
Light-driven ultrafast dual C–C cleavage and coupling of dihydroxyacetone into high-purity carbon monoxide and ethylene glycol†
Fanhao
Kong
,
Hongru
Zhou
,
Zhiwei
Chen
,
Zhaolin
Dou
and
Min
Wang
 *
*
State Key Laboratory of Fine Chemicals, School of Chemistry, Dalian University of Technology, Dalian 116024, Liaoning, China. E-mail: wangmin@dlut.edu.cn
First published on 19th September 2024
Abstract
Bulk chemicals, such as carbon monoxide (CO) and ethylene glycol (EG), derived from biomass feedstocks instead of traditional fossil fuels present a renewable and sustainable energy alternative. However, their direct production under ambient conditions with high purity is challenging. Herein, we report a distinctive photochemical process to produce CO together with EG through the ultrafast C–C cleavage of dihydroxyacetone induced by ultraviolet illumination at ambient temperature and pressure in water. The obtained CO yield was nearly 98% with a high purity above 99.9%, and the EG yield was nearly 80%. The two hydroxyls at the alpha carbons of dihydroxyacetone reduced the energy barriers of photoexcitation and the radical reaction, accounting for fast C–C cleavage to produce CO and EG. A home-built flow reactor achieved the continuous production of CO and EG over 1000 h and maintained efficient operation for 100 h in the sunlight-electricity-ultraviolet light mode. Benefitting from the fast reaction rate and high-purity gas generation, a safe, laboratory-scale, portable, ready-to-use CO generator was designed and assembled. The maximum CO production rate of the CO generator reached 30 mL min−1, and CO purity reached over 99% for 25 L.
Introduction
Chemicals produced from renewable carbon feedstocks, wastes or greenhouse gases under ambient conditions have garnered enormous interest in the past decade as they offer an opportunity to close the gap between carbon emission and conversion.1–6 Carbon monoxide is an important molecule in the chemical industry that can be used to synthesize various chemicals.7–9 Industrial CO is commonly produced through the gasification of fossils and biomass, steam reforming or partial oxidation of oil under harsh conditions (700–1600 °C, 2–8 MPa), and the obtained proportion of CO is about 10–42% of the gas mixture (Fig. S1A†).10–14 Other processes such as CH4 reforming (450–1000 °C, 0.1–10 MPa),15–18 CO2 hydrogenation (200–500 °C, 0.1–5 MPa)19–22 and/or photo-/electrocatalytic CO2 reduction (25 °C, 0.1 MPa)23–25 are alternative ways to produce CO; however, the produced CO gas is commonly of low purity because of either low conversion of feedstocks or low selectivity for CO, thus requiring laborious gas separation and purification.26 Thus, direct high-purity CO production remains a major challenge.Moreover, besides its large-scale use as a bulk chemical in industries, small-scale usage of CO in laboratories is often in the form of high-pressure gas cylinders. Considering the potential security risk of CO leaching and explosion of high-pressure gas cylinders, portable CO generators are more desirable for real-time CO generation and usage. However, the present method is unsuitable for CO generator equipment because of the low purity of CO and the requirement for harsh conditions.
Ethylene glycol is mainly used as an antifreeze agent and feedstock in the synthesis of polyethylene terephthalate.27 Current global annual production of EG is about 42 million metric tons, with the demand growing at the rate of 5–10% per year.28 Industrial EG is traditionally produced via a petroleum process including two steps, namely, ethylene partial oxidation (220–280 °C, 1–3 MPa) and ethylene oxide hydration (130–220 °C, 1–2.5 MPa),29,30 which consume about 1.6 t of CO2 per ton of EG produced31 (Fig. S1B†). Optimization of the raw source, temperature, and pressure for effective ethylene oxidation hydration is under exploration.31,32 Ethylene glycol can also be additionally obtained through syngas hydrogeneration (150–230 °C, 1–5 MPa),30 biomass hydrogeneration (145–245 °C, 4–10 MPa)33 or photocatalytic methanol coupling (25 °C, 0.1 MPa).28,34
Herein, different from the current cracking or catalytic conversion process, we propose a novel photochemical process for high-purity CO and EG production via the direct C–C cleavage of dihydroxyacetone (DHA) under ultraviolet light illumination at ambient temperature and pressure (Fig. S1C†). DHA is listed as one of the most important C3 biomass platform molecules and can be obtained by the catalytic oxidation of glycerol or fermentation of ketose.35,36 This method can be used for continuous CO/EG production, and a flow reactor could operate for 1000 h with stability. More importantly, a laboratory-scale portable CO generator was further designed and assembled given the advantages of this approach, including ultrafast and catalyst-free operation and high-purity CO generation.
Results and discussion
Photolytic activity of dihydroxyacetone and other ketones
We first investigated the optimal reaction conditions for the photolysis of dihydroxyacetone (Fig. 1A). This series of reactions was screened using a home-built lamp set (Fig. S2†) by changing three factors, namely the radiation wavelength, solvent, and atmosphere (Fig. 1B, table). The photolysis of DHA in water at 275 nm under an Ar atmosphere resulted in 100% conversion, with a 98% yield of CO and 80% yield of EG (table, entry 2). Switching the light to 254 nm slightly reduced the yields of CO and EG (table, entry 1), and the reaction did not occur under 365 nm light irradiation (table, entry 3). Sunlight and an Xe lamp afforded 37%–70% conversion of DHA but gave very low yields of CO and EG (table, entries 4 and 5). Directly illuminated solid DHA was almost inactive (table, entry 6), while the dissolution of DHA in a solvent favored its photolysis. Water was more effective than alcoholic solvents, such as methanol and ethanol (table, entries 7 and 8), of which ethanol enabled only an 11% DHA conversion. A solvent mixture of MeCN and water improved DHA conversion and the CO/EG yields, and the photolysis of DHA in MeCN/water mixed at a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio also exhibited 100% DHA conversion and 96% carbon yield for CO and 80% carbon yield for EG similar to those observed in water (table, entries 9 and 10). The atmosphere showed little effect on the photolysis of DHA, and >95% yield of CO and >72% yield of EG were still obtained in the air and pure oxygen atmospheres (table, entries 11 and 12). Therefore, radiation wavelength and solvent are the two main factors affecting the photochemical conversion of DHA to CO and EG. Moreover, the optimal wavelength was near 275 nm, and the optimal solvent was water.
:1 volume ratio also exhibited 100% DHA conversion and 96% carbon yield for CO and 80% carbon yield for EG similar to those observed in water (table, entries 9 and 10). The atmosphere showed little effect on the photolysis of DHA, and >95% yield of CO and >72% yield of EG were still obtained in the air and pure oxygen atmospheres (table, entries 11 and 12). Therefore, radiation wavelength and solvent are the two main factors affecting the photochemical conversion of DHA to CO and EG. Moreover, the optimal wavelength was near 275 nm, and the optimal solvent was water.
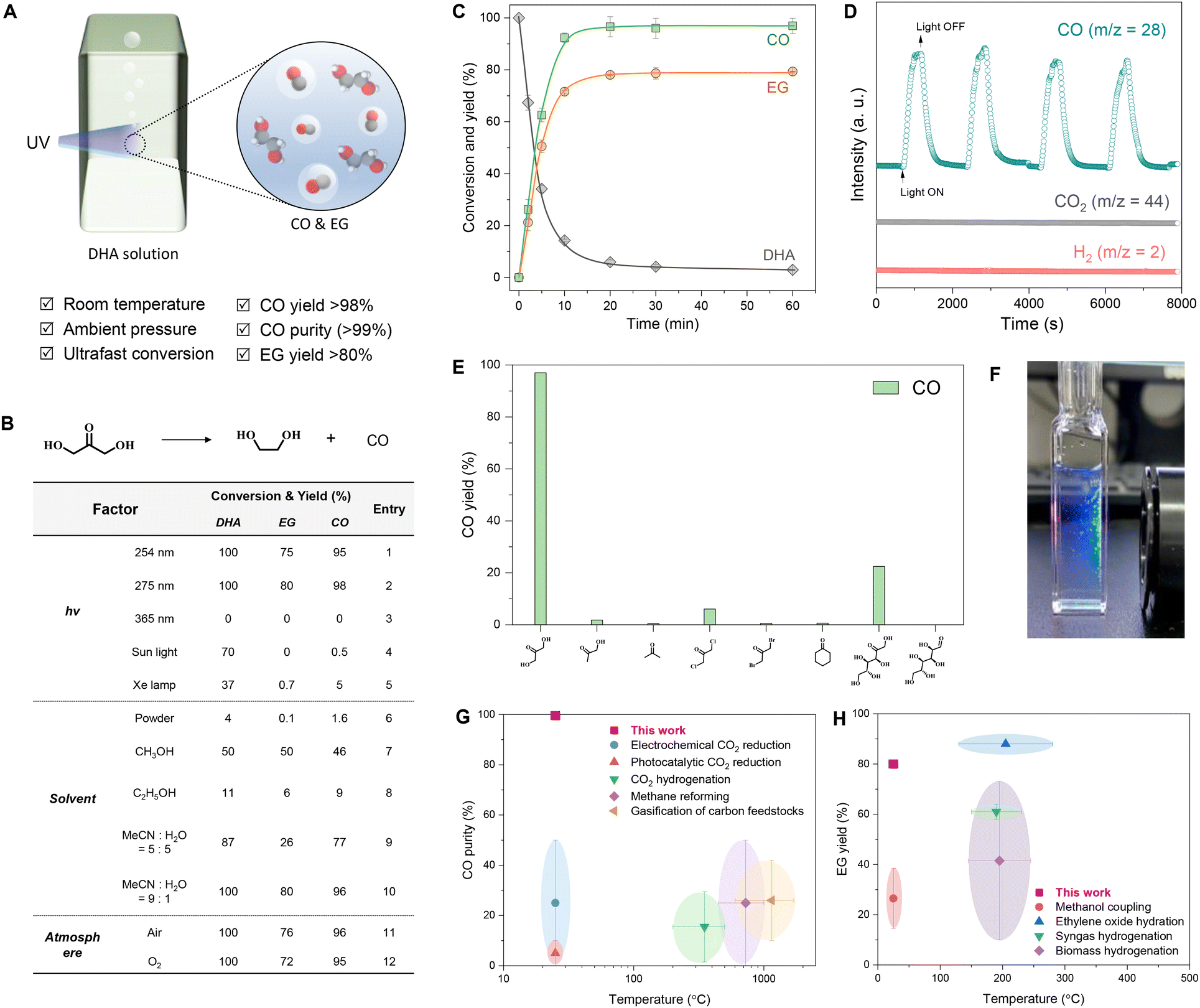 | ||
| Fig. 1 Photolytic activity of dihydroxyacetone and other ketones. (A) Schematic of the photolysis of DHA to CO and EG under ultraviolet light. (B) Condition optimization for the photolysis of DHA to CO and EG. Normal conditions: 0.1 M DHA, 1 mL of H2O, home-made 16 W 275 nm lamp set with a circular arrangement of eight 2 W LED lights in series, irradiation for 1 h, Ar atmosphere, room temperature, stirring at 800 rpm. (C) Time-dependent conversion profile at normal conditions. (D) Online mass spectra of the photolysis of 1.0 M DHA aqueous solution by one-side illumination using a commercial handheld 5 W 275 nm LED with Ar as the carrier gas and stirring at 800 rpm. (E) The CO yield of various ketones and aldehydes after photolysis for 1 h under normal conditions. The solvent for cyclohexanone is acetonitrile. (F) Visualization of the photolysis of 0.1 M DHA aqueous solution in a four-way quartz cuvette by one-side illumination using a commercial handheld 5 W 275 nm LED. (G) CO purity in the outlet gas obtained using different methods based on temperature. (H) EG yield obtained using different methods based on temperature. | ||
We collected the time-dependent conversion profile of the photolysis of the DHA aqueous solution under 275 nm illumination in an Ar atmosphere (Fig. 1C). DHA was rapidly consumed within the first 10 min, with a conversion rate of 2.54 × 10−4 M s−1 accompanied by the formation of CO and EG at generation rates of 2.47 × 10−4 M s−1 and 2.06 × 10−4 M s−1, respectively (Fig. S3A†). Deuterium water had a weak effect on the photolysis of DHA (Fig. S3B†). DHA was completely consumed within 30 min and the carbon yield of CO was 98% and that of EG was 80%. Moreover, CO and EG production was stable even when the reaction time was further extended to 60 min. Over a wide range of concentrations, DHA achieved CO carbon yields >95% and EG carbon yields >70% (Fig. S4†). Online mass spectrometry results showed that the mass spectral signal of CO clearly followed the on–off modes of the light, and signals of H2 and CO2 presented almost no change (Fig. 1D and Fig. S5A†). Trace H2 was detected in the gas chromatograms (Fig. S6A†) and mass spectra (Fig. S6B and S5B†) of the gas collected by ex situ injection, and the H2 proportion was below 0.1% after photolysis for 1 h (Fig. S6C†). The main product in the solution was EG (Fig. S7†). The byproducts were methanol, formaldehyde, formic acid, glycolaldehyde, and other undetected substances (Fig. S6D†). The sum of the carbon yields of CO and EG was >85% (Fig. S6E†). Among the ketones and aldehydes tested, DHA exhibited the fastest photolysis rate and highest yield of CO (Fig. 1E). This ultrafast photolysis could be directly visualized by continuous bubble generation in the DHA solution under UV irradiation (Fig. 1F). In addition, this photolysis method exhibited significant advantages in CO purity (Fig. 1G) and EG yield (Fig. 1H) comparison with current preparation methods.
Reaction mechanism
We investigated the mechanism of the photochemical C–C cleavage of DHA. Carbonyls that absorb ultraviolet light are often involved in two main processes, namely photoexcitation and the radical reaction (Fig. 2A).37–40 The photoexcited triplet (T1) state generated from the singlet (S1) state by intersystem crossing (ISC) brings out the radical reaction.41–43 To probe the pertinent excited state, halide anions (Br− or I−) were employed to quench triplet-state DHA (Fig. S8A†).44 The bromide ions reduced the reaction rate by about seven times, and iodide ions completely deactivated the photolysis of DHA. (Fig. 2B, Fig. S8B and C†). This indicates that triplet DHA is the active excited state of dissociation. TEMPO or styrene were used as radical scavengers to clarify the radical dissociation mechanism. The addition of TEMPO and styrene significantly suppressed the generation of CO and EG (Fig. S9†), and the related acyl radicals of glycolaldehyde and hydroxymethyl radicals were captured, as shown by GC-MS spectrometry (Fig. 2C). Both triplet DHA and the radical dissociation mechanism were further confirmed by the results of DHA photolysis in (deuterated) methanol (Fig. S10†). Triplet DHA activates the C–H(D) bond of (deuterated) methanol39,45 and forms additional (deuterated) hydroxymethyl radicals, resulting in a higher EG yield than that obtained by DHA conversion (Fig. 2D). The activation behavior of the C–H bond in the triplet ketone was also proven by illuminating the methanol solution with added acetone, which produced approximately 40% yield of EG, relative to the amount of acetone added (Fig. S11†). The cross-coupling between the hydroxyacyl radicals from DHA and (deuterated) methyl radicals from (deuterated) acetone generated (deuterated) hydroxyacetone, further confirming the radical mechanism (Fig. S12†).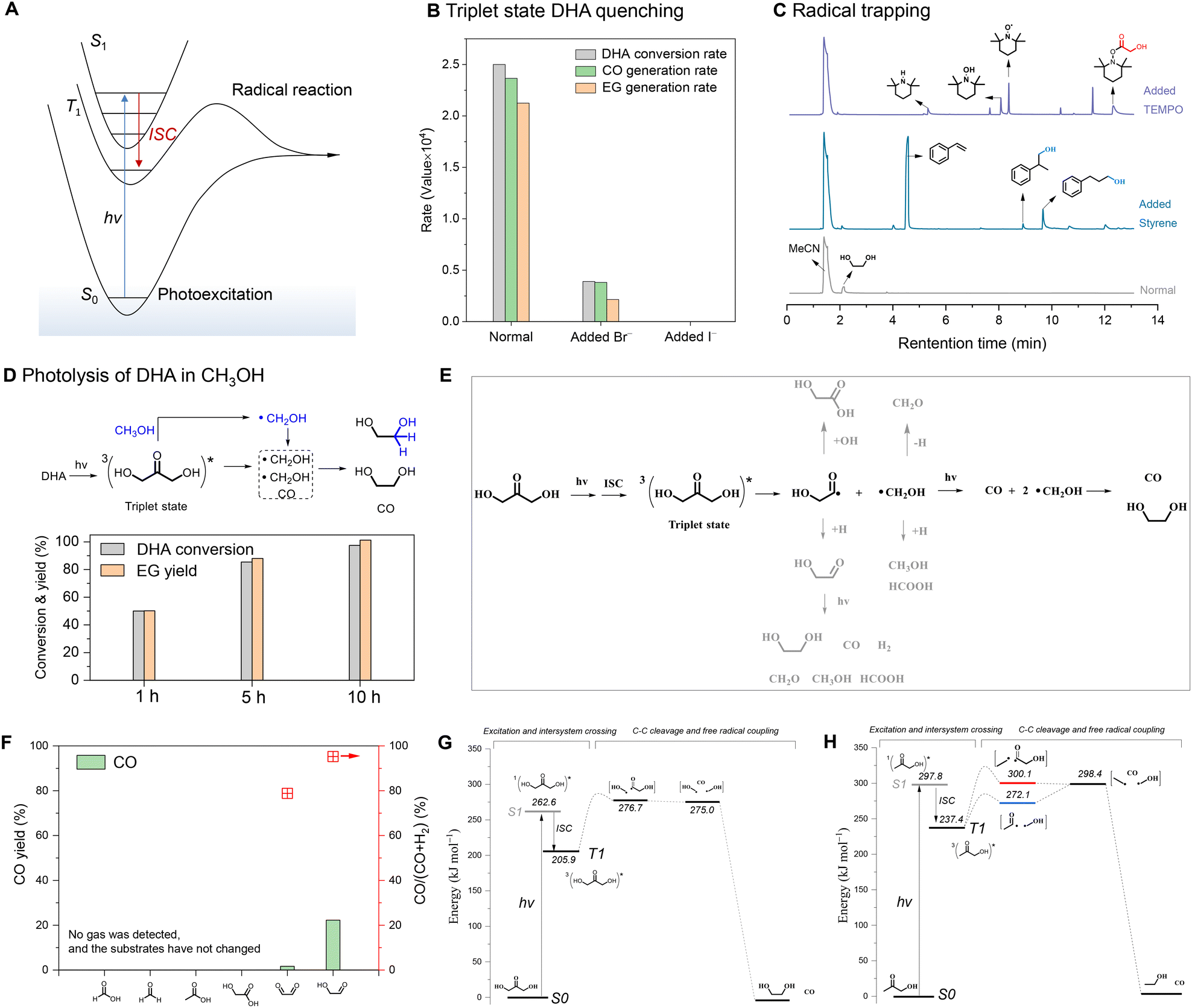 | ||
| Fig. 2 Reaction mechanism. (A) Schematic of carbonyl photochemistry during photoexcitation and the radical reaction. (B) Triplet-state DHA-quenching experiments conducted by adding bromide or iodide ions. (C) GC-MS patterns of the radical-trapping experiments performed by adding TEMPO or styrene. (D) Photolysis of DHA in methanol. (E) The proposed possible reaction mechanism of the photolysis of DHA in water. (F) Photolysis of the detected and possible acid/aldehyde byproducts after 1 h of reaction. Computational studies of the photolysis of (G) DHA and (H) hydroxyacetone. | ||
Based on the above results, a possible reaction mechanism of the photochemical C–C cleavage of DHA is proposed (Fig. 2E). When illuminated, the S0 state of DHA is first excited to the S1 excited state via an n → π* transition, and the excited T1 state is then generated by ISC from the excited S1 state. Triplet DHA undergoes C–C bond cleavage to afford an acyl radical and a hydroxymethyl radical. The acyl radical further rapidly undergoes C–C cleavage to form another hydroxymethyl radical and releases a molecule of CO. The coupling of the two hydroxymethyl radicals forms EG. Besides, the generated hydroxymethyl radicals and acyl radicals can be trapped by water or self-hydrogen to form other byproducts like glycolaldehyde, methanol, formaldehyde, formic acid, etc. Photolysis analysis of the detected (formaldehyde, formic acid, glycolaldehyde) and possible byproducts (glyoxal, acetic acid, hydroxyacetic acid) showed that only glycolaldehyde and glyoxal can undergo photochemical C–C/C–H dissociation to afford CO and H2 (Fig. 2F, Fig. S13†). Both photolysis of DHA and glycolaldehyde in deuterium water produced deuterated hydrogen gas (Fig. S14†), which illustrates the formation of trace hydrogen during the photolysis of DHA is possibly due to the photolysis of the glycolaldehyde byproducts.
We further calculated the energies of photoexcitation and the radical reaction process by comparing DHA (Fig. 2G) and hydroxyacetone (Fig. 2H). The S1 state excitation energy of DHA (262.6 kJ mol−1) was lower than that of hydroxyacetone (297.8 kJ mol−1), illustrating the two alpha carbons of ketone with hydroxyl substitution favor photoexcitation, which was also proven by the red-shift of the absorption spectrum of C3 ketones (Fig. S15†). On the other hand, the C–C bond energy of C(O)–CH2OH (272.1 kJ mol−1) was significantly lower than that of C(O)–CH3 (300.1 kJ mol−1) from hydroxyacetone. When the two alpha carbons of ketone are connected by hydroxyls, both photoexcitation and the radical reaction have low energy barriers, resulting in fast C–C bond cleavage. This is consistent with the above observation that DHA or fructose exhibits a higher CO generation rate than hydroxyacetone or acetone, respectively.
Flow photolysis
Over the past 20 years, continuous flow processes have created a significant impact across various fields, and flow chemistry has been particularly influential in chemical production.46 In this context, we designed and constructed a photochemical flow reaction device to assess the feasibility of continuous flow production of chemicals (Fig. 3A). Specifically, the device pumped a DHA solution into a custom-made spiral quartz tube that encircled an ultraviolet (UV) lamp. The tubular E27 UVC lamp (36 W) produced a wavelength of 254 nm (Fig. 3B). The diameter of each circle of the spiral quartz tubes surrounding the tubular E27 UVC lamp was 49 mm, and the total number of circles was 42, while total length was about 6.5 m. The inner and outer diameters of spiral quartz tubes were 1 mm and 3 mm, respectively. Under UV radiation, the DHA solution was gradually converted to CO and ethylene glycol (Fig. S16†).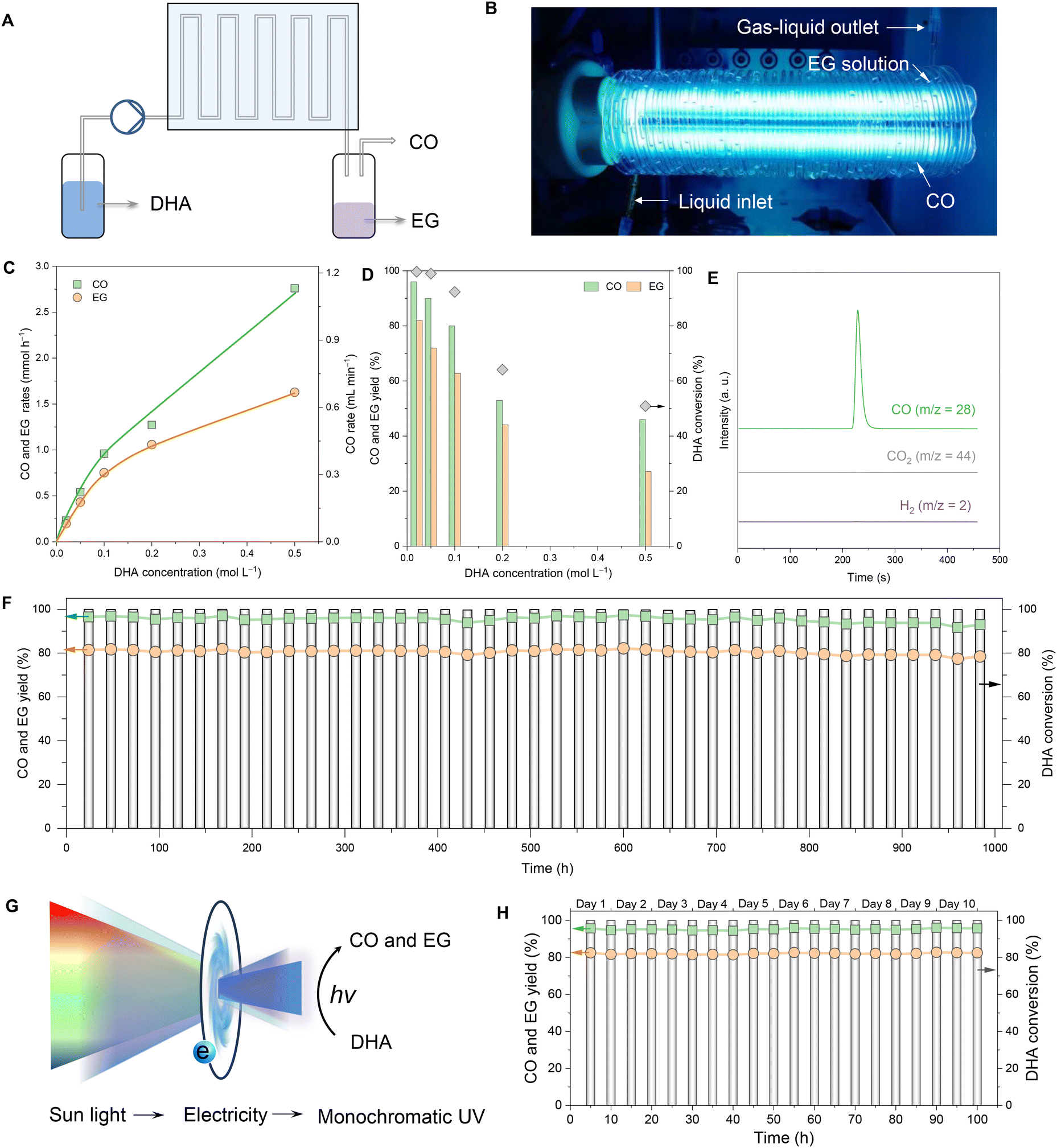 | ||
| Fig. 3 Flow synthesis. (A) Schematic of continuous -flow photolysis of DHA to produce CO and EG. (B) Photograph of the spiral quartz tube surrounding the ultraviolet lamp. (C) Conversion of DHA and the yields of CO and EG obtained with DHA solutions of different concentrations. (D) The production rates of CO and EG in DHA solutions of different concentrations. (E) Online mass spectrometric chromatograms. (F) Photolysis of DHA in the continuous flow mode for 1000 h. (G) Schematic of the sunlight-electricity-ultraviolet light conversion system. (H) DHA conversion and the yields of CO and EG for 100 h using the sunlight-electricity-ultraviolet light conversion system. The experiments at the sunlight-electricity-ultraviolet light mode worked for 10 hours from 10 am to 8 pm every day. | ||
The flow concentration of the DHA solution was initially optimized at a flow rate of 0.2 mL min−1. As the inflow concentration of DHA increased, the production rates of CO and EG also rose; however, the one-way conversion efficiency of DHA decreased (Fig. 3C and D). At the optimal DHA concentration of 0.02 M, the one-way conversion of DHA reached 100%. Under these conditions, the yield of CO was approximately 95%, with purity exceeding 99.9% (Fig. 3E), while the yield of EG was about 80%. The flow device demonstrated robust performance over an extended period when continuously operated for over 1000 hours and maintained nearly a 100% DHA conversion rate, over 93% CO yield, and over 78% EG yield (Fig. 3F). These results clearly confirm the feasibility and efficiency of continuous-flow photolysis of DHA for the production of CO and EG.
To leverage renewable sunlight for effective photolysis of DHA into CO and EG, we developed a Sunlight-Electricity-Ultraviolet light conversion system. This system comprised a photovoltaic panel, electricity storage units, and a photochemical flow device (Fig. 3G). In this setup, sunlight was intermittently captured and converted into continuous, high-intensity monochromatic UV light using electricity as an intermediary. This UV light was used to drive the photochemical flow device, enabling the photolysis of DHA (Fig. S17†). As illustrated in Fig. 3H, the Sunlight-Electricity-Ultraviolet light conversion system achieved stable and continuous production of CO and EG for 100 hours. This innovative approach not only harnesses renewable energy sources but also ensures a steady and efficient production process, demonstrating the practicality of integrating photovoltaic technology with chemical production systems for sustainable and efficient chemical synthesis.
CO generator
The production of high-purity CO in industries generally involves a series of complex processes, which pose potential energy, environmental, and safety risks. They are particularly not convenient to produce CO in small quantities.47 To this end, based on the fast and high-purity CO production characteristics of the photolysis of DHA, we further designed and assembled a portable CO generator that can produce and utilize CO on demand and can be turned on and off (Fig. 4A). The CO generator mainly consisted of a photochemical pool, a gas drying and storage module, display and an intelligent control module (Fig. S18†). The working principle of the CO generator is that the gas pressure sensor controls the power switch of the non-polar submersible UV lamp (Fig. S19†), thereby controlling the photolysis of the DHA solution placed in the photochemical pool. The maximum CO production rate was about 30 mL min−1 (Fig. 4B). The CO purity during the continuous production process could be maintained over 99% for 25 L (Fig. 4C). The palladium-catalyzed carbonylation reactions can effectively employ as-produced CO to produce high-value carbonyl molecules through a simple one-step reaction.48 To verify the direct availability of CO produced by the generator, six types of carbonylation reactions were further performed using CO with ex situ production and aryl iodides, which achieved high yields of carbonylation products in the range from 74% to 95% (Fig. 4D).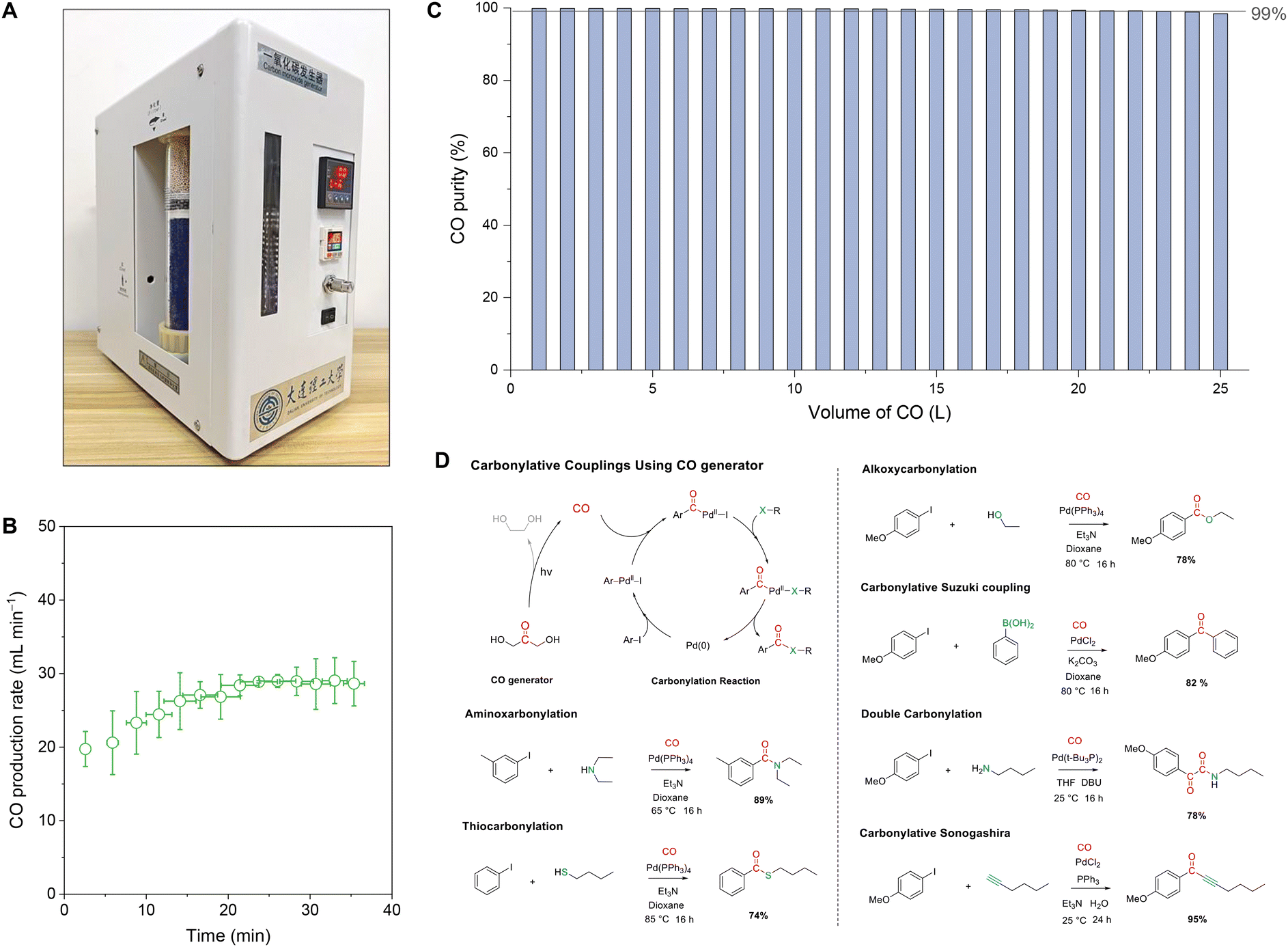 | ||
| Fig. 4 CO Generator. (A) Photograph of the CO generator. (B) Maximum CO production rate of the CO generator. (C) CO purity during the continuous production process. The concentration of DHA added is 2 mol L−1. (D) Carbonylation reactions of CO produced using the generator. | ||
Conclusions
In summary, we propose a novel photochemical method to produce CO and ethylene glycol by ultrafast photolysis of dihydroxyacetone under ultraviolet light irradiation at ambient temperature and pressure. The obtained CO yield was nearly 98% with high purity above 99%, and the yield of ethylene glycol was nearly 80%. Given the fast reaction rate and catalyst-free characteristics of this method, a home-built spiral-type flow reactor achieved CO and ethylene glycol production in the continuous-flow mode over 1000 h, and it also maintained efficient operation in the Sunlight-Electricity-Ultraviolet light conversion mode. Benefitting from the fast reaction rate and high purity of CO produced, a laboratory-scale portable CO generator was designed and assembled to provide the possibility of on-demand CO production with safety. This study not only provides an efficient method for the ultrafast production of high-purity CO and EG but also provides a reference to achieving continuous flow synthesis and instrument assembly.Author contributions
F. Kong conducted most of the experiments of the project, data analysis and wrote the manuscript. H. Zhou, Z. Chen and Z. Dou contributed to the data analysis. M. Wang conceived the project, performed the theoretical calculations, supervised the experiments, and revised the manuscript. All authors reviewed and approved the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22372023), the Natural Science Foundation of Liaoning Province (2022-MS-141) and the Research and Innovation Team Project of Dalian University of Technology (DUT2022TB10).References
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2017, 118, 434–504 CrossRef.
- S. Xie, W. Ma, X. Wu, H. Zhang, Q. Zhang, Y. Wang and Y. Wang, Energy Environ. Sci., 2021, 14, 37–89 RSC.
- Z. Zhang and G. W. Huber, Chem. Soc. Rev., 2018, 47, 1351–1390 RSC.
- W. O. Silva, B. Nagar, M. Soutrenon and H. H. Girault, Chem. Sci., 2022, 13, 1774–1779 RSC.
- F. Jiao, B. Bai, G. Li, X. Pan, Y. Ye, S. Qu, C. Xu, J. Xiao, Z. Jia, W. Liu, T. Peng, Y. Ding, C. Liu, J. Li and X. Bao, Science, 2023, 380, 727–730 CrossRef CAS PubMed.
- J. Kang, S. He, W. Zhou, Z. Shen, Y. Li, M. Chen, Q. Zhang and Y. Wang, Nat. Commun., 2020, 11, 827 CrossRef CAS.
- A. Kaithal, M. Hölscher and W. Leitner, Chem. Sci., 2021, 12, 976–982 RSC.
- J. S. G. Thomas, Nature, 1923, 111, 778–779 CrossRef CAS.
- E. Shafirovich and A. Varma, Ind. Eng. Chem. Res., 2009, 48, 7865–7875 CrossRef CAS.
- F. Wang, N. Gao, C. Quan, H. Liu, W. Li, H. Yuan and X. Yin, Energy Convers., 2024, 300, 117925 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- R. Luque, A. R. de la Osa, J. M. Campelo, A. A. Romero, J. L. Valverde and P. Sanchez, Energy Environ. Sci., 2012, 5, 5186–5202 RSC.
- L. C. Buelens, V. V. Galvita, H. Poelman, C. Detavernier and G. B. Marin, Science, 2016, 354, 449–452 CrossRef CAS.
- D. A. Hickman and L. D. Schmidt, Science, 1993, 259, 343–346 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- R. K. Parsapur, S. Chatterjee and K.-W. Huang, ACS Energy Lett., 2020, 5, 2881–2885 CrossRef CAS.
- X. Wei, G. Johnson, Y. Ye, M. Cui, S.-W. Yu, Y. Ran, J. Cai, Z. Liu, X. Chen, W. Gao, P. J. L. Bean, W. Zhang, T. Y. Zhao, F. A. Perras, E. J. Crumlin, X. Zhang, R. J. Davis, Z. Wu and S. Zhang, J. Am. Chem. Soc., 2023, 145, 14298–14306 CrossRef CAS PubMed.
- Q. Yang, E. A. Fedorova, S. A. Petrov, J. Weiss, H. Lund, A. S. Skrypnik, C. R. Kreyenschulte, V. Y. Bychkov, A. A. Matvienko, A. Brueckner and E. V. Kondratenko, Appl. Catal., B, 2023, 327, 122450 CrossRef CAS.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218167 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- H. Jung, C. Kim, H.-W. Yoo, J. You, J. S. Kim, A. Jamal, I. Gereige, J. W. Ager and H.-T. Jung, Energy Environ. Sci., 2023, 16, 2869–2878 RSC.
- J. Jökel, E. B. Boydas, J. Wellauer, O. S. Wenger, M. Robert, M. Römelt and U.-P. Apfel, Chem. Sci., 2023, 14, 12774–12783 RSC.
- X. Ma, J. Albertsma, D. Gabriels, R. Horst, S. Polat, C. Snoeks, F. Kapteijn, H. B. Eral, D. A. Vermaas, B. Mei, S. de Beer and M. A. van der Veen, Chem. Soc. Rev., 2023, 52, 3741–3777 RSC.
- R. F. Dye, Korean J. Chem. Eng., 2001, 18, 571–579 CrossRef CAS.
- S. Xie, Z. Shen, J. Deng, P. Guo, Q. Zhang, H. Zhang, C. Ma, Z. Jiang, J. Cheng, D. Deng and Y. Wang, Nat. Commun., 2018, 9, 1181 CrossRef PubMed.
- R. Xia, R. Wang, B. Hasa, A. Lee, Y. Liu, X. Ma and F. Jiao, Nat. Commun., 2023, 14, 4570 CrossRef CAS PubMed.
- J. Zheng, L. Huang, C.-H. Cui, Z.-C. Chen, X.-F. Liu, X. Duan, X.-Y. Cao, T.-Z. Yang, H. Zhu, K. Shi, P. Du, S.-W. Ying, C.-F. Zhu, Y.-G. Yao, G.-C. Guo, Y. Yuan, S.-Y. Xie and L.-S. Zheng, Science, 2022, 376, 288–292 CrossRef CAS PubMed.
- Y. Lum, J. E. Huang, Z. Wang, M. Luo, D.-H. Nam, W. R. Leow, B. Chen, J. Wicks, Y. C. Li, Y. Wang, C.-T. Dinh, J. Li, T.-T. Zhuang, F. Li, T.-K. Sham, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 14–22 CrossRef CAS.
- L. Fan, Y. Zhao, L. Chen, J. Chen, J. Chen, H. Yang, Y. Xiao, T. Zhang, J. Chen and L. Wang, Nat. Catal., 2023, 6, 585–595 CrossRef CAS.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- L. Wang, Y. Sun, F. Zhang, J. Hu, W. Hu, S. Xie, Y. Wang, J. Feng, Y. Li, G. Wang, B. Zhang, H. Wang, Q. Zhang and Y. Wang, Adv. Mater., 2023, 35, 2205782 CrossRef CAS PubMed.
- J. Wang, X. Dai, H. Wang, H. Liu, J. Rabeah, A. Brückner, F. Shi, M. Gong and X. Yang, Nat. Commun., 2021, 12, 6840 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2013, 4, 1820–1824 RSC.
- B. R. Heazlewood, A. T. Maccarone, D. U. Andrews, D. L. Osborn, L. B. Harding, S. J. Klippenstein, M. J. Jordan and S. H. Kable, Nat. Chem., 2011, 3, 443–448 CrossRef CAS PubMed.
- A. W. Harrison and S. H. Kable, J. Chem. Phys., 2018, 148, 164308 CrossRef PubMed.
- J. L. Zhu, C. R. Schull, A. T. Tam, Á. Rentería-Gómez, A. R. Gogoi, O. Gutierrez and K. A. Scheidt, J. Am. Chem. Soc., 2023, 145, 1535–1541 CrossRef CAS PubMed.
- J. J. Dotson, S. Perez-Estrada and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2018, 140, 8359–8371 CrossRef CAS PubMed.
- G. S. Hammond, Science, 1964, 145, 1469–1471 CrossRef CAS PubMed.
- D. Cao, M. Ataya, Z. Chen, H. Zeng, Y. Peng, R. Z. Khaliullin and C.-J. Li, Nat. Commun., 2022, 13, 1805 CrossRef CAS PubMed.
- M.-H. Kao, R. K. Venkatraman, M. N. R. Ashfold and A. J. Orr-Ewing, Chem. Sci., 2020, 11, 1991–2000 RSC.
- A. Jammoul, S. Dumas, B. D'Anna and C. George, Atmos. Chem. Phys., 2009, 9, 4229–4237 CrossRef CAS.
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc04180a |
| This journal is © The Royal Society of Chemistry 2024 |