainable, high-performance materials for various industrial and biomedical uses.
5. Limitation of lignin-graft-polyesters
Lignin-graft-polyesters have emerged as promising alternatives in the quest for sustainable materials, leveraging lignin's abundance and eco-friendly attributes. Despite their potential, these copolymers encounter challenges that hinder their widespread adoption in industrial applications.One primary hurdle is the structural complexity of lignin itself. As a natural polymer with a complex molecular structure, achieving a uniform dispersion within polymer matrices remains elusive. This complexity often results in heterogeneous material properties, limiting consistency and reliability in mechanical performance.
Moreover, the reactivity of lignin during polymerization poses challenges. While lignin offers diverse chemical functionalities, ensuring reproducible reactions in a controlled manner proves difficult. This unpredictability can lead to undesirable side reactions, further complicating the manufacturing process and affecting material quality.
In terms of physical properties, some lignin-graft-polyesters exhibit lower mechanical characteristics compared to traditional polymers (e.g., PET, HDPE, and PP). Factors such as reduced strength, hardness, and elongation diminish their suitability for applications requiring robust and high-performance materials. Additionally, lignin's susceptibility to thermal degradation and its high viscosity during processing present obstacles to achieving efficient manufacturing and product consistency.
Aesthetic considerations also play a crucial role in the market acceptance of lignin-graft-polyesters. The inherent dark color of lignin often compromises the desired appearance of the final products, limiting their appeal in consumer-oriented industries where visual appeal is paramount. Generally, white or transparent polymers have much broader applications in industry.
From an economic perspective, the extraction and purification of lignin are resource-intensive processes, adding to production costs. This economic barrier, coupled with the competitive pricing of established polymer alternatives such as petroleum-based polyolefins, challenges the commercial viability of lignin-graft-polyesters.
Furthermore, despite being sourced from renewable biomass, environmental concerns persist. Although the actual amount is not large, the synthesis processes for lignin-graft-polyesters may involve chemicals that raise ecological questions. The unavoidable use of such chemicals is gradually being addressed by developing less environmentally harmful solvents and catalyst systems to perform the same types of chemical reactions that previously required ordinary organic solvents and chemicals.
In conclusion, while lignin-graft-polyester hold promise as sustainable materials, their adoption faces multifaceted challenges, ranging from structural complexities and reactivity issues to economic viability and environmental considerations. Overcoming these obstacles demands continued research efforts aimed at refining processing techniques, enhancing material properties, and ensuring environmental sustainability for broader applications in industrial sectors.
6. Conclusions
This review discusses the synthesis and applications of biodegradable lignin-graft-polyesters. Emphasizing graft copolymerization as an efficient method to seamlessly integrate lignin with other aliphatic polyesters, the study focuses on naturally biodegradable polyesters, with the majority of raw materials sourced from biomass. Beyond their intrinsic sustainability, these lignin-graft-polyesters benefit from lignin, providing mechanical strength, enhanced hydrophobicity and aromaticity, and as a non-food resource. The reviewed polyesters, including PLA, PGA, PCL, PEB, and PHA, and their diverse copolymers, showcase the versatility of lignin-based graft copolymer synthesis. The multitude of presented chemical reaction schemes offers a valuable guide to understanding recent trends in lignin and polyester copolymerization. Apart from synthesis, the review explores the properties and applications of these lignin-graft-polyesters, offering fundamental insights for detailed structure–property relationship studies in closely related biomass-based degradable polymers. The comprehensive examination of these materials not only adds to our understanding of their potential but also positions them as solutions to environmental challenges, with broad applicability across various industrial sectors. This review thus serves as a valuable resource, providing a holistic perspective on biodegradable lignin-graft-polyesters, from synthesis methodologies to their diverse applications and potential environmental contributions.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded at least in part with federal funds awarded to the State of Florida from the United States Department of Agriculture National Institute of Food and Agriculture (Award #2019-70007-30368).References
- J. A. Van Franeker and K. L. Law, Environ. Pollut., 2015, 203, 89–96 CrossRef CAS PubMed.
