
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn isotope dilution mass spectrometry overview: tips and applications for the measurement of radionuclides†‡
Alexandre
Quemet
 a,
Amélie
Hubert
b,
Alkiviadis
Gourgiotis
c,
Ana María
Sánchez Hernández
d,
Marielle
Crozet
a,
Guillaume
Bailly
e,
Andrew
Dobney
f,
Georges
Duhamel
g,
Joe
Hiess
g,
Urska
Repinc
g,
Sébastien
Mialle
h,
Béatrice
Boulet
i,
Raphaëlle
Escoube
c,
Céline
Bouvier-Capely
c,
Fabien
Pointurier
b and
Sébastien
Picart
a
a,
Amélie
Hubert
b,
Alkiviadis
Gourgiotis
c,
Ana María
Sánchez Hernández
d,
Marielle
Crozet
a,
Guillaume
Bailly
e,
Andrew
Dobney
f,
Georges
Duhamel
g,
Joe
Hiess
g,
Urska
Repinc
g,
Sébastien
Mialle
h,
Béatrice
Boulet
i,
Raphaëlle
Escoube
c,
Céline
Bouvier-Capely
c,
Fabien
Pointurier
b and
Sébastien
Picart
a
aCEA, DES, ISEC, DMRC, Univ Montpellier, Marcoule, France. E-mail: alexandre.quemet@cea.fr
bCEA, DAM, DIF, 91297 Arpajon, France
cInstitut de Radioprotection et de Sûreté Nucléaire (IRSN), PSE-ENV/SPDR/LT2S, PSE-SANTE/SESANE/LRSI, Fontenay-aux-Roses, F-92260, France
dEuropean Commission, Joint Research Centre (JRC), Hermann-von-Helmholtz-Platz 1, Eggenstein-Leopoldshafen, Karlsruhe, 76344, Germany
eCEA, DAM, Valduc, Is-sur-Tille, 21120, France
fSCK-CEN, Boeretang 200, Mol, 2400, Belgium
gOffice of Safeguards Analytical Services, Department of Safeguards, International Atomic Energy Agency, IAEA Laboratories, Friedensstrasse 1, A-2444 Seibersdorf, Austria
hUniversité Paris-Saclay, CEA, Service de Physico-Chimie, Gif-sur-Yvette, 91191, France
iInstitut de Radioprotection et de Sûreté Nucléaire (IRSN), PSE-ENV/SAME/LMRE, Orsay, F-91400, France
First published on 6th May 2024
Abstract
This article proposes a practical guide including good laboratory practices followed by a critical review about the application of isotope dilution mass spectrometry (ID-MS) measurement to radionuclide analyses. Several national and international scientific institutions (French Alternative Energies and Atomic Energy Commission, French Radioprotection and Nuclear Safety Institute, European Commission, International Atomic Energy Agency, and Belgian Nuclear Research Centre) participated in this review by sharing their good laboratory practices. ID-MS is one of the primary ratio methods of measurement in the International System of Units. It can produce highly accurate results, as potential sources of bias can be controlled. Many hints are given to obtain the best performance. In addition, a script written with the open source software Octave is provided to calculate the optimal parameters of the {sample–spike} mixture easily. Radionuclide analysis by ID-MS applies to many different fields (environmental, biological, reference materials, irradiated samples, process control, nuclear safeguards and nuclear forensics).
1. Introduction
Radionuclides are unstable radioactive nuclides which emit radiation during their decay to gain more stability. They can be naturally occurring such as 14C or 129I that come from cosmogenic processes or 210Pb or 230Th that are decay products of uranium isotopes. However, the majority of radionuclides are artificial, being end-products or by-products. For example, 99mTc or 131I isotopes are used in medicine. 99mTc, 90Sr and 137Cs are fission products generated by nuclear fission in reactors or during nuclear weapons tests, while Pu and Am isotopes are produced in nuclear reactors.The accurate assay of radionuclides is essential for various reasons. For example, U and Pu are the main elements of interest in the nuclear fuel cycle as some of their isotopes are used as fissile materials. As such, U and Pu accountancy is of prime importance in the nuclear fuel cycle and for safeguards purposes.1,2 Pu is present in common spent fuel at 1% by weight, in Mixed OXide fuel (MOX) at 5–10% by weight for use in pressurized water reactors and at 20–30% by weight for use in fast-neutron reactors.3 Its measurement with high accuracy (i.e. measurement trueness and precision4–6) is essential to manage criticality issues, to verify fuel pellet conformity and for nuclear material accountancy. Am is also an important element in the nuclear fuel cycle as it is the major contributor to the long-term radiotoxicity coming from the waste, once Pu has been removed. Its reliable analysis is required for waste management and is also important for safeguards and nuclear forensics as it acts as a clock since the last separation of Pu.7
Age dating of nuclear materials is also an area of growing interest for safeguards and nuclear forensics purposes. The main radio-chronometers that are subject to analytical developments are 230Th–234U and 231Pa–235U for uranium and 241Am–241Pu, 236U-240Pu and 235U–239Pu for plutonium.8–11 The accurate measurement of radionuclides is necessary to decrease the uncertainty of the calculated material age. Radionuclides are typically measured by radiometric counting methods (alpha, beta and gamma spectrometry) and mass spectrometry techniques (Inductively Coupled Plasma Mass Spectrometry (ICP-MS), accelerator mass spectrometry, Thermal Ionization Mass Spectrometry (TIMS), Resonance Ionization Mass Spectrometry, Secondary Ion Mass Spectrometry or Glow Discharge Mass Spectrometry).12–14
The isotope dilution (ID) method is used for quantification purposes and is based on the internal calibration principle. The origin of ID is not well known. ID is similar to the mark–recapture method used in ecology to estimate animal population size.15 The mark–recapture method is based on reintroducing a small number of marked animals to an original population considered as a closed system. Later a group of animals is captured and the proportion of marked animals helps to estimate the total number of animals. This method was first used in 1896 by Petersen to estimate plaice15 and may have been used to estimate rare bird species living on islands.16 An assumption of this method, fundamental also to ID, is the perfect mixing of the animals.
The principle of ID is also very simple. A sample of an element with a known isotope composition but unknown number of atoms is mixed with a spike solution containing a known number of atoms of the same element enriched in an isotope and with a known isotope composition. After isotope equilibrium, the isotope ratio of the {sample–spike} mixture reflects the sample analyte amount. The application of ID was invented by Hevesy and Paneth in 1913 to determine the solubility of lead sulfide in water.17 In the 1950s, ID was extended to actinides.18,19 As it is based on isotope ratio determination, an accurate technique for isotope ratio measurements must be preferred to reach the best performance for ID. Measurements by TIMS have been mostly used since 1950 for this purpose due to its highly accurate measurement of isotope ratios.20 Since the 1990s, the use of ID applied to mass spectrometric measurement (ID-MS) has grown considerably due to the widespread application of ICP-MS and the availability of certified reference materials (CRMs) from different institutes.
ID-MS has been listed as a primary method, together with coulometry or gravimetry.21 The definition of the Consultative Committee for Amount of Substance (CCQM) of a primary method of measurement in the International System of Units (SI) is a method having the highest metrological qualities, for which a mathematical model and realization are completely described and understood in terms of SI units.22 Two concepts are defined: a primary direct method and a primary ratio method. A primary direct method measures the value of an unknown quantity without reference to a standard of the same quantity, such as coulometry or gravimetry. A primary ratio method measures the value of an unknown to a standard of the same quantity; its operation must be completely described by a measurement equation, such as ID-MS.21 In both cases, the results must be accompanied by a complete uncertainty statement.22 From the list of primary methods, ID-MS is the most widely used one in U and Pu amount determination inter-laboratory comparisons (ILCs).23 Furthermore, the International Atomic Energy Agency (IAEA) considers ID-MS as one of the most accurate methods in terms of the International Target Values (ITVs) for measurement uncertainties applied on safeguarding nuclear materials.24,25 TIMS and ICP-MS are the techniques of choice to measure the isotope ratio of most elements,26 for example radionuclides such as Sr,27 Cs,28–30 Nd,31–34 Th,35,36 U,33,37–41 Pu,3,33,38,40,42–45 Am7,31,38 and Cm.31,46 ID-MS is a versatile tool for radionuclide quantity measurement and the “gold standard” for nuclear safeguards.
Several reviews have described ID-MS for CRM characterization,20 element speciation,47,48 proteomics49 and geochemical analysis.50 Some articles have discussed the general concept of ID-MS.26,51–54 To our knowledge no general reviews on radionuclide measurement by ID-MS have been published.
The Commission for the Establishment of Analytical Methods (CETAMA) is a unit of the French Alternative Energies and Atomic Energy Commission (CEA), which has a goal of measurement quality improvement within the nuclear field.23,55–58 The main activities of CETAMA are to develop and provide CRMs for nuclear analysis laboratories worldwide, to organize ILCs and to participate in the standardization of analytical methods. One of the CETAMA's working groups is dedicated to ID-MS which gathers several national and international scientific bodies (CEA, French Institute for Radiation Protection and Nuclear Safety (IRSN), Joint Research Centres (Europe), IAEA, Belgian Nuclear Research Centre (SCK)) that share their know-how in radionuclide analysis by ID-MS.
This review describes the good laboratory practices of the ID-MS working group members and proposes a critical review about the applications of ID-MS to radionuclide measurement. After a brief theoretical description of ID, the uncertainty budget expression is explained in more detail, including how to minimize it. The implementation and good laboratory practices of ID-MS are further described. The review covers the application of ID in different fields: environment, biology, reference material production, irradiated sample analysis, process control, nuclear safeguards and nuclear forensics. The final chapter is dedicated to the description of different ILCs available for testing the performance of radionuclide measurement by ID-MS.
2. Theory
2.1. General formula
The principle of ID is illustrated in Fig. 1. Isotope “A” is the major isotope of the element “E” in the sample. Isotope “B” is the major isotope of the element “E” in the spike. For clarity and conciseness, the following nomenclature is used throughout the manuscript: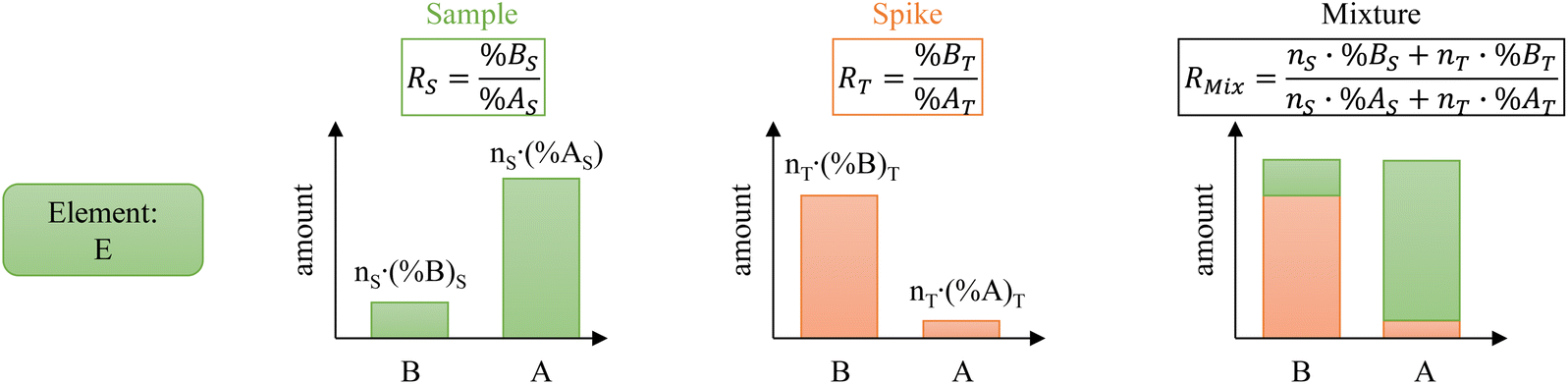 | ||
| Fig. 1 Illustration of the isotope dilution principle. | ||
- Subscripts:
○ Mix refers to the {sample–spike} mixture;
○ T refers to the spike (or tracer);
○ S refers to the sample.
- R is the B/A ratio in mol mol−1.
- R′ is the A/B ratio in mol mol−1.
- %A refers to the isotope abundance in percent of mol mol−1 of A.
- %B refers to the isotope abundance in percent of mol mol−1 of B.
- n refers to the amount of substance (mol).
- w refers to the mass fraction in g g−1.
- M refers to the molar mass in g mol−1.
- m refers to the mass in g.
- u is the standard uncertainty with a coverage factor of k = 1.
- ur is the relative standard uncertainty with a coverage factor of k = 1.
- α refers to the sample amount; spike amount ratio (α = nS/nT) in mol mol−1.
- LOD is the limit of detection.
The mixture isotope ratio (RMix) can be calculated from the atom number or number of moles of an element and the isotope abundance of isotopes A and B.
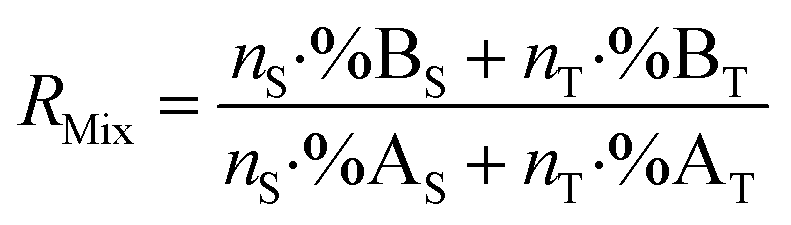 | (1) |
Introducing %BS = %AS·RS and %BT = RT·%AT in eqn (1) leads to the general form of the isotope dilution equation (eqn (2)).
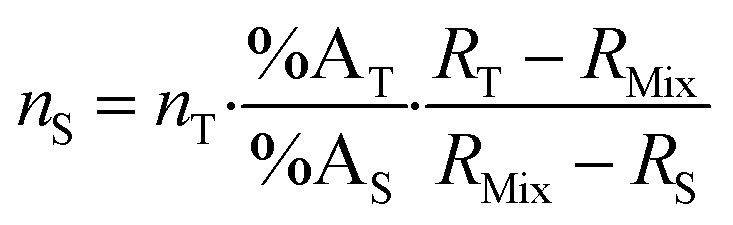 | (2) |
Eqn (2) can be converted into mass fractions by taking into account the masses of the sample and spike, and the atomic masses of the element in the sample and spike (eqn (3)).
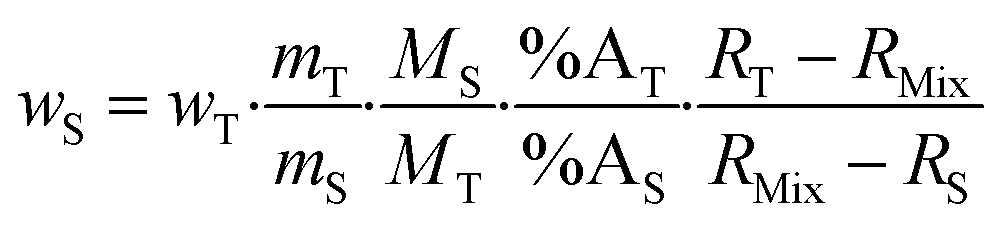 | (3) |
Another form is also commonly used to show the major isotopes in the spike and in the sample (%BT and %AS) in the ID formula (eqn (4)). Eqn (2) and (4) correspond to each other and will give the same results.
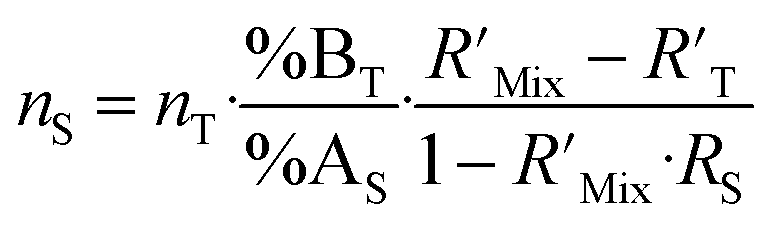 | (4) |
2.2. Formula used for environmental samples
A single (eqn (5)) and a double isotope dilution equation (eqn (6)) are commonly used in environmental samples for the mass fraction determination of 238U using a spike enriched in the 236U isotope and the 230Th/238U ratio using a spike enriched in the 229Th and 236U isotopes. Obviously, these equations can be applied for other target radionuclides by using the appropriate spike and the respective atomic masses.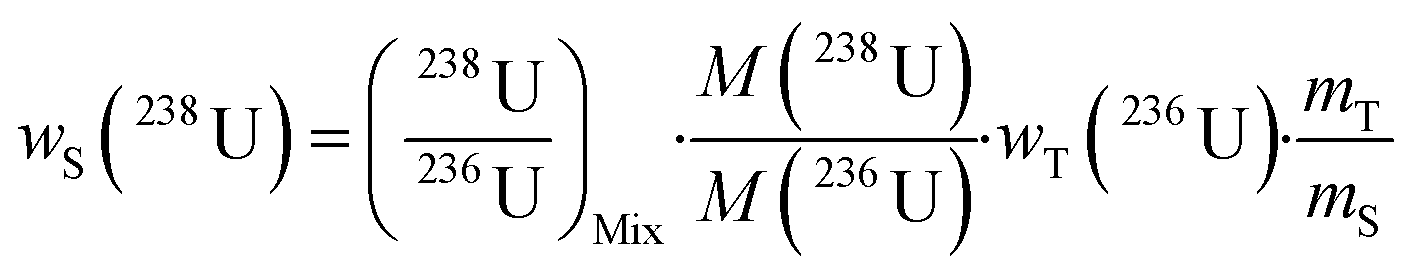 | (5) |
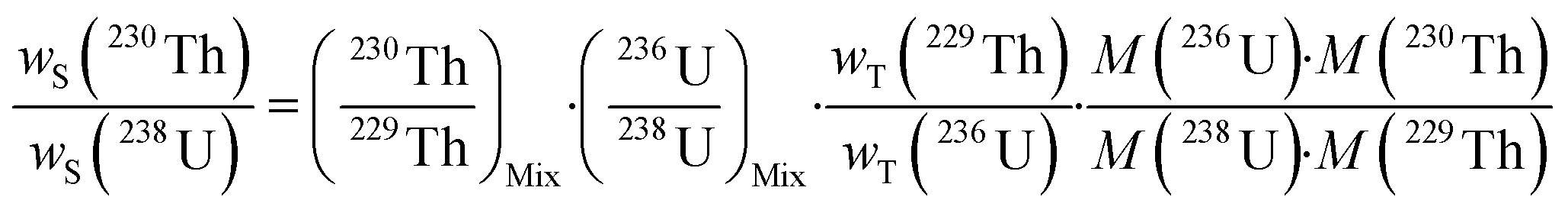 | (6) |
The mass fraction and mass fraction ratio are then converted into activity ratios by considering the radionuclide atomic masses and their respective half-lives (t1/2). It should be noted that the double isotope dilution in combination with a double spike (i.e.229Th–236U) is a powerful tool that helps to minimize weighing errors and eliminate errors due to differential evaporation of spike solutions. Indeed, the spike weight does not play a role in eqn (6).59
It should be noticed that spikes usually contain isotopic impurities (for instance, a 236U spike usually contains traces of 234U, 235U and 238U isotopes). In many cases, especially for the less abundant isotopes at ultra-trace levels, eqn (5) and (6) must be modified to include and correct for the contribution of these isotopic impurities.
2.3. Estimation of the limit of detection
The LOD is the minimum concentration or quantity of an analyte that can be detected with a reasonable certainty for a given analytical procedure.60Eqn (7) can be used to obtain a conservative estimate of the LOD of the element E using ID (LODID(E)) for a specific spike.61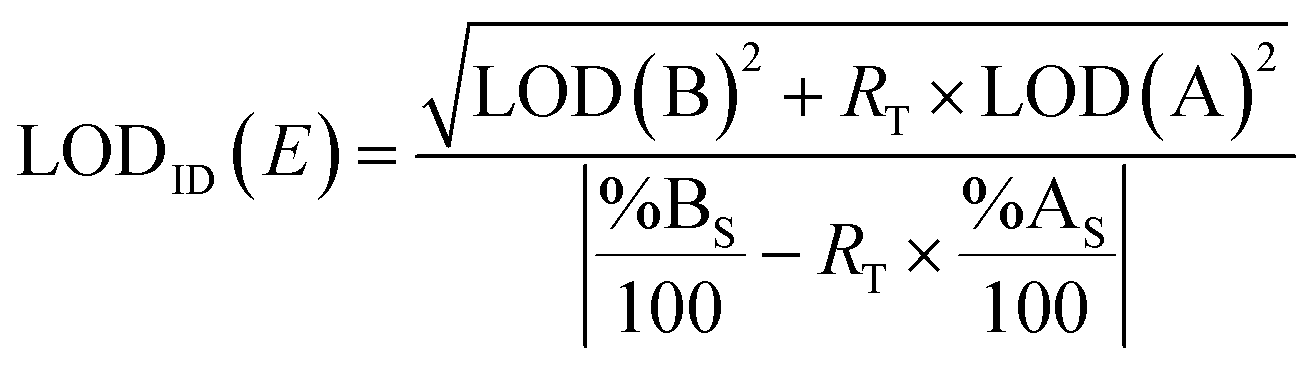 | (7) |
3. Implementing isotope dilution
A careful implementation is needed to obtain the best performance for ID-MS. The highest level of quality can be reached when a weighed aliquot of the sample is mixed with a weighed aliquot of the spike. Volumetric mixing can only achieve a lower level of uncertainty. The uncertainty will also be minimum when an optimal ratio between the sample and the spike is reached.Successful ID requires achieving complete isotope equilibration between the sample and spike. This is practically only possible when the mixture is in solution. Due to isobaric interferences in many instances a separation step to obtain the pure element is needed. Theoretically this separation does not introduce a source of error since after isotope equilibration, losses of the substance have no influence on the final result, an important advantage compared to other methods. In all cases, the appropriate ID-MS spike should be added to the sample aliquots before starting the sample preparation and should be added before the separation protocol. It is therefore also clear that any loss of substance before reaching full equilibrium is a source of error and to be avoided. Another advantage is the compensation of matrix effects as any factor influencing the sensitivity of the analyte is the same for the sample and spike nuclide.
The main disadvantages of the method are its destructive nature and that the preparation steps consume time and resources. The disadvantages are generally balanced out by the advantages. The spike availability is for most elements no issue as the laboratories produce their own spikes or procure them from CRM producers.
The weighing procedure, the preparation of the {sample–spike} mixture, the choice of the spike, spike management, and isotopic homogenization are the main criteria to be taken into consideration when implementing ID-MS. These various influencing parameters are discussed in the ensuing paragraphs. Isotope ratio measurements by MS are also an important parameter to take into account and are largely described in the literature.12,62–68
3.1. Weighing procedure
In the case of solid samples, the material is normally dissolved taking a sample quantity sufficiently large to obtain a representative sample of the material. The mass of the dissolved sample generally exceeds 0.1 g to optimize the uncertainty contribution from mass measurements (see Section 4.2). The solution is diluted, if needed, to match the target sample/spike ratio (see Section 3.2). For mass spectrometric measurements, typically picogram to microgram amounts of spike are used. Therefore, a similar quantity of the sample is required for assay determination by ID-MS. The mass of the sample and of the spike used to prepare the mixture solution should also exceed 0.1 g for the same reasons.
For ID-MS, the amounts of spike and sample are typically small. Thus, the uncertainties in the mass determination contribute significantly to the overall uncertainty budget. Therefore, it is important to consider the different effects that can lead to a significant bias, some of which are related to the weighing itself (buoyancy, electrostatic effects), some to the sample preparation (evaporation, moisture content and stability, type and mass of vials) and others to effects that might happen during aliquoting such as cross-contamination.
The availability of a high-precision analytical balance is a prerequisite, typically with a resolution of at least 0.1 mg. The balance must be assessed before use with appropriate standard weights to ensure proper balance function.
Electrostatic effects can be avoided when needed by using specific tools to prevent or eliminate electrostatic charges. Moreover, properly trained analysts and good analytical practices can improve the mass determination, for instance performing daily check weighings prior to using the balance for critical measurements, and ensuring the correct placement of vials on the balance.69
The weighing procedure can be summarized as follows: the balance is tared with the vial and the cap, then the sample is added and the cap placed on the vial to avoid evaporation, and the mass displayed is the sample aliquot, mS.70 After another tare of the balance, the spike is added and weighed with the cap on the vial. The mass displayed is the spike aliquot, mT.
A different procedure, employing the technique of weighing by difference, was established in the nuclear safeguards laboratories of the Joint Research Centre (JRC) Karlsruhe, the double-weighing method.71 In this procedure, the aliquot mass of the sample, or spike, is determined twice:
1. The mass dispensed from a Pasteur pipette determined by the mass difference between a filled and an emptied pipette (eqn (8)):
| mip = mpf − mpe | (8) |
2. The mass received into a vessel, the mass difference between the spike vial with and without the aliquot (eqn (9)):
| miv = mvf − mve | (9) |
Once the consistency between the two mass determinations has been demonstrated, they should not differ from one another by a laboratory determined limit, and the mass dispensed from the pipette (eqn (8)) is taken as the true aliquot mass. This procedure can minimize the impact of some of the undesirable effects encountered during weighing, and offers a built-in consistency check so that a bad weighing can be detected before mass spectrometric measurements.
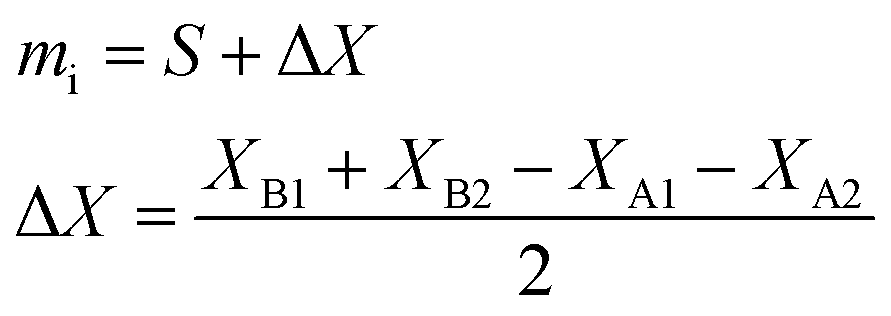 | (10) |
Uncertainties on the order of tens of μg can be achieved with the substitution method.73,74 However, this method requires many standards and is time consuming, and hence is usually restricted to metrological labs and is not routinely used in analytical labs.
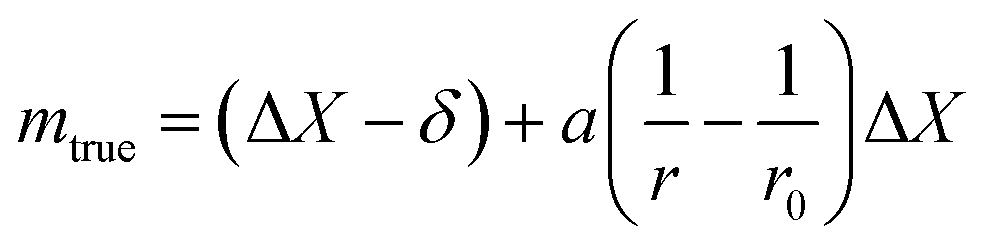 | (11) |
The need to correct for air buoyancy depends on how accurate the results must be and on the density of the samples. It is particularly critical when the target total uncertainty is less than 1% (metrological purposes for example) and for aqueous solutions with a density close to 1000 kg m−3, because such a density is far removed from that (8000–8100 kg m−3) of the stainless steel calibration standards.
The expression for the correction factor, K, for air buoyancy is given in eqn (12)
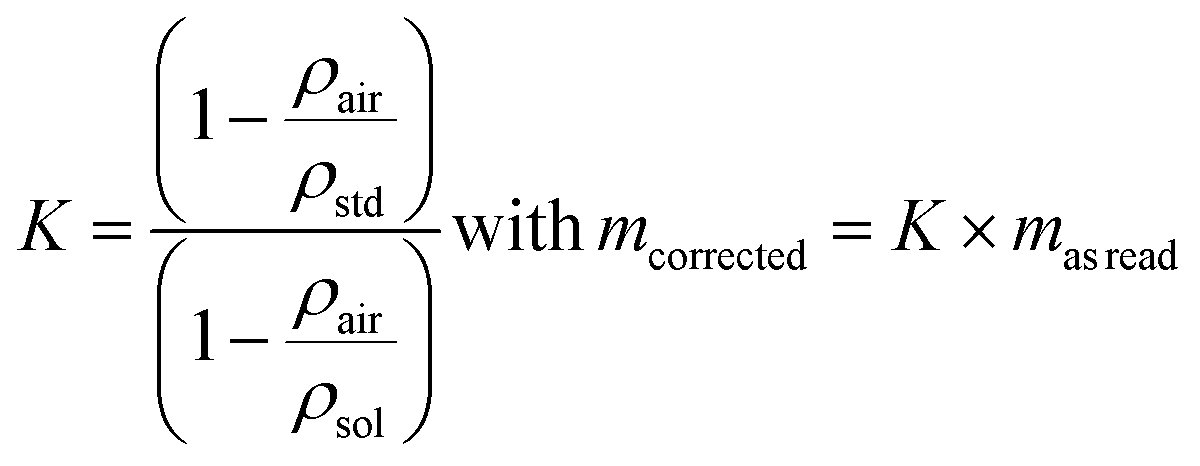 | (12) |
If the requirement in terms of weighing uncertainty is 0.1%, it is not necessary to measure the air density experimentally in order to calculate the K factor for an aqueous sample with a density of 1000 kg m−3. Knowing the density of the sample, the conventional value, 1.2 kg m−3, for the density of air is sufficient to express K and correct the read mass. For solid samples and the same requirement in terms of the uncertainty level, corrections are not necessary.
