
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 catalyzed recycling of polyester and polycarbonate plastics†
Qiao
Zhang
 a,
Nan
Wang
ab,
Chenyang
Hu
a,
Peng-Yuan
Li
c,
Fu-Quan
Bai
cd,
Xuan
Pang
*ab,
Xuesi
Chen
ab and
Xianhong
Wang
a
a,
Nan
Wang
ab,
Chenyang
Hu
a,
Peng-Yuan
Li
c,
Fu-Quan
Bai
cd,
Xuan
Pang
*ab,
Xuesi
Chen
ab and
Xianhong
Wang
a
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, China. E-mail: xpang@ciac.ac.cn; Tel: (+86) 431 85262588
bUniversity of Science and Technology of China, Hefei 230026, China
cInstitute of Theoretical Chemistry, College of Chemistry, Jilin University, Changchun 130023, China
dChongqing Research Institute, Jilin University, Chongqing 401120, China
First published on 31st October 2024
Abstract
Recycling waste polymeric materials is essential for environmental protection and achieving carbon neutrality. This study demonstrates the efficacy of CO2 as a metal-free catalyst for the chemical recycling of common waste polyester and polycarbonate plastics via alcoholysis to yield valuable organic chemicals. CO2 was proposed to act as a Lewis acid–base pair, activating both alcohol and ester (carbonate) functional groups during the catalytic process. The depolymerization mechanism was thoroughly investigated by monitoring conversion rates and changes in Mn values. Pre-treatment of the polymer materials in THF was found to accelerate the depolymerization rate. End-of-life waste materials were completely degraded into valuable organic molecules, irrespective of their physical and chemical properties. Unlike conventional solid and liquid catalysts, CO2 leaves no residue in the final products. Moreover, this work unveils the catalytic role for CO2, expanding its traditional function as a C1 building block in synthetic chemistry.
Introduction
Plastics are indispensable to modern life and serve as crucial engineering materials. Annual global plastic production reached 390 million tons in 2016. This number is projected to climb to 780 million tons by 2036 and 1.2 billion tons by 2060.1,2 Yet approximately 150 million tons of plastic waste are generated each year.3 Regrettably, only about one-fifth of this waste is recycled, with the majority ending up in landfills or oceans.4 As plastic consumption grows, so too does the environmental crisis. Improperly disposed plastic waste is non-biodegradable, contaminating the biosphere. Plastic bags and bottles pose a significant threat to wildlife, causing ingestion and digestive problems. Microplastics, derived from shredded plastic waste, have been linked to human health issues.5 Given these challenges, effective waste plastic management is imperative for individuals, the environment, and the global community.Several methods exist for deconstructing and recycling plastic waste. Incineration, while a simple process for reducing waste volume and recovering energy through combustion, yields worthless and unrecoverable small molecules like H2O, CO2, N2, and NOx. Mechanical recycling, involving sorting, cleaning, shredding, melting, and reforming, is straightforward but struggles with contamination and quality degradation, limiting the applications of recycled materials.6,7 Biodegradation presents a sustainable alternative to traditional plastics.8 Driven by increasing demand, production of biodegradable polymers is expected to soar from 1.09 million tons in 2019 to 1.80 million tons by 2025.9 Polylactide (PLA), a biodegradable aliphatic polyester, exemplifies this category, comprising one-third of global biodegradable polymer production. While biodegradable polymers present an environmentally friendly option compared to traditional plastics like polyolefin, they often require lengthy decomposition times and produce unrecoverable end products e.g. H2O and CO2.
In addition to incineration, mechanical recycling, and biodegradation, chemical recycling offers a promising approach to polymer deconstruction. This process involves controlled reactions to break down polymers into oligomers and monomers.10 For PLA specifically, alcoholysis,11–18 hydrolysis,19–21 aminolysis,22–25 and hydrogenolysis26–30 have been explored. Among these methods, alcoholysis of PLA yields alkyl lactate, a product currently valued at 1.5 to 2 times the price of virgin PLA resins.14 Moreover, alkyl lactate serves as a sustainable solvent in various chemical syntheses.31,32
Our team has previously reported the alcoholysis of PLA, as well as other polyester and polycarbonate materials, facilitated by various catalysts33,34 or aprotic polar solvents.35 We hypothesized that a Lewis acid–base pair is essential for activating both alcohol and ester (or carbonate) groups within these polymers. Building upon our prior work, here we propose that the carbon atom in CO2 acts as a Lewis acid, activating carbonyl groups in polyesters, while the oxygen atom in CO2 serves as a Lewis base, activating alcohols. Based on this hypothesis, we explored the depolymerization of common waste polyester and polycarbonate materials through CO2-catalyzed alcoholysis—an unconventional approach in catalysis (Scheme 1).