- I. Kyrikou and D. Briassoulis, J. Polym. Environ., 2007, 15, 125–150 CrossRef CAS.
- K. L. Law, N. Starr, T. R. Siegler, J. R. Jambeck, N. J. Mallos and G. H. Leonard, Sci. Adv., 2020, 6(44) DOI:10.1126/sciadv.abd0288.
- A.-C. C. Albertsson and M. Hakkarainen, Science, 2017, 358, 872–873 CrossRef CAS PubMed.
- EUPB, European Bioplastics. What are Bioplastics?, https://docs.european-bioplastics.org/2016/publications/fs/EUBP_fs_what_are_bioplastics.pdf.
- C. Matthews, F. Moran and A. K. Jaiswal, J. Cleaner Prod., 2021, 283, 125263 CrossRef.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- M. H. Rahman and P. R. Bhoi, J. Cleaner Prod., 2021, 294, 126218 CrossRef CAS.
- T. J. Gutiérrez, Composites Materials for Food Packaging, Wiley, 2018, pp. 269–296 Search PubMed.
- J. H. Song, R. J. Murphy, R. Narayan and G. B. H. Davies, Philos. Trans. R. Soc., B, 2009, 364, 2127–2139 CrossRef CAS PubMed.
- N. A. A. B. Taib, M. R. Rahman, D. Huda, K. K. Kuok, S. Hamdan, M. K. B. Bakri, M. R. M. B. Julaihi and A. Khan, Polym. Bull., 2023, 80, 1179–1213 CrossRef CAS.
- N. K. Kalita, N. A. Damare, D. Hazarika, P. Bhagabati, A. Kalamdhad and V. Katiyar, Environ. Challenges, 2021, 3, 100067 CrossRef CAS.
- P. T. Martone, J. M. Estevez, F. Lu, K. Ruel, M. W. Denny, C. Somerville and J. Ralph, Curr. Biol., 2009, 19, 169–175 CrossRef CAS PubMed.
- H.-R. Bjørsvik and F. Minisci, Org. Process Res. Dev., 1999, 3, 330–340 CrossRef.
- K. V. Sarkanen and C. H. Ludwig, Lignins: occurrence, formation, structure and reactions, Wiley-Interscience, New York, 1971 Search PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175 RSC.
- C. J. L. Constantino, A. Dhanabalan, A. A. da S. Curvelo and O. N. Oliveira Jr, Thin Solid Films, 1998, 327, 47–51 CrossRef.
- X. Lin, M. Zhou, S. Wang, H. Lou, D. Yang and X. Qiu, ACS Sustainable Chem. Eng., 2014, 2, 1902–1909 CrossRef CAS.
- X. Liu, H. Yin, Z. Zhang, B. Diao and J. Li, Colloids Surf., B, 2015, 125, 230–237 CrossRef CAS PubMed.
- J. Yu, J. F. Wang, C. P. Wang, Y. P. Liu, Y. Z. Xu, C. B. Tang and F. X. Chu, Macromol. Rapid Commun., 2015, 36, 398 CrossRef CAS PubMed.
- Y. S. Kim and J. F. Kadla, Biomacromolecules, 2010, 11, 981–988 CrossRef CAS PubMed.
- H. Liu and H. Chung, Macromolecules, 2016, 49, 7246–7256 CrossRef CAS.
- D. S. Argyropoulos, H. Sadeghifar, C. Cui and S. Sen, ACS Sustainable Chem. Eng., 2014, 2, 264–271 CrossRef CAS.
- A. L. Korich, A. B. Fleming, A. R. Walker, J. Wang, C. Tang and P. M. Iovine, Polymer, 2012, 53, 87–93 CrossRef CAS.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS.
- J. Ruwoldt, Surfaces, 2020, 3, 622–648 CrossRef CAS.
- I. Haq, P. Mazumder and A. S. Kalamdhad, Bioresour. Technol., 2020, 312, 123636 CrossRef CAS PubMed.
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed.
- S. Laurichesse, L. Avérous, L. Averous and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- E. K. Pye and J. H. Lora, Tappi J., 1991, 74, 113–118 CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- J. Sternberg, O. Sequerth and S. Pilla, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS.
- E. Isaac, A. Samson and O. Adeosun, Sustainable Lignin for Carbon Fibers: Principles, Techniques, and Applications, Springer International Publishing, Cham, 2019 Search PubMed.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- H. Liu and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3515–3528 CrossRef CAS.
- H. Liu and H. Chung, ACS Sustainable Chem. Eng., 2017, 5, 9160–9168 CrossRef CAS.
- D. Uhrig and J. Mays, Polym. Chem., 2011, 2, 69–76 RSC.
- S. M. Satti and A. A. Shah, Lett. Appl. Microbiol., 2020, 70, 413–430 CrossRef CAS PubMed.
- C. Zhu, Z. Zhang, Q. Liu, Z. Wang and J. Jin, J. Appl. Polym. Sci., 2003, 90, 982–990 CrossRef CAS.
- T. Tang, T. Moyori and A. Takasu, Macromolecules, 2013, 46, 5464–5472 CrossRef CAS.
- T. Kobayashi, M. Kakimoto and Y. Imai, Polym. J., 1993, 25, 741–746 CrossRef CAS.
- E. N. Zil'berman, A. E. Kulikova and N. M. Teplyakov, J. Polym. Sci., 1962, 56, 417–423 CrossRef.
- H. Feuer and R. Miller, J. Org. Chem., 1959, 24, 118–119 CrossRef CAS.
- R. Okabayashi, Y. Ohta and T. Yokozawa, Polym. Chem., 2019, 10, 4973–4979 RSC.
- N. Cohen-Arazi, J. Katzhendler, M. Kolitz and A. J. Domb, Macromolecules, 2008, 41, 7259–7263 CrossRef CAS.
- Roohi, M. R. Zaheer and M. Kuddus, Polym. Adv. Technol., 2018, 29, 30–40 CrossRef CAS.
- S. A. A. Ghavimi, M. H. Ebrahimzadeh, M. Solati-Hashjin and N. A. A. Osman, J. Biomed. Mater. Res., Part A, 2015, 103, 2482–2498 CrossRef PubMed.
- M. Puchalski, G. Szparaga, T. Biela, A. Gutowska, S. Sztajnowski and I. Krucińska, Polymers, 2018, 10, 251 CrossRef PubMed.
- D. J. A. Cameron and M. P. Shaver, Chem. Soc. Rev., 2011, 40, 1761–1776 RSC.
- V. Lassalle and M. L. Ferreira, Macromol. Biosci., 2007, 7, 767–783 CrossRef CAS PubMed.
- C. Nakafuku and H. Yoshimura, Polymer, 2004, 45, 3583–3585 CrossRef CAS.
- K. Budak, O. Sogut and U. A. Aydemir, J. Polym. Res., 2020, 27, 208 CrossRef CAS.
- Y. Xu, C. S. Kim, D. M. Saylor and D. Koo, J. Biomed. Mater. Res., Part B, 2017, 105, 1692–1716 CrossRef CAS PubMed.
- M. de Geus, I. van der Meulen, B. Goderis, K. van Hecke, M. Dorschu, H. van der Werff, C. E. Koning and A. Heise, Polym. Chem., 2010, 1, 525–533 RSC.
- H. Liu, N. Mohsin, S. Kim and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2121–2130 CrossRef CAS.
- P. Karuppuswamy, J. R. Venugopal, B. Navaneethan, A. L. Laiva and S. Ramakrishna, Mater. Lett., 2015, 141, 180–186 CrossRef CAS.
- L. Cabedo, J. L. Feijoo, M. P. Villanueva, J. M. Lagarón and E. Giménez, Macromol. Symp., 2006, 233, 191–197 CrossRef CAS.
- S. Rahmani, M. Maroufkhani, S. Mohammadzadeh-Komuleh and Z. Khoubi-Arani, in Fundamentals of Bionanomaterials, Elsevier, 2022, pp. 175–215 Search PubMed.
- J. R. Dias, A. Sousa, A. Augusto, P. J. Bártolo and P. L. Granja, Polymers, 2022, 14, 3397 CrossRef CAS PubMed.
- N. Mulchandani, K. Masutani, S. Kumar, S. Sakurai, Y. Kimura and V. Katiyar, Mater. Today Commun., 2022, 33, 104040 CrossRef CAS.
- A. Haryńska, J. Kucinska-Lipka, A. Sulowska, I. Gubanska, M. Kostrzewa and H. Janik, Materials, 2019, 12, 887 CrossRef PubMed.
- S. Müller, H. Uyama and S. Kobayashi, Chem. Lett., 1999, 28, 1317–1318 CrossRef.
- A. Pascual, H. Sardon, A. Veloso, F. Ruipérez and D. Mecerreyes, ACS Macro Lett., 2014, 3, 849–853 CrossRef CAS PubMed.
- A. Pascual, H. Sardón, F. Ruipérez, R. Gracia, P. Sudam, A. Veloso and D. Mecerreyes, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 552–561 CrossRef CAS.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- D. McGinty, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2011, 49, S174–S182 CrossRef CAS PubMed.
- S. Ravi, D. Padmanabhan and V. R. Mamdapur, J. Indian Inst. Sci., 2013, 81, 299 Search PubMed.
- J. Pais, I. Farinha, F. Freitas, L. S. Serafim, V. Martínez, J. C. Martínez, M. Arévalo-Rodríguez, M. A. Prieto and M. A. M. Reis, Enzyme Microb. Technol., 2014, 55, 151–158 CrossRef CAS PubMed.
- A. Bera, S. Dubey, K. Bhayani, D. Mondal, S. Mishra and P. K. Ghosh, Int. J. Biol. Macromol., 2015, 72, 487–494 CrossRef CAS PubMed.
- A. A. Pantazaki, C. P. Papaneophytou, A. G. Pritsa, M. Liakopoulou-Kyriakides and D. A. Kyriakidis, Process Biochem., 2009, 44, 847–853 CrossRef CAS.
- J.-H. Lin, M.-C. Lee, Y.-S. Sue, Y.-C. Liu and S.-Y. Li, Appl. Microbiol. Biotechnol., 2017, 101, 6419–6430 CrossRef CAS PubMed.
- J. Marudkla, W.-C. Lee, S. Wannawilai, Y. Chisti and S. Sirisansaneeyakul, J. Biotechnol., 2018, 268, 12–20 CrossRef CAS PubMed.
- S. J. Park, Y. Jang, W. Noh, Y. H. Oh, H. Lee, Y. David, M. G. Baylon, J. Shin, J. E. Yang and S. Y. Choi, Biotechnol. Bioeng., 2015, 112, 638–643 CrossRef CAS PubMed.
- N. Biglari, M. G. Dashti, P. Abdeshahian, I. Orita, T. Fukui and K. Sudesh, 3 Biotech, 2018, 8, 330 CrossRef PubMed.
- A. F. M. Yatim, I. M. Syafiq, K. H. Huong, A.-A. A. Amirul, A. W. M. Effendy and K. Bhubalan, BioTechnologia, 2017, 2, 141–151 CrossRef.
- R. Das, N. R. Saha, A. Pal, D. Chattopadhyay and A. K. Paul, Front. Biol., 2018, 13, 297–308 CrossRef CAS.
