
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLab-on-chip technologies for exploring the gut–immune axis in metabolic disease
Alexandra E.
Wheeler
,
Verena
Stoeger
 and
Róisín M.
Owens
*
and
Róisín M.
Owens
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, UK. E-mail: rmo37@cam.ac.uk
First published on 16th January 2024
Abstract
The continued rise in metabolic diseases such as obesity and type 2 diabetes mellitus poses a global health burden, necessitating further research into factors implicated in the onset and progression of these diseases. Recently, the gut–immune axis, with diet as a main regulator, has been identified as a possible role player in their development. Translation of conventional 2D in vitro and animal models is however limited, while human studies are expensive and preclude individual mechanisms from being investigated. Lab-on-chip technology therefore offers an attractive new avenue to study gut–immune interactions. This review provides an overview of the influence of diet on gut–immune interactions in metabolic diseases and a critical analysis of the current state of lab-on-chip technology to study this axis. While there has been progress in the development of “immuno-competent” intestinal lab-on-chip models, with studies showing the ability of the technology to provide mechanical cues, support longer-term co-culture of microbiota and maintain in vivo-like oxygen gradients, platforms which combine all three and include intestinal and immune cells are still lacking. Further, immune cell types and inclusion of microenvironment conditions which enable in vivo-like immune cell dynamics as well as host-microbiome interactions are limited. Future model development should focus on combining these conditions to create an environment capable of hosting more complex microbiota and immune cells to allow further study into the effects of diet and related metabolites on the gut–immune ecosystem and their role in the prevention and development of metabolic diseases in humans.
1. Introduction
Obesity is thought to be at epidemic levels1 affecting over 1 billion people globally.2 Besides a reduction in life quality, obesity presents a major burden for healthcare systems and economies, with the cost of overweight and obese populations estimated to reach over $4 trillion by 2035.3 Further, it is a risk factor for the development of other metabolic disorders including type 2 diabetes mellitus (T2D)4 as well as a state known as metabolic syndrome (metS), which is similarly associated with insulin resistance and other metabolic abnormalities.1 Obesity, T2D and metS are therefore interlinked conditions5 which together pose a major global health crisis.The rise of metabolic diseases is likely linked to the modern lifestyle, with contributory factors such as high fat/high sugar diets, limited exercise6 as well as possible exposure to pollution and xenobiotics,7,8 amongst others. Prevention and early stage treatment for these diseases therefore often includes lifestyle and dietary interventions.1,4 Failing this, numerous therapies and medical procedures exist to assist in managing the conditions, however these may suffer with long term efficacy issues9 and side effects.1,10 Further, given that multiple pathophysiological imbalances exist in these diseases, treatment plans may require a combination of multiple therapies and interventions.4 Despite their global scale, much research is still required to better understand factors involved in the onset and progression of these diseases as well as the development of better preventive interventions and treatment options.
Beside other commonalities, a state of chronic low grade systemic inflammation1 affecting multiple organs and tissues is found across these diseases.11 Adipose tissue and pancreatic islets are perhaps the most obvious tissues implicated, however, recently, the gut has also emerged as a possible key role player, with a number of studies identifying a possible causative role for the gut and aberrations in gut microbiota, known as gut dysbiosis, in the development of chronic low-grade inflammation and metabolic diseases.12–16
Animal and 2D cell models have classically been used for metabolic and immune research, however translation and relevance issues limit their applicability to the human condition.17,18 Besides differences in microbiota composition and the intestinal immune system in animal models,17 it is also challenging to untangle how individual factors affect the development or progression of these diseases.19 Further, static 2D models lack physiological tissue morphology, key intestinal functions, and cannot support long-term co-culture of microbes, quickly leading to overgrowth.20–22 Together, these highlight a need for the development of robust in vitro models to study this axis further.
Recent advances in tissue engineering have led to improved 3D tissue models which enable integration of multiple cell types and better mimic the in vivo microenvironment.23,24 The advent of organoid research25 has further improved in vitro models, enabling the complexity of tissues such as the intestinal epithelium to be better recapitulated.17,26 These models, however, generally do not allow for mechanical cues such as fluid flow and peristalsis-like mechanical deformations. Microfluidic lab- and organ-on-chip technology therefore presents a crucial next step, facilitating these cues20,27–32 and supporting co-culture of multiple cell types, including organoid-derived cells and immune cells,29,33 as well as longer-term co-culture of microbes,19 crucial to advancing our understanding of the role the gut and gut microbes play in metabolic diseases.
Given the combined role of intestinal epithelial cells and immune cells in metabolism and inflammation, together with the associated complex set of dynamic interactions and signalling pathways, the rise of lab-on-chip technology offers a promising means to study these interactions and elucidate the role particular cells and signalling cascades play in the onset and progression of metabolic diseases. Thus far however, the incorporation of the immune system has been somewhat limited, despite its central role in maintaining intestinal homeostasis.18 Recently, several “immuno-competent” intestinal lab-on-chip platforms have been developed, however further work is required to capture the complexity and interactions between different cells in these models.
In this review we provide a brief overview of the structure of the gut and associated immune tissue to introduce the complexities involved with the gut–immune axis and how these may be challenging to recapitulate in vitro. We next provide an overview of the primary inflammatory mechanisms identified in the gut–immune axis as playing a role in metabolic diseases and discuss metabolic cross-talk factors, with a focus on diet, which have generally relied on animal or human nutritional and observational studies due to limitations in available in vitro models. Finally, we review the state of lab-on-chip technology for studying this axis and provide an evaluation of its readiness to address the current limitations in in vitro models for metabolic disease research.
2. The metabolic gut–immune axis
2.1. Intestinal structure
The small and large intestine are similar in overall structure34,35 in that both comprise a hollow lumen surrounded by four main layers of tissue known as the mucosa, the submucosa, the muscularis and the serosa.36 The mucosa is the inner layer surrounding the lumen and comprises absorptive epithelium,34 the lamina propria and smooth muscle36 (Fig. 1). | ||
| Fig. 1 Structure of the small and large intestine mucosa. Created with https://BioRender.com. | ||
A dynamic network of proteins known as the extracellular matrix (ECM) provide structural support and modulatory signals throughout the intestine. The ECM includes two separate but linked components, namely the basement membrane and the interstitial matrix,37 components of which are predominantly produced by epithelial and mesenchymal cells, such as fibroblasts, myofibroblasts, and smooth muscle cells, respectively.37 The basement membrane lies between the epithelium and the mesenchyme of the lamina propria and is comprised of various extracellular proteins including type IV collagen and laminin, and plays a role in epithelial cell differentiation, cell attachment and motility.37,38 The interstitial matrix lies below the basement membrane playing a structural role in the lamina propria and submucosa and comprises predominantly of collagen I and III, fibronectin, elastin, decorin and hyaluronan.37
Besides some similarities, there are also several differences between the small and large intestine (Fig. 1), each having distinct features adapted to fulfil their specific functions. For example, the mucosal lining of the small intestine, where digestion and absorption of nutrients predominantly occurs, has a crypt-villus structure39 which creates a large surface area for absorption.34,40 The epithelium of the small intestine is comprised of four main cell types,34,40 commonly known as intestinal epithelial cells (IECs). These include enterocytes, which are the most abundant cell type, exhibit a microvilli brush border and are responsible for nutrient absorption; goblet cells, which secrete mucus; enteroendocrine cells, which secrete various hormones;41 and Paneth cells, which secrete antimicrobial peptides and immunomodulating proteins.42 Besides the main IECs, additional cell types found in intestinal epithelium43 include microfold cells, known as M cells, which overlay immune gut associated lymphoid tissue and are involved in antigen presentation,39 as discussed in section 2.2.
The large intestine includes the caecum and colon, where water and electrolytes are absorbed and where further digestion and absorption takes place; as well as the rectum where waste faeces are stored before egestion.36 The mucosa of the colon also displays crypt architecture but lacks villi,35 while the epithelium comprises predominantly absorptive enterocytes, known in the colon as colonocytes, goblet cells, and enteroendocrine cells.39,44 Additionally, the colon is home to a diverse community of over 1013 bacteria and other microorganisms45 including archaea, bacteriophages, viruses and fungi.46 Collectively known as the gut microbiome, these microorganisms play crucial roles in immunity and immune system development,47 food digestion and the production of bioactive compounds such as short-chain fatty acids (SCFAs), endocrine function,48 neurological signalling,49 drug and xenobiotic metabolism50 as well as protection against toxins and pathogens.51 The bacterial population in the gut is dominated by the phyla Firmicutes and Bacteroidetes, as well as to a lesser extent Proteobacteria, Actinobacteria, Verrucomicrobia, and Fusobacteria, amongst others.50 Perturbations in the microbiome have been linked to various chronic diseases52 including obesity, T2D, metabolic liver disease and malnutrition46 as well as increased susceptibility to infectious diseases.53 While a “healthy” gut microbiome has not been defined, several factors have been identified indicating a state of dysbiosis, such as reduced microbial diversity,46 with reduced bacterial gene richness in the gut linked to inflammation, adiposity, insulin resistance and dyslipidaemia.54
Mucus, a viscoelastic hydrogel, plays an import role in both the small and large intestine as the first line of defence against pathogenic microorganisms,55 in particular in the colon, where the majority of microorganisms reside. There, it is characterised by two mucus layers, namely the inner mucus layer attached to the epithelium; and the outer layer which is looser and more easily removed, similar to the single mucus layer of the small intestine.56 Bacterial adhesion to, and colonisation of, the mucus layer, plays a role in immune and intestinal homeostasis and allows for spatial organization of the microbiome57 which is also linked to oxygen and nutrient gradients from the mucosal surface to the lumen.58 Intestinal motility,59 driven by contraction of smooth muscle in the intestinal wall, plays a role in mucus regulation as well as intestinal homeostasis, moving mucus, microbes and undigested material60 along the intestinal tract.58 Perturbations in the mucus layer leads to increased susceptibility to pathogens and infections and is linked to diseases such as colorectal cancer and inflammatory bowel disease (IBD),57 which collectively refers to two separate conditions namely, Crohn's disease61 and ulcerative colitis,62 the prevalence of which have been increasing globally over the past 20 years.61
2.2. Intestinal immune system
The gut is home to up to 70% of the immune system,63 encompassing a range of innate and adaptive immune cells and compartments, which can be broadly viewed as inductive and effector sites. Inductive sites, where adaptive immune cells are primed, include gut-associated lymphoid tissue (GALT) such as the multi-follicular Peyer's patches found in the small intestine and the isolated lymphoid follicles found throughout the small and large intestine64 as well as intestine-draining mesenteric lymph nodes (MLN). Once primed, the adaptive immune cells move to the effector sites, which include the lamina propria and intestinal epithelium, where they play important roles in immunity and immune modulation, as well as maintaining gut barrier integrity.64,65 Critically, the intestinal immune system must be able to distinguish between commensal microbes and pathogens, as balancing responses to these groups is crucial in maintaining homeostasis, with aberrations leading to inflammation and diseases such as IBD.63,64 Besides microbes, the mucosal immune system must also continuously respond to dietary antigens, playing an important role in oral tolerance; disruption of which can lead to food allergies driven by an adaptive immune response.66Peyer's patches are subepithelial lymphoid follicles found throughout the anti-mesenteric wall of the small intestine, especially in the terminal ileum.67 They comprise up to hundreds of individual lymphoid follicles overlaid on the luminal side by follicle-associated epithelium which is in turn covered by a thin mucus layer and is rich in M cells, while also containing intraepithelial lymphocytes.63,64,67 The subepithelial follicles each include a central B cell follicle or germinal centre, where B cells proliferate, bordered by regions containing T cells (mostly CD4+)68 and overlaid on the luminal side with a subepithelial dome, which is in contact with the basal side of the epithelium and comprises predominantly dendritic and other myeloid cells,64 such as macrophages, as well as B and T cells63 (Fig. 2). Further, antigen-specific T cells are also primed and proliferate in Peyer's patches and play an important role in immunotolerance.63 The specialised basolateral morphology of M cells allows for transcytosis and direct interaction or presentation of luminal antigens, soluble proteins, bacteria and viruses to the underlying immune cells in the subepithelial dome, which modulate, either activating or inhibiting (immune tolerance), immune responses.63 Peyer's patches are therefore critical in intestinal antibody reponses69,70 and act as important immune priming sites.64,71 Isolated lymphoid follicles have a similar structure to Peyer's patches, however comprise only a single B cell follicle or germinal centre, also encompassing dendritic and T cells as well as being overlaid with M-cell rich epithelium.72,73 M cells have also been identified as points of entry for certain pathogens such as Salmonella and other bacteria e.g. Mycobacteria, viruses, and prions.63–65
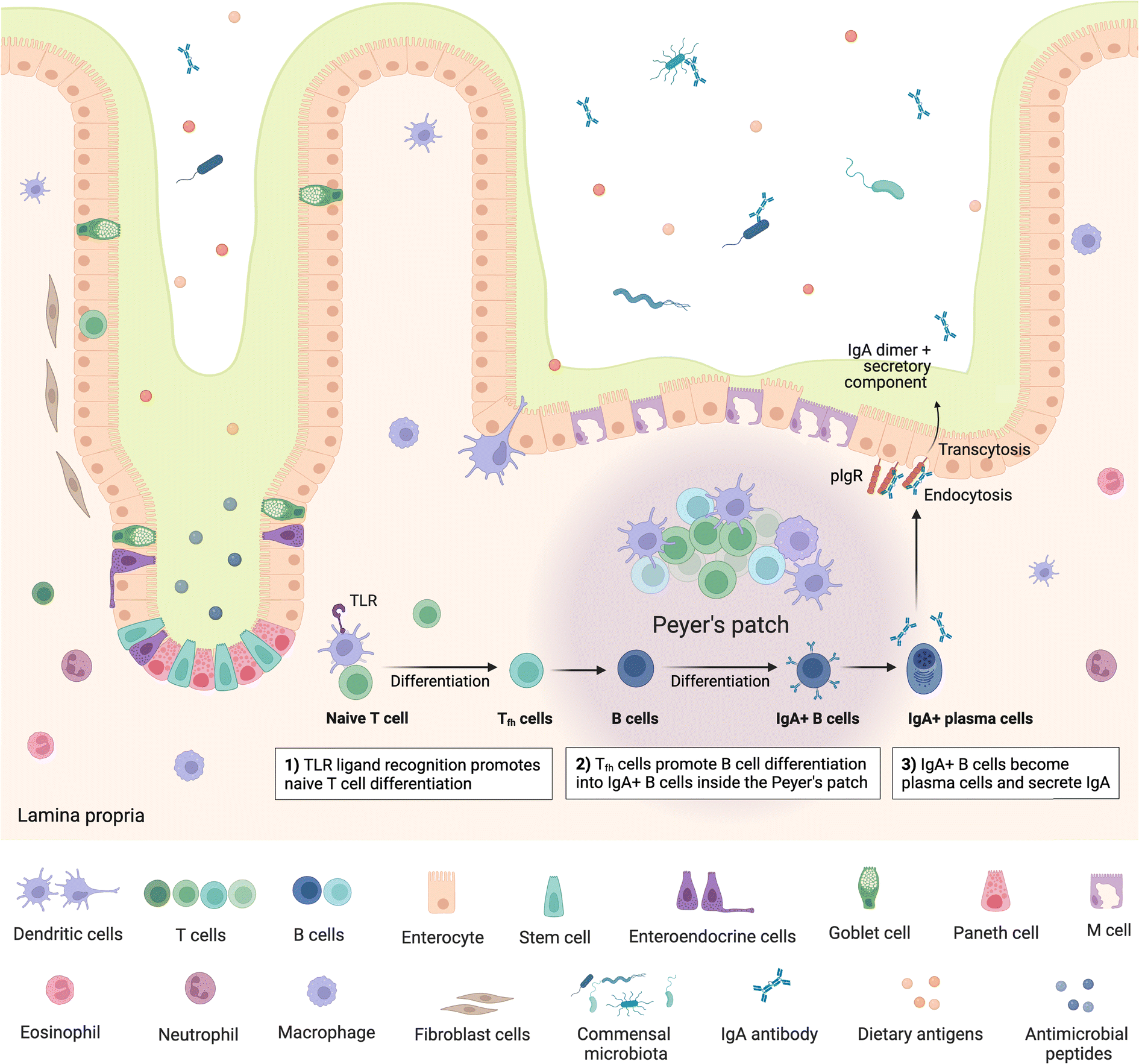 | ||
| Fig. 2 Structure of small intestine Peyer's patches and mechanism of IgA secretion. IgA – immunoglobulin A; TLR – toll-like receptor; polymeric immunoglobulin receptors (pIgR). Created with https://BioRender.com. | ||
Following presentation of antigens by M cells to the underlying dendritic cells, these antigen-presenting cells initiate an adaptive immune response by processing and presenting the antigens to B and T cells. Activation of naïve mature B cells by the antigens causes them to undergo class switching, which refers to changing the type or class of immunoglobulin (Ig) the B cell produces, from IgM to IgA, IgG or IgE,74 stimulating production of the appropriate antibody. Class switching can follow T-cell dependent or T-cell independent routes.75 In the T-cell dependent route, antigen specific T cells, in particular CD4+ T cells/T helper cells (Th cells), provide signals to B cells, stimulating B cell class switching; whereas in the T-cell independent route, antigens can directly trigger B cells specific to the presented antigen to become activated. Some activated B cells differentiate into plasma cells which commence production of the antigen-specific antibodies.74
Overall, there are several key immune cells found in the gut which collectively signal and modulate the immune response. Besides the adaptive B and T cells and the innate macrophages, mentioned above, other innate immune cells include natural killer cells, neutrophils, eosinophils and mast cells which collectively form the first line of defence.18,76 Leukocytes are predominantly found in the lamina propria however T cells are also commonly associated with the epithelium.76 Additionally, stromal fibroblast cells77 and lymphatic endothelial cells also contribute to immune surveillance, with the former playing a role in T cell migration.18
2.3. Gut–immune interactions implicated in metabolic disease
