
Robust biocatalyst for the green continuous flow synthesis of esters from biomass-derived furfuryl alcohol and C8–C18 carboxylic acids†
Anna
Wolny
 a,
Dagmara
Więcławik
a,
Jakub
Zdarta
b,
Sebastian
Jurczyk
c,
Teofil
Jesionowski
b and
Anna
Chrobok
*a
a,
Dagmara
Więcławik
a,
Jakub
Zdarta
b,
Sebastian
Jurczyk
c,
Teofil
Jesionowski
b and
Anna
Chrobok
*a
aDepartment of Chemical Organic Technology and Petrochemistry, Faculty of Chemistry, Silesian University of Technology, Krzywoustego 4, PL-44100 Gliwice, Poland. E-mail: anna.chrobok@polsl.pl
bInstitute of Chemical Technology and Engineering, Faculty of Chemical Technology, Poznan University of Technology, Berdychowo 4, PL-60965 Poznan, Poland
cInstitute for Engineering of Polymer Materials and Dyes, Lukasiewicz Research Network, Sklodowskiej-Curie 55, PL-87100 Torun, Poland
First published on 26th August 2024
Abstract
A sustainable method suitable for industrial-scale continuous flow synthesis of esters from biomass-derived furfuryl alcohol (FA) and C8–C18 carboxylic acids was developed. Under optimized reaction conditions, lipase from Aspergillus oryzae immobilized on an octyl-silane MgO·SiO2 material demonstrated high activity. A conversion of 88.7–90.2% for FA with 100% selectivity to esters using a FA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fatty acid molar ratio of 1:3 and cyclohexane as the solvent at 25 °C in 45 min was achieved in a batch system. The biocatalyst retained its high activity for at least 10 consecutive reaction cycles. Successful upgradation from a batch to continuous flow reactor led to an increased FA conversion of up to 96.8%, with a reagent flow rate of 0.070 mL min−1 and a residence time of 10.5 minutes. The biocatalyst maintained excellent performance for 30 h. The developed method, considered within the framework of green chemistry metrics, ensures a balance between the high activity, stability, recyclability, and biodegradability of the catalyst. This work proposes as a generic approach to green chemistry dedicated to support the biocatalytic continuous flow synthesis of value-added chemicals.
:fatty acid molar ratio of 1:3 and cyclohexane as the solvent at 25 °C in 45 min was achieved in a batch system. The biocatalyst retained its high activity for at least 10 consecutive reaction cycles. Successful upgradation from a batch to continuous flow reactor led to an increased FA conversion of up to 96.8%, with a reagent flow rate of 0.070 mL min−1 and a residence time of 10.5 minutes. The biocatalyst maintained excellent performance for 30 h. The developed method, considered within the framework of green chemistry metrics, ensures a balance between the high activity, stability, recyclability, and biodegradability of the catalyst. This work proposes as a generic approach to green chemistry dedicated to support the biocatalytic continuous flow synthesis of value-added chemicals.
Introduction
Abundant, inexpensive, and renewable lignocellulosic biomass is suitable for the production of value-added biobased chemicals, guiding chemical industry towards sustainable development and circular economy.1 To achieve carbon neutrality, the demand for selective catalysts and technologies for biomass valorization will increase in addition to the requirements for clean production. Biocatalysis fits well into these goals.2 Generally, enzymes serve as renewable catalysts with low toxicity that operate effectively under mild and safe conditions, demonstrating energy efficiency while considerably minimizing waste formation. Despite their excellent catalytic properties, enzymes typically require enhancement before being implemented on an industrial scale, where multiple cycles of high yield processes are desired. One of the properties typically improved through immobilization is enzyme stability. Other critical enzyme properties that should be enhanced for prolonged use in industrial reactors include activity, resistance to inhibition by reaction products, and ease of regeneration. However, the methods for increasing the recyclability or stability of enzymes for continuous flow synthesis remain challenging.3,4Hemicellulose, a component of lignocellulose, undergoes hydrolysis to produce D-xylose, which is subsequently dehydrated to furfural.5 Furfural is next converted into furfuryl alcohol, a multifunctional chemical compound based on furan.6 The majority of furfuryl alcohol is used in the production of furan resins, tetrahydrofurfuryl alcohol and esters and carboxylic acids.7 Esters of long-chain acids and furfuryl alcohol are used mostly as biolubricants, especially esters of oleic acid. A broad range of furfuryl alcohol fatty acid esters are used as surfactants, solvents, plasticizers, biofuel additives, emulsifiers, food additives, and flavors.8–11 Achieving high selectivity in the esterification of furfuryl alcohol presents a significant issue.12,13 Furfuryl alcohol readily undergoes polymerization in the presence of minerals or strong organic acids, resulting in the formation of thermostable polymers or difurfuryl ethers.14–17
Biocatalysis employing enzymes, such as lipases, for the production of furfuryl alcohol esters may bring significant benefits compared to the use of traditional acids. Using alternative biocatalytic approaches employing commercially available Novozym 435 for the esterification of furfuryl alcohol with oleic acid, octanoic acid, or castor oil at temperatures ranging from 55 to 60 °C resulted in nearly complete conversion of oleic acid (99%)18 and castor oil (89%),9 whereas only 77% of octanoic acid underwent esterification after 24 h.19 Novozym 435 is an active lipase derived from Candida antarctica lipase B (CALB) immobilized on macroporous acrylic resin. However, the reuse of Novozym 435 is difficult due to the tendency of the resin to swallow in organic solvents, hindering isolation and recycling efforts, leading to the conclusion that further study is required to meet the challenges of flow processes.20,21
Immobilization is the primary technique for enhancing the activity and stability of lipases. Various types of supports with differing mechanical, thermal, and structural properties, such as porosity, surface activity, and hydrophobicity/hydrophilicity, have frequently been used as carriers for lipases. Inorganic materials, carbon materials, polymers (or biopolymers) and composite materials served as matrixes for adsorption or covalent bonding of lipases, as previously reviewed.22–24 Lipases demonstrated their highest catalytic activity on hydrophobic surfaces, attributed to the protein immobilization in its monomeric, open form, a phenomenon known as interfacial activation.25–28 In the case of hydrophilic supports, such as silica, modification of the surface with hydrophobic groups is necessary to achieve the highest lipase efficiency. Modification of silica surface with alkyl groups (e.g. methyl, octyl, hexadecyl) and used as a support for CALB was tested in Bayer–Villiger oxidation of cyclic ketones to lactones with hydrogen peroxide. High activity of the biocatalyst was reached despite harsh reaction conditions.29 In other studies, a hydrophobic ionic liquid (triethoxysilylpropyl)imidazolium bis(trifluoromethylsulfonyl) imide was attached to the silica-magnesium oxide support to enhance the activity of lipase from Aspergillus oryzae (LAO) and enable its immobilization on the surface. The biocatalyst demonstrated high activity, enantioselectivity and stability in the kinetic resolution of ibuprofen racemate via enantiomeric esterification to (S)-ibuprofen ester (35.2% conversion, 95% enantioselectivity).30
The main aim of this work was to design a sustainable method for the esterification of biomass-derived furfuryl alcohol with C8–C18 carboxylic acids. To face the existing problems in this contribution, we combine our expertise in the design and study of activity of biocatalysts with the potential offered by the possibility of tailoring the surface chemistry of the supports to report the catalytic applications of enzymatic continuous flow synthesis. The novel aspect of this study is represented mainly by the efficient approach to produce heterogeneous catalysts characterized by the stability (thermal, chemical, and mechanical) supported by the ease of enzyme isolation and recycling for flow synthesis that is omitted in the available literature. The goal has been achieved via the implementation of an innovative, novel heterogeneous biocatalyst, which enabled the application of a flow reactor. Therefore, both high activity and, not least, the stability of the biocatalyst are of high importance. Inspired by the activation of two lipases from Candida rugosa and Aspergillus oryzae in the presence of metal cations the tailoring of the silica matrix via the incorporation of Mg2+ was performed. Subsequent introduction of hydrophobic alkyl organosilanes on the surface of silica was used as a strategy to enhance enzyme loading and help to avoid leaching. The sustainability of the presented method was measured using green metrics.
Results and discussion
Biocatalyst preparation
In our quest for a robust biocatalyst dedicated for the esterification of furfuryl alcohol and synthesis of biomass derived biofuel additives, we propose the tailoring of the support surface for lipase immobilization. The expected improvement of the stability of enzyme can facilitate continuous use. This approach is supported by structural studies, which can underpin the rational design and development of biocatalysts and in turn lead to a more sustainable and cost-effective process. Enhanced enzyme performance is reflected in higher biocatalyst productivity, which determines the enzyme cost in the particular process.The selection of the Aspergillus oryzae lipase (LAO) was driven by its superior catalytic efficiency in esterification reactions. The advantage of immobilized lipases is that they operate in nonaqueous media, but they require a certain amount of water. A layer of water, or a water shell, bound to the protein by hydrogen bonds, is crucial for maintaining the three-dimensional structure and activity.25–28 Additionally, LAO is relatively stable in organic solvents and under high substrate concentrations. Notably, LAO was acknowledged as a GRAS (generally regarded as safe) enzyme by the FDA.31 Straightforward adsorption of LAO on the tailor-made solid surface was chosen to simplify the protocol for enzyme immobilization. The hybrid material of MgO with silica (MgO·SiO2) was selected as the carrier for LAO. MgO provides changes in textural and structural properties of the material by introducing besides silanol (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH) magnesil (–Mg–OH) groups.29,31,32 Additionally, the improvement of the activity of lipase from Candida rugosa in solution in the presence of chloride Li+, Na+, K+, Mg2+ and Zn2+ salts was detected.32 Next, to enhance the enzyme-surface affinity the hydrophobicity of the carrier was increased. The hydrophobicity of the silica surface can be introduced by simple modification via physical or chemical bonding of e.g. alkyl organosilanes,29 hydrophobic ionic liquids28,30 or performing reaction in hydrophobic solvents, such as ionic liquids.34,35 In this work chemical modification with triethoxy(octyl)silane or triethoxy(hexadecyl)silane was performed (Scheme 1).
Si–OH) magnesil (–Mg–OH) groups.29,31,32 Additionally, the improvement of the activity of lipase from Candida rugosa in solution in the presence of chloride Li+, Na+, K+, Mg2+ and Zn2+ salts was detected.32 Next, to enhance the enzyme-surface affinity the hydrophobicity of the carrier was increased. The hydrophobicity of the silica surface can be introduced by simple modification via physical or chemical bonding of e.g. alkyl organosilanes,29 hydrophobic ionic liquids28,30 or performing reaction in hydrophobic solvents, such as ionic liquids.34,35 In this work chemical modification with triethoxy(octyl)silane or triethoxy(hexadecyl)silane was performed (Scheme 1).
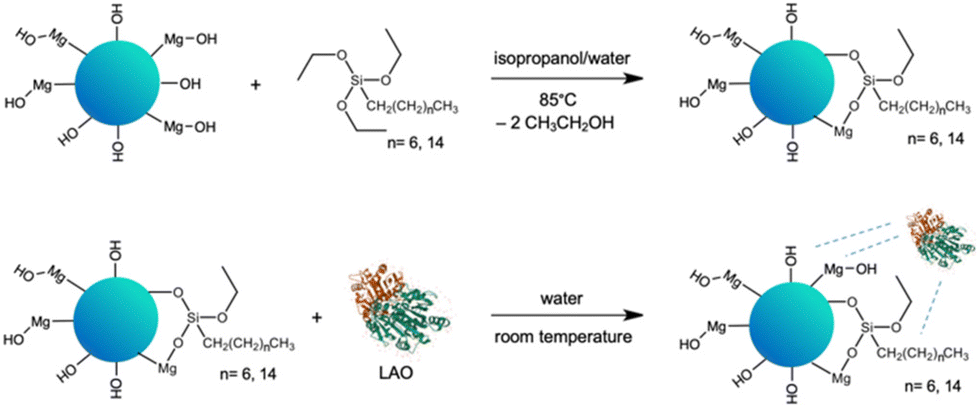 | ||
| Scheme 1 Preparation of MgO·SiO2-C8-LAO; blue dashed lines indicate adsorption interactions. | ||
In the first step of the synthesis of the biocatalyst, a siliceous support MgO·SiO2 (1:1, n:n) was synthesized through the sol–gel method according to the method described in the literature.36 The synthesized oxide material was chemically modified with the alkyl organosilanes, like triethoxy(octyl)silane (C8) or triethoxy(hexadecyl)silane (C16), by stirring the reagents in the isopropanol/water mixture at 85 °C for 24 h. The pristine SiO2 (used as delivered) and MgO·SiO2 materials were applied as references, whereas the calcination process performed at 800 °C was applied to examine how changes in porous structure will affect the properties of the materials towards modification and LAO immobilization.
The chemical grafting of alkyl groups to the siliceous materials (MgO·SiO2-C8, MgO·SiO2-C16, MgO·SiO2-C8(calc.), and SiO2-C8) was proven by FT-IR (ESI, Fig. S1†). From the presented graphs, due to the presence of characteristic bands of methyl and methylene groups from modifier molecules in the range of 2850 to 3000 cm−1, it is clear that the functionalizing agent was deposited efficiently.
The presence and also loading of alkyl groups on MgO·SiO2 was determined by TG/DTG analyses (ESI, Fig. S2–S9†) and is presented in Table 1. Obtained analyses revealed that the longer alkyl chain is the lower amount of alkyl groups was grafted to the material, respectively 8.03 wt% of C8 and 5.86 wt% of C16. Furthermore, the calcination of the MgO·SiO2 support led to a reduction in the chemical immobilization of C8 groups (1.21 wt%), which is likely related to the decreased number of available surface hydroxyl groups capable of binding the modifier. The same effect was observed for the modification of pristine SiO2 which resulted in the introduction of only 5.87 wt% of C8 groups. It is speculated that the presence of magnesium oxide provided additional hydroxyl groups on the surface of the hybrid material, enabling more efficient grafting of the octyl groups.
Additionally, SEM images before and after modification with alkyl groups showed the affinity of support material particles towards agglomeration and formation of irregular structures that is characteristic of silica-based materials after modification (ESI, Fig. S10–S16†). Moreover, the elemental composition of the surface of SiO2, MgO·SiO2, and MgO·SiO2(calc.) materials and their modified forms with alkyl groups was studied with an EDS detector (ESI, Fig. S10–S16†). EDS analyses proved the presence of carbon atoms uniformly distributed on the surface coming from grafted alkyl groups on the surface of modified materials, hence confirming effective incorporation of hydrophobic groups onto the surface of the oxide system.
Finally, in order to prepare the biocatalyst, the aqueous solution of LAO was mixed with siliceous materials MgO·SiO2-C8, MgO·SiO2-C16, MgO·SiO2-C8(calc.), SiO2-C8 and MgO·SiO2 at room temperature for 3 h to perform physical immobilization of lipase on the surface. According to the TG/DTG analyses (ESI, Fig. S17 and S18†) it was observed that longer alkyl chains led to the decrease of the amount of immobilized lipase from 4.24 wt% for MgO·SiO2-C8-LAO to 1.54 wt% for MgO·SiO2-C16-LAO, probably due to the appeared higher steric hindrance in the case of longer alkyl chains. The lower amount of surface functional groups introduced during the modification step (1.21 wt%) resulted in a reduced adsorption of LAO (2.16 wt%) on the calcined material (ESI, Fig. S19†). In comparison with SiO2-C8 (9.25 wt% of LAO, ESI, Fig. S20†), MgO·SiO2-C8 showed a lower sorption capacity due to the higher amount of grafted octyl groups on the MgO·SiO2 surface, which caused steric hindrances in the adsorption of LAO moieties. Lack of LAO on the surface of non-modified MgO·SiO2 according to the TG/DTG thermograms proved that the presence of alkyl groups is crucial (ESI, Fig. S21†). The affinity of the pristine surface of MgO·SiO2 to LAO is too low to enable efficient adsorption.
Modifying the silica surface with MgO increased both the affinity of the enzyme for the matrix and adsorption capacity of the support material. The increased surface porosity, attributed to additional –Mg–OH groups, has also been observed in previous studies.30,32,33 Similarly, modifying silica with alkyl organosilanes had a beneficial impact. Lipases demonstrate higher catalytic performance in hydrophobic environments due to their reduced affinity for essential water molecules. This characteristic is crucial for the interfacial activation.25–27
After the immobilization of lipase, SEM images revealed even higher agglomeration of the particles of the matrix, which is growing shifting from pristine SiO2 and MgO·SiO2via alkyl modified materials to final biocatalysts (ESI, Fig. S22–S26†). The highest particle agglomeration is observed for the SiO2-C8-LAO biocatalyst, where the highest amount of lipase was immobilized (9.25 wt%), clearly showing that immobilization increased the support particles to form aggregates. EDS analyses confirmed the presence of nitrogen atoms coming from the protein structure for all biocatalysts.
It can be expected that the highest loading of lipase may not necessarily result in elevated catalytic activity. This could be attributed to the generally reduced accessibility of the substrate and/or variations in enzyme conformations, including inactive ones, for this particular substrate. Additionally, the presence of crowded lipase agglomerations could contribute to the decline in activity. Hence, conducting further catalytic tests is essential to determine the optimal lipase loading on the surface of the catalyst.37
In the catalytic studies presented below, the most active catalyst was found to be MgO·SiO2-C8-LAO (4.24 wt% of LAO), which was subsequently subjected to adsorption–desorption analysis using the BET method and BJH model for characterization (Table 2). Adsorption–desorption isotherms (ESI, Fig. S27†), the pore size distribution diagram (ESI, Fig. S28†) and SBET micropores examined the high microporosity of the supports and biocatalyst. The characteristics of the MgO·SiO2 material (SBET 421 m2 g−1, Vp 0.06 cm3 g−1, dp 2.1 nm), after chemical modification with alkyl groups C8 (SBET 325 m2 g−1, Vp 0.05 cm3 g−1, dp 2.1 nm), and biocatalyst MgO·SiO2-C8-LAO (SBET 236 m2 g−1, Vp 0.02 cm3 g−1, dp 2.1 nm) were determined. This data clearly shows that the introduction of alkyl groups, as well as lipase adsorption, caused a reduction in the specific surface area and partial blocking of micropores, which results from the successively decreasing specific surface area of the micropores. The decrease in surface area and pore volume is typical for materials upon enzyme immobilization; however, the unchanged pore diameters indicate that the enzyme is primarily deposited on the surface of the support or deep within the pores of the material, leading to a reduction in pore volume after immobilization.
| Material | S BET (m2 g−1) | S BET micropores (m2 g−1) | V p (cm3 g−1) | d p (nm) | C8 loadinga (wt% ± 0.3) | LAO loadinga (wt% ± 0.3) |
|---|---|---|---|---|---|---|
| a Determined using TGA; the standard deviation of 3 replicate experiments. | ||||||
| MgO·SiO2 | 421 | 314 | 0.06 | 2.1 | — | — |
| MgO·SiO2-C8 | 325 | 243 | 0.05 | 2.1 | 8.03 | — |
| MgO·SiO2-C8-LAO | 236 | 169 | 0.02 | 2.1 | 8.03 | 4.24 |
Catalytic activity of MgO·SiO2-C8-LAO in esterification of furfuryl alcohol and C8–C18 carboxylic acids in a batch system
The catalytic activity of MgO·SiO2-C8-LAO was studied in an industrially relevant process of esterification of furfuryl alcohol with fatty acids (C8–C18). In the preliminary studies, the activity of the developed biocatalysts was tested in a model esterification of furfuryl alcohol and caprylic acid (Scheme 2) in a batch system. The reaction was carried out at room temperature with hexane as a solvent and with triple molar excess of caprylic acid to furfuryl alcohol. Samples were collected during the reaction, and the conversion of furfuryl alcohol and selectivity to ester were determined using gas chromatography (GC). In every case, 100% selectivity was observed, making the conversion equal to the yield. The mass of the biocatalyst used for the comparative reactions presented in Table 3 was recalculated in order to contain fixed, the same amount of enzyme in the system (6.36 mg of LAO), e.g. in 150 mg of MgO·SiO2-C8-LAO and other heterogenous biocatalysts.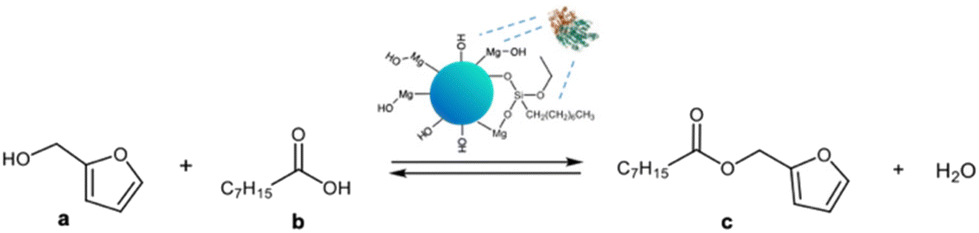 | ||
| Scheme 2 Esterification of furfuryl alcohol and caprylic acid in the presence of the developed catalysts. | ||
| Biocatalyst | Time (min) | Furfuryl alcohol conversiona (%) | Activity (μmol min−1) | Specific activity (μmol mg−1 min−1) | Activity recoveryb (%) |
|---|---|---|---|---|---|
| Reaction conditions: furfuryl alcohol 1.0 mmol, caprylic acid 3.0 mmol, cyclohexane 0.5 mL, biocatalyst containing 6.36 mg of LAO (146 μL of LAO solution (43.7 mg mL−1); 530 μL of CALB solution (12 mg mL−1); 118 mg of Novozym 435; 300 mg of MgO·SiO2-LAO; 69 mg of SiO2-C8-LAO; 150 mg of MgO·SiO2-C8-LAO; 294 mg of MgO·SiO2(calc.)-C8-LAO; 413 mg of MgO·SiO2-C16-LAO), 25 °C, 250 rpm.a Determined using GC.b Activity recovery = (activity of immobilized enzyme/activity of native enzyme)·100%.c 300 mg of biocatalyst (maximum possible loading). | |||||
| LAO | 120 | 83.5 | 7.3 | 1.1 | — |
| CALB | 120 | 74.7 | 6.1 | 1.0 | — |
| Novozym 435 | 30 | 92.4 | 30.1 | 4.7 | 493 |
| MgO·SiO2-LAOc | 120 | 6.1 | — | — | — |
| SiO2-C8-LAO | 90 | 87.1 | 10.0 | 1.6 | 137 |
| MgO·SiO2-C8-LAO | 45 | 90.2 | 20.2 | 3.2 | 277 |
| MgO·SiO2(calc.)-C8-LAO | 45 | 89.4 | 20.3 | 3.2 | 277 |
| MgO·SiO2-C16-LAO | 45 | 87.3 | 20.4 | 3.2 | 277 |
As shown in Table 3 and Fig. S30 and S31 (ESI†), the heterogenization of native LAO resulted in significantly higher catalytic performance of lipase compared to the native form (87.1–90.2% conversion versus 83.5%), reaching 277% activity recovery and 90.2% conversion of furfuryl alcohol in 45 min for the reaction with MgO·SiO2-C8-LAO. Definitely it is a fundamental achievement for the isolation and recycling of enzyme. The commercially available native form of CALB used in the same amount as LAO is slightly less active (74.7%).
The unique interfacial activation of LAO on the hydrophobic support MgO·SiO2-C8 resulted in an improved activity of the immobilized lipase. Lipases exhibit two distinct conformations which are in equilibrium: an inactive closed form where the active site is shielded from the reaction medium by a polypeptide chain called a lid, and an open form where the lid is displaced, fully exposing the active site to the reaction medium.25–28 The protein strongly adsorbed on the surface after immobilization, affecting the equilibrium and fixing the open form of the lipase.
Additionally, the porous form of the matrix permits an “operational stabilization” of the enzyme, stabilizing the enzyme against interaction with other molecules, e.g. from the enzymatic extract and limiting the contact with any external hydrophobic interface or the effects of vigorous stirring.
As expected, the highest activity was observed for the benchmark Novozym 435 (92.4% conversion of furfuryl alcohol after 30 min). The recycling test of Novozym 435 revealed issues with filtering off the biocatalyst after the reaction due to the swelling of the resin. As previously mentioned, the objective of this work was to find an alternative to Novozym 435 due to technical issues related to its use, such as filtration, operation as a fixed bed in a flow reactor, and regeneration difficulties.
The crucial parameter of the developed heterogeneous biocatalysts is the structure and hydrophilicity of the surface of the carrier. An additional calcination of the MgO·SiO2 matrix did not affect the stabilization of active conformation of LAO as suspected (89.4% conversion in 45 min).
Changes in crystalline structure influenced the amount of grafted C8 groups (for MgO·SiO2-C8: 8.03 wt% of C8, for MgO·SiO2(calc.)-C8: 1.21 wt% of C8) and adsorbed LAO on the surface (for MgO·SiO2-C8-LAO: 4.24 wt% of LAO, for MgO·SiO2(calc.)-C8-LAO: 2.16 wt% of LAO), that is lower as in the case of uncalcined materials. It is worth underlining that lower enzyme loading resulted in high activity recovery and conversion of the substrate probably due to uniform enzyme deposition and escaped the trap of enzyme overloading. Hence, to avoid an additional high energy-consuming step in catalyst preparation that does not improve lipase performance, matrix calcination is not recommended.
Introducing a longer alkyl group C16 instead of C8 to oxide materials influenced the immobilization step (MgO·SiO2-C16 1.54 wt% versus 4.24 wt% on MgO·SiO2-C8-LAO) and slightly decreased the activity of the enzyme (87.3% conversion in 45 min) and longer alkyl chains caused larger steric hindrances, decreasing the capacity of protein to adsorb on the surface. Additionally, the presence of alkyl hydrophobic groups on the MgO·SiO2 surface is crucial to maintain LAO immobilization and high activity, which was proven by TG/DTG analyses (ESI, Fig. S21†). Only 6.1% conversion of furfuryl alcohol after 120 minutes of reaction in the presence of MgO·SiO2-LAO was detected, even with the maximum possible loading of biocatalyst (300 mg). A higher amount of biocatalyst prevented effective stirring.
The important aspect of improving the LAO activity was to introduce Mg species to silica and create MgO·SiO2 oxide material. The presence of Mg particles in the siliceous material likely improved the activation and stabilization of the active conformation of the enzyme through electrostatic interactions between Mg2+ ions and LAO amino acids by providing additional spatial rearrangements in the α-helix and β-sheet orientation.38 This effect led to higher performance of the immobilized biomolecules. A slightly different approach based on Mg2+ doped-Candida rugosa lipase (CRL) which was encapsulated into a zeolitic imidazolate framework (ZIF) structure was reported.38 The presence of Mg2+ changed the conformation of CRL, confirmed by circular dichroism and fluorescence spectroscopy. This effect improved the structural stability of the enzyme and an increase in catalytic performance, up to two-fold higher, was observed compared to materials lacking metal ions. Additionally, as mentioned above, Mg2+ also provided more efficient grafting of the octyl groups. The increase in reaction rate (90.2% conversion of alcohol for MgO·SiO2-C8-LAO in 45 min, versus 87.1% for SiO2-C8-LAO in 90 min) and 2 times higher enzyme specific activity (3.2 μmol mg−1 min−1 for MgO·SiO2-C8-LAO and 1.6 μmol mg−1 min−1 for SiO2-C8-LAO, respectively) confirmed the significant influence of MgO on enzyme activity. Additionally, the presence of advanced microporous channel systems enhances the flow of the reaction mixture through the biocatalyst, once again resulting in excellent lipase catalytic performance.
Key reaction parameters