In the case of ID-TIMS, the weights are expressed in the equation in the form of a ratio, 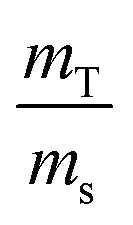 . If the solutions are alike (e.g. same nitric acid concentrations), their densities and K factors will be identical and the ratio of the masses as read will correspond to the ratio of true masses (corrected masses). No corrections will be needed. The same conclusions can be drawn if serial dilutions are made using solutions with similar characteristics.
. If the solutions are alike (e.g. same nitric acid concentrations), their densities and K factors will be identical and the ratio of the masses as read will correspond to the ratio of true masses (corrected masses). No corrections will be needed. The same conclusions can be drawn if serial dilutions are made using solutions with similar characteristics.
If higher accuracy is required, corrections for air buoyancy should be considered throughout the entire preparation of sample. For example, when a solid is dissolved, the K factor for the solid is 1; whereas for a solution is greater than 1. By considering a density solution of 1100 kg m−3 (≈3 mol L−1 nitric acid solution), the K factor for this solution can be written as in eqn (13)
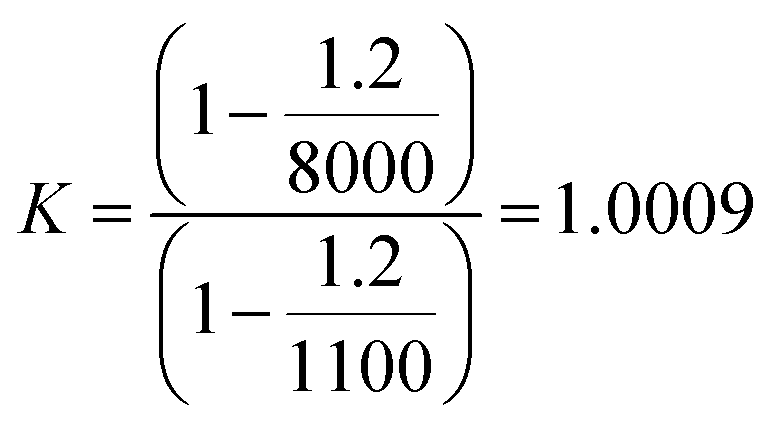 | (13) |
The relative mass correction of the solution is approximately +0.1%. The mass fraction for the dissolved solid should then be corrected inversely by −0.1%. In other words, not taking the air buoyancy into account would lead to an overestimation of the mass fraction of the sample.
3.2. Optimization of the {sample–spike} mixture
ID-MS requires an optimum spiking ratio to obtain the best accuracy (i.e. the minimum relative uncertainty of nS, eqn (2)). Several studies have been reported in the literature.37,52,76–80 Here, three different methods are detailed. The first derives the error multiplication factor (F) as a function of the mixture isotope ratio, leading to a very simple formula to derive the optimal isotope ratio for the mixture. The second derives the F factor as a function of the ratio between the amount of sample and the amount of spike which makes it straightforward to prepare the mixture experimentally. The third method is broader, in that it considers all isotope ratio uncertainties. Lastly, the choice of the spike isotope and examples are discussed.The three methods have been implemented in a script written with the open source software Octave,81,82 version 8.3.0. The method calculating the LOD as a function of the spike's isotope ratio (eqn (7)) was also implemented. To start the script, download the “startup_ID.m” file available in the ESI.‡ Start the Octave software and set the browser directory to the appropriate folder where the file “startup_ID.m” is located. Starting the script is performed by typing “startup_ID” in the Octave command window. The script then prompts the user to select an optimization method. All parameters must be selected. Once the calculation has been completed, the optimum parameters are displayed in the Octave command windows. The plot is also displayed. The plot can be saved as an 8 cm × 8 cm “.png” file. The results and the raw data can be saved as a “.txt” file.
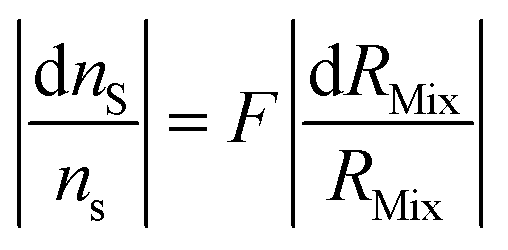 | (14) |
The F factor is obtained from eqn (15).
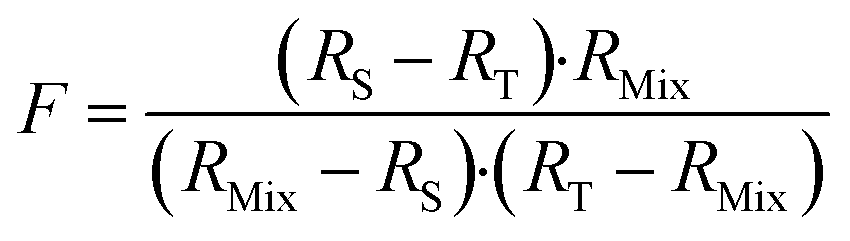 | (15) |
Finding the optimal value of RMix is simply a matter of locating the minimum F factor in the function F = f(RMix). The minimum of a function is obtained when the derivative is equal to zero (eqn (16)).
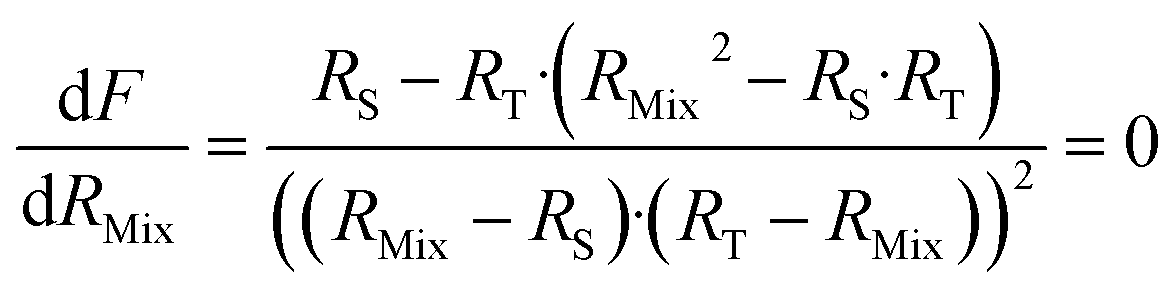 | (16) |
Eqn (16) is equal to zero if the numerator is equal to zero. After rearrangement, eqn (17) is obtained. The optimum mixture ratio (RMix,opt) can be calculated using the square root of the product of RS and RT.
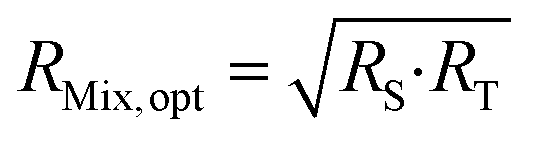 | (17) |
Eqn (17) is a very simple equation for deriving the optimum mixture isotope ratio. However, one prerequisite is the lower RS and RT uncertainties compared to RMix uncertainties. This condition is generally true: RT uncertainty is usually very low as coming from the CRM certificate or can be accurately measured in the case of an in-house spike; RS can be measured several times to decrease its uncertainty.
The blue curve in Fig. 2 shows the F factor as a function of RMix (eqn (15)) for a sample with 3.0% of 235U and 97% of 238U (%AS = 97%, %BS = 3.0%, RS = 0.0309) using a spike enriched with the 235U isotope such as IRMM-054 CRM (93.2% of 235U and 5.4% of 238U: RT = 17.3). Using this raw data, the optimum isotope ratio of the mixture is RMix,opt = 0.73. In this example, the parameters are robust since the F factor varies little in the region around the optimum value.
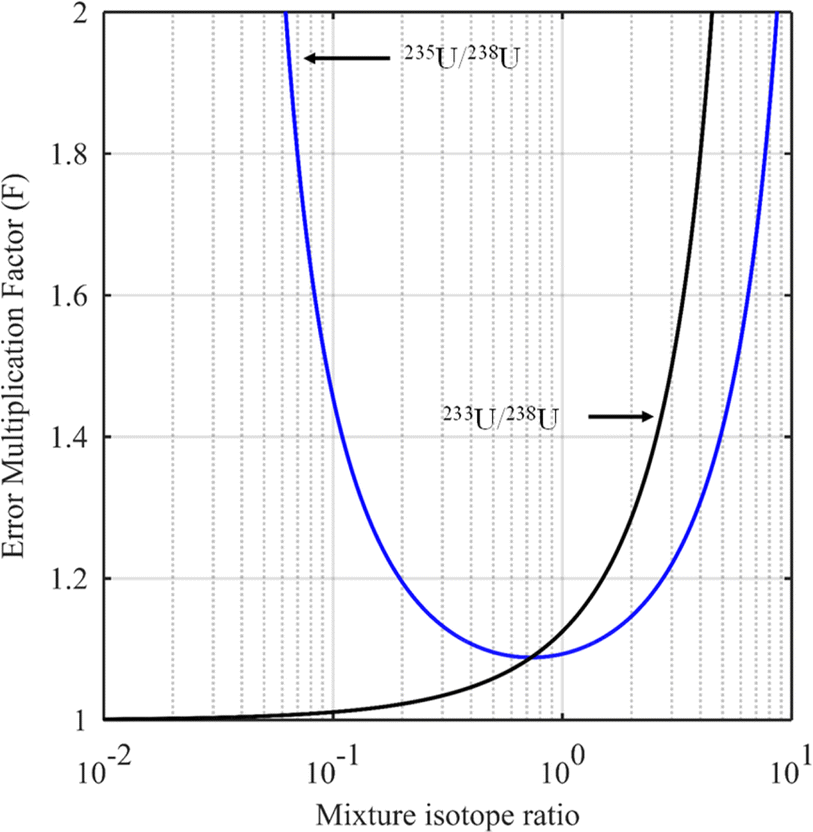 | ||
| Fig. 2 Error multiplication factor as a function of the 235U/238U mixture isotope ratio (blue curve) for a sample with 235U/238U = 0.0309 and for a spike enriched with the 235U isotope (235U/238U = 17.3), and as a function of the 233U/238U mixture isotope ratio (black curve) for a sample with 233U/238U = 0 and for a spike enriched with a 233U isotope (233U/238U = 9). | ||
When the major spike isotope is absent from the sample (RS = 0), eqn (17) can be simplified (eqn (18)).
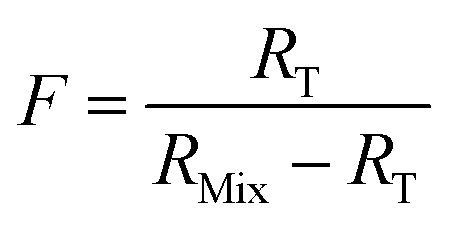 | (18) |
Evaluation of the optimum isotope ratio of the mixture can be done graphically. An example is shown in Fig. 2 (black curve), for a uranium sample with 233U/238U = RS = 0 and a spike enriched at 90% in the 233U isotope (233U/238U = RT = 9). The F factor tends to 1 as RMix gets closer to RS and becomes larger as Rmix approaches RT. The best choice would appear to be RMix = RS (i.e. when no spike is used), but this makes no sense for an ID measurement. A compromise needs to be reached between the minimum F factor and spike addition. In the example in Fig. 2 (black curve), mixture isotope ratios in the range 0.1 to 1 seem to provide an interesting compromise between a low F factor and needing to add a spike.
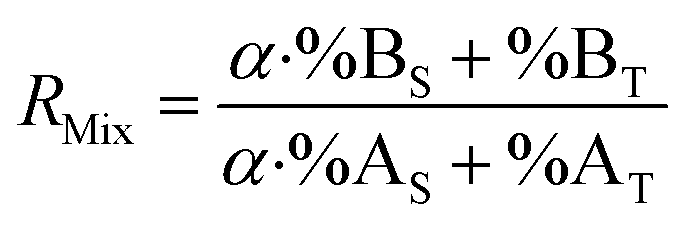 | (19) |
By combining eqn (15) and (19), the F factor as a function of α can be calculated (eqn (20)).
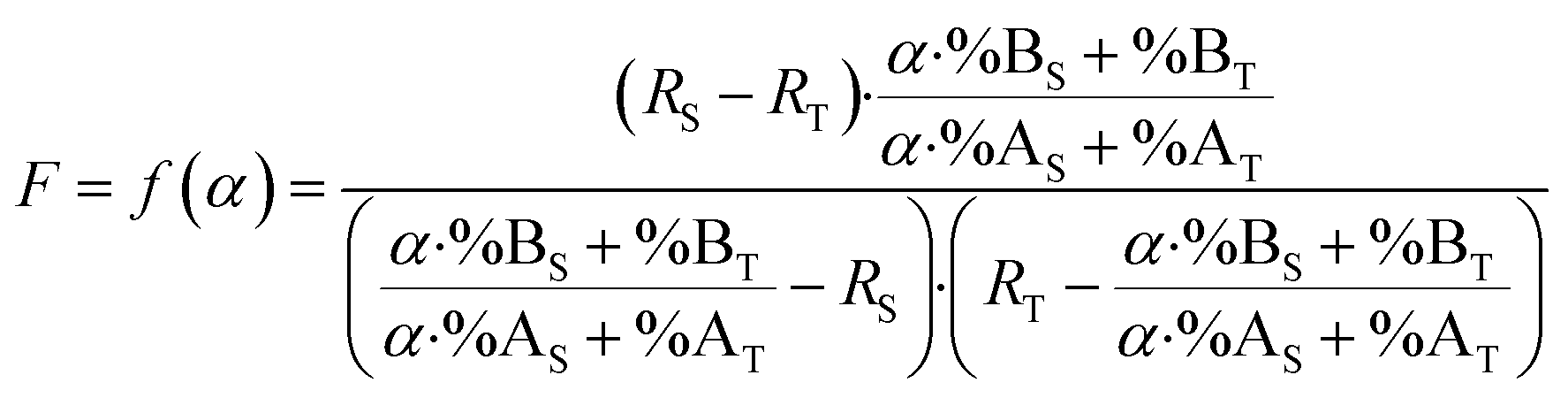 | (20) |
Determining the α optimum value is simply a matter of locating the minimum F value. To understand how the F factor varies around the optimum α value, the F factor can be plotted as a function of α (eqn (20)). This plot is useful for illustrating how robust the optimal value is. In practice, the sample amount is generally not accurately known. It is therefore difficult to mix the sample and spike in the exact optimal proportions.
Combining eqn (4) and (17) helps to quickly obtain the optimal sample amount over spike amount ratio (αopt, eqn (21)).
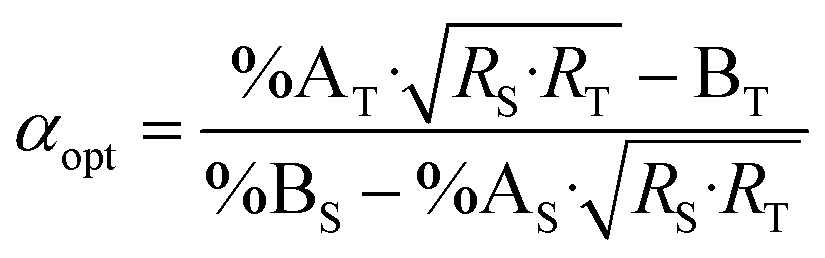 | (21) |
This way of expressing the F factor provides the analyst with a simple way to derive the {sample–spike} mixture. As for the previous method, one prerequisite is that the uncertainties of RS and RT are lower than that of RMix. In cases where the major isotope of the spike is absent from the sample (RS = 0), eqn (20) cannot be applied. In this case, the optimum ratio of sample amount to spike amount can be determined graphically.
The curves in Fig. 3 show the F factor as a function of α (eqn (20)) for a sample with 3.0% of 235U and 97.0% of 238U using a spike enriched with the 235U isotope such as IRMM-054 CRM (blue curve, 93.2% of 235U and 5.4% of 238U, same example as for Fig. 2) and for a sample with 70.0% of 239Pu and 1.0% of 242Pu using a spike enriched with a 242Pu isotope like the IRMM-049d CRM (black curve, 0.2% of 239Pu and 94.7% of 242Pu). Using these raw data, the optimal α values are 1.3 and 0.5 for uranium and plutonium samples respectively. In these examples, the F factor varies little in the region around the optimum value of α.
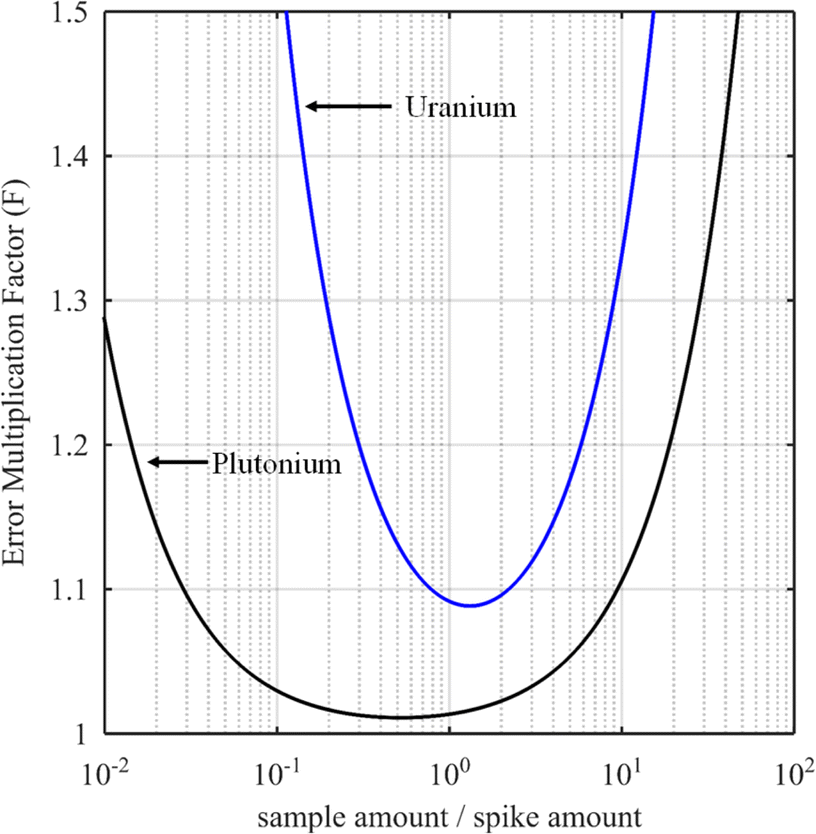 | ||
| Fig. 3 Error multiplication factor as a function of the sample over spike amount for uranium (blue curve, sample with 3% of 235U and 97% of 238U using a spike enriched with 93.2% of 235U) and plutonium (black curve, sample with 70% of 239Pu and 1% of 242Pu using a spike enriched with 94.7% 242Pu). | ||
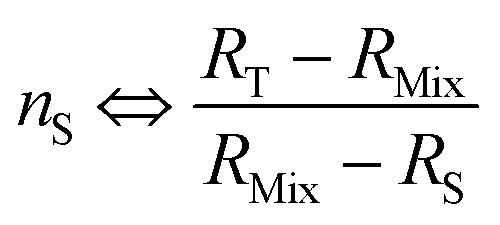 | (22) |
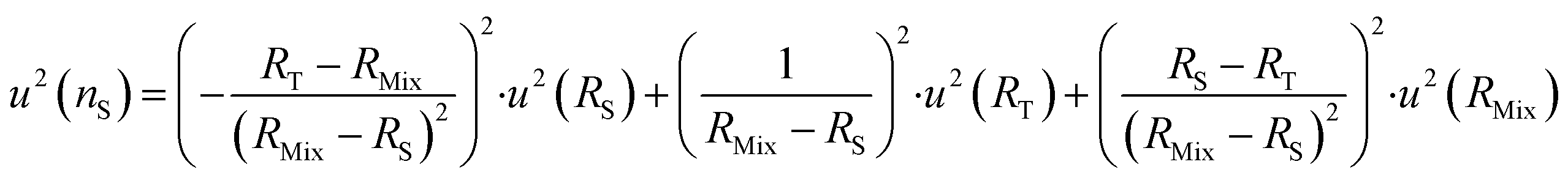 | (23) |
The optimum spiking should minimize the relative uncertainty of nS, which can be expressed as a function of RMix (eqn (24)).
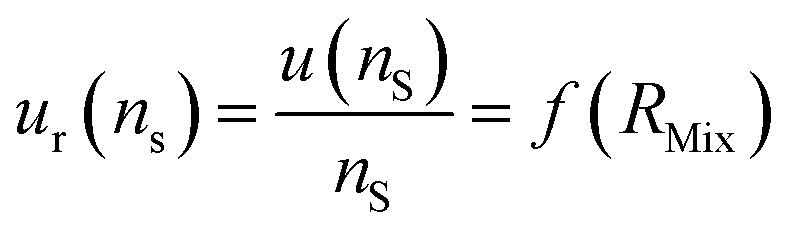 | (24) |
Determining the optimal value of RMix is simply a matter of locating the minimum relative uncertainty of nS (ur,min(nS)). To understand how the F factor varies around the RMix optimal value, the error of the relative uncertainty as a function of RMix can be plotted (eqn (25)).
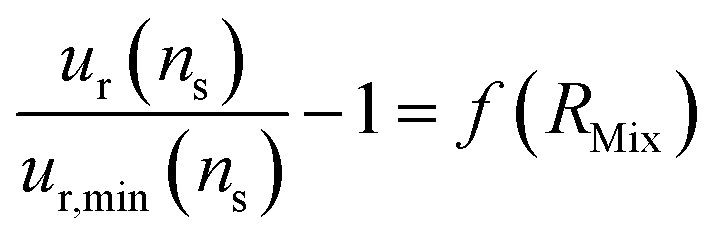 | (25) |
If the uncertainties of RS and RT are equal to zero, then this solution is identical to that obtained by using method 1 (eqn (15)).
To illustrate this method, a sample with 3% of 235U and 97% of 238U was considered: %AS = 97%, %BS = 3%, RS = 0.0309 and uR(RS) = 0.5%. A spike, enriched in the 235U isotope (such as the IRMM-054 CRM), was used: RT = 17.3 and uR(RT) = 0.03%. The RMix relative uncertainty was fixed at 0.07%, corresponding to the ITV value.24 In this example, the optimum isotope ratio of the mixture is RMix,opt = 1.03. It should be noted that the optimum isotope ratio of the mixture (1.03) determined by taking into account all uncertainties is slightly different from, but not dissimilar to, the value (0.73) when not taking into account uncertainties of the isotope ratio RS and RT (eqn (15)).
Just as for the previous two methods, in cases where the major isotope of the spike is absent from the sample (RS = 0), the optimum isotope ratio of the mixture can be determined graphically.
- It is better if the spike isotope is absent from the sample or has the lowest isotopic abundance in the sample.
- A spike enrichment higher than 90% is recommended, allowing the accuracy of the ID to be practically independent of the mixture preparation.
- It is better if the spike isotope has no isobaric interference.
For uranium, the most common spikes are 233U, 235U or 236U, since the main uranium isotope in most samples is generally 238U (excluding highly enriched uranium material). For plutonium, 240Pu, 242Pu or 244Pu spike isotopes are commonly used, since 239Pu is usually the main major isotope in most samples. For americium, since the samples have 241Am as the major isotope, a spike enriched in the 243Am isotope is a good option.56 In the case of U, Pu and Am, the choice is easy as the sample has one major isotope and spikes highly enriched in minor isotopes are commercially available. It is worth noting, for the radioactive spike, the isotope abundance and mass fraction must be updated for the day of analysis to correct for the radioactive decay.
For certain other elements, the choice of the spike isotope is not as straightforward. For example, in the case of neodymium measurement with natural isotopic composition, several choices are possible. In theory, the best choice of reference isotope in the sample is the 142Nd isotope since it is the most abundant isotope in such a sample. However, since 142Ce can be an isobaric interference on the 142Nd isotope, the 144Nd isotope is generally preferred as a sample isotope.31Fig. 4 shows the F factor as a function of the sample/spike amount ratio for different Nd isotopes as the major isotope in the spike. The curves for 148Nd and 150Nd lie close together and overlap each other, as shown in Fig. 4. Since 148Nd and 150Nd are the two least abundant isotopes, in a sample with natural neodymium isotopic composition, a spike enriched in either 148Nd or 150Nd is the best choice to obtain the lowest uncertainty (i.e. minimum F factor) and the easiest to implement (because the plot of the F factor is almost flat in the region around the optimum value). Since all other Nd isotopes in such a sample have higher abundances, choosing an isotope other than 148Nd or 150Nd as the spike isotope will generate a higher minimum F factor and a narrower range around the optimal F factor. Fig. 5 shows the F factor as a function of the sample/spike amount ratio for different spike enrichments when using a spike enriched in the 150Nd isotope. The higher the spike enrichment is, the smaller the optimal F factor is. Moreover, the region around the optimal F factor value is relatively flat for spikes with RT > 9 (i.e. spike enrichment higher than 90%), which makes it easier to implement ID. The LOD, in arbitrary units, as a function of the spike isotope ratio is presented in Fig. 6 for different major isotopes of the spike, assuming the LOD of A and B isotopes is equal to 1 in arbitrary units (eqn (7)). The curves for 148Nd and 150Nd isotopes overlap in Fig. 6. The LOD decreases from RT = RS, tending towards lower LOD values at higher spike enrichments. It should be noted that the choice of spike isotope has no significant influence on the LOD when RT is high enough. In this example, all LODs are similar irrespective of the spike isotope chosen when RT > 6. In this way, for the measurement of natural Nd by ID-MS, a judicious choice is a spike enriched in 148Nd or 150Nd isotopes with an enrichment greater than 90%. The optimal sample/spike amount ratio in the mixture is then calculated as a function of the spike enrichment (Fig. 5).
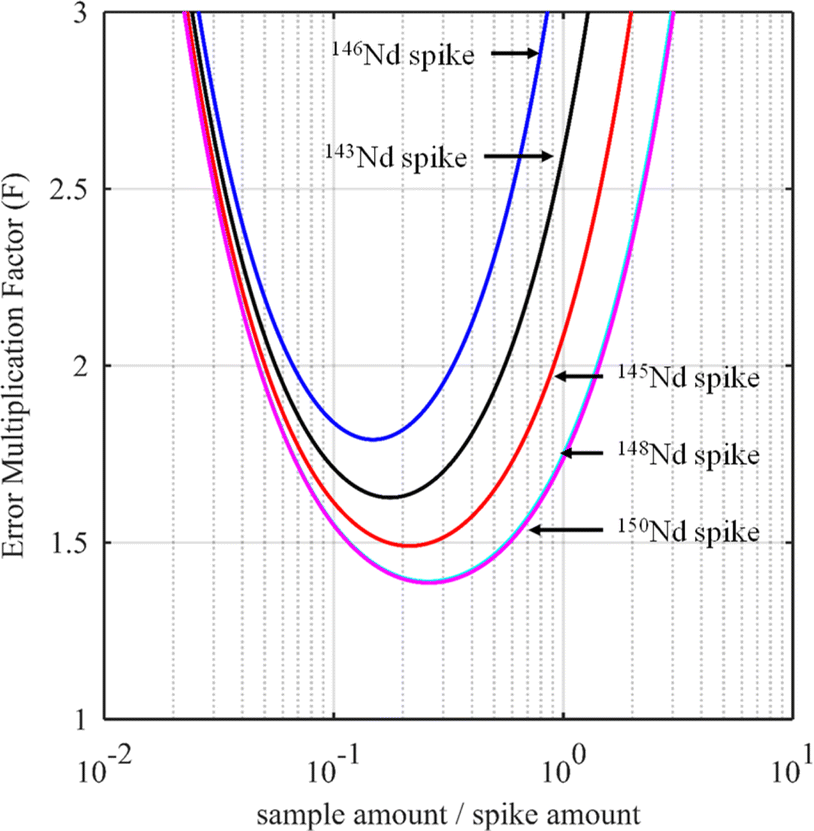 | ||
| Fig. 4 Error multiplication factor as a function of the sample/spike amount ratio for 143Nd (black line), 145Nd (red line), 146Nd (blue line), 148Nd (cyan line) and 150Nd (purple line) spikes (RT = 9 for all spikes) with a neodymium sample of natural isotopic composition. The 144Nd isotope is used as a reference sample isotope. | ||
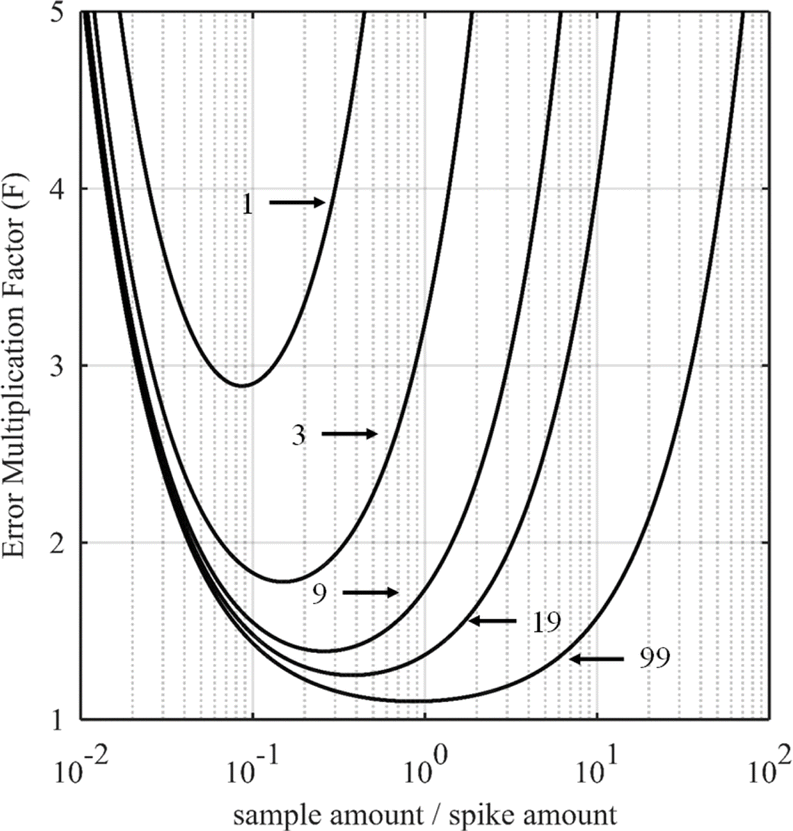 | ||
| Fig. 5 Error multiplication factor as a function of the sample/spike amount ratio for different 150Nd/144Nd spike isotope ratios (RT = 1, 3, 9, 19 or 99) for measuring natural neodymium. | ||
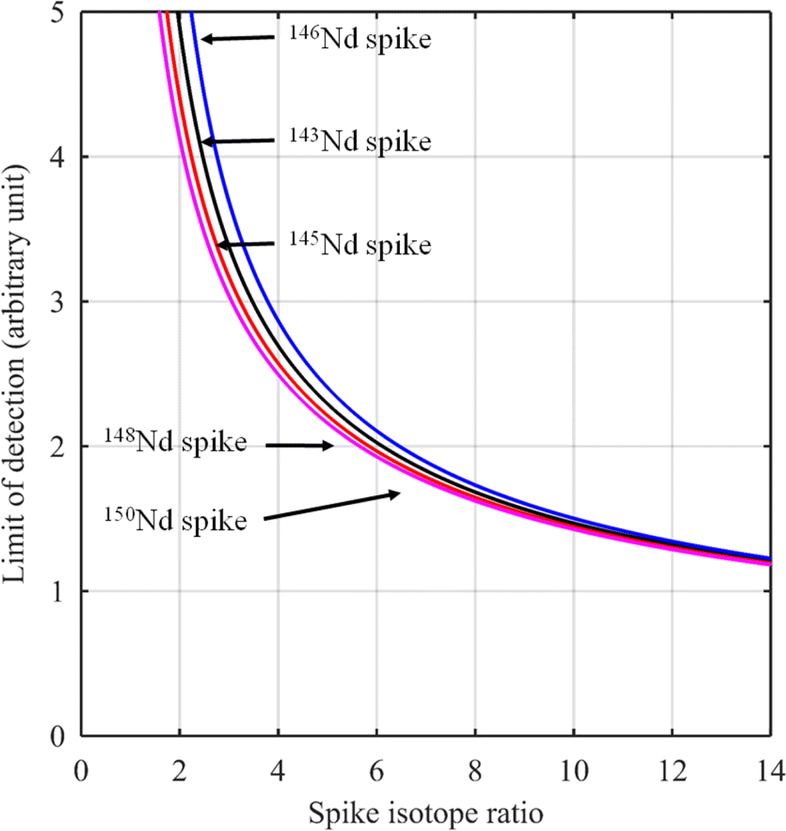 | ||
| Fig. 6 Limit of detection (arbitrary unit) for a natural sample of neodymium as a function of the spike isotope ratio for spikes enriched in 143Nd (black line), 145Nd (red line), 146Nd (blue line), 148Nd (cyan line) and 150Nd (purple line) isotopes. | ||
Some laboratories do not calculate the optimal parameters for every new sample when measuring U or Pu. Instead, the mixture is prepared in such a way as to obtain a 1/1 ratio between the amounts of spike and sample. This method works well if the conditions for the selection of the spike are fulfilled: the spike isotope is absent from the sample or has the lowest isotopic abundance in the sample and the spike enrichment is high enough (Fig. 3). In these examples, the optimal nS/nT ratio is found to be 1.3 (F factor = 1.088) for the uranium sample and 0.5 (F factor = 1.011) for the plutonium sample. However, at values around nS/nT = 1, the F factor (1.093 for uranium and 1.014 for plutonium) is very similar to that at the optimum ratio. In these cases, employing a 1/1 ratio between the amount of the spike and the sample instead of the optimal amount ratio will have no impact on the ID-MS accuracy.
3.3. Spike monitoring
Assessment of the spike stability and homogeneity of liquid spike materials is crucial to ensure the quality of the ID analysis results and their reliability. The behavior of spike materials is difficult to predict reliably solely from stability studies over short periods. Due to evaporation, the observed differences in element mass fraction can be higher than 0.1% after several weeks, which is generally larger than the element mass fraction uncertainty of the spike. To overcome this issue, several solutions are presented in this section.When using liquid spikes in the form of ampoules, it is recommended to consume the spike solution immediately after opening, or alternatively to dry the prepared aliquots as small-size dried spikes for later use, eliminating the need to correct evaporation effects and additional uncertainty due to dilution. However, due to the scarcity and costs of commercially available spikes, this option is not often applied.
IRMM-1027 Large-Sized Dried (LSD) and LSD spikes produced by the IAEA, prepared from U and Pu metal CRMs, are applied to measure the uranium and plutonium content of dissolved fuels in reprocessing plants.73,84–86 LSD spike vials have certified masses of uranium (235U and 238U) and 239Pu and isotopic composition with associated uncertainty. The main advantage is that they are to be used on an undiluted sample solution containing mg amounts of U and Pu. A further advantage is that cross-contamination is limited precisely because of the large amounts of the analyte in both the spike and the sample. However, they are prepared for a specific and single use, and until the spikes were in a dried nitrate form, it was not easy to maintain their integrity over longer periods. Nowadays, organic stabilizing substances are used to prolong shelf-life and guarantee stability during shipment.73
Liquid spikes are often used after dilution to prolong the operational lifetime of the spike and to adjust the spike solution mass fraction. In this case, not only spike solution evaporation but also the associated uncertainty of performing gravimetrical dilution needs to be considered. Spike solutions in glass ampoules with a screw-cap were introduced by JRC-Geel to replace the flame sealing of ampoules, and are a good choice to reduce the evaporation effects during use. The storage in glass ampoules for U, Pu or Am spikes shows glass–solution interactions have no effect on the spike concentration. Due to safety reasons ampoule sealing by a flame or laser is not commonly applied in analytical laboratories, but with the use of screw-capped ampoules it is possible to verify the evaporation of the spike solution by weighing.
The Atalante Analysis Laboratory of the CEA (CEA/LAAT) and the JRC-Karlsruhe adapted a similar system,87 where the CRM spike is first gravimetrically diluted to a known mass fraction suitable for mass spectrometry, at the μg g−1 level. Approximately 10 mL aliquots of this diluted spike solution are then stored in sealed borosilicate glass ampoules. Upon opening a sealed ampoule of a diluted spike, the diluted spike solution is divided into several vials with caps, each containing a known amount (i.e. mass) of spike. The vials can be stored in racks for up to several weeks before use. Any evaporation of this solution will affect the mass fraction of the spike in the vials but not the amount (i.e. mass) of spike in the vials. The sample is added to the vials to produce the {sample–spike} mixture. This entire mixture is then evaporated to dryness, thereby eliminating any influence of potential changes in the spike mass fraction due to evaporation during storage. This step ensures the trueness of the isotope ratio in the mixture. The dried {sample–spike} mixture is then dissolved in nitric acid medium for ID-MS measurement (e.g. mass spectrometric measurement or separation step). The dilution of the CRM spike is verified annually by means of inter-laboratory comparison exercises.
3.4. Isotopic homogenization of the sample–spike mixture
Essential for correct ID analysis is the isotopic equilibrium in the mixture which may be defined as the homogeneous distribution between the isotopes coming from the sample and the spikes in the mixtures.88In the nuclear field, most of the samples and spikes are in the same strong acidic media. The isotope homogenization is therefore quite easy and fast for most of the radionuclides (U, Th, Am, Cm, lanthanides…). Manual or mechanical shaking of the mixture is carried out for several seconds or minutes to ensure the homogenization.54 Some laboratories may also heat overnight under reflux to homogenize the mixture or dry the mixture and re-dissolve it in nitric acid.
The analyte in the sample and the spike should be in the same form or species to reach an isotopic equilibrium. For example, in case of Pu ID-MS measurements, the Pu contained in the spike and the Pu coming from the sample may not have the same oxidation state due to the multiple possible redox states of Pu in aqueous solution (+III, +IV, +V, +VI). The Pu in the mixture may be discriminated if a redox cycle is not performed before a separation step to adjust the Pu to a unique valency. Different redox reagents can be used: hydrogen peroxide, an iron II solution, sodium nitrite, potassium metabisulfite or titanium(III) chloride.89–93 The optimum time to reach the Pu valency adjustment is variable: the Fe(II)/NaNO2 method requires a waiting time of 20 min whereas the hydrogen peroxide requires a minimum waiting time of 2 h.58
4. Uncertainty evaluation
4.1. Weighing uncertainty
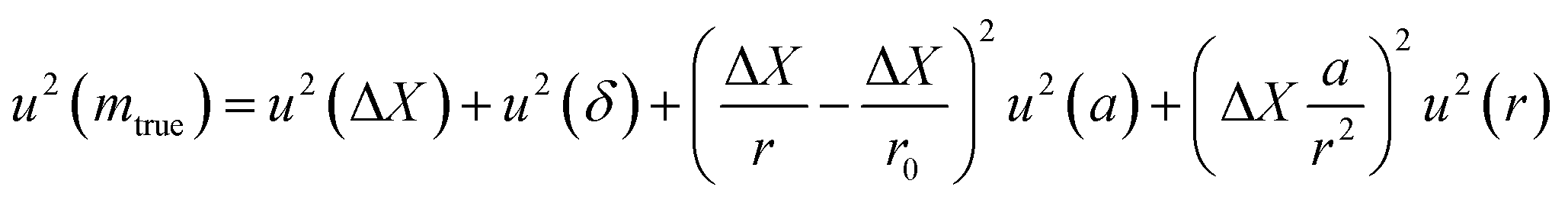 | (26) |
The term u2(ΔX), described as the weighing result component, depends on parameters such as the resolution of the balance and the repeatability of the measurement. The term u2(δ) is the trueness component, which is also dependent on the resolution and repeatability of the balance, but also on the uncertainty of the standard used for calibration and the uncertainty of its durability. It also takes into account (i) the effect of the air density variation, (ii) the effect of temperature variation and (iii) the effect of eccentricity on the trueness of measurement during calibration.72 The final terms of the equation, 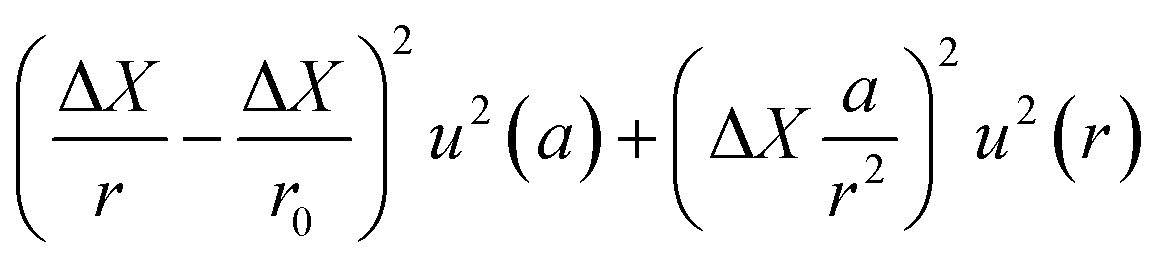 , concern the uncertainty on the corrections for air buoyancy and the effects of variation in air density and that of the sample density under the measurement conditions. It is evident that, even for one measurement, evaluating the uncertainty of a weighing is quite tedious and time consuming.
, concern the uncertainty on the corrections for air buoyancy and the effects of variation in air density and that of the sample density under the measurement conditions. It is evident that, even for one measurement, evaluating the uncertainty of a weighing is quite tedious and time consuming.
A metrological confirmation of the balance is then carried out (calibration + verification) to ensure the following inequality after calibration of the balance (eqn (27)).
| |δ| + 2u(δ) ≤ MPE(ΔX) | (27) |
If this condition is respected over the entire considered mass range, the balance is metrologically confirmed and the MPE can be used for the expression of the weighing uncertainty in a simpler manner.
Assuming that the statistical distribution of weighing measurements follows a continuous uniform law with a span of 2 MPE, the standard uncertainty equals half the span of the distribution (MPE) divided by the square root of 3 (eqn (28)).
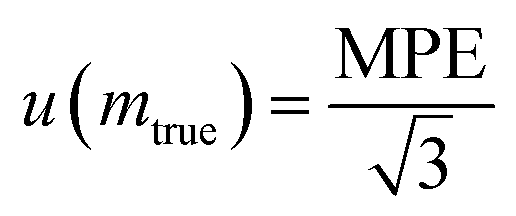 | (28) |
For example, a class 1 balance having a range of 200 g and a resolution of 0.1 mg would traditionally be assigned an MPE of 1 mg, resulting in a standard uncertainty of 0.6 mg according to eqn (28). The mass weight of 0.5 g would then be associated with an expanded uncertainty of 1.2 mg at a confidence level of 95%, corresponding to a relative uncertainty of 0.24%. For such a balance, in order to guarantee a weighing uncertainty on the order of 0.1%,69 it is recommended to weigh only masses greater than 1 g.
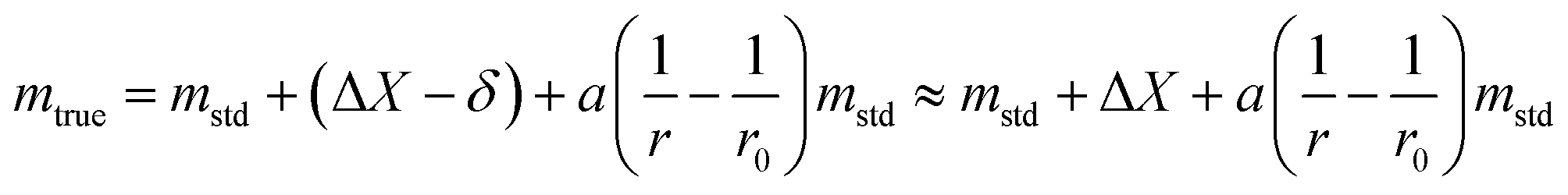 | (29) |
The uncertainty of mtrue (eqn (30)) is derived from eqn (29).
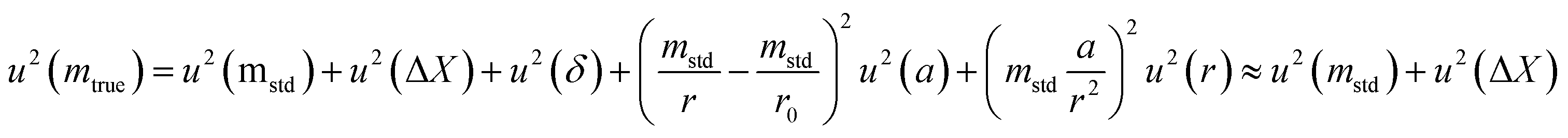 | (30) |
This method is not sensitive to balance drift if weighing measurements are spaced regularly over time and if the drift is linear. The trueness error δ is then reduced drastically, because ΔX – the difference between the sample and the standard mass – is very small. The trueness error can then be neglected, as well as its uncertainty contribution when calculating the weighing uncertainty. In practice, only the contributions from standard mass uncertainty and from repeatability are retained, and hence very low uncertainties on the order of tens of μg can be achieved.73,74
4.2. Isotope dilution uncertainty
The various ways of estimating measurement uncertainty can be divided into two main categories: inter-laboratory and intra-laboratory approaches.5 The result of an ID-MS measurement (i.e. ws) can be expressed as a function of various parameters which makes it well suited to be used as an intra-laboratory approach with measurement modelling. Uncertainty estimation of the measurement model must be taken into account, at all the stages of the measurement process and for all input parameters coming from certified values or measured values. Here, we apply this approach to an example: the measurement result for the U mass fraction from CEA/LAAT during the 2022 “Nuclear Material Round Robin” (NMRoRo) ILCs organized by the IAEA. For this measurement LAAT used the IRMM-054 spike, which was first diluted in nitric acid to obtain a known mass fraction of approximately 10 μg g−1. The NMRoRo ILC sample (a uranium oxide powder) was dissolved, after which the solution was diluted three times consecutively to produce a diluted sample. Three {diluted sample–spike} mixtures were gravimetrically prepared. The measurement model is reported in eqn (31). The different steps of spike dilution, sample dissolution and dilutions have been taken into account and only measured or certified data are used as inputs.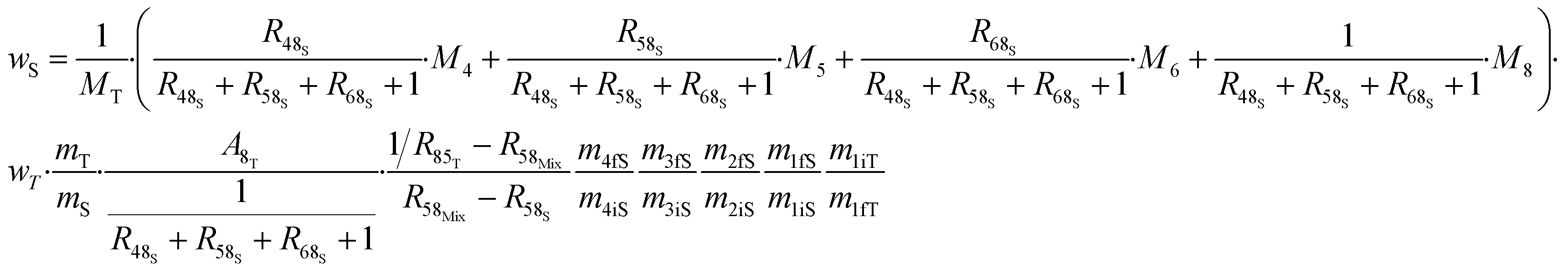 | (31) |
From the measurement process model, the uncertainty is estimated by propagating the uncertainty of the input data. This can be done using the Monte Carlo method (MCM), as recommended in supplement 1 of the GUM.96 This approach requires no assumptions to be made about the distribution of the result, nor about any correlation between the input parameters. However, for each of the input parameters in the NMRoRo example, a particular distribution needs to be chosen, a most probable value and a standard deviation are required before performing the Monte Carlo simulation on the model (eqn (31)).
Using the information provided by the LAAT about the parameter values and their standard uncertainties, and assuming a uniform distribution for each of the masses and a normal distribution for the other parameters, a standard measurement uncertainty of 0.15%§ (probability density function (pdf) of ws in Fig. 7) is obtained.
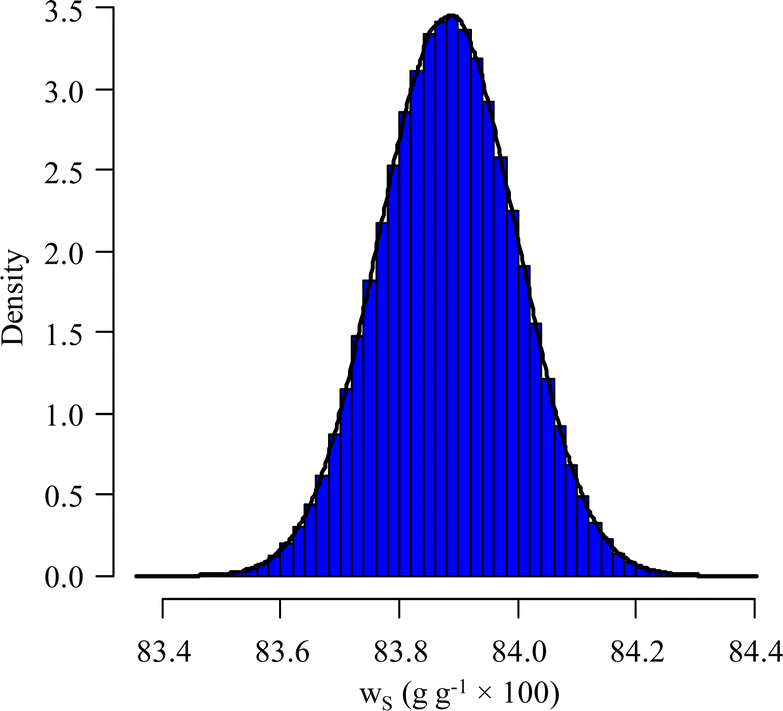 | ||
| Fig. 7 Distribution law of ws by MCM – LAAT measurement on the NMRoRo 2022 sample. | ||
The simulated pdf of wS is not significantly different from a normal law. The uncertainty budget can be established using the sensitivity indices (first-order and total Sobol indices):97 the budget obtained for this example is illustrated in Fig. 8.
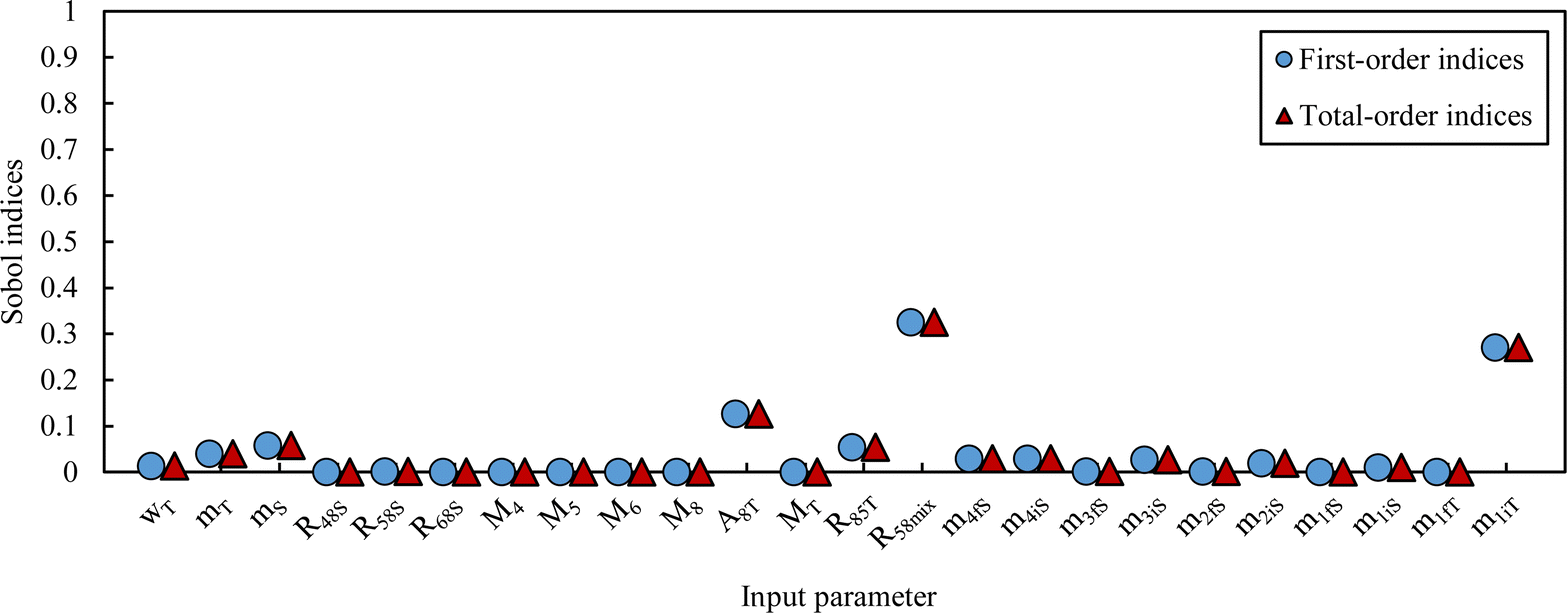 | ||
| Fig. 8 Sobol indices – LAAT measurement on the NMRoRo 2022 sample. | ||
The contributions to the uncertainty of parameters R48S, R58S, R68S, M4, M5, M6,M8, and MT are not significant and therefore the uncertainty calculation can be simplified here by treating these inputs as constants. Although some contributions from masses weighed during dissolution or dilution are small, they have been retained in order to maintain the overall dissolution/dilution process, the total contribution of which is not negligible. The measurement model (eqn (31)) then becomes a simplified model (eqn (32)).
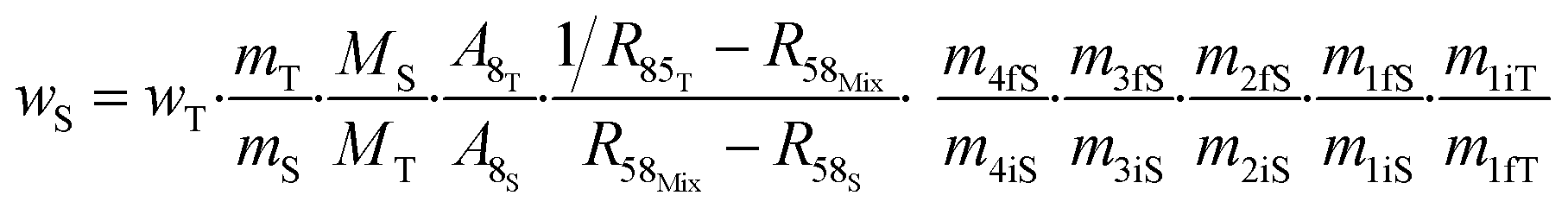 | (32) |
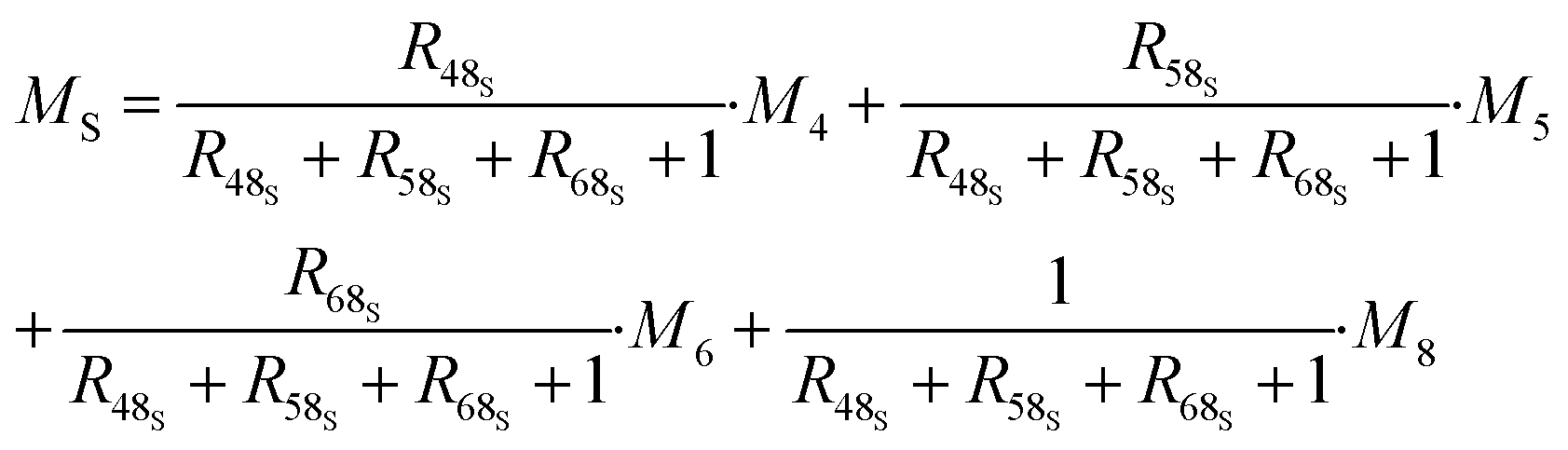 | (33) |
From this equation, we can conclude that (1) the result distribution is not significantly different from a normal distribution; (2) several influencing parameters contribute to the uncertainty, and (3) the first-order and total Sobol indices are very similar, meaning there is no correlation between the influencing parameters (Fig. 8). These findings mean that, if desired, the GUM approach98 (quadratic cumulation method) without covariance can also be applied to the simplified model (eqn (32)). This approach leads to the same standard uncertainty on wS as the MCM approach, i.e. 0.15%. The similarity between uncertainty budgets estimated with the MCM (recommended in GUM-S1) and by the quadratic cumulation method (recommended in GUM) for the NMRoRo sample at LAAT is illustrated in Fig. 9. For this measurement, the main contributors to the uncertainty are R58Mix (33%), m1iT (28%), A8T (13%), mS (6%), R85T (5%), and mT (4%).
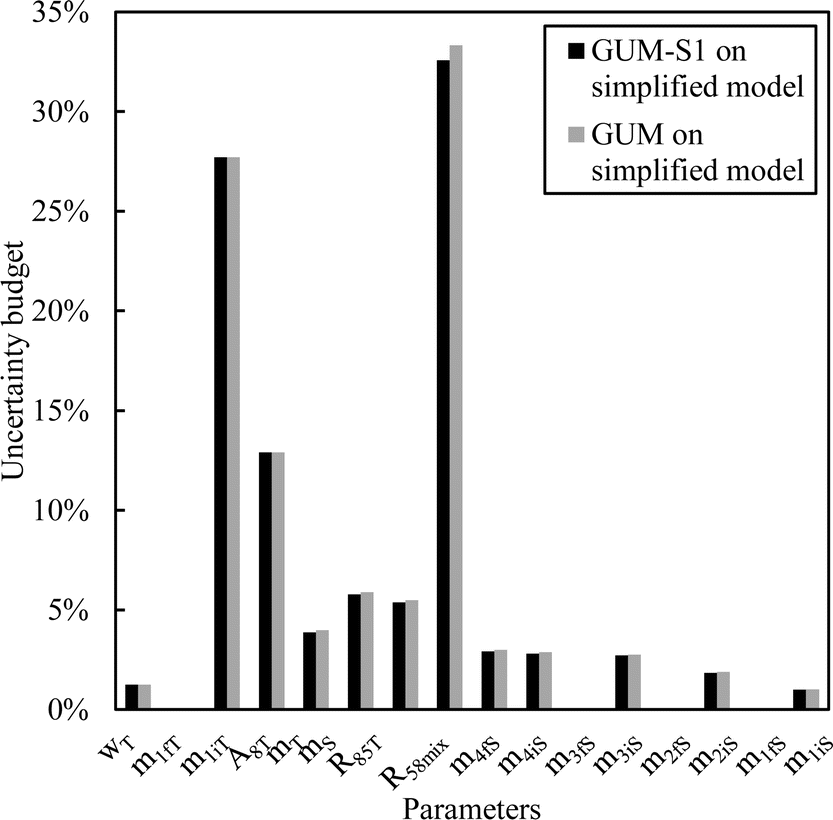 | ||
| Fig. 9 Sensitivity indices obtained by the MCM or quadratic cumulation method – LAAT measurement on the NMRoRo 2022 sample. | ||
In conclusion, the measurement uncertainty of ID-MS is estimated in several stages:
(1) The measurement process is modelled on the basis of eqn (31) or taking into account all the stages in the process (dissolution, dilution, etc.) and using input parameters with certified values or measured values (avoiding input parameters that are mathematical expressions).
(2) The propagation of the uncertainty by using the MCM, preferably, and assess the uncertainty budget (sensitivity indices).
(3) If needed or desired, simplify the measurement model by reducing the number of variables and considering other parameters as constants.
(4) If needed or desired, in the case where (i) the result distribution does not differ significantly from a normal distribution, (ii) more than one parameter contributes to the uncertainty, and (iii) the first-order and total Sobol indices are very similar, we can apply the quadratic cumulation method (GUM) without covariance terms to the model.
Note: As calculations are easy and quick with R,99 we recommend using the MCM approach (GUM-S1).
5. Application for radionuclide measurement
5.1. Environmental applications
5.1.1.1. U-series disequilibrium. The ID-MS technique plays a key role in the U-series disequilibrium studies in environmental and Earth sciences. U-series geochemistry provides a profound understanding of fundamental processes in widespread disciplines as diverse as geomorphology,100 weathering and erosion of geological formations,101 oceanography,102 and environmental pollution science.103,104 A wide range of U-series geochemistry applications to the Earth sciences was thoroughly summarized in “Uranium-series Disequilibrium, Applications to Earth, Marine and Environmental Sciences”.105 The uranium and thorium decay-series contain radioactive isotopes of many elements (in particular, 234, 235, 238U, 230, 232Th, 231Pa, 226Ra and 227Ac) with varied physicochemical properties leading to a fractionation (mainly elemental fractionation) within the chain in different geological environments during geochemical processes. The accurate measurement of these radionuclides and more specifically their elemental ratios (i.e., 230Th/238U, 238U/232Th, 231Pa/235U, 226Ra/230Th) allows the investigation of processes occurring on time scales from a hundred years to ∼600
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years. To this end, radionuclide concentrations are accurately determined in environmental samples by ID-MS. It is worth mentioning that isotopic compositions (i.e., 234U/238U and 230Th/232Th) are also mandatory for disequilibrium studies, however this topic will not be addressed in this section, as it does not fall under the scope of ID-MS.
000 years. To this end, radionuclide concentrations are accurately determined in environmental samples by ID-MS. It is worth mentioning that isotopic compositions (i.e., 234U/238U and 230Th/232Th) are also mandatory for disequilibrium studies, however this topic will not be addressed in this section, as it does not fall under the scope of ID-MS.
Sample preparation is a crucial step that strongly influences the quality of analytical results. Generally, natural solid samples undergo total dissolution in Teflon bombs or beakers. The digestion protocol is adapted according to the sample matrix. Usually for rocks, sediments and soils, concentrated HNO3, HF, or HClO4 (ref. 106 and 107) or alkaline fusion108 is used. After sample digestion, separation protocols for U/Th,106,109–114 Ra,111,112,115–121 Pa111,113,122,123 and Ac124,125 extraction are used. An exhaustive review for Ra separation protocols can be found in Boudias et al.126 Radionuclide separation helps concentrating the radionuclides of interest, minimizing matrix effects and reducing or removing polyatomic interferences.
For the determination of U concentrations in natural samples two kinds of spikes are commonly used: a single spike (233U or 236U) or a double spike (233U–236U) certified in both concentration and isotope ratio. The 233U–236U double spike method was first introduced by Dietz et al.127 and thanks to this technique it is possible to proceed to ID by internally correcting the instrumental mass fractionation using the certified ratio 233U/236U (i.e. to correct the 238U/236U ratio).106,128,129 If a single isotope is used for spiking, the standard-bracketing method should be employed to externally correct for the instrumental mass fractionation or the total evaporation method may be employed in the case of TIMS measurement to overcome the isotope fractionation.37,130 With a new generation multi-collector-ICP-MS (MC-ICP-MS) equipped with high-efficiency desolvation systems, it is possible to achieve LODs of ∼10−7 and ∼10−8 ng g−1 for 233U and 236U, respectively.131
The 230Th dating (based on the radioactive decay: 238U ⇒ 234U ⇒ 230Th) method has become one of the most important dating methods for quaternary research,132 radiocarbon calibration133 and also for weathering and erosion studies.134 A major source of uncertainty for the 230Th/238U ratio is the accurate measurement of the amount of 230Th. More commonly, 230Th is measured by using an isotopically enriched 229Th material as a spike.135–138 This 229Th spike can be an “in-house” standard or certified 229Th standards such as the standard reference material (SRM) 4328C139,140 and reference materials provided by the National Physical Laboratory (NPL)141 or the Eckert and Ziegler company. However, to use an “in-house” spike, the analyst must expend considerable effort in producing, calibrating, and, if possible, establishing traceability. Recently, a new high purity 229Th (230Th/229Th = 5.18 × 10−5 and 232Th/229Th = 3.815 × 10−4) reference solution was commercialized by the National Institute of Standards and Technology (NIST).36 This spike is ideal for measuring samples with very low 230, 232Th content (i.e. young carbonates, etc.). For the correction of the instrumental mass fractionation on the 230Th/229Th or 232Th/229Th (for 232Th determination) ratios, the IRMM-035 CRM142 and the standard-bracketing method are used. Thanks to the double isotope dilution method (eqn (6)), the 230Th/238U concentration ratio can also be accurately determined by using a double (229Th–236U or 233U) or a triple spike (229Th–223U–236U). A triple spike was used in U migration studies in marine sediments143 and for environment pollution purposes.103 The LODs of 230Th for seawater samples are close to 0.15 fg kg−1.144 For analytical purposes, it is preferable in certain cases to measure the 230Th/235U ratio (as 230Th is closer in terms of abundance to 235U compared to 238U), and subsequently calculate the 230Th/238U ratio by taking into account the natural 238U/235U ratio (137.818 ± 0.045).145
For 226Ra determination in natural samples, a spike such as 228Ra is used. It is worth mentioning that the natural 228Ra contribution is considered negligible due to its very low natural abundance and its very short half-life (t1/2 = 5.75 years146) compared to that of 226Ra (t1/2 = 1600 years146). However, in some specific cases for solid samples containing a high concentration of 232Th, a pre-analysis before spiking for the determination of the 228Ra/226Ra ratio is mandatory to correct for the contribution of natural 228Ra. The 228Ra spike is commercially provided (i.e. NIST-SRM 4339B, NPL, etc.), but generally in geosciences, an enriched 228Ra spike is prepared by milking the Ra fraction from an old pure thorium nitrate material (i.e. Th(NO3)4). The 228Ra fraction can be used directly after its separation from 232Th120,121,147 or the 232Th solution is purified from Ra (226,228Ra) and then the purified thorium solution is set aside, and 228Ra grows in through 232Th decay.148 As 226Ra (impurities of 230Th ⇒ 226Ra) grows much more slowly than 228Ra, at the beginning of 232Th decay (some months or years) a high 228Ra/226Ra ratio suitable for ID-MS can be obtained. The 228Ra spike concentrations were calibrated against certified 226Ra solutions (i.e. provided by NIST and NPL) or by using the Harwell Uraninite (HU-1)119 that is known to be close to secular equilibrium.128 Typical values of LOD of 226Ra are close to ∼0.1 fg g−1 (3 mBq kg−1).149,150
The 231Pa/235U radiochronometer is complementary to that of 230Th/238U and can be used to obtain more reliable age estimates, especially for samples with complex histories. Moreover, the 231Pa/230Th ratio is a very useful paleoproductivity and scavenging ocean tracer.151,152 The measurement of 231Pa concentration is performed by ID-MS with 233Pa as the spike isotope.153,154 Due to the short half-life of 233Pa (t1/2 = 26.98 days155), no certified 233Pa spike exists. Rather, the 233Pa spike is prepared by milking the Pa fraction from a 237Np solution immediately prior to use and calibrated for 233Pa concentration for its working-lifetime of approximately 3–4 months. The 233Pa spike is calibrated using commercially available156 or “in-house” 231Pa standard solutions calibrated against the HU-1. Recent developments allow the measurement of quantities as low as 0.1 fg of 231Pa by MC-ICP-MS.102,157 TIMS protocols for 231Pa analysis allow the determination of as little as a few tens of fg to a few ag.153,158 Moreover, high resolution ICP-MS (HR-ICP-MS) reported LOD values were close to 0.02 fg kg−1.144
Recently, the analysis of 227Ac by ID-MS was developed for nuclear forensic purposes.125 However, this technique has found important applications in environmental studies mainly in oceanography.124 As there is no commercial spike for Ac, the method for isotope dilution measurement of 227Ac uses a 229Th standard to provide 225Ac, which is used as the spike. 227Ac determination by MC-ICP-MS in seawater makes reliable measurements with 10 L samples with 227Ac concentrations of ∼2 ag kg−1 with a LOD of around 0.7 ag kg−1.124
5.1.1.2. Other radiogenic elements: 87Rb–87Sr method. The 87Rb–87Sr method stands out as one of the earliest dating techniques in geology. It has been widely used, particularly since the introduction of the Sr evolution diagram which enabled the determination of both the age and initial 87Sr/86Sr ratio for sets of co-genetic samples.159,160 Contrary to U-series applications where non-naturally occurring tracers could be used, in this case isotopically enriched tracers of natural isotopes (e.g., 84Sr or 86Sr, 87Rb at purities higher than 96%) are commonly used.161 It should be noted that before analysis, Rb and Sr are chemically separated to avoid isobaric interference at m/z 87.
A good candidate spike for ID-MS has a half-life large enough to limit the activity handled, be readily available and contain few isotopic impurities. If the isotopic composition of the radioelement that is suspected to be present as a contaminant in the environmental sample is not known, it is important to ensure that the radioactive tracer added for ID is not already present in the sample. In such a case, the measured elemental concentration of the radioelement will be wrong. If suspected, an analysis without the addition of the spike is mandatory to correct for the presence of the isotope used as an ID spike in the environmental sample. If ID-MS is done with an isotope naturally present in the environmental sample, an analysis of the sample without and with the spike needs to be done. In any case, the purity of the spike has to be verified before use, to check that it does not contain the isotopes of interest in the sample, and if present to carry out the appropriate corrections. In many cases, the isotope composition of the spike, including impurities, is mentioned in the certificate. Otherwise, abundances of isotope impurities must be quantified with an appropriate technique, for instance by direct measurement of a significant amount of spike with the most accurate mass spectrometer available.
In the following, we describe the implementation of ID to the most common radioelements that can be measured by mass spectrometry and that may be the subject of environmental monitoring of nuclear facilities.
5.1.2.1. Uranium. The isotopes of interest for the survey of uranium in the environment are 234U, 235U, 236U and 238U. The measurement of the abundances of these isotopes (or isotope ratios with respect to 238U) helps to distinguish an anthropogenic contribution from naturally occurring uranium. In particular, the abundance of 235U is characteristic of the industrial origin of uranium and shows a significant difference with respect to that of natural uranium. Indeed, the abundance of 235U in man-modified uranium ranges from ∼0.2% in depleted U to more than 20% for highly enriched U, reaching abundances higher than 90% in nuclear weapons.165 By contrast, 235U abundance in naturally occurring uranium shows very small variations around an average value of 0.725% (isotopic abundance). The 236U isotope is also an excellent fingerprint of anthropogenic nuclear activities. Indeed, this isotope does not exist in measurable abundance in the environment (236U/238U < 10−12), but it is produced in a nuclear reactor through neutron capture by 235U nuclei. For instance, 236U/238U isotope ratios can reach 10−3 in spent fuel. So the detection of a significant abundance of 236U by mass spectrometry is also characteristic of environmental marking through nuclear activities.166 As a consequence, use of 236U as an ID spike is not recommended when the presence of irradiated uranium is suspected. By contrast, 233U is generally not present in nuclear materials (except in a few cases, for instance when 232Th is irradiated) and so is absent in environmental samples. Thus, the spike recommended for ID-MS is 233U.162 It can be provided with a good purity by NPL or JRC (IRMM-057) for example. Mass bias factor correction, and gain calibration of the detectors in the case of measurements with a multiple-collector mass spectrometer, can be performed with certified reference materials whose isotope ratios are certified (for example NBL CRM U030-A or IRMM-186).
5.1.2.2. Plutonium. Plutonium isotopes are produced in nuclear reactors through successive neutron captures: 239Pu by capture of a neutron by a 238U nuclei, 240Pu by capture of a neutron by a 239Pu nuclei, etc. The most common plutonium isotopes measured in environmental samples by ID-MS are 239Pu, 240Pu, 241Pu and 242Pu. It should be mentioned that extremely low concentrations of plutonium are present in soil samples, although they are measurable by combining thorough radiochemical separations with the most sensitive mass spectrometry techniques.167 This is a worldwide marking which originates from the global fallouts of thermonuclear atmospheric nuclear weapons tests. So quantification of plutonium is not sufficient to identify a recent, man-made origin: measurement of isotope ratios is mandatory for that. Indeed, isotope ratio compositions of plutonium from global fallout and from various nuclear activities (separation of military or civilian plutonium, reprocessing, etc.) show significant differences. For example, the 240Pu/239Pu and 241Pu/239Pu atomic ratios of the global fallout of atmospheric nuclear weapons tests are respectively 0.182 and 9.7 × 10−4,168 whereas the ratio of 240Pu/239Pu in environmental samples from nuclear weapons test sites at the atolls of Mururoa is 0.018 ± 0.005 (at./at. ±2σ).169 In the fuel cycle facilities, isotope ratios of Pu are slightly different. In the pressured water reactor using oxide enriched uranium fuel, the isotope ratios of 240Pu/239Pu and 241Pu/239Pu are around 0.4 and 0.2 (wt/wt) respectively, and in MOX fuel the ratios are around 0.9 and 0.4 (wt/wt) respectively.170 The 240Pu/239Pu ratio is the easiest ratio to measure in the environment as 239Pu and 240Pu are generally the most abundant isotopes. 241Pu is a short-lived isotope (t1/2 = 14.325 years171) whose abundance can be correlated with that of its decay product 241Am.172 However, 238Pu is generally measured by alpha spectrometry because of the non-negligible interference of 238U at the same mass and its relatively short radioactive period (t1/2 = 87.74 years173). The spike commonly used for ID-MS is 242Pu in water163 and solid matrices,172 but 244Pu can also be chosen if 242Pu is expected in the sample.163,174 A 242Pu spike can be provided with a very high purity by NIST (SRM 4334) or JRC (IRMM-049) and a 244Pu spike by IRMM (IRMM-042a). 242Pu can also be used as a spike for alpha spectrometry measurements.164 Mass bias can be corrected with a 235U/238U certified ratio.
5.1.2.3. Americium. Traditionally, 241Am (t1/2 = 432.6 years146) was measured by alpha spectrometry. In the past few decades, ICP-MS has been more and more widely used as a fast and highly sensitive technique for the measurement of 241Am.175 Indeed, new generations of ICP-MS/MS are more sensitive and help to better remove the interferences. For example, for 1 g of ash sediment sample, the measurement of 241Am with an ICP-MS/MS coupled with the APEX Ω sample introduction system and the addition of O2/He gas to remove the 241Pu interference, allows a LOD of 241Am of 20 mBq kg−1 to be reached.175 The most suitable long-lived americium isotope suitable for the determination of the 241Am mass fraction in environmental samples by ID-MS is 243Am (t1/2 = 7370 years146).176 The 243Am spike can be provided by NPL (A14063), CETAMA (STAM CRM) or JRC (IRMM-0243) with good purity.56
5.1.2.4. Technetium-99. 99Tc is a long-life fission product (t1/2 = 2.115 × 105 years146). For 99Tc, the best candidates for ID-MS are 97Tc (t1/2 = 2.6 × 106 years177) and 98Tc (t1/2 = 4.2 × 106 years178) due to their long half-life. Unfortunately, they are not commercially available.179 However, natural 185,187Re is commonly used, as an analog of Tc, to estimate the radiochemical recovery, or the short-lived 99mTc (t1/2 = 6.00660 h180) measured by gamma spectrometry. Another point to consider is that interferences such as 98,99,100Ru and 97,98,100Mo may still be present even after a high decontamination factor of the sample. Thus, even with the use of 97Tc for ID-MS, interferences have to be corrected by calculation or eliminated by the introduction of an appropriate gas into the collision–reaction cell of an ICP-MS. For instance, Yang et al. implemented an ICP-MS/MS with O2, as a reaction gas, to remove Mo.179
5.1.2.5. Radio-cesium. The 135Cs/137Cs ratio is a new relevant anthropogenic signature in environmental samples that emerged after the Fukushima accident, thanks to the new possibilities offered by ICP-MS/MS. Both are fission products with half-lives of t1/2(135Cs) = 2.3 × 106 years and t1/2(137Cs) = 30.018 years.146 The 135Cs/137Cs ratio is useful for identifying the origin of radio-cesium. It can be determined after a high purification of the sample and the use of gas in the collision–reaction cell for ICP-MS measurement.28,29 However, no radioactive spike is available for ID-MS and no certified solutions of the 135Cs/137Cs ratio are available commercially to correct the mass bias effect. Then, the chemical yield can be estimated by gamma measurement of the intrinsic 137Cs of the sample before and after purification of the sample as an internal isotope dilution standard. Moreover, a solution qualified by a more accurate method (TIMS for example) can be used to correct for the mass bias effect. In addition, reference materials such as IAEA-330, IAEA-375 and IAEA-385 can be analyzed to validate the analysis.28,29
5.1.2.6. Iodine-129. Recent publications present the possibility of measuring 129I, a very long-life decay product (t1/2 = 1.614 × 107 years181), by mass spectrometry in environmental samples with a lower LOD than γ-spectrometry.182 However, even if its long half-life is favorable for MS measurement, its volatility and the numerous interferences can be challenging. Also, an important memory effect of I in ICP-MS has been reported.183 The quantification of 129I is usually based on the isotopic dilution technique using stable 127I as an internal standard and the mass shift mode of the ICP-MS/MS. The initial amount of 127I is then quantified with an external calibration.182 The use of 125I present in certified NIST SRMs 4407H and 4407L is also mentioned to quantify 129I by ID because the abundance of 129I in the CRMs is negligible.183
5.1.2.7. Neptunium-237. The measurement of Np by ID-MS is difficult. In radioactive samples originating from the nuclear fuel cycle, the 237Np isotope is the only isotope of Np with a long half-life. The only other isotope of Np with a long half-life is the 236Np isotope (t1/2 = 1.55 × 105 years146) which might permit the determination of 237Np by ID-MS. However, 236Np is not commercially available and its production is difficult.184,185 If a laboratory happens to have a 236Np solution, it is possible to create an in-house spike by measuring the 236Np amount fraction by ID-MS using a commercially available 237Np material. The in-house 236Np spike can then be used to measure the 237Np in a sample by ID-MS.186 In the absence of a 236Np spike, a 242Pu spike can be used to measure the 237Np by ID-MS.184,187 In that case, measurements must be made by ICP-MS, and not by TIMS, due to the different behaviors to sublimation between Pu and Np during the measurements.
5.2. Nuclear forensics
In the framework of nuclear forensic investigations, seized materials are characterized as completely as possible by complementary techniques, to carry out physical characterization (size and weight) and determine the chemical, elemental, and isotopic compositions, as well as concentrations of non-radioactive and radioactive impurities. It is the combination of all these parameters that provides clues to determine the origin or the processing history of the nuclear material intercepted outside regulatory control.188–190 Of all these analyses, age dating measurement gives an indication of the time elapsed since a nuclear material last underwent chemical purification. The date of the purification (also called “model age”) is obtained by measuring the ratio between a nuclide and its daughter product. The “model age” is calculated by using the equation derived from Bateman's equations (eqn (34)). This equation is valid provided that all the daughter products have been removed during the last chemical operation on the material (at the production stage), and the system has been kept closed ever since (no contamination, no elemental segregation).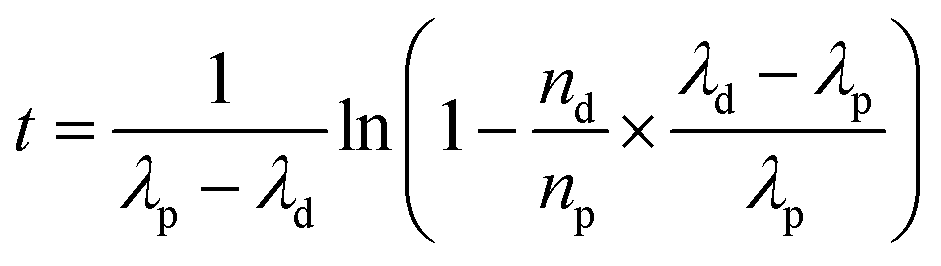 | (34) |
This nd/np ratio is derived from the concentration measurement of each nuclide in the nuclear material. The accuracy of the age calculation depends directly on the accuracy of these concentrations and the accuracy of the half-lives involved in the calculation. To this end, ID-MS is the method that can achieve the best results, provided there is rigor in the choice of the isotopic spikes, in the chemical purification of the material and in the mass spectrometric measurements. The uncertainties of the half-lives of the parent nuclides and of the daughter products (mainly 234U–230Th, 235U–231Pa, 234U–238Pu, 235U–239Pu, 236U–240Pu, 238U–242Pu and 241Pu–241Am) are very low (between less than 0.01% and 0.4% (for 231Pa)) and their contribution to the global uncertainty is generally negligible.
In the preceding decade, there has been a large international effort made to improve the methods for uranium age dating measurements, through the development of reference materials or round robin exercises.8,10,191,192 Uranium has two commonly used chronometers: 230Th/234U and 231Pa/235U. To quantify the different uranium isotopes, most laboratories use a 233U spike. For 230Th quantification, a spike enriched in the 229Th isotope can be used.36 The method used to calibrate an ID spike can influence the uncertainty. For example, 229Th spikes are often calibrated by means of alpha spectrometry, which leads to a large relative uncertainty (>1%) in the determination of the 230Th concentration and the necessity to convert the certified 229Th specific activity to molality using the 229Th half-life. Bias in this half-life value will affect the accuracy of the results. Production of a new 229Th reference material certified by mass spectrometric measurements36 in combination with the publication of coherent values for 229Th half-life193–196 have enabled the development of uranium age dating with the 230Th–234U chronometer.
The 231Pa–235U chronometer is not as widely used. One of the main reasons for this is the difficulty in obtaining a calibrated isotopic dilution spike. 233Pa is obtained by milking a 237Np solution. The short half-life of 233Pa (t1/2 = 26.98 days155) makes it difficult to have a commercially available certified spike. Laboratories must prepare it, when required, for immediate use. The 233Pa solution can be calibrated by various methods: gamma measurement, ID-MS with a rock at secular equilibrium,197 ID-MS with a 231Pa solution or after decay of the 233Pa, by measuring the concentration of the decay product (233U).198233Pa and its decay product (233U) are isobars. The chemical purification has to be optimized to eliminate U from the Pa fraction, and the Pa measurements have to be performed immediately after purification. If the measurement is delayed, a correction for ingrowth of 233U has to be included in the calculation, which increases the measurement uncertainty.
As illustrated with Pa, the decay of the spike is important for age dating measurement. This is also the case for thorium, where 229Th is the daughter product of 233U. If 233U is used as a spike for U ID-MS, the aliquot for Th ID-MS must be prepared separately.
Another difficulty with U age dating is the variability in the atomic abundance of the parent nuclide compared to its daughter product (Fig. 10). The amount of sample to be analyzed has to be carefully defined before spiking the different aliquots and starting the analyses. The laboratory environment must be considered. The use of a large amount of spike or sample will affect the laboratory and instrumental background, thereby leading to biases in the measurement. When the chemical purification is optimized and if the instrument is very sensitive, it is possible to date small masses of nuclear materials,9 thereby economizing on reagents and the amount of precious spike.
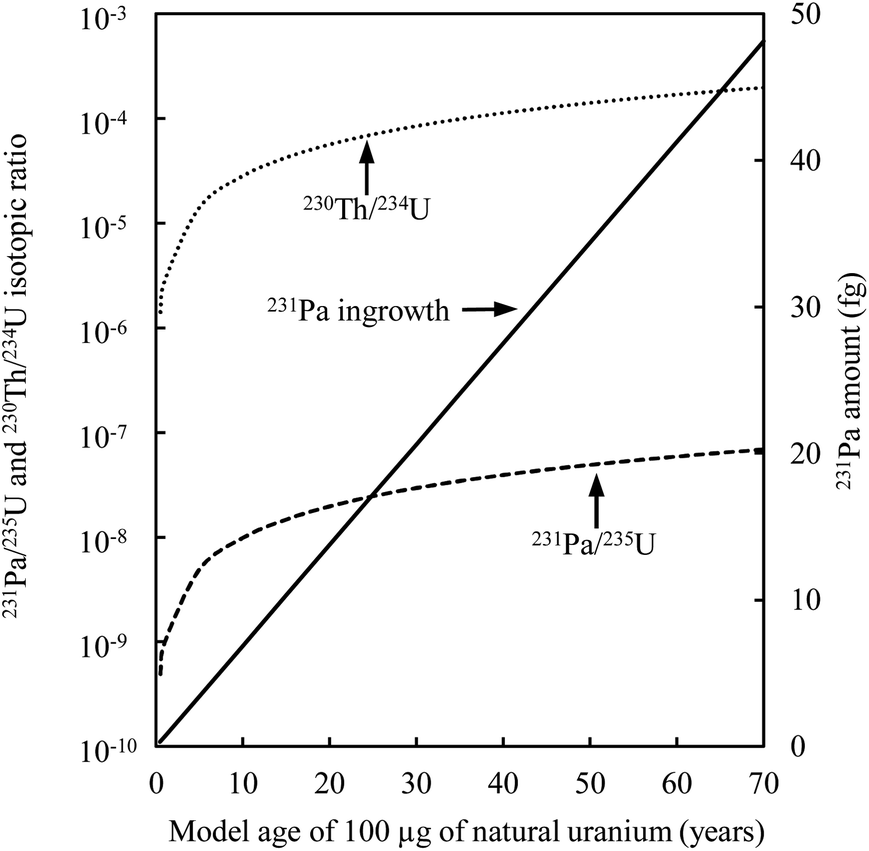 | ||
| Fig. 10 Temporal evolution of the daughter/parent isotopic ratio for 100 μg of natural uranium. The dotted line is for the 230Th–234U chronometer, dashed line is for the 231Pa–235U chronometer and solid line is for the ingrowth of 231Pa in fg. | ||
It should be noted that, in the case of plutonium, although five radio-chronometers are potentially measurable, 234U–238Pu, 235U–239Pu, 236U–240Pu, 241Am–241Pu and 238U–242Pu, only three different spikes are required, 233U for U ID-MS, 244Pu for Pu ID-MS and 243Am for Am ID-MS. The use of the 244Pu isotope enables one to determine the concentration of all the Pu isotopes from 239Pu to 242Pu. Due to the isobaric interference at mass 238 from 238U, it is preferable to measure the 238Pu by alpha spectrometry. Also simultaneous measurements of 238U and 238Pu are reportedly possible by TIMS.199,200 The impact of the minor isotopes in the 244Pu spike depends on the composition of such spike. A new 244Pu reference material has been released recently,43 which is really pure (the main impurity is 242Pu with 242Pu/244Pu < 10−4). With such spike the ID-MS has no impact on the spike's minor Pu isotopes on the final uncertainties. If the level of minor isotopes is around the percent level, it is then recommended to do an independent isotopic measurement of the material, and to use ID-MS to quantify only the major Pu isotope.
For completeness, it should also be mentioned that granddaughter–parent chronometers (such as 226Ra–234U and 227Ac–235U) can be used for age dating determination. However, this is not very common.125
5.3. Safeguards
Destructive analysis of safeguards samples is used to independently verify fuel cycle facility operators' nuclear material accountancy declarations and the quality of the operator's measurement systems. ID-MS is one of the most accurate analytical technique to determine the uranium/plutonium content in safeguards samples. Furthermore, the ITVs for measurement uncertainties in safeguarding nuclear materials provide state-of-practice uncertainty limits for ID-MS measurements.24,25 The safeguards laboratories of the IAEA and the JRC-Karlsruhe use ID-MS for analysis of nuclear safeguards samples. These laboratories use ID-MS for the production of reference materials, for external verification activities in the production of LSD spikes,84,201 for the verification of spike solutions and in-house working standards, as well as for the preparation of reference solutions that are needed for the calibration of instruments.LSD spikes are used for fissile material control of irradiated nuclear spent fuel by operators, as well as regulatory and safeguards authorities, for instance the IAEA safeguards laboratories at the Rokkasho On-Site laboratory (OSL) and at the EURATOM OSL in the Orano nuclear fuel reprocessing facilities at La Hague. LSD spikes are provided by JRC-Geel,84,201 the IAEA86 and the Japan Atomic Energy Agency (JAEA)202,203 and they are typically prepared from Pu and U metal CRMs. They are verified using independent analysis techniques and ID-MS. LSD spikes typically contain 2 mg Pu (90–98 wt% 239Pu) and 10–50 mg of enriched uranium (20–93 wt% 235U). The IAEA LSD spikes are prepared as glassy dried nitrates deposited on the bottom of penicillin vials. The JRC-Geel IRMM-1027 series are commercially available84 and they are also prepared as dried nitrates deposited on the bottom of penicillin vials, although a stabilizing matrix is used to improve the mechanical stability of the spike, carboxymethyl cellulose sodium salt (CMC) or cellulose acetate butyrate (CAB).73,204,205 The stability of spikes, which can be affected by transportation or storage conditions in the OSLs, is essential for the quality of the ID-MS analysis. In analogy to the IRMM-1027 spikes,206 the IAEA is investigating the use of CMC to create foam-like LSD-type spike materials to increase stability and extend the life-time of the LSD spikes. For quality control and instrument calibrations for primary assay measurement techniques applied in the Rokkasho OSL (HKED or Spectrophotometry), with the current status of operations of a reprocessing facility, the Office of Safeguards Analytical Services Nuclear Material Laboratory (SGAS-NML) prepares approximately 500 units of LSD spike vials every two years. For analysis of highly active liquid waste materials, small size 233U and 242Pu spikes are used containing mg amounts of U and μg amounts of 242Pu.
In SGAS-NML and in the safeguards laboratory at the JRC-Karlsruhe there is a wider range of spike materials available for analysis of safeguards samples. At the SGAS-NML milligram-sized dried U/Pu samples are spiked directly in the vials as received. Mono-element spike solutions, containing major isotopes 238U, 235U, or 233U, and 239Pu or 242Pu, are selected, depending on the declared isotopic composition of the samples that are used for ID-MS analysis. High abundance 240Pu spike solutions have been used in the past, but their use has been abandoned due to safety concerns (pressure build-up in ampoules during storage), with 242Pu spikes being used instead. In general, most of the spike materials used for analysis of those samples are liquid solutions, produced in-house from metal CRMs, commercially available spike solutions, or custom-made materials provided to the IAEA through the Member States Support Programmes (MSSPs), that allow us to establish the traceability to reference materials produced by NBL, CETAMA or JRC-Geel. At the JRC-Karlsruhe small spike mono-elemental solutions containing major isotopes 238U or 233U and 239Pu or 242Pu are used for ID-MS analyses.87 These small spikes are prepared with a concentration suitable for mass spectrometry and they are characterized against standards prepared from CRMs.87
For analysis of environmental samples in the environmental sample laboratory (ESL) of SGAS (mostly swipe and hot cotton swipe samples), two categories are distinguished, low-level and high-level samples, with total 10 pg Pu and 1000 ng U considered to be high-level samples. After sample digestion and pre-screening, sample splitting is done followed by spiking with enriched 242Pu, 244Pu, 233U and 243Am spikes for isotope dilution analysis, using 242Pu/Pu or 244Pu/Pu amount ratios between 1 and 10, and 233U/U of around 100. Spike solutions used are prepared by step-wise dilutions from mother solutions, and therefore the concentration needs to be corrected for evaporation loss during storage. U and Pu analysis is performed by MC-ICP-MS following the methods outlined in ref. 45,131 and 207.
ID-MS is performed on nuclear material samples by TIMS applying the total evaporation method.208,209 After purification when needed,58 appropriate amounts of uranium and plutonium solutions are deposited onto a filament for each measurement of safeguards samples, CRMs and process control materials. By making duplicate analyses of both the spiked aliquots and unspiked sample aliquots the isotopic composition and elemental concentrations can be calculated and compared with defined control and warning limits.
As an alternative to traditional ID-MS when both pure and spiked sample aliquots are measured by TIMS, a combination of techniques can be used. In the OSL, High Resolution Gamma Spectrometry (HRGS) is combined with TIMS to determine the isotopic composition210 of samples that are subsequently subjected to spiking to perform the plutonium assay in pure plutonium solutions or mixed U and Pu solutions (free of strong beta gamma emitters, U/Pu ratio greater than two). For timeliness considerations, this method has the merit of skipping the chemical separation of the {sample–spike} fractions for isotope and ID analysis. In this case, values for isotope ratios of 241Pu/239Pu and 238Pu/239Pu in the ID-MS assay equation are replaced by ones obtained by HRGS Multi-Group Analysis Software (MGA) codes. For U containing samples with complex matrices and unknown declarations (also Pu materials containing traces of U), acceptable performance is achievable by using over-spiking (high spike-to-sample amounts) with highly enriched 233U spikes to determine the uranium amount and isotopic composition of the material from TIMS measurements of spiked sample aliquots. This so-called isotope stripping approach has also been used for the determination of traces of U in Pu materials.211
5.4. Irradiated samples
Irradiated samples, for which ID-MS is commonly applied, typically originate from the nuclear fuel cycle (definition of NFC in ref. 212). Nuclear fuels, for generation of electricity in a Nuclear Power Plant (NPP), are based predominantly on uranium (e.g. uranium oxide (UOX) fuels) or mixtures of uranium with other elements (e.g. Pu in MOX fuels). The so-called major actinide elements, U and Pu, possess fertile isotopes. These are isotopes which can be converted into a fissile isotope by neutron absorption: for example, the 238U isotope can be converted into 239Pu or 242Pu can be converted into 243Am. The major actinide elements also possess fissile isotopes, so it is possible that an actinide nucleus splits into two smaller nuclei, which are referred to as fission products: most of the lanthanides, cesium, and metallic fission products. Therefore, a nuclear fuel can contain a wide range of chemical elements once it has been irradiated. The isotopic composition of a fission product element can differ substantially from the naturally occurring isotopic composition of that same element: certain stable isotopes of an element might not be formed by fission. The amount of particular fission products (e.g. specific isotopes of Cs, Ce and Nd) present in an irradiated fuel can be very indicative of how much fission has taken place, and hence such isotopes can be used as fission burn-up monitors.213 Information about the extent of fission in a particular fuel (rod) is very useful knowledge for the safe and correct operation of an NPP. In the case of non-power reactors, which are used for material testing, R&D, and training purposes for example, the trend to switch from highly enriched uranium (HEU) to low enriched uranium (LEU) fuels (an anti-proliferation measure) has involved the testing of a range of fuels that differ quite substantially, in chemical and physical form, and enrichment, from those used in NPPs. Irradiated samples received by a radiochemical analysis laboratory can therefore differ widely in terms of matrices, their chemical composition (oxide based fuels to metallic or silicide fuels) and homogeneity, their enrichment (from LEU to HEU), and their physical form (a segment from an oxide fuel or a segment from a plate fuel).Despite the variety of irradiated samples, the determination of the U, Pu and Nd isotopic and elemental mass fractions by means of ID-MS can already provide considerable information. This is a common analysis as part of post irradiation examination. This gives information about the residual heavy metal atoms (the major actinides) and the concentration of the burn-up monitor (in this case the Nd fission product) can be used in the estimation of the fuel burn-up. Such information can be fed back into areas such as the assessment of fuel performance/qualification, validation of nuclear codes for criticality or for long-term storage of nuclear waste.31,38,130,214,215 Crucially, these rely on the low measurement uncertainties ID-MS can provide.
The irradiated sample first has to be brought into solution, before spiking can occur. Dissolution of spent fuels for analysis is detailed elsewhere,216–221 but in general terms an irradiated fuel is often dissolved quantitatively by heating (under reflux) under strong acidic conditions over several days inside a shielded hot cell. Only then can spikes be added to an aliquot of the dissolved sample, but crucially before any separations are started. Separations of lanthanides and actinides are discussed elsewhere.31–33,58,222–228 Van Winckel et al.220 provide a useful summary of the situations in which off-line and on-line separations are used for spent nuclear fuels.
Many practical details of the radiochemical analyses of the major actinides and Nd (amongst other actinides and fission products) to enable determination of the burn-up of an irradiated fuel are described elsewhere.229–232 The methodology reported by Roach et al.231 relies on on-line separation hyphenated to an MC-ICP-MS for isotopic vectors and Pu and Nd concentration or Davies & Gray titration for U concentration, all with calibration by ID-MS to provide results much more rapidly than off-line separations combined with an analytical technique such as TIMS.
The ASTM E321-20 method233 uses the 148Nd concentration as the fission burn-up monitor, due to the small difference between the fission yield of the two dominant fissile isotopes (235U and 239Pu). Govers et al.234 reported that this approach works well for NPPs, but that in high-flux reactors (non-power reactors) the 147Nd(n)148Nd (i.e. short form for the 147Nd + n → 148Nd formula) capture reaction may no longer be insignificant. In such cases, a correction needs to be applied to avoid an overestimation of the burn-up when using the 148Nd concentration. If it is not possible to correct the 148Nd for such capture by 147Nd an alternative is to measure the entire Nd vector. Data from the other Nd isotopes can provide estimates of the burn-up value.
The isotopic composition of stable or long-lived Nd isotopes formed during fission in irradiated fuels, including the absence of 142Nd, differs from that of natural Nd. Data from almost 40 different irradiated fuel samples at the SCK laboratories indicate the isotopic abundances of 146Nd and 150Nd are rather similar to their natural abundances whilst those of 145Nd and 148Nd are approximately doubled. For 143Nd and 144Nd the abundances can deviate more widely from their natural isotopic abundances. Roach et al.231 give examples of such data from three samples. The choice of the spike isotope for the quantification of Nd formed during fission is different than for Nd with natural isotopic composition due to their different isotopic compositions. A spike enriched in the 146Nd isotope can be an alternative to 148Nd and 150Nd isotopes33,38 if the entire Nd vector is to be measured. The amount of spike to be added depends on the concentration in the sample, and to some extent this depends on the laboratory's operating license. Some laboratories perform spiking inside a glovebox and others under a fumehood, the difference being the amount of U, Pu or Nd in the sample to be spiked. To perform spiking under a fumehood, an aliquot of diluted mother solution is required as the mother solution is too active, which in turn requires an appropriately lower spike concentration to avoid overspiking and to avoid spike masses with too high an uncertainty.
Cesium can also be used as an alternative fission burn-up monitor to neodymium.235–239 After irradiation of the fuel, the sample contains 133Cs, 134Cs, 135Cs and 137Cs. Cesium is naturally monoisotopic (133Cs), which makes it a good spike for such sample. The difficulties of cesium measurement by ID-MS come from the lack of reference materials to estimate the mass fractionation. Different methods can be used, such as a combination of TIMS and gamma spectrometry or intertechnique comparison.235,240
5.5. Biological applications
Radionuclide analysis in biological samples became important considering their intensive use in industry and medicine, and for some of them, their natural presence in the environment, such as Th and U (also named NORM, for Naturally Occurring Radioactive Material). Therefore, monitoring human exposure is crucial for evaluating the risks to the wider population and to workers. In addition, in case of a nuclear crisis or radiological exposure, such analyses are crucial to identify each individual's contamination. The most common matrices in which radionuclides are analyzed are urine and feces, but others biological tissues can also be considered in the research field.The ICP-MS technique has become increasingly popular in recent years for measuring those radionuclides with a sufficiently long half-life (t1/2 > 10000 years).241 The performance of ICP-MS, in terms of limits of detection and analysis time (several minutes), offers considerable advantages, even for complex matrices, over nuclear techniques such as alpha spectrometry.242,243 Furthermore, in addition to activity concentration determination, the isotopic composition can be determined by means of ICP-MS, which enables the origin of a contamination to be traced.242,244
For urine samples, diluted samples can be measured directly by means of ICP-MS with external calibration, if their concentrations are high enough.245,246 However, for urine samples with extremely low radionuclide concentrations and/or for other biological matrices such as feces, blood, nail and hair, a radiochemical purification and pre-concentration may be necessary before measurement by ICP-MS (urinary U,247,248 urinary Pu249). The ID technique is, in this case, highly recommended: (i) to evaluate the chemical yield and (ii) to use as an internal standard to measure the concentrations accurately by means of ICP-MS. Therefore, ID coupled to ICP-MS permits proper characterization by more accurate matrix effect corrections in case of high matrix or low concentration measurements.
ICP-MS is widely used for actinide (U, Pu, Am) analyses of biological samples241,246,247,249–270 with calibration methods that rely on an actinide isotope that is absent from the sample, such as 233U for U isotopes (234U, 235U, 236U and 238U), 242Pu for Pu isotopes (239Pu, 240Pu, and 241Pu) or 243Am for 241Am analysis. Ni et al. developed a protocol for the rapid determination of ultra-trace plutonium isotopes in small volumes of urine.249 This method is based on a chemical treatment of the urine samples (acid digestion, co-precipitation, extraction chromatography) before plutonium measurement by means of HR-ICP-MS. A 242Pu spike is used as a yield tracer to characterize the Pu isotopes. The method allows a high 238U decontamination factor (3.8 × 106), which is essential to limit the polyatomic interference of 238UH+ during 239Pu measurement. An average value of 72.7 ± 5.5% was achieved for 242Pu recovery for 20 mL or 100 mL urine bioassays. For 100 mL urine samples, the limits of detection (LODs) of 239Pu, 240Pu and 241Pu were 0.003 fg mL−1, 0.002 fg mL−1, and 0.003 fg mL−1, corresponding to 0.006 mBq L−1, 0.015 mBq L−1 and 9.8 mBq L−1, respectively. The current LODs of 239Pu and 240Pu by means of ICP-MS are more than 100 times lower than those usually achieved by means of the alpha spectrometry method (1 mBq L−1). In another study, Wang et al. developed an online separation and pre-concentration method coupled with ICP-MS/MS for measurement of 90Sr, 234U, 239Pu and 241Am isotopes using 233U, 242Pu and 86Sr tracers to correct for column recovery and matrix effects.271 This method required small sample volumes (10 mL) and offered rapid analysis times (46 min per sample for the full process) and was applied to various liquid matrices (lake water, seawater, and urine samples). The limits of detection were 1.48 pg L−1 (8257 mBq L−1) for 90Sr, 1.75 pg L−1 (0.40 mBq L−1) for 234U, 0.65 pg L−1 (77.65 mBq L−1) for 241Am, and 0.56 pg L−1 (1.25 mBq L−1) for 239Pu. In this work, no specific spike was used for 241Am analysis. The authors mentioned that high recoveries were obtained for 241Am in samples analyzed by means of this protocol. Therefore, it may be suspected that no recovery correction was applied for the determination of 241Am. To a limited extent, spike addition has been used to trace another radioelement, e.g. the use of 242Pu to trace 237Np, for which no suitable tracer is available, during quantification of 237Np by means of ICP-MS.254 In this study, the authors showed that 242Pu can serve as the recovery monitor for 237Np in the separation and pre-concentration procedure developed.
It should be noted that, in all these publications, no ID equation was detailed. Furthermore, the use of spikes is often mentioned in these papers as a yield tracer. Consequently, it may be suspected that these spikes are used more as internal tracers for recovery correction than as spikes for ID.
Clases et al.272 suggested an “isobaric dilution analysis” (IBDA) method as a “new quantitative method for long-lived radionuclides” by means of ICP-MS. The technique relies on using a spike of a different element, but which contains an isobaric isotope and has similar chemical properties to those of the analyte. Demonstration of IBDA has been performed on the determination of gadolinium concentration using a certified dysprosium solution spike by measuring the 157Gd/160Dy ratio. The same authors applied IBDA for technetium, for which no safe standard, in terms of radioprotection, is available. They demonstrated that the IBDA technique can also be applied on an element with monoisotopic abundance such as 99Tc using a ruthenium solution as a spike solution. The determination of 99Tc in the 99Tc-MAG3 drug is therefore obtained using the 99Tc/101Ru ratio. This technique could be extended to 41Ca, 60Co and 90Sr using K, Ni and Zr, respectively and applied in various application fields such as industry, environment, cosmochemistry and medicine.272
Furthermore, understanding the behavior, biokinetics and excretion of radionuclides from the human body increases even more in priority due to the increase of their medical applications. Therefore, radionuclide quantification by ID-ICP-MS is a promising tool for investigating the partitioning and behavior of radionuclides in organ tissues, such as the use of 242Pu and 243Am spikes to determine 239Pu, 240Pu and 241Am isotopes in brain tissues.273
Enriched spikes have also been used to trace specific speciation into the organism.47 However, this technique has not yet been applied to radionuclides.
ID-MS is a promising technique for quantifying low radionuclide concentration and/or in complex matrices for radiation protection purposes and also for investigating the behavior of radionuclides in biological tissues. However, this technique has not been used widely with well-described ID equations. This is perhaps due to the limited number of laboratories performing radioisotope quantification in biological samples by means of ICP-MS. Nevertheless, developments in ID-MS can be anticipated in the coming years.
5.6. Reference material
For CRM certification, reference measurement techniques that provide metrological traceability are required. ID-MS is a very accurate technique for the determination of element mass fraction and provides results with low uncertainties: it can be applied as a primary ratio method when suitable CRMs and calibrators are available. The challenges associated with the development of reference measurement systems for clinical analytes, and in particular for ID-MS, are highlighted in ref. 274. The role of ID-MS in the certification of CRMs is presented in ref. 20 and 21. ID-MS is commonly used by nuclear CRM producers either for characterization or for verification of the assigned certified values determined by means of other techniques.43,56,71,201 Facility operator laboratories and other laboratories performing nuclear material analyses use ID-MS for the characterization of laboratory CRMs for quality management, process or instrument control and preparation of spikes.86,201,275 ID-MS was used in the last few years to certify several CRMs: 243Am spike,7,56,276 LSD spike,73,201229Th spike,36231Pa spike,156242Pu spike,277244Pu spike43 or uranium oxide particle CRM.278On the other hand, CRMs play a crucial role in the performance of ID-MS measurements. Using high quality CRMs in ID-MS provides accurate and traceable measurement results. For instance, in nuclear safeguards spikes are a metrological quality tool to meet the existing requirements indicated by the ITVs for reliable accountancy and verification measurements.24,25 Considering modern instrumentation and state-of-the-practice methods, uncertainties of the CRM isotope ratios often significantly dominate the uncertainty budgets for the assay values of U and Pu. Several studies conducted by the IAEA for evaluating the fitness for purpose of CRMs in use279 have concluded that the certified uncertainties of some CRMs were larger than the estimated intermediate precision of the TIMS measurements. Thus, the need for re-certification for better uncertainty is to be addressed by CRM producers. For this important metrology mission, the IAEA and the JRC-Geel organize technical meetings among the safeguards community to discuss the current requirements of the CRMs.
5.7. Process control
ID is a method of choice for process control applications that require an especially high accuracy, granted that appropriate spikes are available for the measured elements. ID comes with a significant advantage over almost all other methods, as it is not affected by sample losses, as long as the loss occurs after the preparation of the {sample–spike} mixture. This is a great feature of ID for trace and ultra-trace analysis that is very sensitive to any sample loss during preparation steps such as separation, dilution, evaporation, etc. Another actual advantage of ID is its very low sensitivity to matrix effects, because the spike and sample both share the same matrix and valency after mixture homogenization.However, it comes with a rather complex setup that complicates and extends the sample preparation, which may be an issue for routine analysis. This is mainly due to the spiking step, where each sample is carefully spiked with an accurately weighed amount of spike. Automated systems based on highly accurate volumetric dispensers have been developed to solve this issue, helping to prepare a definite number of containers loaded with a known, accurately weighed amount of spike.280 These automated systems are particularly effective for process control, where the analyst has generally access to a decent estimation of expected sample concentrations.
For applications where speciation is important, various reports have demonstrated the compatibility of the ID method with techniques such as laser ablation (LA)-ICP-MS, gas chromatography (GC)-MS, high-performance liquid chromatography (HPLC)-ICP-MS, gamma spectrometry, etc.281–285
6. Inter-laboratory comparisons involving ID-MS
ILCs can be divided into three categories: those involving expert laboratories participating in the certification of a CRM (such as that organized for STAM CRM 243Am certification276), those involving laboratories validating a new measurement method, and finally those testing the proficiency of laboratories/methods for measurement (Proficiency Test, PT). These last two types of ILC are efficient tools to monitor the quality of measurement of analytical laboratories and, in particular, for those using ID-MS. Usually, PTs are organized on a regular basis to evaluate the ability of the laboratory to measure one or several quantities called measurands.286 PTs also play an important role in a laboratory quality system and are increasingly recognized by national accreditation bodies and are part of the ISO17025 standard requirements for the competence of analytical laboratories.287 The benefit for a laboratory of participating in PTs is to evaluate the accuracy of its measurements and to take corrective actions if necessary. The evaluation procedure consists of providing performance scores, such as the z-score or the zeta-score.The z-score (eqn (35)) compares the difference between the laboratory measurement result (ylab) and the PT assigned value (yPT, certified or consensual value) to the PT standard deviation (SPT, usually determined by robust statistics).
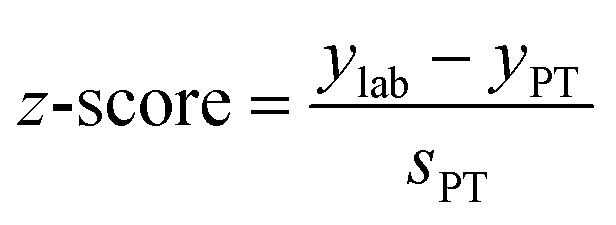 | (35) |
The zeta-score compares the same numerator to the uncertainty of the difference between the laboratory measurement value (ulab) and assigned value (uPT, eqn (36)).
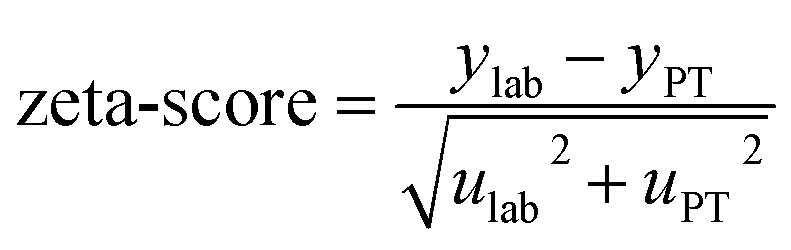 | (36) |
According to z or zeta-score values, results are classified as:
- Satisfactory if |z| or |zeta| < 2.
- Questionable if 2 < |z| or |zeta| < 3.
- Unsatisfactory if |z| or |zeta| > 3.
The z-score indicates the number of PT standard deviations between the laboratory measurement value and the PT assigned value. The zeta-score determines whether the measurement uncertainty value claimed by the laboratory allows the laboratory's measurement value and the assigned value to be considered identical. Action is then encouraged if unsatisfactory performance is indicated.
In the field of radionuclide analysis, several international PTs are organized annually by different institutions (IAEA, CETAMA, NBL, JRC, etc.) in different fields ranging from environmental concerns to safeguards or nuclear mining/enrichment/reprocessing activities.
Laboratories using ID techniques are generally concerned with PTs for the measurement of radionuclide mass fractions (or amount content) in solids or solutions. For example, CETAMA offers PTs, called EQRAIN U, EQRAIN Pu and EQRAIN (U + Pu), for the determination of U or/and Pu mass fractions in solution. These PTs are typically organized every 18 months.23 The content values lie in the ranges of 1 to 6 g kg−1 for Pu and 50 to 200 g kg−1 for U, and are designed for laboratories connected with the mining or reprocessing industries, institutions for safeguards or institutions dealing with reference materials.
The IAEA regularly organizes NMRoRo PTs and ILCs to assist the members of the IAEA network analytical laboratories (NWAL), as well as nuclear facility operators and other relevant laboratories, in the assessment of their nuclear material analyses (PTs) and environmental sample analyses (ILCs). During the last two exercises, the goal of NMRoRo was to determine uranium mass and uranium isotopic ratios in uranium oxide powder, and uranium and plutonium masses and uranium and plutonium isotopic ratios in a dried mixed Pu–U nitrate sample.288 The goals of another ILC organized by the IAEA every two years are to measure the uranium and plutonium masses and isotopic abundances in cotton wipe samples (referred to as “swipe sample”) thanks to “bulk analysis” methods (swipe dissolution, uranium and plutonium purifications and ID-MS measurements).
7. Conclusion
This article reviews isotope dilution analysis and its application for radionuclide measurements in different contexts. ID-MS is a primary ratio method that helps to measure radionuclide mass fraction, in solids or liquids, with high accuracy. The ID-MS analysis process is well understood and the ID-MS measurement uncertainty is well controlled and quantified. An uncertainty estimation is calculated considering all stages in the ID-MS process. MCM or quadratic cumulation methods can be used for uncertainty calculation: both approaches yield identical results. To obtain the most accurate measurement, implementation is needed for all stages: weighing process, mixture preparation, spike monitoring, isotopic homogenization, and mass spectrometry measurements.The excellent performance of ID-MS for radionuclide characterization has been demonstrated in a wide variety of fields: environmental, biological analysis, irradiated sample characterization, certified reference material production, safeguards and nuclear forensics. The ability to evaluate ID-MS measurement performance by using a wide range of ILCs is essential for guaranteeing measurement reliability and demonstrates that ID-MS has the potential to satisfy current and future needs in radioanalytical chemistry.
List of acronyms
| CAB | Cellulose Acetate Butyrate |
| CCQM | Consultative Committee for Amount of Substance |
| CEA | French Alternative Energy and Atomic Energy Commission |
| CETAMA | Commission for the Establishment of Analytical Methods |
| CMC | CarboxyMethyl Cellulose sodium salt |
| CRM | Certified Reference Material |
| F | Error Multiplication Factor |
| HEU | Highly Enriched Uranium |
| HKED | Hybrid K-Edge Densitometry |
| HRGS | High Resolution Gamma Spectrometry |
| HR-ICP-MS | High Resolution-Inductively Coupled Plasma Mass Spectrometry |
| HU-1 | Harwell Uraninite |
| IAEA | International Atomic Energy Agency |
| IBDA | Isobaric Dilution Analysis |
| ICP-MS | Inductively Coupled Plasma Mass Spectrometry |
| ID | Isotope Dilution |
| ID-MS | Isotope Dilution Mass Spectrometry |
| ILCs | Inter-Laboratory Comparisons |
| IRSN | French Institute for Radiation Protection and Nuclear Safety |
| ITVs | International Target Values |
| JAEA | Japan Atomic Energy Agency |
| JRC | Joint Research Centre |
| LAAT | Atalante Analysis Laboratory |
| LEU | Low Enriched Uranium |
| LOD | Limit Of Detection |
| LSD | Large-Sized Dried |
| MC-ICP-MS | Multi-Collector-Inductively Coupled Plasma Mass Spectrometry |
| MCM | Monte Carlo Method |
| MOX | Mixed OXide |
| MPE | Maximum Permissible Deviation |
| MSSP | Member States Support Programme |
| MGA | Multi-Group Analysis |
| NBL | New Brunswick Laboratory |
| NFC | Nuclear Fuel Cycle |
| NIST | National Institute of Standards and Technology |
| NMRoRo | Nuclear Material Round Robin |
| NORM | Naturally Occurring Radioactive Material |
| NPL | National Physical Laboratory |
| NPP | Nuclear Power Plant |
| NWAL | IAEA Network Analytical Laboratories |
| OSL | On-Site Laboratory |
| PT | Proficiency Test |
| R | Isotope Ratio |
| SCK | Belgian Nuclear Research Centre |
| SGAS | Office of Safeguards Analytical Services |
| SGAS-ESL | Environmental Sample Laboratory of SGAS |
| SGAS-NML | Nuclear Material Laboratory of SGAS |
| SI | International System of Units |
| SRM | Standard Reference Material |
| t 1/2 | Half-life |
| TIMS | Thermal Ionization Mass Spectrometry |
| u | Standard Uncertainty |
| UOX | Uranium Oxide |
| u r | Relative Standard Uncertainty |
Author contributions
Alexandre Quemet: conceptualization, writing – original draft (Introduction, Theory, Optimization of the {sample–spike} mixture, Spike monitoring, and Conclusion parts) and review, supervision. Amélie Hubert: conceptualization, writing – original draft (Nuclear forensics part). Alkiviadis Gourgiotis: conceptualization, writing – original draft (Environmental applications: natural radionuclides: U-series disequilibrium part). Ana María Sánchez Hernández: writing – original draft (Weighing procedure and Reference material parts) and review. Marielle Crozet: writing – original draft (Isotope dilution uncertainty part) and review. Guillaume Bailly: writing – original draft (Process control part). Andrew Dobney: writing – original draft (Irradiated samples part) and review. Georges Duhamel: writing – original draft (Safeguards part). Joe Hiess: writing – original draft (Safeguards part) and review. Urska Repinc: writing – original draft (Safeguards part) and review. Sébastien Mialle: writing – original draft (Isotopic homogenization of the sample–spike mixture and Irradiated samples part). Béatrice Boulet: writing – original draft (Application to environmental monitoring of nuclear facilities part). Raphaelle Escoube: writing – original draft (Biological applications part). Céline Bouvier-Capely: writing – original draft (Biological applications part). Fabien Pointurier: writing – review & editing. Sébastien Picart: writing – original draft (Weighing uncertainty and Interlaboratory comparison involving ID-MS parts) and review, supervision.Conflicts of interest
There are no conflicts to declare.References
- M. L. D. P. Godoy, J. M. Godoy, L. A. Roldão and L. Tauhata, Determination of total content and isotopic compositions of plutonium and uranium in environmental samples for safeguards purposes by ICP-QMS, J. Environ. Radioact., 2009, 100, 613–625 CrossRef CAS PubMed.
- S. Boulyga, S. Konegger-Kappel, S. Richter and L. Sangély, Mass spectrometric analysis for nuclear safeguards, J. Anal. At. Spectrom., 2015, 30(7), 1469–1489 RSC.
- A. Quemet, E. Buravand, B. Catanese, P. Huot, V. Dalier and A. Ruas, Monitoring the plutonium depletion in dissolution residues of a spent fuel solution using a surrogate and plutonium isotope ratio measurements, J. Radioanal. Nucl. Chem., 2020, 326(1), 255–260, DOI:10.1007/s10967-020-07311-5.
- BIPM, International vocalulary of metrology – Basic and general concepts and associated terms (VIM), 3rd edn, 2008, vol. 3, Available from: http://www.bipm.org/utils/common/documents/jcgm/JCGM_200_2008.pdf Search PubMed.
- M. Désenfant and M. Priel, Road map for measurement uncertainty evaluation, Measurement, 2006, 39(9), 841–848 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0263224106000789.
- ISO 5725-1:2023, Accuracy (Trueness and Precision) of Measurement Methods and Results — Part 1: General Principles and Definitions Search PubMed.
- A. Quemet, A. Ruas, V. Dalier and C. Rivier, Americium isotope analysis by Thermal Ionization Mass Spectrometry using the Total Evaporation Method, Int. J. Mass Spectrom., 2018, 431, 8–14 CrossRef CAS.
- A. M. Gaffney, A. Hubert, W. S. Kinman, M. Magara, A. Okubo and F. Pointurier, et al., Round-robin 230Th–234U age dating of bulk uranium for nuclear forensics, J. Radioanal. Nucl. Chem., 2016, 307(3), 2055–2056, DOI:10.1007/s10967-015-4334-8.
- F. Pointurier, A. Hubert and G. Roger, A method for dating small amounts of uranium, J. Radioanal. Nucl. Chem., 2013, 296(2), 593–598, DOI:10.1007/s10967-012-2198-8.
- Z. Varga, K. Mayer, C. E. Bonamici, A. Hubert, I. Hutcheon and W. Kinman, et al., Validation of reference materials for uranium radiochronometry in the frame of nuclear forensic investigations, Appl. Radiat. Isot., 2015, 102, 81–86, DOI:10.1016/j.apradiso.2015.05.005.
- P. van Calsteren and L. Thomas, Uranium-series dating applications in natural environmental science, Earth Sci. Rev., 2006, 75(1–4), 155–175 CrossRef CAS . https://linkinghub.elsevier.com/retrieve/pii/S0012825205001418.
- X. Hou and P. Roos, Critical comparison of radiometric and mass spectrometric methods for the determination of radionuclides in environmental, biological and nuclear waste samples, Anal. Chim. Acta, 2008, 608(2), 105–139 CrossRef CAS PubMed.
- P. P. Povinec, Ultra-sensitive radionuclide analyses: new frontiers in radioanalytics, J. Radioanal. Nucl. Chem., 2019, 322(3), 1247–1255, DOI:10.1007/s10967-019-06707-2.
- S. Diez-Fernández, H. Isnard, A. Nonell, C. Bresson and F. Chartier, Radionuclide analysis using collision–reaction cell ICP-MS technology: a review, J. Anal. At. Spectrom., 2020, 35(12), 2793–2819 RSC . Available from: http://xlink.rsc.org/?DOI=D0JA00211A.
- T. R. E. Southwood and P. A. Henderson, Ecological Methods, Blackwell, 2015 Search PubMed.
- E. Roth, Critical evaluation of the use and analysis of stable isotopes (Technical Report), Pure Appl. Chem., 1997, 69(8), 1754–1813 CrossRef.
- G. V. Hevesy and F. Paneth, Die Löslichkeit des Bleisulfids und Bleichromats, Zeitschrift für Anorg Chemie., 1913, 82(1), 323–328 CrossRef.
- P. Goris, W. E. Duffy and F. H. Tingey, Uranium Determination by the Isotope Dilution Technique, Anal. Chem., 1957, 29(11), 1590–1592 CrossRef CAS.
- G. R. Tilton, L. T. Aldrich and M. G. Inghram, Mass Spectrometric Determination of Thorium, Anal. Chem., 1954, 26(5), 894–898, DOI:10.1080/18811248.1967.9732709.
- J. Vogl, Characterisation of reference materials by isotope dilution mass spectrometry, J. Anal. At. Spectrom., 2007, 22(5), 475–492 RSC.
- M. J. T. Milton and T. J. Quinn, Primary methods for the measurement of amount of substance, Metrologia, 2001, 38(4), 289–296 CrossRef CAS.
- BIPM, Consultative Committee for Amount of Substance (CCQM) — 5th Meeting (February 1999), 1999 Search PubMed.
- M. Crozet, D. Roudil, C. Rigaux, C. Bertorello, S. Picart and C. Maillard, EQRAIN: uranium and plutonium interlaboratory exercises from 1997 to 2016—comparison to ITVs-2010, J. Radioanal. Nucl. Chem., 2019, 319(3), 1013–1021, DOI:10.1007/s10967-018-6399-7.
- International Atomic Energy Agency, International Target Values 2010 for Measurement Uncertainties in Safeguarding Nuclear Materials - STR368, Vienna, Austria, 2010 Search PubMed.
- International Atomic Energy Agency, International Target Values for Measurement Uncertainties in Safeguarding Nuclear Materials - STR368 (Revision1.1), Vienna, Austria, 2022 Search PubMed.
- J. Irrgeher, J. Vogl, J. Santner and T. Prohaska, Sector Field Mass Spectrometry for Elemental and Isotopic Analysis, ed. T Prohaska, J. Irrgeher, A. Zitek and N. Jakubowski, Royal Society of Chemistry, Cambridge, 2014. pp. 126–151, New Developments in Mass Spectrometry, Available from: 10.1039/9781849735407.
- G. Cavazzini, D. Roccato and A. Fasson, The determination of the isotopic composition of strontium, Int. J. Mass Spectrom., 2022, 471, 116757, DOI:10.1016/j.ijms.2021.116757.
- L. Zhu, C. Xu, X. Hou, J. Qiao, Y. Zhao and G. Liu, Determination of Ultratrace Level 135 Cs and 135 Cs/137 Cs Ratio in Small Volume Seawater by Chemical Separation and Thermal Ionization Mass Spectrometry, Anal. Chem., 2020, 92, 6709–6718, DOI:10.1021/acs.analchem.0c00688.
- A. Magre, B. Boulet, H. Isnard, S. Mialle, O. Evrard and L. Pourcelot, Innovative ICP-MS/MS Method To Determine the 135 Cs/137 Cs Ratio in Low Activity Environmental Samples, Anal. Chem., 2023, 95(17), 6923–6930, DOI:10.1021/acs.analchem.3c00207.
- Y. Shibahara, S. Nakamura, A. Uehara, T. Fujii, S. Fukutani and A. Kimura, et al., Measurement of cesium isotopic ratio by thermal ionization mass spectrometry for neutron capture reaction studies on 135Cs, J. Radioanal. Nucl. Chem., 2020, 325(1), 155–165, DOI:10.1007/s10967-020-07198-2.
- A. Quemet, M. Angenieux and A. Ruas, Nd, Am and Cm isotopic measurements after simultaneous separation in transmutation irradiated samples, J. Anal. At. Spectrom., 2021, 36(8), 1758–1767 RSC . Available from: http://xlink.rsc.org/?DOI=D1JA00165E.
- N. N. Wanna, K. Van Hoecke, A. Dobney, M. Vasile, T. Cardinaels and F. Vanhaecke, Determination of the lanthanides, uranium and plutonium by means of on-line high-pressure ion chromatography coupled with sector field inductively coupled plasma-mass spectrometry to characterize nuclear samples, J. Chromatogr. A, 2020, 1617, 460839 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S002196731931310X.
- N. N. Wanna, A. Dobney, K. Van Hoecke, M. Vasile and F. Vanhaecke, Quantification of uranium, plutonium, neodymium and gadolinium for the characterization of spent nuclear fuel using isotope dilution HPIC-SF-ICP-MS, Talanta, 2021, 221(September 2020), 121592, DOI:10.1016/j.talanta.2020.121592.
- L. Vio, G. Crétier, F. Chartier, V. Geertsen, A. Gourgiotis and H. Isnard, et al., Separation and analysis of lanthanides by isotachophoresis coupled with inductively coupled plasma mass spectrometry, Talanta, 2012, 99, 586–593 CrossRef CAS PubMed.
- A. Quemet, J. Sevilla and R. Vauchy, A combined TIMS and ICP-MS study for Th0.5Np0.5O2 thorium neptunium mixed oxide analysis, Int. J. Mass Spectrom., 2021, 460, 116479, DOI:10.1016/j.ijms.2020.116479.
- R. M. Essex, J. L. Mann, R. W. Williams, W. S. Kinman, A. Hubert and M. E. Bennett, et al., A new thorium-229 reference material, Appl. Radiat. Isot., 2018, 134(July 2017), 23–31, DOI:10.1016/j.apradiso.2017.07.050.
- A. Quemet, A. Ruas, E. Esbelin, V. Dalier and C. Rivier, Development and comparison of two high accuracy methods for uranium concentration in nuclear fuel: ID-TIMS and K-edge densitometry, J. Radioanal. Nucl. Chem., 2019, 321(3), 997–1004, DOI:10.1007/s10967-019-06670-y.
- A. Quemet, E. Buravand, J.-G. Peres, V. Dalier and S. Bejaoui, Irradiated UAmO2 transmutation discs analyses: from dissolution to isotopic analyses, J. Radioanal. Nucl. Chem., 2022, 331(2), 1051–1061, DOI:10.1007/s10967-021-08156-2.
- H. Jaegler, A. Gourgiotis, P. Steier, R. Golser, O. Diez and C. Cazala, Pushing Limits of ICP-MS/MS for the Determination of Ultralow 236U/238U Isotope Ratios, Anal. Chem., 2020, 92(11), 7869–7876 CrossRef CAS PubMed.
- A. Gourgiotis, M. Granet, H. Isnard, A. Nonell, C. Gautier and G. Stadelmann, et al., Simultaneous uranium/plutonium separation and direct isotope ratio measurements by using CO2 as the gas in a collision/reaction cell based MC-ICPMS, J. Anal. At. Spectrom., 2010, 25(12), 1939–1945 RSC.
- C. Venchiarutti, G. Stadelmann, R. Middendorp, Z. Macsik and A. Venzin, Determination of picogram amounts of uranium in micrometre-sized particles by isotope dilution mass spectrometry, J. Anal. At. Spectrom., 2021, 36(3), 548–560 RSC.
- P.-A. Chaboche, F. Pointurier, P. Sabatier, A. Foucher, T. Tiecher and J. P. G. Minella, et al., 240Pu/239Pu signatures allow refining the chronology of radionuclide fallout in South America, Sci. Total Environ., 2022, 843(April), 156943 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0048969722040402.
- R. M. Essex, R. W. Williams, K. C. Treinen, A. Hubert, M. A. Humphrey and J. D. Inglis, et al., A highly-enriched 244Pu reference material for nuclear safeguards and nuclear forensics measurements, J. Radioanal. Nucl. Chem., 2020, 324(1), 257–270, DOI:10.1007/s10967-020-07075-y.
- A. M. Sánchez Hernández, J. Horta Domenech, D. Wojnowski and E. Zuleger, Improvement of the 241Pu/239Pu isotope ratio measurement by thermal ionisation mass spectrometry - An approach to account for the 241Am in-growth after plutonium purification, Int. J. Mass Spectrom., 2021, 468, 116660 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380621001408.
- G. R. Eppich, Z. Mácsik, R. Katona, S. Konegger-Kappel, G. Stadelmann and A. Köpf, et al., Plutonium assay and isotopic composition measurements in nuclear safeguards samples by inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2019, 34(6), 1154–1165 RSC . Available from: http://xlink.rsc.org/?DOI=C9JA00047J.
- A. Gourgiotis, H. Isnard, M. Aubert, E. Dupont, I. AlMahamid and G. Tiang, et al., Accurate determination of Curium and Californium isotopic ratios by inductively coupled plasma quadrupole mass spectrometry (ICP-QMS) in 248Cm samples for transmutation studies, Int. J. Mass Spectrom., 2010, 291(3), 101–107, DOI:10.1016/j.ijms.2010.02.002.
- P. Rodríguez-González, J. M. Marchante-Gayón, J. I. García Alonso and A. Sanz-Medel, Isotope dilution analysis for elemental speciation: A tutorial review, Spectrochim. Acta Part B At. Spectrosc., 2005, 60(2), 151–207 CrossRef.
- P. Rodríguez-González and J. I. García Alonso, Recent advances in isotope dilution analysis for elemental speciation, J. Anal. At. Spectrom., 2010, 25(3), 239–259 RSC.
- J. Villanueva, M. Carrascal and J. Abian, Isotope dilution mass spectrometry for absolute quantification in proteomics: Concepts and strategies, J. Proteonomics, 2014, 96, 184–199, DOI:10.1016/j.jprot.2013.11.004.
- C. Pin and B. Le Fèvre, Isotope Dilution with Matrix Element Removal: A Key for High-Precision, High-Accuracy Trace Analysis of Geological Samples Using Inductively Coupled Plasma-Mass Spectrometry, Geostand. Geoanal. Res., 2002, 26(2), 135–148, DOI:10.1111/j.1751-908X.2002.tb00882.x.
- J. D. Fassett and P. J. Paulsen, Isotope Dilution Mass Spectrometry for Accurate Elemental Analysis, Anal. Chem., 1989, 61(10), 643A–649A CrossRef CAS.
- F. Vanhaecke and P. Degryse, Isotopic Analysis, ed. F. Vanhaecke and P. Degryse, Wiley, 2012, Available from: DOI:10.1002/9783527650484.
- P. J. De Bièvre and G. H. Debus, Precision mass spectrometric isotope dilution analysis, Nucl. Instrum. Methods, 1965, 32(2), 224–228 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/0029554X65905161.
- P. Rodríguez-González and J. Ignacio García Alonso, Mass spectrometry | Isotope dilution mass spectrometry, Encycl Anal Sci, 2019, 6, 411–420 Search PubMed.
- D. Roudil, C. Rigaux, C. Rivier, J. C. Hubinois and L. Aufore, CETAMA Contribution to Safeguards and Nuclear Forensic Analysis based on Nuclear Reference Materials, Procedia Chem., 2012, 7, 709–715, DOI:10.1016/j.proche.2012.10.108.
- R. Jakopič, A. Fankhauser, Y. Aregbe, S. Richter, M. Crozet and C. Maillard, et al., 243Am certified reference material for mass spectrometry, J. Radioanal. Nucl. Chem., 2021, 327(1), 495–504, DOI:10.1007/s10967-020-07521-x.
- G. Canciani, Y. Davrain, M. Crozet, D. Roudil and S. Picart, Controlled Potential Coulometry for the accurate determination of plutonium in the presence of uranium: The role of sulfate complexation, Talanta, 2021, 222, 121490, DOI:10.1016/j.talanta.2020.121490.
- C. Maillard, D. Maloubier, O. Boulay, V. Savigny and A. Quemet, U and Pu separation with U/TEVA resin : Influence of some parameters on chromatographic cycle performances, J. Radioanal. Nucl. Chem., 2021, (0123456789, DOI:10.1007/s10967-021-07986-4.
- A. Quemet and S. Baghdadi, Optimization of double isotope dilution, J. Anal. At. Spectrom., 2022, 37(2), 240–248 RSC . Available from: http://rsc.66557.net/en/Content/ArticleLanding/2022/JA/D1JA00322D.
- IUPAC. The IUPAC Compendium of Chemical Terminology, The IUPAC Compendium of Chemical Terminology, ed. V. Gold, NC: International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, 2019, p. 1670, Available from: https://goldbook.iupac.org/terms/view/L03540 Search PubMed.
- L. L. Yu, J. D. Fassett and W. F. Guthrie, Detection Limit of Isotope Dilution Mass Spectrometry, Anal. Chem., 2002, 74(15), 3887–3891, DOI:10.1021/ac011254l.
- L. Yang, Accurate and precise determination of isotopic ratios by MC-ICP-MS: A review, Mass Spectrom. Rev., 2009, 28(6), 990–1011 CrossRef CAS PubMed . Available from: https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf?sequence=1%0Ahttp://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf;jsessionid=F9A3B620D10ECFAF8498AC0E08EFCBC8?sequence=1%0Ahttp://apps.who.int/.
- M. E. Wieser, D. Buhl, C. Bouman and J. Schwieters, High precision calcium isotope ratio measurements using a magnetic sector multiple collector inductively coupled plasma mass spectrometer, J. Anal. At. Spectrom., 2004, 19(7), 844 RSC . Available from: http://xlink.rsc.org/?DOI=b403339f.
- L. Yang and R. E. Sturgeon, Comparison of mass bias correction models for the examination of isotopic composition of mercury using sector field ICP-MS, J. Anal. At. Spectrom., 2003, 18(12), 1452 RSC . Available from: http://xlink.rsc.org/?DOI=b307973b.
- P. Lindahl, G. Olszewski and M. Eriksson, Performance and optimisation of triple quadrupole ICP-MS for accurate measurement of uranium isotopic ratios, J. Anal. At. Spectrom., 2021, 14–16 Search PubMed.
- S. K. Aggarwal, Thermal ionisation mass spectrometry (TIMS) in nuclear science and technology – a review, Anal. Methods, 2016, 8(1996), 942–957 RSC.
- T. Prohaska, J. Irrgeher, A. Zitek and N. Jakubowski, Sector Field Mass Spectrometry for Elemental and Isotopic Analysis, New Developments in Mass Spectrometry, ed. T. Prohaska, J. Irrgeher, A. Zitek and N. Jakubowski, The Royal Society of Chemistry, 2014. Available from: https://books.rsc.org/books/book/996/Sector-Field-Mass-Spectrometry-for-Elemental-and Search PubMed.
- F. Albarède, P. Telouk, J. Blichert-Toft, M. Boyet, A. Agranier and B. Nelson, Precise and accurate isotopic measurements using multiple-collector ICPMS, Geochim. Cosmochim. Acta, 2004, 68(12), 2725–2744 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0016703703008871.
- C. E. Mathew, V. K. Rai, C. Ottenfeld and R. Keller, Effect of weighing uncertainty on assay values by isotope dilution mass spectrometry, J. Radioanal. Nucl. Chem., 2021, 328(1), 235–243, DOI:10.1007/s10967-021-07646-7.
- ASTM C1380-04 - Standard Test Method for Determination of Uranium Content and Isotopic Composition by Isotope Dilution Mass Spectrometry Search PubMed.
- P. van Belle, E. Zuleger and L. Duinslaeger, An improved self-checking weighing practice and a method for contamination detection for use in IDMS nuclear material assay, INMM Rep, 2008 Search PubMed.
- OIML, International Recommendation OIML R 111-1 Edition 2004 (E), Int. Organ. Leg. Metrol., 2004, 2004, 1–78 Search PubMed . Available from: https://www.oiml.org/en/files/pdf_r/r111-1-e04.pdf.
- K. Toth, R. Jakopic, J. Bauwens, C. Hennessy, F. Kehoe and U. Jakobsson, et al., Preparation and Certification of Large-Sized Dried (LSD) Spike : IRMM-1027w, JRC Ref. Mater. Rep., 2022 DOI:10.2760/817757.
- S. Belz, I. Bianchi, C. Cella, H. Emteborg, F. Fumagalli, O. Geiss, et al., Current Status of the Quantification of Microplastics in Water - Results of a JRC/BAM Inter-laboratory Comparison Study on PET in Water, EUR 30799 EN, Publications Office of the European Union, 2021 Search PubMed.
- OIML R 33, 1979 International Organization of Legal Metrology - Conventional Value of the Result of Weighing in Air, 1979 Search PubMed.
- M. Ohata, T. Ichinose, N. Furuta, A. Shinohara and M. Chiba, Isotope Dilution Analysis of Se in Human Blood Serum by Using High-Power Nitrogen Microwave-Induced Plasma Mass Spectrometry Coupled with a Hydride Generation Technique, Anal. Chem., 1998, 70(13), 2726–2730 CrossRef CAS PubMed.
- R. Hoelzl, C. Hoelzl, L. Kotz and L. Fabry, The optimal amount of isotopic spike solution for ultratrace analysis by isotope dilution mass spectrometry, Accredit. Qual. Assur., 1998, 3(5), 185–188 CrossRef CAS.
- P. Bedson, Guidelines for Achieving High Accuracy in Isotope Dilution Mass Spectrometry (IDMS), Guidelines for Achieving High Accuracy in Isotope Dilution Mass Spectrometry (IDMS), Royal Society of Chemistry, Cambridge, 2002, pp. 1–34, Available from: 10.1039/9781847559302.
- B. N. Colby, A. E. Rosecrance and M. E. Colby, Measurement parameter selection for quantitative isotope dilution gas chromatography/mass spectrometry, Anal. Chem., 1981, 53(12), 1907–1911 CrossRef CAS.
- C. J. Park and J. K. Suh, Determination of Trace Elements in Rice Flour by Isotope Dilution Inductively Coupled Plasma Mass Spectrometry, J. Anal. At. Spectrom., 1997, 12(5), 573–577 RSC . Available from: http://xlink.rsc.org/?DOI=a607393j.
- J. W. Eaton, GNU Octave and reproducible research, J. Process Control, 2012, 22(8), 1433–1438, DOI:10.1016/j.jprocont.2012.04.006.
- J. W. Eaton, D. Bateman, S. Hauberg and R. Wehbring, {GNU Octave} version 8.3.0 manual: a high-level interactive language for numerical computations, 2023, Available from: https://www.gnu.org/software/octave/doc/v8.3.0/.
- J. Irrgeher, J. Vogl, J. Santner and T. Prohaska, Chapter 8 Measurement Strategies, In Sector Field Mass Spectrometry for Elemental and Isotopic Analysis, The Royal Society of Chemistry, 2015, pp. 126–151 Search PubMed.
- R. Jakopič, K. Toth, J. Bauwens, R. Buják, C. Hennessy and F. Kehoe, et al., 30 years of IRMM-1027 reference materials for fissile material accountancy and control: development, production and characterisation, J. Radioanal. Nucl. Chem., 2021, 330(1), 333–345, DOI:10.1007/s10967-021-07927-1.
- N. Surugaya, T. Hiyama, A. Verbruggen and R. Wellum, Preparation, Certification and Validation of a Stable Solid Spike of Uranium and Plutonium Coated with a Cellulose Derivative for the Measurement of Uranium and Plutonium Content in Dissolved Nuclear Fuel by Isotope Dilution Mass Spectrometry, Anal. Sci., 2008, 24(2), 247–252, DOI:10.2116/analsci.24.247.
- G. Bagliano, J. Cappis, N. Doubek, G. Jammet, W. Raab and A. Zoigner, Preparation and Validation of a Large Size Dried Spike: Batch SAL-9924, 1989, IAEA/AL/02 Search PubMed.
- A.-M. Sanchez-Hernandez, C.-L. Andor, R. Buda, L. Commin, F. D'Amati, J. Domenech, et al., Optimising the use of reference materials for destructive analysis in nuclear safeguards, In Proceedings of 41st ESARDA Symposium, 2019, pp. 188–199 Search PubMed.
- J. Meija and Z. Mester, Paradigms in isotope dilution mass spectrometry for elemental speciation analysis, Anal. Chim. Acta, 2008, 607(2), 115–125 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0003267007019253.
- W. Men, J. Zheng, H. Wang, Y. Ni, Y. Kumamoto and M. Yamada, et al., Pu isotopes in the seawater off Fukushima Daiichi Nuclear Power Plant site within two months after the severe nuclear accident, Environ. Pollut., 2019, 246(July 2013), 303–310, DOI:10.1016/j.envpol.2018.12.007.
- H. Wang, Y. Ni, W. Men, Z. Wang, M. Liu and D. Xiao, et al., Distributions of fallout 137Cs, 239+240Pu and 241Am in a soil core from South Central China, J. Environ. Radioact., 2022, 251–252, 106971, DOI:10.1016/j.jenvrad.2022.106971.
- Y. Fan, H. Jiang, J. Ren, X. Liu, J. Lan and P. Cheng, et al., Plutonium-based radiometric dating of rapidly accumulated sediments in the Sanyuan sinkhole, southern Chinese Loess Plateau, Sci. Total Environ., 2023, 864(December 2022), 160937, DOI:10.1016/j.scitotenv.2022.160937.
- C. Maillard and J.-M. Adnet, Plutonium(IV) peroxide formation in nitric medium and kinetics Pu(VI) reduction by hydrogen peroxide, Radiochim. Acta, 2001, 89(8), 485–490, DOI:10.1524/ract.2001.89.8.485/html.
- A. Brunstad, Oxidation of Plutonium (III) by Sodium Nitrite, Niskayuna, NY (United States), 1957, Available from: http://www.osti.gov/servlets/purl/4345806/.
- J. Meija, T. B. Coplen, M. Berglund, W. A. Brand, P. De Bièvre and M. Gröning, et al., Atomic weights of the elements 2013 (IUPAC Technical Report), Pure Appl. Chem., 2016, 88(3), 265–291 CrossRef CAS.
- EC-JRC, IRMM-054 Uranium-235 spike, nitrate solution, [cited 2023 Jul 21], Available from: https://crm.jrc.ec.europa.eu/p/40454/40475/By-application-field/Nuclear/IRMM-054-URANIUM-235-SPIKE-NITRATE-SOLUTION/IRMM-054.
- Joint Committee for Guides in Metrology, JCGM 101:2008 Evaluation of Measurement Data — Supplement 1 to the “Guide to the Expression of Uncertainty in Measurement” — Propagation of Distributions Using a Monte Carlo Method, Jcgm, 2008, vol. 101:2008 Search PubMed.
- A. Saltelli, Making best use of model evaluations to compute sensitivity indices, Comput. Phys. Commun., 2002, 145(2), 280–297 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0010465502002801.
- Joint Committee for Guides in Metrology, JCGM 100:2008 GUM 1995 Evaluation of Measurement Data — Guide to the Expression of Uncertainty in Measurement, Jcgm, 2008, vol. 100, 2008 Search PubMed.
- R Core Team, R: A Language and Environment for Statistical Computing, Vienna, Austria, 2023, https://www.R-project.org Search PubMed.
- K. W. W. Sims, S. Pichat, M. K. Reagan, P. R. Kyle, H. Dulaiova and N. W. Dunbar, et al., On the Time Scales of Magma Genesis, Melt Evolution, Crystal Growth Rates and Magma Degassing in the Erebus Volcano Magmatic System Using the 238U, 235U and 232Th Decay Series, J. Petrol., 2013, 54(2), 235–271, DOI:10.1093/petrology/egs068.
- F. Chabaux, E. Blaes, P. Stille, R. di Chiara Roupert, E. Pelt and A. Dosseto, et al., Regolith formation rate from U-series nuclides: Implications from the study of a spheroidal weathering profile in the Rio Icacos watershed (Puerto Rico), Geochim. Cosmochim. Acta, 2013, 100, 73–95 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0016703712005431.
- S. Gdaniec, M. Roy-Barman, M. Levier, O. Valk, M. R. van der Loeff and L. Foliot, et al., 231Pa and 230Th in the Arctic Ocean: Implications for boundary scavenging and 231Pa230Th fractionation in the Eurasian Basin, Chem. Geol., 2020, 532(April 2019), 119380 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0009254119305091.
- A. Beaumais, A. Mangeret, D. Suhard, P. Blanchart, M. Neji and C. Cazala, et al., Combined U-Pb isotopic signatures of U mill tailings from France and Gabon: A new potential tracer to assess their fingerprint on the environment, J. Hazard Mater., 2022, 430(February), 128484 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0304389422002722.
- A. Gourgiotis, A. Mangeret, G. Manhès, P. Blanchart, L. Stetten and G. Morin, et al., New Insights into Pb Isotope Fingerprinting of U-Mine Material Dissemination in the Environment: Pb Isotopes as a Memory Dissemination Tracer, Environ. Sci. Technol., 2020, 54(2), 797–806, DOI:10.1021/acs.est.9b04828.
- M. Ivanovich and R. S. Harmon, Uranium-series disequilibrium: applications to earth, Marine & Envirol. Sci., 1992, 2, 910 Search PubMed.
- J. H. Chen and G. J. Wasserburg, Isotopic Determination of Uranium in Picomole and Subpicomole Quantities, Anal. Chem., 1982, 54(2), 350, DOI:10.1021/ac00239a601.
- T. Yokoyama, A. Makishima and E. Nakamura, Evaluation of the coprecipitation of incompatible trace elements with fluoride during silicate rock dissolution by acid digestion, Chem. Geol., 1999, 157(3–4), 175–187 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S000925419800206X.
- P. Lindahl, G. Olszewski and M. Eriksson, Total dissolution of environmental samples for the determination of uranium and thorium by ICP-MS and alpha-particle spectrometry, Appl. Radiat. Isot., 2022, 181(October 2021), 110103, DOI:10.1016/j.apradiso.2022.110103.
- E. Douville, E. Sallé, N. Frank, M. Eisele, E. Pons-Branchu and S. Ayrault, Rapid and accurate U–Th dating of ancient carbonates using inductively coupled plasma-quadrupole mass spectrometry, Chem. Geol., 2010, 272(1–4), 1–11, DOI:10.1016/j.chemgeo.2010.01.007.
- G. Yang, H. Tazoe and M. Yamada, Determination of 236 U in environmental samples by single extraction chromatography coupled to triple-quadrupole inductively coupled plasma-mass spectrometry, Anal. Chim. Acta, 2016, 944, 44–50, DOI:10.1016/j.aca.2016.09.033.
- O. Alhassanieh, A. Abdul-Hadi, M. Ghafar and A. Aba, Separation of Th, U, Pa, Ra and Ac from natural uranium and thorium series, Appl. Radiat. Isot., 1999, 51(5), 493–498 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804399000688.
- C. Dalencourt, M. N. Chabane, J.-C. Tremblay-Cantin and D. Larivière, A rapid sequential chromatographic separation of U- and Th-decay series radionuclides in water samples, Talanta, 2020, 207(May 2019), 120282, DOI:10.1016/j.talanta.2019.120282.
- A. W. Knight, E. S. Eitrheim, A. W. Nelson, S. Nelson and M. K. Schultz, A simple-rapid method to separate uranium, thorium, and protactinium for U-series age-dating of materials, J. Environ. Radioact., 2014, 134, 66–74, DOI:10.1016/j.jenvrad.2014.02.010.
- G. Manhes, C. Allegre and A. Provost, U-Th-Pb systematics of the eucrite “Juvinas”: Precise age determination and evidence for exotic lead, Geochim. Cosmochim. Acta, 1984, 48(11), 2247–2264 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/0016703784902217.
- M. Verlinde, J. Gorny, G. Montavon, S. Khalfallah, B. Boulet and C. Augeray, et al., A new rapid protocol for 226Ra separation and preconcentration in natural water samples using molecular recognition technology for ICP-MS analysis, J. Environ. Radioact., 2019, 202(February), 1–7, DOI:10.1016/j.jenvrad.2019.02.003.
- D. Larivière, V. N. Epov, R. D. Evans and R. J. Cornett, Determination of radium-226 in environmental samples by inductively coupled plasma mass spectrometry after sequential selective extraction, J. Anal. At. Spectrom., 2003, 18(4), 338–343 RSC . Available from: http://xlink.rsc.org/?DOI=b212361d.
- D. Larivière, V. N. Epov, K. M. Reiber, R. J. Cornett and R. D. Evans, Micro-extraction procedures for the determination of Ra-226 in well waters by SF-ICP-MS, Anal. Chim. Acta, 2005, 528(2), 175–182 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0003267004013030.
- C. Dalencourt, A. Michaud, A. Habibi, A. Leblanc and D. Larivière, Rapid, versatile and sensitive method for the quantification of radium in environmental samples through cationic extraction and inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2018, 33(6), 1031–1040 RSC . Available from: http://xlink.rsc.org/?DOI=C8JA00060C.
- B. Ghaleb, E. Pons-Branchu and P. Deschamps, Improved method for radium extraction from environmental samples and its analysis by thermal ionization mass spectrometry, J. Anal. At. Spectrom., 2004, 19(7), 906 RSC . Available from: http://xlink.rsc.org/?DOI=b402237h.
- A. S. Cohen and R. K. O'Nions, Precise determination of femtogram quantities of radium by thermal ionization mass spectrometry, Anal. Chem., 1991, 63(23), 2705–2708, DOI:10.1021/ac00023a008.
- F. Chabaux, D. B. Othman and J. L. Birck, A new Ra-Ba chromatographic separation and its application to Ra mass-spectrometric measurement in volcanic rocks, Chem. Geol., 1994, 114(3–4), 191–197 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/0009254194900523.
- A. P. Fleer and M. P. Bacon, In Notes on Some Techniques of Marine Particle Analysis Used at Whoi, 2013, pp. 223–226, Available from: DOI:10.1029/GM063p0223.
- R. A. Mortlock, R. G. Fairbanks, T. Chiu and J. Rubenstone, 230Th/234U/238U and 231Pa/235U ages from a single fossil coral fragment by multi-collector magnetic-sector inductively coupled plasma mass spectrometry, Geochim. Cosmochim. Acta, 2005, 69(3), 649–657 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0016703704005216.
- M. Levier, M. Roy-Barman, C. Colin and A. Dapoigny, Determination of low level of actinium 227 in seawater and freshwater by isotope dilution and mass spectrometry, Mar. Chem., 2021, 233(March), 103986, DOI:10.1016/j.marchem.2021.103986.
- T. M. Kayzar and R. W. Williams, Developing 226Ra and 227Ac age-dating techniques for nuclear forensics to gain insight from concordant and non-concordant radiochronometers, J. Radioanal. Nucl. Chem., 2016, 307(3), 2061–2068, DOI:10.1007/s10967-015-4435-4.
- M. Boudias, A. Gourgiotis, G. Montavon, C. Cazala, V. Pichon and N. Delaunay, 226Ra and 137Cs determination by inductively coupled plasma mass spectrometry: state of the art and perspectives including sample pretreatment and separation steps, J. Environ. Radioact., 2022, 244–245, 106812 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0265931X22000029.
- L. A. Dietz, C. F. Paghugki and G. A. Land, Internal Standard Technique for Precise Isotopic Abundance Measurments in Thermal Ionization Mass Spectrometry, Anal. Chem., 1962, 34(6), 709–710, DOI:10.1021/ac60186a001.
- H. Cheng, R. Edwards, J. Hoff, C. Gallup, D. Richards and Y. Asmerom, The half-lives of uranium-234 and thorium-230, Chem. Geol., 2000, 169(1–2), 17–33 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0009254199001576.
- S. Richter, A. Alonso-Munoz, R. Eykens, U. Jacobsson, H. Kuehn and A. Verbruggen, et al., The isotopic composition of natural uranium samples—Measurements using the new n(233U)/n(236U) double spike IRMM-3636, Int. J. Mass Spectrom., 2008, 269(1–2), 145–148 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380607003855.
- A. Quemet, V. Dalier and C. Rivier, Americium isotope analysis by TIMS using the total evaporation method: investigation of method accuracy using a new CETAMA/EC-JRC certified reference material, In International Conference on Radioanalytical and Nuclear Chemistry, 5-10 Mai 2019, Budapest, Hongrie, 2019 Search PubMed.
- S. F. Boulyga, A. Koepf, S. Konegger-Kappel, Z. Macsik and G. Stadelmann, Uranium isotope analysis by MC-ICP-MS in sub-ng sized samples, J. Anal. At. Spectrom., 2016, 31(11), 2272–2284 RSC . Available from: http://xlink.rsc.org/?DOI=C6JA00238B.
- T.-Y. Li, X. Wang, C.-J. Chen, M. Tan and Y. Wu, Testing the initial 230Th/232Th for “Known Age Carbonate” and its significance for 230Th dating and paleoclimate research, Quat. Int., 2022, 607(2021), 113–119, DOI:10.1016/j.quaint.2021.07.020.
- T.-C. Chiu, R. G. Fairbanks, R. A. Mortlock, L. Cao, T. W. Fairbanks and A. L. Bloom, Redundant 230Th/234U/238U, 231Pa/235U and 14C dating of fossil corals for accurate radiocarbon age calibration, Quat. Sci. Rev., 2006, 25(17–18), 2431–2440 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0277379106000667.
- F. Chabaux, U-Th-Ra Fractionation During Weathering and River Transport, Rev. Mineral. Geochem., 2003, 52(1), 533–576 CrossRef CAS . Available from: https://pubs.geoscienceworld.org/rimg/article/52/1/533-576/87464.
- K. R. Ludwig, K. R. Simmons, B. J. Szabo, I. J. Winograd, J. M. Landwehr and A. C. Riggs, et al., Mass-Spectrometric 230 Th- 234 U- 238 U Dating of the Devils Hole Calcite Vein, Science, 1992, 258(5080), 284–287, DOI:10.1126/science.258.5080.284.
- G. Hall, Uranium age determination by measuring the 230Th/234U ratio, J. Radioanal. Nucl. Chem., 2005, 264(2), 423–427, DOI:10.1007/s10967-005-0732-7.
- R. W. Williams and A. M. Gaffney, 230 Th- 234 U model ages of some uranium standard reference materials, Proc. Radiochem., 2011, 1(1), 31–35, DOI:10.1524/rcpr.2011.0005/html.
- S. Rihs, E. Lascar, L. Pourcelot, P. Calmon, P. O. Redon and C. Galy, et al., U- and Th- series disequilibria in separated soil mineral fractions: Insight into the mechanism and timescale of U, Th and Ra redistribution, Chem. Geol., 2021, 583, 120455 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0009254121003983.
- R. Fitzgerald, R. Collé, L. Laureano-Pérez, L. Pibida, M. M. Hammond and S. Nour, et al., A new primary standardization of 229Th, Appl. Radiat. Isot., 2010, 68(7–8), 1303–1308 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804310000230.
- NIST, Standard Reference Material 4328C Thorium-229 Radioactivity Standard, National Institute of Standards and Technology, Gaithersburg, 2013 Search PubMed.
- P. de Lavison, L. J. Husband, S. M. Jerome, J. D. Keightley, A. P. Woodman and D. H. Woods, et al., The standardisation of for an environmental yield tracer, Appl. Radiat. Isot., 2000, 53(1–2), 243–249 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804300001779.
- K. W. W. Sims, J. B. Gill, A. Dosseto, D. L. Hoffmann, C. C. Lundstrom and R. W. Williams, et al., An Inter-Laboratory Assessment of the Thorium Isotopic Composition of Synthetic and Rock Reference Materials, Geostand. Geoanal. Res., 2008, 32(1), 65–91, DOI:10.1111/j.1751-908X.2008.00870.x.
- A. Gourgiotis, J. L. Reyss, N. Frank, A. Guihou and C. Anagnostou, Uranium and radium diffusion in organic-rich sediments (sapropels), Geochem., Geophys., Geosyst., 2011, 12(9), 1–15, DOI:10.1029/2011GC003646.
- M. Choi, R. Francois, K. Sims, M. Bacon, S. Brown-Leger and A. Fleer, et al., Rapid determination of 230Th and 231Pa in seawater by desolvated micro-nebulization Inductively Coupled Plasma magnetic sector mass spectrometry, Mar. Chem., 2001, 76(1–2), 99–112 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0304420301000500.
- J. Hiess, D. J. Condon, N. McLean and S. R. Noble, 238 U/235 U Systematics in Terrestrial Uranium-Bearing Minerals, Science, 2012, 335(6076), 1610–1614 CrossRef CAS PubMed . Available from: https://www.cambridge.org/core/product/identifier/CBO9781139167291A041/type/book_part.
- LNHB, Atomic and nuclear data - Table of radionuclides, [cited 2023 Nov 14], Available from: http://www.lnhb.fr/nuclear-data/nuclear-data-table/.
- D. Larivière, D. K. Brownell, V. N. Epov, R. J. Cornett and R. D. Evans, Determination of 226Ra in sediments by ICP-MS: A comparative study of three sample preparation approaches, J. Radioanal. Nucl. Chem., 2007, 273(2), 337–344, DOI:10.1007/s10967-007-6870-3.
- A. M. Volpe, J. A. Olivares and M. T. Murrell, Determination of radium isotope ratios and abundances in geological samples by thermal ionization mass spectrometry, Anal. Chem., 1991, 63(9), 913–916, DOI:10.1021/ac00009a015.
- Z. Varga, Ultratrace-level radium-226 determination in seawater samples by isotope dilution inductively coupled plasma mass spectrometry, Anal. Bioanal. Chem., 2008, 390(2), 511–519, DOI:10.1007/s00216-007-1394-9.
- Y.-T. Hsieh and G. M. Henderson, Precise measurement of 228Ra/226Ra ratios and Ra concentrations in seawater samples by multi-collector ICP mass spectrometry, J. Anal. At. Spectrom., 2011, 26(7), 1338 RSC . Available from: http://xlink.rsc.org/?DOI=c1ja10013k.
- L. I. Bradtmiller, J. F. McManus and L. F. Robinson, 231Pa/230Th evidence for a weakened but persistent Atlantic meridional overturning circulation during Heinrich Stadial 1, Nat. Commun., 2014, 5(1), 5817 CrossRef CAS PubMed . Available from: https://www.nature.com/articles/ncomms6817.
- M. Levier, M. Roy-Barman, L. Foliot, A. Dapoigny and F. Lacan, Distribution of Pa in the Atlantic sector of the Southern Ocean: Tracking scavenging during water mass mixing along neutral density surfaces, Deep-Sea Res. Part I Oceanogr. Res. Pap., 2023, 194, 103951 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0967063722002643.
- D. A. Pickett, M. T. Murrell and R. W. Williams, Determination of Femtogram Quantities of Protactinium in Geologic Samples by Thermal Ionization Mass Spectrometry, Anal. Chem., 1994, 66(7), 1044–1049, DOI:10.1021/ac00079a020.
- M. Regelous, S. Turner, T. Elliott, K. Rostami and C. Hawkesworth, Measurement of Femtogram Quantities of Protactinium in Silicate Rock Samples by Multicollector Inductively Coupled Plasma Mass Spectrometry, Anal. Chem., 2008, 80(1), 344, DOI:10.1021/ac702316r.
- V. P. Chechev and N. K. Kuzmenko, Decay Data Evaluation Project (DDEP): Evaluation of the main 233Pa decay characteristics, Appl. Radiat. Isot., 2006, 64(10–11), 1403–1411 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804306001072.
- R. M. Essex, R. W. Williams, K. C. Treinen, R. Collé, R. Fitzgerald and R. Galea, et al., Preparation and calibration of a 231Pa reference material, J. Radioanal. Nucl. Chem., 2019, 322(3), 1593–1604, DOI:10.1007/s10967-019-06711-6.
- M. Roy-Barman, L. Foliot, E. Douville, N. Leblond, F. Gazeau and M. Bressac, et al., Contrasted release of insoluble elements (Fe, Al, rare earth elements, Th, Pa) after dust deposition in seawater: a tank experiment approach, Biogeosciences, 2021, 18(8), 2663–2678 CrossRef CAS . Available from: https://bg.copernicus.org/articles/18/2663/2021/.
- C.-C. Shen, H. Cheng, R. L. Edwards, S. B. Moran, H. N. Edmonds and J. A. Hoff, et al., Measurement of Attogram Quantities of 231 Pa in Dissolved and Particulate Fractions of Seawater by Isotope Dilution Thermal Ionization Mass Spectroscopy, Anal. Chem., 2003, 75(5), 1075–1079, DOI:10.1021/ac026247r.
- A. Beranoaguirre, C. Pin, M. E. Sanchez-Lorda, S. García de Madinabeitia and J. I. Gil Ibarguchi, An evaluation of Rb-Sr isotope dilution analyses with a 86Sr-enriched tracer and Multiple Collection-ICP-MS, Int. J. Mass Spectrom., 2019, 435, 234–240 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380618302574.
- L. O. Nicolaysen, Graphic interpretation of discordant age measurements on metamorphic rocks, Ann. N. Y. Acad. Sci., 1961, 91(2), 198–206, DOI:10.1111/j.1749-6632.1961.tb35452.x.
- C. Pin, S. Joannon, C. Bosq, B. Le Fèvre and P.-J. Gauthier, Precise determination of Rb, Sr, Ba, and Pb in geological materials by isotope dilution and ICP-quadrupole mass spectrometry following selective separation of the analytes, J. Anal. At. Spectrom., 2003, 18(2), 135–141 RSC . Available from: http://xlink.rsc.org/?DOI=b211832g.
- ISO 17294-2:2023 “Water Quality - Application of Inductively Coupled Plasma Mass Spectrometry (ICP-MS) - Part 2: Determination of Selected Elements Including Uranium Isotopes, 2023 Search PubMed.
- ISO 20899:2018 “Water Quality - Plutonium and Neptunium - Test Method Using ICP-MS, 2018 Search PubMed.
- ISO 18589-4:2019 “Measurement of Radioactivity in the Environment-Soil - Part 4: Plutonium 238 and Plutonium 239 + 240, 2019 Search PubMed.
- L. Pourcelot, O. Masson, L. Saey, S. Conil, B. Boulet and N. Cariou, Empirical calibration of uranium releases in the terrestrial environment of nuclear fuel cycle facilities, J. Environ. Radioact., 2017, 171, 74–82 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0265931X16305586.
- Y. Shao, J. Zhang, M. Luo, D. Xu and L. Ma, A review of anthropogenic radionuclide 236U: Environmental application and analytical advances, J. Environ. Radioact., 2022, 251–252 Search PubMed :106944.Available from: https://linkinghub.elsevier.com/retrieve/pii/S0265931X22001357.
- J. M. Kelley, L. A. Bond and T. M. Beasley, Global distribution of Pu isotopes and 237Np, Sci. Total Environ., 1999, 237–238, 483–500 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0048969799001606.
- United Nations Scientific Committe on the Effects of Atomic Radiation, Sources and Effects of Ionizing Radiation Volume I: Source, United Nations Scientific Committee on the Effects, 2000, vol. I, pp. 1–654 Search PubMed.
- E. Hrnecek, P. Steier and A. Wallner, Determination of plutonium in environmental samples by AMS and alpha spectrometry, Appl. Radiat. Isot., 2005, 63(5–6), 633–638 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804305001417.
- H. Yamana, T. Yamamoto and H. Moriyama, Isotopic ratio of Pu released from fuel cycle facilities — importance of radiochemically pure 236Pu as a tracer, Radioact. Environ., 2001, 1, 31–46 CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S156948600180005X.
- R. Wellum, A. Verbruggen and R. Kessel, A new evaluation of the half-life of 241Pu, J. Anal. At. Spectrom., 2009, 24(6), 801 RSC . Available from: http://xlink.rsc.org/?DOI=b819584f.
- P. Bouisset, M. Nohl, C. Cossonnet, B. Boulet, S. Thomas and N. Cariou, et al., Contribution of close-in fallout from the French atmospheric tests in inventories of 137Cs, 241Am and plutonium (238, 239, 240) in Gambier Islands (French Polynesia) – Signatures of stratospheric fallout in the Southern Hemisphere, J. Environ. Radioact., 2021, 235–236, 106624 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0265931X21000965.
- V. P. Chechev, Standard Reference Data on the Nuclear Characteristics of the Alpha-Radiating Radionuclides 226Ra, 233U, 238Pu, and 239Pu, Meas. Tech., 2001, 44(11), 1134–1137 CrossRef CAS.
- H. Jaegler, F. Pointurier, Y. Onda, A. Hubert, J. P. Laceby and M. Cirella, et al., Plutonium isotopic signatures in soils and their variation (2011-2014) in sediment transiting a coastal river in the Fukushima Prefecture, Japan, Environ. Pollut., 2018, 240, 167–176 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0269749117347875.
- K. Xiong, W. Bu, Y. Ni, X. Liu, J. Zheng and T. Aono, et al., Rapid monitoring of 241Am in small amount of sediment samples by combining extraction chromatography for highly efficient separation of interfering and matrix elements and ICP-MS/MS measurement, Microchem. J., 2023, 190, 108581 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0026265X23001996.
- H. Zhang, X. Hou, J. Qiao and J. Lin, Determination of 241Am in Environmental Samples: A Review, Molecules, 2022, 27(14), 4536 CrossRef CAS PubMed . Available from: https://www.mdpi.com/1420-3049/27/14/4536.
- B. Salbu and E. Holm. Radiochemical methods | Technetium, in Encyclopedia of Analytical Science, Elsevier, 2005, pp. 24–32. Available from: https://linkinghub.elsevier.com/retrieve/pii/B0123693977005197 Search PubMed.
- T. Hayakawa, H. Ko, M.-K. Cheoun, M. Kusakabe, T. Kajino and M. D. Usang, et al., Short-Lived Radioisotope 98Tc Synthesized by the Supernova Neutrino Process, Phys. Rev. Lett., 2018, 121(10), 102701, DOI:10.1103/PhysRevLett.121.102701.
- L. Yang, W. Bu, K. Xiong, Y. Yang and T. Yang, Determination of 99Tc in solid environmental samples by combining alkali fusion and rapid separation using TRans Uranium elements resin in alkali medium to triple quadrupole inductively coupled plasma mass spectrometry, Spectrochim. Acta Part B At. Spectrosc., 2022, 198, 106564 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0584854722002087.
- S. Pommé, J. Paepen and R. Van Ammel, Linearity check of an ionisation chamber through 99 mTc half-life measurements, Appl. Radiat. Isot., 2018, 140, 171–178 CrossRef PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804318302148.
- E. García-Toraño, T. Altzitzoglou, P. Auerbach, M.-M. Bé, C. Bobin and P. Cassette, et al., The half-life of 129I, Appl. Radiat. Isot., 2018, 140, 157–162 CrossRef PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804318303348.
- C. Carrier, H. Azza, A. Michelle, A. Celine, B. Didier and M. Denis, et al., Mass-shift mode to quantify low level 129 I in environmental samples by ICP-MS/MS, J. Anal. At. Spectrom., 2022, 37(6), 1309–1317 RSC . Available from: http://xlink.rsc.org/?DOI=D2JA00128D.
- J. Kim, J. Y. Kim, S. E. Bae, K. Song and J. H. Park, Review of the development in determination of 129I amount and the isotope ratio of 129I/127I using mass spectrometric measurements, Microchem. J., 2021, 169, 106476, DOI:10.1016/j.microc.2021.106476.
- B. S. Matteson, S. K. Hanson, J. L. Miller and W. J. J. Oldham, Concurrent determination of 237Np and Pu isotopes using ICP-MS: Analysis of NIST environmental matrix standard reference materials 4357, 1646a, and 2702, J. Environ. Radioact., 2015, 142, 62–67, DOI:10.1016/j.jenvrad.2015.01.007.
- S. Zhang, Z. Liu, G. Yang, J. Zheng, S. Pan and T. Aono, et al., Rapid Method To Determine 137Cs, 237Np, and Pu Isotopes in Seawater by SF-ICP-MS, Anal. Chem., 2023, 95(46), 16892–16901 CrossRef CAS PubMed.
- S. J. Goldstein, A. A. Price, K. A. Hinrichs, S. P. Lamont, A. J. Nunn and R. S. Amato, et al., High-precision measurement of U-Pu-Np-Am concentrations and isotope ratios in environmental reference materials by mass spectrometry, J. Environ. Radioact., 2021, 237(June), 106689, DOI:10.1016/j.jenvrad.2021.106689.
- N. Qin, J. Zheng, G. Yang and K. Tagami, A comprehensive survey of reference materials for their use in quality assurance for the determination of Np-237 in environmental samples, J. Environ. Radioact., 2024, 271(August 2023), 107328, DOI:10.1016/j.jenvrad.2023.107328.
- K. Mayer, M. Wallenius and Z. Varga, Nuclear Forensic Science: Correlating Measurable Material Parameters to the History of Nuclear Material, Chem. Rev., 2013, 113(2), 884–900, DOI:10.1021/cr300273f.
- M. J. Kristo, A. M. Gaffney, N. Marks, K. Knight, W. S. Cassata and I. D. Hutcheon, Nuclear Forensic Science: Analysis of Nuclear Material Out of Regulatory Control, Annu. Rev. Earth Planet Sci., 2016, 44(1), 555–579 CrossRef CAS . Available from: https://www.annualreviews.org/doi/10.1146/annurev-earth-060115-012309.
- Z. Varga, M. Wallenius, M. Krachler, N. Rauff-Nisthar, L. Fongaro and A. Knott, et al., Trends and perspectives in Nuclear Forensic Science, Trac. Trends Anal. Chem., 2022, 146, 116503, DOI:10.1016/j.trac.2021.116503.
- Z. Varga, C. Venchiarutti, A. Nicholl, J. Krajkó, R. Jakopič and K. Mayer, et al., IRMM-1000a and IRMM-1000b uranium reference materials certified for the production date. Part I: methodology, preparation and target characteristics, J. Radioanal. Nucl. Chem., 2016, 307(2), 1077–1085, DOI:10.1007/s10967-015-4227-x.
- C. Venchiarutti, Z. Varga, S. Richter, A. Nicholl, J. Krajko and R. Jakopič, et al., IRMM-1000a and IRMM-1000b: uranium reference materials certified for the production date based on the 230Th/234U radiochronometer. Part II: certification, J. Radioanal. Nucl. Chem., 2016, 308(1), 105–111, DOI:10.1007/s10967-015-4368-y.
- S. J. Goldstein, M. T. Murrell and R. W. Williams, Half-life of 229Th, Phys. Rev. C, 1989, 40(6), 2793–2795, DOI:10.1103/PhysRevC.40.2793.
- H. Kikunaga, T. Suzuki, M. Nomura, T. Mitsugashira and A. Shinohara, Determination of the half-life of the ground state of 229Th by using 232U and 233U decay series, Phys. Rev. C, 2011, 84(1), 014316, DOI:10.1103/PhysRevC.84.014316.
- Z. Varga, A. Nicholl and K. Mayer, Determination of the 229Th half-life, Phys. Rev. C, 2014, 89(6), 064310, DOI:10.1103/PhysRevC.89.064310.
- R. M. Essex, J. L. Mann, R. Collé, L. Laureano-Perez, M. E. Bennett and H. Dion, et al., New determination of the 229Th half-life, J. Radioanal. Nucl. Chem., 2018, 318(1), 515–525 CrossRef CAS PubMed.
- G. R. Eppich, R. W. Williams, A. M. Gaffney and K. C. Schorzman, 235U–231Pa age dating of uranium materials for nuclear forensic investigations, J. Anal. At. Spectrom., 2013, 28(5), 666 RSC . Available from: http://xlink.rsc.org/?DOI=c3ja50041a.
- K. C. Treinen, A. M. Gaffney, J. M. Rolison, K. M. Samperton, K. C. McHugh and M. L. Miller, et al., Improved protactinium spike calibration method applied to 231Pa–235U age-dating of certified reference materials for nuclear forensics, J. Radioanal. Nucl. Chem., 2018, 318(1), 209–219, DOI:10.1007/s10967-018-6149-x.
- S. K. Aggarwal and D. Alamelu, A novel approach for the determination of 238Pu by thermal ionization mass spectrometry (TIMS) using interfering element correction methodology, Int. J. Mass Spectrom., 2005, 241(1), 83–88 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380604004300.
- D. Alamelu, P. S. Khodade, P. M. Shah and S. K. Aggarwal, Investigations on atomic and oxide ion formation of plutonium and uranium in thermal ionization mass spectrometry (TIMS) for determination of 238Pu, Int. J. Mass Spectrom., 2004, 239(1), 51–56 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380604003756.
- R. Jakopič, Y. Aregbe, S. Richter, E. Zuleger, S. Mialle and S. D. Balsley, et al., Verification measurements of the IRMM-1027 and the IAEA large-sized dried (LSD) spikes, J. Radioanal. Nucl. Chem., 2017, 311(3), 1781–1791, DOI:10.1007/s10967-016-5123-8.
- M. Sumi, K. Abe, T. Kageyama, H. Nakazawa, A. Kurosawa, M. Yamamoto, et al., Verification of LSD spikes prepared in Japan from a MOX source material, in Proceedings of INMM 51st Annual Meeting (CD-ROM), Baltimore, USA, 2010 Search PubMed.
- M. Sumi, K. Abe, T. Kageyama and P. Mason, Pu standard material preparation in Japan, In INMM 55th Annual Meeting, Atalante, USA, 2014 Search PubMed.
- A.-M. Sanchez-Hernandez, K. Toth, Y. Aregbe, N. Banik, J. Bauwens, R. Buda, et al., Alternative nuclear certified reference materials for safeguards and industry, In ESARDA 39th Annual Meeting 2017 Symposium, Duesseldorf, Germany, 2017, Available from: https://publications.jrc.ec.europa.eu/repository/handle/JRC110509 Search PubMed.
- K. Toth, A.-M. S. Hernández, J. Bauwens, R. Buják, R. C. Marquez and R. Jakopič, et al., Embedding matrices to extend the shelf life of reference materials 1: Cellulose acetate butyrate, Polym. Degrad. Stabil., 2022, 202, 110024 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0141391022002075.
- K. Toth, R. Buják, A. M. S. Hernández, J. Bauwens, R. C. Marquez and R. Buda, et al., Research on long-term stability of mixed U and Pu large-sized dried (LSD) spikes for fissile material control, Prog. Nucl. Sci. Technol., 2018, 5, 48–51 CrossRef . Available from: https://www.aesj.net/document/pnst005/data/048_051.pdf.
- S. Konegger-Kappel, A. Koepf, R. Katona, M. R. Kilburn and S. F. Boulyga, A new era of plutonium assay and isotope ratio measurements at ultra-low levels in IAEA safeguards environmental swipe samples by isotope dilution MC-ICP-MS using a 244Pu spike, J. Radioanal. Nucl. Chem., 2023, 332(7), 2887–2904, DOI:10.1007/s10967-023-08880-x.
- ASTM C1672-17 - Standard Test Method for Determination of Uranium or Plutonium Isotopic Composition or Concentration by the Total Evaporation Method Using a Thermal Ionization Mass Spectrometer Search PubMed.
- S. Bürger, S. D. Balsley, S. Baumann, J. Berger, S. F. Boulyga and J. A. Cunningham, et al., Uranium and plutonium analysis of nuclear material samples by multi-collector thermal ionisation mass spectrometry: Quality control, measurement uncertainty, and metrological traceability, Int. J. Mass Spectrom., 2012, 311, 40–50 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380611004702.
- C.-K. Kim, A. Bosko, D. Nakazawa, G. Duhamel, K. Mise, S. Hara, et al., A Combined Method of TIMS and HRGS for Rapid Determination of Pu Concentrations in Safeguards Samples at the On-Site Laboratory in the Rokkasho Reprocessing Plant, Symposium on International Safeguards: Building Future Safeguards Capabilities, In Symposium on International Safeguards: Building Future Safeguards Capabilities, Vienna, Austria, 2018, vol. 5–8 Search PubMed.
- U. Repinc, Application of strip isotope dilution analysis to safeguards samples containing low levels of uranium and plutonium, In International Conference on Radioanalytical and Nuclear Chemistry (RANC-2023), Budapest from 7 – 12 May 2023 Search PubMed.
- M. H. A. Piro and K. Lipkina, Mining and milling, In Advances in Nuclear Fuel Chemistry, Elsevier, 2020, pp. 315–329. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780081025710000094 Search PubMed.
- A. Beaumais, A. Nonell, C. Caussignac, S. Mialle, G. Stadelmann and M. Janin, et al., Determination of the 144 Ce/238 U atomic ratio in spent nuclear fuel using double spike isotope dilution mass spectrometry, J. Anal. At. Spectrom., 2022, 37, 1288–1297, 10.1039/D2JA00052K.
- A. Quemet, C. Maillard and A. Ruas, Determination of zirconium isotope composition and concentration for nuclear sample analysis using Thermal Ionization Mass Spectrometry, Int. J. Mass Spectrom., 2015, 392, 34–40 CrossRef CAS.
- S. Baghdadi, A. Quémet, E. Esbelin, Y. Manidren, S. Gracia and V. Dalier, et al., Zr precipitation kinetics in irradiated fuel dissolution solution by TIMS and ICP-MS: a combined study, J. Radioanal. Nucl. Chem., 2017, 1–6 Search PubMed.
- A. Inoue, Mechanism of the oxidative dissolution of UO2 in HNO3 solution, J. Nucl. Mater., 1986, 138(1), 152–154 CrossRef CAS . Available from: https://linkinghub.elsevier.com/retrieve/pii/0022311586902710.
- N. Desigan, N. Bhatt, M. A. Shetty, G. K. P. Sreekumar, N. K. Pandey and U. Kamachi Mudali, et al., Dissolution of nuclear materials in aqueous acid solutions, Rev. Chem. Eng., 2019, 35(6), 707–734, DOI:10.1515/revce-2017-0063/html.
- V. N. Momotov, E. A. Erin and D. E. Tikhonova, Dissolution of Spent Nuclear Fuel Samples for Analytical Purposes, Radiochemistry, 2022, 64(5), 551–580, DOI:10.1134/S1066362222050010.
- F. Liu, T. H. Yan, B. Li and G. A. Ye, Dissolution behavior of irradiated fuels in nitric acid and characteristics of insoluble residue, J. Radioanal. Nucl. Chem., 2020, 326(1), 337–341, DOI:10.1007/s10967-020-07350-y.
- S. Van Winckel, R. Alvarez-Sarandes, D. Serrano Purroy and L. Aldave de las Heras, Assay data of spent nuclear fuel: the lab-work behind the numbers, Front. Energy Res., 2023, 11 DOI:10.3389/fenrg.2023.1168460.
- M. Crozet and C. Rivier, Impact of dissolution on the uncertainty of spent fuel analysis, J. Radioanal. Nucl. Chem., 2013, 298(1), 325–336, DOI:10.1007/s10967-013-2518-7.
- S. Röllin, Z. Kopatjtic, B. Wernli and B. Magyar, Determination of lanthanides and actinides in uranium materials by high-performance liquid chromatography with inductively coupled plasma mass spectrometric detection, J. Chromatogr. A, 1996, 739(1–2), 139–149 CrossRef.
- M. Betti, Use of ion chromatography for the determination of fission products and actinides in nuclear applications, J. Chromatogr. A, 1997, 789(1–2), 369–379 CrossRef CAS.
- S. F. Wolf, D. L. Bowers and J. C. Cunnane, Analysis of high burnup spent nuclear fuel by ICP-MS, J. Radioanal. Nucl. Chem., 2005, 263(3), 581–586 CrossRef CAS.
- A. Morgenstern, C. Apostolidis, R. Carlos-Marquez, K. Mayer and R. Molinet, Single-column extraction chromatographic separation of U, Pu, Np and Am, Radiochim. Acta, 2002, 90(2), 81–85 CrossRef CAS.
- J. W. Grate, M. J. O'Hara, A. F. Farawila, M. Douglas, M. M. Haney and S. L. Petersen, et al., Extraction chromatographic methods in the sample preparation sequence for thermal ionization mass spectrometric analysis of plutonium isotopes, Anal. Chem., 2011, 83, 9086–9091 CrossRef CAS PubMed.
- K. Van Hoecke, J. Bussé, M. Gysemans, L. Adriaensen, A. Dobney and T. Cardinaels, Isolation of lanthanides from spent nuclear fuel by means of high performance ion chromatography (HPIC) prior to mass spectrometric analysis, J. Radioanal. Nucl. Chem., 2017, 314(3), 1727–1739 CrossRef CAS.
- H. Isnard, M. Aubert, P. Blanchet, R. Brennetot, F. Chartier and V. Geertsen, et al., Determination of 90Sr/238U ratio by double isotope dilution inductively coupled plasma mass spectrometer with multiple collection in spent nuclear fuel samples with in situ 90Sr/90Zr separation in a collision-reaction cell, Spectrochim. Acta Part B At. Spectrosc., 2006, 61(2), 150–156 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0584854705003435.
- P. Regge and R. Boden, Determination of neodymium isotopes as burnup indicator of highly irradiated (U, Pu)O2 lmfbr fuel, J. Radioanal. Chem., 1977, 35(1), 173–184, DOI:10.1007/BF02518224.
- E. A. Erin, V. N. Momotov, A. Y. Volkov, M. I. Khamdeev, V. N. Kupriyanov and A. Y. Shadrin, et al., Radiochemical analysis of a sample of mixed uranium–plutonium nitride fuel, Radiochemistry, 2017, 59(4), 372–378, DOI:10.1134/S1066362217040099.
- B. D. Roach, K. T. Rogers, N. A. Zirakparvar, J. S. Delashmitt, S. C. Metzger and B. T. Manard, et al., The need for speed – Burnup determination of spent nuclear fuel, Talanta Open, 2022, 6, 100152 CrossRef . Available from: https://linkinghub.elsevier.com/retrieve/pii/S2666831922000698.
- J. S. Kim, Y. S. Jeon, S. D. Park, Y. K. Ha and K. Song, Analysis of high burnup pressurized water reactor fuel using uranium, plutonium, neodymium, and cesium isotope correlations with burnup, Nucl. Eng. Technol., 2015, 47(7), 924–933, DOI:10.1016/j.net.2015.08.002.
- ASTM International, ASTM E321-20 - Standard Test Method for Atom Percent Fission in Uranium and Plutonium Fuel (Neodymium-148 Method), West Conshohocken, PA, USA, 2021 Search PubMed.
- K. Govers, L. Adriaensen, A. Dobney, M. Gysemans, C. Cachoir and M. Verwerft, Evaluation of the irradiation-averaged fission yield for burnup determination in spent fuel assays, EPJ Nucl. Sci. Technol., 2022, 8 DOI:10.1051/epjn/2022018.
- H. Isnard, M. Granet, C. Caussignac, E. Ducarme, A. Nonell and B. Tran, et al., Comparison of thermal ionization mass spectrometry and Multiple Collector Inductively Coupled Plasma Mass Spectrometry for cesium isotope ratio measurements, Spectrochim. Acta Part B At. Spectrosc., 2009, 64(11–12), 1280–1286, DOI:10.1016/j.sab.2009.10.004.
- V. Bulovič, J. Krtil, F. Sus, Z. Maksimovic and E. Klosova, Determination of the balance of the nuclear reactor fuel burn-out process by gamma-spectrometry of fission products III, J. Radioanal. Chem., 1983, 80(1–2), 199–207, DOI:10.1007/BF02517663.
- J. R. Phillips, B. K. Barnes and T. R. Bement, Correlation of the Cesium-134/Cesium-137 Ratio to Fast Reactor Burnup, Nucl. Technol., 1979, 46(1), 21–29, DOI:10.13182/NT46-21.
- L. Pibida, C. A. McMahon and B. A. Bushaw, Laser resonance ionization mass spectrometry measurements of cesium in nuclear burn-up and sediment samples, Appl. Radiat. Isot., 2004, 60(2–4), 567–570 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804303002975.
- A. P. Smirnov-Averin, V. I. Galkov, I. G. Sheinker, V. P. Meshcheryakov, L. A. Stabenova and B. S. Kir’yanov, Determining burn-up in spent fuel elements, Sov. J. At. Energy, 1962, 11(5), 1102–1104, DOI:10.1007/BF01490500.
- M. Granet, A. Nonell, G. Favre, F. Chartier, H. Isnard and J. Moureau, et al., Cs-Ba separation using N2O as a reactant gas in a Multiple Collector-Inductively Coupled Plasma Mass Spectrometer collision-reaction cell: Application to the measurements of Cs isotopes in spent nuclear fuel samples, Spectrochim. Acta Part B At. Spectrosc., 2008, 63(11), 1309–1314 CrossRef.
- J. S. Becker, R. S. Soma, K. L. Sutton, J. A. Caruso and H.-J. Dietze, Determination of long-lived radionuclides by inductively coupled plasma quadrupole mass spectrometry using different nebulizers, J. Anal. At. Spectrom., 1999, 14(6), 933–937 RSC . Available from: http://xlink.rsc.org/?DOI=a809252d.
- S. F. Boulyga, Mass spectrometric analysis of long-lived radionuclides in bio-assays, Int. J. Mass Spectrom., 2011, 307(1–3), 200–210, DOI:10.1016/j.ijms.2011.02.012.
- C. Bouvier-Capely, J. Ritt, N. Baglan and C. Cossonnet, Potentialities of mass spectrometry (ICP-MS) for actinides determination in urine, Appl. Radiat. Isot., 2004, 60(5), 629–633 CrossRef CAS PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969804304000132.
- I. T. Platzner, J. S. Becker and H. Dietze, Stability Study of Isotope Ratio Measurements for Uranium and Thorium by ICP-QMS, At. Spectr., 1999, 20(February), 6–12 CAS.
- W. S. Kinman, S. P. Lamont and R. E. Steiner, A rapid isotope dilution inductively coupled plasma mass spectrometry procedure for uranium bioassay, J. Radioanal. Nucl. Chem., 2009, 282(3), 1027–1030, DOI:10.1007/s10967-009-0204-6.
- J. G. Arnason, C. N. Pellegri and P. J. Parsons, Determination of uranium isotope ratios in human urine by sector field inductively coupled plasma mass spectrometry for use in occupational and biomonitoring studies, J. Anal. At. Spectrom., 2013, 28(9), 1410 RSC . Available from: http://xlink.rsc.org/?DOI=c3ja50151e.
- C. Li, K. Benkhedda, S. Tolmachev, L. Carty, R. Ko and D. Moir, et al., Measurement of 236U in human tissue samples using solid phase extraction coupled to ICP-MS, J. Anal. At. Spectrom., 2010, 25(5), 730 RSC . Available from: http://xlink.rsc.org/?DOI=b923909j.
- H. Kurosaki, A. A. Tucker, S. E. Iden, S. M. Sexton, B. D. Gonzalez and G. R. Rao, Use of chromatographic pre-concentration for routine uranium bioassay analysis by ICP-MS, J. Radioanal. Nucl. Chem., 2013, 298(2), 1017–1022, DOI:10.1007/s10967-013-2437-7.
- Y. Ni, J. Zheng, Q. Guo, W. Men, K. Tagami and S. Uchida, Rapid determination of ultra-trace plutonium isotopes (239Pu, 240Pu and 241Pu) in small-volume human urine bioassay using sector-field inductively coupled plasma mass spectrometry, Anal. Chim. Acta, 2018, 1000, 85–92, DOI:10.1016/j.aca.2017.10.012.
- E. Werner, P. Roth, I. Wendler, P. Schramel, H. Hellmann and U. Kratzel, Feasibility of ICP-MS for the assessment of uranium excretion in urine, J. Radioanal. Nucl. Chem., 1997, 226(1–2), 201–203, DOI:10.1007/BF02063647.
- E. J. Wyse, J. A. MacLellan, C. W. Lindenmeier, J. P. Bramson and D. W. Koppenaal, Actinide bioassays by ICPMS, J. Radioanal. Nucl. Chem., 1998, 234(1–2), 165–170, DOI:10.1007/BF02389766.
- Y. Shi, X. Dai, R. Collins and S. Kramer-Tremblay, Rapid determination of uranium isotopes in urine by inductively coupled plasma-mass spectrometry, Health Phys., 2011, 101(2), 148–153 CrossRef CAS PubMed . Available from: https://journals.lww.com/00004032-201108000-00008.
- Y. Shi, R. Collins and C. Broome, Determination of uranium, thorium and plutonium isotopes by ICP-MS, J. Radioanal. Nucl. Chem., 2013, 296(1), 509–515, DOI:10.1007/s10967-012-2128-9.
- Y. Shi, X. Dai, C. Li, R. Collins, S. Kramer-Tremblay and R. Riopel, et al., ICP-MS method for Pu and Np isotopes in population monitoring by a micro-flow injection sample introduction system, J. Anal. At. Spectrom., 2014, 29(9), 1708–1713 RSC . Available from: http://xlink.rsc.org/?DOI=C4JA00105B.
- P. J. Gray, L. Zhang, H. Xu, M. McDiarmid, K. Squibb and J. A. Centeno, Determination of 236U/238U and 235U/238U isotope ratios in human urine by inductively coupled plasma mass spectrometry, Microchem. J., 2012, 105, 94–100, DOI:10.1016/j.microc.2012.07.004.
- H. Hernández-Mendoza, E. Chamizo, A. Delgado, M. García-León and A. Yllera, Comparison of methods and application of alpha spectrometry and mass spectrometry techniques for 239Pu determination in biological samples, J. Anal. At. Spectrom., 2011, 26(7), 1509 RSC . Available from: http://xlink.rsc.org/?DOI=c0ja00093k.
- A. A. Reinhard, J. D. Inglis, R. E. Steiner, S. P. LaMont, A. R. Cardon and A. M. Wende, et al., An improved method for uranium separation from urine for rapid bioassay by inductively coupled plasma mass spectrometry, J. Radioanal. Nucl. Chem., 2020, 326(1), 611–619, DOI:10.1007/s10967-020-07335-x.
- D. Clases and R. Gonzalez de Vega, Facets of ICP-MS and their potential in the medical sciences—Part 1: fundamentals, stand-alone and hyphenated techniques, Anal. Bioanal. Chem., 2022, 414(25), 7337–7361, DOI:10.1007/s00216-022-04259-1.
- D. Clases and R. Gonzalez de Vega, Facets of ICP-MS and their potential in the medical sciences—Part 2: nanomedicine, immunochemistry, mass cytometry, and bioassays, Anal. Bioanal. Chem., 2022, 414(25), 7363–7386, DOI:10.1007/s00216-022-04260-8.
- Y. Liu, G. Xiao and R. L. Jones, High-Throughput Determination of Ultratrace Actinides in Urine by In-Line Extraction Chromatography Combined with Quadrupole Inductively Coupled Plasma Mass Spectrometry (EC-ICP-MS), Anal. Chem., 2022, 94(51), 18042–18049, DOI:10.1021/acs.analchem.2c04458.
- G. Xiao and J. Button, Rapid determination of 235U/238U in urine using Q-ICP-MS by a simple dilute-and-shoot approach, J. Radioanal. Nucl. Chem., 2023, 332(1), 185–191, DOI:10.1007/s10967-022-08713-3.
- B. C. Russell, P. E. Warwick, H. Mohamud, O. Pearson, Y. Yu and H. Thompkins, et al., Development of a single method for direct measurement of multiple radionuclides using ICP-MS/MS, J. Anal. At. Spectrom., 2023, 38(1), 97–110 RSC . Available from: http://xlink.rsc.org/?DOI=D2JA00174H.
- V. N. Epov, K. Benkhedda, R. J. Cornett and R. D. Evans, Rapid determination of plutonium in urine using flow injection on-line preconcentration and inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2005, 20(5), 424–430 RSC.
- P. Schramel, I. Wendler, P. Roth and E. Werner, Method for the determination of thorium and uranium in urine by ICP-MS, Mikrochim. Acta, 1997, 126(3–4), 263–266, DOI:10.1007/BF01242331.
- R. S. Pappas, B. G. Ting, J. M. Jarrett, D. C. Paschal, S. P. Caudill and D. T. Miller, Determination of uranium-235, uranium-238 and thorium-232 in urine by magnetic sector inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2002, 17(2), 131–134 RSC . Available from: http://xlink.rsc.org/?DOI=b108414c.
- J. W. Ejnik, T. I. Todorov, F. G. Mullick, K. Squibb, M. A. McDiarmid and J. A. Centeno, Uranium analysis in urine by inductively coupled plasma dynamic reaction cell mass spectrometry, Anal. Bioanal. Chem., 2005, 382(1), 73–79, DOI:10.1007/s00216-005-3173-9.
- J. Qiao, X. Hou, P. Roos and M. Miró, Bead injection extraction chromatography using high-capacity lab-on-valve as a front end to inductively coupled plasma mass spectrometry for urine radiobioassay, Anal. Chem., 2013, 85(5), 2853–2859 CrossRef CAS PubMed.
- S. Baghdadi, G. Cote, C. Beyaert, B. Mane, A. Peroux and L. Masclet, et al., Coupling between a calix[6]arene-based chromatography column and ICP-MS for on-line actinide analysis, J. Anal. At. Spectrom., 2016, 31(7), 1406–1413 RSC . Available from: http://xlink.rsc.org/?DOI=C6JA00063K.
- G. Yang, J. Zheng, E. Kim, S. Zhang, H. Seno and M. Kowatari, et al., Rapid analysis of 237Np and Pu isotopes in small volume urine by SF-ICP-MS and ICP-MS/MS, Anal. Chim. Acta, 2021, 1158, 338431, DOI:10.1016/j.aca.2021.338431.
- D. Larivière, T. A. Cumming, S. Kiser, C. Li and R. J. Cornett, Automated flow injection system using extractionchromatography for the determination of plutonium in urine by inductively coupled plasma mass spectrometry, J. Anal. At. Spectrom., 2008, 23(3), 352–360 RSC . Available from: http://xlink.rsc.org/?DOI=B714135A.
- H. Wang, Y. Ni, J. Zheng, Z. Huang, D. Xiao and T. Aono, Low-temperature fusion using NH4HSO4 and NH4HF2 for rapid determination of Pu in soil and sediment samples, Anal. Chim. Acta, 2019, 1050, 71–79, DOI:10.1016/j.aca.2018.10.065.
- D. Clases, M. Birka, M. Sperling, A. Faust and U. Karst, Isobaric dilution analysis as a calibration tool for long lived radionuclides in ICP-MS, J. Trace Elem. Med. Biol., 2017, 40, 97–103, DOI:10.1016/j.jtemb.2017.01.002.
- D. L. Arbova, S. Y. Tolmachev and J. D. Brockman, ICP-MS analysis of actinides in brain tissue of an occupationally exposed individual, J. Radioanal. Nucl. Chem., 2022, 331(12), 5147–5151, DOI:10.1007/s10967-022-08460-5.
- D. M. Bunk, Reference materials and reference measurement procedures: an overview from a national metrology institute, Clin. Biochem. Rev., 2007, 28(4), 131–137 Search PubMed . Available from: http://www.ncbi.nlm.nih.gov/pubmed/18392127%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2282405.
- C. Venchiarutti, R. Jakopič, C. Hennessy and K. Toth, Preparation and characterisation of uranium and plutonium quality control samples for isotope dilution mass spectrometry measurements and uncertainty estimation, J. Radioanal. Nucl. Chem., 2021, 327(3), 1305–1316, DOI:10.1007/s10967-020-07593-9.
- M. Crozet, D. Roudil, C. Maillard, C. Rivier, R. Jakopič and A. Fankhauser, et al., Contribution of an interlaboratory comparison to the certification of the STAM/IRMM-0243 243Am reference material, J. Radioanal. Nucl. Chem., 2019, 319(3), 717–725, DOI:10.1007/s10967-018-06404-6.
- C. Venchiarutti, C. Hennessy, R. Jakopič, Y. Aregbe, S. Richter and U. Jacobsson, Preparation and certification of a new batch of 242Pu spike IRMM-049e, JRC Ref. Mater. Rep., 2017 DOI:10.2760/115196.
- S. Richter, J. Truyens, C. Venchiarutti, Y. Aregbe, R. Middendorp and S. Neumeier, et al., Certification of the First Uranium Oxide micro-particle reference materials for Nuclear Safety and Security, IRMM-2329P and IRMM-2331P, J. Radioanal. Nucl. Chem., 2022, 1–5, DOI:10.1007/s10967-022-08255-8.
- K. J. Mathew, F. E. Stanley, M. R. Thomas, K. J. Spencer, L. P. Colletti and L. Tandon, Critical need for plutonium and uranium isotopic standards with lower uncertainties, Anal. Methods, 2016, 8, 7289–7305 RSC.
- A. A. Specht and D. Beauchemin, Automated On-Line Isotope Dilution Analysis with ICP-MS Using Sandwich Flow Injection, Anal. Chem., 1998, 70(5), 1036–1040, DOI:10.1021/ac9710691.
- K. G. Heumann, S. M. Gallus, G. Raedlinger and J. Vogl, Accurate determination of element species by on-line coupling of chromatographic systems with ICP-MS using isotope dilution technique, Spectrochim. Acta Part B At. Spectrosc., 1998, 53(2), 273–287 CrossRef.
- G. Centineo, P. Rodríguez-González, E. B. González, J. I. García Alonso, A. Sanz-Medel and N. F. Cardona, et al., Isotope dilution GC-MS routine method for the determination of butyltin compounds in water, Anal. Bioanal. Chem., 2006, 384(4), 908–914, DOI:10.1007/s00216-005-0061-2.
- N. Tretyakova, M. Goggin, D. Sangaraju and G. Janis, Quantitation of DNA Adducts by Stable Isotope Dilution Mass Spectrometry, Chem. Res. Toxicol., 2012, 25(10), 2007–2035, DOI:10.1021/tx3002548.
- S. F. Boulyga, D. Desideri, M. A. Meli, C. Testa and J. S. Becker, Plutonium and americium determination in mosses by laser ablation ICP-MS combined with isotope dilution technique, Int. J. Mass Spectrom., 2003, 226(3), 329–339 Search PubMed . Available from: https://linkinghub.elsevier.com/retrieve/pii/S1387380603000241.
- S. Paul, A. Sarkar, D. Alamelu, R. V. Shah and S. K. Aggarwal, Isotope dilution gamma spectrometry for Pu using low energy photons, Radiochim. Acta, 2012, 100(5), 291–296, DOI:10.1524/ract.2012.1919/html.
- M. H. Ramsey, S. L. R. Ellison, H. Czichos, W. Hässelbarth, H. Ischi, W. Wegscheider, et al., Quality in Measurement and Testing, In Springer Handbook of Metrology and Testing, Berlin, Heidelberg, Springer Berlin Heidelberg, 2011, pp. 39–141. Available from: DOI:10.1007/978-3-642-16641-9_3.
- ISO/CASCO Committee on conformity assessment, General Requirements for the Competence of Testing and Calibration Laboratories, ISO/IEC 17025:2017, 2017 Search PubMed.
- IAEA, Technical Meeting on Nuclear Material Round Robin, [cited 2023 Oct 3], Available from: https://nucleus.iaea.org/sites/SGAS-TM/SGASTMS/NMRORO Search PubMed.
Footnotes |
| † In memoriam: René Brennetot |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00029c |
| § All the calculations presented in this paragraph were made using R software (free to use).99 |
| This journal is © The Royal Society of Chemistry 2024 |