 | ||
| Scheme 1 (a) CO2 catalyzed depolymerization and recycling waste polyesters and polycarbonates; (b) CO2 as a Lewis acid–base pair for activating MeOH and polyester. | ||
CO2 is often recognized as a sustainable C1 building block capable of replacing toxic carbon sources like CO and phosgene in organic and polymer synthesis.36,37 However, employing CO2 as a metal-free catalyst remains challenging.38 For instance, List et al. described CO2-assisted α-allylation of ketones using palladium and a chiral phosphoric acid catalyst.39 Young et al. explored CO2-mediated C–H arylation of amines by utilizing CO2 as an amine protecting and directing group.40,41 Rovis, Schoenebeck, and colleagues investigated the direct α-alkylation of primary aliphatic amines, where CO2 forms an in situ carbamate to accelerate C–H activation.42 Additionally, Li, Wang, and co-workers reported Cl2 evolution through NaCl electrolysis using an amide-containing organocatalyst and CO2.43 While these outstanding examples showcase CO2 involvement, they necessitate additional catalysts, reagents, or external stimuli. In contrast, our research focuses on utilizing CO2 as a standalone catalyst for a specific reaction, eliminating the need for catalyst removal as the gaseous CO2 can be readily released.44–47 Beyond PLA, we extended our investigation to the depolymerization of widely used polyethylene terephthalate (PET)48,49 and bisphenol A based polycarbonate (BPA-PC).50 Our work not only explores the fundamental catalytic role of CO2 for activating alcohol and polymers, but also translates this knowledge into practical applications for recycling waste plastics, offering a sustainable approach to materials management.
Results and discussion
Initial investigations focused on evaluating the catalytic reactivity of CO2. PLA resin R190 (Mn = 98.1 kg mol−1, Đ = 1.66) served as the starting material. Control experiments confirmed negligible methanolysis without a catalyst at 140 °C in methanol (Table 1, entry 1). However, under 1 MPa CO2 pressure in a stainless-steel autoclave, 90% of PLA resins were degraded within 6 hours. Complete degradation achieved within 8 hours (Table 1, entry 2). 1H NMR spectroscopy identified methyl lactate as the sole product, and 88% isolated yield was obtained by distillation. This unexpected result highlighted the efficacy of CO2 as a catalyst for PLA alcoholysis. The ethanolysis of PLA required a longer reaction time due to a higher activation energy (Table S1†).51 The influence on CO2 pressure on the depolymerization rate was listed in Table S2.† Note that the reaction conditions employed were below the supercritical point of CO2 (Tc = 31 °C and pc = 7.4 MPa).52 Importantly, CO2 consistently catalyzed the degradation of various commercial PLA resins irrespective of grade, size, or manufacturer (Table 1, entries 3 and 4). Unlike traditional solid or liquid catalysts, CO2 leaves no catalyst contamination in the final product, eliminating the need for post-reaction catalyst removal. This advantage may also enable the recycling of waste PLA into novel PLA-based polymer materials.53,54 Chiral stationary phase gas chromatography determined the chirality of the depolymerized product from PLLA. The product was confirmed as methyl L-lactate (Fig. S1†). Moreover, green metrics were calculated for the depolymerization process.55,56 The atom economy (AE) was 100%. Environmental factor (e-factor) was 7.38, and process mass intensity (PMI) was 8.38, primarily due to the excess amount of MeOH used (Table S3†).| Entry | PLA material | Cat. | t (h) | Conv.b (%) | Methyl lactate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimer ratiob :dimer ratiob |
|---|---|---|---|---|---|
| a Reaction conditions: each reaction was carried out in a 25 mL stainless-steel autoclave. PLA material (288 mg, containing 4 mmol ester bonds), MeOH (3.2 g, 100 mmol, 25 equivalents relative to PLA ester bonds), CO2 (1 MPa), T = 140 °C. b Conversion and percentage of dimers were determined by 1H NMR spectroscopy (300 MHz, in CDCl3). | |||||
| 1 | R190 resin | N/A | 6 | Trace | N/A |
| 2 | R190 resin | CO2 | 6 | 90 | 91:9 |
| 8 | 100 | 100:0 |
|||
| 3 | R290 resin | CO2 | 6 | 43 | 54:46 |
| 12 | 100 | 100:0 |
|||
| 4 | 4032D resin | CO2 | 6 | 100 | 100:0 |
To elucidate the mechanism underlying PLA alcoholysis, we employed methyl lactate as a simplified model of the polymeric chain, reacting it with methanol-d4 (CD3OD) in the presence of CO2. Fig. 1 presents the 1H NMR spectroscopic characterization of the transesterification between methyl lactate and CD3OD. In the absence of CO2, only 21% conversion was observed after a 6-hour reaction at 140 °C (Fig. 1b). In contrast, CO2 catalysis significantly enhanced the reaction, achieving a 92% conversion rate under identical conditions (Fig. 1c). The organic mimic reactions strongly suggest that CO2 catalyzes PLA depolymerization through a transesterification mechanism. Furthermore, 2H NMR spectroscopy revealed a signal increase at 3.13 ppm, corresponding to deuterated methyl lactate (Fig. S2†).
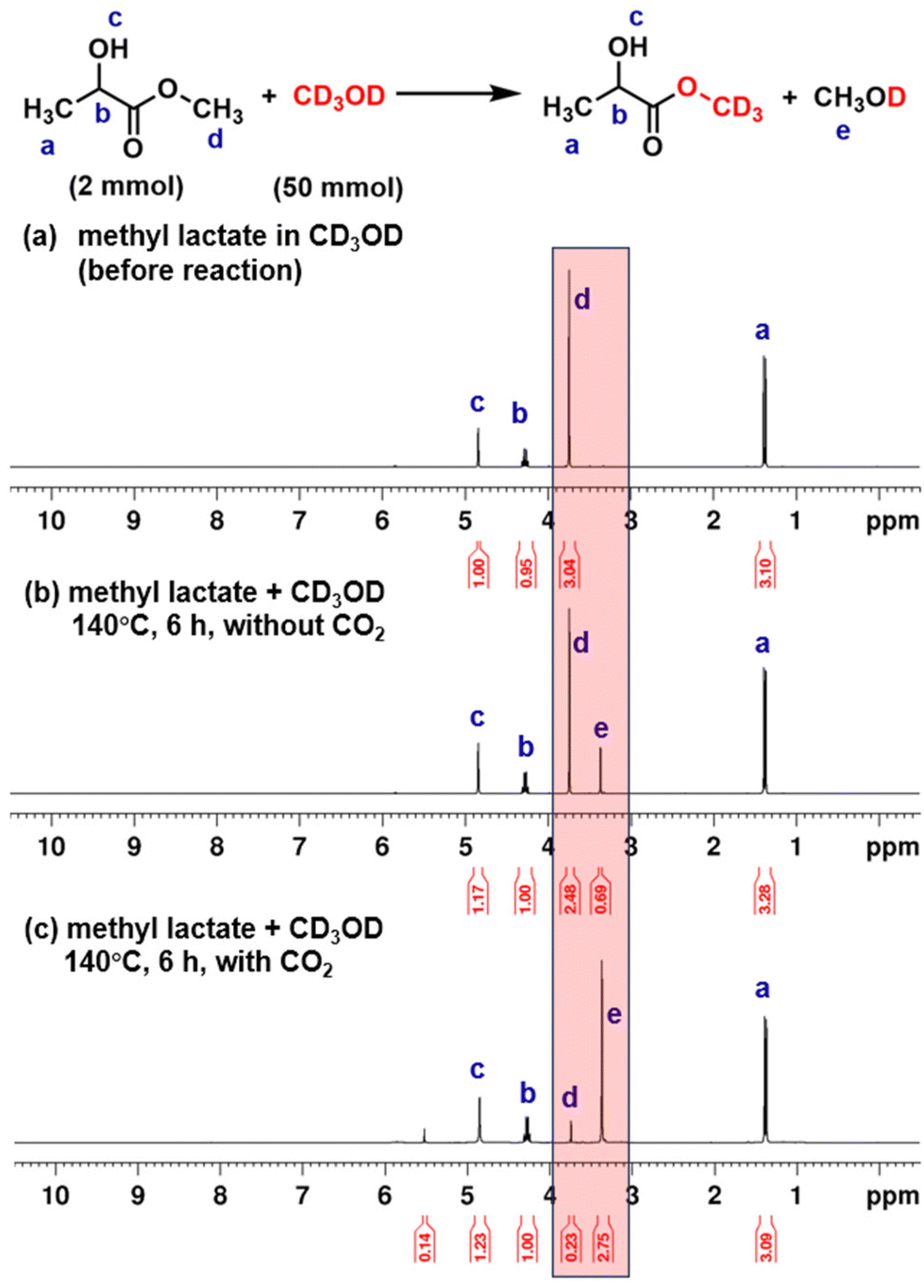 | ||
| Fig. 1 1H NMR spectra of transesterification between methyl lactate and CD3OD. | ||
Transesterification between ethyl acetate and methanol serves as a model for PLA alcoholysis. Computational analysis reveals the catalytic role of CO2 in this process (Fig. 2). In pathway 1, CO2 catalyzes the reaction by forming a six-membered ring intermediate. The carbon in CO2 acts as a Lewis acid, binding to the ester carbonyl, while the oxygen in CO2 functions as a Lewis base, interacting with the hydrogen in methanol. This dual activation by CO2 lowers the energy barrier to ΔG = 13.5 kcal mol−1 in transition state 1 (TS-1) and 8.7 kcal mol−1 in TS-2. Pathway 2, proceeding without a catalyst, involves two methanol molecules forming a six-membered ring transition state, requiring higher activation energy with ΔG = 15.8 kcal mol−1 and 18.3 kcal mol−1 in TS-1 and TS-2, respectively. Pathway 3, also catalyst-free, involves only one methanol molecule in a four-membered ring transition state with ΔG = 27.3 kcal mol−1 and 26.8 kcal mol−1 in TS-1 and TS-2, respectively, resulting in the highest energy barrier among the three pathways.
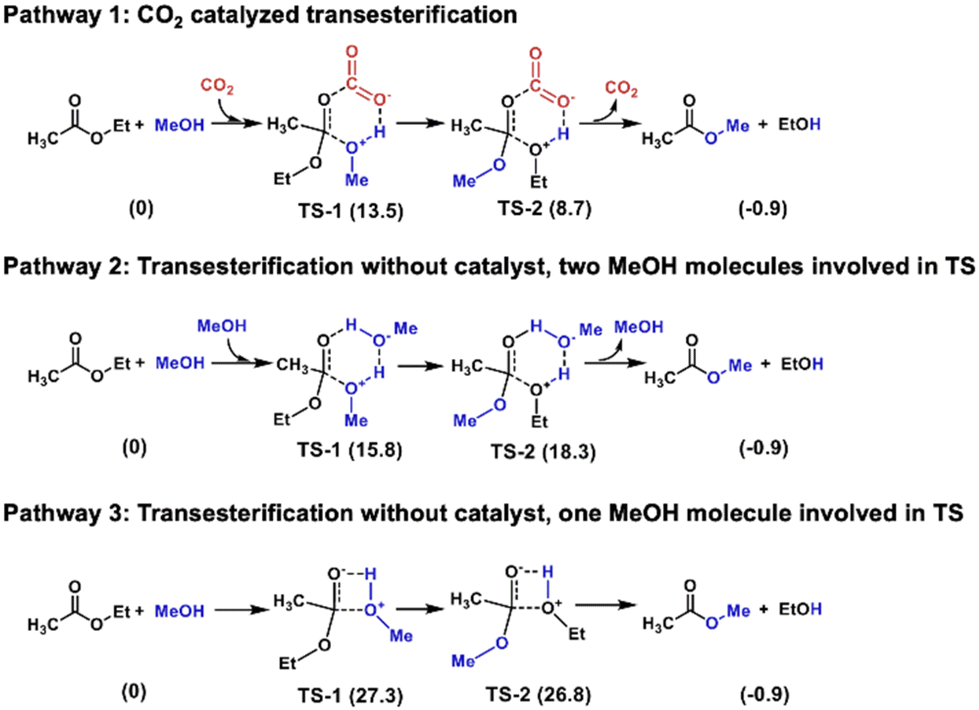 | ||
| Fig. 2 Computational analysis of the transesterification between ethyl acetate and methanol. Numbers in parentheses represent energy changes in kcal mol−1. | ||
The kinetics of PLA methanolysis were investigated by monitoring methyl lactate formation and changes in Mn values during the depolymerization process (Fig. 3). Methyl lactate conversion was determined via1H NMR spectroscopy. Initially, the depolymerization rate was slow, likely due to the reaction being confined to the surface of PLA resin. After 4 hours, the rate accelerated as the resin was progressively degraded into shorter polymer chains and oligomers. Gel permeation chromatography (GPC) was employed to track Mn changes. In contrast to the initial slow depolymerization, Mn values decreased rapidly within the first 2 hours, dropping from 98.1 to 2.6 kg mol−1. This dramatic decrease indicates a random chain scission mechanism rather than a stepwise depolymerization from the polymer chain end.
 | ||
| Fig. 3 Changes in (a) conversion and (b) Mn values for methanolysis of PLA resin R190 catalyzed by CO2. (c) A scheme for the depolymerization mode of PLA (random chain scission). | ||
A plausible mechanism for CO2-catalyzed depolymerization is outlined in Scheme 2. CO2 functions as a Lewis acid–base pair, with the carbon atom acting as a Lewis acid to activate ester carbonyls and the oxygen atom acting as a Lewis base to activate the proton in methanol. In the transesterification process, methanol is thought as a nucleophile attacking the activated carbonyl group in the polyester. Subsequent ester bond cleavage leads to depolymerization, ultimately yielding methyl lactate as the final product.
 | ||
| Scheme 2 A plausible mechanism for CO2 catalyzed methanolysis of PLA. | ||
Beyond PLA, we extended our research to the depolymerization of PET, the most consumed polyester in the world.57 With excellent thermal and tensile properties,58 PET finds extensive applications in textiles, packaging, and electronics. While chemical recycling of PET typically involves hydrolysis, alcoholysis, or glycolysis. These processes often require high temperatures due to PET's aromatic nature. Previous studies on PET methanolysis have relied on metal catalysts e.g., Zn59 and Ti60 based catalyst. In this work, we explored the potential of CO2 as a catalyst for converting PET to dimethyl terephthalate (DMT) via methanolysis. In the absence of CO2, PET pellets remained unchanged (Table 2, entry 1). However, under CO2 catalysis, complete depolymerization of PET pellets was achieved (Table 2, entry 3). Unlike the insoluble PET pellets, the product exhibited solubility in methanol and was amenable to removing solvent in vacuo with 83% isolated yield. 1H NMR spectroscopy confirmed the formation of DMT (Fig. S9†).
| Entry | Material | Cat. | T (°C) | Conv. (%) | Selec. (%) |
|---|---|---|---|---|---|
| a Reaction conditions: each reaction was carried out in a 25 mL stainless-steel autoclave. PET pellets (384 mg, containing 4 mmol ester bonds), MeOH (3.2 g, 100 mmol, 25 equivalents relative to PET ester bonds) or BPA-PC pellets (508 mg, containing 2 mmol carbonate units), MeOH (3.2 g, 100 mmol, 50 equivalents relative to PC carbonate units), CO2 (if any, 1 MPa), 6 h. b PET pellets could be observed visually. c Conversion and selectivity were determined by 1H NMR spectroscopy (300 MHz, in CDCl3). | |||||
| 1 | PET | N/A | 200 | N/Ab | N/Ab |
| 2 | PET | CO2 | 200 | 100c | 100c |
| 3 | PET | CO2 | 170 | N/Ab | N/Ab |
| 4 | BPA-PC | N/A | 140 | 14c | 92c |
| 5 | BPA-PC | CO2 | 140 | 89c | 90c |
| 6 | BPA-PC | CO2 | 120 | Tracec | N/Ab |
Our third objective was to employ CO2 catalysis for the depolymerization of BPA-PC, another widely consumed plastic. Renowned for its toughness, strength, and transparency, BPA-PC finds extensive applications in data storage, automotive and aircraft components, electronics, and construction.57 Although the repeating unit is a carbonate rather than an ester, we hypothesized that the methanolysis mechanism for polycarbonate would resemble that of polyester. Indeed, we observed significant differences in reaction rates with and without CO2 catalysis. Without CO2 as catalyst, only 14% conversion was achieved at 140 °C after 6 hours (Table 2, entry 4). In contrast, CO2 catalysis boosted conversion to 89% under identical conditions (Table 2, entry 5). Lowering the temperature to 120 °C virtually eliminated depolymerization (Table 2, entry 6). Consequently, we investigated CO2-catalyzed methanolysis of BPA-PC pellets, monitoring the process hourly via1H NMR spectroscopy. Fig. 4 revealed minimal BPA formation during the initial 2 hours, likely due to the reaction being confined to the pellet surface. At the 3-hour mark, a significant increase in BPA signals was observed as the pellets disintegrated into powder, substantially increasing the surface area. Some incomplete degradation products were also detected.61 After 6 hours, most of the BPA-PC had been converted to BPA. The isolated yield was 82% by removing solvent in vacuo.
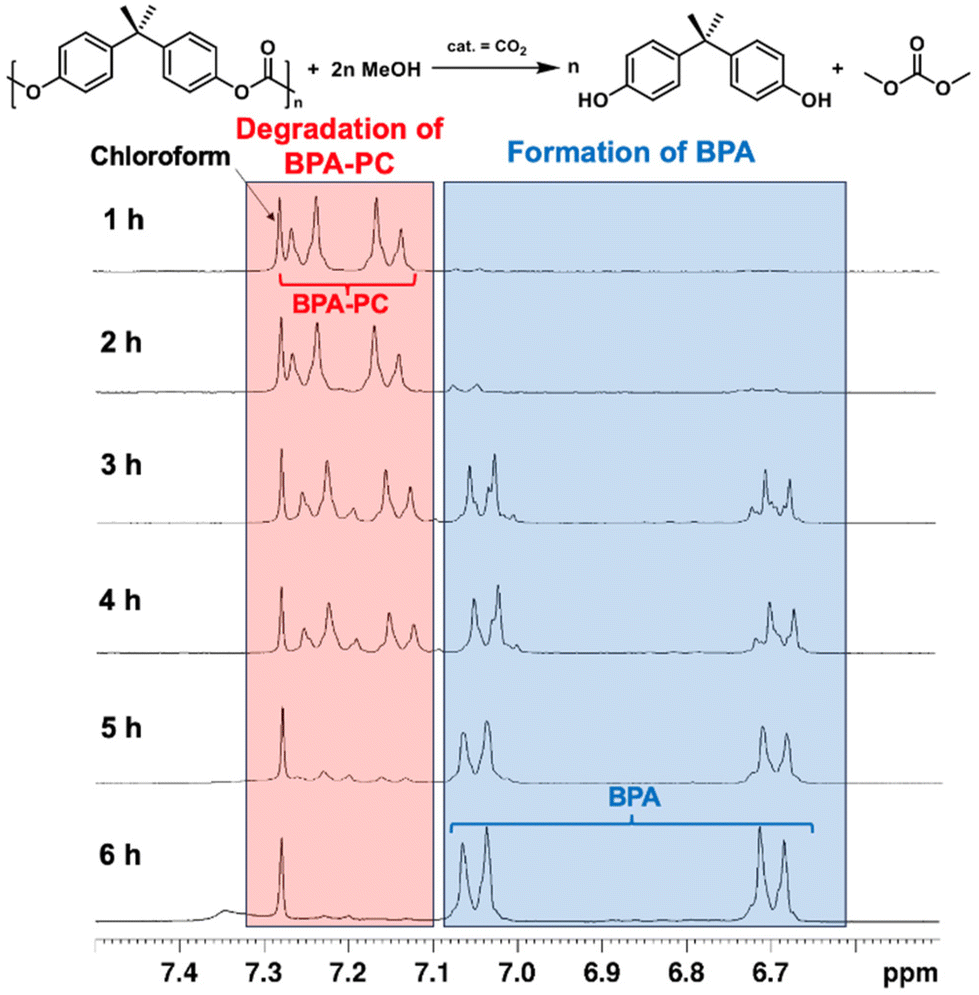 | ||
| Fig. 4 CO2 catalyzed methanolysis of the BPA-PC pellets monitored by 1H NMR spectroscopy (300 MHz, CDCl3, 6.5–7.5 ppm). Reaction condition: BPA-PC (508 mg, 2 mmol carbonate units), MeOH (3.2 g, 100 mmol, 50 equivalents relative to carbonate units), CO2 (1 MPa), T = 140 °C. | ||
To expedite the depolymerization process, we explored the effects of pre-treatment of polymeric materials with a solvent like tetrahydrofuran (THF).62 Dissolution or swelling with THF can facilitate greater interaction with methanol. In our experiments, polymeric materials, including PLA, PET, and BPA-PC, were initially treated with THF for 1 hour at 65 °C to induce either dissolution (for PLA and BPA-PC) or swelling (for PET) prior to CO2-catalyzed methanolysis (Fig. 5). We observed that pre-treatment significantly accelerated depolymerization compared to non-treatment materials. For instance, PLA methanolysis without solvent treatment yielded only 6% methyl lactate conversion after 2 hours, whereas pre-treatment increased conversion to 80%. Similarly, PET and BPA-PC required at least 6 hours for complete degradation without pre-treatment, but were fully depolymerized within 1 hour with pre-treatment. To sum up, pre-treatment polymeric materials with THF can largely enhance the rate of methanolysis.
 | ||
| Fig. 5 Comparison of PLA, PET, and BPA-PC methanolysis under non-treatment and pre-treatment in THF conditions. | ||
Our protocol was also effective in depolymerizing end-of-life polyester and polycarbonate products. We selected 3D printing material (PLA), cup lids (PLA), digital disc (BPA-PC), goggles (BPA-PC), drinking bottles (PET), and food package (PET) as representative samples for CO2-catalyzed methanolysis under both non-swelling and swelling conditions (Fig. 6). Complete conversion of these waste materials to the corresponding organic molecules were confirmed by 1H NMR spectroscopy. The successful depolymerization of end-of-life plastics demonstrates the broad applicability of our CO2 catalytic approach. Unlike conventional solid or liquid catalysts, CO2 is a gaseous catalyst that readily dissipates upon autoclave opening, leaving no residue in the product. This eliminates the need for catalyst removal, streamlining the overall process.
 | ||
| Fig. 6 Depolymerization of end-of-life materials. | ||
Conclusions
In summary, this study explored the unconventional use of CO2 as a standalone catalyst for the depolymerization of waste polyester and polycarbonate materials. Experimental and computational investigations, employing small molecule transesterification as a model, elucidated the catalytic mechanism of CO2. The depolymerization process was determined to proceed via random scission of polymer chains. PLA, PET, and BPA-PC materials were successfully depolymerized through methanolysis, regardless of their molecular weight, shape, or other physical and chemical properties. Notably, CO2 offers advantages over traditional solid or liquid catalysts by being inexpensive, non-toxic, metal-free, and residue-free. Beyond its established role as a C1 building block in synthetic chemistry, this research introduces CO2 as a promising catalyst for sustainable chemistry, particularly in the context of waste polymer recycling and upcycling.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China, Fund for Distinguished Young Scholars (No. 52325301), CAS Project for Young Scientists in Basic Research (YSBR-094), and the National Natural Science Foundation of China, Basic Science Center Program (No. 51988102).References
- T. H. Epps, III, L. T. J. Korley, T. Yan, K. L. Beers and T. M. Burt, JACS Au, 2022, 2, 3–11 CrossRef PubMed.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L. W. Majumder and G. W. Huber, Chem. Rev., 2023, 123, 9915–9939 CrossRef PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef PubMed.
- A. D. Vethaak and J. Legler, Science, 2021, 371, 672–674 CrossRef PubMed.
- J. M. Millican and S. Agarwal, Macromolecules, 2021, 54, 4455–4469 CrossRef.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef PubMed.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef PubMed.
- C. Sun, S. Wei, H. Tan, Y. Huang and Y. Zhang, Chem. Eng. J., 2022, 446, 136881 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef PubMed.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- P. McKeown, M. Kamran, M. G. Davidson, M. D. Jones, L. A. Román-Ramírez and J. Wood, Green Chem., 2020, 22, 3721–3726 RSC.
- J. M. Payne, G. Kociok-Köhn, E. A. C. Emanuelsson and M. D. Jones, Macromolecules, 2021, 54, 8453–8469 CrossRef CAS.
- F. Santulli, M. Lamberti and M. Mazzeo, ChemSusChem, 2021, 14, 5470–5475 CrossRef CAS PubMed.
- R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860–9871 CrossRef CAS.
- M. Fuchs, P. M. Schäfer, W. Wagner, I. Krumm, M. Walbeck, R. Dietrich, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2023, 16, e202300192 CrossRef CAS PubMed.
- A. J. Spicer, A. Brandolese and A. P. Dove, ACS Macro Lett., 2024, 13, 189–194 CrossRef CAS PubMed.
- M. Brió Pérez, M. A. Hempenius, S. de Beer and F. R. Wurm, Macromolecules, 2023, 56, 8856–8865 CrossRef.
- M. Pérez-Venegas, T. Friščić and K. Auclair, ACS Sustainable Chem. Eng., 2023, 11, 9924–9931 CrossRef.
- W. Wu, H. Zhai, K. Wu, X. Wang, W. Rao, J. Ding and L. Yu, Chem. Eng. J., 2024, 480, 148131 CrossRef CAS.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu, M. Wang and D. Ma, J. Am. Chem. Soc., 2021, 143, 16358–16363 CrossRef CAS PubMed.
- L. Shao, Y.-C. Chang, C. Hao, M.-e. Fei, B. Zhao, B. J. Bliss and J. Zhang, Green Chem., 2022, 24, 8716–8724 RSC.
- F. Wu, Y. Wang, Y. Zhao, M. Tang, W. Zeng, Y. Wang, X. Chang, J. Xiang, B. Han and Z. Liu, Sci. Adv., 2023, 9, eade7971 CrossRef CAS PubMed.
- Y. Zhao, H. Zhang, F. Wu, R. Li, M. Tang, Y. Wang, W. Zeng, B. Han and Z. Liu, Chem. Sci., 2024, 15, 10892–10899 RSC.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- R. Mi, L. Zeng, M. Wang, S. Tian, J. Yan, S. Yu, M. Wang and D. Ma, Angew. Chem., Int. Ed., 2023, 62, e202304219 CrossRef CAS PubMed.
- Y. Hu, S. Zhang, J. Xu, Y. Liu, A. Yu, J. Qian and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202312564 CrossRef CAS PubMed.
- W. Zeng, Y. Zhao, F. Zhang, R. Li, M. Tang, X. Chang, Y. Wang, F. Wu, B. Han and Z. Liu, Nat. Commun., 2024, 15, 160 CrossRef PubMed.
- M. Tang, J. Shen, Y. Wang, Y. Zhao, T. Gan, X. Zheng, D. Wang, B. Han and Z. Liu, Nat. Commun., 2024, 15, 5630 CrossRef PubMed.
- C. S. M. Pereira, V. M. T. M. Silva and A. E. Rodrigues, Green Chem., 2011, 13, 2658–2671 RSC.
- Y. Gu and F. Jérôme, Chem. Soc. Rev., 2013, 42, 9550–9570 RSC.
- Q. Zhang, C. Hu, R. Duan, Y. Huang, X. Li, Z. Sun, X. Pang and X. Chen, Green Chem., 2022, 24, 9282–9289 RSC.
- Q. Zhang, C. Hu, X. Pang and X. Chen, ChemSusChem, 2024, 17, e202300907 CrossRef PubMed.
- Q. Zhang, C. Hu, P.-Y. Li, F.-Q. Bai, X. Pang and X. Chen, ACS Macro Lett., 2024, 13, 151–157 CrossRef PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- G. A. Bhat and D. J. Darensbourg, Coord. Chem. Rev., 2023, 492, 215277 CrossRef CAS.
- P. K. Sahoo, Y. Zhang and S. Das, ACS Catal., 2021, 11, 3414–3442 CrossRef CAS.
- G. Pupo, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 6099–6102 CrossRef CAS.
- M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818–6822 CrossRef CAS PubMed.
- M. Kapoor, P. Chand-Thakuri and M. C. Young, J. Am. Chem. Soc., 2019, 141, 7980–7989 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- J. Yang, W.-H. Li, H.-T. Tang, Y.-M. Pan, D. Wang and Y. Li, Nature, 2023, 617, 519–523 CrossRef CAS PubMed.
- T. Roy, M. J. Kim, Y. Yang, S. Kim, G. Kang, X. Ren, A. Kadziola, H.-Y. Lee, M.-H. Baik and J.-W. Lee, ACS Catal., 2019, 9, 6006–6011 CrossRef CAS.
- M. Juhl, A. R. Petersen and J.-W. Lee, Chem. – Eur. J., 2021, 27, 228–232 CrossRef.
- Y. Yang, J. Liu, F. S. Kamounah, G. Ciancaleoni and J.-W. Lee, J. Org. Chem., 2021, 86, 16867–16881 CrossRef PubMed.
- H. Wang, Y. Li, S. Liu, M. Makha, J.-F. Bai and Y. Li, ChemSusChem, 2022, 15, e202200227 CrossRef PubMed.
- A. Kulkarni, G. Quintens and L. M. Pitet, Macromolecules, 2023, 56, 1747–1758 CrossRef.
- T. El Darai, A. Ter-Halle, M. Blanzat, G. Despras, V. Sartor, G. Bordeau, A. Lattes, S. Franceschi, S. Cassel, N. Chouini-Lalanne, E. Perez, C. Déjugnat and J.-C. Garrigues, Green Chem., 2024, 26, 6857–6885 RSC.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- T. Becker, A. Hermann, N. Saritas, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2024, 17, e202400933 CrossRef PubMed.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef CAS.
- S. Liu, L. Hu, J. Liu, Z. Zhang, H. Suo and Y. Qin, Macromolecules, 2024, 57, 4662–4669 CrossRef CAS.
- Z.-X. Luo, G.-Q. Tian, S.-C. Chen, G. Wu and Y.-Z. Wang, Macromolecules, 2024, 57, 6828–6837 CrossRef CAS.
- N. Fantozzi, J.-N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650 CrossRef CAS PubMed.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS.
- X. Bai, D. R. Aireddy, A. Roy and K. Ding, Angew. Chem., Int. Ed., 2023, 62, e202309949 CrossRef CAS PubMed.
- B. Ye, R. Zhou, Z. Zhong, S. Wang, H. Wang and Z. Hou, Green Chem., 2023, 25, 7243–7252 RSC.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- E. Luna, I. Olazabal, M. Roosen, A. Müller, C. Jehanno, M. Ximenis, S. de Meester and H. Sardon, Chem. Eng. J., 2024, 482, 148861 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, etc. See DOI: https://doi.org/10.1039/d4gc04782f |
| This journal is © The Royal Society of Chemistry 2024 |