- D. Kalaiyezhini and K. B. Ramachandran, Prep. Biochem. Biotechnol., 2015, 45, 69–83 CrossRef CAS PubMed.
- P. C. Sabapathy, S. Devaraj, A. Parthiban and P. Kathirvel, Environ. Technol., 2018, 39, 1430–1441 CrossRef CAS PubMed.
- C. F. P. Malacara, A. G. Romero, M. M. Ponce and T. C. Marenco, Microbial Factories, Springer, India, New Delhi, 2015 Search PubMed.
- J. Valdés, G. Kutralam-Muniasamy, B. Vergara-Porras, R. Marsch, F. Pérez-Guevara and M. R. López-Cuellar, Nat. Biotechnol., 2018, 40, 200–206 Search PubMed.
- M. C. Eremia, I. Lupescu, M. Vladu, M. Petrescu, G. Savoiu, A. Stefaniu and M. Spiridon, Ovidius Univ. Ann. Chem., 2016, 27, 44–47 CrossRef.
- J. Fernández, H. Amestoy, H. Sardon, M. Aguirre, A. L. Varga and J. R. Sarasua, J. Mech. Behav. Biomed. Mater., 2016, 64, 209–219 CrossRef PubMed.
- S. Tiptipakorn, N. Keungputpong, S. Phothiphiphit and S. Rimdusit, J. Appl. Polym. Sci., 2015, 132(18), 41915 CrossRef.
- J. Fernández, H. Amestoy, H. Sardon, M. Aguirre, A. L. Varga and J.-R. Sarasua, J. Mech. Behav. Biomed. Mater., 2016, 64, 209–219 CrossRef PubMed.
- J. Fernández, M. Montero, A. Etxeberria and J.-R. Sarasua, Polym. Degrad. Stab., 2017, 137, 23–34 CrossRef.
- G. Cama, D. E. Mogosanu, A. Houben and P. Dubruel, in Science and Principles of Biodegradable and Bioresorbable Medical Polymers, Elsevier, 2017, pp. 79–105 Search PubMed.
- Z. Luo, Y. L. Wu, Z. Li and X. J. Loh, Biotechnol. J., 2019, 14, 1900283 CrossRef CAS PubMed.
- K. Sudesh, H. Abe and Y. Doi, Prog. Polym. Sci., 2000, 25, 1503–1555 CrossRef CAS.
- M. Koller, Bioengineering, 2019, 6, 34 CrossRef PubMed.
- Z. A. Raza, S. Noor and S. Khalil, Biotechnol. Prog., 2019, 35, e2855 CrossRef CAS PubMed.
- A. V. Samrot, S. K. Samanvitha, N. Shobana, E. R. Renitta, P. Senthilkumar, S. S. Kumar, S. Abirami, S. Dhiva, M. Bavanilatha, P. Prakash, S. Saigeetha, K. S. Shree and R. Thirumurugan, Polymers, 2021, 13, 3302 CrossRef CAS PubMed.
- Q. Gao, H. Yang, C. Wang, X.-Y. Xie, K.-X. Liu, Y. Lin, S.-Y. Han, M. Zhu, M. Neureiter, Y. Lin and J.-W. Ye, Front. Bioeng. Biotechnol., 2022, 10, 966598 CrossRef PubMed.
- J. Rydz, W. Sikorska, M. Kyulavska and D. Christova, Int. J. Mol. Sci., 2015, 16, 564–596 CrossRef PubMed.
- S. R. Andersson, M. Hakkarainen, S. Inkinen, A. Södergård and A.-C. Albertsson, Biomacromolecules, 2010, 11, 1067–1073 CrossRef CAS PubMed.
- A. A. Shah, S. Kato, N. Shintani, N. R. Kamini and T. Nakajima-Kambe, Appl. Microbiol. Biotechnol., 2014, 98, 3437–3447 CrossRef CAS PubMed.
- R.-J. Müller, I. Kleeberg and W.-D. Deckwer, J. Biotechnol., 2001, 86, 87–95 CrossRef PubMed.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L.-W. Majumder and G. W. Huber, Chem. Rev., 2023, 123, 9915–9939 CrossRef CAS PubMed.
- T. Hiraishi and S. Taguchi, Mini-Rev. Org. Chem., 2009, 6, 44–54 CrossRef CAS.
- A. Banerjee, K. Chatterjee and G. Madras, Mater. Sci. Technol., 2014, 30, 567–573 CrossRef CAS.
- S.-J. Zhou, H.-M. Wang, S.-J. Xiong, J.-M. Sun, Y.-Y. Wang, S. Yu, Z. Sun, J.-L. Wen and T.-Q. Yuan, ACS Sustainable Chem. Eng., 2021, 9, 12017–12042 CrossRef CAS.
- Y. L. Chung, J. V. Olsson, R. J. Li, C. W. Frank, R. M. Waymouth, S. L. Billington and E. S. Sattely, ACS Sustainable Chem. Eng., 2013, 1, 1231–1238 CrossRef CAS.
- L. Dai, Q. Cao, K. Wang, S. Han, C. Si, D. Liu and Y. Liu, Ind. Crops Prod., 2020, 143, 111954 CrossRef CAS.
- R. Liu, L. Dai, Z. Zou and C. Si, Int. J. Biol. Macromol., 2018, 119, 1129–1136 CrossRef CAS PubMed.
- W. Ren, X. Pan, G. Wang, W. Cheng and Y. Liu, Green Chem., 2016, 18, 5008–5014 RSC.
- L. E. Chile, S. J. Kaser, S. G. Hatzikiriakos and P. Mehrkhodavandi, ACS Sustainable Chem. Eng., 2018, 6, 1650–1661 CrossRef CAS.
- M. Gallego-García, A. D. Moreno, P. Manzanares, M. J. Negro and A. Duque, Bioresour. Technol., 2023, 369, 128397 CrossRef PubMed.
- S. P. Makri, E. Xanthopoulou, M. A. Valera, A. Mangas, G. Marra, V. Ruiz, S. Koltsakidis, D. Tzetzis, A. Z. Karathanasis, I. Deligkiozi, N. Nikolaidis, D. Bikiaris and Z. Terzopoulou, Polymers, 2023, 15, 2386 CrossRef CAS PubMed.
- S. P. Makri, P. A. Klonos, G. Marra, A. Z. Karathanasis, I. Deligkiozi, M. Á. Valera, A. Mangas, N. Nikolaidis, Z. Terzopoulou, A. Kyritsis and D. N. Bikiaris, Soft Matter, 2024, 20, 5014–5027 RSC.
- S. Y. Park, J. Y. Kim, H. J. Youn and J. W. Choi, Int. J. Biol. Macromol., 2019, 138, 1029–1034 CrossRef CAS PubMed.
- Q. Zong, A. Xu, K. Chai, Y. Zhang and Y. Song, Polym. Adv. Technol., 2020, 31, 2239–2249 CrossRef CAS.
- R. Liu, L. Dai, L.-Q. Hu, W.-Q. Zhou and C.-L. Si, Mater. Sci. Eng., C, 2017, 80, 397–403 CrossRef CAS PubMed.
- X.-F. Wei, R.-Y. Bao, Z.-Q. Cao, W. Yang, B.-H. Xie and M.-B. Yang, Macromolecules, 2014, 47, 1439–1448 CrossRef CAS.
- M. Li, Y. Pu, F. Chen and A. J. Ragauskas, Nat. Biotechnol., 2021, 60, 189–199 CAS.
- R. A. Pérez-Camargo, G. Saenz, S. Laurichesse, M. T. Casas, J. Puiggalí, L. Avérous and A. J. Müller, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 1736–1750 CrossRef.
- M. Abdollahi, R. B. Habashi and M. Mohsenpour, Ind. Crops Prod., 2019, 130, 547–557 CrossRef CAS.
- S. Laurichesse and L. Avérous, Polymer, 2013, 54, 3882–3890 CrossRef CAS.
- J. Wang, L. Tian, B. Luo, S. Ramakrishna, D. Kai, X. J. Loh, I. H. Yang, G. R. Deen and X. Mo, Colloids Surf., B, 2018, 169, 356–365 CrossRef CAS PubMed.
- I.-K. Park, H. Sun, S.-H. Kim, Y. Kim, G. E. Kim, Y. Lee, T. Kim, H. R. Choi, J. Suhr and J.-D. Nam, Sci. Rep., 2019, 9, 7033 CrossRef PubMed.
- A. Duda, S. Penczek, A. Kowalski and J. Libiszowski, Macromol. Symp., 2000, 153, 41–53 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 2000, 33, 689–695 CrossRef CAS.
- M. Ryner, K. Stridsberg, A. C. Albertsson, H. Von Schenck and M. Svensson, Macromolecules, 2001, 34, 3877–3881 CrossRef CAS.
- W. Punyodom, W. Limwanich, P. Meepowpan and B. Thapsukhon, Des. Monomers Polym., 2021, 24, 89–97 CrossRef CAS PubMed.
- M. C. Najarro, M. Nikolic, J. Iruthayaraj and I. Johannsen, ACS Appl. Polym. Mater., 2020, 2, 5767–5778 CrossRef CAS.
- Y. Zhang, J. Liao, X. Fang, F. Bai, K. Qiao and L. Wang, ACS Sustainable Chem. Eng., 2017, 5, 4276–4284 CrossRef CAS.
- J. Tian, Y. Yang and J. Song, Int. J. Biol. Macromol., 2019, 141, 919–926 CrossRef CAS PubMed.
- J. J. C. Lee, S. Sugiarto, P. J. Ong, X. Y. D. Soo, X. Ni, P. Luo, Y. Y. K. Hnin, J. S. Y. See, F. Wei, R. Zheng, P. Wang, J. Xu, X. J. Loh, D. Kai and Q. Zhu, J. Energy Storage, 2022, 56, 106118 CrossRef.
- D. Xie, Y. Pu, X. Meng, N. D. Bryant, K. Zhang, W. Wang, A. J. Ragauskas and M. Li, ACS Sustainable Chem. Eng., 2022, 10, 16882–16895 CrossRef CAS.
- S. H. Jang, D. H. Kim, D. H. Park, O. Y. Kim and S. H. Hwang, Prog. Org. Coat., 2018, 120, 234–239 CrossRef CAS.
- A. Lendlein, A. M. Schmidt and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 842–847 CAS.
- P. Deshmukh, H. Yoon, S. Cho, S. Y. Yoon, O. V. Zore, T. Kim, I. Chung, S. Ahn and R. M. Kasi, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3424–3433 CrossRef CAS.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- Q. Zhao, H. J. Qi and T. Xie, Prog. Polym. Sci., 2015, 49, 79–120 CrossRef.
- S. Kim and H. Chung, ACS Sustainable Chem. Eng., 2021, 9, 14766–14776 CrossRef CAS.
- B. McAdam, M. B. Fournet, P. McDonald and M. Mojicevic, Polymers, 2020, 12, 2908 CrossRef CAS PubMed.
- I. Vroman and L. Tighzert, Materials, 2009, 2, 307–344 CrossRef CAS.
- F. M. García-Valle, V. Tabernero, T. Cuenca, M. E. G. Mosquera, J. Cano and S. Milione, Organometallics, 2018, 37, 837–840 CrossRef.
- S. M. Guillaume, L. Annunziata, I. del Rosal, C. Iftner, L. Maron, P. W. Roesky and M. Schmid, Polym. Chem., 2013, 4, 3077–3087 RSC.