Gut–immune interactions have been identified as contributing to the inflammatory state seen in metabolic diseases, initiating inflammatory signalling and cytokine release.78 Cani et al. (2007)15 identified metabolic endotoxemia, which refers to translocation of bacterial endotoxins, namely lipopolysaccharides (LPS), which are components of Gram-negative bacterial cell walls, into the blood stream, as a causative factor for inflammation, weight gain and diabetes. This included observations in mice that a high-fat diet increased endotoxemia and that mimicking metabolic endotoxemia through subcutaneous infusion of LPS in control mice resulted in increased inflammation as well as increased fasting glycemia and weight gain.15 In a follow up study, Cani et al. (2008)16 showed that antibiotic-induced changes in gut microbiota reduced LPS and metabolic endotoxemia which correlated with improved glucose tolerance as well as reduced fat gain, inflammation and oxidative stress, further supporting a causative role of the gut microbiome in inflammation.16 It is now commonly thought that reduced gut barrier integrity and metabolic endotoxemia causes immune-mediated release of pro-inflammatory cytokines and a state of low-grade systemic inflammation, which, as mentioned previously, is correlated with obesity, T2D and MetS.34 Further to this mechanism, other gut–immune associated links to metabolic diseases identified include reduced immunoglobulin A (IgA), which is also associated with increased inflammation, impaired glucose homeostasis and reduced intestinal barrier integrity;79 as well as perturbations in gut hormone secretion.80 Further, oxidative stress due to the release of reactive oxygen species has also been implicated in intestinal inflammation.27 It should however be noted that the role of the gut and gut microbiome is contentious, with arguments for and against its causative role in metabolic diseases.41 Therefore, especially given the complexity of these diseases, more research is required to better understand their development and progression as well as disentangle the cause-effect relationship of intestinal inflammation in these diseases. Further details regarding metabolic-gut–immune interactions can be found in different reviews.81–843. Gut–immune crosstalk in metabolic disease
3.1. Metabolic crosstalk between the gut and the immune system
Diet and its derived metabolites are key regulators of the metabolic gut–brain axis since it is well understood that it regulates the human immune system thereby influencing the development of irritable bowel syndrome (IBS), IBD and metabolic diseases. Due to limitations in currently available study models and study designs, our understanding of the mechanisms behind these effects is incomplete. For example, controlled feeding trials, whose design corresponds to drug discovery studies, can deliver insights into cause–effect relationships. However, controlled nutritional studies influence the dietary habits of humans thereby limiting the translation of outcomes to a whole population. In addition, controlled studies can only be conducted over the short term, which complicates understanding of the development of metabolic diseases as they are commonly developed over a longer-term basis. Observational studies allow for a long-term approach, but data obtained cannot be used for generating cause–effect relationships. The complexity between the interplay of food and host physiology is further demonstrated by considering that processing (e.g. baking, boiling, frying) and seasonal variations due to climate conditions, for example pH and temperature, influence the nutrient composition and availability of substrates for microbial metabolite synthesis. The hosts' gender and age also influence digestion and absorption of nutrients.85 This evidence clearly demonstrates an urgent need for better study models to unravel mechanisms of diet along the metabolic gut–immune axis, aiding the prevention of metabolic disease development.85 So far, it is well understood that the commonly consumed Western lifestyle diet, characterized by a high fat and sugar content, but low in fibre-rich foods (vegetables and fruits), promotes an impaired immune system, dysbiosis and the development of gut and metabolic diseases.86–88 Complex and insoluble fibres are suggested as the main beneficial diet component for human health since gut bacteria synthesise SCFAs within a fermentation reaction in the colon. For the main bacterial metabolites, butyrate, propionate, and acetate, beneficial effects along the metabolic gut–immune axis have been described in vitro and in vivo.89–91 A mediterranean diet, known for its high amount of fruit, vegetables and unsaturated fatty acids is recommended for maintaining a healthy overall state. It was demonstrated that a mediterranean diet shifts the gut microbiome pattern towards an increase in SCFA-producing strains like Bacteroides, Prevotella, Roseburia, Ruminococcus, and Faecalibacterium prausnitzii.92 Although the mediterranean diet induces several beneficial health effects in humans, adherence to this diet is low, especially in non-Mediterranean countries, as dietary habits often need to be adapted.92 A fact that further emphasizes the need for study tools which enable the development of personal diet compositions that promote functional gut–immune interactions. For the specific treatment of the gut diseases IBS and IBD, the FODMAP (fermentable oligo-, di-, and monosaccharide and polyol) diet is commonly advised for patients. This diet is split into two phases, the first strictly reduces the intake of slowly absorbable carbohydrates (e.g. vegetables, fruits, legumes and cereals) and the second one integrates them according to the patients' tolerance.93,94 Up to now, it has been widely reported that the first phase induces relief of gastrointestinal symptoms in most patients, assumed to be mediated via a suppression of a histamine-mediated inflammatory response. Subsequent changes reported in the microbiome composition have been controversial, which occur again due to the limitations of conducting controlled randomized longer-term trials on food intake. Similar to drugs, the consumption of the right “dosage” of foods is crucial, demonstrated by the fact that some non-digestible carbohydrates can cause IBS/IBD-like symptoms such as bloating and diarrhoea.Findings of recent studies95,96 clearly indicate that shaping the gut microbiome with specific dietary interventions could be a promising and easily accessible tool in combating the pandemic extent of obesity. Nonetheless, limitations need to be considered, for example, a small number of study participants only allows for limited population-wide translation of these findings. Moreover, it is not clear if a change in the energy balance is a cause or an effect of the gut–microbiome interactions. For understanding the influence of diet, food pattern analysis including frequency and quantity, but also interaction and cumulative effects of nutrients, need to be counted.97
The latest findings on diet and single nutrients along the metabolic gut–immune axis are summarized in Table 1.
| Diet/nutrient | Study model | Marker | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gut/microbiome | Immunea | Metabolic | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a IL-1 – interleukin-1, IL-1β – interleukin-1β, IL-6 – interleukin-6, IL-10 – interleukin-10, IL-17 – interleukin-17, TNFα – tumour necrosis factor alpha (cytokines); ILC2/3 – type 2/3 innate lymphoid cells; ZO-1 - zonula occludens-1 (tight junction protein). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Microbiome enhancer diet98 | Healthy humans (n = 17) | SCFA ↑ | GLP-1 ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (Prevotella copri, Lachnospira pectinoschiza, Lachnospira pectinoschiza, Eubacterium eligans) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Strict vegetarian diet96 | Obese humans w/o diabetes (n = 6) | SCFA ↓ | Bodyweight ↓ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Firmicutes and Bacteroides | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Caloric restriction diet ± inulin/resistant starch99 | Healthy humans (n = 39–80) | Parabacteroides/Bifidobacterium ↑ | Alanine aminotransferase in women ↓ | Visceral adipose tissue in men ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bilophila ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Malnutrition100 | Male mice (n = 3–40) | Permeability ↑ | Neutrophil gelatinase associated lipocalin ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ZO-1 ↓ | IgA ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Peyer's patch, Th17 cells ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Eicosapentaenoic acid (2 g d−1)101 | UC patients (n = 19) | Bacteroidaceae family ↓ | IL-1 ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mucosal inflammation ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Glucose or fructose (15% solution)102 | Male mice (n = 5–7) | Glucose:Permeability ↑ | Fructose: IL-6 ↑ | Glucose: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body weight ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Proteobacteria ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Visceral adiposity ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Firmicutes ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fructose: | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Firmicutes ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bacteroidaceae ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Erythritol (5% solution)103 | Obese male mice (n = 6) | SCFA ↑ | ILC2 & 3 ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Advanced glycation end (AGEs) products104 | Male mice (n = 15) | Lactobacillus, Prevotella, Anaerostipes, Candidatus Arthromitus ↓ | IL-1β, IL-17, TNFα ↑ | Insulin ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Parabacteroides, Ruminococcus Lawsonia ↑ | IL6, IL10 ↓ | GLP-1, GIP, ghrelin ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
3.2. Microbial substrates and derived metabolites
According to the International Scientific Association for Probiotics and Prebiotics (ISAPP), prebiotics are defined as “selectively fermented ingredient that results in specific changes in the composition and/or activity of the gastrointestinal microbiota, thus conferring benefit(s) upon host health”.105 Prebiotics mainly consist of dietary non-digestible oligosaccharides like fibre, but due to increasing evidence, further food constituents have recently been included like polyphenols.106,107 In contrast to prebiotics, which serve as a substrate for the gut microbiome, probiotics are defined as the “actual administration of live organisms for health-promoting effects”. These organisms are able to live on foods, supplements and most importantly in the intestine.105 Along the metabolic gut–immune axis, probiotics are able to stimulate immune cells and interact with the residing gut microbiome for stimulating immunomodulatory effects108 (Table 2). In addition, metabolites, molecules synthesized within these interactions, are crucial in the communication between the host and microbiome. Besides that, metabolite synthesis is dictated by substrate (e.g. diet, nutrients, prebiotics) availability and the generated type can either induce beneficial (e.g. anti-inflammatory), or negative health effects (e.g. TMAO).109 Considering that metabolites are not only synthesized by the microbiome but also by the host, the complexity of these interactions is demonstrated.110 Moreover, the commonly used study models allow only limited insights into the mechanism of metabolites. For example, some metabolites are absorbed so quickly by the gut barrier that they cannot be properly studied, especially if faecal samples are used for this purpose.111 It is assumed that 95% of synthesized SCFAs in the large intestine are absorbed by the gut cells.112 Subsequently, the question arises how well faeces samples represent the different regions of the gut. This was recently studied in a human study using non-invasive digestible devices that allow for the collection of samples from the different regions. Authors of this study observed a dramatic difference between metabolites in the stool and the intestinal samples.113 Thus, whether metabolites are derived from the host or from the microbiome remains to be elucidated as well as if these are modified or de novo synthesized molecules. This review summarizes latest findings on the main metabolites of the SCFAs. In-depth discussions on mechanisms of different metabolites can be read in several other published review papers.90,114–116| Microbial substrate/metabolite | Study model | Marker | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gut/microbiome | Immunea | Metabolic | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a IL-4 – interleukin-4, IL-8 – interleukin-8; IL-10 – interleukin-10, IL-17A – interleukin-17A, IL-18 – interleukin-18; IFN-γ – interferon gamma; TGF-β – transforming growth factor beta, TNFα – tumour necrosis factor alpha (cytokines); Ccr4 – C–C chemokine receptor type 4; Irf7 – interferon regulatory factor 7; NF-κB – nuclear factor-κB; Nfkbia – nuclear factor-κB inhibitor alpha; Rorc – RAR related orphan receptor C; Tyk2 – tyrosine kinase 2. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prebiotic: Cranberry polyphenols (200 mg kg−1 of body weight)125 | Male mice (n = 12) | Mucous thickness ↑ | Tyk2, Rorc, Nfkbia, Irf7, Ccr4 | Glucose homeostasis ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Akkermansiaceae, Bacteroidaceae ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Barrier markers claudin-1 and muc2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prebiotic: insulin (50 & mg)126 | Female mice (n = 4–12) | Low dose: barrier function ↑ | Low dose: intestinal inflammation ↓ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| High dose: barrier function ↓ | High dose: inflammation response ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prebiotic: nanoparticles of broccoli extracts127 | Colitis mice (n = 5) | TNF-α, IL-17A, and IFN-γ ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IL-10↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Probiotic: Lactobacillus acidophilus, Bifidobacterium breve, and Bifidobacterium infantis128 | TNF-α stimulated HT29 cells | IL-8 ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Probiotic: Lactobacillus rhamnosus MTCC-5897 (ref. 129) | Male mice (n = 9) | Barrier function ↑ | IL-4, TNF-α ↓ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TGF-β, IgA ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Metabolite: propionate (2 M solution)130 | Piglets (n = 8) | Bacteroidetes, Firmicutes, | NF-κB, IL-18 ↑ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prevotella ↑ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Turicibacter ↓ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Acetate, propionate, and butyrate make up the majority of the microbial-derived SCFAs. Based on evidence, SCFAs are one of the most abundant and studied metabolite classes demonstrating positive effects on combating inflammation and metabolic diseases, but also promoting a functional gut barrier.117 After binding of the SCFA to its cognate receptors GPR41/43 or 109A, these are the key proteins for mediating the beneficial health effects. Thus, a change in the abundance of SCFA concentrations hints also at an impaired metabolic gut–immune axis. This has been found in IBD patients, in whom perturbed carbohydrate metabolism of the gut microbiome is assumed to result in decreased synthesis of SCFAs thereby promoting dysbiosis. Consequently, IBD patients do not obtain benefits from SCFAs to the same extent as healthy individuals.118,119
Overall, SCFAs are assumed to induce mainly positive effects, but there are studies that raise doubts about inducing only health-promoting effects and point out the need for a more critical discussion.112,120 A recent systematic meta-analysis review, including 43 original articles on glycaemic control in obese people, revealed no effect of SCFAs on glycaemic control in humans with or without a metabolic disease.120 Authors of this review suggest that a better understanding of acute and chronic SCFAs interventions is crucial.120 Moreover, methodological approaches for understanding mechanisms of SCFAs have been critically discussed by Sakata in 2019.121 This article already pointed out that the in vitro study of SCFAs is limited by physiological conditions such as high viscosity of gut contents as well as existence of bacterial biofilms and of the mucus layer at the mucosal surface.121 Thus, these findings further emphasize the need for developing improved in vitro study models with improved physiology. Recently an improved in vitro platform called GuMI (gut microbiome), was developed by Zhang et al. (2021), using colon organoids co-cultured long-term with the main butyrate-producer F. prausnitzii.122 Considering that the structure of the metabolites is often modified within the host–microbiome interactions, multi-organ chips would be highly valuable in understanding structure–activity relationships. In addition, whether a metabolite is derived from the host or the microbiome is challenging since there are also bidirectional processes involved. This has been recently studied in newly developed microphysiological systems integrating the gut and liver123 as well as the gut, liver and brain124 allowing for better insights into the casualty of metabolites on gut–immune interactions to be elucidated.
3.3. Hormonal crosstalk
There is increasing evidence that the activity of the gut microbiome influences satiety via a modification of hormone release. However, studies examining the functionality and composition of the gut microbiota in relation to satiety are lacking.131 Satiety hormones are mainly released by enteroendocrine cells in the gut. Indeed, 95% of peripheral serotonin is released after the chyme enters the intestine for promoting gut peristalsis and nutrient absorption.132 Furthermore, serotonin has been reported to induce anti-inflammatory effects in animal models of IBD and colitis.133 A mechanistic approach demonstrated that administration of dietary tryptophan, a precursor of serotonin, protects the gut function.134 The satiety hormone GLP-1 has not only been shown to be beneficial for obese or diabetic people, but also for people suffering from IBD. In a nationwide cohort study, GLP-1 administration to IBD patients improved their disease course. It was concluded that this was not only due to the GLP-1 regulating effect on blood glucose concentrations, but also by its positive effects on intestinal barrier function as well as gut inflammation.135,136 Since metabolic diseases often go along with an altered sensitivity to satiety hormones, their study is further complicated.137,138 This was recently found by Sundaresan et al. (2023),137 when an intraduodenally applied glucose solution induced a delayed response in PYY and GLP-1 release in obese humans.137 In addition, a desensitization to the satiety hormone leptin was found to alter gut microbiome activity.1393.4. Lab-on-chip technology as a new tool to study the gut–immune axis
The recent evidence in this review on the metabolic gut–immune axis mainly derives from human and animal intervention studies that deliver the most significant insights for understanding this axis. Thus, the question arises why do we need lab-on-chip technology for studying the influence of diet? Time and money play a crucial role, for example obtaining the ethics approval, receiving food-/pharmaceutical grade compounds, or recruiting study participants that adhere to the study design are just some of these factors. In addition, the statistical power of the study outcomes is highly dependent on the number of study participants which can vary greatly over the study time as shown in Table 2. Moreover, studying mechanisms in humans is crucial for understanding specific use of nutrients in prevention and treatment of diseases. However, investigations of their bioavailability and transformations along the metabolic gut–immune axis is limited due to accessibility and a lack of non-invasive methods available for humans. This becomes clear, by considering the intense chemical and physical digestion process in the main digestive organs before some nutrients can reach the colon. Nonetheless, if single nutrients are considered to be used as a therapeutic, understanding of their bioavailability and metabolization is crucial. This could be achieved by studying the pharmacodynamic principles of ADME characteristics – absorption, distribution, metabolism, and excretion.140,141 Special environmental conditions (e.g. oxygen, pH) in the colon allow the hosting of microbes that further digest these nutrients to metabolites. It is widely understood that faeces samples are limited in reflecting the different gut regions, but also in which of the generated metabolites are available for the host organism. The use of plasma concentrations further provides only limited insights in bioavailability of metabolites, unless expensive stable-isotope labelling is conducted beforehand. Challenges in conducting animal studies are similar to human studies, since special expensive animal facilities and licenses are needed. Although a high-throughput approach can be more easily done in animals, translation to humans is still challenging due to differences in physiology.Thus, lab-on-chip technologies provide a highly valuable tool by keeping costs and time lower in comparison to animal and human intervention studies. In particular, multi-organ platforms that integrate digestion and absorption processes will be highly beneficial for understanding generation of metabolites and if they derive from the host or the microbiome and how their synthesis can be provoked.
4. Gut–immune lab-on-chip technologies for metabolic disease research
Current static in vitro models fail to reflect the complex human intestinal physiology especially in terms of mechanical and biophysical cues.23 Lab- and organ-on-chip platforms have therefore become important technologies over the past two decades facilitating perfusion, delivery of nutrients and removal of waste products, as well as offering enhanced control over cell microenvironment including physiochemical gradients such as pH and oxygen.142 Further, designs can be made that are compatible with ECM-mimicking biomimetic and biocompatible matrices or scaffolds on or in which cells can be grown33,143–145 as well as to offer mechanical cues such as peristalsis-like motion.19,31Intestine-on-chip studies have already been important tools in furthering our understanding of physiological processes and diseases. Many of these have however been devoid of an immune component.143,146–148 Immune cell incorporation and improved immune modelling therefore represents an important next step in improving intestinal models, especially given the important role the immune systems plays in intestinal homeostasis,63 as outlined in section 2 above. This has already begun, and several lab-on-chip studies incorporating both intestinal and immune cells were identified. The majority of these have been conducted in the context of IBD,31–33,123 small intestinal bacterial overgrowth (SIBO)19 and COVID-19,29,149 while none appear to have been conducted specifically in the context of metabolic diseases. Results from the aforementioned studies are however translatable, given certain commonalities such as endotoxemia and inflammation between these conditions, as introduced in section 2.3 above. In this review we focus on lab-on-chip platforms incorporating both intestinal and immune cells. While other lab-on-chip studies on intestinal inflammation were noted, many were based on the direct addition of cytokines150–154 rather than the inclusion of immune cells and were therefore omitted.
4.1. Gut–immune lab-on-chip model designs and materials
Broadly speaking, four different approaches to model design were noted in gut–immune lab-on-chip studies, with the most common being stacked microfluidic channels separated by a porous membrane. A total of seven different models based on this stacked membrane-channel approach were noted, utilising varying chip and membrane materials19,27–32,143,149,155 as summarised in Table 3. Other approaches noted include hydrogel-based channel separation,33,144,145 tissue culture-insert based22,123,156,157 and multi-chamber based designs.158,159| Designs | Chip/channel material | Membrane material | Physical/biological cues | Cell types | Application | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| Intestinal cells | Immune cells | Other | |||||||
| Single organ | |||||||||
| Stacked membrane-separated microfluidic channels | Chip-S1 (emulate) | - PDMS | - PDMS | - Cyclic mechanical deformation to mimic peristalsis | - Caco2 (Caco-2BBE) | - Peripheral blood mononuclear cells (PBMCs) – including monocytes, lymphocytes, granulocytes | - Endothelial – human capillary microvascular endothelial cells (capillary or lymphatic) | Small intestinal bacterial overgrowth (SIBO) and intestinal inflammation | Kim et al. (2016)19 |
| - Caco2 (Caco-2BBE) | - PBMCs | Mechanistic study to identify factors which trigger or contribute to the onset of inflammation and gastrointestinal disease | Shin and Kim (2018)27 | ||||||
| - Human intestinal organoid derived IECs (duodenal) | - PBMCs | - Endothelial – human large intestine microvascular endothelial cells (HIMECs) | Enteric coronavirus infection and drug efficacy testing | Bein et al. (2021)29 | |||||
| - MKN45 (human gastric adenocarcinoma) | - PBMCs | - Endothelial – human Large intestine microvascular endothelial cells (cHIMEC) | Cancer immunotherapy safety testing – T-cell bispecific antibodies (TCBs) | Kerns et al. (2021)30 | |||||
| - Colonoids | |||||||||
| HuMix | - Silicone rubber gaskets | - PC | - Oxygen gradient | - Caco2 | - Primary CD4+ T cells | Development of a model capable of supporting facultative and obligate anaerobe growth interfaced with intestinal cells | Shah et al. (2016)160 | ||
| - CCD-18Co (non-cancerous colonic cell line) | |||||||||
| - Polycarbonate (PC) enclosures | |||||||||
| MOTiF biochip | - Polysterol | - Polyethylene terephthalate (PET) | - None | - Caco2 | - PBMCs and derived Macrophages & Dendritic Cells | - Endothelial – Human umbilical cord vein endothelial cells (HUVECs) | Intestinal homeostasis, inflammation, barrier function and immunotolerance; host–microbe interactions in inflammation, intestinal disease and infection | Maurer et al. (2019)155 | |
| Peristaltic intestine on a chip | - PDMS | - PDMS | - Pressure difference to mimic peristalsis | - Caco2 | - Macrophage U937 | - Endothelial – HUVECs | Host–microbe interaction, intestinal damage and inflammation in the context of IBD through E. coli infection. Evaluation of anti-inflammatory effects of probiotic bacteria and antibiotics | Jing et al. (2020)31 | |
| - Caco2 | - Primary human macrophage cells | - Endothelial – HUVECs | Regulatory effects of chitosan oligosaccharides (COS), thought to have anti-inflammatory and prebiotic properties, in enteritis such as IBD | Jing et al. (2022)32 | |||||
| Gut-on-a-chip | - PDMS | - PDMS | - None | - Caco2 | - PBMCs | - Endothelial – HUVECs | Effects of SARS-CoV-2 infection in the intestine | Guo et al. (2021)149 | |
| - HT-29 | |||||||||
| Embedded membrane microfluidic chip | - PDMS | - PC | - None | - Caco2 | - PBMCs | - Endothelial – HUVECs | Host–microbe interaction in the context of enteritis and device suitability for drug-resistance screening | Zhao et al. (2022)161 | |
| Microfluidic organotypic device (MOD) | - Nitex mesh | - Cyclic olefin copolymer (COC) | - Oxygen gradient | - Mouse proximal colon tissue explant including mucus layer, mucosa, submucosa and muscularis externa | - Mast cells (within tissue) | - Multiple cell types present in tissue | Model of leaky gut – the effect of collagen breakdown, using bacterial collagenase, on intestinal barrier disruption | Cherwin et al. (2023)162 | |
| - Polyurethane gaskets | |||||||||
| Hydrogel-based channel separation | OrganoPlate (Mimetas) | - Polystyrene, glass | - None | - None | - Caco2 | - THP-1 cells differentiated into macrophages | – | Intestinal inflammation and suitability of the model for screening of anti-inflammatory agents | Gijzen et al. (2020)144 |
| - MUTZ-3 (acute myelomonocytic leukemia cell line/dendritic cells) | |||||||||
| - HT-29-MTX-E12 | |||||||||
| - Proprietary polymers | |||||||||
| - Caco2 | -THP-1/macrophages | Intestinal barrier disruption and inflammation; neutrophil dynamics and infiltration; crosstalk between resident and circulating immune cells | Gjorevski, et al. (2020)145 | ||||||
| - Neutrophils isolated from fresh human peripheral blood | |||||||||
| - Human intestinal organoid derived IECs | - Macrophages (differentiated from PBMC-isolated monocytes) | Inflammation in the context of IBD, monitoring of macrophage activation and inflammatory cytokine release as well as the effects of anti-inflammatory agent exposure | Beaurivage et al. (2020)33 | ||||||
| Multi-chamber design | Nutrichip | - PDMS | - Polyester (from Transwell® insert) | - None | - Caco2 | - Macrophages – differentiated from monocyte cell line U937 (histiocytic lymphoma cell line with monoblastic characteristics) | Proof of concept inflammation studies | Ramadan et al. (2013)158 | |
| Nutrichip iteration | - PMMA | - PET | - None | - Caco2 | - U937 (monocyte like) – differentiated to macrophages | Inflammation and immune response modulation | Ramadan and Jing, (2016)159 | ||
| - Polystyrene | |||||||||
| - PDMS sheets | |||||||||
| Tissue-culture insert-based | 3D-hI | - Gelatine microscaffold | - PC | - Oxygen gradient | - Caco2 | - PBMCs | Development of a model capable of supporting a complex serosal microenvironment incl. ECM, oxygen gradient and vertical distribution of bacteria; barrier dysfunction and inflammation | De Gregorio et al. (2022)22 | |
| - Human intestinal subepithelial myofibroblasts (hISEMFs) | |||||||||
| - PDMS | |||||||||
| - PMMA | |||||||||
| LiverChip™/Physiomimix (CN Bio Innovations) and 3XGL and 3XGLB iterations | Multi-organ | ||||||||
| - Acrylic pneumatic plates | - Polyurethane membrane | - None | - Caco2 | - Dendritic cells derived from PBMC isolated monocytes | - Liver – human primary hepatocytes and Kupffer cells | Inflammation and related gut–liver crosstalk | Chen et al. (2017)156 | ||
| - Polystyrene scaffolds | - HT29-MTX | ||||||||
| - Polysulfone fluidic plates | |||||||||
| - Polyester (Transwell® insert) | - Caco-2 (4-MPS) or C2BBe1 (7- and 10-MPS) | - Dendritic cells | - Liver/immune – human primary hepatocytes and Kupffer cells | LiverChip™ platform flexibility – three different configurations comprising a mixing chamber with four, seven, or 10 MPSs respectively tested, MPS viability confirmation and potential of the platform for pharmacological applications | Edington et al. (2018)157 | ||||
| - HT29-MTX-E21 | |||||||||
| - Plus a combination of lung, endometrial, brain, heart, pancreatic, kidney, skin & skeletal muscle cells | |||||||||
| - Human intestinal organoids (colon) | - Dendritic cells | - Liver – human primary hepatocytes and Kupffer cells | IBD and its connection with liver disease; investigation into the effects of SCFA on ulcerative colitis | Trapecar et al. (2020)123 | |||||
| - Macrophages | |||||||||
| - Treg cells | |||||||||
| - Primary ulcerative colitis epithelium | |||||||||
| - Th17 cells | |||||||||
| - Human intestinal organoids (colon) | - Dendritic cells | - Liver – human primary hepatocytes and Kupffer cells | Gut/microbiome–liver–brain interactions in Parkinson's disease including investigation of immune interactions, microbial metabolites, in particular SCFAs, and inflammation | Trapecar et al. (2021)124 | |||||
| - Brain – neurons/astrocytes and microglia | |||||||||
| - Macrophages | |||||||||
| - Treg cells | |||||||||
| Th17 cells | |||||||||
Kim et al. (2016)19 was one of the first studies to incorporate immune cells in an intestinal lab-on-chip model and relied on a now commercially available device previously developed by Kim and colleagues28,163 in the early 2010's (Fig. 3A). Known as Chip-S1, this device is poly(dimethylsiloxane) (PDMS)-based and comprises two microfluidic channels separated by a porous ECM-coated PDMS membrane. The device allows for perfusion and application of cyclic strain, to mimic shear stress and peristalsis respectively, in an effort to better recapitulate the gut microenvironment and support long-term co-culture of bacteria.28 It additionally allows for on-chip monitoring of barrier integrity through transepithelial electrical resistance (TEER) using Ag/AgCl electrode wires coupled with a Voltage–Ohm meter.28
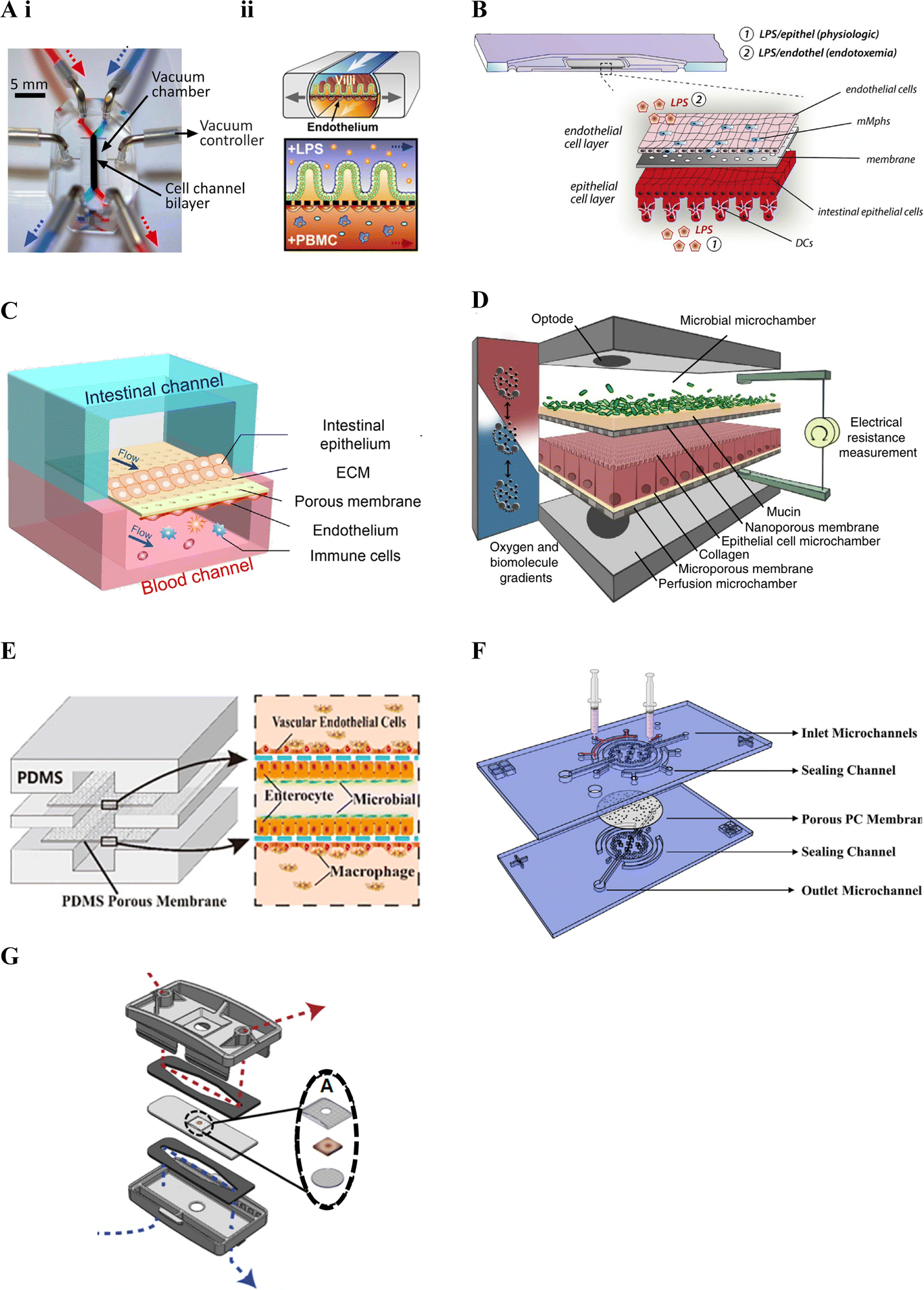 | ||
| Fig. 3 Membrane-based gut–immune microfluidic lab-on-chip designs. A. i. Image of Chip-S1 indicating vacuum control used to induce mechanical deformation of the membrane (reprinted from ref. 28 with permission, copyright 2012, Royal Society of Chemistry) and ii. A schematic of an experimental set up for gut–immune applications, showing introduction of lipopolysaccharides (LPS) into the apical compartment housing intestinal epithelial cells, and introduction of immune (peripheral blood mononuclear cells (PBMCs)) into the basal compartment housing endothelial cells (reprinted from ref. 19 under the Creative Commons CC BY license). B. Schematic of the MOTiF biochip showing immune cell seeding, namely mucosal macrophages (mMphs) and dendritic cells (DC), in the apical and basal compartment respectively; as well as LPS exposure conditions155 (reprinted from ref. 155 with permission, copyright 2019, Elsevier Science & Technology Journals). C. Schematic of the gut-on-a-chip device showing an apical epithelial channel (blue) and basal endothelial channel incorporating immune cells, namely PBMCs (red)149 (reprinted from ref. 149 with permission, copyright 2021, Elsevier Science & Technology Journals). D. Schematic of the HuMix model showing its three-microchamber design including microbial, epithelial and perfusion microchambers; as well as incorporation of oxygen and electrical monitoring through inclusion of optodes and electrode ports. For the gut–immune model set-up, CD4+ T cells were introduced into the lower perfusion chamber160 (reprinted from ref. 160 under the Creative Commons CC BY license). E. Schematic of the peristaltic intestine on a chip design showing a gut–immune study set up with immune cells, namely macrophages, seeded in upper and lower endothelial chambers, together with epithelial cells and enterobacteria in the central chamber31 (reprinted from ref. 31 under the Creative Commons CC BY license). F. Schematic of the embedded membrane microfluidic chip showing individual components of the model including the sealing channel into which the polycarbonate (PC) membrane, which separates epithelial (basal) from endothelial plus immune (PBMCs) (apical) compartments, is secured161 (reprinted from ref. 161 under the Creative Commons CC BY license). G. Schematic of the microfluidic organotypic device (MOD) showing the outer and central cyclic olefin copolymer layers separated by two polyurethane gaskets. Fluid flow in the upper/lower channel can be seen in red/blue. In the centre, seen in the exploded view, the tissue explant is supported by two mesh pieces (reprinted from ref. 162 with permission, copyright 2023, Royal Society of Chemistry). | ||
Other gut–immune lab-on-chip models relying on membrane-based channel separation with similar designs to Chip-S1 include that in Guo et al. (2021)149 (Fig. 3C), comprised of PDMS with a PDMS membrane, and that in Maurer et al. (2019),155 known as the MOTiF biochip (Fig. 3B), comprised of a polysterol-based chip with a polyethylene terephthalate (PET) membrane. Neither of these platforms however incorporated mechanical stimulation as provided by Chip-S1, or employed electrical barrier integrity monitoring (i.e. TEER).
Since the majority of host–microbiome interactions take place under anaerobic or microaerophillic conditions, Shah et al. (2016)160 followed a somewhat different design approach, yet still relying on stacked membrane-based separation, to develop a model capable of facilitating maintenance of varying oxygen conditions to enable the study of anaerobic and/or microaerophilic bacteria interfaced with intestinal cells. The model, known as the HuMix (human–microbial crosstalk) model (Fig. 3D), incorporated stacked elastomeric gaskets with spiral microchannels separated by semi-permeable polycarbonate (PC) membranes and secured between two PC enclosures. This set up resulted in the formation of three microchambers, each with their own inlets and outlets; 1) for the perfusion of media for the intestinal cells (perfusion microchamber), 2) for intestinal epithelial cell growth and maintenance (epithelial cell microchamber; Caco2) and 3) for the microbial cells (microbial microchamber) respectively. Further, this model included integrated oxygen sensors (optodes) to allow for real-time dissolved oxygen concentration monitoring in different parts of the model, with media oxygen conditions optimised until an oxygen gradient similar to that found in vivo was achieved. A specialised version was additionally designed to accommodate conventional chopstick electrodes for TEER measurement. The HuMix model was shown to be capable of supporting facultative and obligate anaerobe growth while maintaining intestinal cell viability and allowing epithelial-microbial cell crosstalk. Direct interactions between the bacteria and epithelial cells were however limited by the presence of the membrane separating the compartments.
Jing et al. (2020)31 developed a model with a similar design to the HuMix160 model above and with the ability to provide peristalsis-like mechanical cues in a simpler manner compared to Kim et al. (2016).19 Their three-channel microfluidic device incorporated stacked PDMS plates separated by porous ECM-coated PDMS membranes, secured between poly(methyl methacrylate) (PMMA) plates (Fig. 3E). A pneumatic pump was employed to facilitate culture media flow in the channels as well as to create cyclic pressure changes in the central intestinal chamber. These pressure changes were used to generate periodic pressure differences between the top/bottom chambers and the central one, resulting in a peristalsis-like movement of the cell-laden membranes. Besides the use of different materials, Jing et al. (2020)'s model included a round central cell compartment fed by straight microchannels, compared to the spiral microchannel design of the HuMix model.160 Benefits of this and the HuMix160 model include that they can be disassembled and reassembled.
Besides design, material choice is another key factor in lab-on-chip technologies, with considerations including biocompatibility, mechanical and optical properties, ease of fabrication and sterilization, and cost, amongst others.164 Much of the early microfluidics research employed the polymer PDMS, due to it possessing many of these properties.165 It is therefore no surprise that many of the platforms in Table 3 have also relied on this material.19,27,29–32,149,150,158,161 PDMS is however associated with some drawbacks including that it absorbs certain drugs, metabolites and hormones, compromising its use for drug and metabolite screening.164 To avert this, some devices have employed other polymers including the MOTiF biochip155 and the HuMix platform160 introduced above. Further, manufacture of PDMS membranes can be challenging,161 leading to other materials such as polycarbonate (PC) being used in some studies.22,160,161 Challenges associated with PC membranes have however also been found relating to their adhesion to PDMS chips, affecting sealing of the channels. Zhao et al. (2022)161 therefore set out to overcome this challenge with a sealing channel design.161 Their device comprised a PDMS chip with sealing channels into which liquid PDMS was injected to embed a collagen-coated porous PC membrane (Fig. 3F).161
Recently, developments in integrating tissue explants with microfluidic models, also known as tissue-on-a-chip devices,166 offer a potential new frontier in modelling the complexity of the intestinal tract. While a number of microfluidic-explant models were noted, some of which mention that immune cells are present,166–168 identification of the types of immune cells has generally been lacking. Recently, Cherwin et al. (2023)162 reported the presence of mast cells in the lamina propria layer of a mouse colon explant. In this study, the explant was shown to include the mucus layer, mucosa, submucosa and muscularis externa and was maintained in a microfluidic device for over 72 h. The microfluidic organotypic device (MOD) used in the study was modified from a previous version developed in Richardson et al. (2020)166 and comprised of three layers of cyclic olefin copolymer (COC) separated by polyurethane gaskets (Fig. 3G). Porous Nitex mesh was integrated on the central COC layer to support the tissue. Separate media flow was enabled by the microfluidic channels on the mucosal and serosal side of the central tissue layer allowing for a differential oxygen gradient across the tissue through use of low and ambient oxygen conditions.162 Oxygen sensor spots were further incorporated to allow for oxygen level monitoring and low oxygen conditions were noted to increase bacterial presence.166
Besides the membrane-based approaches above, other commercially available models relying on hydrogel-based channel separation and standard tissue-culture inserts include the OrganoPlate® platform169 made by Mimetas170 and the LiverChip™171 developed by CN Bio, respectively.
The 3-lane 40 OrganoPlate® platform169 incorporates 40 chambers, each with a three-lane microfluidic chip, and comprises of a top and bottom plate made of virgin polystyrene and glass respectively, with integrated microfluidics. Each chip in the top plate incorporates nine reservoirs, in a 3 × 3 matrix, on top of the 3-lane microchannel system, with the six side reservoirs acting as inlets and outlets for the different microchannels (Fig. 4A). The OrganoPlate® uses a membrane free approach, employing Phaseguide® technology developed in Vulto and colleagues172–174 which exploits the meniscus pinning effect at an liquid–air interface172 to fill the central channel with ECM, generally collagen I rat tail.33,151,175 Cells can then be seeded in one or both of the side flow channels. Plate positioning and flow are used to ensure cells proliferate around the microchannel forming a confluent tubular structure with polarised epithelial cells surrounding a hollow lumen.175 To simulate perfusion, media is placed in the four corner reservoirs and the plate placed on a rocker; providing a gravity driven and pump free approach. Different rocking angles, resulting in different intermittent shear stress forces, are recommended for each plate.170 Feasibility of the OrganoPlate® for building intestinal models was validated prior to incorporation of immune cells, such as in Beaurivage et al. (2019),151 where cytokine-stimulated inflammation was considered.151 Incorporation of immune cells in the OrganoPlate® has included addition to the side flow channels, as demonstrated in Gijzen et al. (2020),144 embedded in the central ECM channel, as demonstrated in Beaurivage et al. (2020)33 as well as both in a side channel and embedded in the central ECM channel, as demonstrated in Gjorevski et al. (2020).145
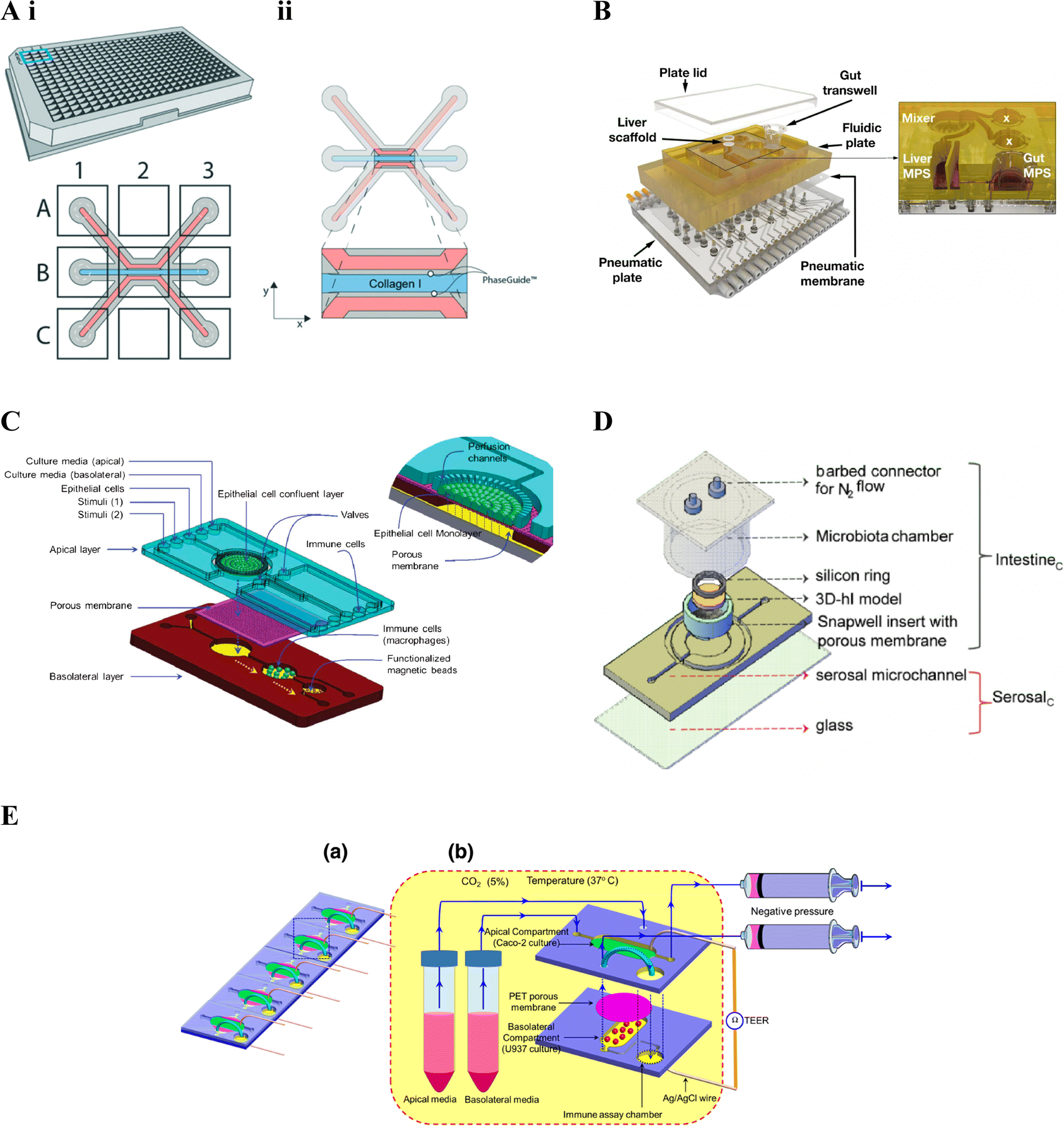 | ||
| Fig. 4 Gut–immune lab-on-chip designs. A. OrganoPlate® design. i. Schematic of the 3-lane 40 OrganoPlate® incorporating 40 chambers each with a three-lane microfluidic chip. ii. Schematic of the three-lane microfluidic chip, the central blue channel is initially filled with type I collagen, delineated by PhaseGuides, forming a membrane-free interstitial matrix separating fluidic channels on either side. In gut–immune studies, epithelial cells have been seeded in one of the side fluidic channels and immunes cells embedded into the central collagen channel, seeded in the opposite microfluidic channel, or both.145 (reprinted from ref. 145 with permission, copyright 2020, Royal Society of Chemistry). B. LiverChip™ design. Schematic of the multi-microphysiological systems (MPS) platform showing a fluidic plate with compartments for different MPSs (yellow) and a bottom plate fitted with microchannels and pumps for media circulation; with the exploded view showing three Transwell-based MPS compartments (the gut MPS and two disconnected compartments), a scaffold-based MPS (liver MPS) and a mixer chamber176 (reprinted from ref. 176 under the Creative Commons CC BY license). C. Schematic of the NutriChip showing a connected three chamber system incorporating epithelial cells, immune cells and functionalised magnetic beads for immunomagnetic assays, respectively158 (reprinted from ref. 158 with permission, copyright 2013, Royal Society of Chemistry). D. Schematic of the three-dimensional microbiota human intestine on chip (MihI-OC) model showing a central chamber housing a Snapwell® insert in which intestinal epithelial cells, grown on stromal equivalents, are housed; a fluidic serosal microchannel for culture media flow, and into which immune cells (PBMCs) were introduced; and a microbiota chamber where bacteria can be grown under different oxygen conditions, regulated by the flow of N2 through the connectors at the top of the chamber22 (reprinted from ref. 22 with permission, copyright 2022, Elsevier Science & Technology Journals). E. a. Schematic of an iteration on C, showing a five-chip approach, each with a two chamber-design. b. The first chamber, seeded with intestinal epithelial cells in the apical compartment and immune cells in the basal compartment; connects to the second immune assay chamber159 (reprinted from ref. 159 with permission, copyright 2016, Springer Nature BV). | ||