Optimization of the reaction parameters for a model reaction in the presence of the MgO·SiO2-C8-LAO biocatalyst was studied. First, the influence of the type of solvent (cyclohexane, toluene, hexane, and isooctane) used for the reaction was tested (Fig. 1). The choice of a proper solvent in biocatalysis is necessary to avoid the fast deactivation of the enzyme. The biocatalyst demonstrated superior catalytic performance in cyclohexane (86.2% conversion in 60 min), and next in toluene (85.7% conversion in 90 min), achieving 100% selectivity in both cases (see ESI, Fig. S29†). Using hexane and isooctane, furfuryl alcohol conversions reached 52.6% and 37.4%, respectively, in 180 min. Acetonitrile, being a more hydrophilic solvent, was also tested and showed no conversion of furfuryl alcohol. This can be attributed to the strong affinity of acetonitrile to the essential water required by the lipase. Solvents such as methanol, ethanol or alkyl carbonates as greener alternatives were not suitable due to their reactivity with caprylic acid. Additionally, hydrophilic solvents tend to attract the essential water in lipase, leading to its deactivation.25,39 Unfortunately, using less than 0.50 mL of cyclohexane per 1 mmol of furfuryl alcohol for the synthesis resulted in lower biocatalyst activity due to problems with mixing of the reagents in the presence of heterogeneous MgO·SiO2-C8-LAO, making the solventless process impossible. Higher amounts of cyclohexanone, such as 0.75 and 1.0 mL, resulted in slower reaction rates due to the lower concentration of reagents.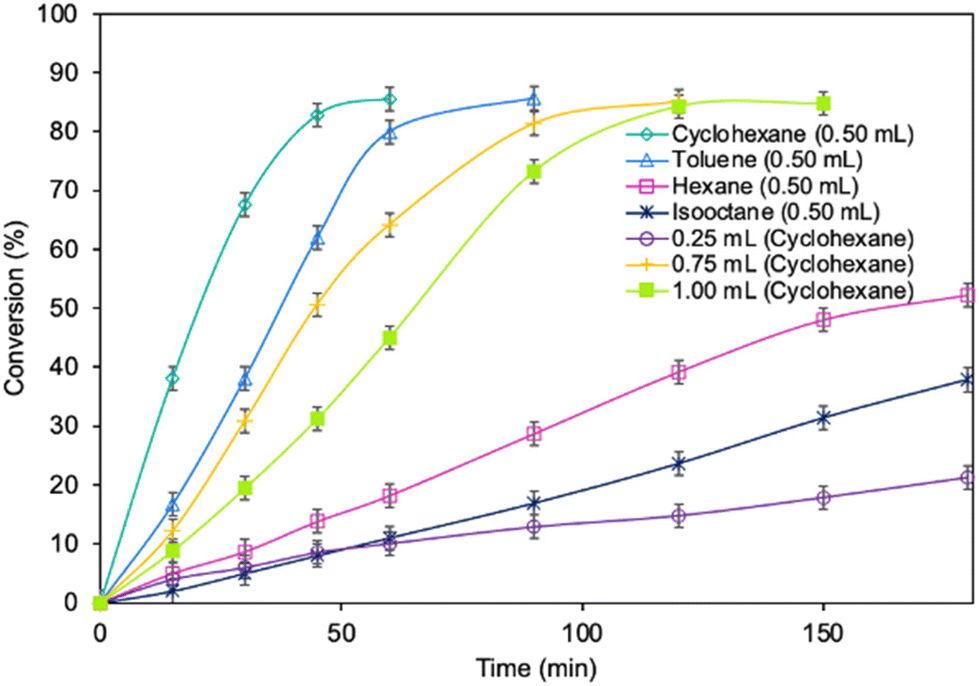 | ||
| Fig. 1 The influence of type of solvent and its amount on furfuryl alcohol conversion. Reaction conditions: furfuryl alcohol 1.0 mmol, caprylic acid 3.0 mmol, solvent, MgO·SiO2-C8-LAO 100 mg, 25 °C, 250 rpm; conversion was determined using GC. | ||
Subsequently, the influence of the amount of MgO·SiO2-C8-LAO biocatalyst was studied (Fig. 2). Using 150 mg of the biocatalyst per 1 mmol of furfuryl alcohol resulted in an even higher furfuryl alcohol conversion (90.2%) within a shorter reaction time of 45 min. Increasing the amount of biocatalyst to 175 mg did not lead to a higher conversion or reaction rate (90.4% in 45 min). As observed, using only 25 mg of biocatalyst still enabled a high conversion of furfuryl alcohol (84.8%), similar to the reaction with 150 mg, but in longer time, after 180 min. This indicates lower robustness of the system due to the reduced amount of immobilized LAO. This observation confirms the exceptionally high activity of the developed MgO·SiO2-C8-LAO biocatalyst. For further studies 150 mg of MgO·SiO2-C8-LAO was used.
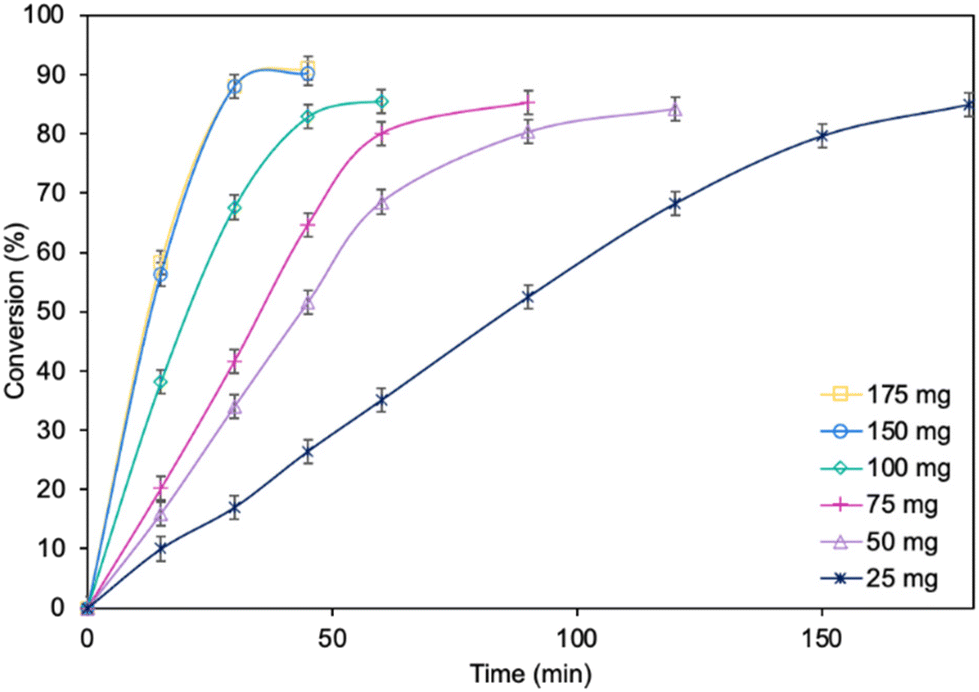 | ||
| Fig. 2 The influence of MgO·SiO2-C8-LAO content on furfuryl alcohol conversion. Reaction conditions: furfuryl alcohol 1.0 mmol, caprylic acid 3.0 mmol, cyclohexane 0.5 mL, MgO·SiO2-C8-LAO, 25 °C, and 250 rpm; conversion was determined using GC. | ||
The influence of molar ratio of the furfuryl alcohol to caprylic acid on the catalytic performance of the biocatalyst in furfuryl alcohol esterification was also examined (Fig. 3). The best outcome was obtained for 3-fold molar excess of caprylic acid, achieving 90.2% conversion of furfuryl alcohol in 45 min. Increasing the molar excess of acid further did not affect the conversion. The studies concerning the influence of temperature (Fig. 4) revealed the similar catalytic performance of MgO·SiO2-C8-LAO at 25 °C, 30 °C and 35 °C. As expected, lower temperature (20 °C) caused a slight decrease in reaction rate, designating 25 °C as the most favourable temperature. Higher temperature (40 °C) resulted in retardation of the reaction, probably due to partial deactivation of the lipase-based biocatalyst. According to the literature, LAO maintains its optimum activity at around 30 °C and pH 7, with a significant drop in activity observed at 40 °C, which is consistent with the obtained results.40
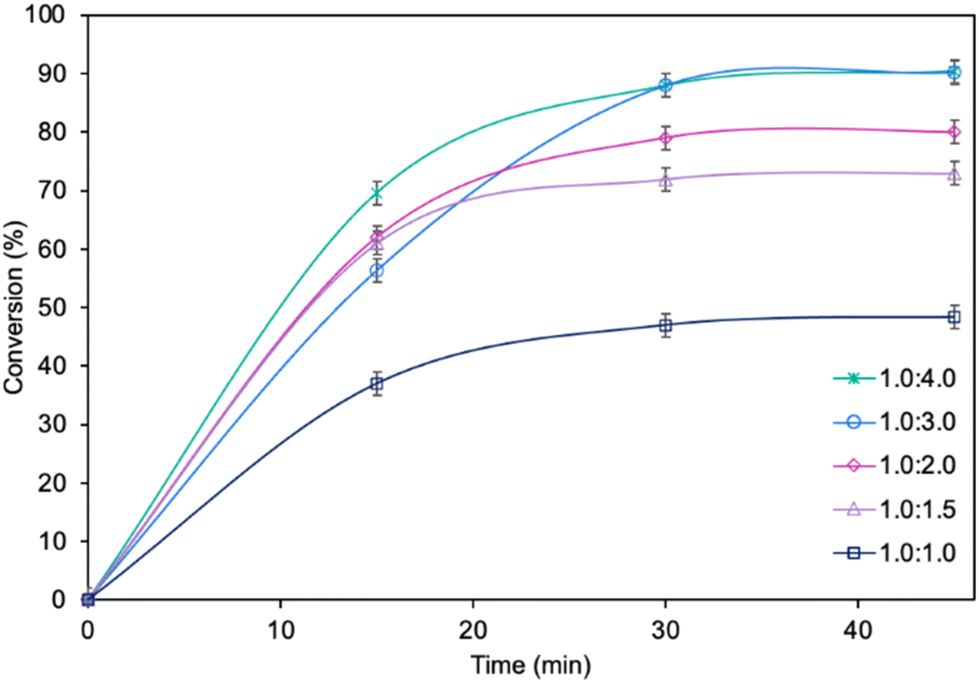 | ||
| Fig. 3 The influence of the furfuryl alcohol to caprylic acid molar ratio on furfuryl alcohol conversion. Reaction conditions: furfuryl alcohol 1.0 mmol, caprylic acid, cyclohexane 0.5 mL, MgO·SiO2-C8-LAO 150 mg, 25 °C, and 250 rpm; conversion was determined using GC. | ||
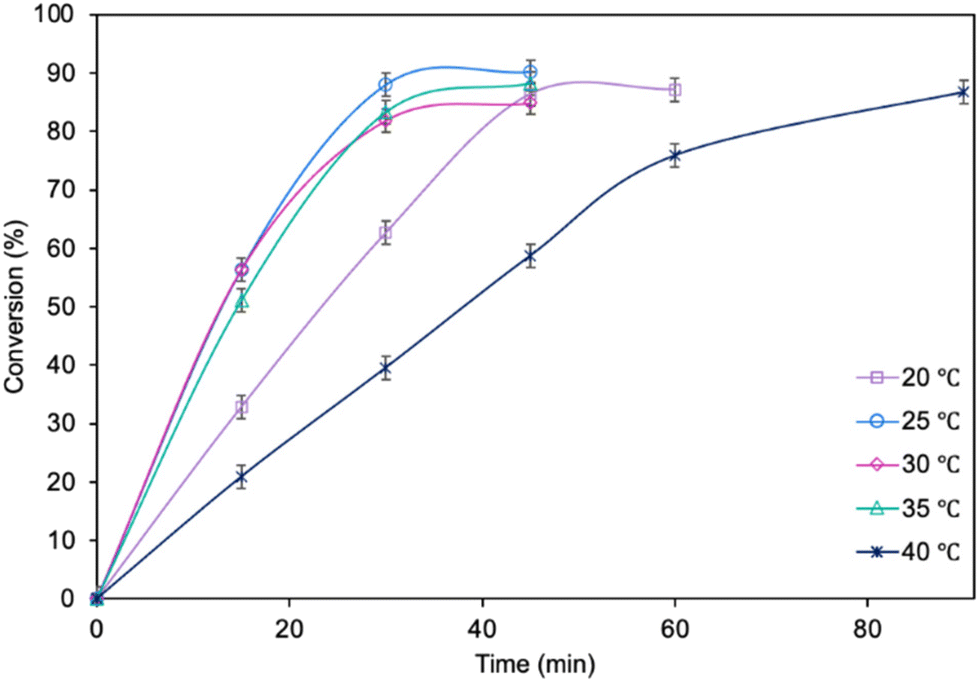 | ||
| Fig. 4 The influence of temperature on furfuryl alcohol conversion. Reaction conditions: furfuryl alcohol 1.0 mmol, caprylic acid 3.0 mmol, cyclohexane 0.5 mL, MgO·SiO2-C8-LAO 150 mg, 250 rpm; conversion was determined using GC. | ||
Finally, the potential for easy and safe biocatalyst reuse is essential for the advancement of environmentally friendly and effective catalytic systems. The recycling ability of MgO·SiO2-C8-LAO was studied in the esterification of furfuryl alcohol by upscaling the process twofold to 2 mmol of furfuryl alcohol, which consequently required 300 mg of biocatalyst in batch mode. As presented in Fig. 5, the developed MgO·SiO2-C8-LAO retained its catalytic performance for 10 consecutive runs in furfuryl alcohol esterification but a progressive drop was observed. A reduction in conversion to 84.6% was observed after the 10th cycle. TG/DTG analyses of the reused biocatalyst showed no changes in LAO loading, which indicates the slow deactivation of LAO (ESI, Fig. S33†).
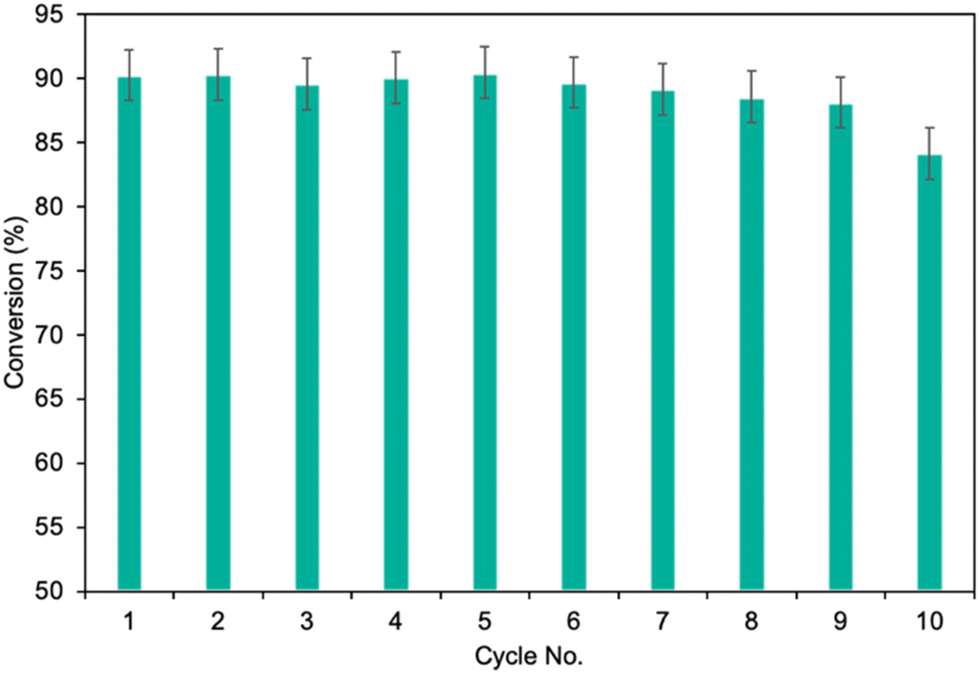 | ||
| Fig. 5 The recycling study of MgO·SiO2-C8-LAO in furfuryl alcohol esterification. Reaction conditions: furfuryl alcohol 2.0 mmol, caprylic acid 6.0 mmol, cyclohexane 1 mL, MgO·SiO2-C8-LAO 300 mg, 25 °C, 250 rpm, and 45 min; conversion was determined using GC. | ||
Additionally, the ‘reaction stop’ experiment, which involved the rapid removal of the biocatalyst from the reaction mixture, demonstrated no further conversion of furfuryl alcohol in the filtrate (ESI, Fig. S33†), which confirms that there was no leaching of LAO into the reaction system. Hence, the observed drop in biocatalytic properties is mainly related to enzyme inhibition by the substrate or products, or due to enzyme inactivation by denaturation.
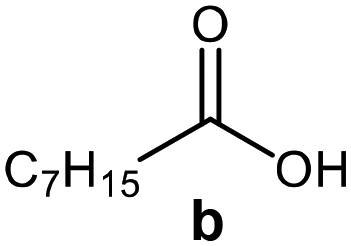
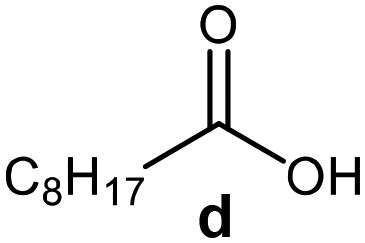
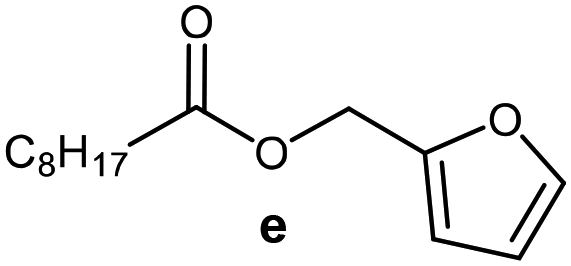
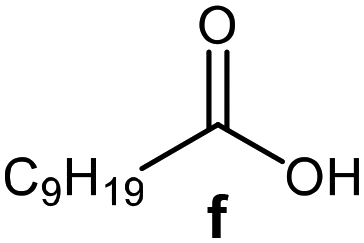
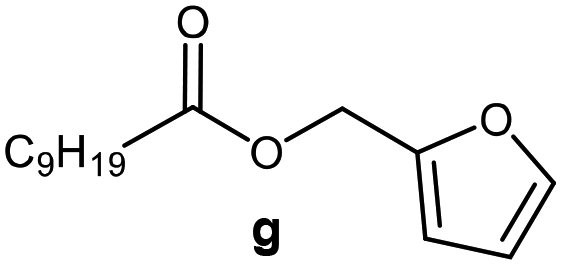
Further, to use the developed biocatalytic system for the synthesis of other fatty esters of furfuryl alcohol, the following acids were used: caprylic acid, pelargonic acid, capric acid, lauric acid and oleic acid (Table 4). In all cases high conversion of alcohol (88.7–90.2%) was achieved. As expected, the longer alkyl chain of fatty acid caused steric hindrance and prolonged reaction times (from 45 min for C7 to 90 min for C17). Nevertheless, in all cases esters were isolated with high yields, confirming that the biocatalyst is universal for the synthesis of esters from biomass-derived furfuryl alcohol and C8–C18 carboxylic acids. The selectivity of the esterification reached 100% for each ester.
| Fatty acid | Furfuryl ester | Time (min) | Furfuryl alcohol conversiona (%) | Isolated yield of ester (%) |
|---|---|---|---|---|
| Reaction conditions: furfuryl alcohol 1.0 mmol, fatty acid 3.0 mmol, cyclohexane 0.5 mL, MgO·SiO2-C8-LAO 150 mg, 25 °C, 250 rpm.a Conversion was determined using GC. | ||||
|

|
45 | 90.2 | 88 |
|
|
60 | 89.8 | 86 |
|
|
60 | 89.4 | 88 |
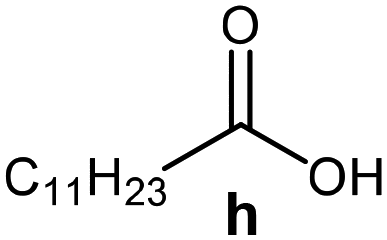
|
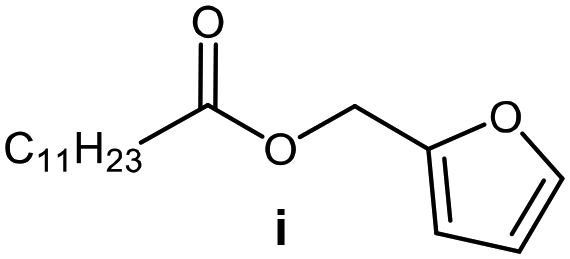
|
60 | 88.7 | 85 |
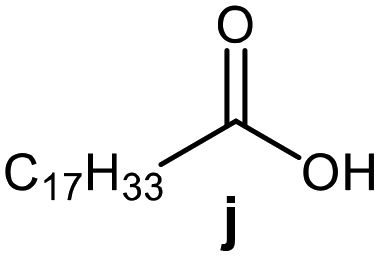
|
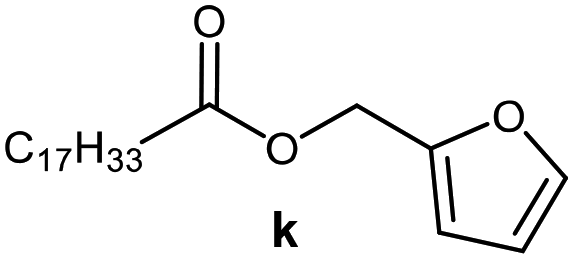
|
90 | 89.9 | 87 |
Catalytic activity of MgO·SiO2-C8-LAO in esterification of furfuryl alcohol and C8–C18 carboxylic acids in a flow system
Continuous flow synthesis of fine chemicals and pharmaceuticals is expected to improve the selectivity, safety, and sustainability of chemical production while also reducing costs. Encouraged by the high activity and stability of the designed biocatalysts, the upgrade of the batch esterification to a flow process was studied. Experiments were carried out in a fully automated Syrris Asia column packed-bed type flow microreactor filled with 300 mg of MgO·SiO2-C8-LAO biocatalyst. Before all experiments a fresh mixture of furfuryl alcohol (6.2 g), octanoic acid (27.4 g) and cyclohexane (31.7 mL) was prepared and then pumped to the column reactor filled with the biocatalyst. To keep the stable reagent flow through the column reactor, the system was equipped with a backpressure regulator and set to 2 bar (Fig. 6). Experiments were carried out under the conditions optimised in the batch system. Total reagent flow rates ranging from 0.04 to 0.20 mL min−1 were tested (Table 5).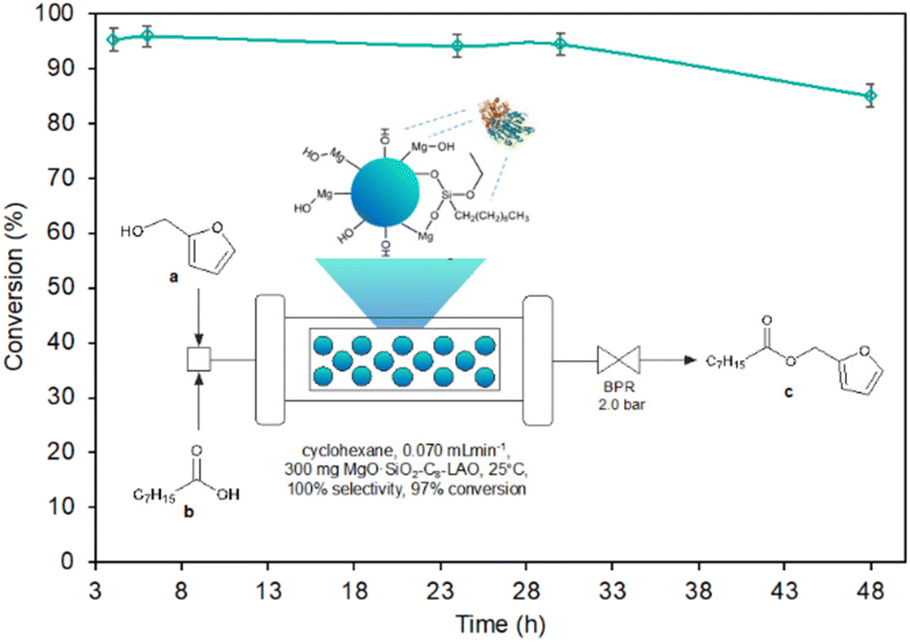 | ||
| Fig. 6 Conversion of furfuryl alcohol under continuous flow conditions catalysed by MgO·SiO2-C8-LAO over time. Reaction conditions: furfuryl alcohol in cyclohexane (2.0 M), caprylic acid (1:3, n:n, furfuryl alcohol:caprylic acid), MgO·SiO2-C8-LAO 300 mg, and 25 °C. Conversion of furfuryl alcohol was determined using GC. | ||
| Reagents flow (mL min−1) | Residence timea (min) | Furfuryl alcohol conversion (%) | SPb (g h−1 mg−1) | STYc (g h−1 L−1) |
|---|---|---|---|---|
| Reaction conditions: furfuryl alcohol in cyclohexane (2.0 M), caprylic acid (1:3, n:n, furfuryl alcohol:caprylic acid), MgO·SiO2-C8-LAO 300 mg (volume 0.735 mL), 25 °C. Furfuryl alcohol conversion was determined using GC.a τ = Vbiocatalyst/Freagents (mL min−1).b Specific productivity SP = mproduct/τmprotein (g h−1 mg−1).c Space–time–yield STY = mproduct/τVbiocatalyst (g h−1 L−1). |
||||
| 0.04 | 18.4 | 1 h, 96.7 | 0.13 | 2173 |
| 2 h, 96.7 | ||||
| 3 h, 96.6 | ||||
| 0.06 | 12.3 | 1 h, 96.5 | 0.28 | 4867 |
| 2 h, 96.2 | ||||
| 3 h, 96.6 | ||||
| 0.07 | 10.5 | 1 h, 96.5 | 0.38 | 6651 |
| 2 h, 96.8 | ||||
| 3 h, 96.6 | ||||
| 0.08 | 9.2 | 1 h, 91.7 | 0.96 | 8244 |
| 2 h, 92.3 | ||||
| 3 h, 91.8 | ||||
| 0.10 | 7.4 | 1 h, 85.2 | 0.69 | 11904 |
| 2 h, 84.5 | ||||
| 3 h, 84.8 | ||||
| 0.20 | 3.7 | 1 h, 78.4 | 2.53 | 43814 |
| 2 h, 79.1 | ||||
| 3 h, 78.3 | ||||
The use of reagent residence times in the catalytic zone, ranging from 10.5 to 18.4 minutes, associated with reagent flow rates of 0.04 mL min−1, 0.06 mL min−1, and 0.07 mL min−1 (τ = 18.4 min, 12.3 min, and 10.5 min) resulted in a high conversion of furfuryl alcohol, reaching 96.8% and remaining stable for at least 3 h, while the reduction of residence time below 10.5 min led to a reduction in the conversion of furfuryl alcohol. For 0.08 mL min−1, 0.1 mL min−1, and 0.2 mL min−1 reagent flow a significant deterioration of alcohol conversion to 92.3%, 85.2%, and 79.1% respectively was noticed. The optimal flow reaction conditions, considering the highest conversion of furfuryl alcohol (96.8%) achieved using 0.07 mL min−1 (τ = 10.5 min) with the space–time–yield SPY of 6651 (g h−1 L−1), were determined and used for stability tests of MgO·SiO2-C8-LAO over the long-term experiment. The biocatalyst exhibited excellent stability over 30 h, with furfuryl alcohol conversion reaching 96.8%. A slight drop to 85.7% was observed after 48 h of conducting the process.
The productivity of this process of 0.376 g h−1 (1.67 mmol h−1) is quite efficient, especially considering the reactor size (bed volume 0.735 mL) with a space–time–yield SPY of 6651 (g h−1 L−1). Additionally, the stability of the catalyst bed up to 30 h can ensure the production of 11.28 g of product. The esterification of furfuryl alcohol represented in this work can be considered as a relatively slow reaction that requires a residence time of 10.5 min; however, rapid mixing and heat transfer bring safety and quality advantages. Moreover, 21 times higher productivity compering to batch process was reached (6651 versus 320 (g h−1 L−1)) recalculating for the volume of used biocatalyst.
The STY is a reasonable and important quantity for the comparability of continuously operated flow processes. Chemical reactions can generally be categorized by reaction time into three groups: rapid reactions that are completed within seconds, intermediate reactions that take between 1 second and 10 minutes, and slow reactions that require more than 10 minutes to complete. According to examples in the literature, a residence time of approximately 10 minutes in continuous flow biocatalysis is deemed favourable.41 In some cases, enzyme-catalysed flow reactions can last up to 20 minutes or even more for micro and macroreactors with the flow rates ranging from 0.05 to 1.45 mL min−1 with the most often used rates being around 0.1–0.2 mL min−1. Reactor volumes generally range from 0.3 to 12 mL, with an average volume of around 1 mL. For example, hydroxynitrile lyases covalently immobilized in a siliceous monolithic microreactor were applied for the fast production of chiral cyanohydrins in 4 min at 0.1 mL min−1 in a 0.96 mL rector with STY 1229 [g L−1 h−1].42 Other examples described much lower parameters of STY, around 10–60 [g L−1 h−1]. For example, the formylglycine generating enzyme from Thermomonospora curvata immobilized on epoxy-activated Sepharose beads (1.0 mL packed-bed reactor, residence time 20 min, flow 0.5 mL min−1) used for aldehyde tag conversion showed 10 times higher productivity (STY = 21.6) compared to the batch process.43
For the upscaling of the process described in this work for large-scale production of esters scaling up to a packed-bed macroreactor with higher reactor volume should be tested first. Next, probably the numbering up increasing the number of channels in the macroreactors what is a common strategy in the scale-up of flow chemistry enabling the retention of the transfer properties, such as mixing, heat transfer associated with the micro-environment.44
Summing up, under flow conditions, a higher conversion of furfuryl alcohol 96.8% was achieved, compared to the 90.2% in a batch reactor, along with a shortened reaction residence time to 10.5 min versus 45 min in the batch process. These outstanding results for the continuous flow process might be explained by effective enzyme adsorption and the provision of a suitable hydrophobic microenvironment, which resulted in high enzyme activity over a prolonged period.
Green chemistry metrics
To emphasize the sustainability of the developed method for the synthesis of furfuryl esters, a green metrics analysis of all literature data was conducted in accordance with green chemistry principles using J. Clark's Green Chemistry Metrics toolkit (Appendix 2, ESI†).46The MgO·SiO2-C8-LAO biocatalyst used in the batch and flow synthesis of furfuryl caprylate was compared to metal-based catalysts (Y2O3-ZrO2 and Fe-DTP-ZIF-8), commercial biocatalyst (Novozym 435), and coupling reagent (EDC) described in the literature for the synthesis of furfuryl acetate, furfuryl oleate, furfuryl caprylate, furfuryl ricinoleate, and furfuryl 2-furoate (Table 6). No by-products were observed in almost all cases, reaching 100% selectivity of the process, except the lower esterification selectivity of 86.3% obtained in the presence of the Fe-DTP-ZIF-8 catalyst (iron-exchanged heteropolyacid encapsulated inside ZIF-8). A yield of ester higher than 90% was achieved only in two instances: with the biocatalysts described in this work and with Novozym 435 (entries 3, 7 and 8), highlighting the significance of our research. Once again, it is worth underlining that the lack of or difficulties with recycling of Novozym 435 could be a significant obstacle for its application in the flow process.
| Catalyst/reagent | Y2O3-ZrO2a |
Fe-DTP-ZIF-8b | Novozym 435c | Novozym 435d | Novozym 435e | EDCf | MgO·SiO2-C8-LAO batchg | MgO·SiO2-C8-LAO flowh |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: furfuryl alcohol 10 mmol, acetic acid 50 mmol, Y2O3-ZrO2 0.2 g, 110 °C, 7 h. b Reaction conditions: furfuryl alcohol 46 mmol, acetic acid 28 mmol, Fe-DTP-ZIF-8 0.006 g, 100 °C, 6 h. c Reaction conditions: furfuryl alcohol 71 mmol, oleic acid 71 mmol, Novozym 435 1 g, 60 °C, 533.29 Pa, 6 h. d Reaction conditions: furfuryl alcohol 1 mmol, caprylic acid 1 mmol, Novozym 435 (0.01 g), 55 °C, 24 h. e Reaction conditions: furfuryl alcohol 3 mmol, caprylic acid 1 mmol, Novozym 435 0.06 g, 60 °C, 28 mmHg vacuum, 5 h. f Reaction conditions: furfuryl alcohol 2 mmol, 2-furoic acid 6 mmol, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide – EDC 0.06 g, dichloromethane 3 mL, 90 °C, microwave 200 W, 30 min. g Reaction conditions: furfuryl alcohol 1 mmol, caprylic acid 3 mmol, cyclohexane 0.50 mL, MgO·SiO2-C8-LAO 0.15 g, 25 °C, 45 min. h Reaction conditions: furfuryl alcohol 19 mmol, caprylic acid 57 mmol, cyclohexane 95 mL, MgO·SiO2-C8-LAO 0.30 g, 0.070 mL min−1 reagent flow, 25 °C, 10.5 min (residence time), 48 h (to consume all furfuryl alcohol). i Solventless conditions, no data concerning work-up; flag system: the green flag denotes preferred, amber is acceptable, and red is undesirable. Appendix 2, ESI.† | ||||||||
| Entry | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Acid | Acetic acid | Acetic acid | Oleic acid | Caprylic acid | Ricinoleic acid | Furoic acid | Caprylic acid | Caprylic acid |
| Yield of product (%) | 88 | 75.7 | 99.9 | 73 | 88.6 | 70.8 | 90.2 | 96.8 |
| Selectivity of the esterification (%) | 100 | 86.3 | 100 | 100 | 100 | 100 | 100 | 100 |
| Atom economy (%) | 88.6 | 88.6 | 95.3 | 92.6 | 95.5 | 91.4 | 92.6 | 92.6 |
| RME (%) | 31 | 47.9 | 95.1 | 68.7 | 56.1 | 31.3 | 38.1 | 40.9 |
| Solvent choice |

|
—i | —i | —i | —i |

|

|

|
| Catalyst or reagent |

|

|

|

|

|

|

|

|
| Recoverable catalyst |

|
No data | No data | No data |

|

|

|
|
| Critical elements |

|

|

|

|

|

|

|

|
| Energy |

|

|

|

|

|

|

|

|
| Reaction run below solvent boiling point |

|

|

|

|

|

|

|

|
| Batch/flow |

|

|

|

|

|

|

|

|
| Work-up |

|
—i | —i | —i | —i |

|

|

|
| Health and safety |

|

|

|

|

|

|

|

|
| Ref. | 13 | 12 | 18 | 19 | 9 | 45 | This work | This work |
Biocatalysts are considered as biodegradable and environmentally friendly alternatives to metal-based and traditional acidic catalysts, which enhances the green factor of the developed methods (entries 3, 4, 5, 7, and 8). The possibility of catalyst recycling and its stability are among the most important aspects when considering the environmental impact and applicability of the developed method. Recyclability of designed catalytic systems was proved only for Fe-DTP-ZIF-8 for at least 5 cycles (entry 2) versus at least 10 cycles obtained for MgO·SiO2-C8-LAO in a batch system and high STY = 6651 (g h−1 L−1) for a flow system (entries 7 and 8, respectively, this work). The possibility of recycling of Y2O3–ZrO2 (entry 1) and Novozym 435 (entry 4) was only postulated. Atom economy for esterification reactions is lower than 100% due to the formation of water molecules during the synthesis. On the other hand, water can be beneficial during work-up through extraction and can also enhance the catalytic performance of lipase.25,39
Reaction mass efficiency (RME) for furfuryl ester synthesis typically ranges from 31% to 69%, except for entry 3 (95.1%). This efficiency is largely dependent on the yield and the excess of reagents used in each method. The synthesis of furfuryl esters can be performed under solventless conditions, but at high temperature, under pressure and long reaction time (110 °C, 7 h, entry 1; 100 °C, 6 h, entry 2; 60 °C, 533.29 Pa, 6 h, entry 3; 55 °C, 24 h, entry 4; 60 °C, 28 mmHg vacuum, entry 5) or with solvents like dichloromethane (90 °C, microwave 200 W, 30 min, entry 6) and cyclohexane (25 °C, 45 min, entry 7; 25 °C, 10.5 min residence time, entry 8). It is noteworthy that the majority of existing examples of the synthesis of furfuryl esters were performed in batch systems (entries 1–7). However, continuous flow synthesis demonstrated in this work not only enhances production efficiency but also improves the environmental sustainability of the developed method.
The work-up procedure was described only for entries 1,13 6,45 and 7, 8.This work Furfuryl esters can be isolated via distillation under reduced pressure or high temperature, or extraction with organic solvents, such as heptane,This work dichloromethane,13,45 and column chromatography.13,45 The red flags in the Health and safety row for our study resulted from the need for the use of hydrophobic organic solvents to extract the furfuryl ester from the post-reaction mixture. The developed method of continuous flow synthesis of furfuryl esters in the presence of the MgO·SiO2-C8-LAO biocatalyst considered under green chemistry metrics in this work strikes a balance between a highly active, stable, recyclable, and biodegradable catalyst, and an environmentally friendly and sustainable approach to chemical production. Future research should focus on developing more eco-friendly isolation methods to enhance the environmental sustainability of the process.
The sustainable character of the developed method of production of furfuryl octanoate in continuous flow in the presence of the MgO·SiO2-C8-LAO biocatalyst has been already assessed using the Green Chemistry Metrics Analysis. Next, another approach was considered by evaluating not only the ester synthesis method but also the preceding life cycle stages.
To evaluate the method in terms of life cycle thinking,47 a synthesis tree was drawn (Fig. 7).
 | ||
| Fig. 7 Synthesis tree for furfuryl octanoate under continuous flow conditions catalysed by MgO·SiO2-C8-LAO. | ||
All necessary substrates to produce ester, furfuryl alcohol, and octanoic acid can be obtained from natural sources, such as lignocellulosic biomass,48–50 coconut and palm kernel oil51 or microbial production from renewable carbon sources.52 In the context of waste management and closed-loop economy principles, it is especially important to use waste as a resource for producing other substances. Various strategies of furfuryl alcohol and octanoic acid have been employed including continuous flow production, the use of deep eutectic solvents, ionic liquids, and enzymes, all aimed at achieving the most environmentally sustainable technologies.48–52 The production of the MgO·SiO2-C8-LAO biocatalyst includes the use of LAO, which is considered generally safe, whereas several critical factors can be observed in the process of the synthesis of the MgO·SiO2-C8 support. The primary issue arises from the inclusion of volatile fluorohydrocarbons in the synthesis of silicon compounds, which poses environmental risks such as acid rain, ozone layer depletion, and global warming. However, some alternative methods for preparing valuable organosilicon molecules using enzymes have already been reported.53
Experimental
Materials and methods
All reagents and lipases were supplied by Sigma-Aldrich (Merck Group, Poland). MgO·SiO2 was synthesized according to the literature (Ciesielczyk et al., 2014; Kołodziejczak-Radzimska et al., 2018).33,36 GC was performed on a SHIMADZU GC-2010 Plus equipped with a Zebron ZB-5MSi column (30 m × 0.32 mm; 0.25 μm). TG/DTG analyses of all materials were performed using a TGA/SDTA 851 Mettler-Toledo thermogravimeter in the temperature range 25 °C–800 °C at a heating rate of 10 °C min−1 in a stream of nitrogen (60 mL min−1). BET surface area (SBET), average pore size (dp) and average pore volume (Vp) of the MgO·SiO2 based materials were determined on an ASAP 2020 Plus Version 2.00 using low-temperature nitrogen adsorption–desorption (−196 °C) according to the BET method and BJH model. SEM-EDX images of all materials were recorded on a Phenom Pro Desktop SEM coupled with an EDS detector (15 kV) (Thermo Fischer Scientific). FTIR analyses of modified materials were performed on a Vertex 70 apparatus over 4000–420 cm−1 (at a resolution of 0.5 cm−1) (Bruker, Germany) by mixing 2.0 mg of the material and 200 mg of anhydrous KBr. 1H NMR and 13C NMR spectra of furfuryl esters were recorded on a Varian system (400 MHz and 101 MHz, respectively). The Lowry test was utilized to determine the protein concentration in the commercial solutions of Aspergillus oryzae and CALB. A standard curve was generated employing bovine serum albumin. A mixture comprising 0.5 mL of prepared Lowry solution and 0.5 mL of diluted sample was created (20 min incubation), followed by the addition of 0.25 mL of Folin solution (30 min incubation), and the absorbance measurement using a spectrophotometer.:water mixture (4 mL; 4:1, v:v) in a 25 mL round-bottom flask, then triethoxy(octyl)silane or triethoxy(hexadecyl)silane (0.2 mmol) was added, and the reaction mixture was stirred at 85 °C for 24 h. After that, the material was filtered off, washed with isopropanol (3 × 20 mL) and dried under vacuum on a Schlenk line (rt, 4 h).
:3, n:n) was pumped with a flow of 0.070 mL min−1 through the column reactor filled with 300 mg of MgO·SiO2-C8-LAO biocatalyst at 25 °C for 48 h. Reaction progress was monitored by GC.
Conclusions
Green chemistry is essential for achieving Sustainable Development Goals. Innovative chemical methodologies must focus on consolidating high-yield reactions into a minimal number of unit operations, by using green solvents and integrating advanced starting materials. At every early stage of research, assessing developing technologies using green metrics is necessary. Despite their excellent catalytic properties, enzymes typically require enhancement before being implemented on an industrial scale, where multiple cycles of high yield processes are desired. One of the properties typically improved through immobilization is enzyme stability. Other critical enzyme properties that should be enhanced for prolonged use in industrial reactors include activity, resistance to inhibition by reaction products, and ease of regeneration. The strategy to improve enzyme properties during the performance via a tailor-made enzyme immobilization protocol is the goal of this work.The novelty of this work lies in the innovative approach to a catalyst matrix (MgO·SiO2) and modifying its surface with alkyl groups for LAO immobilization, which brings a high enhancement in the efficiency of the enzyme during 30 h of performance in the synthesis of esters from biomass-derived furfuryl alcohol and C8–C18 carboxylic acids in a flow system while maintaining low environmental hazard.
The sustainability of the developed method was assessed through green metrics and life cycle thinking, which are effective tools for designing environmentally friendly chemical processes.
Author contributions
Anna Wolny: writing – original draft, conceptualization, methodology, validation, data curation, investigation, formal analysis, project administration, funding acquisition. Dagmara Więcławik: investigation. Jakub Zdarta: writing – review & editing, resources. Sebastian Jurczyk: formal analysis. Teofil Jesionowski: conceptualization, writing – review & editing. Anna Chrobok: conceptualization, methodology, data curation, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Science Centre, Poland. Grant no. UMO-2023/49/N/ST8/01633 (PRELUDIUM-22).References
- A. Saravanan, P. R. Yaashikaa, P. S. Kumar, P. Thamarai, V. C. Deivayanai and G. Rangasamy, Ind. Crops Prod., 2023, 200, 116822, DOI:10.1016/j.indcrop.2023.116822.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838, DOI:10.1021/acs.chemrev.7b00203.
- J. M. Bolivar, J. M. Woodley and R. Fernandez-Lafuente, Chem. Soc. Rev., 2022, 51, 6251–6290, 10.1039/D2CS00083K.
- P. Lozano and E. García-Verdugo, Green Chem., 2023, 25, 7041–7057, 10.1039/D3GC01878D.
- A. Racha, C. Samanta, S. Sreekantan and B. Marimuthu, Energy Fuels, 2023, 37, 11475–11496, DOI:10.1021/acs.energyfuels.3c01174.
- N. Li and M. H. Zong, ACS Catal., 2022, 12, 10080–10114, DOI:10.1021/acscatal.2c02912.
- Q. Li, C. Ma, J. Di, J. Ni and Y. C. He, Bioresour. Technol., 2022, 34, 126376, DOI:10.1016/j.biortech.2021.126376.
- V. Antón, J. Muñoz-Embid, I. Gascón, M. Artal and C. Lafuente, Energy Fuels, 2017, 31, 4143–4154, DOI:10.1021/acs.energyfuels.7b00304.
- S. Mukherjee and M. Ghosh, Carbohydr. Polym., 2017, 157, 1076–1084, DOI:10.1016/j.carbpol.2016.10.075.
- M. E. Fortunato, F. Taddeo, R. Vitiello, R. Turco, R. Tesser, V. Russo and M. Di Serio, ACS Sustainable Chem. Eng., 2023, 11, 12406–12413, DOI:10.1021/acssuschemeng.3c02882.
- A. L. Raley, J. P. Beldin and R. J. Franks, J. Chem. Biochem., 2019, 7 DOI:10.15640/jcb.v7n1a3.
- R. S. Malkar, H. Daly, C. Hardacre and G. D. Yadav, React. Chem. Eng., 2019, 4, 1790–1802, 10.1039/C9RE00167K.
- P. Kumar, R. K. Pandey, M. S. Bodas, S. P. Dagade, M. K. Dongare and A. V. Ramaswamy, J. Mol. Catal. A: Chem., 2002, 181, 207–213, DOI:10.1016/S1381-1169(01)00365-X.
- T. T. Mokoena, W. A. A. Ddamba and B. M. Keikotlhaile, S. Afr. J. Chem., 1999, 52, 73–78 CAS.
- E. M. Wewerka, J. Appl. Polym. Sci., 1968, 12, 1671–1681, DOI:10.1002/app.1968.070120716.
- R. González, R. Martínez and P. Ortiz, Makromol. Chem., 1992, 193, 1–9, DOI:10.1002/macp.1992.021930101.
- H. E. Hoydonckx, D. E. De Vos, S. A. Chavan and P. A. Jacobs, Top. Catal., 2004, 27, 83–96, DOI:10.1023/B:TOCA.0000013543.96438.1a.
- A. Sengupta, T. Dey, M. Ghosh, J. Ghosh and S. Ghosh, J. Inst. Eng. (India): Ser. E, 2012, 93, 31–36, DOI:10.1007/s40034-013-0008-7.
- Y. Satyawali, V. Akemeier, W. Dejonghe, H. De Wever and W. Van Hecke, Waste Biomass Valorization, 2019, 10, 311–317, DOI:10.1007/s12649-017-0060-5.
- M. Markiton, S. Boncel, D. Janas and A. Chrobok, ACS Sustainable Chem. Eng., 2017, 5, 1685–1691, DOI:10.1021/acssuschemeng.6b02433.
- A. Szelwicka, S. Boncel, S. Jurczyk and A. Chrobok, Appl. Catal., A, 2019, 574, 41–47, DOI:10.1016/j.apcata.2019.01.030.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2017, 40, 1451–1463, DOI:10.1016/j.enzmictec.2007.01.018.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904, DOI:10.1002/adsc.201100534.
- E. P. Cipolatti, A. Valério, R. O. Henriques, D. E. Moritz, J. L. Ninow, D. M. G. Freire, E. A. Manoel, R. Fernandez-Lafuente and D. De Oliveira, RSC Adv., 2016, 6, 104675–104692, 10.1039/C6RA22047A.
- R. Fernandez-Lafuente, P. Armisén, P. Sabuquillo, G. Fernández-Lorente and J. M. Guisán, Chem. Phys. Lipids, 1998, 93, 185–197, DOI:10.1016/S0009-3084(98)00042-5.
- G. Fernandez-Lorente, J. Rocha-Martín and J. M. Guisan, Methods Mol. Biol., 2020, 2100, 143–158, DOI:10.1007/978-1-0716-0215-7_9.
- S. Arana-Peña, N. S. Rios, D. Carballares, L. R. B. Gonçalves and R. Fernandez-Lafuente, Catal. Today, 2021, 362, 130–140, DOI:10.1016/j.cattod.2020.03.059.
- A. Wolny and A. Chrobok, Nanomaterials, 2021, 11, 2030, DOI:10.3390/nano11082030.
- A. Drozdz, A. Chrobok, S. Baj, K. Szymańska, J. Mrowiec-Białoń and A. B. JarzÈ©bski, Appl. Catal., A, 2013, 467, 163–170, DOI:10.1016/j.apcata.2013.07.009.
- A. Wolny, A. Siewniak, J. Zdarta, F. Ciesielczyk, P. Latos, S. Jurczyk, L. D. Nghiem, T. Jesionowski and A. Chrobok, Environ. Technol. Innovation, 2022, 28, 102936, DOI:10.1016/j.eti.2022.102936.
- J. J. Shangguan, Y. Q. Liu, F. J. Wang, J. Zhao, L. Q. Fan, S. X. Li and J. H. Xu, Appl. Biochem. Biotechnol., 2011, 165, 949–962, DOI:10.1007/s12010-011-9311-2.
- S. Salgin and S. Takac, Chem. Eng. Technol., 2007, 30, 1739–1743, DOI:10.1002/ceat.200700285.
- A. Kołodziejczak-Radzimska, J. Zdarta, F. Ciesielczyk and T. Jesionowski, Korean J. Chem. Eng., 2018, 35, 2220–2231, DOI:10.1007/s11814-018-0146-1.
- R. A. Sheldon, Green Chem., 2021, 23, 8406–8427, 10.1039/D1GC03145G.
- P. Lozano, E. Alvarez, J. M. Bernal, S. Nieto, C. Gomez and G. Sanchez-Gomez, Curr. Green Chem., 2017, 4, 116–129, DOI:10.2174/2213346104666171115160413.
- F. Ciesielczyk, M. Przybysz, J. Zdarta, A. Piasecki, D. Paukszta and T. Jesionowski, J. Sol-Gel Sci. Technol., 2014, 71, 501–513, DOI:10.1007/s10971-014-3398-1.
- R. M. Blanco, P. Terreros, M. Fernández-Pérez, C. Otero and G. Diaz-Gonzalez, J. Mol. Catal. B: Enzym., 2004, 30, 83–93, DOI:10.1016/j.molcatb.2004.03.012.
- J. Lin, Y. Cheng, O. M. Lai, C. P. Tan, W. Panpipat, C. Shen and L. Z. Cheong, ChemistrySelect, 2022, 7, e202202721, DOI:10.1002/slct.202202721.
- A. Kumar, K. Dhar and S. S. Kanwar, Biol. Proced. Online, 2016, 18 DOI:10.1186/s12575-016-0033-2.
- J. Toida, K. Kondoh, M. Fukuzawa, K. Ohnishi and J. Sekiguchi, Biosci., Biotechnol., Biochem., 1995, 59, 1199–1203, DOI:10.1271/bbb.59.1199.
- P. De Santntis, L. E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184, 10.1039/D0RE00335B.
- J. Coloma, Y. Guiavarc'h, P. L. Hagedoorn and U. Hanefeld, Catal. Sci. Technol., 2020, 10, 3613–3621, 10.1039/D0CY00604A.
- Q. Peng, B. Zang, W. Zhao, D. Li, J. Ren, F. Ji and L. Jia, Catal. Sci. Technol., 2020, 10, 484–492, 10.1039/C9CY01856E.
- L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247, 10.1039/D3SC00992K.
- Ł. Janczewski, D. Zieliński and B. Kolesińska, Open Chem., 2021, 19, 265–280, DOI:10.1515/chem-2021-0034.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121, 10.1039/C5GC00340G.
- P. Jessop, Green Chem., 2020, 22, 13–15, 10.1039/C9GC90119A.
- V. K. Vaidyanathan, K. Saikia, P. S. Kumar, A. K. Rathankumar, G. Rangasamy and G. D. Saratale, Bioresour. Technol., 2023, 378, 128975, DOI:10.1016/j.biortech.2023.128975.
- Y. Bao, Z. Du, X. Liu, H. Liu, J. Tang, C. Qin, C. Liang, C. Huang and S. Yao, Green Chem., 2024, 26, 6318–6338, 10.1039/D4GC00883A.
- K. J. Yong, T. Y. Wu, C. B. T. Loong Lee, Z. J. Lee, Q. Liu, J. Md Jahim, Q. Zhou and L. Zhang, Biomass Bioenergy, 2022, 161, 106458, DOI:10.1016/j.biombioe.2022.106458.
- Y. Chen, Y. She, J. Lei, D. Wang, S. Wu and K. Men, IOP Conf. Ser.: Earth Environ. Sci., 2021, 705, 012013, DOI:10.1088/1755-1315/705/1/012013.
- J. H. Ahn, K. H. Jung, E. S. Lim, S. M. Kim, S. O. Han and Y. Um, Bioresour. Technol., 2023, 381, 129147, DOI:10.1016/j.biortech.2023.129147.
- N. S. Sarai, B. J. Levin, J. M. Roberts, D. E. Katsoulis and F. H. Arnold, ACS Cent. Sci., 2021, 7, 944–953, DOI:10.1021/acscentsci.1c00182.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, TGA/DTG, SEM-EDS, NMR, graphs, appendix 2 (green metrics analysis). See DOI: https://doi.org/10.1039/d4gc03821e |
| This journal is © The Royal Society of Chemistry 2024 |