- J. Dobrogojski, M. Spychalski, R. Luciński and S. Borek, Acta Physiol. Plant., 2018, 40, 1–17 CrossRef CAS.
- R. Handrick, S. Reinhardt, P. Kimmig and D. Jendrossek, J. Bacteriol., 2004, 186, 7243–7253 CrossRef CAS PubMed.
- D. Kai, K. Zhang, S. S. Liow and X. J. Loh, ACS Appl. Bio Mater., 2019, 2, 127–134 CrossRef CAS PubMed.
- Y. Sun, L. Yang, X. Lu and C. He, J. Mater. Chem. A, 2015, 3, 3699–3709 RSC.
- D. Kai, K. Zhang, L. Jiang, H. Z. Wong, Z. Li, Z. Zhang and X. J. Loh, ACS Sustainable Chem. Eng., 2017, 5, 6016–6025 CrossRef CAS.
- W. Yang, G. Qi, H. Ding, P. Xu, W. Dong, X. Zhu, T. Zheng and P. Ma, Compos. Commun., 2020, 22, 100497 CrossRef.
- A. L. Rivera-Briso and Á. Serrano-Aroca, Polymers, 2018, 10(7), 732 CrossRef PubMed.
- M. Larsson, O. Markbo and P. Jannasch, RSC Adv., 2016, 6, 44354–44363 RSC.
- Y.-X. Weng, Y. Wang, X.-L. Wang and Y.-Z. Wang, Polym. Test., 2010, 29, 579–587 CrossRef CAS.
- E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, eXPRESS Polym. Lett., 2014, 8, 791–808 CrossRef.
- T. Erceg, S. Rackov and P. Terek, Polymers, 2023, 15, 4694 CrossRef CAS PubMed.
- T. Ahmed, H. Marçal, M. Lawless, N. S. Wanandy, A. Chiu and L. J. R. Foster, Biomacromolecules, 2010, 11, 2707–2715 CrossRef CAS PubMed.
- B. M. P. Ferreira and E. A. R. Duek, J. Appl. Biomater. Biomech., 2005, 3, 50–60 CAS.
- M. L. Tebaldi, A. L. C. Maia, F. Poletto, F. V. de Andrade and D. C. F. Soares, J. Drug Delivery Sci. Technol., 2019, 51, 115–126 CrossRef CAS.
- F. Türesin, I. Gürsel and V. Hasirci, J. Biomater. Sci., Polym. Ed., 2001, 12, 195–207 CrossRef PubMed.
- M. L. Tebaldi, A. L. C. Maia, F. Poletto, F. V. de Andrade and D. C. F. Soares, J. Drug Delivery Sci. Technol., 2019, 51, 115–126 CrossRef CAS.
- S. Luo, J. Cao and A. G. McDonald, ACS Sustainable Chem. Eng., 2016, 4, 3465–3476 CrossRef CAS.
- D. Kai, H. M. Chong, L. P. Chow, L. Jiang, Q. Lin, K. Zhang, H. Zhang, Z. Zhang and X. J. Loh, Compos. Sci. Technol., 2018, 158, 26–33 CrossRef CAS.
- E. L. Young and A. G. McDonald, Molecules, 2021, 26, 2437 CrossRef CAS PubMed.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. (C) Xu, Renewable Sustainable Energy Rev., 2020, 123, 109768 CrossRef CAS.
- J. Kautto, M. J. Realff, A. J. Ragauskas and T. Kässi, BioResources, 2014, 9, 6041–6072 CrossRef.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- H. Ludmila, J. Michal, Š. Andrea and H. Aleš, Wood Res., 2015, 60, 973–986 Search PubMed.
- Y. Han, L. Yuan, G. Li, L. Huang, T. Qin, F. Chu and C. Tang, Polymer, 2016, 83, 92–100 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |