
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemodivergent alkylation of trifluoromethyl alkenes via photocatalytic coupling with alkanes†
Pol
Martínez-Balart‡
 ,
Álvaro
Velasco-Rubio‡
,
Sergio
Barbeira-Arán
,
Hugo
Jiménez-Cristóbal
and
Martín
Fañanás-Mastral
*
,
Álvaro
Velasco-Rubio‡
,
Sergio
Barbeira-Arán
,
Hugo
Jiménez-Cristóbal
and
Martín
Fañanás-Mastral
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: martin.fananas@usc.es
First published on 4th October 2024
Abstract
gem-Difluoroalkenes and trifluoromethyl alkanes are prominent structures in biologically active compounds. Radical alkylation of α-trifluoromethyl alkenes represents a useful strategy to access these structures. However, reported methods have relied on the use of pre-functionalized radical precursors and examples involving the use of simple hydrocarbons as coupling partners are elusive. Here we report a chemodivergent methodology based on the direct activation of C(sp3)–H bonds enabled by HAT photoredox catalysis. This protocol provides an efficient platform for preparing both gem-difluoroalkenes and trifluoromethyl alkanes from ubiquitous hydrocarbon feedstocks, including gaseous alkanes. Importantly, chemoselectivity is easily achieved by simple modification of reaction conditions and/or additives.
Introduction
Fluorinated molecules play a prominent role in modern drug discovery and agrochemical development given the ability of several fluorinated moieties to mimic common functional groups found in bioactive molecules, while altering medicinally relevant properties, such as lipophilicity, metabolic stability, conformation, and pKa.1 Among these fluorinated motifs, gem-difluoroalkenes and trifluoromethyl alkanes are of particular interest being important metabolically stable bioisosteres of several functional groups (Scheme 1a).2 Thus, synthetic transformations that can selectively generate both structures are in high demand.3 α-Trifluoromethyl alkenes have served as efficient substrates for the formation of gem-difluoroalkenes, typically via SN2′ reaction with organometallic reagents4 or transition-metal catalyzed addition–elimination processes.5 Recently, methods for the synthesis of gem-difluoroalkenes have been described based on radical chemistry by means of either photocatalysis or nickel catalysis (Scheme 1b, left).6–12 In these transformations an alkyl radical generated from a radical precursor reacts with the α-trifluoromethyl alkene to provide an α-trifluoromethyl alkyl radical intermediate that is easily reduced to generate an anion (or an organonickel species) which undergoes facile β-F elimination to afford the product. The typically employed radical precursors are organic halides,6 carboxylic acid derivatives,7 organoborons,8 alkylbis(catecholato)silicates,8a sulfonyl salts,9 Hantzsch esters,10 acetals11 or activated C–H bonds.12 Despite these notable advances, methods based on the more atom efficient use on simple alkanes as radical precursors are elusive. Moreover, the development of α-trifluoromethyl alkene radical hydroalkylation processes in which the CF3 group is preserved represents a major challenge (Scheme 1b, right),13–18 mainly due to the high tendency of α-trifluoromethyl alkyl radicals to undergo reduction followed by β-fluoride elimination, rather than protonation to furnish the trifluoromethyl alkane. This issue has seriously limited the number of chemodivergent methods that allow access to both gem-difluoroalkenes and trifluoromethyl alkanes from a common α-trifluoromethyl alkene. To achieve this chemodivergency, those methods require the use of different photocatalysts,15 change in radical precursor16,17 or the use of stoichiometric reducing agents.18 Notably, all the aforementioned methods rely on the use of pre-functionalized radical precursors, something that inevitably leads to the formation of stoichiometric waste, and results in low atom and step economy.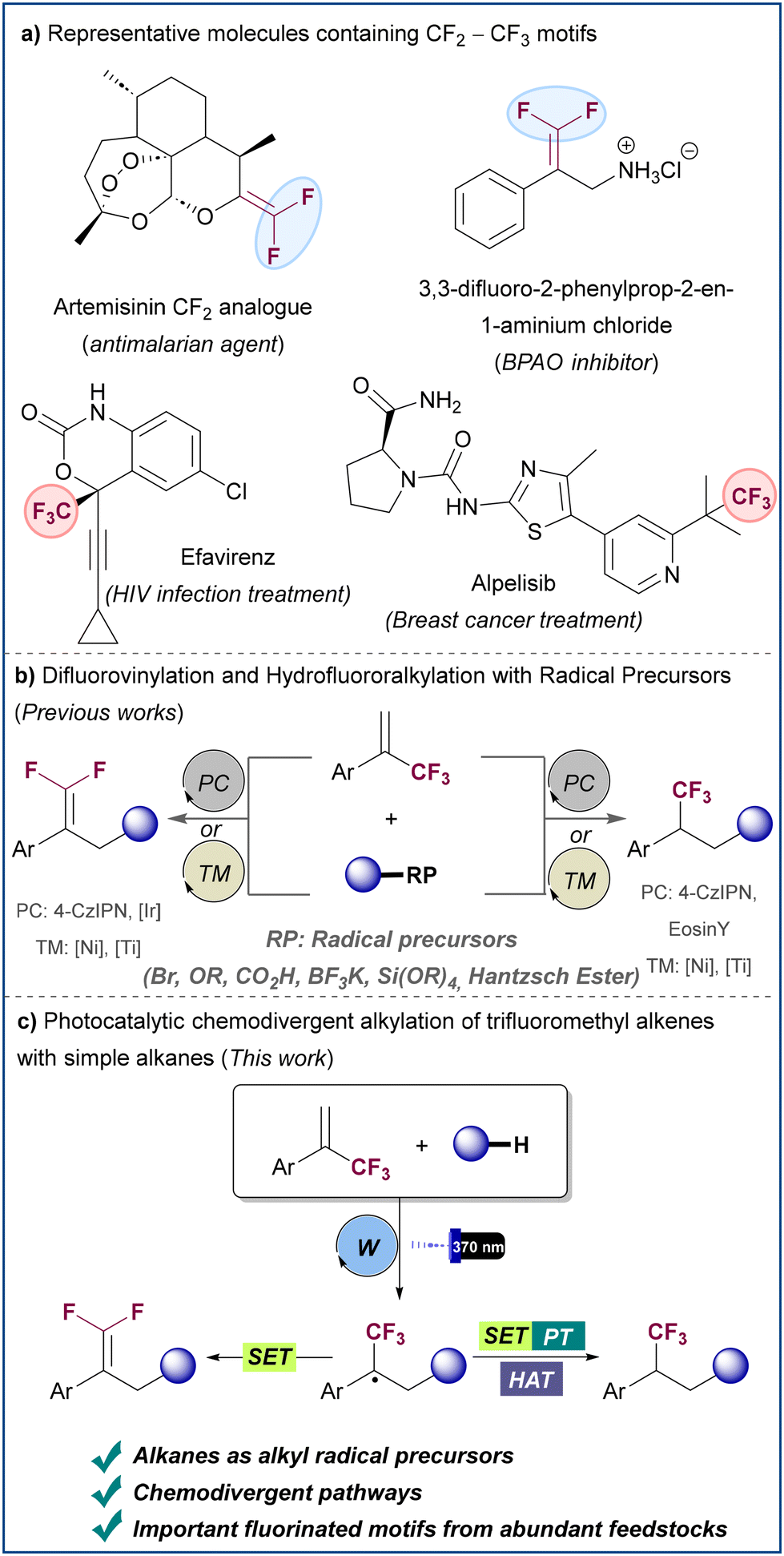 | ||
| Scheme 1 Examples of bioactive compounds featuring gem-difluoroalkene and trifluoromethyl groups and photocatalytic methods for the radical functionalization of α-trifluoromethyl alkenes. | ||
Alkanes represent a class of very abundant compounds with a great potential to be used as sustainable feedstocks. Indeed, the direct functionalization of alkane C–H bonds offers a more atom-efficient and straightforward way to build organic frameworks when compared to pre-made organometallic compounds or functionalized organic substrates. However, the direct functionalization of C(sp3)–H bonds in alkanes still represents a challenge in synthetic chemistry. In this context, photoredox-mediated processes in which saturated hydrocarbons are activated by hydrogen atom transfer (HAT) have emerged as a useful tool for the direct functionalization of alkanes.19 Within this line, we envisaged a potential scenario where the generation of alkyl radicals from simple unactivated alkanes could be engaged in the selective defluorinative alkylation and in the hydroalkylation of α-trifluoromethyl alkenes to achieve the chemodivergent synthesis of gem-difluoroalkenes and trifluoromethyl alkanes from abundant commodity chemicals. To address this challenge, we report herein a general platform for the divergent synthesis of these important fluorine-containing molecules based on HAT photoredox catalysis (Scheme 1c). The transformations are enabled by the use of tetrabutylammonium decatungstate (TBADT),20 which serves as a universal photocatalyst for the synthesis of both gem-difluoroalkenes and trifluoromethyl alkanes. The strategy relies on the ability of the photoexcited decatungstate anion to generate a carbon-centered radical by hydrogen atom abstraction from a range of alkanes, while slight modifications in reaction conditions and/or additives allow to control the divergent evolution of the radical intermediate that results from alkyl radical attack to the trifluoromethyl olefin, either by SET or HAT mechanisms with the reduced photocatalyst.
Results and discussion
Defluorinative alkylation
We began our investigation by exploring the reaction between cyclohexane (1) and (3,3,3-trifluoroprop-1-en-2-yl)benzene (2). After some experimentation (see Table 1 and ESI† for details), we found that the combination of TBADT as photocatalyst and LiOH as base under near-ultraviolet light irradiation (Kessil 43 W, 370 nm LED) in acetonitrile was a proper system for the selective formation of gem-difluoroalkene 3 (Table 1, entry 1). Acetonitrile, despite the relatively low bond dissociation energy of its C–H bonds, is inert under these HAT reaction conditions. This is due to a polarity mismatch between the electrophilic reactive oxygen atoms in the photoexcited TBADT and the hydrogen bonds in acetonitrile that have also electrophilic chatacter.20c|
|
||
|---|---|---|
| Entrya | Deviation from standard conditions | 3 yieldb (%) |
| a Reactions performed on a 0.2 mmol scale. b Yields were determined by 19F-NMR analysis using α,α,α-trifluorotoluene as internal standard. c Yield of isolated product. d CuCl (5 mol%) + PPh3 (6 mol%) with collidine (1.1 equiv.) instead of LiOH. e Irreproducibility issues were found. | ||
| 1 | None | 32 |
| 2 | 3 mol% of TBADT | 81(81) |
| 3 | 5 mol% [Cu]d as co-catalyst | 11 |
| 4 | Collidine, lutidine or DMAP instead of LiOH | <11 |
| 5 | K3PO4 instead of LiOH | 17 |
| 6 | KHCO3 instead of LiOH | 23 |
| 7 | Li2CO3 instead of LiOH, 3 mol% of TBADT | 20–75e |
| 8 | 5 equiv. of 1, 3 mol% of TBADT | 65 |
| 9 | 390 nm instead of 370 nm, 3 mol% of TBADT | 63 |
| 10 | No TBADT, light or base | — |
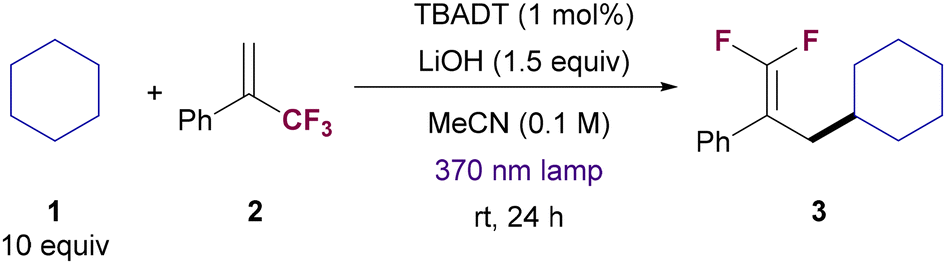
Optimal conditions required the use of 3 mol% of TBADT, 10 equiv. of cyclohexane and 1.5 equiv. of LiOH. Under those conditions 3 was obtained in 81% yield (entry 2). The employment of our previous reported conditions for the C–H allylation of alkanes with allylic chlorides using TBADT and Cu/PPh3 as co-catalyst21 provided 3 in diminished yield (entry 3). Notably, the use of organic bases instead of LiOH was detrimental to access 3 (entry 4). Inorganic bases generally performed better, with Li-based salts providing 3 in higher yields (entries 5–7). This effect is probably due to a Lewis acid-type Li–F activation22 which might facilitate the β-fluoride elimination step. Li2CO3 proved to be an equally efficient base than LiOH (entry 7), although it was discarded based on observed irreproducible results which were likely due to solubility issues. Interestingly, the reaction could be performed with a smaller amount of cyclohexane 1 (5 equiv., entry 8) or with different irradiation wavelength (390 nm, entry 9), albeit with slight erosion in reaction yield. Finally, control experiments revealed that the reaction does not proceed in the absence of TBADT, light or base (entry 10).
Having established optimized conditions for the defluorinative alkylation of trifluoromethyl alkenes with alkanes, we set out to explore the scope of this transformation (Scheme 2). Cycloalkanes with different ring sizes proved to be efficient coupling partners leading to the corresponding products 3–5 in very good yields. Notably, this methodology could be applied to the defluorinative alkylation of more complex aliphatic natural products such as camphene (6), fenchone (7) and eucalyptol (8). The late-stage modification of these compounds was accomplished in good yield (49–61%) with moderate regioselectivity (up to 2.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr), and with excellent diastereoselectivity (>20:1 dr). These examples show the capability of this transformation to install the gem-difluoroalkene group onto complex aliphatic substrates without the need for directing groups.
:1 rr), and with excellent diastereoselectivity (>20:1 dr). These examples show the capability of this transformation to install the gem-difluoroalkene group onto complex aliphatic substrates without the need for directing groups.
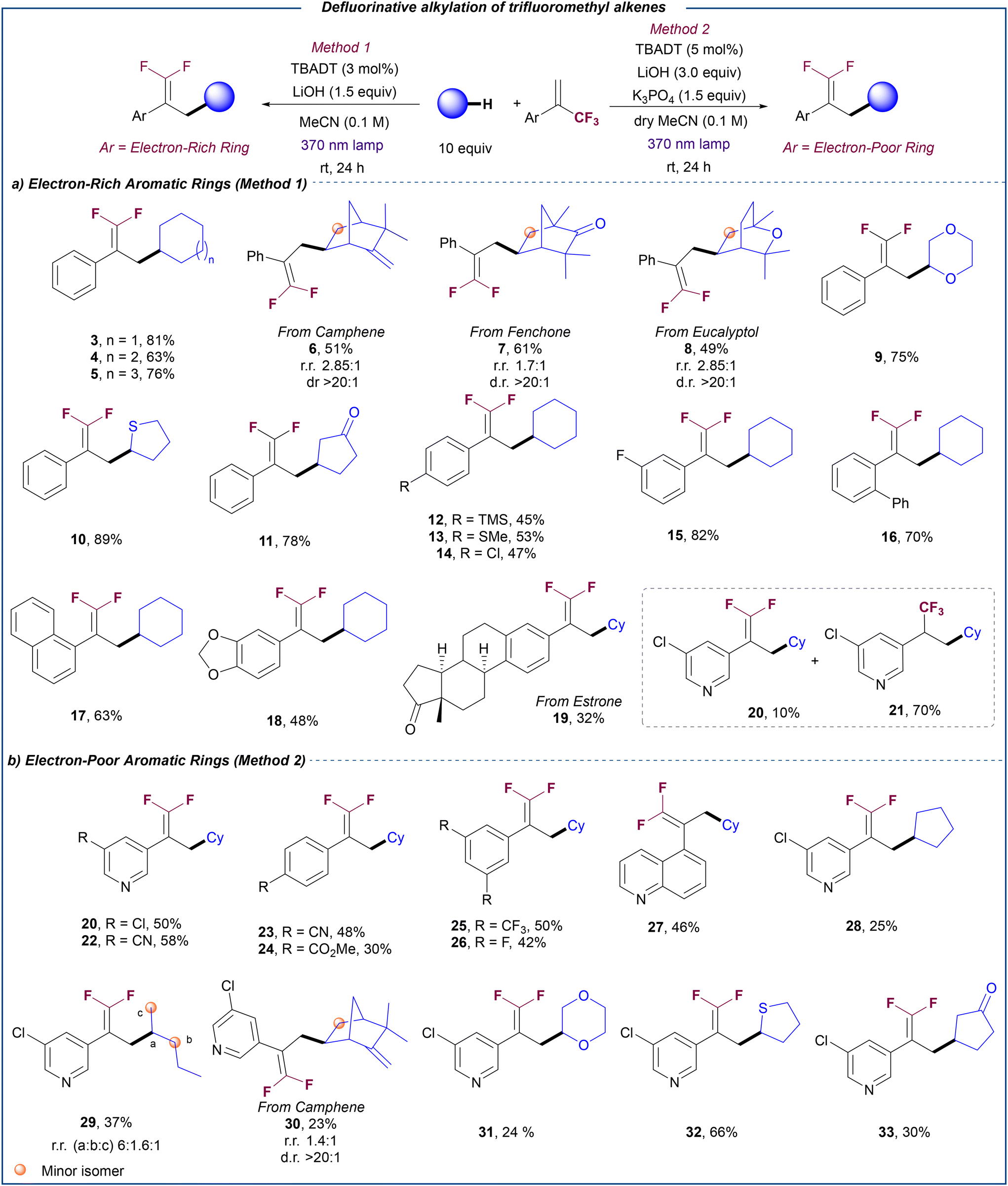 | ||
| Scheme 2 Scope for the defluorinative alkylation of (hetero)aromatic trifluoromethyl alkenes with alkanes. Reactions performed on a 0.2 mmol scale. Yields refer to the isolated product. | ||
This photocatalytic defluorinative alkylation could also be extended to non-electron-neutral C–H donors. Cyclic ether (9) and thioether (10) could be selectively functionalized at the α-position, while cyclopentanone was alkylated at the β-position (11). The observed regiocontrol is in good agreement with the preference of the electrophilic excited state of decatungstate anion for the most hydridic C–H bonds.20c
We then focused on the evaluation of a diverse range of α-trifluoromethyl alkenes. Aryl-substituted substrates bearing trimethylsilyl (12), thioether (13), chloride (14), fluoride (15) and phenyl (16) were coupled with cyclohexane in good yields regardless of the position of the substituent on the aromatic ring. Substrates bearing more sterically encumbered substituents such as naphthalene (17) or 1,3-benzodioxole (18) bicyclic rings or even an α-trifluoromethyl alkene derived from estrone (19) were also compatible for this transformation. Notably, when a substrate bearing an electron-poor 3-chloropyridine ring was reacted with cyclohexane under optimized conditions (Table 1, entry 2), a mixture of the corresponding gem-difluoroalkene 20 and trifluoromethyl alkane 21 was obtained. We reasoned that the presence of an electron poor substituent at the α position of the trifluoromethyl alkene may stabilize the carbanion intermediate thus facilitating the competitive protonation pathway that provides the hydroalkylation product versus the β-fluoride elimination. Thus, we decided to explore alternative conditions to render the defluorinative alkylation of electron-poor substrates selective by securing the efficient quenching of any protic traces. In line with our hypothesis, the use of a more basic medium involving a combination of LiOH (3 equiv.) and K3PO4 (1.5 equiv.) led to the chemoselective formation of gem-difluoroalkene 20 (see ESI,† section 5.2, for optimization details). Under these modified conditions, different α-trifluoromethyl alkenes bearing other 3-substituted pyridine (22), electron poor para- and meta-(di-) substituted aryl rings (23–26) and quinoline (27) could be reacted with cyclohexane to selectively yield the corresponding gem-difluoroalkenes in moderate to good yields. Similarly, other C–H donors such as cyclopentane (28), n-pentane (29), camphene (30), dioxane (31), tetrathiophene (30), and cyclopentanone (32) proved efficient for the defluorinative coupling with an electron-deficient substrate.
Hydroalkylation
Based on the different reaction outcome observed for the 3-chloropyridine-substituted α-trifluoromethyl alkene depending on the reaction conditions, we decided to explore a general methodology for the selective hydroalkylation of these fluorinated olefins with alkanes. Opposite to the formation of the gem-difluoroalkene, we reasoned that the absence of basic additives may favour the protonation step that leads to the formation of the hydroalkylation product. Indeed, when this substrate was reacted with cyclohexane in the absence of base under TBADT photocatalysis, hydroalkylation product 21 was exclusively obtained in 73% yield (Scheme 3). Importantly, reaction could be scaled up to 5 mmol without significant erosion in reaction yield. A variety of α-trifluoromethyl alkenes bearing other pyridine groups (34) and electron-poor aryl rings (35–38) reacted with cyclohexane, furnishing the corresponding trifluoromethyl alkanes in good yields. In some cases, products were obtained together with small amounts of the defluoroalkylation products. Fortunately, the use of hexafluoroisopropanol (HFIP) as additive restored the chemoselectivity towards the hydroalkylation product (see the ESI,† section 5.3.2, for details). As described for the defluorinative alkylation, other C–H donors such as cyclopentane (39), camphene (40), fenchone (41), cyclopentanone (42), tetrahydrothiophene (43) and 1,4-dioxane (44) also proved efficient for the hydroalkylation reaction. Despite proving unsuccessful for the defluorinative coupling (see ESI,† section 9), toluene could be efficiently used for the hydroalkylation reaction as illustrated with the synthesis of product 45 in 53% yield. Moreover, this photocatalytic hydroalkylation protocol could be extended to the use of acyclic alkanes. The reaction with 2,3-dimethylbutane (46) resulted in the site-selective functionalization of the methine C–H bond, while trifluoromethylalkylation of methylene C–H bonds was preferred over the methyl C–H bonds for n-hexane (47), in accordance with the site-selectivity typically displayed by the photoexcited decatungstate anion in other transformations with these saturated hydrocarbons.20c,22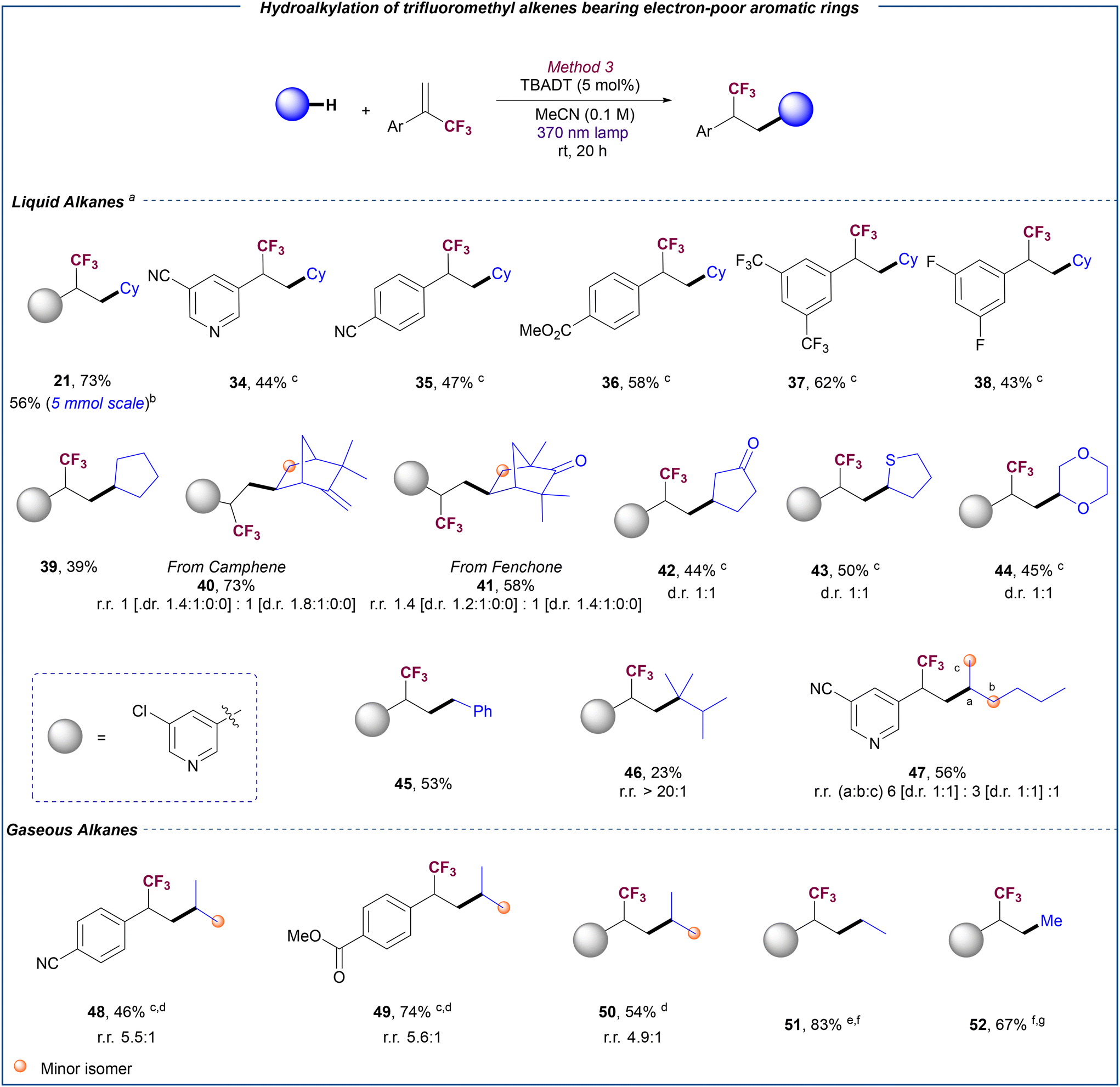 | ||
| Scheme 3 Scope for the hydroalkylation of (hetero)aromatic trifluoromethyl alkenes bearing electron-poor substituents with liquid and gaseous alkanes. (a) Reactions conditions: trifluoromethyl alkene (0.2 mmol), liquid alkane (10 equiv.), TBADT (5 mol%), MeCN (0.1 M) at rt over 20 h and irradiation by a 370 nm 43 W Kessil LED lamp. Yields refer to the isolated product. (b) TBADT 1.0 mol%. (c) HFIP (1.0 equiv.) used. (d) Reactions performed at 5.0 bar of propane over 14 h, MeCN (0.05 M). (e) Reactions performed at 30 bar of ethane in MeCN (0.0125 M) over 14 h. (f) FeCl3·6H2O (5 mol%) used instead of TBADT and irradiation by a 390 nm 52 W Kessil LED lamp. (g) Reactions performed at 50 bar of methane in MeCN-d3 (0.0125 M) over 48 h. | ||
To further demonstrate the versatility of our developed hydroalkylation process, we next explored its application in the functionalization of gaseous alkanes. The use of these feedstocks as sustainable, inexpensive and atom/step economical reagents in chemical synthesis is highly attractive given their vast abundancy in our planet. However, the intrinsic inertness of the C–H bonds in these alkanes (high bond dissociation energy BDE = 99–105 kcal mol−1, ionization potential ∼12.5 eV, pKa ∼ 45–50), has rendered their use in organic synthesis extremely challenging.23 Moreover, their gaseous nature adds the extra challenge of achieving a proper gas/liquid exchange regime that enables efficient alkyl radical formation. Notably, the reaction conditions optimized for the hydroalkylation of liquid alkanes could be successfully applied to propane just by using 5 bar pressure of this gas. Products 48–50 were obtained in good yield with a preferential functionalization of the secondary C–H bond over the primary ones. With ethane, our optimized conditions using TBADT as photocatalyst provided product 51 in low yield (see ESI,† section 5.3.3 for details). Nevertheless, by changing the HAT photocatalyst to FeCl3·6H2O, which can serve as a chlorine radical source upon photoinduced LMCT,24 a significant improvement of reaction efficiency was observed and product 51 could be isolated in a satisfactory 83% yield. Importantly, these new conditions could be applied to the efficient functionalization of methane, the most abundant alkane and main component of natural gas,25 but an extremely challenging feedstock to activate under mild conditions.26 Using FeCl3·6H2O as photocatalyst, and after minimal optimization (see ESI,† section 5.3.3), methane could be coupled with 3-chloro-5-(3,3,3-trifluoroprop-1-en-2-yl)pyridine to furnish 52 in a remarkable 67% yield.
An interesting observation was done when α-trifluoromethyl alkenes bearing electron-neutral or electron-rich substituents were employed for the hydroalkylation reaction. Under optimized conditions (method 3), no reaction was observed between cyclohexane and substrate 2. Control experiments in the presence of difference Li-based Lewis acid additives showed a similar lack of reactivity (see ESI,† section 5.4.1). Given the successful behaviour of substrate 2 under photocatalytic defluorinative alkylation conditions (see Table 1 and Scheme 2), these results suggest that the electron transfer between the reduced TBADT and the trifluoromethyl alkyl radical generated from 2 is inefficient in the absence of base (see below for mechanism proposals). Based on this hypothesis, we reasoned that the use of a thiol as HAT co-catalyst27 may facilitate the hydrogen transfer from the reduced TBADT to the trifluoromethyl alkyl radical via a complementary pair of HAT events (see below for mechanistic proposal). Screening of several thiols and disulfides (see ESI,† section 5.4.2 for details) revealed that thiophenols 53 and 54 in combination with TBADT efficiently promote the selective hydroalkylation of 2 by using MeCN-d3 to avoid solvent functionalization (Scheme 4). Under these cooperative catalysis conditions, different C–H donors could be coupled with α-trifluoromethyl alkenes bearing an electron-neutral (55–59), or electron-rich substituent (60–62) and even with a complex Estrone derivative (63). Note that choice of the thiol catalyst was done depending on the polarity of the product to facilitate purification.
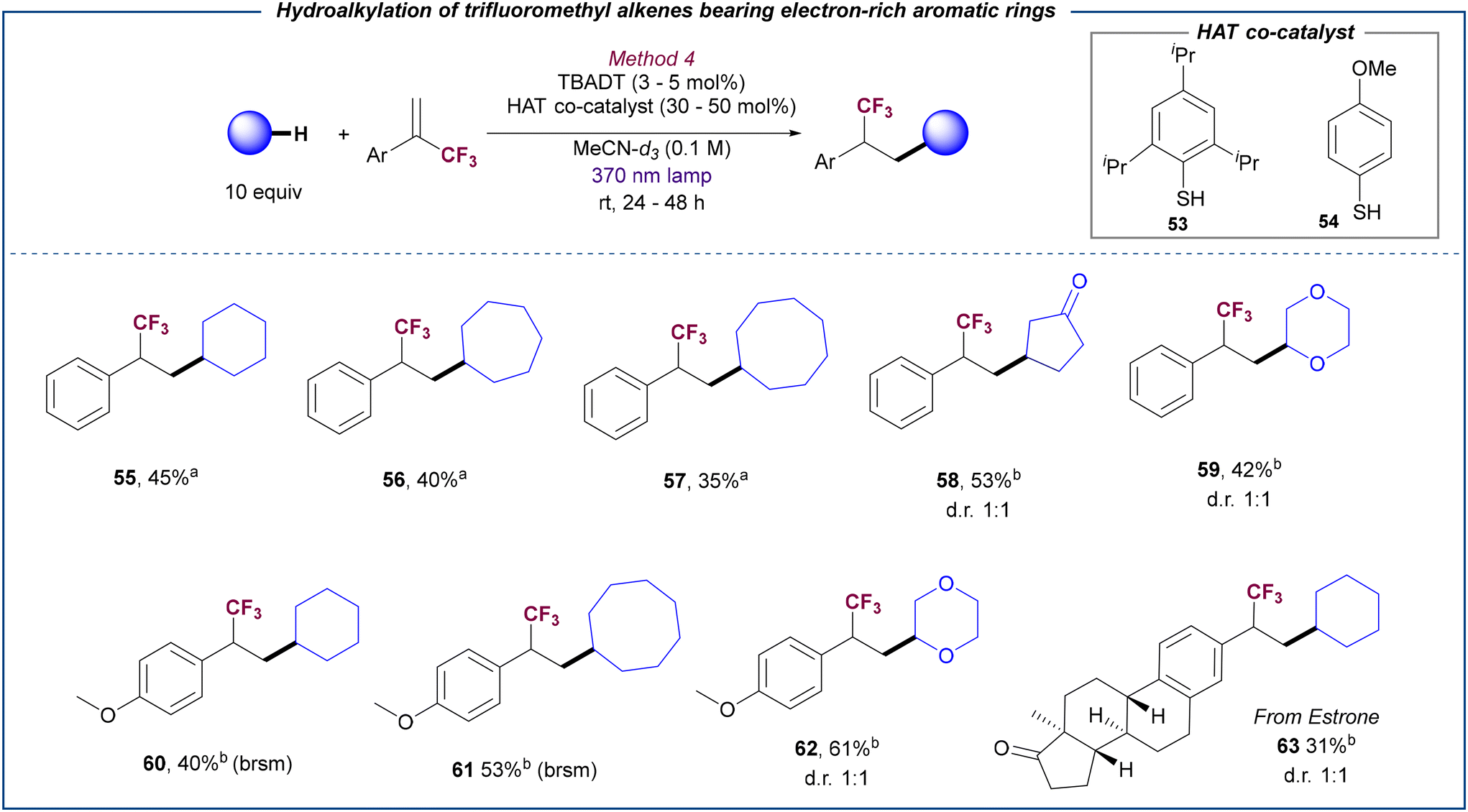 | ||
| Scheme 4 Scope for the hydroalkylation of trifluoromethyl alkenes bearing electron-rich aromatic rings. Reactions performed on a 0.2 mmol scale. Yields refer to the isolated product. (a) 54 as HAT co-catalyst (30 mol%) and TBADT (5 mol%) in 24 h. (b) 53 as HAT co-catalyst (50 mol%) and TBADT (3 mol%) in 48 h. brsm = based on recovered starting material. | ||
Mechanistic considerations
To gain insight into the mechanism of these transformations, a set of deuteration labelling experiments were performed (Scheme 5a). First, we run the reaction between 3-chloropyridyl-substitued trifluoromethyl alkene 64 and deuterated cyclohexane, observing a 15% of deuterium incorporation at the benzylic position of product 21-d. Interestingly, 40% of deuterium incorporation was observed at positions 2 and 6 of the pyridine ring. This result may suggest that formation of 21-d occurs through the generation of a benzylic carbanion which is stabilized by resonance along the pyridine ring. A similar experiment with trifluoromethyl alkene 65 that bears a 3,5-(bis)trifluoromethyl-substituted benzene ring in which the delocalization of the carbanion is less favored resulted in the formation of product 37-d with 30% of deuterium incorporation at the benzylic position. Albeit deuteration might arise from a direct HAT process between the reduced TBADT and the trifluoromethyl alkyl radical intermediate, the low percentage of deuteration observed for these two results suggest that the hydroalkylation of α-trifluoromethyl alkenes bearing electron-poor aryl rings likely occurs by SET reduction of the radical intermediate to the carbanion. When deuterated cyclohexane is used, subsequent protonation may occur by reaction with a deuteron coming from the reduced TBADT or with any adventitious protic agent present in the reaction medium. Indeed, when acetonitrile was not dried prior to the reaction between 64 and deuterated cyclohexane, no deuteration was observed in the product (see ESI† for details).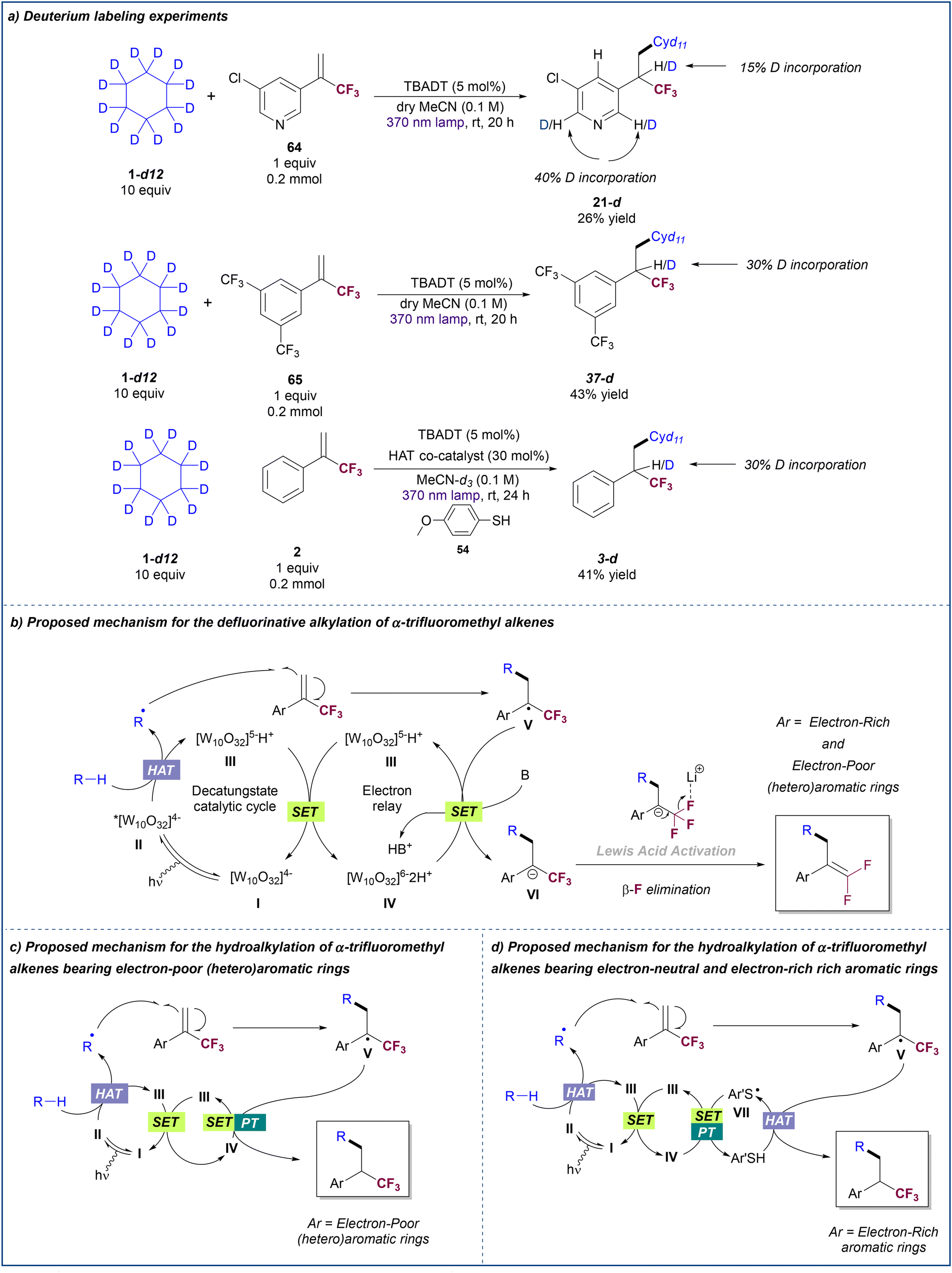 | ||
| Scheme 5 (a) Deuterium labelling experiments and proposed mechanism for (b) the photocatalytic defluorinative alkylation of α-trifluoromethyl alkenes with alkanes, (c) the photocatalytic hydroalkylation of electron-poor α-trifluoromethyl alkenes with alkanes and (d) the photocatalytic hydroalkylation of electron-neutral and electron-rich α-trifluoromethyl alkenes under dual TBADT/thiol catalysis. | ||
The reaction between trifluoromethyl alkene 2 and deuterated cyclohexane run under TBADT/thiol cooperative catalysis conditions was also explored. The fact that a significant degree of deuteration was observed at the benzylic position of product 3-d, despite using a non-deuterated thiol, supports the complementarity of the proposed paired catalytic events.
On the basis of these experimental results and previous reports on TBADT photocatalysis,20,28 we propose the following mechanisms for the defluorinative alkylation (Scheme 5b) and the hydroalkylation (Scheme 5c and d) of α-trifluoromethyl alkenes with alkanes. Light irradiation of the decatungstate anion I produces the excited-state decatungstate II which promotes the hydrogen atom abstraction from the alkane. This process generates an alkyl radical and singly reduced decatungstate III which undergoes disproportionation into doubly reduced decatungstate IV and ground state decatungstate I. Addition of the alkyl radical to the trifluoromethyl alkene would form radical intermediate V. In the presence of base (Scheme 5b), single electron transfer between this radical species and the doubly reduced decatungstate IV would regenerate singly reduced decatungstate III and would form carbanion VI which undergoes β-fluoride elimination to furnish the gem-difluoroalkene product. Given the observed absence of reactivity of α-trifluoromethyl styrene 2 in the absence of base, the SET event between IV and V would be likely facilitated by the base.
When using α-trifluoromethyl alkenes bearing electron-poor (hetero)aryl rings in the absence of base (Scheme 5c), single electron transfer between IV and V is efficient and subsequent proton transfer furnishes the hydroalkylation product. This does not occur for substrates bearing electron-neutral and electron-rich aryl rings where the use of a paired thiol HAT catalysis cycle is necessary to promote this reaction (Scheme 5d). In this case, radical intermediate V undergoes HAT with the thiol yielding the hydroalkylated product and a sulphur-centred radical VII. Both catalytic cycles are completed by a SET process between IV and VII (Ered1/2 ([W10O32]5−/[W10O32]6−) = −1.56 V vs. SCE in MeCN28aEred1/2[ArS˙/ArS−] = −1.26 V vs. SCE in MeCN29) that regenerates the singly reduced decatungstate III and the thiol after proton transfer.
Conclusions
In summary, we have developed a chemodivergent methodology for the conversion of α-trifluoromethyl alkenes into gem-difluoroalkenes and trifluoromethyl alkanes based on a HAT photoredox catalysis strategy. Saturated hydrocarbons, including gaseous alkanes, can be directly used as alkylating agents without the requirement of pre-activated radical precursors, making this strategy more attractive from a step- and atom-economy point of view. Activation of alkanes was achieved by using TBADT (or FeCl3 for methane and ethane) as photocatalyst, while chemoselectivity can be easily tuned by simple modification of reaction conditions and/or additives. Both defluorinative alkylation and hydroalkylation reactions proceed under mild conditions and can be applied to electron-poor and electron-rich substrates, though we found that the hydroalkylation of the latter requires the use of cooperative TBADT/thiol catalysis. Further studies on the use of gaseous alkanes as sustainable and atom-efficient feedstocks for the synthesis of fluorine-containing molecules are currently underway.Author contributions
P. M.-B., A. V.-R., S. B.-A. and H. J.-C. performed the experiments. All the authors analysed the data. M. F.-M. supervised the project and wrote the manuscript, with input of all the authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Research Council (ERC-CoG 863914-BECAME), the Agencia Estatal de Investigación (TED2021-132449B-I00, PID2023-151875NB-I00), the Xunta de Galicia (ED431C 2022/27; Centro de investigación do Sistema universitario de Galicia accreditation 2023-2027, ED431G 2023/03) and the European Regional Development Fund (ERDF) is gratefully acknowledged.This work is dedicated to the memory of our beloved friend Dr Alejandro Gutiérrez-González.
References
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (b) M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- (a) G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon and J.-P. Bégué, J. Fluorine Chem., 2006, 127, 637–642 CrossRef CAS; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (c) C.-C. Tseng, G. Baillie, G. Donvito, M. A. Mustafa, S. E. Juola, C. Zanato, C. Massarenti, S. Dall'Angelo, W. T. A. Harrison, A. H. Lichtman, R. A. Ross, M. Zanda and I. R. Greig, J. Med. Chem., 2019, 62, 5049–5062 CrossRef CAS.
- (a) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef CAS PubMed; (b) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS; (c) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC; (d) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC; (e) L. Zhou, Molecules, 2021, 26, 7051 CrossRef CAS PubMed; (f) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (g) K.-J. Bian, Y.-C. Lu, D. Nemoto Jr, S.-C. Kao, X. Chen and J. G. West, Nat. Chem., 2023, 15, 1683–1692 CrossRef CAS.
- (a) S. Watanabe, K. Sugahara, T. Fujita, M. Sakamoto and T. Kitazume, J. Fluorine Chem., 1993, 62, 201–206 CrossRef CAS; (b) J.-P. Bégué, D. Bonnet-Delpon and M. H. Rock, Tetrahedron Lett., 1995, 36, 5003–5006 Search PubMed.
- (a) T. Ichitsuka, T. Fujita and J. Ichikawa, ACS Catal., 2015, 5, 5947–5950 CrossRef CAS; (b) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2016, 138, 12340–12343 CrossRef CAS; (c) Y. J. Jang, D. Rose, B. Mirabi and M. Lautens, Angew. Chem., Int. Ed., 2018, 57, 16147–16151 CrossRef CAS; (d) M. Wang, X. Pu, Y. Zhao, P. Wang, Z. Li, C. Zhu and Z. Shi, J. Am. Chem. Soc., 2018, 140, 9061–9065 CrossRef CAS PubMed; (e) R. Kojima, S. Akiyama and H. Ito, Angew. Chem., Int. Ed., 2018, 57, 7196–7199 CrossRef CAS; (f) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Chem, 2018, 4, 2201–2211 CrossRef CAS; (g) P. H. S. Paioti, J. del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917–19934 CrossRef CAS.
- (a) Y. Lan, F. Yang and C. Wang, ACS Catal., 2018, 8, 9245–9251 CrossRef CAS; (b) R. J. Wiles, J. P. Phelan and G. A. Molander, Chem. Commun., 2019, 55, 7599–77602 RSC; (c) C. Zhu, Z. Y. Liu, L. N. Tang, H. Zhang, Y. F. Zhang, P. J. Walsh and C. Feng, Nat. Commun., 2020, 11, 4860 CrossRef CAS; (d) Y. Guo, Y. Cao, H. Song, Y. Liu and Q. Wang, Chem. Commun., 2021, 57, 9768–9771 RSC.
- (a) T. Xiao, L. Li and L. Zhou, J. Org. Chem., 2016, 81, 7908–7916 CrossRef CAS; (b) X. Lu, X.-X. Wang, T.-J. Gong, J.-J. Pi, S.-J. He and Y. Fu, Chem. Sci., 2019, 10, 809–814 RSC; (c) Y.-Q. Guo, R. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2020, 22, 709–713 CrossRef CAS.
- (a) S. B. Lang, R. J. Wiles, C. B. Kelly and G. A. Molander, Angew. Chem., Int. Ed., 2017, 56, 15073–15077 CrossRef CAS; (b) Y. Chen, N. Ni, D. Cheng and X. Xu, Tetrahedron Lett., 2020, 61, 152425 CrossRef CAS.
- L.-H. Wu, J.-K. Cheng, L. Shen, Z.-L. Shen and T.-P. Loh, Adv. Synth. Catal., 2018, 360, 3894–3899 CrossRef CAS.
- (a) J. P. Phelan, S. B. Lang, J. Sim, S. Berritt, A. J. Peat, K. Billings, L. Fan and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 3723–3732 CrossRef CAS PubMed; (b) H. Chen, D. Anand and L. Zhou, Asian J. Org. Chem., 2019, 8, 661–664 CrossRef CAS.
- Z. Lin, Y. Lan and C. Wang, ACS Catal., 2019, 9, 775–780 CrossRef CAS.
- (a) W. J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS; (b) Y.-Q. Guo, Y. Wu, R. Wang, H. Song, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 2353–2358 CrossRef CAS PubMed; (c) J. Tian and L. Zhou, Chem. Sci., 2023, 14, 6045–6051 RSC.
- S. O. Badir, A. Lipp, M. Krumb, M. J. Cabrera-Afonso, L. M. Kammer, V. E. Wu, M. Huang, A. Csakai, L. A. Marcaurelle and G. A. Molander, Chem. Sci., 2021, 12, 12036–12045 RSC.
- C. Zhu, H. Zhang, Q. Liu, K. Chen, Z.-Y. Liu and C. Feng, ACS Catal., 2022, 12, 9410–9417 CrossRef CAS.
- Y. Zhang, Y. Zhang, Y. Guo, S. Liu and X. Shen, Chem Catal., 2022, 2, 1380–1393 CrossRef CAS.
- H. Tian, R. Zhang, L. Shi, C. Zhao and X. Wang, Chin. J. Chem., 2023, 41, 1783–1790 CrossRef CAS.
- P. Lei, B. Chen, T. Zhang, Q. Chen, L. Xuan, H. Wang, Q. Yan, W. Wang, J. Zeng and F. Chen, Org. Chem. Front., 2024, 11, 458–465 RSC.
- J.-X. Wang, M.-C. Fu, L.-Y. Yan, X. Lu and Y. Fu, Adv. Sci., 2024, 11, 2307241 CrossRef CAS.
- (a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS; (b) L. Chang, S. Wang, Q. An, L. Liu, H. Wang, Y. Li, K. Feng and Z. Zuo, Chem. Sci., 2023, 14, 6841–6859 RSC; (c) A. Velasco-Rubio, P. Martínez-Balart, A. M. Álvarez-Constantino and M. Fañanás-Mastral, Chem. Commun., 2023, 59, 9424–9444 RSC.
- (a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC; (b) V. D. Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef CAS; (c) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS; (d) Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS; (e) S. Xu, H. Chen, Z. Zhou and W. Kong, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed; (f) J. Zhang and A. Studer, Nat. Commun., 2022, 13, 3886 CrossRef CAS.
- P. Martínez-Balart, B. L. Tóth, Á. Velasco-Rubio and M. Fañanás-Mastral, Org. Lett., 2022, 24, 6874–6879 CrossRef PubMed.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (c) K. Lyea and R. D. Young, Chem. Sci., 2024, 15, 2712–2724 RSC.
- (a) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS; (b) G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed; (c) A. Pulcinella, D. Mazzarella and T. Noël, Chem. Commun., 2021, 57, 9956–9967 RSC.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- (a) J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2020, 11, 1026–1031 RSC; (b) Y. Kuang, H. Cao, H. Tang, J. Chew, W. Chen, X. Shi and J. Wu, Chem. Sci., 2020, 11, 8912–8918 RSC; (c) C. J. Oswood and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 93–98 CrossRef CAS PubMed; (d) Y.-A. Zhang, V. Palani, A. E. Seim, Y. Wang, K. J. Wang and A. E. Wendlandt, Science, 2022, 378, 383–390 CrossRef CAS.
- (a) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS; (b) X. Hu, I. Cheng-Sánchez, W. Kong, G. A. Molander and C. Nevado, Nat. Catal., 2024, 7, 655–665 CrossRef CAS.
- R. Chang, Y. Pang and J. Ye, Angew. Chem., Int. Ed., 2023, 62, e202309897 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, optimization studies, experimental procedures and compound characterization. See DOI: https://doi.org/10.1039/d4gc04176c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |