
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sustainable lecithin-based ligand for the bio-functionalization of iron and hybrid metal organic frameworks (MOFs) nanoparticles with the sugar mannose†
Camilla M.
Cova
 a,
Víctor
Ramos
a,
Alberto
Escudero
ab,
Juan P.
Holgado
bc,
Noureddine
Khiar
*a and
Alessio
Zuliani
*a
a,
Víctor
Ramos
a,
Alberto
Escudero
ab,
Juan P.
Holgado
bc,
Noureddine
Khiar
*a and
Alessio
Zuliani
*a
aAsymmetric Synthesis and Functional Nanosystems Group (Art&Fun), Institute for Chemical Research (IIQ), CSIC-University of Seville, 41092 Seville, Spain. E-mail: khiar@iiq.csic.es; a.zuliani@csic.es
bDepartment of Inorganic Chemistry, Faculty of Chemistry, University of Seville, 41012 Seville, Spain
cInstitute of Materials Science (ICMS), CSIC-University of Seville, 41092 Seville, Spain
First published on 15th October 2024
Abstract
The functionalization of nanoparticles with specific ligands, such as antibodies, peptides, and small molecules, plays a critical role in achieving targeted delivery, enhancing biocompatibility, and controlling drug release. However, to date, practically no attention has been paid to the design of green ligands. Herein, an innovative approach to develop a sustainable ligand for nanoparticle functionalization is reported. Its synthesis involved a photochemical thio–ene “click” reaction between the natural compounds phosphatidylcoline, the main component of lecithin, and cysteine, followed by a reductive amination with mannose, a sugar of growing interest for biomedical targeting, in a continuous flow hydrogenation reactor. Comprehensive characterization techniques, including nuclear magnetic resonance (NMR), mass spectrometry (MS), Fourier-transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and elemental analysis, confirmed the structure and properties of the novel ligand. The environmental sustainability of the ligand was evaluated determining some green metrics using the EATOS software. The obtained E-factor was compared with a conventional PEG-based ligand. The newly developed lecithin-derived ligand was successfully used to functionalize diverse NP platforms, including the MOFs MIL-101(Fe), PCN-222, UiO-66, and iron nanoparticles (in the form of akaganeite), demonstrating its potential in nanomedicine applications.
1. Introduction
Nanomedicine has the potential to unlock precise, targeted treatments and diagnostics at the molecular level, enhancing therapeutic efficacy and safety while minimizing side effects, thereby transforming the future of healthcare.1–3 In this context, the development of ligands for the functionalization of nanoparticles (NPs) for biomedical applications, such as metal organic frameworks (MOFs), metallic nanoparticles, nanogels, or carbon nanotubes, has emerged as crucial due to their ability to provide targeted delivery, enhanced biocompatibility, and controlled release of therapeutic agents.4–7 Ligands can be designed presenting antibodies,8,9 peptides,10 and small molecules, such as carbohydrates,11,12 in their structures to target specific cellular receptors or molecular markers that are overexpressed in certain disease states, such as cancer, or to direct these particles to accumulate preferentially in diseased tissues while minimizing their distribution to healthy ones. This approach not only enhances the therapeutic efficacy of the NPs, or the eventual drugs loaded into them, but also reduces off-target effects and associated toxicities.13,14 For instance, Wang et al. investigated the use of a PEG-folate ligand for the functionalization of a liposome carrying doxorubicin for the treatment of ovarian cancer. The ligand allowed for targeting of folate-receptor-alpha, overexpressed on the surface of ovarian cancer cells, resulting in reduced off-target cytotoxicity compared to non-targeted nanoparticles.15 Ligands can also play a pivotal role in controlling the release kinetics of drugs loaded into the NPs. By utilizing stimuli-responsive ligands, such as those sensitive to pH, temperature, or enzymatic activity, it is possible to design nanoparticles that release their therapeutic payloads in response to the unique microenvironment of diseased tissues.16,17 This smart release mechanism ensures that drugs are released in a controlled manner at the site of action, thereby maximizing their therapeutic potential and minimizing systemic side effects.18 The surface modification of NPs with biocompatible ligands can also significantly improve their circulation time in the bloodstream and reduce their recognition and clearance by the immune system.19 Among all, polyethylene glycol (PEG) and other hydrophilic polymers are commonly used ligands that provide a hydrophilic “stealth” coating to NPs, reducing opsonization and subsequent phagocytosis by macrophages. This prolongation of circulation time increases the likelihood that NPs will reach their target sites and enhances the overall efficacy of the treatment. There is also significant interest in ligands that impart multiple functionalities to NPs, such as those facilitating both targeting and imaging for simultaneous diagnosis and treatment, i.e., theragnostic.20According to the current literature, researchers follow two main approaches for the design of ligands for the functionalization of NPs. The most common and widely employed method involves using synthetic petroleum-derived ligands. In this case, advanced synthetic techniques are employed to create novel ligands with high specificity and affinity for target molecules. However, this classic approach normally entails the consumption of large volumes of solvents, often toxic, the use of petroleum-derived chemicals, the production of large volumes of waste, the need for extensive purification steps and the exploitation of low energy efficiency techniques. As a consequence, the sustainability of the synthetic processes is also a significant concern. In this scenario, it has to be mentioned that the E-factor (i.e., ratio between the mass of waste and mass of product) for the chemicals of the pharma industry, to which these ligands belong to, is normally between 25 and 100, indicating a high ratio of waste produced to the amount of product obtained.21
The other recently born and still barely explored approach for the design of ligands is based on the use of naturally occurring compounds, such as cell membrane coatings, small molecules such as sugars or natural biomolecules. For example, NPs coated with red blood cell membranes can evade the immune system and prolong circulation time, mimicking the natural behaviour of red blood cells.22 Besides, NPs covered with hyaluronic acid, a naturally occurring polysaccharide, can target CD44 receptors overexpressed in many tumours.23
However, despite their potential, the use of naturally-derived ligands is still at an early stage and more effort should be paid to investigate this strategy. Especially considering that natural ligands are normally employed without adapting their molecular structure for specific biomedical applications. Furthermore, the design of environmentally friendly processes to modify and upgrade these ligands remains practically unexplored. This is due to the fact that the application of the principles of green chemistry and, more broadly, considerations regarding the sustainability of chemical and compound productions for biomedical applications has not been given much attention in recent years. Nevertheless, green approaches in biomedicine, and especially in methods for the preparation of functionalized NPs, are in a germination phase and the research has started giving some results, also at an industrial level, as evidenced by a few patents. For example, US16735009B2 describes the green synthesis of tellurium nanowires with anticancer properties using starch as a coating agent to improve their cytocompatibility.
Herein, aiming to take a step forward in this field and promote the expansion of green chemistry in biomedicine, we introduce an innovative sustainable approach for the synthesis of a natural-derived ligand for the functionalization of different NPs used in nanomedicine. More in detail, we designed a lecithin-derived ligand composed by phosphatidylcholine, the main component of lecithin, cysteine and D-Mannose (Fig. 1). Phosphatidylcholine, a natural phospholipid abundantly found in cell membranes, serves as the core structure of the ligand due to its intrinsic biocompatibility and amphiphilic nature, containing a polar hydrophilic phosphate head group and a non-polar lipophilic hydrocarbon tail. Its seamless interaction with biological systems makes it an ideal starting material for the conception of functionalized NPs. We further extended the versatility of the ligand by the inclusion of cysteine, an amino acid bearing a thiol (–SH) group, through a photochemical thio–ene “click” reaction. Building upon the functionalized phosphatidylcholine-cysteine structure, we thus introduce D-Mannose, a sugar renowned for its targeting capabilities in biological systems, by a reductive amination reaction performed in continuous flow (c.f.).
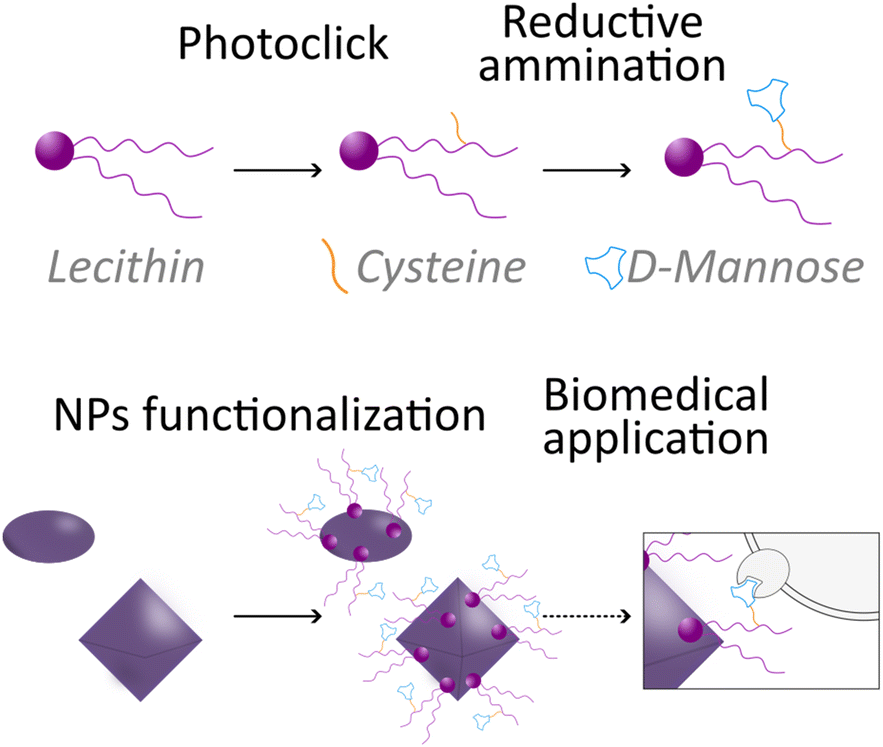 | ||
| Fig. 1 Schematic illustration of the novel lecithin-derived ligand and its use to functionalize nanoparticles for potential targeting. | ||
We characterized the ligand by different techniques, including NMR, mass-spectroscopy, Fourier-transform infrared spectroscopy (FTIR), elemental analysis, X-ray photoelectron spectroscopy (XPS), and Dynamic Light Scattering (DLS). In addition, we calculated the sustainable features of the novel ligand in terms of some green metrics using EATOS software. We compared the obtained E-factor and additional Reaction Mass Efficiency% (RME%) with those of a PEG-phosphate ligand.
Finally, we successfully used the new lecithin-based ligand for the functionalization of different NPs, including the MOFs MIL-101(Fe), PCN-222, UiO-66, and iron NPs (in the specific form of akageneite rods), demonstrating its capability for potential biomedical application.
2. Methods
Details of the materials and methods are reported in the ESI Sections S1 and S2.†2.1 Synthesis of the ligand
As shown in Fig. 2, the first step for the preparation of the novel ligand involved a photochemical click reaction. For this, we dissolved 2.35 g (3 mmol, basing on average MW 783 g mol−1) of commercially available L-α-phosphatidylcholine and 0.36 g (3 mmol, 121.16 g mol−1, [α]20D = 5.19°) of L-cysteine in the minimal amount of ethanol (EtOH, 20 mL). We thus used UV light (254 nm) to promote the reaction using 76 mg (0.3 mmol, 256.30 g mol−1, 0.1 eq.) of DMPA (2,2-dimethoxy-2-phenylacetophenone) as a photo initiator.24 We carried out the reaction at room temperature for 48 h under stirring. Then, we removed the solvent by vacuum evaporation. We obtained the phosphatidylcholine-cysteine, denoted “Phos-Cys”, in the form of a yellow/brown solid paste (average of 2.57 g of product, MW ca. 904 g mol−1, 95% isolated yield).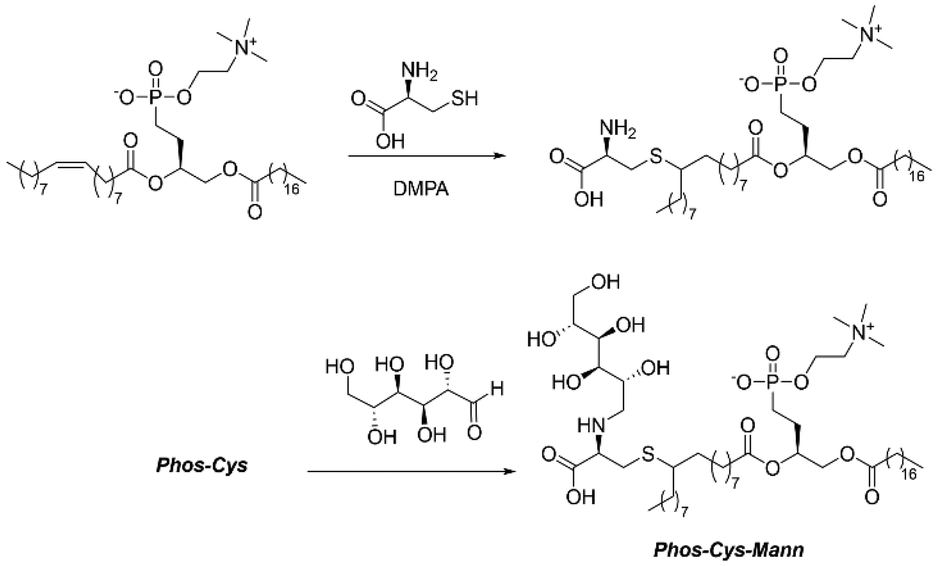 | ||
| Fig. 2 Scheme for the preparation of the novel Phos-Cys-Man ligand. | ||
Sequentially, we mixed 1.81 g (2 mmol, MW 904 g mol−1) of Phos-Cys, without performing any prior purification, with 0.36 g of D-Mannose (2 mmol, MW 180.16 g mol−1, [α]20D = −8.34°) into 300 mL of EtOH. We carried out the reductive amination under liquid phase c.f. conditions in an H-Cube Mini Plus™ flow hydrogenation reactor. We employed a Pd heterogeneous catalyst (Pd/C 10 wt%) loaded into a 30 mm-long cartridge (ThalesNano CatCart). After optimizing the reaction conditions based on reported literature of similar reagents,25 we obtained the best results operating at 70 °C, 25 bar, 0.3 mL min−1 rate flow. Hydrogen was generated in situ during the reaction by water electrolysis in the same H-Cube equipment. In order to react all the solution containing the Phos-Cys, we carried out the reaction for a total of 17 hours. We removed the solvent by vacuum evaporation, obtaining the final ligand in the form of a viscous brownish paste named as “Phos-Cys-Man” (average of 2.57 g of product, MW ca. 1068 g mol−1 97% isolated yield, [α]20D = 9.04°). Additional details for the synthesis of the lecithin-based ligand can be find in the ESI Section S2.†
2.2 Green metrics
We utilized the EATOS software for the evaluation of the environmental features of the synthesis of Phos-Cys-Man. EATOS is a software capable of computing four key parameters: the well-known mass index (MI), the environmental factor (E-factor), and two additional parameters, EI_in and EI_out.26 These latter two parameters represent the environmental impacts associated with the inward and outward material flows, respectively. The mass of each chemical used was multiplied by its total weighting factor, Qtot, which was calculated using data from the Safety Data Sheets (SDS) of each chemical taken from Sigma-Aldrich or directly from the library of EATOS. For the compounds without SDS, i.e., Phos-Cys and Phos-Cys-Man, we referred to the SDS of phosphatidylcholine. We further analyse the environmental impact of the synthesis of Phos-Cys-Man by making a comparison with the synthesis of a PEG-phosphate ligand reported in the literature.27 Since we haven't access to the full data regarding the synthesis of this PEG-phosphate ligand, we opted to compare the E-factor, the MI, and the RME (%) of the two synthesis (i.e., of the Phos-Cys-Man and of the PEG-phosphate) since we couldn't perform the analysis with EATOS.We calculated RME (%) based on eqn (1):28
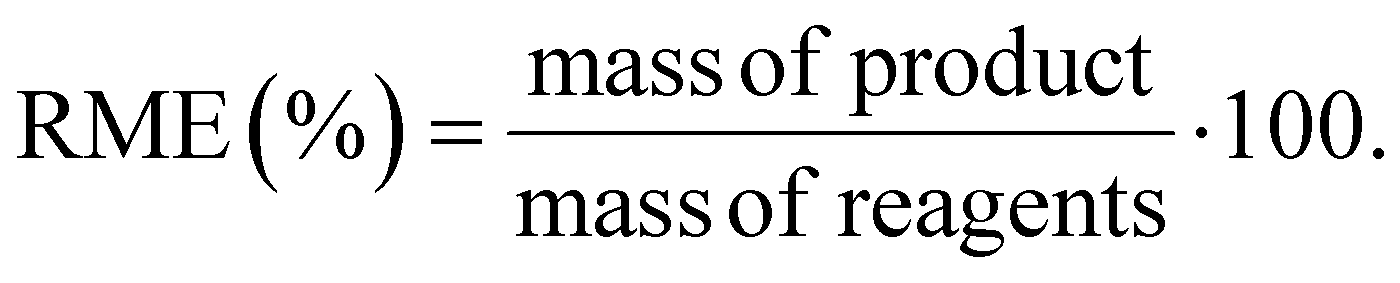 | (1) |
3 Results and discussion
3.1 Design of the sustainable synthesis of the ligand
As previously shown in Fig. 1, our aim was to design an innovative sustainable ligand characterized by high versatility for the functionalization of different NPs, also incorporating targeting molecules in its structure to demonstrate its potential for biomedical applications. Consequently, we opted for a ligand derived from natural sources, ensuring that it adheres to the principles of green chemistry and reduces the environmental impact of the final compound. In addition, we also focused on the sustainable features of the production process of the ligand, in contrast with the traditional synthetic approaches normally used to prepare ligands for NPs functionalization, characterized by the generation of high volume of waste, low energy efficiency, etc.1,29,30To achieve all these objectives, and thus establish a new green strategy for NPs functionalization, we firstly investigated the different types of ligands reported in the literature. We noticed that commonly used ones, which typically form either covalent bonds or robust coordination with organic groups (e.g., ligands with –NH2, –COOH, –N3),31 are often associated with limited biodegradability and/or complex ligand synthesis. On the contrary, ligands with functionalities based on the latest literature advancements, i.e., sulfate (–OSO3H),32 and phosphate (–OPO3H),27 can be degraded by human enzymes, such as phosphatase and sulfatase, overexpressed in some types of cancer, facilitating the liberation of NPs, and the eventual loaded drugs. At the same time, the interaction of phosphates and sulfates with metals occur at milder conditions, leading to easier preparation of functionalized NPs. For example, phosphates are negatively charged in a large pH range, while metal ions are positively charged.33 We thus look for natural compounds having these types of functionalities. Phosphates, naturally occurring in many living organisms, are the most abundant and accessible. Among them, lecithin, mixture of phospholipids (60–80 wt%), triglycerides/other lipids (10–20 wt%), glycolipids (5–10 wt%), carbohydrates (5–10 wt%) and water (<5 wt%) commonly found in cellular membranes and other biological tissues, attracted our attention for its large availability. Indeed, lecithin is a cheap industrial emulsifier and stabilizer generally recognized as a safe (GRAS) food substance by FDA used in various industries such as food, cosmetics, and pharmaceuticals.34 Lecithin is produced on a large scale through extraction from different natural sources, mainly crude vegetable oils. The most common extraction process involves several key steps: (1) degumming the oil by heating to 70–82 °C and adding water, followed by centrifugation to isolate the lecithin gums; (2) dehydration to reduce the moisture content to below 0.5 wt% by vacuum evaporation or using a thin film evaporator; (3) de-oiling by mixing with ethanol; (4) additional processing as bleaching with hydrogen peroxide or benzoyl peroxide to improve colour; (5) drying.35
While soy is the most common source, lecithin can also be extracted from sunflower, rapeseed, and other vegetable oils.36 Notably, the environmental impact of the current industrial process is quite low, and new sustainable methods have already been reported, some of them on a pilot scale.36 Alongside with its natural abundance and easy extraction process, lecithin has also exceptional biocompatibility. As a natural component of cellular membranes, it is well-tolerated by the human body, reducing the risk of adverse reactions. This biocompatibility makes lecithin especially suitable for biomedical applications. For example, lecithin has been used to for the preparation of micelles for the delivery of curcumin, and of microspheres, for the delivery of resveratrol.37,38
Aiming at exploring the possibilities of functionalizing lecithin with compounds for targeting, we observed that the phospholipids composing lecithin are primarily phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, and phosphatidylserine (Fig. 3). As suggested by the name, phosphatidylcholine (20–25 wt% relative to lecithin), shows in its structure a choline molecule, i.e., a quaternary ammonium compound, and a glycerophosphoric acid composed of a variety of different fatty acids. Since we cannot modify the phosphate group as it was the group needed to link the ligand to the NPs, and the quaternary ammonium cation has low reactivity and was too close to the phosphate, implying possible steric hindrances if linked to other molecules, we opted to focus on the different fatty acids composing the glycerophosphoric acid. These are mainly monounsaturated and polyunsaturated fatty acids, presenting large numbers of double bonds that can be modified. We then selected phosphatidylcholine extracted from soy lecithin, due to its commercial abundance (approx. 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000 metric tons produced annually). Importantly, using edible soybean oil or other lecithin-rich edible oils such as sunflower or rapeseed oils for biomedical applications does not represent an ethical issue, since their utilization aligns with sustainability goal.39 At the same time, it has also to be considered that alternative non-edible sources of lecithin are currently under study, such as the production from algae.40 Furthermore, phosphatidylcholine can be extracted from lecithin trough some easy solvent extractions and purification processes.
000–300000 metric tons produced annually). Importantly, using edible soybean oil or other lecithin-rich edible oils such as sunflower or rapeseed oils for biomedical applications does not represent an ethical issue, since their utilization aligns with sustainability goal.39 At the same time, it has also to be considered that alternative non-edible sources of lecithin are currently under study, such as the production from algae.40 Furthermore, phosphatidylcholine can be extracted from lecithin trough some easy solvent extractions and purification processes.
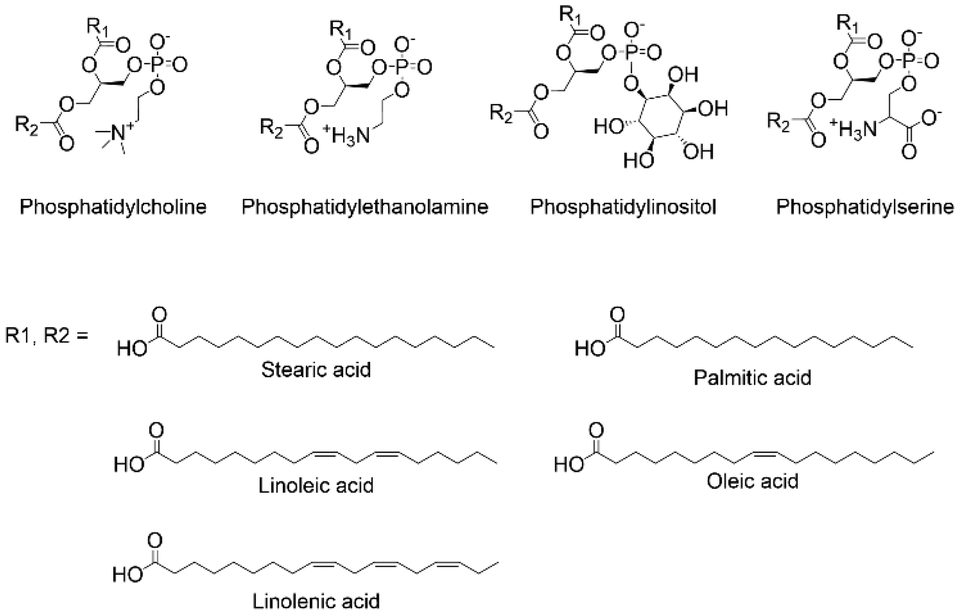 | ||
| Fig. 3 Structures of the phospholipids composing lecithin. | ||
Soy-phosphatidylcholine is composed of 10–16 wt% palmitic acid (16:0), 2–5 wt% stearic acid (18:0), 15–20 wt% oleic acid (18:1), 50–60 wt% linoleic acid (18:2), and 5–10 wt% linolenic acid (18:3), resulting in abundant presence of double bonds (see ESI Section S1† “Materials” for the specific composition of the phosphatidylcholine used in this work). Thus, at least 20 wt% of the fatty acids present unsaturated bonds, meaning that statistically more than 97 wt% of the phospholipids present at least one double bond (see ESI Section S3† for calculations). Based on our expertise in the targeting properties of carbohydrates,41,42 we decided to modify these double bonds accordingly. Nevertheless, reacting an alcohol or aldehyde (i.e., the functional groups of carbohydrates) with double bonds cannot be performed in an environmentally friendly manner. Processes such as hydroalkoxylation, oxymercuration, epoxidation, Wittig reaction, and ozonolysis have substantial environmental drawbacks, which are contrary to our aim of achieving a sustainable process. Therefore, we decided to introduce an additional molecule to serve as a building block for further modification with carbohydrates. Once again, we opted to use a natural compound, selecting cysteine. Cysteine, an amino acid used in different industries, has a global market production of 10000–15000 metric tons per year and its produced mainly by fermentation methods. This method is considered environmentally friendly, since it uses bacteria or yeast to produce high-purity cysteine as a metabolic by-product.43 Cysteine bears a thiol (–SH) group offering a rich platform for functionalization due to its ability to undergo diverse chemical transformations, including oxidation, reduction, and metal coordination. One particularly fascinating reaction that can be performed with thiol is the photochemical thiol–ene reaction, a powerful synthetic methodology that enables rapid and selective bond formation under mild conditions typically employing a photoactive catalyst and UV or visible light irradiation as the driving force for bond formation.44,45 According to the literature, the exploitation of cysteine for thiol–ene reaction is practically unexplored,46–48 and completely new for the formation of ligand for NPs functionalization. After reacting phosphatidylcholine with cysteine, we obtained phospholipids presenting amine functionalities in their fatty acid chains (Fig. 2). Due to the practically complete conversion of cysteine (checked by TLC), and also considering the abundance of double bonds in phosphatidylcholine as previously described, but above all considering that only chemicals bearing phosphate groups anchor to the NPs, we opted to not perform any purification step, diminishing the produced waste (although chromatographic purification was performed merely to characterize the compound, as described in methods in the ESI Section S2 and in the HPLC and MS analysis in ESI Section S4†).
We denoted the phosphatidylcholine-cysteine structure as “Phos-Cys”, and we sequentially modified this compound with D-Mannose. D-Mannose is well-known for its high affinity and specific binding towards cells and pathogens expressing mannose receptors, such as macrophages and dendritic cells and bacteria, making it a potent components of ligand for targeted drug delivery and imaging applications in biomedical research.49–51 Furthermore, D-Mannose is also acquiring more and more interest as a promising target ligand in cancer therapy.52 For example, Gonzalez et al.53 showed that mannose induces growth retardation in various tumour types and increases cell death when combined with major forms of chemotherapy. On our side, recently, we have shown, contrary to what is reported in some works in the literature, that mannose is a better ligand than galactose for targeting hepatocarcinoma cells.54 We covalently attach D-Mannose to the Phos-Cys through reductive amination, drawing inspiration from recent research on modifying the MOF UiO-66-NH2 with D-Mannose for targeted treatment of pulmonary fibrosis.55 However, unlike the reported study, which, despite its significant advancements in biomedicine, did not address the sustainability aspects of the synthesis, we chose to replace the toxic sodium triacetoxyborohydride with H2 and to operate in a continuous flow (c.f.) mode rather than batch processing. We also observed that the reaction can be performed in EtOH instead of using a buffer solution. We opted for flow chemistry since it allows for precise control over critical reaction parameters, such as temperature, pressure, and residence time, which are crucial for achieving optimal reaction conditions and outcomes, enhancing the efficiency and reproducibility of the reaction.56–58 Furthermore, flow chemistry enables scalability, which is vital for producing larger quantities of functionalized compounds while maintaining high quality, and also supports more environmentally friendly conditions by reducing the generation of waste and the consumption of energy, as well as by minimizing the use of hazardous reagents compared to conventional batch methods. After the reductive amination in c.f., we finally obtain the novel phosphatidylcholine-cysteine-mannose ligand, denoted as “Phos-Cys-Man”. D-Mannose was present in the structure of the ligand in the form of 1-amino-1-deoxy-D-mannitol. Again, to reduce the environmental impact of the synthesis, the final ligand was not purified (chromatographic purification was merely performed to characterize the compound by HPLC and MS analysis, as described in the ESI Sections S2 and S4†). This because only the molecules bearing phosphates sequentially anchored onto the NPs, leaving the unreacted D-Mannose (and other impurities) in solution, while the functionalized NPs were separated by centrifugation.
3.2 Characterization of the ligand
The new ligand was fully characterized using mass spectroscopy (MS) analysis, Fourier-transform infrared spectroscopy (FTIR), elemental analysis (CHNS%), X-ray photoelectron spectroscopy (XPS) and NMR (1H, 13C and 31P NMR). Moreover, green metrics were calculated for the synthetic process of the ligand in comparison with a PEG-phosphate one.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1737 cm−1 corresponding to the ester carbonyl groups in the lecithin structure; PO stretching at 1231 cm−1 and P–O–C stretching at 1064 cm−1.59 When we reacted cysteine with phosphatidylcholine forming Phos-Cys, additional peaks appeared in the FTIR spectrum. Cysteine contains a thiol (–SH) observable through a S–H stretching (2566 cm−1). This peak disappeared after the thiol–ene reaction. We assigned the peak around 3200–3400 cm−1 to N–H stretching, while C–N stretching corresponded to the peak around 1217 cm−1, visible both on cysteine and on Phos-Cys.60
O stretching at 1737 cm−1 corresponding to the ester carbonyl groups in the lecithin structure; PO stretching at 1231 cm−1 and P–O–C stretching at 1064 cm−1.59 When we reacted cysteine with phosphatidylcholine forming Phos-Cys, additional peaks appeared in the FTIR spectrum. Cysteine contains a thiol (–SH) observable through a S–H stretching (2566 cm−1). This peak disappeared after the thiol–ene reaction. We assigned the peak around 3200–3400 cm−1 to N–H stretching, while C–N stretching corresponded to the peak around 1217 cm−1, visible both on cysteine and on Phos-Cys.60
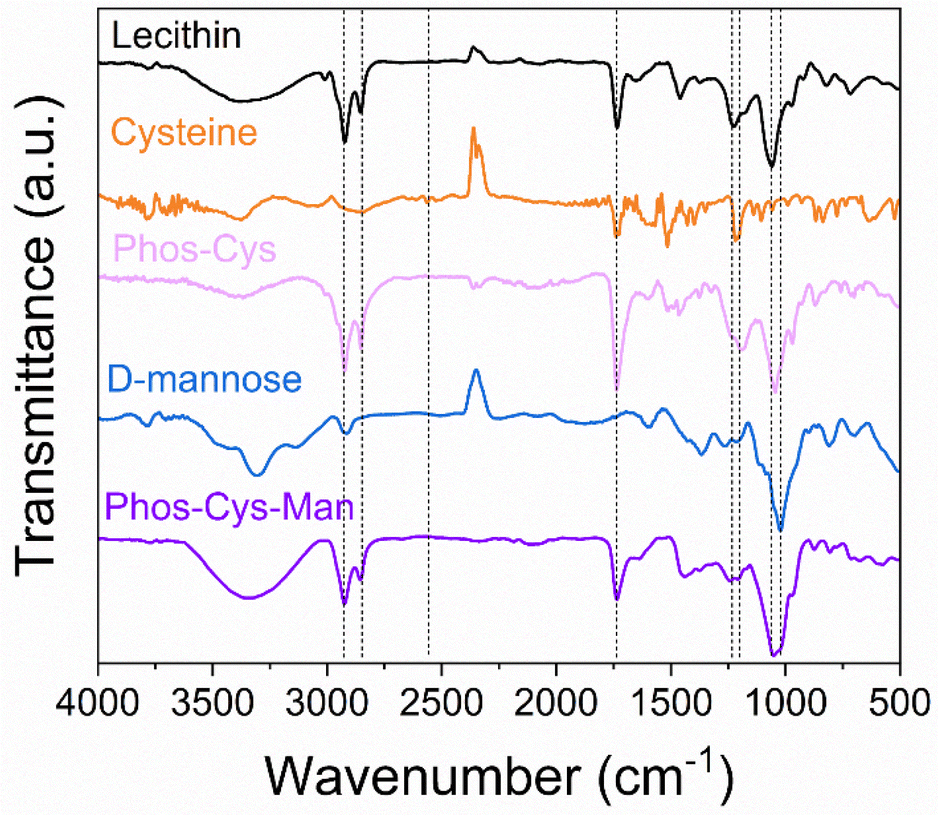 | ||
| Fig. 4 FTIR analysis of phosphatidylcholine, cysteine, Phos-Cys, D-Mannose and Phos-Cys-Man. | ||
By adding D-Mannose to Phos-Cys-Man we further introduced additional hydroxyl groups to the system, resulting in enhanced O–H stretching (around 3400 cm−1) and the addition of a band at 1022 cm−1 corresponding to C–OH bond stretching of D-Mannose.
We identified the peaks of the spectrum of phosphatidylcholine and compared against the main ion assignments reported in the literature.61 We identified specific peaks such as: m/z 279, as linoleic acid; m/z 476 as lysophosphatidylethanolamine 18:2; m/z 518 as lysophosphatidylethanolamine acetylated 18:2; m/z 714 as phosphatidylethanolamine 16:0/18:2; m/z 738 as phosphatidylethanolamine 18:2/18:2 and/or 18:1/18:3; and m/z 833 as phosphatidylcholine, the main component (with MW 788 g mol−1 as a result of subtracting the 45 units of formate ion from m/z 833).
The spectrum of Phos-Cys, showed a peak at m/z 883. We interpreted this peak as the sum of the major lecithin peak (m/z 833) and an additional 49 mass units corresponding to the cysteine moiety. Cysteine has a molecular weight of 121 g mol−1, but the typical fragment of cysteine has a m/z of 75, leaving a residue of m/z 46. As a conclusion, the presence of the peak of m/z 883 was explained by the conjugation of cysteine to lecithin.
We then observed that the spectrum of Phos-Cys-Man featured a peak at m/z 829, corresponding to the presence of phosphatidylcholine. We noticed also a peak at m/z 926 demonstrating the integration of the mannose moiety, as the difference between this value and m/z 883 (m/z of Phos-Cys) was 43 mass units. This was the residual mass of the amino-1-deoxy-D-mannitol, i.e., the D-Mannose (with a typical fragment of 74 units) reacted with cysteine, summed with the residual mass of the cysteine after losing the formate ion (m/z 45). The spectra and schemes for the fragmentation of the Phos-Cys and Phos-Cys-Man are reported in the ESI Section S4.†
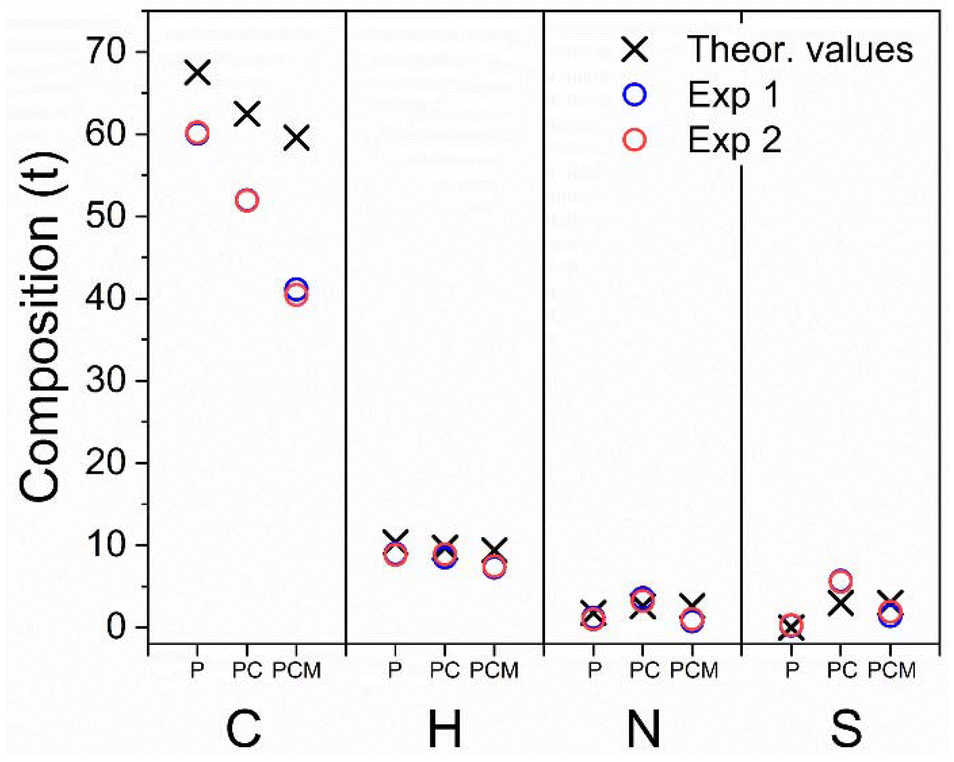 | ||
| Fig. 5 Graphical representation of CHNS% elemental analysis of: phosphatidylcoline (“P”); Phos-Cys (“PC”) and Phos-Cys-Man (“PCM”). | ||
We observed experimental values of N, S and H close to the theoretical expectations for all the three compounds, indicating a reasonably accurate synthesis (values are reported in the ESI, Section S5†). We found some discrepancies in the amount of C, due to experimental errors or the presence of impurities.
We also observed insignificant differences with the other elements due to experimental conditions, minor deviations in the synthesis process, or the presence of traces of impurities, as confirmed by the presence of a small amount of sulfur in phosphatidylcholine, which theoretically should not be observed.
CH– 128 and 129 ppm). We also observed changes in the positions of the peaks in the 31P NMR spectrum (Fig. S9†) consistent with this observation.
By 13C NMR data we also confirmed the successful incorporation of D-Mannose to the Phos-Cys-Man, since six new peaks corresponding to the carbohydrate appear on the spectrum (see ESI Fig. S10,†δ from 63.4 to 96.3 ppm), as well as the previously observed peaks ascribed to cysteine (three singlets centred on δ 28.1, 55.6 and 170.7 ppm) and original lecithin. We noticed that, analogously to the Phos-Cys, the slightly shifts in the 31P signals observed in the 31P NMR spectra (Fig. S11†) were compatible with the formation of the Phos-Cys-Man.
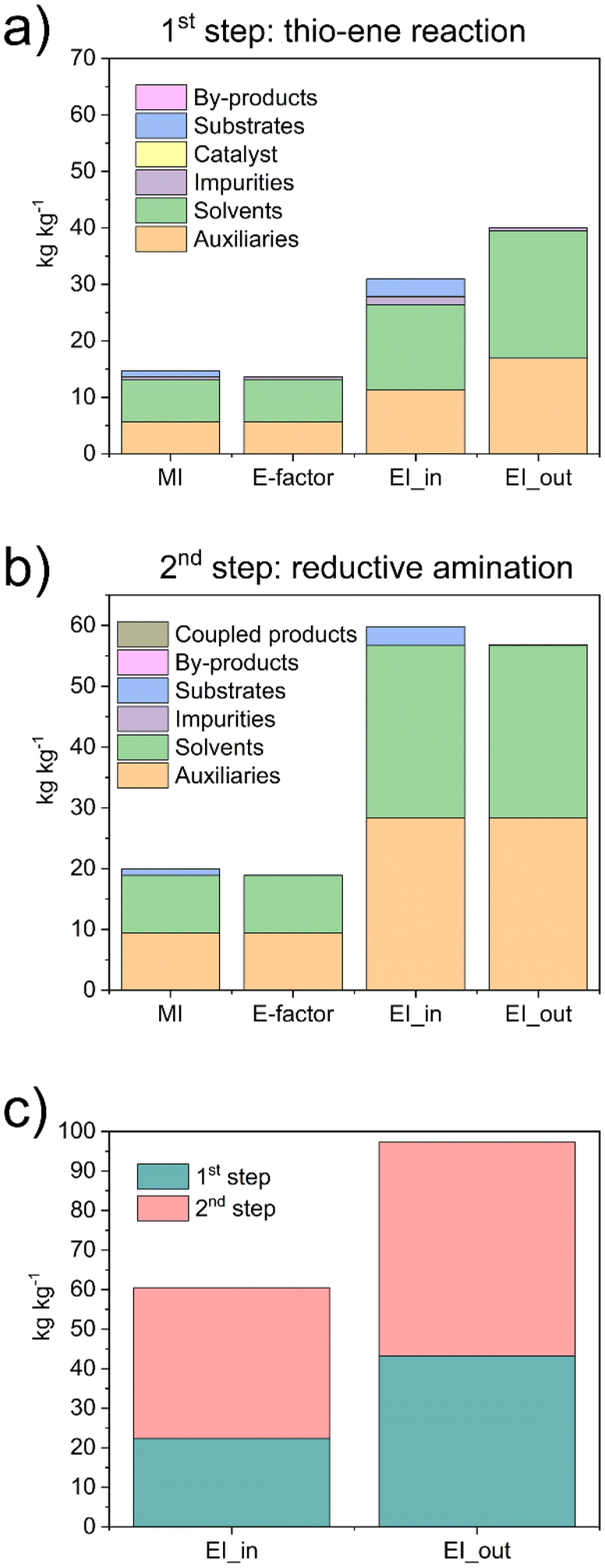 | ||
| Fig. 6 Green metrics calculated by EATOS software for the synthesis of: (a) Phos-Cys from phosphatidylcholine and (b) Phos-Cyst-Man from Phos-Cys. (c) Comparison of the EI_in and EI_out of the two synthetic steps. | ||
We observed that the main contributors to the metrics were the solvents and auxiliaries, i.e., the solvent used to clean the reactors. We observed E-factors of 14.7 and 19.9 for the first and second steps, respectively, with MI values being similarly close. Regarding the EI_in values, we observed a substantial impact from solvents and auxiliaries. In the first step, 10% of the contribution was due to the substrates and approximately 5% to impurities, resulting in a total of 31 Potential Environmental Impact (PEI) per kg. In the second step, more than 95% of the contribution was due to the solvents and auxiliaries, totalling 59 PEI per kg. Similar results were observed with the EI_out, with values of 40 and 57 PEI per kg for the first and second steps, respectively. We also compared the environmental loads associated with the two steps, reporting both EI_in and EI_out parameters (Fig. 6c). Although the values were similar, we noticed that the main impact was due to step 2. The results highlighted that the synthetic protocol has significant potential to be easily upgraded to an even greener process. Indeed, by reducing the volume of ethanol used as an auxiliary to clean the reactors, the different metrics can be substantially reduced.
Overall, the values of the metrics were relatively low. We also highlight the main green characteristics of our procedure: (I) it uses only natural products as reagents, i.e., phosphatidylcholine, cysteine, and D-Mannose; (II) it uses only ethanol, considered a green solvent, for both reactions and for cleaning the reactors; (III) it avoids the use of hydrogen precursors by producing H2in situ; (IV) it is based on efficient synthetic methods, including a “click” reaction and a reaction in c.f.
We also performed a calculation of the RME (%), obtaining a values of ca. 95% for both Phos-Cys, and for Phos-Cys-Man (considering as separate steps), with an overall RME (%) of the process of 79%.
As a comparison, we calculated the E-factor and the RME (%) for the synthesis of a PEG-phosphate ligand.27 It is important to note that the reported procedure did not specify the amount of water used for dialysis, a crucial step in the process, neither the amount of auxiliaries used to clean the reactors, etc. To perform the calculations, we thus estimated the volume of water for dialysis to the minimum amount used for similar quantity of materials based on our experience (3 L), but we did not include any solvents for the cleaning up of the reactor to not overestimate the green metrics. Thus, we calculated the E-factor as 17.7 and the RME as 77%. Despite the values were close to our ligand, it is important to highlight that the PEG-phosphate was not functionalized with targeting molecules. Furthermore, the synthesis of the PEG ligand involves the use of high volume (3 eq.) of toxic phosphoric acid.
3.3 Functionalization of NPs
We thus functionalized various NPs with the novel ligand to demonstrate its broad versatility and improved functionality. To this end, we selected diverse MOFs and an iron-based NPs that have been largely utilized in nanomedicine research, including PCN-222, UiO-66, MIL-101(Fe), and akageneite NPs. PCN-222 is a zirconium-based MOF (Zr4+) characterized by large pores and porphyrin-based linkers. It is used in photodynamic therapy and can also be used for imaging and drug delivery, such as doxorubicin to cancer cells.27 UiO-66 is a zirconium-based MOF (Zr4+) known for its high thermal and chemical stability. Its structure consists of Zr6 clusters connected by terephthalate linkers and is used as a platform for biomolecule immobilization and for drug delivery, such as in the case of cisplatin to tumour tissues.62 MIL-101(Fe) is an iron-based MOF featuring mesoporous cages and a high surface area. Its structure is composed of Fe3+ complexes linked by terephthalate ligands, and is suitable for drug encapsulation and work as catalyst for biomedical applications, for example for promoting reactive oxygen species (ROS) evolution. Iron NPs are widely utilized for magnetic resonance imaging (MRI), drug delivery hyperthermia treatment, and as contrast agents.63 Among the different types of Fe-NPs we specifically selected akageneite nanorods (Fe3+), due to their simple preparation. Furthermore, akageneite nanorods are currently employed in the commercially available iron deficiency drug Ferinject® (Vifor, Switzerland).We prepared UiO-66 and MIL-101(Fe) following previously reported methods, while we synthesized PCN-222 and akageneite by optimizing reported procedures (synthetic protocols are reported in the ESI, Section S2†). Specifically, we prepared PCN-222 based on our previous efforts to produce this MOF in controlled nanometric size using green solvents, substituting toxic DMF with DMSO. Furthermore, we found that the preparation of the Zr6 nodes could be optimized to improve the final yield. Unlike our previous work and literature referring to the preparation of Zr6 nodes for PCN-222 and similar MOFs,27,32,64 we carried out the synthesis under an argon atmosphere. The absence of oxygen prevented the formation of ZrO nanoparticles, which are inevitable when Zr(OBu)4, i.e., the commonly used Zr-precursor, is heated under ambient atmosphere.65 Indeed, the oldest references for Zr6-clusters (but not referring to PCN-222 synthesis) described the preparation under an argon atmosphere,66 but this was subsequently unreported,64 resulting in a form of error propagation.27 We also observed that 1-propanol can be substituted with 2-propanol in the preparation of the clusters. We preferred isopropanol since it is more sustainable as it can be produced by microbial biosynthesis.67 As a result of our optimization, we were able to increase the yield of PCN-222 from ca. 29%32 to 73% (after purification).
We also prepared akaganeite Fe-NPs by slightly modifying the synthetic procedure reported in the literature. Specifically, standard synthesis use NaOH or HCl and/or purification by centrifugation. Our approach involved using only the Fe precursor and acetic acid, employing a simple purification by filtration. This latter time- and waste-saving procedure, compared to centrifugation, was made possible thanks to DLS analysis. Indeed, we observed that the particles agglomerated in water, allowing for filtration, while after redispersion in EtOH, the particles were monodispersed.
We thus functionalized the different NPs with the novel Phos-Cys-Man ligand, exploiting the strong binding affinity of the phosphate group to the metal ions, which are abundant on the surfaces of MOFs and Fe-NPs. More in details, we firstly dispersed the NPs in EtOH, followed by the addition of an aqueous solution of the ligand. The mixture was thus heated under stirring at 60 °C for 24 h followed by purification by centrifugation and washing with water and EtOH, and final redispersion in MeOH. Additional details regarding the functionalization of the NPs can be found in the ESI Section S2.†
3.4 Characterization of functionalized nanostructures
We characterized the ligand-NPs structures using various techniques including FTIR, Raman spectroscopy, DLS, ζ-potential, ICP-MS, SEM images and X-Ray photoelectron spectroscopy (XPS).O stretching vibrations, indicating the coordination between –COOH and Zr atoms. Additionally, we associated the peak near 1400–1600 cm−1 with CC stretching vibrations from aromatic rings, and the band in the range of 500–700 cm to the asymmetric vibrations of the CN, NH, CC and C–H of the pyrrole ring in tetrakis (4-carboxyphenyl) porphyrin (TCPP).32
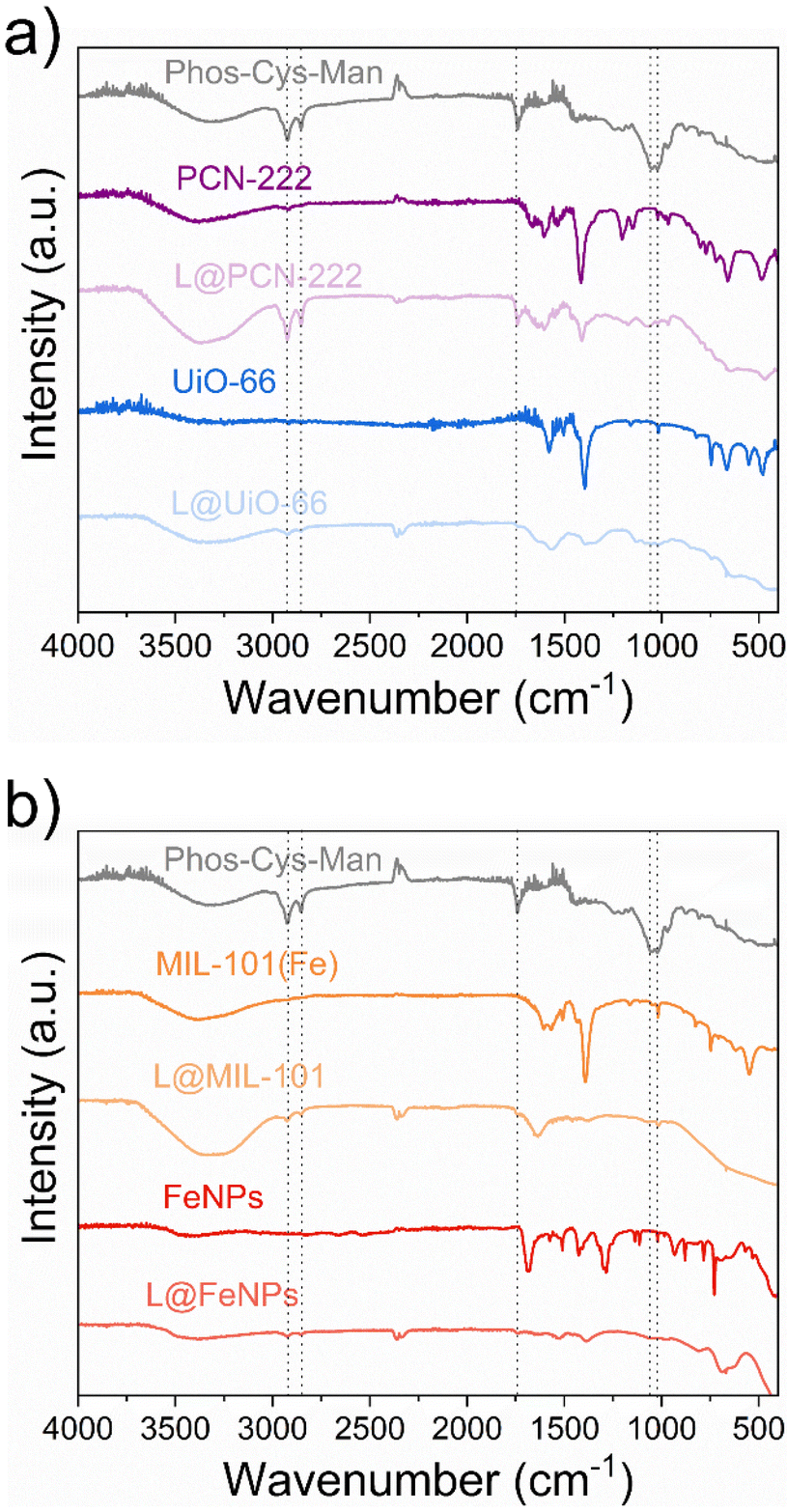 | ||
| Fig. 7 FTIR analysis of the pristine and functionalized (“L@NPs”) nanoparticles: (a) PCN-222 and UiO-66; (b) MIL-101(Fe) and FeNPs. | ||
The FTIR spectrum of UiO-66 (Fig. 7a) also showed distinct characteristic bands. The broad band around 3300–3500 cm−1 indicated O–H stretching vibrations, while the sharp peak at 1650–1700 cm−1 was due to CO stretching vibrations. We also observed two intense bands around 1600 and 1400 cm−1, associated to the OCO asymmetric and symmetric stretch in the carboxylate group in terephthalic acid, respectively Additionally, we confirmed the presence of Zr–O stretching vibrations by the bands around 550 cm−1, while we observed the C–H bending, CC stretch, OH bend and OCO bend in terephthalic acid in the range of 700–900 cm−1.68
Iron nanoparticles demonstrated characteristic FTIR absorption bands as well (Fig. 7b). We observed a broad band around 3200–3500 cm−1 indicating O–H stretching vibrations. We attributed the peaks around 550–600 cm−1 to Fe–O stretching vibrations. Moreover, we assigned the peak near 1650 cm−1 to presence of surface-adsorbed organic molecules due to CO stretching vibrations.69
Lastly, we observed a broad band around 3300–3500 cm−1 indicating O–H stretching vibrations in the FTIR spectrum of MIL-101(Fe) (Fig. 7b). We assigned the strong peak at approximately 1650–1700 cm−1 to CO stretching vibrations, while we observe the Fe–O stretching vibrations bands around 500–600 cm−1, and C–H bending vibrations in the range of 700–900 cm−1.70
We thus confirmed the loading of the ligands by the presence of the characteristic peaks previously described in Section 3.2.2. In particular, we could observe the peaks for the asymmetric and symmetric stretching vibrations of the –CH2− at ca. 2700 cm−1 and 2900 cm−1 and we attributed the peak at 1739 cm−1 to the stretching vibrations of the CO group. Moreover, we attributed the peak at 1064 cm−1 to P–O–C stretching while peak at 1022 cm−1 to the C–OH bond stretching of D-Mannose.
We further analysed the functionalization of NPs with Phos-Cys-Man by Raman spectroscopy (see ESI Section S8†). For MIL-101(Fe) we observed the vibrational bands at 874, 1140, 1303, 1522 and 1618 cm−1 indicating aromatic and dicarboxylate groups in the terephthalate ligands. We also observed C–S stretching vibrations of Phos-Cys-Man between 613 and 668 cm−1. We didn't observe other relevant peaks, due to the abundance of terephthalate respect to Phos-Cys-Man.
As shown in Table 1, we did not observe aggregation after the NP functionalization with the Phos-Cys-Man ligand, while we observed that the hydrodynamic diameter (HD) of increased slightly. By 3D modelling of the structure of the ligand, we noticed that the maximum length of the ligand would be 4–5 nm, consequently causing the HD to theoretically increase by a maximum of 10 nm in the functionalized NPs. Interestingly, all the functionalized NPs could be dispersed in both methanol and water, the latter being especially relevant for Fe NPs. However, the values of the standard deviation did not allow us to make general conclusions on the effect of the ligand on the HD, although the increase was indicative of the ligand being placed on the surfaces of the particles. We thus measured the ζ potentials, observing that the pristine NPs showed positive or neutral values, whereas when the ligand was loaded, the charges decreased in all the samples except FeNPs. The positive values of the pristine NPs indicated that the negatively charged phosphates of the ligand could anchor onto them. Conversely, the presence of the ligand decreased the charge of the particles since mannose is negatively charged at pH 7. The variation of the charge was more evident with PCN-222 and UiO-66, but less so in MIL-101(Fe) and practically absent in FeNPs. This was due to the stronger charge of iron ions. Indeed, iron ions, particularly in the form of Fe3+, can dominate the surface charge characteristics due to their high charge density. This could lead to a situation where the functionalization does not result in a noticeable change in ζ potential, as the existing charge from the iron ions overshadows the contributions from the newly introduced ligands.71,72
| DLS | ζ potentials | ||
|---|---|---|---|
| MeOH | H2O | H2O | |
| PCN-222 | 155 ± 52 | 161 ± 39 | +3.19 ± 0.7 |
| UiO-66 | 1131 ± 64 | 1129 ± 146 | +1.1 ± 1.7 |
| MIL-101(Fe) | 518 ± 112 | 583 ± 105 | +23.1 ± 0.2 |
| FeNPs | 244 ± 78 | Agglomerates | +0.9 ± 0.6 |
| L@PCN-222 | 167 ± 63 | 175 ± 45 | −7.7 ± 1.6 |
| L@UiO-66 | 1198 ± 133 | 1191 ± 193 | −14.7 ± 1.1 |
| L@MIL-101(Fe) | 540 ± 55 | 530 ± 133 | +16.2 ± 1.1 |
| L@FeNPs | 287 ± 91 | 299 ± 101 | −0.2 ± 0.5 |
 | ||
| Fig. 8 SEM images of the pristine and functionalized (“L@NPs”). Scale bar (white bar) is valid for all the figures and corresponds to 1 μm. | ||
| Element (wt%) | |||
|---|---|---|---|
| S | Zr | Fe | |
| PCN-222 | — | 19.93 | — |
| UiO-66 | — | 32.55 | — |
| FeNPs | — | — | 10.14 |
| MIL101(Fe) | — | — | 18.09 |
| L@PCN-222 | 1.08 | 17.00 | — |
| L@UiO-66 | 0.99 | 35.35 | — |
| L@FeNPs | 0.83 | — | 14.56 |
| L@MIL101(Fe) | 1.12 | — | 21.53 |
We observed the presence of sulfur exclusively in the functionalized nanostructures, and not in the pristine forms, validating the ligand attachment process. This was a crucial finding as it not only demonstrated the successful functionalization but also allowed us for quantification of ligand binding. In details, we calculated the amount of anchored ligand based on the MW of the Phos-Cys-Man (MW 1068 g mol−1), obtaining similar values for all the nanostructures: L@PCN-222: 36%; L@UiO-66: 33%; L@FeNPs: 27.67%; L@MIL101(Fe): 37.38%.
4. Conclusions
We have successfully developed and characterized a novel green Phos-Cys-Man ligand for nanoparticles functionalization. This ligand, leveraging the natural properties of phosphatidylcholine merged with cysteine and D-Mannose, demonstrated exceptional versatility for advanced biomedical applications. In particular, the presence of a specific carbohydrate on the external part of the ligand allows for potential active targeting to different receptors. This characteristic can be exploited by substituting D-Mannose with numerous other carbohydrates or small molecules, such as N-acetylgalactosamine for the targeting of overexpressed asialoglycoprotein receptors in hepatic cancer,32 or carbohydrate-based NK1R antagonists for the treatment of different types of cancer.42 The streamlined synthetic route for the preparation of the ligand not only facilitates its practical application but also enhances its scalability, making it feasible for large-scale production and industrial use. Simplified synthesis processes often translate to cost-effectiveness and reduced resource consumption, which are essential considerations in both research and commercial settings.The ligand showed a strong affinity for metal ions, enabling effective functionalization of various nanostructures including the MOFs MIL-101(Fe), PCN-222, UiO-66, and iron nanoparticles (herein in the form of akaganeite). The functionalization process proved to be straightforward and efficient, utilizing simple synthetic routes and environmentally friendly solvents, thereby ensuring scalability and sustainability. Notably, the new synthetic method avoids the use of toxic reagents, aligning with green chemistry principles.
Our findings highlight the potential of the Phos-Cys-Man ligand for broad applications in nanomedicine, including drug delivery, imaging, and diagnostics but might also find other applications such as catalysis, agriculture, electronics, electrical, energy, environmental and sustainable technology, etc.76
Author contributions
Conceptualization, C. M. C., A. Z., N. K.; methodology, C. M. C., A. Z., N. K.; validation, A. Z., N. K.; formal analysis, C. M. C., A. E., A. Z., N. K., J. P. H; investigation, C. M. C., V. R., A. E., A. Z., N. K.; resources, N. K.; data curation, C. M. C., A. E., A. Z., N. K., J. P. H.; writing—original draft preparation, C. M. C., A. Z.; writing—review and editing, C. M. C., V. R., A. E., J. P. H., A. Z., N. K.; supervision, A. Z., N. K.; funding acquisition, N. K. All authors have read and agreed to the published version of the manuscript.Data availability
All data needed to evaluate this work are available in the main manuscript and/or the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Andalusian Ministry of Economy, Science and Innovation in the framework of the Plan Andaluz de Investigación, Desarrollo e Innovación (PAIDI 2020, programme 54ª “Investigación científica e innovación”, “POSTDOC_21_00594”), the Spanish Ministry of Science and Innovation (Ref: PID2020-119949RB-I00), the Andalusian Ministry of Economy, Science and Innovation cofinanced by the European Regional Development Fund (ERDF) from FEDER and the European Social Fund (ESF) (PY20_00882 and CV20-04221). The COST action CA-18132 “Functional Glyconanomaterials for the Development of Diagnostic and Targeted Therapeutic Probe” is also acknowledged. The COST action CA-22147 “European metal–organic framework network: combining research and development to promote technological solutions” (EU4MOFs) is also acknowledged.References
- S. Hejmady, R. Pradhan, A. Alexander, M. Agrawal, G. Singhvi, B. Gorain, S. Tiwari, P. Kesharwani and S. K. Dubey, Drug Discovery Today, 2020, 25, 2227–2244 Search PubMed
.
- S. Malik, K. Muhammad and Y. Waheed, Molecules, 2023, 28, 661 Search PubMed
- M. Chehelgerdi, M. Chehelgerdi, O. Q. B. Allela, R. D. C. Pecho, N. Jayasankar, D. P. Rao, T. Thamaraikani, M. Vasanthan, P. Viktor, N. Lakshmaiya, M. J. Saadh, A. Amajd, M. A. Abo-Zaid, R. Y. Castillo-Acobo, A. H. Ismail, A. H. Amin and R. Akhavan-Sigari, Mol. Cancer, 2023, 22, 169 Search PubMed
- H. Y. Yang, Y. Li and D. S. Lee, Adv. NanoBiomed Res., 2021, 1, 2000043 Search PubMed
- S. G. Arkhipov, P. S. Sherin, A. S. Kiryutin, V. A. Lazarenko and C. Tantardini, CrystEngComm, 2019, 21, 5392–5401 Search PubMed
- A. Y. Fedorov, T. N. Drebushchak and C. Tantardini, Comput. Theor. Chem., 2019, 1157, 47–53 Search PubMed
- C. Tantardini, S. G. Arkhipov, K. A. Cherkashina, A. S. Kil'Met'Ev and E. V. Boldyreva, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 1856–1859 Search PubMed
- A. Juan, F. J. Cimas, I. Bravo, A. Pandiella, A. Ocaña and C. Alonso-Moreno, Pharmaceutics, 2020, 12, 802 Search PubMed
- S. Yan, J. Na, X. Liu and P. Wu, Pharmaceutics, 2024, 16, 248 Search PubMed
- S. T. Hong, H. Lin, C. S. Wang, C. H. Chang, A. M. Y. Lin, J. C. H. Yang and Y. L. Lo, J. Nanobiotechnol., 2019, 17, 89 Search PubMed
- N. Muhamad, T. Plengsuriyakarn and K. Na-Bangchang, Int. J. Nanomed., 2018, 13, 3921–3935 Search PubMed
- M. M. Agwa, H. Elmotasem, H. Elsayed, A. S. Abdelsattar, A. M. Omer, D. T. Gebreel, M. S. Mohy-Eldin and M. M. G. Fouda, Int. J. Biol. Macromol., 2023, 239, 124294 Search PubMed
- K. Elumalai, S. Srinivasan and A. Shanmugam, Biomed. Technol., 2024, 5, 109–122 Search PubMed
- S. Puri, M. Mazza, G. Roy, R. M. England, L. Zhou, S. Nourian and J. Anand Subramony, Adv. Drug Delivery Rev., 2023, 200, 114962 Search PubMed
- L. Wang, J. C. Evans, L. Ahmed and C. Allen, Sci. Rep., 2023, 13, 3226 Search PubMed
- M. A. Rahim, N. Jan, S. Khan, H. Shah, A. Madni, A. Khan, A. Jabar, S. Khan, A. Elhissi, Z. Hussain, H. C. Aziz, M. Sohail, M. Khan and H. E. Thu, Cancers, 2021, 13, 670 Search PubMed
- Q. Tang, B. Yu, L. Gao, H. Cong, N. Song and C. Lu, Curr. Med. Chem., 2018, 25, 1837–1866 Search PubMed
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–104 Search PubMed
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 Search PubMed
- A. Bandyopadhyay, T. Das, S. Nandy, S. Sahib, S. Preetam, A. V. Gopalakrishnan and A. Dey, Naunyn-Schmiedeberg's Arch. Pharmacol., 2023, 396, 2417–3441 Search PubMed
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233–1246 Search PubMed
- Q. Xia, Y. Zhang, Z. Li, X. Hou and N. Feng, Acta Pharm. Sin. B, 2019, 9, 675–689 Search PubMed
- Z. Luo, Y. Dai and H. Gao, Acta Pharm. Sin. B, 2019, 9, 1099–1112 Search PubMed
- M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J. P. Habas and J. J. Robin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2434–2444 Search PubMed
- J. M. Orduña, N. del Río and M. J. Pérez-Pérez, Org. Biomol. Chem., 2023, 21, 5457–5468 Search PubMed
- M. Eissen and J. O. Metzger, Chem. – Eur. J., 2002, 8, 3580–3585 Search PubMed
- X. Chen, Y. Zhuang, N. Rampal, R. Hewitt, G. Divitini, C. A. O'Keefe, X. Liu, D. J. Whitaker, J. W. Wills, R. Jugdaohsingh, J. J. Powell, H. Yu, C. P. Grey, O. A. Scherman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2021, 143, 13557–13572 Search PubMed
- A. Zuliani, S. Chen and R. Giorgi, Appl. Mater. Today, 2023, 30, 101716 Search PubMed
- L. He, Y. Liu, J. Lau, W. Fan, Q. Li, C. Zhang, P. Huang and X. Chen, Nanomedicine, 2019, 14, 1343–1365 Search PubMed
- Z. Sun, T. Li, T. Mei, Y. Liu, K. Wu, W. Le and Y. Hu, J. Mater. Chem. B, 2023, 11, 3273–3294 Search PubMed
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 Search PubMed
- A. Zuliani, M. Carmen Castillejos and N. Khiar, Green Chem., 2023, 25, 10569–10610 Search PubMed
- R. Jastrzab, M. Nowak, M. Zabiszak, A. Odani and M. T. Kaczmarek, Coord. Chem. Rev., 2021, 435, 213810 Search PubMed
- M. J. Alhajj, N. Montero, C. J. Yarce and C. H. Salamanca, Cosmetics, 2020, 7, 87 Search PubMed
- F. Bot, D. Cossuta and J. A. O'Mahony, Trends Food Sci. Technol., 2021, 111, 261–270 Search PubMed
- F. Zhao, R. Li, Y. Liu and H. Chen, Front. Nutr., 2023, 9, 1082671 Search PubMed
- L. C. Chen, Y. C. Chen, C. Y. Su, W. P. Wong, M. T. Sheu and H. O. Ho, Sci. Rep., 2016, 6, 37122 Search PubMed
- L. Wang, C. Lai, D. Li, Z. Luo, L. Liu, Y. Jiang and L. Li, Antioxidants, 2022, 11, 1666 Search PubMed
- T. E. Clemente and E. B. Cahoon, Plant Physiol., 2009, 151, 1030–1040 Search PubMed
- I. M. Maliki, M. Misson, P. L. Teoh, K. F. Rodrigues and W. T. L. Yong, Mar. Drugs, 2022, 20, 102 Search PubMed
- E. Romero-Ben, M. C. Castillejos, C. Rosales-Barrios, M. Expósito, P. Ruda, P. M. Castillo, S. Nardecchia, J. de Vicente and N. Khiar, J. Mater. Chem. B, 2023, 11, 10189–10205 Search PubMed
- R. Recio, P. Lerena, E. Pozo, J. M. Calderón-Montaño, E. Burgos-Morón, M. López-Lázaro, V. Valdivia, M. Pernia Leal, B. Mouillac, J. Á. Organero, N. Khiar and I. Fernández, J. Med. Chem., 2021, 64, 10350–10370 Search PubMed
- W. Li, Z. Zhou and D. Wang, Fermentation, 2023, 9, 802 Search PubMed
- H. Choi, M. Kim, J. Jang and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22514–22522 Search PubMed
- Y. Liu, W. Hou, H. Sun, C. Cui, L. Zhang, Y. Jiang, Y. Wu, Y. Wang, J. Li, B. S. Sumerlin, Q. Liu and W. Tan, Chem. Sci., 2017, 8, 6182–6187 Search PubMed
- M. D. Nolan and E. M. Scanlan, Front. Chem., 2020, 8, 583272 Search PubMed
- X. Wang, H. Liang, J. Jiang, Q. Wang, Y. Luo, P. Feng and C. Zhang, Green Chem., 2020, 22, 5722–5729 Search PubMed
- M. Kuhlmann, O. Reimann, C. P. R. Hackenberger and J. Groll, Macromol. Rapid Commun., 2015, 36, 472–476 Search PubMed
- A. Sharma, J. E. Porterfield, E. Smith, R. Sharma, S. Kannan and R. M. Kannan, J. Controlled Release, 2018, 283, 175–189 Search PubMed
- F. Huo, Y. Zhang, Y. Li, H. Bu, Y. Zhang, W. Li, Y. Guo, L. Wang, R. Jia, T. Huang, W. Zhang, P. Li, L. Ding and C. Yan, Chem. – Asian J., 2022, 17, e202200342 Search PubMed
- J. J. Cid, M. Assali, E. Fernández-García, V. Valdivia, E. M. Sánchez-Fernández, J. M. Garcia Fernández, R. E. Wellinger, I. Fernández and N. Khiar, J. Mater. Chem. B, 2016, 4, 2028–2037 Search PubMed
- M. Paurević, M. Šrajer Gajdošik and R. Ribić, Int. J. Mol. Sci., 2024, 25, 1370 Search PubMed
- P. S. Gonzalez, J. O'Prey, S. Cardaci, V. J. A. Barthet, J. ichi Sakamaki, F. Beaumatin, A. Roseweir, D. M. Gay, G. Mackay, G. Malviya, E. Kania, S. Ritchie, A. D. Baudot, B. Zunino, A. Mrowinska, C. Nixon, D. Ennis, A. Hoyle, D. Millan, I. A. McNeish, O. J. Sansom, J. Edwards and K. M. Ryan, Nature, 2018, 563, 719–723 Search PubMed
- M. Negrete, E. Romero-Ben, A. Gutiérrez-Valencia, C. Rosales-Barrios, E. Alés, T. Mena-Barragán, J. A. Flores, M. C. Castillejos, P. De La Cruz-Ojeda, E. Navarro-Villarán, C. Cepeda-Franco, N. Khiar and J. Muntané, ACS Appl. Bio Mater., 2021, 4, 4789–4799 Search PubMed
- J. Cui, C. Zhang, H. Liu, L. Yang, X. Liu, J. Zhang, Y. Zhou, J. Zhang and X. Yan, ACS Appl. Mater. Interfaces, 2023, 15, 11520–11535 Search PubMed
- C. M. Cova, A. Zuliani, R. Manno, V. Sebastian and R. Luque, Green Chem., 2020, 22, 1414–1423 Search PubMed
- A. Zuliani, C. M. Cova, R. Manno, V. Sebastian, A. A. Romero and R. Luque, Green Chem., 2020, 22, 379–387 Search PubMed
- V. Hessel, S. Mukherjee, S. Mitra, A. Goswami, N. Nghiep Tran, F. Ferlin, L. Vaccaro, F. Malekpour Galogahi, N. T. Nguyen and M. Escribà-Gelonch, Green Chem., 2024, 26, 9503–9528 Search PubMed
- S. Setiadi and N. Hidayah, Int. J. Technol., 2018, 9, 380–389 Search PubMed
- S. Devi, B. Singh, A. K. Paul and S. Tyagi, Anal. Methods, 2016, 8, 4398–4405 Search PubMed
- G. D. Fernandes, R. M. Alberici, G. G. Pereira, E. C. Cabral, M. N. Eberlin and D. Barrera-Arellano, Food Chem., 2012, 135, 1855–1860 Search PubMed
- S. X. Lin, W. L. Pan, R. J. Niu, Y. Liu, J. X. Chen, W. H. Zhang, J. P. Lang and D. J. Young, Dalton Trans., 2019, 48, 5308–5314 Search PubMed
- R. K. Chandunika, V. Rajagopalan and N. K. Sahu, IET Nanobiotechnol., 2020, 14, 823–829 Search PubMed
- H. Noh, C. W. Kung, T. Islamoglu, A. W. Peters, Y. Liao, P. Li, S. J. Garibay, X. Zhang, M. R. Destefano, J. T. Hupp and O. K. Farha, Chem. Mater., 2018, 30, 2193–2197 Search PubMed
- H. Kumazawa, Y. Hori and E. Sada, Chem. Eng. J., 1993, 51, 129–133 Search PubMed
- G. Kickelbick, P. Wiede and U. Schubert, Inorg. Chim. Acta, 1999, 284, 1–7 Search PubMed
- H. Thi Quynh Le and E. Y. Lee, Bioresour. Technol., 2022, 362, 127835 Search PubMed
- Y. Han, M. Liu, K. Li, Y. Zuo, Y. Wei, S. Xu, G. Zhang, C. Song, Z. Zhang and X. Guo, CrystEngComm, 2015, 17, 6434–6440 Search PubMed
- P. Ramalingam, P. Priyadharsini, G. Chandrasekaran, R. Parameshwari and P. Priyadarshini, Am. J. Mater. Sci., 2011, 1, 18–25 Search PubMed
- A. Pangestu, W. W. Lestari, F. R. Wibowo and L. Larasati, J. Inorg. Organomet. Polym. Mater., 2022, 32, 1828–1839 Search PubMed
- J. J. Carlson and S. K. Kawatra, Miner. Process. Extr. Metall., 2013, 34, 269–303 Search PubMed
- X. Meng, J. Ryu, B. Kim and S. Ko, Clin. Nutr. Res., 2016, 5, 172–179 Search PubMed
- A. T. Kozakov, A. G. Kochur, K. A. Googlev, A. V. Nikolskii, V. I. Torgashev, V. G. Trotsenko and A. A. Bush, J. Alloys Compd., 2015, 647, 947–955 Search PubMed
- A. T. Kozakov, A. G. Kochur, K. A. Googlev, A. V. Nikolskii, I. P. Raevski, V. G. Smotrakov and V. V. Yeremkin, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 16–23 Search PubMed
- A. G. Kochur, T. M. Ivanova, A. V. Shchukarev, R. V. Linko, A. A. Sidorov, M. A. Kiskin, V. M. Novotortsev and I. L. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2010, 180, 21–26 Search PubMed
- K. Bhattacharjee and B. L. V. Prasad, Chem. Soc. Rev., 2023, 52, 2573–2595 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials and methods; calculation of the probability of double bonds in phosphatidylcholine; details of the MS, the elemental (CHNS%) and the NMR analysis; green metric values; additional data regarding the DLS analysis, SEM mapping. See DOI: https://doi.org/10.1039/d4gc03743j |
| This journal is © The Royal Society of Chemistry 2024 |