
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of the La-doped CuCoAl hydrotalcite catalyst for selective hydrogenation of furfuryl alcohol to 1,5-pentanediol†
Jingjing
Tan
*a,
Hailong
Huang
 ab,
Yuanna
Zhang
ab,
Jinglei
Cui
*c,
Jing
Zhang
d,
Long
Huang
e,
Yongzhao
Wang
a and
Yulei
Zhu
*fg
ab,
Yuanna
Zhang
ab,
Jinglei
Cui
*c,
Jing
Zhang
d,
Long
Huang
e,
Yongzhao
Wang
a and
Yulei
Zhu
*fg
aEngineering Research Center of Ministry of Education for Fine Chemicals, Shanxi University, Taiyuan 030006, Shanxi Province, PR China. E-mail: tanjingjing@sxu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shanxi University, Taiyuan 030006, Shanxi Province, PR China
cInstitute of Resources and Environmental Engineering, Shanxi University, Taiyuan 030006, China. E-mail: cuijl@sxu.edu.cn
dInstitute of Applied Chemistry, Shanxi University, Taiyuan, 030006, P. R. China
eBeijing Key Laboratory of Fuels Cleaning and Advanced Catalytic Emission Reduction Technology, Beijing Institute of Petrochemical Technology, Beijing 102617, China
fState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, PR China. E-mail: zhuyulei@sxicc.ac.cn
gSynfuels China Co. Ltd., Beijing, PR China
First published on 5th October 2024
Abstract
1,5-Pentanediol (1,5-PeD) is an important raw material for the preparation of degradable polyesters, polyurethanes and pharmaceutical intermediates. Efficient synthesis of 1,5-PeD from biomass-derived furfuryl alcohol (FFA) by hydrogenation is a green synthetic route instead of using fossil raw material production. Nevertheless, it suffers from great challenges as the various adsorption configurations of FFA on the catalyst surface induce diverse product distributions and low selectivity for 1,5-PeD. Herein, a CuCoAl hydrotalcite catalyst modified by La was fabricated and applied in the hydrogenation of FFA to 1,5-PeD. The results demonstrated that in the catalyst doped with La via deposition–precipitation methods (La/CuCoAl-DP) there appeared a strong Cu–La interaction, and it exhibited superior activity compared with other catalysts. A near 60% yield of 1,5-PeD was achieved under 160 °C, 4 MPa H2 within 2 h. Extensive characterizations including XRD, HRTEM, N2O-TPD and CO2-TPD demonstrated that the doping of La improved markedly the dispersion of Cu and the concentration of strong basic sites. Furthermore, HRTEM and the in situ XPS characterization verified that the addition of La species promoted the formation of a Cu–La interface with a stable Cun+–O–La(OH)3 structure on the catalyst surface. Such Cun+–O–La(OH)3 sites can simultaneously activate the furan ring and the –OH group in FFA with an intermediate six-membered ring transition state, leading to high selective cleavage of the C2–O1 bond in the furan ring to 1,5-PeD. Meanwhile, the DFT calculation results corroborated that the modifying by La species remarkably promoted the C2-end tilted adsorption of FFA on the catalyst surface and enhanced the ability of the catalyst to activate hydrogen. This study provided a new strategy for the high-value utilization of biomass resources and the development of multi-center catalysts.
1. Introduction
1,5-Pentanediol (1,5-PeD) is an important monomer for the preparation of biodegradable polyesters, unsaturated polyesters, polyurethanes and pharmaceutical intermediates.1–3 Moreover, it is expected to replace 1,4-butanediol and 1,6-hexanediol in the polyester market due to its excellent thermal stability. At present, 1,5-PeD is in great demand, while the production capacity cannot meet the required levels. In industry, 1,5-PeD has been mainly synthesized from non-renewable petroleum-based C5 hydrocarbons, likely glutaric acid and its derivatives. The production process is complex and has high energy consumption, causing the high price and limited utilization of 1,5-PeD. According to the statistics, its global economic output reached approximately $133 million, and the price soared to $9700 per ton in 2020.3 Thus, it is of great significance and high economic value to explore a green, sustainable and efficient process for the synthesis of 1,5-PeD.Biomass is a renewable organic carbon with the advantages of having a wide variety of sources and being highly renewable, which makes it one of the most explored potential new energy sources.4–9 Compared with fossil materials, biomass and its derivatives are more suitable as raw materials for the production of diols as they possessed high oxygen content and possess C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.10 Furfural is an important biomass platform compound, and its annual production exceeds 400
O bonds.10 Furfural is an important biomass platform compound, and its annual production exceeds 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons.11,12 More than 75% of furfural was used for the production of furfuryl alcohol (FFA). FFA can be converted to 1,5-PeD by the hydrogenation and highly selective breakage of the C2–O1 bond in the furan ring (Fig. 1). This is a green and sustainable process, which is promising for the synthesis of 1,5-PeD.
000 tons.11,12 More than 75% of furfural was used for the production of furfuryl alcohol (FFA). FFA can be converted to 1,5-PeD by the hydrogenation and highly selective breakage of the C2–O1 bond in the furan ring (Fig. 1). This is a green and sustainable process, which is promising for the synthesis of 1,5-PeD.
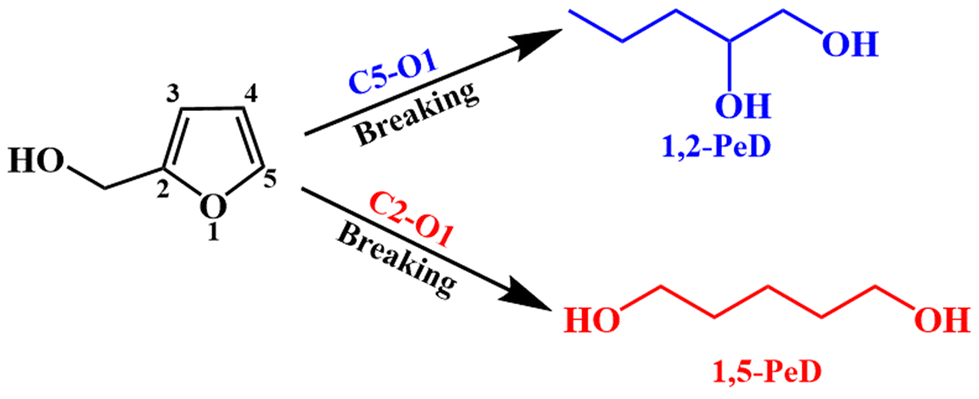 | ||
| Fig. 1 The route of furfuryl alcohol to 1,5-PeD and 1,2-PeD. | ||
It is a great challenge to synthesise 1,5-PeD from FFA with high selectivity due to the diverse adsorption configuration of FFA on the catalyst surface. The product distribution is complex and the selectivity of 1,5-PeD is low. As shown in Fig. 2, FFA can be adsorbed on the catalyst surface by planar (Fig. 2A) and tilted adsorption configurations, in which tilted adsorption includes C5-end and C2-end adsorption (Fig. 2B and C).13,14 Tetrahydrofurfuryl alcohol (THFA) generated easily when FFA was adsorbed by planar (furan ring) adsorption (Fig. 2A) on the catalyst surface. In contrast, pentanediols can be obtained via cleavage of the C–O bond in the furan ring when FFA was adsorbed by tilted adsorption configurations (Fig. 2B and C). Typically, C5-end oblique adsorption of FFA was prone to occur due to the large steric hindrance of the –OH group and the high reaction energy barrier of the C2-end, and 1,2-pentanediol (1,2-PeD) will be the dominant product by C5–O1 bond cleavage (Fig. 2B).13,14 Therefore, the surface active site in the catalyst is required to possess the ability to adsorb and anchor the –OH group outside the furan ring, which will reduce the reaction energy barrier and eliminate the steric hindrance. Subsequently, the C2-end of the furan ring would close to the catalyst surface, and the oblique adsorption of FFA can occur (Fig. 2C). Then, the active H on the metal site attacks the C2C3 bond, the C2–O1 bond will be weakened and broken to form 1,5-PeD.
 | ||
| Fig. 2 The adsorption configurations of FFA on the catalyst surface. | ||
Recently, researchers have developed a few precious metal and non-precious metal catalysts to modulate the adsorption performance of FFA on the catalyst surface and enhance the selectivity of 1,5-PeD. Meanwhile, metal–acid or metal–base synergistic catalytic mechanisms were proposed. For instance, Chen et al. explored the metal–acid bifunctional Cu/Al2O3 catalysts for FFA conversion.15 The conversion of FFA was 85.8% together with an 18.9% yield of 1,5-PeD at 140 °C, 8 MPa H2 within 8 h. They proposed that the –OH group of FFA can be anchored by Lewis-acid sites derived from Al2O3, inducing C2-end oblique adsorption of FFA and promoting the cleavage of the C2–O1 bond to produce 1,5-PeD. Peng et al. claimed that a 42.5% yield of 1,5-PeD was obtained from FFA hydrogenation on a Ni–Co–Al mixed metal oxides catalyst at 160 °C, 4 MPa H2 for 4 h.16 They considered that the –OH group of FFA was absorbed by CoOx species due to its oxophilicity. The tilted adsorption of FFA occurred on the catalyst surface, thereby promoting the cleavage of the C2–O1 bond to create 1,5-PeD. These acid sites in the catalyst can promote FFA to show C2-end oblique adsorption, which is favored for the production of 1,5-PeD. However, such acid sites also boosted the generation of 2-methylfuran and n-pentanol by the hydrogenolysis of FFA and 1,5-PeD, respectively.17 Therefore, metal–basic dual-site catalysts and their catalytic mechanism have gained more attention. For example, basic support, likely CeO2, MnOx and hydrotalcite supported Ru or Pt were used to transform FFA to 1,5-PeD at 150–170 °C and 1–2 MPa H2.13,18–20 The authors considered that the surface basic site in the catalyst can anchor the –OH group of FFA to form an alkoxide structure, making FFA absorb on the catalyst in the form of C2-end oblique adsorption. However, 1,2-PeD was the principal product with a yield of 1,5-PeD (<10%) due to the insufficient alkaline strength. Meanwhile, such noble metal catalysts are difficult to use for large-scale industrial production due to their high price. Thus, researchers have paid more attention to the non-precious metal catalysts, like Cu, Ni and Co-based catalysts.14,21,22 Liu et al. designed a CuMgAl hydrotalcite catalyst for the conversion of FFA to 1,5-PeD. A 25.5% yield of 1,5-PeD was gained at 140 °C, 6 MPa H2 within 24 h. They claimed that the –CH2OH of FFA interacted with the basic sites of hydrotalcite to form an alkoxide complex, inducing the C2-end oblique adsorption to generate 1,5-PeD.21 Our group explored CuMgAl and CuCoAl hydrotalcite catalysts with basic sites to catalyze the transformation of FFA to 1,5-PeD, respectively. The catalyst CuMgAl displayed a 21% yield of 1,5-PeD at 140 °C, 4 MPa H2 for 8 h.22 Notably, the yield of 1,5-PeD raised to 45% on CuCoAl hydrotalcite catalyst in a shorter reaction time under the same reaction conditions.14 Besides the C2-end tilted adsorption of FFA being promoted, it was observed that the CoOx species derived from a Co3O4 reduction facilitated the adsorption and activation of the C2C3 bond in FFA, resulting in the weakening and cleavage of C2–O1 bond to 1,5-PeD.23,24 However, the yield of the main by-product THFA was more than 20%, which can be assigned to the inadequate strength of the basic sites. Meanwhile, the Co0 and CoOx were uniformly dispersed on the catalyst surface, resulting in the parallel adsorption of FFA for THFA production (Fig. 2A). Moreover, THFA is more stable than FFA as the furan ring is saturated, and it would not convert to 1,5-PeD over the CuCoAl catalyst. To further enhance the selectivity of 1,5-PeD, stronger basic sites in the catalyst were required, which will regulate the adsorption configuration of FFA with C2-end tilted adsorption rather than parallel adsorption. Furthermore, this catalyst can catalyze THFA conversion to 1,5-PeD.
Rare earth metal oxides and hydroxides, especially La2O3 and La(OH)3, are widely used in catalysis due their strong basicity and good chemical stability.2,3,25 It is reported that the strong basic sites of La(OH)3 in the Ni–Ln (Ln = La, Pr and Sm) catalyst can extract H from the –OH group of THFA, making it deprotonate to form alkoxide species, thereby promoting the C2-end inclined adsorption for THFA.3 Then, the C2–O1 bond was cleaved and hydrogenated to 1,5-PeD. However, the variation of La species, the relationship between several active sites and the cooperative catalysis are not clear.
Herein, La-doped CuCoAl catalysts were explored and used in the synthesis of 1,5-PeD from FFA for the first time. The impact of La doping on the catalyst structure and catalytic activity was studied. The results showed that the catalyst doped with La by deposition–precipitation methods exhibited superior catalytic activity. The yield of 1,5-PeD reached about 60% at 160 °C, 4 MPa H2 within 2 h. Moreover, THFA can be transformed to 1,5-PeD under the same conditions when it was the reaction substrate. Extensive characterizations, including XRD, H2-TPR, H2-TPD, in situ XPS, N2O-TPD, HRTEM, CO2-TPD and DFT calculations were executed to understand the active sites and their synergistic catalysis. This study will provide a new strategy for the high-value utilization of biomass resources and the development of multi-center catalysts.
2. Experimental
2.1 Synthesis of the catalysts
The CuCoAl hydrotalcite-like catalyst precursors were synthesized by a co-precipitation method with a fixed molar ratio of metals (Cu:Co:Al = 1:29:10). The La/CuCoAl-DP, La/CuCoAl-CP and La/CuCoAl-IM catalyst were prepared by the deposition–precipitation method, co-precipitation method and incipient wetness impregnation method, respectively. The details are shown in the ESI.†
2.2 Catalyst characterization and DFT calculation
The catalysts’ structure and properties were measured by XRD, BET, ICP-OES, H2-TPR, H2-TPD, TEM, in situ XPS and CO2-TPD. The adsorption behaviors of furfuryl alcohol and the energy values for dissociative H2 adsorption over CuCoAl and La/CuCoAl catalysts were investigated by DFT calculation. More details are shown in the ESI.†2.3 Catalyst evaluation and product analysis
The catalytic performance of the catalysts for the conversion of FFA to 1,5-PeD was performed in a 100 ml stainless-steel autoclave (MSG-100-P5, AnhuKemi Machinery Technology Co., Ltd, Hefei, China) with a gas input system and mechanical agitation. Typically, 0.1 g catalyst (reduced at 400 °C for 1 h within an H2/N2 mixed atmosphere), 0.5 g FFA and 39.5 g solvent were added into the autoclave. Afterward, the reactor was sealed and flushed with N2 and H2 5 times, respectively. Then, H2 pressure was added to reach the required pressure. The control program temperature was set. Next, the reaction system was heated (5 °C min−1) to the required temperature. After the reactions, the reactor was cooled to normal temperature, and the H2 inside the reactor was vented. The mixture was centrifuged to remove the catalyst, a clear solution filtered using a 0.45 μm needle-type filter was obtained. The Agilent 7890 gas chromatograph with a hydrogen flame detector was used to analyze the reaction products. The chromatographic column was HP-INNOWAX (30 m × 0.32 mm × 0.5 μm). The catalyst was recycled by the following procedure: After finishing the first hydrogenation run, the catalyst was separated from the reaction mixture by a high rate of centrifugation. Then, it was washed with ethanol 3 times for the next run under identical conditions.3. Results and discussion
3.1 Evaluation of catalytic performance
Fig. 3a shows the preparation of 1,5-PeD from FFA on different catalysts. The activity of the catalyst and the selectivity of 1,5-PeD were enhanced apparently over the La/CuCoAl catalyst compared with the CuCoAl catalyst. Moreover, the catalyst synthesized by the deposition–precipitation method (La/CuCoAl-DP) exhibited higher FFA conversion and 1,5-PeD selectivity than that prepared by the impregnation (La/CuCoAl-IM) and co-precipitation (La/CuCoAl-CP) method. Specifically, the conversion of FFA increased markedly from 53% to 82%, and the 1,5-PeD selectivity reached 50.4% over the La/CuCoAl-DP catalyst. This phenomenon implied that more effective La active species can be formed by the deposition–precipitation method. Such La species have high basicity and can act as adsorption sites to adsorb the –OH group in FFA. Then, FFA was absorbed on the catalyst surface by C2-end oblique adsorption, forming an alkoxide structure and facilitating the generation of 1,5-PeD.3,25 Additionally, such La species can promote the activating of H2 to form active hydrogen by metal Cu sites. Generally, the La species in the catalyst might exist in the form of La(OH)3, LaOOH or La2O3, in which La(OH)3 can transform to LaOOH by dehydration, and LaOOH was further dehydrated to obtain La2O3 with an elevated calcination temperature in air (typically high than 590 °C).26 The order of basic sites for these La species were La(OH)3 > LaOOH > La2O3.27 To prevent the conversion of La(OH)3 into La2O3 during the calcination process, the catalysts were reduced with H2/N2 at 400 °C without calcination in air. The catalyst activity and the selectivity of 1,5-PeD were at the same level compared with that of the calcined catalyst (Table S1,† entries 1 and 2), indicating that the current calcination and reduction condition did not change the structure of the La species. Meanwhile, multiple complex metal oxides were generated and the interactions between Co and other metal oxides were enhanced by calcination. In comparison, this interaction was weak in the catalyst without calcination, leading to the Co species being relatively easier to reduce, and producing more Co0 sites. The Co0 sites have been identified to be more favorable in the hydrogenation of the CC bond, leading to a higher selectivity of THFA.28 To further verify the role of La(OH)3 species, the catalysts mixed mechanically with different content of pure La(OH)3 and reduced CuCoAl were applied to transform FFA to 1,5-PeD (Table S1,† entries 3–5). A significant improvement in FFA conversion and 1,5-PeD selectivity, together with the decrease in THFA selectivity occurred over La(OH)3 (content 1.5%–3%) & CuCoAl mixed catalyst compared with CuCoAl catalyst. The conversion of FFA reached 87.6% with a 52.9% selectivity of 1,5-PeD. Meanwhile, as expected, the selectivity of THFA was reduced from 22.6% to 17.5%. These results further indicated that La(OH)3 species play a crucial role in the conversion of FFA to 1,5-PeD. However, both the FFA transformation and the 1,5-PeD selectivity decreased with further increasing of the content of La(OH)3. The reason might be that the excessive La(OH)3 encapsulated the Cu sites, reducing the amount of surface Cu0 and leading to a great decrease in activity. Notably, the catalytic activity and 1,5-PeD selectivity over La/CuCoAl-DP were much higher than those over the catalyst with physically-mixed La species. This was ascribed to the stronger interaction between La sites and active metals (Cu or CoOx) for doping La species by the deposition–precipitation method than that with doped La species via physical mixing. Furthermore, THFA was used as the substrate to verify the effect of La (Table S2†). 1,5-PeD was detected in the product over the La/CuCoAl-DP catalyst, while it was not detected on the CuCoAl catalyst and the main product was 2-MTHF. This result confirmed that the doping of La promoted the ring cleavage of THFA.
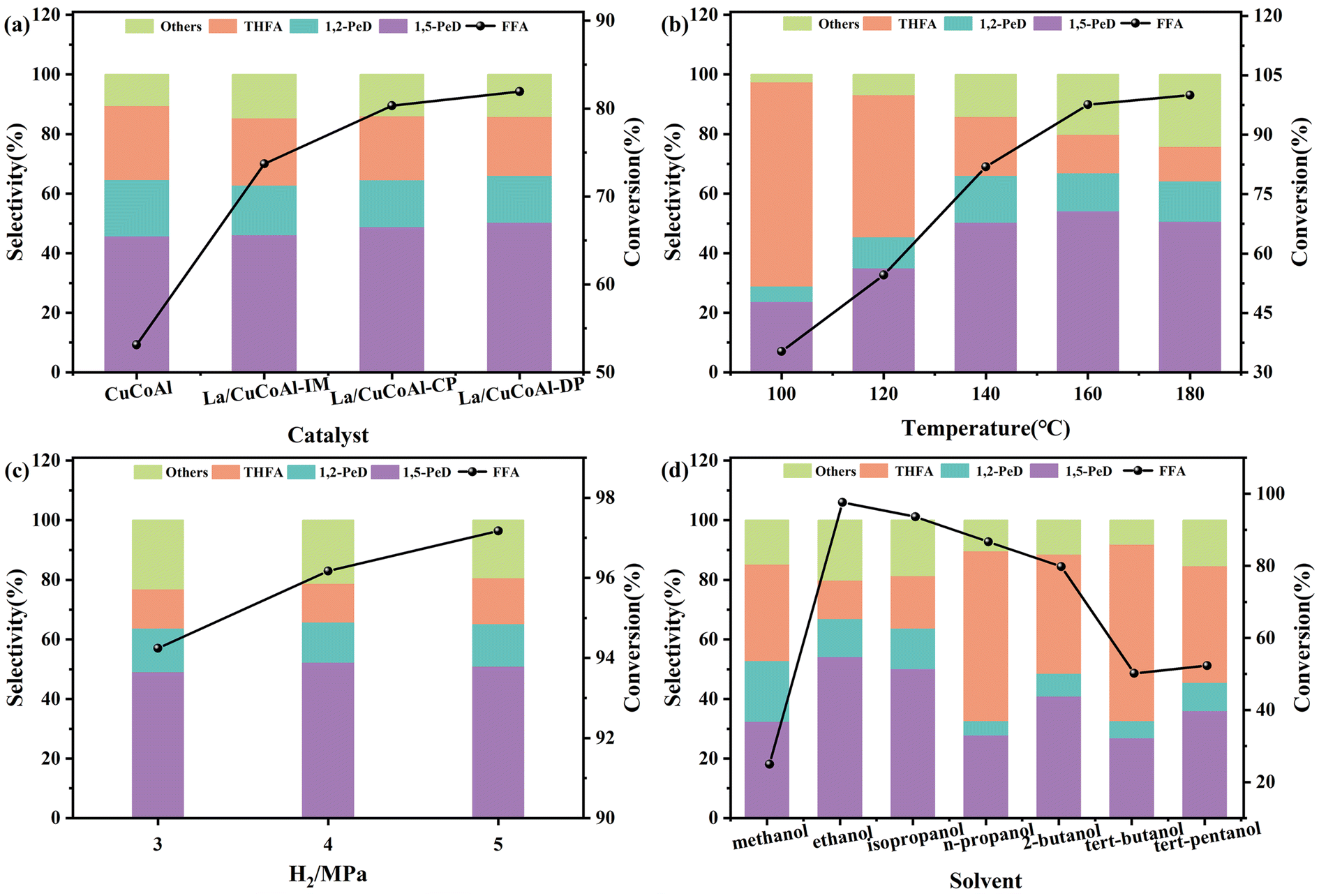 | ||
| Fig. 3 The test for the hydrogenation of FFA. (a) Effect of catalyst, (b) Effect of temperature, (c) Effect of H2 pressure, (d) Effect of solvent. Reaction conditions: FFA 0.5 g, catalyst 0.1 g, ethanol 39.5 g, reaction time 2 h, carbon balance in all reactions ≥ 97%. THFA: tetrahydrofurfuryl alcohol; FFA: furfuryl alcohol; 1,2-PeD: 1,2-pentanediol; 1,5-PeD: 1,5-pentanediol, others: n-pentanol, 2-pentanol and 1-butanol, 2-MF: 2-methylfuran. | ||
Fig. 3b depicts the impact of reaction temperature on the transformation of FFA to 1,5-PeD on the La/CuCoAl-DP catalyst. The conversion of FFA improved monotonically from 35.3% to 100%, while the selectivity of 1,5-PeD displayed a “volcano pattern” with rising the temperature from 100 °C to 180 °C. The 1,5-PeD selectivity improved initially until achieving the highest value (54.2%) at 160 °C, accompanied by the visible decline of THFA selectivity. However, the 1,5-PeD selectivity gradually decreased when the temperature further increased to 180 °C as more over-hydrogenated products (n-pentanol, 2-pentanol and 1-butanol) were formed. The product distribution changed evidently with the varying of the temperature, which might be related to the variation of FFA adsorption performance on the catalyst surface due to its heat-sensitive nature.16,29,30 It has been reported that higher temperatures can promote the weak bond cleavage between the substrate and the surface metal sites (–CC–metal complex). Therefore, the adsorption configuration of the reactant will change to that linked by a stronger bond, likely the –OH-basic site complex on the catalyst surface.31 At lower temperatures (below 140 °C), the selectivity of 1,5-PeD is low, even much lower than that of THFA. This might result from the strong affinity between the catalyst and the furan ring with parallel adsorption for FFA without steric hindrance and the reaction energy barrier being much lower. Such an adsorption performance of FFA facilitated the generation of THFA by furan ring hydrogenation,16 which was too stable to allow ring-opening for 1,5-PeD production.30 However, the strong basic sites on the catalyst surface can anchor the –OH groups in FFA to form an alkoxide structure when the reaction temperature is elevated. This resulted in a C2-end tilted adsorption configuration, while it reduced the affinity of the C2C3 bond on the catalyst surface. The hydrogenolysis of –CH3OH and the hydrogenation of the furan ring can be prevented with this adsorption configuration, thereby enhancing the selectivity of 1,5-PeD. Thus, appropriate reaction temperature and suitable adsorption structure are indispensable for the conversion of FFA to 1,5-PeD.
Other reaction conditions, including H2 pressure (Fig. 3c) and the solvents used (Fig. 3d) were examined to ensure their influence on FFA conversion. With an increase in H2 pressure from 3 MPa to 5 MPa, the conversion of FFA grew linearly, while the selectivity of 1,5-PeD showed a tendency to the normal distribution curve. The selectivity of 1,5-PeD reached the maximum value of 54.2% at 4 MPa H2. Hence, the optimal H2 pressure was 4 MPa, which was used for further investigation.
Fig. 3d illustrates the effect of various alcohols on the synthesis of 1,5-PeD from FFA on the La/CuCoAl-DP catalyst. Methanol, ethanol, n-propanol, isopropanol, isobutanol, 2-butanol, tert-butanol and tert-pentanol were selected as the solvents. Compared to tertiary alcohol solvents (tert-butanol and tert-pentanol), secondary alcohol (isopropanol and isobutanol) solvents exhibited higher activity during the reaction. In particular, the conversion of FFA and the selectivity of 1,5-PeD achieved were 93.6% and 50.2% in isopropanol solvent, respectively. The lower activity of tertiary alcohols with branched chains was likely due to the lack of α-H, resulting in an insufficient hydrogen donation ability. Both the conversion of FFA and the selectivity of 1,5-PeD were relatively low when primary alcohol (methanol) was used as the solvent. This was ascribed to the difficulty of deprotonation for methanol.32–34 Interestingly, the superior activity and higher selectivity of 1,5-PeD were achieved with the solvent of ethanol (the other primary alcohol) compared to isopropanol. This can be attributed to the lower reduction potential of ethanol. Meanwhile, ethanol, derived from biomass, is a green and renewable solvent. Considering economic factors and the toxicity of alcohol compounds, ethanol is an ideal solvent compared to isopropanol. Additionally, the effects of water, which can promote the ring-opening of furan and hydrogenation,18 on the catalyst activity and the product distribution were investigated. As shown in Table S3,† the selectivity of 1,5-PeD was maintained, while the conversion of FFA decreased significantly when a small amount of water was added. Furthermore, besides the conversion, the selectivity of 1,5-PeD declined obviously in the aqueous solution. This can be attributed to the change of the catalyst structure as the “memory effect of the hydrotalcite structures” in the presence of water.35–37 Meanwhile, FFA can be rearranged and hydrogenated to cyclopentanone and cyclopentanol in an aqueous solution with weak acidity,38,39 thereby lowering the selectivity of 1,5-PeD.
The hydrogenation and ring-cleavage of FFA is a complex reaction. The product distribution was influenced remarkably by the reaction time (Fig. S1†). The transformation of FFA raised gradually while the selectivity of 1,5-PeD enhanced initially and then decreased with the increase of reaction time. The conversion of FFA attained 100% together with the highest selectivity of 54.1% for 1,5-PeD within 3 h. However, the selectivity of 1,5-PeD, 1,2-PeD and THFA decreased gradually, while the over-hydrogenated products, such as n-pentanol, 2-pentanol and 1-butanol were formed with further extending the reaction time. It can be inferred that THFA undergoes ring-opening to produce 1,5-PeD, and 1,5-PeD can be further hydrogenated to n-pentanol, 2-pentanol and 1-butanol, which was corroborated by the results illustrated in Tables S2 and S4.†
3.2 Catalysts characterization
Fig. S2† presents the N2 adsorption–desorption isotherms and the pore distribution curves of the calcined catalysts. All catalysts were mesoporous materials as each of them showed a type IV isotherm shape with an H3 hysteresis loop.40Table 1 lists the surface area, pore volume and average pore diameter of the catalyst. The surface area improved evidently after doping La onto the CuCoAl catalyst. The highest specific surface area was obtained for the La/CuCoAl-DP catalyst prepared by the deposition–precipitation method (149.1 m2 g−1), which was conducive to the distribution of active metal sites. The pore size decreased from 30.8 nm to 14.9 nm, and the pore volume decreased from 0.64 cm3 g−1 to 0.56 cm3 g−1. This phenomenon might result from the strong interaction between La sites and Cu sites, and Cu–O–La bonds were formed. Such interaction inhibited grain growth and reduced the crystallite size, thereby enhancing the surface area. The content of each metal in the catalysts was determined by ICP-OES analysis, and each of them is close to the theoretical value. Moreover, some physical properties of Cu, likely its dispersion (DCu) and surface area (SCu) were measured by N2O chemisorption. The dispersion of Cu and its surface area were improved with the doping of La onto CuCoAl. Compared with the catalyst of La/CuCoAl-DP and La/CuCoAl-IM, the La/CuCoAl-CP catalyst showed the highest Cu dispersion and surface area. This can be ascribed to the confinement effect of the hydrotalcite-like structure. However, the catalytic performance of the La/CuCoAl-CP catalyst was lower than that of the La/CuCoAl-DP catalyst. This implied that the excellent activity of the La/CuCoAl-DP catalyst might mainly derive from the strong interaction between Cu and La rather than the high dispersion and surface area of the active metal.| Catalyst | wt%a |
S
BETb (m2 g−1) |
V
pb (cm3 g−1) |
D
Pb (nm) |
D
Cuc (%) |
S
Cuc (m2 g−1) |
||
|---|---|---|---|---|---|---|---|---|
| Cu | Co | La | ||||||
| a Determined by ICP-OES analysis. b Determined by N2-adsorption–desorption, SBET: specific surface area. Vp: pore volume, Dp: pore size. c Determined by N2O chemisorption, DCu: dispersion of metallic Cu, SCu: Cu metallic surface area per gram of catalyst. d The catalyst that had been reused 8 times. | ||||||||
| CuCoAl | 2.8 | 73.8 | — | 83.3 | 0.64 | 30.8 | 10.0 | 1.3 |
| La-CuCoAl-CP | 2.3 | 66.1 | 1.3 | 94.4 | 1.0 | 42.3 | 21.9 | 2.5 |
| La/CuCoAl-IM | 2.6 | 65.3 | 1.9 | 87.8 | 0.87 | 39.7 | 11.0 | 1.4 |
| La/CuCoAl-DP | 2.7 | 68.0 | 1.8 | 149.1 | 0.56 | 14.9 | 16.7 | 2.2 |
| La/CuCoAl-DPd |
2.3 | 66.6 | 1.7 | — | — | — | — | — |
The microstructure and the elemental distribution of the reduced catalysts were analyzed using TEM (Fig. 4). The TEM images (Fig. 4a, c, e and g) reveal the formation of metal Cu nanoparticles (NPs) after reduction and they dispersed uniformly on the catalyst surface. Lattice fringes can be observed from the HRTEM images (Fig. 4b, d, f and h), which can be used to identify the crystalline phases of different metals or metal oxides on the catalyst surface. For instance, the interplanar spacing of 0.21 nm, 0.23 nm and 0.28 nm appeared in each sample, which correspond to Cu (111), CoO (200) and Co3O4 (220), respectively. This implied that Co3O4 was partly reduced during the process of reduction. The interplanar spacing of 0.19 nm assigned to La(OH)3 (002) appeared in La/CuCoAl-CP and La/CuCoAl-DP, while that of 0.16 nm ascribed to La2O3(111) crystal planes was observed in the La/CuCoAl-IM catalyst. The alkalinity of La(OH)3 was stronger than that of La2O3, which likely improved the catalytic efficiency of La/CuCoAl-DP and La/CuCoAl-CP compared to that of La/CuCoAl-IM for the conversion of FFA to 1,5-PeD. Notably, the La(OH)3 species and Cu species in the La/CuCoAl-DP catalyst were close. The structural distortions and poor resolution of lattice fringes with high defect concentration at Cu–La(OH)3 interfaces were observed (Fig. 4h, dashed red circle), displaying that there was a strong interaction between Cu and La (OH)3 species.41,42
 | ||
| Fig. 4 TEM images of the reduced catalyst. (a–b) CuCoAl, (c–d) La/CuCoAl-CP, (e–f) La/CuCoAl-IM, (g–h) La/CuCoAl-DP. | ||
Additionally, HAADF-STEM-EDS was employed to analyze the elemental distribution of the reduced La/CuCoAl-DP catalyst (Fig. 5 and Fig. S3†). The Cu, Co, Al and La species coexisted on the catalyst surface, displaying the successful modification of the CuCoAl catalyst by the La species. Moreover, the La species dispersed uniformly on the catalyst surface and overlapped with Cu and Co species, further verifying a strong interaction exists between La and Cu or Co species.
 | ||
| Fig. 5 The HAADF-STEM-EDS image of the reduced La/CuCoAl-DP catalyst. | ||
Fig. 6 depicts the XRD patterns and H2-TPR for CuCoAl and La/CuCoAl catalysts. As shown in Fig. 6a, all catalyst precursors exhibited diffraction peaks at 2θ 11.7°, 23.7°, 34.7°, 39.5°, 47.0°, 60.5° and 61.8°, corresponding to the (003), (006), (009), (015), (018), (110) and (113) crystal planes of hydrotalcite, respectively.14,35 These characteristic peaks almost disappeared after calcination at 500 °C (Fig. 6b). The emerging peaks centered at 31.3°, 36.8°, 44.8°, 59.4° and 65.2° were attributed to the (220), (311), (400), (511) and (440) crystal planes of Co3O4 (JCPDS 78-1970), respectively. The Co3O4 species is still present in the catalysts after reduction (Fig. 6c), while the peaks became wide and weak compared to those of the calcined catalyst, indicating that part of Co3O4 was reduced to CoOx during the reduction procedure. No Cu species were detected in each sample, implying the smaller NPs of Cu, which were uniformly dispersed on the catalyst surface. Meanwhile, the La species in the catalyst of La/CuCoAl were not observed in the patterns. This can be attributed to the high dispersion of La on the CuCoAl hydrotalcite or the fact that their concentration is below the detection limit of XRD.
 | ||
| Fig. 6 XRD pattern of the catalyst precursor (a); the calcined catalysts (b); the reduced catalysts (c). (d) H2-TPR of the catalysts. | ||
H2-TPR was conducted to understand the redox behavior of the catalyst and the interactions between mixed metal oxides. As shown in Fig. 6d, all catalysts exhibited two H2 consumption peaks within the temperature scope of 150 °C–250 °C and 300 °C–550 °C, respectively. The peak centered at low temperatures (150 °C–250 °C) was related to Cu species reduction, while the peak at high temperatures (300 °C–550 °C) was ascribed to Co species reduction.14,43,44 Compared with the CuCoAl catalyst, the H2 consumption peaks assigned to Cu reduction for each La/CuCoAl were shifted to higher temperatures, suggesting that a strong interaction between La and Cu species existed and inhibited the reduction of Cu sites. Particularly, such a reduction peak in La/CuCoAl-DP was split into two peaks; a new shoulder peak appeared at 230 °C, signifying there are two kinds of Cu species in the catalyst. The main peak at about 180 °C was ascribed to the reduction of Cu species with good dispersion and small NPs, while the shoulder peak at 230 °C was assigned to the reduction of Cu species that strongly interacted with La. For Co species in the La/CuCoAl-DP catalyst, the reduction peak shifted to higher temperatures. This might result from the strong interaction between Cu and La weakening the interaction between Cu and Co, reducing the H spillover from Cu, and making the Co species difficult to reduce.
XPS characterization was carried out to provide further insight into the interaction between the active sites and the electronic structure of Cu, Co and La sites in the catalysts (Fig. 7). As shown in Fig. 7a, a weak “shakeup” satellite peak centered at 940 eV–950 eV assigned to Cu2+ was observed for the La/CuCoAl catalyst, confirming that the Cu2+ were not fully reduced due to the strong interaction between La and Cu species. However, it disappeared in the CuCoAl catalyst, suggesting that Cu2+ was entirely reduced to Cu0 or Cu+. The XANS spectra of Cu were executed to identify Cu0 and Cu+, because they are overlapped in Cu 2p XPS and difficult to distinguish from each other. As depicted in Fig. 7b, three peaks can be deconvoluted in the Cu LMM spectra for all catalysts. Among them, the peak at around 918.8 eV and 916.2 eV matched with the kinetic energy of Cu0 and Cu+ within the limit of accuracy, respectively.45 The new peak at about 911.0 eV intensified after doping La to the catalyst, which was ascribed to the Cun+ in the La/CuCoAl catalyst.46–48 For each sample, the content of Cun+ species increased from 20.6% (CuCoAl) to 42.4% (La/CuCoAl-DP), further verifying the speculation of a strong interaction between the Cu species and La (OH)3. The strong basicity of La (OH)3 and its geometric effect promote the electron transfer from Cu species to La species,47,48 facilitating the formation of a Cun+–O–La(OH)3 structure at the Cu–La interface, corroborated by HRTEM results.2,48,49 The structure–activity relationship between Cun+ and the selectivity of 1,5-PeD demonstrated that the content of Cun+ was positively correlated with the selectivity of 1,5-PeD (Fig. S4†). This indicated that the Cun+ at the Cu–La interface was responsible for the C2–O1 bond cleavage. Additionally, Cun+ can promote the adsorption and activation of H2 to generate more active hydrogen species on the catalyst surface as the unoccupied d-orbitals of Cu can bond with H2, which weakened the H–H bond and facilitated the bond breaking.50,51 As displayed in Fig. 3a, the conversion of FFA accelerated after doping La to the catalyst, which might relate to the formation of Cun+ at the Cu–La interface.
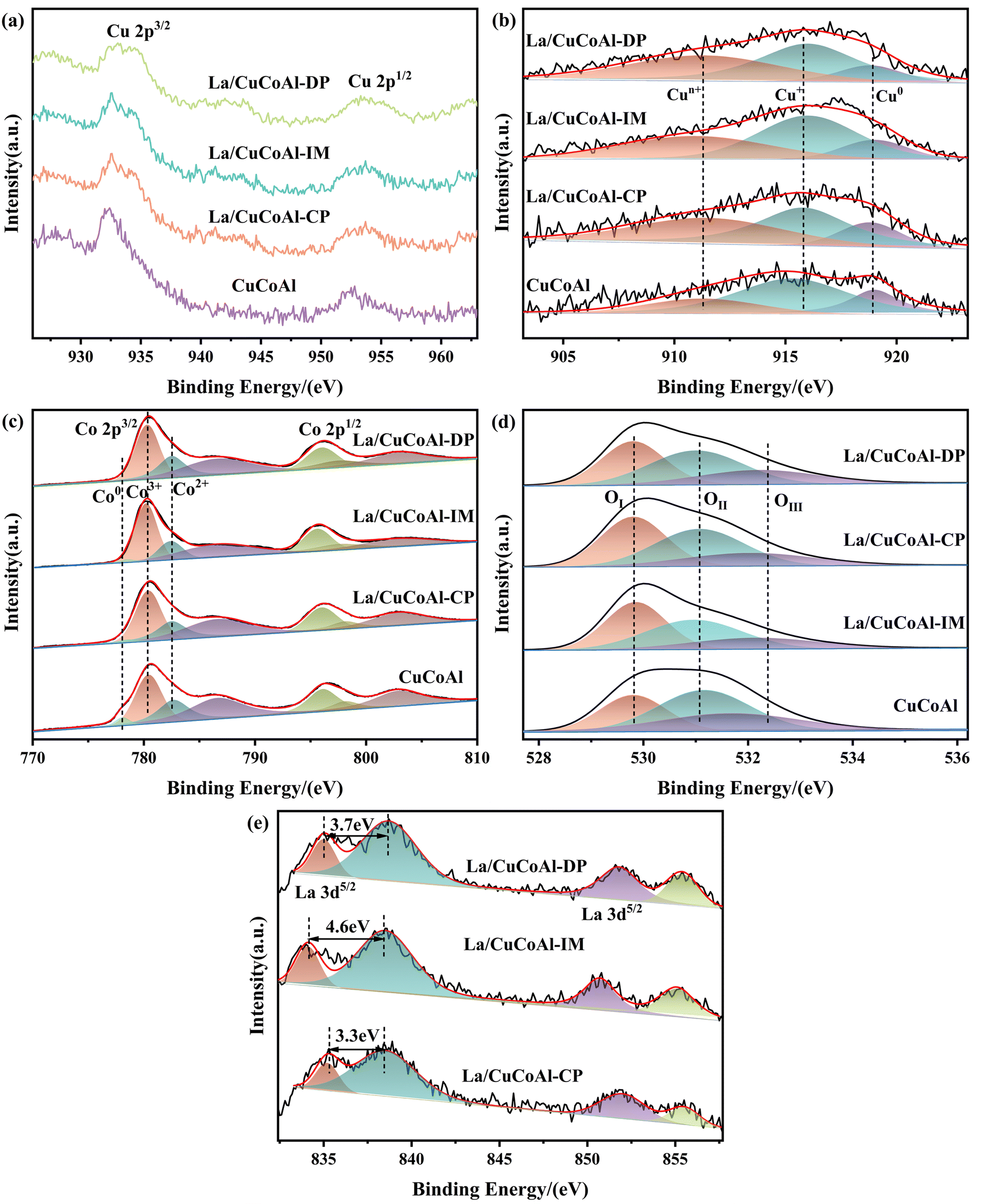 | ||
| Fig. 7 XPS patterns of reduced catalysts. (a) Cu 2p XPS; (b) Cu LMM XANS; (c) Co 2p XPS; (d) O 1s XPS; (e) La 3d XPS. | ||
Fig. 7c shows the XPS spectra of Co 2p for the catalysts. Two broad and asymmetric peaks were observed at around 780.5 eV and 796.0 eV, respectively, corresponding to Co 2p3/2 and Co 2p1/2 of Co3+ in Co3O4. The peaks centered around 782.5 eV and 798.1 eV belong to Co 2p3/2 and Co 2p1/2 of Co2+ in Co3O4, and a small peak at about 778.5 eV belongs to Co0.1,14,52,53 In addition, there were two satellite peaks of Co 2p at near 786.6 eV and 802.9 eV, which were related to the Co 2p3/2 and Co 2p1/2 of Co2+ species in CoO, respectively.14,54,55 These results demonstrated that part of the Co3O4 species in the catalyst was reduced to Co0 and CoOx (mainly Co2+) accompanied by the generation of an oxygen vacancy,14,16,43,54,55 which was consistent with the XRD and HRTEM results. As listed in Table S5,† the content of Co0 and CoOx decreased obviously with the doping of La to CuCoAl, together with the increase for Co3+species. The weak interaction between Cu and Co3O4 reduced the hydrogen spillover effect between them, thereby lowering the reduction degree of Co3O4, which was also revealed by the H2-TPR results. Combined with the results in Fig. 3a, the content of Co0 is positively correlated with the selectivity of THFA due to the higher hydrogenation activity of Co0 species towards the CC bond. Additionally, the content of CoOx (mainly Co2+) and the selectivity of 1,5-PeD present a correlational dependence over the La/CuCoAl catalyst. The oxygen vacancies in CoOx were also beneficial to the adsorption of –OH groups of FFA, promoting the C2-end tilted absorption on the surface of the catalyst.16 Furthermore, the low coordinated CoOx species have unoccupied orbitals that can hybridize with CC bonds, promoting the adsorption and activation of C2C3 bonds near the –OH group,23,53 thereby enhancing 1,5-PeD selectivity. However, the lower selectivity of 1,5-PeD was gained over CuCoAl with a higher content of CoOx. This can be attributed to the high content of Co0 in CuCoAl, which was beneficial to the production of THFA.
Fig. 7d displays the O 1s spectra of the reduced catalysts. The O 1s peak can be deconvoluted into three Gaussian peaks positioned at 529.8 eV, 531.0 eV and 532.2 eV, which were attributed to lattice oxygen (OI), surface defect oxygen (OII) and other oxygen species (OIII, including –OH, H2O and CO32−), respectively.56–59 As exhibited in Table S5,† the content of surface defect oxygen (OII) in the La-doped catalyst was in the order La/CoCuAl-DP > La/CoCuAl-CP > La/CoCuAl-IM, corresponding to the content of CoOx.
Fig. 7e depicts the spectra of La 3d for the reduced catalyst and the details are shown in Table S6.† The La 3d spectra generally split two distinct peaks because of the spin-splitting effect and its well-separated spin–orbit components. Four obvious satellite peaks were observed. Among them, the peaks at around 835.0 eV and 838.7 eV were ascribed to the multiplet splitting at La 3d5/2, while the peaks centered at about 851.9 and 855.4 eV were assigned to the multiplet splitting at La 3d3/2.2,3,60 The binding energy gap between La 3d3/2 and La 3d5/2 was about 16.8 eV, confirming the presence of La3+ species. It has been reported that the La3+ species is in the form of La(OH)3 if the splitting distance (ΔE) is 3.9 eV in the La 3d5/2 multiplet, while a ΔE value of 4.6 eV is for La2O3.61,62Fig. 7f shows that the value of ΔE in the La 3d5/2 multiplet for La/CuCoAl-DP and La/CuCoAl-CP catalysts were 3.7 eV and 3.3 eV, respectively. These ΔE values match with that of La(OH)3 within the error limit, indicating that the La sites in these catalysts were La(OH)3. However, the ΔE value was 4.6 eV for the La/CuCoAl-IM catalyst, implying that the La sites were La2O3 in this catalyst, which is consistent with the HRTEM results.
To understand the ability to activate H2, H2-TPD was carried out for the reduced catalysts. As demonstrated in Fig. 8a, each sample exhibited three H2 desorption peaks, marked as α, β and γ, respectively, indicating that three types of H2 desorption active sites existed. The peak centered at 95 °C (α) was ascribed to the H2 desorption that adsorbed on oxygen vacancies.50,63–66 The peak located at 120 °C (β) is related to the weak adsorption and dissociation of H2 species at the metal–support interface, which is closely adjacent to the oxygen vacancies on the support.63–66 In the high-temperature region (400 °C–600 °C), all catalysts exhibit a broad H2 desorption peak γ, which was ascribed to the dissociated hydrogen anchored on Cu and CoOx sites.50,63–66 Compared to the CuCoAl catalyst, the integrated peak area of α + β and γ increased notably after La addition, resulting in a substantial increase in the amount of desorbed hydrogen (Table S7†). The H2 desorption amounts for each catalyst were in the order of La-CuCoAl-DP > La/CuCoAl-CP > La/CuCoAl-IM > CuCoAl, suggesting that the doping of La promoted the formation of active H2. Such enhancement can be explained by two factors. Firstly, the introduction of La increased the dispersion and the surface area of Cu0, which favored the H2 adsorption and activation. Secondly, as indicated by the Cu 2p XPS, electronic effects existed between Cu species and La species due to the geometric effect of La(OH)3 and the strong interaction between them.50,67 The d-electrons of Cu species can easily flow to the d-orbitals of La species, creating unoccupied d-orbitals for Cu (Cun+). Cun+ can bond with the H2 molecule and weaken the H–H bond, leading to the formation of active H.50,67 It is thus speculated that the electronic effect between La species and Cu species was the other key factor for the improved active H absorption on the La/CuCoAl-DP catalyst, since the dispersion and the surface area of Cu0 was lower than that of the La/CuCoAl-CP catalyst. Consequently, an appropriate amount of La incorporated into the CuCoAl catalysts enhanced the H2 adsorption and activation capacity, thereby improving the conversion of FFA, which is in accordance with the results in Fig. 3a.
 | ||
| Fig. 8 (a) H2-TPD and (b) CO2-TPD patterns of the reduced catalysts. | ||
Fig. 8b demonstrates the basicity of the reduced catalysts evaluated by CO2-TPD characterization. Each catalyst displayed the CO2 desorption peak centered at 50 °C–300 °C, 300 °C–500 °C and 500 °C–800 °C, indicating that each catalyst owns weak, medium and strong basic sites.68,69 As indicated by the integral area of CO2-TPD curves and the capacity of CO2 desorption (Tables S7 and S8†), the total basic density of the La-doped catalyst increased compared with the CuCoAl catalyst. This implied that the introduction of La significantly enhanced its basicity, particularly for the strong basic sites. Moreover, the La/CuCoAl-DP catalyst possessed more basic sites, followed by La/CuCoAl-CP and La/CuCoAl-IM. This indicated that more strongly basic La(OH)3 species were formed by doping La via the deposition–precipitation method, which was also verified by the results of HRTEM and XPS. As observed in Fig. 3a and Fig. S4,† the incorporation of La enhanced remarkably the conversion of FFA and 1,5-PeD selectivity, together with the decrease in THFA selectivity. The selectivity of 1,5-PeD was in the order of La/CuCoAl-DP > La/CuCoAl-CP > La/CuCoAl-IM > CuCoAl, which was positively correlated with the concentration of strongly basic La(OH)3 on the catalyst surface. Furthermore, as corroborated by HRTEM and XPS, the Cun+–O–La(OH)3 structure was formed at the Cu–La interface because of the strong basicity of La (OH)3 and its geometric effect. Cun+–O–La(OH)3 sites can simultaneously activate the furan ring and the –OH group in FFA. In detail, Cun+ adsorbed the O atom in the furan ring as it is highly electron-deficient, while La(OH)3 attacked the H in the –OH group outside the furan ring. Then, the –OH group deprotonated and generated an alkoxide complex, inducing the C2-end tilted adsorption on the catalyst surface, thereby enhancing the selectivity of 1,5-PeD. These results demonstrated that the presence of strongly basic La(OH)3 species and the Cu–La interface played a vital role in boosting the activity and 1,5-PeD selectivity over the La/CuCoAl-DP catalyst.
3.3 DFT calculations
To gain further insight into the effect of La on the catalytic activity and 1,5-PeD selectivity, DFT calculations were carried out. Fig. S5† shows the catalyst models for CuCo and CuCoLa. These models were constructed based on the results provided by XRD and HRTEM characterizations. Specific computational parameters can be found in the ESI.†To investigate the influence of La on H2 activation on the catalyst surface, the energy values for dissociative H2 adsorption on both models were calculated. Fig. S6† illustrates the adsorption reaction pathways for H2 dissociation on the CuCo and CuCoLa models. The H2 molecules were favorably attached to Cu atoms in both the CuCo model and the CuCoLa model. After H2 dissociation, the H atoms are forcefully adsorbed on Cu and Co atoms in the CuCo model, whereas they were strongly adsorbed on Cu and La atoms in the CuCoLa model. The activation energy barriers required for H2 dissociation on CuCoLa (0.84 eV) were lower than that on CuCo (1.21 eV), confirming that the catalyst doped by La is more favorable for the dissociation of H2 to active H atoms (Fig. 9), which was consistent with the H2-TPD results. This result further verified that the incorporation of La into the catalyst promoted the activation of H2 at adjacent active Cu sites, resulting in a higher content of activated H atoms on the catalyst surface, thereby enhancing the catalytic activity.
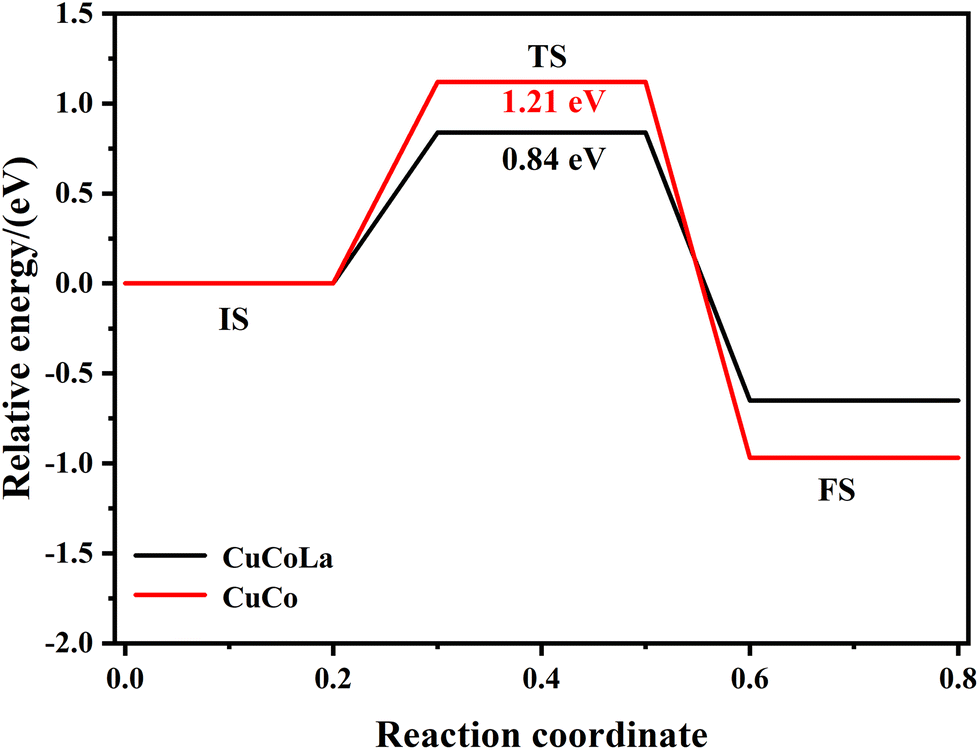 | ||
| Fig. 9 Reaction coordinates of dissociated H2 adsorption on CuCo and CuCoLa surfaces. | ||
The adsorption energies of FFA with different adsorption configurations on the pristine CuCo and CuCoLa models were calculated by DFT (Fig. 10 and Fig. S7†). As shown in Fig. 10, the adsorption energies of FFA on the CuCo and CuCoLa surfaces appeared in three modes: C2-end tilted adsorption (C2), C5-end tilted adsorption (C5) and planar adsorption (pa). According to the results in Fig. 10a and Fig. S7(a–c),† the order of adsorption energy for FFA on CuCo catalyst surface with different adsorption configurations was parallel (−2.18 eV) > C2 (−1.44 eV) > C5 (−1.28 eV). After modification by La (Fig. 10b and Fig. S7(e, f)†), the parallel adsorption energy decreased obviously (from −2.18 eV to −1.92 eV), the adsorption energy of C5-end oblique adsorption reduced slightly (from −1.28 eV to −1.24 eV), while the adsorption energy of C2-end oblique adsorption increased remarkably (from −1.44 eV to −1.76 eV). This result supported the speculation that the addition of La species regulated the adsorption configuration of FFA on the catalyst surface. The C2-end tilted adsorption was more favorable, while the possibility of parallel adsorption and C5-end tilted adsorption was reduced on the CuCoLa catalyst surface. It is worth noting that FFA was more prone to parallel adsorption both on CuCo or CuCoLa surface according to the adsorption energies, implying that the main product might be THFA. This was not consistent with the experiment results under the optimal conditions (160 °C, 4 MPa H2), where 1,5-PeD was the main product with a yield exceeding 50%. This was likely due to the fact that the DFT calculation models were carried out under normal temperatures and pressure. Except for the adsorption, a variety of factors, including temperature and H2 pressure, played an important role on the catalytic activity and product distribution during the catalytic reaction. As indicated in Fig. 3, the main product was THFA when the reaction temperature was below 140 °C, in line with the DFT calculation results, and 1,5-PeD was the main product when raising the reaction temperature. This phenomenon can be ascribed to the difficulty of overcoming the large steric hindrance and the high reaction energy barrier of oblique adsorption at low temperatures. However, the surface strong basic sites can overcome the large steric hindrance and high reaction energy barrier by adsorbing and anchoring the –OH group in FFA, leading to an alkoxide structure under the higher temperatures. Then, the C2-end tilted adsorption of FFA on the catalyst surface was more favorable, thereby improving the selectivity of 1,5-PeD.
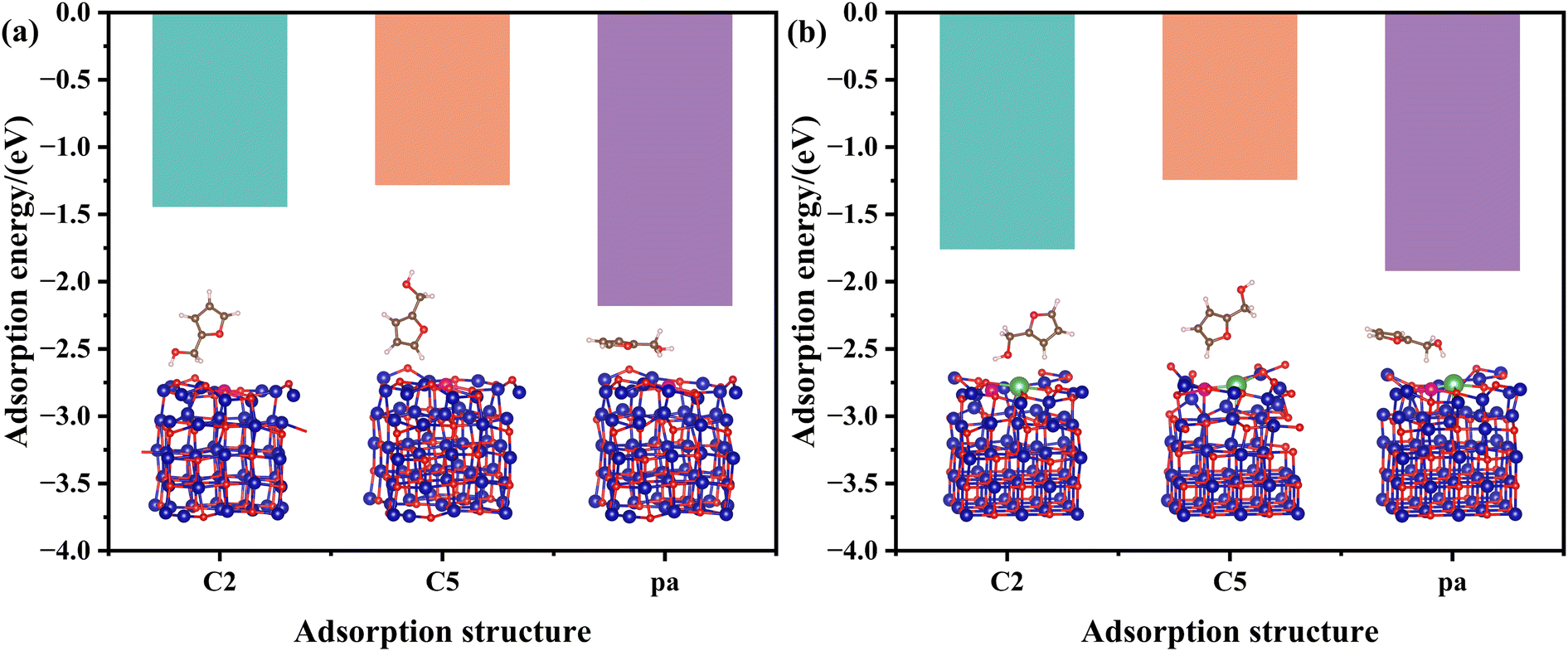 | ||
| Fig. 10 Optimized adsorption geometries and adsorption energies of FFA on (a) CuCo and (b) CuCoLa surfaces. Cu: magenta, Co: dark blue, La: green, O: red, H: light pink; C: brown. | ||
3.4 The reusability of the La/CuCoAl-DP catalyst
To explore the stability and reusability of the La/CuCoAl-DP catalyst, eight consecutive evaluations were conducted under optimal conditions. As shown in Fig. 11, the conversion of FFA remained high level after 8 cycles, whereas the selectivity of 1,5-PeD decreased slowly (from 54.2% to 46.9%) accompanied by an increased THFA selectivity (from 12.9% to 25.9%) with the increased recycling numbers. To investigate the reason for the decrease of 1,5-PeD selectivity in the recycling experiments, the catalyst after 8 cycles was characterized by ICP, HRTEM, XRD and XPS (Fig. S8 and S9†). As illustrated in Table 1 and Fig. S8,† the content of each element and the phase structure did not change significantly after the catalyst was used for 8 times, demonstrating that the catalyst possessed good structural stability. However, the concentration of Cun+ decreased from 42.4% to 35.5% together with an increase of Cu0 from 11.6% to 17.2% (Fig. S9a and Table S5†). This result indicated that the Cun+ sites were gradually reduced to Cu0 during the reaction process under the hydrogen and ethanol environment. Additionally, the content of Co0 in the spent catalyst also increased obviously with a slight improvement for CoOx (Fig. S9b and Table S5†), further verifying the reduction of the catalyst in the reaction process. The Co0 species were more favorable for the formation of THFA because of its high capability of adsorbing and activating furan ring. It is thus that the selectivity of 1,5-PeD decreased accompanied by an increasing of THFA after 8 recycling runs. Moreover, compared with the fresh catalyst, the chemical environment of the O element and the structure of La(OH)3 did not change obviously (Fig. S9c, d and Tables S5, S6†). Consequently, it was concluded that the decrease of 1,5-PeD selectivity was mainly derived from the change in the electronic structure of Cu and Co species due to the in situ reduction during the reaction.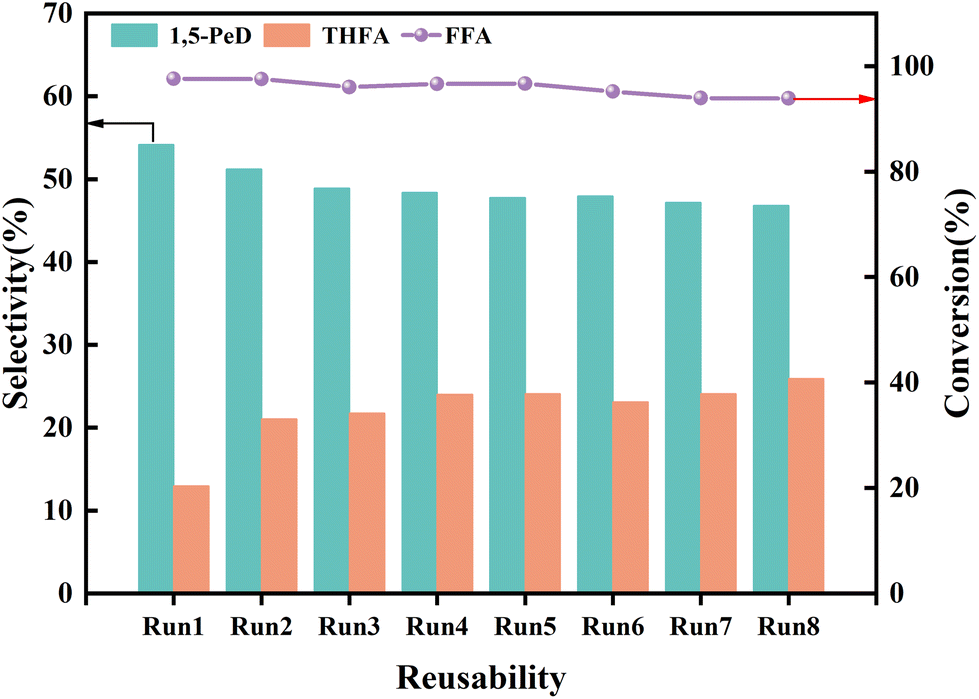 | ||
| Fig. 11 The reusability of the La/CuCoAl-DP catalyst. Reaction conditions: FFA 0.5 g, catalyst 0.1 g, ethanol 39.5 g, 160 °C, 4 MPa H2, 2 h. | ||
3.5 The impact of pretreatment conditions on the catalytic performance
To further improve the selectivity of 1,5-PeD, the pretreatment conditions for La/CuCoAl-DP catalyst were optimized. As shown in Table 2, a 59.5% yield of 1,5-PeD was achieved on the catalyst treated by calcination at 250 °C for 8 hours in air and reduction at 250 °C for 6 hours. To compare the catalytic ability of the La/CuCoAl-DP catalyst in this work, the results compared to non-noble catalysts reported in the literature are summarized in Fig. 12 and Table S9.† It was observed that the yield of 59.5% for 1,5-PeD in this work was higher than that reported in the literature, displaying the superior ability of the La/CuCoAl-DP catalyst for the conversion of FFA to 1,5-PeD. The La 3d spectrum in Fig. S10a† showed a splitting distance ΔE of 3.9 eV, indicating the presence of La(OH)3 in the catalyst, which was also verified by the results of HRTEM (Fig. S11a†). The CO2-TPD results (Fig. S11c and Tables S7, S8†) demonstrated that the total basic sites increased remarkably in the catalyst pretreated under lower temperatures due to the abundant surface La(OH)3. Particularly, a significant enhancement of medium and strong basic site concentration (from 0.27 mmol g−1 to 0.84 mmol g−1) was observed. The surface La(OH)3 was beneficial to the C2-end tilted absorption of FFA on the catalyst surface, thus enhancing the selectivity of 1,5-PeD. The XPS spectra of Cu LMM (Fig. S10b†) shows that the amount of Cun+ was enhanced obviously by optimization of pretreatment conditions. This was attributed to the increased surface La(OH)3 promoting the generation of the Cu–La interface, which corresponds with the results of HRTEM and CO2-TPD (Fig. S11†). Meanwhile, the content of CoOx and OII also increased markedly (Fig. S10(c, d) and Table S5†), which might result from the difficulty in reducing Co3O4 to Co0 under low temperatures. The H2-TPR results (Fig. S12a†) of the catalyst calcined at 250 °C for 8 h showed that the H2 consumption peak (around 150 °C–300 °C, assigned to Cu species reduction) shifted to a higher temperature compared with the catalyst calcined at 500 °C for 3 h. This phenomenon indicated that a stronger interaction between Cu and La, resulting from more La(OH)3, was generated with lower calcination temperature. Furthermore, the H2-TPD of reduced catalysts was performed. As illustrated in Fig. S12,† the desorption peak area of H2 at high temperatures increased. The total desorption of H2 (nH) of the catalyst raised from 0.42 mmol g−1 to 0.79 mmol g−1 (Table S7†). This increase suggests that the catalyst pretreated under lower calcination and reduction temperature has a stronger ability to adsorb and store H2, which would enhance the hydrogenation activity. Consequently, it was judged that the excellent activity of the La/CuCoAl-DP catalyst was derived from the cooperation catalysis of Cu0, CoOx, strongly basic La (OH)3 and the Cu–La interface (Cun+–O–La(OH)3). | ||
| Fig. 12 The comparison of the catalytic ability of La/CuCoAl-DP with the non-noble metal catalysts reported in the literature (refer to Table S9† for details). | ||
| Entry | Calcination | Reduction | Conversion % | Yield % | |||||
|---|---|---|---|---|---|---|---|---|---|
| T 1/°C | t 1/h | T 2/°C | t 2/h | 1,5-PeD | 1,2-PeD | THFA | Others | ||
| Reaction conditions: FFA 0.5 g, catalyst 0.1 g, ethanol 39.5 g; temperature 160 °C, reaction time 2 h, 4 MPa H2; carbon balance in all reactions ≥ 97%, THFA: tetrahydrofurfuryl alcohol; FFA: furfuryl alcohol; 1,2-PeD: 1,2-pentanediol; 1,5-PeD: 1,5-pentanediol; others: 1-pentanol, 2-pentanol, 1-butanol, 2-methylfuran. | |||||||||
| 1 | 500 | 3 | 400 | 1 | 97.6 | 52.9 | 12.5 | 12.7 | 19.6 |
| 2 | 350 | 3 | 400 | 1 | 98.8 | 54.6 | 13.2 | 12.6 | 18.7 |
| 3 | 250 | 3 | 400 | 1 | 99.3 | 52.9 | 13.4 | 13.4 | 19.6 |
| 4 | 250 | 5 | 400 | 1 | 99.0 | 53.9 | 14.1 | 12.8 | 18.3 |
| 5 | 250 | 8 | 400 | 1 | 99.3 | 53.7 | 13.0 | 13.8 | 18.7 |
| 6 | 250 | 8 | 250 | 4 | 91.2 | 52.4 | 14.3 | 9.7 | 14.8 |
| 7 | 250 | 8 | 250 | 5 | 96.3 | 55.1 | 15.9 | 11.0 | 14.4 |
| 8 | 250 | 8 | 250 | 6 | 100.0 | 59.5 | 15.5 | 10.4 | 14.6 |
| 9 | 250 | 8 | 270 | 6 | 97.2 | 53.1 | 15.2 | 14.4 | 14.5 |
Based on the above analysis, a rational reaction mechanism for FFA transformation to 1,5-PeD on La/CuCoAl-DP catalyst was presented (Fig. 13). Initially, H2 molecules are dissociatively adsorbed on Cu species. FFA adsorbed on the sites of Cu–La interface (Cun+–O–La(OH)3) with an intermediate six-membered ring transition state. The furan ring and the –OH group in FFA can be simultaneously activated. Specifically, the strong basic site La (OH)3 at the Cu–La interface attacked the H in –OH group outside the furan ring. The –OH group deprotonated and generated an alkoxide complex, promoting the C2-end tilted adsorption of FFA on the surface of the catalyst. Meanwhile, the O atom in the furan ring was adsorbed by Cun+ with a strong oxygen affinity, resulting in a polarizing of the C2–O1 bond. Then, the low-coordination CoOx activated the C2C3 bond in the furan ring through coordination adsorption, and it was hydrogenated by the active H formed on the Cu species. Finally, the C2–O1 bond was further weakened and cleaved to form 1,5-PeD with high selectivity.
 | ||
| Fig. 13 Reaction mechanism of FFA to 1,5-PeD over the La/CuCoAl-DP catalyst. | ||
4. Conclusions
In this work, the effect of La doping into CuCoAl catalysts on the conversion of FFA to 1,5-PeD was systematically evaluated. It was demonstrated that the catalyst doped with La by deposition–precipitation methods (La/CuCoAl-DP) displayed excellent activity compared with other catalysts. A nearly 60% yield of 1,5-PeD was obtained at 160 °C, 4 MPa H2 within 2 h. Extensive catalyst characterization, including H2-TPD, CO2-TPD and N2O-TPD demonstrated that the addition of La in the catalyst significantly improved the dispersion of Cu, the ability to activate H2 and the concentration of strong basic sites on the catalyst surface. Furthermore, HRTEM and XPS results corroborated that the doping of La favored the formation of the Cu–La interface. A Cun+–O–La(OH)3 structure was generated due to the strong electronic interaction between Cu and La(OH)3. Such Cun+–O–La(OH)3 sites can simultaneously activate the furan ring and the –OH group in FFA with an intermediate six-membered ring transition state. Typically, the strong basic site La (OH)3 anchored the –OH group to form an alkoxide complex. The tilted adsorption of FFA at the C2-end on the catalyst surface was promoted. At the same time, Cun+ with strong oxygen affinity adsorbed the O atom in the furan ring, the C2–O1 bond was polarized and cleaved to 1,5-PeD. The DFT calculations results verified that the doping of La to the catalyst enhanced the possibility of C2-end tilted adsorption for FFA on the surface of the catalyst, and improved the ability of the catalyst to activate H2. Consequently, it was summarized that the superior catalytic activity of the catalyst was attributed to the synergistic catalysis between Cu0, CoOx, La (OH)3 and the Cu–La interface on the catalyst surface.Author contributions
Jingjing Tan: conceptualization, writing – review & editing, supervision, resources. Hailong Huang: investigation, data curation, formal analysis, methodology, writing – original draft. Yuanna Zhang: methodology, investigation, data curation. Jinglei Cui: writing – review & editing, supervision. Jing Zhang: writing – review & editing, supervision, Long Huang: writing – review & editing, supervision. Yongzhao Wang: writing – review & editing, supervision. Yulei Zhu: software, writing – review & editing, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22005182, 22378243 and 22178202) and Fundamental Research Program of Shanxi Province, China (202103021224008).References
- R. G. Kurniawan, N. Karanwal, J. Park, D. Verma, S. K. Kwak, S. K. Kim and J. Kim, Appl. Catal., B, 2023, 320, 121971–121988 CrossRef CAS.
- W. Zhao, X. Bai, X. Lin, Y. Tur sun, M. Zhong, Z. Dai and J. Li, Fuel, 2023, 354, 129312–129321 CrossRef CAS.
- M. Al-Yusufi, N. Steinfeldt, R. Eckelt, H. Atia, H. Lund, S. Bartling, N. Rockstroh and A. Köckritz, ACS Sustainable Chem. Eng., 2022, 10, 4954–4968 CrossRef CAS.
- J. Tan, J. He, K. Gao, S. Zhu, J. Cui, L. Huang, Y. Zhu and Y. Zhao, Appl. Surf. Sci., 2022, 604, 154472–154484 CrossRef CAS.
- Y. Wu, H. Wang, J. Peng, J. Zhang and M. Ding, Chem. Eng. J., 2023, 454, 140156–140165 CrossRef CAS.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- D.-J. Tao, X. Zhao, Y. Wang, X. Liu, H.-P. Li, Z.-M. Li, Y. Zhou, Z. Yuan and Z. Zhang, Green Energy Environ., 2022, 7, 1084–1092 CrossRef CAS.
- X. Liu, L. Huang, Y. Ma, G. She, P. Zhou, L. Zhu and Z. Zhang, Nat. Commun., 2024, 15, 7012–7026 CrossRef CAS PubMed.
- S. Xu, P. Zhou, Z. Zhang, C. Yang, B. Zhang, K. Deng, S. Bottle and H. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS PubMed.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- J. Zhu and G. Yin, ACS Catal., 2021, 11, 10058–10083 CrossRef CAS.
- N. Enjamuri and S. Darbha, Catal. Rev., 2020, 62, 566–606 CrossRef CAS.
- B. Zhang, Y. Zhu, G. Ding, H. Zheng and Y. Li, Green Chem., 2012, 14, 3402–3409 RSC.
- J. Tan, Y. Su, X. Hai, L. Huang, J. Cui, Y. Zhu, Y. Wang and Y. Zhao, Mol. Catal., 2022, 526, 112391–112400 CrossRef CAS.
- H. Liu, Z. Huang, H. Kang, C. Xia and J. Chen, Chin. J. Catal., 2016, 37, 700–710 CrossRef CAS.
- J. Peng, D. Zhang, Y. Wu, H. Wang, X. Tian and M. Ding, Fuel, 2023, 332, 126261–126271 CrossRef CAS.
- B. Wang, P. Zhou, X. Yan, H. Li, H. Wu and Z. Zhang, J. Energy Chem., 2023, 79, 535–549 CrossRef CAS.
- R. Ma, X.-P. Wu, T. Tong, Z.-J. Shao, Y. Wang, X. Liu, Q. Xia and X.-Q. Gong, ACS Catal., 2017, 7, 333–337 CrossRef CAS.
- T. Tong, X. Liu, Y. Guo, M. Norouzi Banis, Y. Hu and Y. Wang, J. Catal., 2018, 365, 420–428 CrossRef CAS.
- T. Mizugaki, T. Yamakawa, Y. Nagatsu, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustainable Chem. Eng., 2014, 2, 2243–2247 CrossRef CAS.
- H. Liu, Z. Huang, F. Zhao, F. Cui, X. Li, C. Xia and J. Chen, Catal. Sci. Technol., 2016, 6, 668–671 RSC.
- C. Wei, J. Tan, X. Xia and Y. Zhao, CIESC J., 2019, 70, 1409–1419 CAS.
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924–3396 RSC.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- H. W. Wijaya, T. Sato, H. Tange, T. Hara, N. Ichikuni and S. Shimazu, Chem. Lett., 2017, 46, 744–746 CrossRef CAS.
- W. Xi, P. Jian, X. Mao, D. Ning, X. Xia, G. Liu, D. Shi and Y. Lan, Mater. Res. Express, 2021, 8, 105002–105011 CrossRef CAS.
- A. Singh, V. Palakollu, A. Pandey, S. Kanvah and S. Sharma, RSC Adv., 2016, 6, 103455–103462 RSC.
- E. A. Cepeda, U. Iriarte-Velasco, B. Calvo and I. Sierra, J. Am. Oil Chem. Soc., 2016, 93, 731–741 CrossRef CAS.
- Y. Nakagawa, M. Tamura and K. Tomishige, ACS Catal., 2013, 3, 2655–2668 CrossRef CAS.
- G. R. Jenness, W. Wan, J. G. Chen and D. G. Vlachos, ACS Catal., 2016, 6, 7002–7009 CrossRef CAS.
- P. Mäki-Arvela, J. Hájek, T. Salmi and D. Y. Murzin, Appl. Catal., A, 2005, 292, 1–49 CrossRef.
- M. J. Gilkey and B. Xu, ACS Catal., 2016, 6, 1420–1436 CrossRef CAS.
- Y. Deng, R. Gao, L. Lin, T. Liu, X.-D. Wen, S. Wang and D. Ma, J. Am. Chem. Soc., 2018, 140, 14481–14489 CrossRef CAS PubMed.
- R. Ghosh, N. C. Jana, S. Panda and B. Bagh, ACS Sustainable Chem. Eng., 2021, 9, 4903–4914 CrossRef CAS.
- X. Guo, F. Zhang, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 5119–5120 Search PubMed.
- D. Kwon, J. Y. Kang, S. An, I. Yang and J. C. Jung, J. Energy Chem., 2020, 46, 229–236 CrossRef.
- J. Pérez-Ramírez, S. Abelló and N. M. vanderPers, Chem. – Eur. J., 2007, 13, 870–878 CrossRef PubMed.
- M. Hronec, K. Fulajtárová, I. Vávra, T. Soták, E. Dobročka and M. Mičušík, Appl. Catal., B, 2016, 181, 210–219 CrossRef CAS.
- Y. Yang, Z. Du, Y. Huang, F. Lu, F. Wang, J. Gao and J. Xu, Green Chem., 2013, 15, 1932–1940 RSC.
- Y. Zheng, J. Zang, Q. Zhang, X. Wu, S. Qiu, Q. Meng and T. Wang, Green Chem., 2023, 25, 1128–1136 RSC.
- C. Li, S. Zhang, B. Zhang, D. Su, S. He, Y. Zhao, J. Liu, F. Wang, M. Wei, D. G. Evans and X. Duan, J. Mater. Chem. A, 2013, 1, 2461–2467 RSC.
- J. Tan, J. Cui, T. Deng, X. Cui, G. Ding, Y. Zhu and Y. Li, ChemCatChem, 2015, 7, 508–512 CrossRef CAS.
- J. Wu, G. Gao, P. Sun, X. Long and F. Li, ACS Catal., 2017, 7, 7890–7901 CrossRef CAS.
- K. Sun, X. Gao, Y. Bai, M. Tan, G. Yang and Y. Tan, Catal. Sci. Technol., 2018, 8, 3936–3947 RSC.
- K. Chen, H. Fang, S. Wu, X. Liu, J. Zheng, S. Zhou, X. Duan, Y. Zhuang, S. Chi Edman Tsang and Y. Yuan, Appl. Catal., B, 2019, 251, 119–129 CrossRef CAS.
- G. Bonura, M. Cordaro, C. Cannilla, F. Arena and F. Frusteri, Appl. Catal., B, 2014, 152–153, 152–161 CrossRef CAS.
- K. Chen, X. Duan, H. Fang, X. Liang and Y. Yuan, Catal. Sci. Technol., 2018, 8, 1062–1069 RSC.
- X. Zheng, H. Lin, J. Zheng, X. Duan and Y. Yuan, ACS Catal., 2013, 3, 2738–2749 CrossRef CAS.
- Z. Zhao, G. Gao, Y. Xi, J. Wang, P. Sun, Q. Liu, W. Yan, Y. Cui, Z. Jiang and F. Li, Chem, 2022, 8, 1034–1049 CAS.
- Z. Ren, M. N. Younis, C. Li, Z. Li, X. Yang and G. Wang, RSC Adv., 2020, 10, 5590–5603 RSC.
- J. Shan, Y. Shi, H. Li, Z. Chen, C. Sun, Y. Shuai and Z. Wang, Chem. Eng. J., 2022, 433, 133769–133776 CrossRef CAS.
- Z. Zhao, W. Lu, R. Yang, H. Zhu, W. Dong, F. Sun, Z. Jiang, Y. Lyu, T. Liu, H. Du and Y. Ding, ACS Catal., 2018, 8, 228–241 CrossRef CAS.
- S. Yao, X. Wang, Y. Jiang, F. Wu, X. Chen and X. Mu, ACS Sustainable Chem. Eng., 2014, 2, 173–180 CrossRef CAS.
- S. Li, H. Wang, W. Li, X. Wu, W. Tang and Y. Chen, Appl. Catal., B, 2015, 166–167, 260–269 CrossRef CAS.
- M. G. Dohade and P. L. Dhepe, Green Chem., 2017, 19, 1144–1154 RSC.
- Q. Yang, X. Wang, W. Luo, J. Sun, Q. Xu, F. Chen, J. Zhao, S. Wang, F. Yao, D. Wang, X. Li and G. Zeng, Bioresour. Technol., 2018, 247, 537–544 CrossRef CAS PubMed.
- Q. Dai, Z. Zhang, J. Yan, J. Wu, G. Johnson, W. Sun, X. Wang, S. Zhang and W. Zhan, Environ. Sci. Technol., 2018, 52, 13430–13437 CrossRef CAS PubMed.
- Z. Hu, Z. Wang, Y. Guo, L. Wang, Y. Guo, J. Zhang and W. Zhan, Environ. Sci. Technol., 2018, 52, 9531–9541 CrossRef CAS PubMed.
- L. Liu, C. Zhang, S. Chen, L. Ma, Y. Li and Y. Lu, Chemosphere, 2022, 286, 131773–131779 CrossRef CAS PubMed.
- J.-G. Kang, Y.-I. Kim, D. Won Cho and Y. Sohn, Mater. Sci. Semicond. Process., 2015, 40, 737–743 CrossRef CAS.
- M. F. Sunding, K. Hadidi, S. Diplas, O. M. Løvvik, T. E. Norby and A. E. Gunnæs, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 399–409 CrossRef CAS.
- X. Wang, Y. Tong, W. Feng, P. Liu, X. Li, Y. Cui, T. Cai, L. Zhao, Q. Xue, Z. Yan, X. Yuan and W. Xing, Nat. Commun., 2023, 14, 3767–3777 CrossRef PubMed.
- X. Liao, Y. Zhang, M. Hill, X. Xia, Y. Zhao and Z. Jiang, Appl. Catal., A, 2014, 488, 256–264 CrossRef CAS.
- Z. He, X. Wang, R. Liu, S. Gao and T. Xiao, Appl. Petrochem. Res., 2016, 6, 235–241 CrossRef CAS.
- F. Hou, H. Zhao, J. Zhao, J. Yang, L. Yan, H. Song and L. Chou, J. Nanopart. Res., 2016, 18, 66–82 CrossRef.
- Z. Wang, X. Wang, C. Zhang, Y. Yang, L. Zhou, H. Cheng and F. Zhao, Catal. Today, 2022, 402, 79–87 CrossRef CAS.
- J. Shan, Y. Shi, H. Li, Z. Chen, C. Sun, Y. Shuai and Z. Wang, Chem. Eng. J., 2022, 433, 133769–133776 CrossRef CAS.
- S. Wang, L. Zhao, W. Wang, Y. Zhao, G. Zhang, X. Ma and J. Gong, Nanoscale, 2013, 5, 5582–5588 RSC.
- P. Kumar, V. C. Srivastava and I. M. Mishra, Energy Fuels, 2015, 29, 2664–2675 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc03974b |
| This journal is © The Royal Society of Chemistry 2024 |