The LiverChip™171 also facilitates incorporation of more than one cell compartment however through reconfigurable incorporation of multiple micro-physiological systems (MPSs). The platform includes a top polysulfone plate which can incorporate multiple MPS chambers, capable of housing either scaffolds (flow-through module) or Transwell® inserts; a mixing chamber (mixer) representing systemic circulation (Fig. 4B); and an on-board pneumatic fluidics system which facilitates flow of culture media through the platform.157 The LiverChip™ platform had previously been used to study multi-organ pharmacokinetics176 however gut-liver inflammatory cross-talk was first considered in Chen et al. (2017).156 Gut-liver cross-talk is an important factor in physiological homeostasis and disease, with the liver linked to the gut via portal blood circulation which carries many metabolites, microbial antigens and inflammatory mediators from the gut. In this case, the platform was set up to connect a Transwell®-style gut MPS, a flow-through scaffold-based liver MPS and a mixing chamber (mixer).176 The flexibility of the LiverChip™ platform was later demonstrated by Edington et al. (2018)157 where three different configurations were explored, comprising a mixing chamber with four, seven, or 10 MPSs respectively and termed the “physiome-on-a-chip”. More recently, Trapecar et al. (2020)123 used a similar gut-liver platform, termed the 3 module gut–liver (3XGL) platform, to study IBD and its connection with liver disease. To achieve this they used a model of Ulcerative colitis, developed in a Transwell(R) format, similar to the above gut MPS, however using primary Ulcerative colitis epithelium, as well as dendritic cells and macrophages; together with the same flow-through Liver MPS. Further, Treg and Th17 cells were introduced in circulation. They investigated the effects of SCFA on Ulcerative colitis in different versions of the model, showing that the presence of T cells altered inflammatory responses. Trapecar et al. (2021)124 built on the model in Trapecar et al. (2020), including a cerebral MPS, termed the 3X gut–liver–brain (3XGLB) physiomimetic platform, to study this axis in the context of Parkinson's disease (PD). The study sought to investigate how immune pathways and SCFAs affect a PD phenotype. Trapecar et al. (2020, 2021) were the only studies of those relevant to this review found to consider gut–immune–SCFA interactions,123,124 the relevance of which have been introduced in section 3 above.
In comparison with the other platforms mentioned in this review, the OrganoPlate® represents the highest throughput approach, enabling formation and real-time TEER monitoring of 40 fluidic chips.151 Further, given its membrane-free design enhanced immune cell migration and epithelial-immune cell interactions are possible, compared to the membrane-based models above.145,151 For both the OrganoPlate® and LiverChip™ or 3XGL(B) studies in Table 3, inclusion of bacteria and mechanical cues was not seen the and potentially the membrane and pump-free approach of the former and Transwell-based approach of the latter may limit their longer-term inclusion.
Ramadan et al. (2013)158 were one of, if not the, first to include immune cells in an intestinal lab-on-chip model, using a three chamber system; the first housing a monolayer of intestinal (Caco2) cells grown on the apical side of a polyester membrane; the second chamber, downstream of the basolateral side of the first, containing immune cells (macrophages); and the third, downstream of the second, containing antibody-functionalised magnetic beads for immunomagnetic assays for cytokine detection (Fig. 4C). Ramadan and Jing (2016)159 iterated on the above, developing a more stacked model allowing higher throughput, namely five identical microfluidic chips in parallel, each comprising two vertically stacked compartments separated by a Polyethylene terephthalate (PET) membrane; as well as a third immune assay compartment downstream of the basolateral compartment. The apical and basolateral compartments were formed by two patterned PMMA sheets separated by the PET membrane (Fig. 4E); AG/AgCl electrodes were additionally inserted into each compartment and connected to a Volt-Ohm meter to enable TEER measurement.
De Gregorio et al. (2022)22 combined more advanced tissue engineering with microfluidic chip technology to develop an immune-responsive three-dimensional human intestine (3D-hI) model which could be combined with microfluidic lab-on-chip technology. The 3D-hI model comprised of a 3D cell-synthetized stromal equivalent (3D-CSSE) which was developed in previous papers,38,177 whereby cells were used to produce and assemble their own ECM-like matrix in vitro.38 Following three weeks of intestinal cell (Caco2) growth on 3D-CSSEs, 3D-hIs were moved to commercial Snapwell® inserts and positioned in the central chamber of a PDMS microfluidic device, now termed the Human intestine on chip (hI-oC), with the intestinal compartment on the top side of the chip and the serosal compartment on the lower basal side of the hI-oC. A microbiota chamber, comprising a cylinder with a port for an oxygen probe and a lid fitted with connectors for nitrogen (N2) flow, was then secured around the upper intestinal compartment where bacteria could be anaerobically grown, then termed the MihI-OC (Fig. 4D). N2 was flushed into the cylindrical chamber to create an oxygen gradient similar to that seen in vivo, showing that a physiological near-anoxic microenvironment could be generated at the midpoint of the intestinal lumen, a feature critical for obligate anaerobe colonisation. The ability of this set up to mimic the oxygen gradients and bacterial vertical distribution was confirmed through the introduction of anaerobic and microaerophilic bacteria to the MihI-OC, which localised as expected.
Incorporation of the 3D-CSSE provides a more representative and complex extracellular microenvironment for the intestinal cells in comparison to cell growth on planar 2D membranes and represents the only model of those under review here to feature this. To further mimic in vivo conditions, the authors could consider incorporating immune cells into the 3D-CSSE in future. Additionally, besides the HuMix160 and MOD models above, it is the only other gut–immune model identified that incorporates an oxygen gradient. Limitations of this approach however include that, similar to the LiverChip™, the set-up was only combined with microfluidic technology following formation of the 3D-hI under static conditions, requiring 21 days, in addition to the time required to form the 3D-CSSE, and further TEER measurements were taken off-platform. This architecture may also limit the ability to incorporate mechanical cues and longer-term bacterial co-culture as well as complicate higher throughput applications.
4.2. Intestinal epithelial cell types
As noted, the role of the gut in metabolism and metabolic diseases is very complex, with multiple interlinked processes and pathways, making in vitro modelling thereof very challenging. Cell lines, including enterocyte and enteroendocrine, have been widely used to study nutrient transport processes and incretin hormone release due to their reproducibility, availability, low cost and ease of use. However, given that cell lines have a cancerous phenotype, often displaying divergent features compared to the original tissue, and represent limited cell types, they fail to recapitulate in vivo complexity.23 Primary cells have also been used, and while more representative compared to cell lines, they come with numerous challenges including limited life span, slow proliferation and supply issues.178 Overcoming some of these limitations, stem cells and organoids have emerged as valuable alternatives. Organoids, derived from stem or progenitor cells, more accurately mimic the multiple in vivo cell types and organ structures, with Sato et al. (2009) pioneering the development of intestinal organoids.25 Challenges related to batch-to-batch consistency and reproducibility, complexities around culture conditions and the need for ECM however complicate their use.23 Further, long-term culture of 3D organoids is limited due to their cystic shape, leading to a build-up of toxins and cell debris in their centre, necessitating regular passaging.179 Organoid dissociation and subsequent growth as monolayers or within microfluidic lab-on-chip platforms overcomes some of these challenge.23,161 Organoids are however still limited in that they generally do not include other physiological components such as the immune system. Tissue explants offer enhanced tissue-like features180 and recent studies have demonstrated incorporation thereof in microfluidic models, such as Richardson et al. (2020), Martinez et al. (2022) and Cherwin et al. (2023) which, using mouse explants, showed the presence of microbiota,166 studied SCFA profiles as a function of oxygen conditions181 and showed presence of immune (mast) cells162 respectively. Explants are however constrained by limited life-spans, a need for suitable microenvironment conditions and lower throughput compared to cell-monolayer approaches.166 Currently many of the microfluidic explant-based studies rely on mouse tissue reverting to reliance on animals and related inconsistencies with human intestinal tracts. While some studies have incorporated human explants,168 transition to tissue from larger mammals, such as pigs, or humans is complicated by increased thickness and stronger muscle contractions,162 as well as possible challenges and ethical complexities in obtaining samples.In terms of the intestinal epithelial components of the non-explant lab-on-chip studies in this review (Table 3) which incorporate intestinal epithelial and immune components, many of the studies, especially the earlier ones, have relied on commercially available cell lines (Caco2, HT-29). Caco2 are known to differentiate into enterocyte-like cells over a period of 21 days in conventional static culture, however lab-on-chip studies have more recently shown accelerated growth and differentiation, with three to seven, but most commonly five, days reported28,31,32,149,151,155,160,161,175 Further, microfluidic conditions have also been shown in multiple studies to improve polarization, three-dimensional crypt-villi morphology149,155 and differentiation. Under perfusion and strain conditions, Caco2 cells have further been shown to differentiate into the four major intestinal epithelial cell types namely, absorptive, mucus-secretory, enteroendocrine, and Paneth cells, which had not previously been seen in static cultures,28,33,163 while also exhibiting improved glucose reuptake and mucus production.163 While the aforementioned are encouraging improvements compared to static culture, the use of cells lines is still a limiting factor given their cancerous phenotype. Progress has been made in replacing these cell lines with organoid derived cells29,33 which more faithfully represent the cell diversity found in the intestine and further can be derived from specific parts of the intestine for more targeted research.23 Recent progress in integration of tissue explants on chip offers potential for even more in vivo-relevant models as explants have been shown to include the mucus layer, mucosa, submucosa and muscularis externa.162 While numerous papers, both Caco2 and organoid-based, have indicated the presence of enteroendocrine cells, none of the papers in this review have focused on their role in disease. Given the interplay and cross-communication between enteroendocrine and immune cells, this could be an interesting future line of study using lab-on-chip technology.
As introduced in section 2, mucus plays an important role in intestinal homeostasis,55,182 yet it has not been an area of focus thus far in gut–immune lab-on-chip models. Given that Caco2 cells were shown to differentiate into all four major epithelial cell types, presence of mucus production was confirmed in Caco2 only models22,31,32,161 and some limited mucus-focused studies were noted. For example, Jing et al. (2022) investigated dextran sodium sulfate (DSS) induced mucus layer injury together with the ability of chitosan oligosaccharides (COS) to regulate mucus secretion and reduce injury;32 while De gregorio et al. (2022) monitored bacterial localisation within the mucus layer.22 Further, several cell line-based studies included HT29 cells144,149 to increase mucus production.182 Mucus has additionally been seen using the explant approach162 and other lab-on-chip models of the gut, not covered in this review due to lack of immune components, have also confirmed mucus secretion when using intestinal organoid derived cells, such as Kasendra et al. (2018) where mucus production by duodenal derived organoid cells in Chip-S1 was reported.183 More recently, Sontheimer-Phelps et al. (2023) conducted a mucus-focused study using primary patient-derived colonic epithelial cells grown in Chip-S1, showing that inner and outer layers of mucus could be seen along with changes in hydration state, such as mucus swelling when stimulated with prostaglandin E2.184 These studies demonstrate the potential for more physiologically relevant mucus modelling to be incorporated in gut–immune lab-on-chip models in future to further our understanding of how mucus is affected or implicated in metabolic diseases.
4.3. Intestine-relevant immune cell types
Immune-specific screening has historically involved studying immune cells in isolation, yet given the complex interactions between immune cells and their 3D microenvironment in vivo, the accuracy of results from this approach may be limited. More complex models which aim to incorporate this microenvironment are therefore being developed in an effort to better recapitulate the in vivo situation.185,186The cell types incorporated in the lab-on-chip models discussed above can be seen in Table 3. As introduced above, the intestinal immune system involves complex interactions between the epithelium and multiple different innate and adaptive immune cells. Incorporation of the immune system in intestinal lab-on-chip models still appears to be in its infancy with many of the models employing simplified incorporation of a limited number of immune cells, lacking recapitulation of the complexity associated with the in vivo situation. The models considered as part of this review have primarily focused on macrophages and monocytes as well as blood-derived peripheral blood mononuclear cells (PBMCs) which, as demonstrated in Kim et al. (2016) contain a mixed population of both innate and adaptive immune cells, including monocytes and granulocytes; and lymphocytes respectively.19 A limited number of the studies presented here also specifically included T cells, dendritic cells and neutrophils. In terms of the former, Shah et al. used primary CD4 + T cells purified from healthy blood donors which they activated with IL-2, CD3 antibodies and CD28 antibodies,160 while Gjorevski et al. (2020) used monocytes which were subsequently differentiated toward macrophages together with neutrophils isolated from fresh human peripheral blood.145 A number of studies considered dendritic cells including, Maurer et al. (2019),155 derived from PBMCs, and Gijzen et al. (2020), using the acute myelomonocytic leukemia cell line MUTZ-3.144 Incorporation of multiple immune cells, increasing the complexity of immune interactions studied, has more recently emerged, such as the studies by Gjorevski et al. (2020)145 and Trapecar et al. (2020, 2021).123,124 The former represents the first model to include mucosal epithelium with both resident (macrophages) and infiltrating (neutrophils) immune cells which could migrate in the 3D ECM, allowing for functional cross-talk between the cells;145 while the latter studies combined dendritic cells and macrophages with circulating Treg and Th17 cells.123,124 Further work is however required to improve immune cell complexity and cross-talk, allowing for adaptive and innate as well as intraepithelial and mucosal immune interaction modelling. While associated with other challenges, tissue explant potentially offer a route for studying some of these more complex resident immune cell interactions, with studies indicating that immune cells should be present166 and Cherwin et al. (2023) confirming presence of mast cells.162
Incorporation of immune cells in non-explant models has further generally appeared to be during end point-type assays, being transiently introduced into one of the compartments rather than seeded early-on for long-term co-culture with the epithelial and/or endothelial cells.19,160 Highlighting limitations of these models, which fail to provide a more in vivo like immune microenvironment capable of supporting enhanced immune cell diversity and facilitating immune cell dynamics. Maurer et al. (2019) showed, through co-culture of macrophages with endothelial cells for a week, that longer-term immune cell incorporation in lab-on-chip models is possible and observed that immune cells (macrophages and dendritic cells) could adapt and self-organise in response to LPS stimuli.155 Later, using the 3XGL(B) platform, Trapecar et al. (2020, 2021)123,124 conducted interaction studies over four days and using the OrganoPlate® platform, Gjorevski et al. (2020)145 demonstrated the feasibility of longer-term immune-epithelial cell co-culture, embedding THP-1 cells, subsequently differentiated into macrophages, within the central ECM channel before seeding Caco2 cells and neutrophils in the two side channels respectively. Gjorevski et al. (2020) observed neutrophil movement and infiltration - for which ECM composition and mechanical properties required optimisation – as well as crosstalk between resident and circulating immune cells.145 Numerous static models have also been developed using collagen hydrogels to support immune-epithelial co-culture including Hinman et al. (2022) and Moysidou et al. (2022) using neutrophil-like cells187 and macrophages or monocytes188 respectively. Studies have additionally found that the presence of macrophages in in vitro models improved enteroid-derived barrier maturity and function.189
While numerous human immune cell lines exist including for T-cells (i.e. Jurkat),190 monocytes and macrophages (i.e. THP-1, U937)144,145,158,159,191 and B cells (i.e. Raji, Daudi, Ramos),192 amongst others, only a limited number of these have been used in the gut–immune lab-on-chip models under consideration. To improve models, not only is the type of immune cell or cells relevant to the research question important, whether innate or adaptive, the source of the immune cells is also an important consideration. Primary immune cells may therefore be preferred however in vitro culture thereof can be challenging, especially given that several immune cell types, including naïve lymphocytes, do not naturally proliferate in culture, requiring either fresh harvesting or mitogen treatment to induce proliferation. Complicating this further, is that significant donor-to-donor variability can occur, affecting outcomes and relevant controls.18 Besides primary or donor-derived origins, some immune cells can also be derived from embryonic or induced pluripotent stem cells (iPSC), with a variety of immune cell types including macrophages,193 natural killer cells194 and T cells195,196 derived through this method. It should however be noted that multiple other immune-focused lab-on-chip applications,185,186,197,198 lacking the intestinal component, do exist, using cells lines and primary cells; for example multiple studies have developed lymph nodes on chip,186 such as Moura Rosa et al. (2016) who developed a lymph node-on-chip to study intercell dynamics of T cells and dendritic cells.199 Recently, advances have been made in modelling immune tissues, with Goyal et al. (2022) developing a lab-on-chip model (Chip-S1) of ectopic lymphoid follicles using primary human blood B- and T-lymphocytes which were able to self-assemble in a 3D extracellular matrix gel within one of the fluidic channels, offering a new way to explore how the human immune system responds to infections and vaccinations.200 Further, Delong and Ross (2023) developed a multi-organ system to allow for communication between ex vivo Peyer's patches and mesenteric lymph node slices.201 The advances in immune models will hopefully aid in developing more advanced gut–immune models in future, allowing us to discover more about the intestinal immune system and its role in the development and progression of various diseases.
4.4. Biophysical and biochemical cues
Tissue microenvironment together with biophysical and biochemical cues play an important role in cellular processes and cell function.18 Mechanical forces such as peristalsis, intestinal smooth muscle contraction and relaxation, and fluid sheer stress, which is exerted on the apical surface of the intestinal epithelium due to luminal flow, are key actions which aid the gut in performing its primary function of nutrient absorption.202 Intestinal cells are therefore adapted to sense and respond to these mechanical cues, converting the mechanical forces into biochemical signals which regulate gene expression and cell behaviour, modulating gut function and barrier integrity.203 Aberrations in these movements can therefore lead to bacterial overgrowth and inflammation and may play a role in, amongst others, Crohn's disease and IBS.204,205 Of the lab-on-chip models relevant to this review, only two designs incorporate mechanical cues.19,31 Chip-S1 allows for mechanical stretching of the porous PDMS membrane, achieved through more complex application of a vacuum in chambers on either side of the central channel,19 while the chip in Jing et al. (2020) employs a simpler method relying on a pneumatic pump to periodically alter the pressure difference between the fluids in the lumen and vessel.31 Kim et al. (2016) and Jing et al. (2020) both showed, in line with the in vivo findings, that the cyclic stretching was key to preventing bacterial overgrowth19 and allowing longer-term co-culture.31 Peristalsis-like cues with shear stress from luminal fluid flow was further found to promote epithelial cell growth, glycocalyx and microvilli formation as well as affect absorption and metabolic functions.31Beside mechanical cues, a number of other unique and important conditions exist in the gut including oxygen, nutrient and pH gradients which also play a role in regulating cellular processes.23 In particular, oxygen levels in the gut, which vary along the length of the intestine and along the crypt-villi structures of the small intestine mucosa,206,207 are important regulators of gene expression and barrier function, mediated by hypoxia-inducible factors (HIFs).206 HIF pathways have been shown to play an important role in metabolism and inflammation and have therefore been implicated in numerous metabolic diseases including obesity and T2D.208 Intestinal microbiota play a role in this oxygen gradient, with vertical stratification of facultative anaerobic and microaerophilic bacteria across the gradient.22 Of the lab-on-chip models relevant to this review, only the HuMix,160 3D-hI and explant-based MOD models22 incorporated oxygen gradients, highlighting a need of further development of gut–immune models to more readily incorporate these cues. Technologies exist to monitor these conditions, such as the optical oxygen sensor spots (optodes) used in the HuMix and MOD models,160 however again, incorporation of sensors has been limited in the models under consideration. While not seen in the models in this review, Chip-S1 has been shown to be compatible with oxygen monitoring and longer-term bacterial co-culture as demonstrated in Jalili-Firoozinezhad et al. (2019) where a physiologically relevant oxygen gradient was maintained, enabling longer-term (days) co-culture of intestinal epithelium with aerobic and anaerobic human gut microbiota.209
As demonstrated by Gjorevski et al. (2020) engineering the microenvironment in which immune cells are maintained is another critical yet potentially challenging aspect to consider. They showed that the mechanical properties of the central ECM in which they seeded neutrophils was critical for neutrophil movement and subsequent infiltration.145 This is not unique, with numerous other studies showing how mechanical properties and physical cues affect different immune cells.18,210–215 For example, ECM or substrate stiffness has been found to affect sensitivity and responses of primary T cells isolated for immunotherapies,210,211 shown to affect macrophage polarisation, function and migration,212–214 and B cell activation, proliferation and class switching,215 amongst others. Engineering or selecting appropriate materials, such as natural or synthetic biomaterials, in which to maintain immune cells, as well as scaffolds for intestinal modelling, is therefore critical, including optimisation of physical, chemical and biological conditions and cues.18
Together, these studies highlight the importance of incorporating and optimizing mechanical cues and microenvironment conditions, as summarised in Fig. 5, to better recapitulate the in vivo conditions. No studies relevant to this review were found that incorporate gut and immune cells, oxygen gradients and mechanical deformation, highlighting the need for further development of lab-on-chip technology for gut–immune interaction studies.
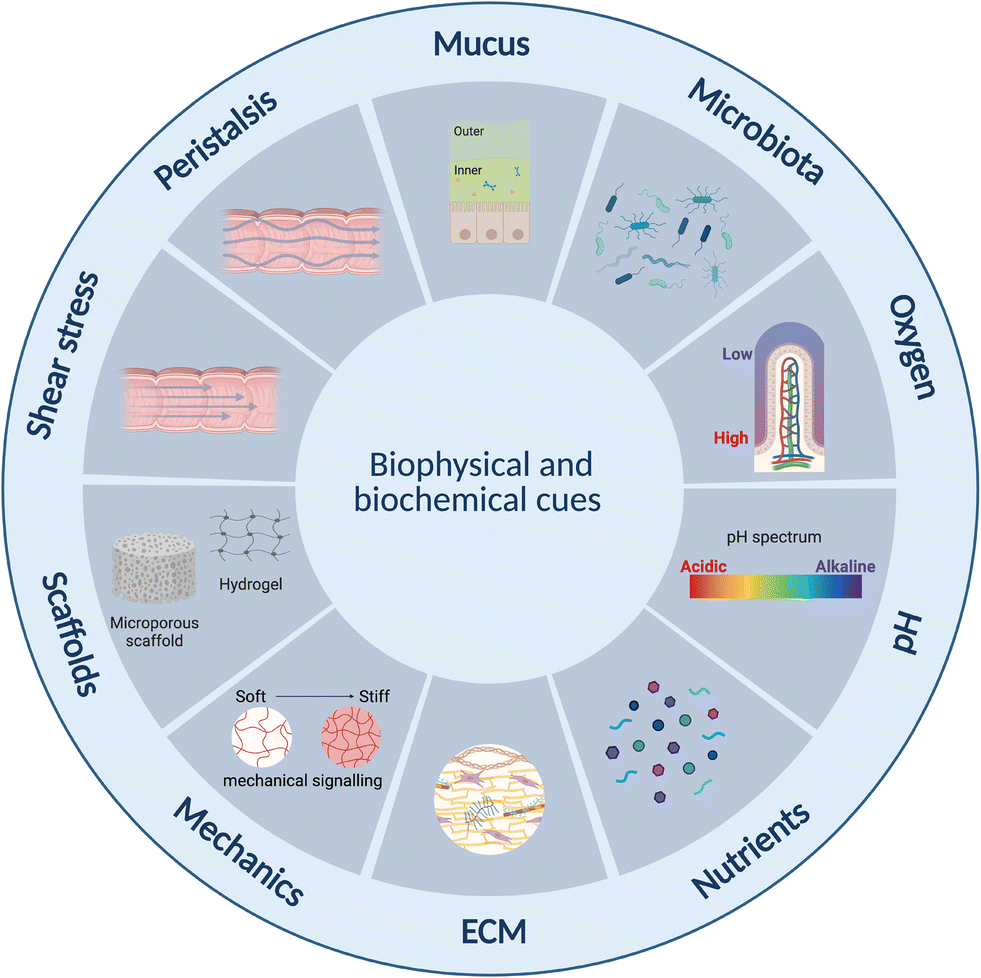 | ||
| Fig. 5 Summary of biophysical and biochemical cues affecting intestinal and immune cells for consideration in gut–immune lab-on-chip model development. ECM: extracellular matrix. Created with https://BioRender.com. | ||
5. Conclusions and future directions
Unarguably, a growing body of evidence demonstrates a significant role of the gut–immune axis in the development of metabolic disease in humans. Due to the current pandemic extent of metabolic diseases, associated with a high burden for individuals but also the national health system, a better understanding of the mechanisms regulating this axis is urgently needed. It is well understood that diet is a key regulator of gut–immune interactions and the development of metabolic diseases. However, mechanisms are still elusive, for example due to unknown mechanisms of nutrient interactions, cumulative effects and gut microbiome activities in humans. In addition, outcomes of controlled feeding studies are limited since they address only short-term effects but also change the study participants' dietary habits. Thus, the development of personalized dietary recommendations and the integration of gut microbes will be highly beneficial for maintaining functional gut–immune interaction.Emerging lab-on-chip technologies aim to offer better in vitro models capable of delivering predictions of human physiologies. The models discussed above highlight the recent progress in modelling the gut–immune axis using this technology, showing proof-of-concept for integration of important compartments and cues for studying the gut–immune axis, including integration of peristalsis-like contractions, shear stress, oxygen gradients and immune and organoid-derived cells, amongst others. Incorporation of mechanical and biophysical cues is however not present across all models and combination of these cues has not been noted in a single design, opportunities therefore still exist for improvement.
Generally, many of these lab-on-chip models still suffer from low through-put, high cost, limited read-out techniques and in some cases the need for specialised equipment.146 Model improvements required however strongly depend on the research question and therefore must be considered on a case-by-case basis, in particular as there is classically a trade-off between model complexity, experimental control and throughput.151,216 Careful consideration is therefore required to ensure models developed are fit-for-purpose without being needlessly complex.
In terms of metabolism-related applications, material choice remains an important consideration given limitations associated with the widely used PDMS related to absorption of hydrophobic molecules such as metabolites, cytokines and target drugs217 making it potentially unsuitable for metabolic studies. Improvements in material choice and design may additionally assist in overcoming other limitations around scalability, standardisation of devices, to improve comparability amongst studies, as well as issues related to miniaturisation.146 Further, in terms of metabolic diseases, more research is in particular required to better understand the immune-microbiota axis as mechanistic studies have thus far been limited by the lack of suitable experimental models.155 Inclusion of more complex microbiota is therefore a key future development. Until now, only limited strains have been included in the lab-on-chip models discussed above. Further, the studies failed to consider IgA or hormone-related factors, identified as possible links to metabolic diseases. Incorporation of more complex microbiota however necessitates development of improved approaches for oxygen gradient maintenance and monitoring, coupled with microfluidic approaches capable of providing mechanical cues, for longer-term microbial co-culture, and higher-throughput designs.
Opportunities also exist to improve incorporation of in situ read-out methods such as TEER, and other biosensors relevant to the research questions, such as those related to metabolic, biochemical and biophysical modification217 (pH, oxygen, cytokines218). Incorporation of more complex tissue cultures presents another area of improvement to allow for a more in vivo-like microenvironment and improved cell–cell interaction, in particular for immune cell migration which is strongly influenced by microenvironment mechanical properties.185 Expansion of models to include stroma and basement membrane-like features would be useful inclusions.38 In terms of this, recent development of electrically active scaffolds such as those fabricated from the biocompatible conducting polymer poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) open a new frontier for growth and in situ monitoring of 3D cell cultures,24 work however remains to confirm compatibility with immune cells and their dynamics as well as provision of mechanical cues.
Lastly, with the dawn of patient derived organoids and microfluidic explant models, opportunities exist to more faithfully study different parts of the intestine as well as explore genetic, epigenetic, immune and microbiome variations in the population. Further, as gut microbiome composition has been linked to treatment efficacy,219 such as for cancer immunotherapy;220 development of robust personalised gut–immune-microbiome models could be key resources for predicting efficacy and improving treatments across different conditions.
Author contributions
Conceptualization, AEW, VS, RMO; writing – original draft, AEW, VS; writing – review & editing, AEW, VS, RMO.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. E. W. acknowledges funding from the Cambridge Commonwealth, European & International Trust and Newnham College at the Cambridge University.References
- P. González-Muniesa, M.-A. Mártinez-González, F. B. Hu, J.-P. Després, Y. Matsuzawa, R. J. F. Loos, L. A. Moreno, G. A. Bray and J. A. Martinez, Nat. Rev. Dis. Primers, 2017, 3, 17034 CrossRef PubMed.
- World Health Organization, World Obesity Day 2022 – Accelerating action to stop obesity, https://www.who.int/news/item/04-03-2022-world-obesity-day-2022-accelerating-action-to-stop-obesity, (accessed July 17, 2023) Search PubMed.
- E. Mahase, BMJ, 2023, 380, 523 CrossRef PubMed.
- R. A. DeFronzo, E. Ferrannini, L. Groop, R. R. Henry, W. H. Herman, J. J. Holst, F. B. Hu, C. R. Kahn, I. Raz, G. I. Shulman, D. C. Simonson, M. A. Testa and R. Weiss, Nat. Rev. Dis. Primers, 2015, 1, 15019 CrossRef PubMed.
- A. R. Saltiel and J. M. Olefsky, J. Clin. Invest., 2017, 127, 1–4 CrossRef PubMed.
- W. Kopp, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2019, 12, 2221–2236 CrossRef CAS PubMed.
- M. J. Bailey, N. N. Naik, L. E. Wild, W. B. Patterson and T. L. Alderete, Gut Microbes, 2020, 11, 1188–1202 CrossRef CAS PubMed.
- A. Vaiserman and O. Lushchak, Ageing Res. Rev., 2019, 55, 100957 CrossRef CAS PubMed.
- S. E. Kahn, S. M. Haffner, M. A. Heise, W. H. Herman, R. R. Holman, N. P. Jones, B. G. Kravitz, J. M. Lachin, M. C. O'Neill, B. Zinman, G. Viberti and ADOPT Study Group, N. Engl. J. Med., 2006, 355, 2427–2443 CrossRef CAS PubMed.
- M. Bouchoucha, B. Uzzan and R. Cohen, Diabetes Metab., 2011, 37, 90–96 CrossRef CAS PubMed.
- M. Y. Donath and S. E. Shoelson, Nat. Rev. Immunol., 2011, 11, 98–107 CrossRef CAS PubMed.
- V. K. Ridaura, J. J. Faith, F. E. Rey, J. Cheng, A. E. Duncan, A. L. Kau, N. W. Griffin, V. Lombard, B. Henrissat, J. R. Bain, M. J. Muehlbauer, O. Ilkayeva, C. F. Semenkovich, K. Funai, D. K. Hayashi, B. J. Lyle, M. C. Martini, L. K. Ursell, J. C. Clemente, W. Van Treuren, W. A. Walters, R. Knight, C. B. Newgard, A. C. Heath and J. I. Gordon, Science, 2013, 341, 1241214 CrossRef PubMed.
- L. Garidou, C. Pomié, P. Klopp, A. Waget, J. Charpentier, M. Aloulou, A. Giry, M. Serino, L. Stenman, S. Lahtinen, C. Dray, J. S. Iacovoni, M. Courtney, X. Collet, J. Amar, F. Servant, B. Lelouvier, P. Valet, G. Eberl, N. Fazilleau, V. Douin-Echinard, C. Heymes and R. Burcelin, Cell Metab., 2015, 22, 100–112 CrossRef CAS PubMed.
- C. Pomié, V. Blasco-Baque, P. Klopp, S. Nicolas, A. Waget, P. Loubières, V. Azalbert, A. Puel, F. Lopez, C. Dray, P. Valet, B. Lelouvier, F. Servant, M. Courtney, J. Amar, R. Burcelin and L. Garidou, Mol. Metab., 2016, 5, 392–403 CrossRef PubMed.
- P. D. Cani, J. Amar, M. A. Iglesias, M. Poggi, C. Knauf, D. Bastelica, A. M. Neyrinck, F. Fava, K. M. Tuohy, C. Chabo, A. Waget, E. Delmée, B. Cousin, T. Sulpice, B. Chamontin, J. Ferrières, J.-F. Tanti, G. R. Gibson, L. Casteilla, N. M. Delzenne, M. C. Alessi and R. Burcelin, Diabetes, 2007, 56, 1761–1772 CrossRef CAS PubMed.
- P. D. Cani, R. Bibiloni, C. Knauf, A. Waget, A. M. Neyrinck, N. M. Delzenne and R. Burcelin, Diabetes, 2008, 57, 1470–1481 CrossRef CAS PubMed.
- T. T. Roh, Y. Chen, S. Rudolph, M. Gee and D. L. Kaplan, Trends Biotechnol., 2021, 39, 274–285 CrossRef CAS PubMed.
- J. H. Hammel, J. M. Zatorski, S. R. Cook, R. R. Pompano and J. M. Munson, Adv. Drug Delivery Rev., 2022, 182, 114111 CrossRef CAS PubMed.
- H. J. Kim, H. Li, J. J. Collins and D. E. Ingber, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7–E15 CAS.
- A. Bein, W. Shin, S. Jalili-Firoozinezhad, M. H. Park, A. Sontheimer-Phelps, A. Tovaglieri, A. Chalkiadaki, H. J. Kim and D. E. Ingber, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 659–668 CrossRef PubMed.
- Y. Belkaid and T. W. Hand, Cell, 2014, 157, 121–141 CrossRef CAS PubMed.
- V. De Gregorio, C. Sgambato, F. Urciuolo, R. Vecchione, P. A. Netti and G. Imparato, Biomaterials, 2022, 286, 121573 CrossRef CAS PubMed.
- C.-M. Moysidou, C. Barberio and R. M. Owens, Front. Bioeng. Biotechnol., 2021, 8, 620962 CrossRef PubMed.
- C. Pitsalidis, D. Van Niekerk, C.-M. Moysidou, A. J. Boys, A. Withers, R. Vallet and R. M. Owens, Sci. Adv., 2022, 8, eabo4761 CrossRef CAS PubMed.
- T. Sato, R. G. Vries, H. J. Snippert, M. Van De Wetering, N. Barker, D. E. Stange, J. H. Van Es, A. Abo, P. Kujala, P. J. Peters and H. Clevers, Nature, 2009, 459, 262–265 CrossRef CAS PubMed.
- T. Zietek, E. Rath, D. Haller and H. Daniel, Sci. Rep., 2015, 5, 16831 CrossRef CAS PubMed.
- W. Shin and H. J. Kim, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(45), E10539–E10547 CrossRef CAS PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165 RSC.
- A. Bein, S. Kim, G. Goyal, W. Cao, C. Fadel, A. Naziripour, S. Sharma, B. Swenor, N. LoGrande, A. Nurani, V. N. Miao, A. W. Navia, C. G. K. Ziegler, J. O. Montañes, P. Prabhala, M. S. Kim, R. Prantil-Baun, M. Rodas, A. Jiang, L. O'Sullivan, G. Tillya, A. K. Shalek and D. E. Ingber, Front. Pharmacol., 2021, 12, 718484 CrossRef CAS PubMed.
- S. J. Kerns, C. Belgur, D. Petropolis, M. Kanellias, R. Barrile, J. Sam, T. Weinzierl, T. Fauti, A. Freimoser-Grundschober, J. Eckmann, C. Hage, M. Geiger, P. R. Ng, W. Tien-Street, D. V. Manatakis, V. Micallef, R. Gerard, M. Bscheider, E. Breous-Nystrom, A. Schneider, A. M. Giusti, C. Bertinetti-Lapatki, H. S. Grant, A. B. Roth, G. A. Hamilton, T. Singer, K. Karalis, A. Moisan, P. Bruenker, C. Klein, M. Bacac, N. Gjorevski and L. Cabon, eLife, 2021, 10, e67106 CrossRef CAS PubMed.
- B. Jing, Z. A. Wang, C. Zhang, Q. Deng, J. Wei, Y. Luo, X. Zhang, J. Li and Y. Du, Front. Bioeng. Biotechnol., 2020, 8, 272 CrossRef PubMed.
- B. Jing, K. Xia, C. Zhang, S. Jiao, L. Zhu, J. Wei, Z. A. Wang, N. Chen, P. Tu, J. Li and Y. Du, Front. Cell Dev. Biol., 2022, 10, 877892 CrossRef PubMed.
- C. Beaurivage, A. Kanapeckaite, C. Loomans, K. S. Erdmann, J. Stallen and R. A. J. Janssen, Sci. Rep., 2020, 10, 21475 CrossRef CAS PubMed.
- S. Mohammad and C. Thiemermann, Front. Immunol., 2021, 11, 594150 CrossRef PubMed.
- S. Siri, Y. Zhao, F. Maier, D. M. Pierce and B. Feng, Bioengineering, 2020, 7, 130 CrossRef CAS PubMed.
- M. E. B. FitzPatrick and S. Keshav, in Oxford Textbook of Medicine, Oxford Academic, Oxford, 6th edn, 2020, p. C15.1.P42 Search PubMed.
- S. Pompili, G. Latella, E. Gaudio, R. Sferra and A. Vetuschi, Front. Med., 2021, 8, 610189 CrossRef PubMed.
- V. De Gregorio, G. Imparato, F. Urciuolo and P. A. Netti, Biotechnol. Bioeng., 2018, 115, 1062–1075 CrossRef CAS PubMed.
- J. M. Allaire, S. M. Crowley, H. T. Law, S.-Y. Chang, H.-J. Ko and B. A. Vallance, Trends Immunol., 2018, 39, 677–696 CrossRef CAS PubMed.
- S. W. Crawley, M. S. Mooseker and M. J. Tyska, J. Cell Biol., 2014, 207, 441–451 CrossRef CAS PubMed.
- F. M. Gribble and F. Reimann, Nat. Rev. Endocrinol., 2019, 15, 226–237 CrossRef CAS PubMed.
- S. R. Lueschow and S. J. McElroy, Front. Immunol., 2020, 11, 587 CrossRef CAS PubMed.
- F. Gerbe, C. Legraverend and P. Jay, Cell. Mol. Life Sci., 2012, 69, 2907–2917 CrossRef CAS PubMed.
- A. Humphries and N. A. Wright, Nat. Rev. Cancer, 2008, 8, 415–424 CrossRef CAS PubMed.
- R. Sender, S. Fuchs and R. Milo, PLoS Biol., 2016, 14, e1002533 CrossRef PubMed.
- Y. Fan and O. Pedersen, Nat. Rev. Microbiol., 2021, 19, 55–71 CrossRef CAS PubMed.
- T. Gensollen, S. S. Iyer, D. L. Kasper and R. S. Blumberg, Science, 2016, 352, 539–544 CrossRef CAS PubMed.
- M. Rastelli, P. D. Cani and C. Knauf, Endocr. Rev., 2019, 40, 1271–1284 CrossRef PubMed.
- T. C. Fung, C. A. Olson and E. Y. Hsiao, Nat. Neurosci., 2017, 20, 145–155 CrossRef CAS PubMed.
- I. J. Dikeocha, A. M. Al-Kabsi, M. Miftahussurur and M. A. Alshawsh, FASEB J., 2022, 36(6), e22350 CrossRef CAS PubMed.
- A. J. Bäumler and V. Sperandio, Nature, 2016, 535, 85–93 CrossRef PubMed.
- A. Vijay and A. M. Valdes, Eur. J. Clin. Nutr., 2022, 76, 489–501 CrossRef CAS PubMed.
- J. Libertucci and V. B. Young, Nat. Microbiol., 2018, 4, 35–45 CrossRef PubMed.
- E. Le Chatelier, T. Nielsen, J. Qin, E. Prifti, F. Hildebrand, G. Falony, M. Almeida, M. Arumugam, J.-M. Batto, S. Kennedy, P. Leonard, J. Li, K. Burgdorf, N. Grarup, T. Jørgensen, I. Brandslund, H. B. Nielsen, A. S. Juncker, M. Bertalan, F. Levenez, N. Pons, S. Rasmussen, S. Sunagawa, J. Tap, S. Tims, E. G. Zoetendal, S. Brunak, K. Clément, J. Doré, M. Kleerebezem, K. Kristiansen, P. Renault, T. Sicheritz-Ponten, W. M. De Vos, J.-D. Zucker, J. Raes, T. Hansen, MetaHIT consortium, E. Guedon, C. Delorme, S. Layec, G. Khaci, M. Van De Guchte, G. Vandemeulebrouck, A. Jamet, R. Dervyn, N. Sanchez, E. Maguin, F. Haimet, Y. Winogradski, A. Cultrone, M. Leclerc, C. Juste, H. Blottière, E. Pelletier, D. LePaslier, F. Artiguenave, T. Bruls, J. Weissenbach, K. Turner, J. Parkhill, M. Antolin, C. Manichanh, F. Casellas, N. Boruel, E. Varela, A. Torrejon, F. Guarner, G. Denariaz, M. Derrien, J. E. T. Van Hylckama Vlieg, P. Veiga, R. Oozeer, J. Knol, M. Rescigno, C. Brechot, C. M'Rini, A. Mérieux, T. Yamada, P. Bork, J. Wang, S. D. Ehrlich and O. Pedersen, Nature, 2013, 500, 541–546 CrossRef CAS PubMed.
- R. Cai, C. Cheng, J. Chen, X. Xu, C. Ding and B. Gu, Gut Microbes, 2020, 11, 680–690 CrossRef PubMed.
- G. C. Hansson, Curr. Opin. Microbiol., 2012, 15, 57–62 CrossRef CAS PubMed.
- R. Donahue, J. K. Sahoo, S. Rudolph, Y. Chen and D. L. Kaplan, Adv. Healthcare Mater., 2023, 2300301 CrossRef CAS PubMed.
- M. Herath, S. Hosie, J. C. Bornstein, A. E. Franks and E. L. Hill-Yardin, Front. Cell. Infect. Microbiol., 2020, 10, 248 CrossRef CAS PubMed.
- K. M. Sanders, S. D. Koh, S. Ro and S. M. Ward, Nat. Rev. Gastroenterol. Hepatol., 2012, 9, 633–645 CrossRef CAS PubMed.
- B. Waclawiková, A. Codutti, K. Alim and S. El Aidy, Gut Microbes, 2022, 14, 1997296 CrossRef PubMed.
- G. Roda, S. Chien Ng, P. G. Kotze, M. Argollo, R. Panaccione, A. Spinelli, A. Kaser, L. Peyrin-Biroulet and S. Danese, Nat. Rev. Dis. Primers, 2020, 6, 22 CrossRef PubMed.
- T. Kobayashi, B. Siegmund, C. Le Berre, S. C. Wei, M. Ferrante, B. Shen, C. N. Bernstein, S. Danese, L. Peyrin-Biroulet and T. Hibi, Nat. Rev. Dis. Primers, 2020, 6, 74 CrossRef PubMed.
- C. Jung, J.-P. Hugot and F. Barreau, Int. J. Inflammation, 2010, 2010, 1–12 CrossRef PubMed.
- U. M. Mörbe, P. B. Jørgensen, T. M. Fenton, N. Von Burg, L. B. Riis, J. Spencer and W. W. Agace, Mucosal Immunol., 2021, 14, 793–802 CrossRef PubMed.
- A. M. Mowat and W. W. Agace, Nat. Rev. Immunol., 2014, 14, 667–685 CrossRef CAS PubMed.
- L. Steele, L. Mayer and M. Cecilia Berin, Immunol. Res., 2012, 54, 75–82 CrossRef CAS PubMed.
- R. L. Owen and A. L. Jones, Gastroenterology, 1974, 66, 189–203 CrossRef CAS.
- S. Fagarasan, S. Kawamoto, O. Kanagawa and K. Suzuki, Annu. Rev. Immunol., 2010, 28, 243–273 CrossRef CAS PubMed.
- F. Barone, A. Vossenkamper, L. Boursier, W. Su, A. Watson, S. John, D. K. Dunn–Walters, P. Fields, S. Wijetilleka, J. D. Edgeworth and J. Spencer, Gastroenterology, 2011, 140, 947–956 CrossRef CAS PubMed.
- Y. Zhao, M. Uduman, J. H. Y. Siu, T. J. Tull, J. D. Sanderson, Y.-C. B. Wu, J. Q. Zhou, N. Petrov, R. Ellis, K. Todd, K.-M. Chavele, W. Guesdon, A. Vossenkamper, W. Jassem, D. P. D'Cruz, D. J. Fear, S. John, D. Scheel-Toellner, C. Hopkins, E. Moreno, N. L. Woodman, F. Ciccarelli, S. Heck, S. H. Kleinstein, M. Bemark and J. Spencer, Nat. Commun., 2018, 9, 3857 CrossRef PubMed.
- T. M. Fenton, P. B. Jørgensen, K. Niss, S. J. S. Rubin, U. M. Mörbe, L. B. Riis, C. Da Silva, A. Plumb, J. Vandamme, H. L. Jakobsen, S. Brunak, A. Habtezion, O. H. Nielsen, B. Johansson-Lindbom and W. W. Agace, Immunity, 2020, 52, 557–570.e6 CrossRef CAS PubMed.
- B. R. Glaysher and N. A. Mabbott, Immunology, 2007, 120, 336–344 CrossRef CAS PubMed.
- J. A. Layhadi and M. H. Shamji, Allergy, 2021, 76, 1292–1293 CrossRef PubMed.
- A. Fleming, T. Castro-Dopico and M. R. Clatworthy, Scand. J. Immunol., 2022, 95, e13139 CrossRef CAS PubMed.
- A. Cerutti, Nat. Rev. Immunol., 2008, 8, 421–434 CrossRef CAS PubMed.
- L. Lutter, D. P. Hoytema Van Konijnenburg, E. C. Brand, B. Oldenburg and F. Van Wijk, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 637–649 CrossRef CAS PubMed.
- B. M. J. Owens and A. Simmons, Mucosal Immunol., 2013, 6, 224–234 CrossRef CAS PubMed.
- C. L. Boulangé, A. L. Neves, J. Chilloux, J. K. Nicholson and M.-E. Dumas, Genome Med., 2016, 8, 42 CrossRef PubMed.
- H. Luck, S. Khan, J. H. Kim, J. K. Copeland, X. S. Revelo, S. Tsai, M. Chakraborty, K. Cheng, Y. Tao Chan, M. K. Nøhr, X. Clemente-Casares, M.-C. Perry, M. Ghazarian, H. Lei, Y.-H. Lin, B. Coburn, A. Okrainec, T. Jackson, S. Poutanen, H. Gaisano, J. P. Allard, D. S. Guttman, M. E. Conner, S. Winer and D. A. Winer, Nat. Commun., 2019, 10, 3650 CrossRef PubMed.
- T. Zietek and E. Rath, Front. Immunol., 2016, 7, 154 Search PubMed.
- D. A. Winer, H. Luck, S. Tsai and S. Winer, Cell Metab., 2016, 23, 413–426 CrossRef CAS PubMed.
- D. A. Winer, S. Winer, H. J. Dranse and T. K. T. Lam, J. Clin. Invest., 2017, 127, 33–42 CrossRef PubMed.
- N. Zmora, S. Bashiardes, M. Levy and E. Elinav, Cell Metab., 2017, 25, 506–521 CrossRef CAS PubMed.
- S. Khan, H. Luck, S. Winer and D. A. Winer, Nat. Commun., 2021, 12, 2598 CrossRef CAS PubMed.
- M. Z. Vitolins and T. L. Case, Diabetes Spectr., 2020, 33, 113–117 CrossRef PubMed.
- A. Christ, M. Lauterbach and E. Latz, Immunity, 2019, 51, 794–811 CrossRef CAS PubMed.
- D. Statovci, M. Aguilera, J. MacSharry and S. Melgar, Front. Immunol., 2018, 8, 838 CrossRef PubMed.
- M. J. Butler, Brain, Behav., Immun.: Health, 2021, 16, 100298 CAS.
- D. J. Morrison and T. Preston, Gut Microbes, 2016, 7, 189–200 CrossRef PubMed.
- P. Gonçalves, J. R. Araújo and J. P. Di Santo, Inflammatory Bowel Dis., 2018, 24, 558–572 CrossRef PubMed.
- J. Tan, C. McKenzie, M. Potamitis, A. N. Thorburn, C. R. Mackay and L. Macia, The Role of Short-Chain Fatty Acids in Health and Disease, Elsevier Inc., 1st edn, 2014, vol. 121 Search PubMed.
- R. Nagpal, C. A. Shively, T. C. Register, S. Craft and H. Yadav, F1000Res., 2019, 8, 1–18 Search PubMed.
- M. Bellini, S. Tonarelli, A. G. Nagy, A. Pancetti, F. Costa, A. Ricchiuti, N. de Bortoli, M. Mosca, S. Marchi and A. Rossi, Nutrients, 2020, 12, 1–21 Search PubMed.
- K. H. Allin, V. Tremaroli, R. Caesar, B. A. H. Jensen, M. T. F. Damgaard, M. I. Bahl, T. R. Licht, T. H. Hansen, T. Nielsen, T. M. Dantoft, A. Linneberg, T. Jørgensen, H. Vestergaard, K. Kristiansen, P. W. Franks, T. Hansen, F. Bäckhed and O. Pedersen, Diabetologia, 2018, 61, 810–820 CrossRef PubMed.
- K. D. Corbin, E. A. Carnero, B. Dirks, D. Igudesman, F. Yi, A. Marcus, T. L. Davis, R. E. Pratley, B. E. Rittmann, R. Krajmalnik-Brown and S. R. Smith, Nat. Commun., 2023, 14, 3161 CrossRef CAS PubMed.
- M. S. Kim, S. S. Hwang, E. J. Park and J. W. Bae, Environ. Microbiol. Rep., 2013, 5, 765–775 CrossRef CAS PubMed.
- E. M. Cespedes and F. B. Hu, Am. J. Clin. Nutr., 2015, 101, 899–900 CrossRef CAS PubMed.
- K. D. Corbin, E. A. Carnero, B. Dirks, D. Igudesman, F. Yi, A. Marcus, T. L. Davis, R. E. Pratley, B. E. Rittmann, R. Krajmalnik-Brown and S. R. Smith, Nat. Commun., 2023, 14, 3161 CrossRef CAS PubMed.
- A. Benítez-Páez, A. L. Hess, S. Krautbauer, G. Liebisch, L. Christensen, M. F. Hjorth, T. M. Larsen and Y. Sanz, Mol. Nutr. Food Res., 2021, 65, 1–13 CrossRef PubMed.
- F. Hidalgo-Villeda, M. Million, C. Defoort, T. Vannier, L. Svilar, M. Lagier, C. Wagner, C. Arroyo-Portilla, L. Chasson, C. Luciani, V. Bossi, J. P. Gorvel, H. Lelouard and J. Tomas, iScience, 2023, 26(6) DOI:10.1016/j.isci.2023.106910.
- A. Prossomariti, E. Scaioli, G. Piazzi, C. Fazio, M. Bellanova, E. Biagi, M. Candela, P. Brigidi, C. Consolandi, T. Balbi, P. Chieco, A. Munarini, M. Pariali, M. Minguzzi, F. Bazzoli, A. Belluzzi and L. Ricciardiello, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- X. Zhang, M. Monnoye, M. Mariadassou, F. Beguet-Crespel, N. Lapaque, C. Heberden and V. Douard, Front. Immunol., 2021, 12 DOI:10.3389/fimmu.2021.742584.
- R. Kawano, T. Okamura, Y. Hashimoto, S. Majima, T. Senmaru, E. Ushigome, M. Asano, M. Yamazaki, H. Takakuwa, R. Sasano, N. Nakanishi, M. Hamaguchi and M. Fukui, Int. J. Mol. Sci., 2021, 22(11), 5558 CrossRef CAS PubMed.
- R. Mastrocola, D. Collotta, G. Gaudioso, M. Le Berre, A. S. Cento, G. A. Ferreira, F. Chiazza, R. Verta, I. Bertocchi, F. Manig, M. Hellwig, F. Fava, C. Cifani, M. Aragno, T. Henle, L. Joshi, K. Tuohy and M. Collino, Nutrients, 2020, 12, 1–20 CrossRef PubMed.
- C. Hill, F. Guarner, G. Reid, G. R. Gibson, D. J. Merenstein, B. Pot, L. Morelli, R. B. Canani, H. J. Flint, S. Salminen, P. C. Calder and M. E. Sanders, Nat. Rev. Gastroenterol. Hepatol., 2014, 11, 506–514 CrossRef PubMed.
- X. Tzounis, A. Rodriguez-Mateos, J. Vulevic, G. R. Gibson, C. Kwik-Uribe and J. P. E. Spencer, Am. J. Clin. Nutr., 2011, 93, 62–72 CrossRef CAS PubMed.
- S. Anand, L. Mohan and N. Bharadvaja, ECS Trans., 2022, 107, 13505–13514 CrossRef.
- C. Mazziotta, M. Tognon, F. Martini, E. Torreggiani and J. C. Rotondo, Probiotics Mechanism of Action on Immune Cells and Beneficial Effects on Human Health, Cells, 2023, 12(1), 184 CrossRef CAS PubMed.
- A. Agus, K. Clément and H. Sokol, Gut, 2021, 70, 1174–1182 CrossRef CAS PubMed.
- K. A. Krautkramer, J. Fan and F. Bäckhed, Nat. Rev. Microbiol., 2021, 19, 77–94 CrossRef CAS PubMed.
- D. J. Morrison and T. Preston, Gut Microbes, 2016, 7, 189–200 CrossRef PubMed.
- W. Von Engelhardt, K. Rönnau, G. Rechkemmer and T. Sakata, Anim. Feed Sci. Technol., 1989, 23, 43–53 CrossRef CAS.
- J. Folz, R. N. Culver, J. M. Morales, J. Grembi, G. Triadafilopoulos, D. A. Relman, K. C. Huang, D. Shalon and O. Fiehn, Nat. Metab., 2023, 5, 777–788 CrossRef CAS PubMed.
- K. A. Krautkramer, J. Fan and F. Bäckhed, Nat. Rev. Microbiol., 2021, 19, 77–94 CrossRef CAS PubMed.
- J. E. Lee, K. S. Kim, H. Koh, D. W. Lee and N. J. Kang, Curr. Dev. Nutr., 2022, 6, nzac110 CrossRef PubMed.
- M. N. Read and A. J. Holmes, Front. Immunol., 2017, 8, 1–9 Search PubMed.
- J. Tan, C. McKenzie, M. Potamitis, A. N. Thorburn, C. R. Mackay and L. Macia, The Role of Short-Chain Fatty Acids in Health and Disease, Elsevier Inc., 1st edn, 2014, vol. 121 Search PubMed.
- H. Nagao-Kitamoto, A. B. Shreiner, M. G. Gillilland, S. Kitamoto, C. Ishii, A. Hirayama, P. Kuffa, M. El-Zaatari, H. Grasberger, A. M. Seekatz, P. D. R. Higgins, V. B. Young, S. Fukuda, J. Y. Kao and N. Kamada, Cell. Mol. Gastroenterol. Hepatol., 2016, 2, 468–481 CrossRef PubMed.
- M. Kim, Y. Qie, J. Park and C. H. Kim, Cell Host Microbe, 2016, 20, 202–214 CrossRef CAS PubMed.
- A. Cherta-Murillo, J. E. Pugh, S. Alaraj-Alshehhi, D. Hajjar, E. S. Chambers and G. S. Frost, Am. J. Clin. Nutr., 2022, 116, 335–361 CrossRef CAS PubMed.
- T. Sakata, Anim. Sci. J., 2019, 90, 3–13 CrossRef PubMed.
- J. Zhang, Y.-J. Huang, J. Y. Yoon, J. Kemmitt, C. Wright, K. Schneider, P. Sphabmixay, V. Hernandez-Gordillo, S. J. Holcomb, B. Bhushan, G. Rohatgi, K. Benton, D. Carpenter, J. C. Kester, G. Eng, D. T. Breault, O. Yilmaz, M. Taketani, C. A. Voigt, R. L. Carrier, D. L. Trumper and L. G. Griffith, Med, 2021, 2, 74–98.e9 CrossRef CAS PubMed.
- M. Trapecar, C. Communal, J. Velazquez, C. A. Maass, Y.-J. Huang, K. Schneider, C. W. Wright, V. Butty, G. Eng, O. Yilmaz, D. Trumper and L. G. Griffith, Cell Syst., 2020, 10, 223–239.e9 CrossRef CAS PubMed.
- M. Trapecar, E. Wogram, D. Svoboda, C. Communal, A. Omer, T. Lungjangwa, P. Sphabmixay, J. Velazquez, K. Schneider, C. W. Wright, S. Mildrum, A. Hendricks, S. Levine, J. Muffat, M. J. Lee, D. A. Lauffenburger, D. Trumper, R. Jaenisch and L. G. Griffith, Sci. Adv., 2021, 7, eabd1707 CrossRef CAS PubMed.
- A. S. Medina-Larqué, M. C. Rodríguez-Daza, M. Roquim, S. Dudonné, G. Pilon, É. Levy, A. Marette, D. Roy, H. Jacques and Y. Desjardins, Front. Immunol., 2022, 13, 1–21 Search PubMed.
- Q. Xie, K. Mu, C. Chen, S. Gu, D. Luo, W. Fu and W. Xue, Int. J. Biol. Macromol., 2023, 230, 123234 CrossRef CAS PubMed.
- Z. Deng, Y. Rong, Y. Teng, J. Mu, X. Zhuang, M. Tseng, A. Samykutty, L. Zhang, J. Yan, D. Miller, J. Suttles and H. G. Zhang, Mol. Ther., 2017, 25, 1641–1654 CrossRef CAS PubMed.
- M. Anwar, S. Mros, M. McConnell and A. E.-D. A. Bekhit, Nutrients, 2018, 14(10), 2128 CrossRef PubMed.
- H. Kaur, T. Gupta, S. Kapila and R. Kapila, Food Funct., 2021, 12, 6102–6116 RSC.
- Y. Zhang, K. Yu, H. Chen, Y. Su and W. Zhu, Microb. Biotechnol., 2018, 11, 859–868 CrossRef CAS PubMed.
- J. J. A. J. Bastings, K. Venema, E. E. Blaak and T. C. Adam, Trends Endocrinol. Metab., 2023, 34, 243–255 CrossRef CAS PubMed.
- J. M. Yabut, J. D. Crane, A. E. Green, D. J. Keating, W. I. Khan and G. R. Steinberg, Endocr. Rev., 2019, 40, 1092–1107 CrossRef PubMed.
- N. Terry and K. G. Margolis, Handb. Exp. Pharmacol., 2017, 239, 319–342 CAS.
- B. Wang, S. Sun, M. Liu, H. Chen, N. Liu, Z. Wu, G. Wu and Z. Dai, J. Nutr., 2020, 150, 1966–1976 CrossRef PubMed.
- A. N. Anbazhagan, M. Thaqi, S. Priyamvada, D. Jayawardena, A. Kumar, T. Gujral, I. Chatterjee, E. Mugarza, S. Saksena, H. Onyuksel and P. K. Dudeja, Nanomedicine, 2017, 13, 659–665 CrossRef CAS PubMed.
- J. J. Meier, Nat. Rev. Endocrinol., 2012, 8, 728–742 CrossRef CAS PubMed.
- S. Sundaresan, C. Johnson, K. B. Dixon, M. Dole, D. Kilkelly, J. Antoun, C. R. Flynn, N. N. Abumrad and R. Tamboli, Am. J. Clin. Nutr., 2023, 118(3), 646–656 CrossRef PubMed.
- A. Shehzad, R. Rabail, S. Munir, H. Jan, D. Fernández-Lázaro and R. M. Aadil, Curr. Nutr. Rep., 2023, 12, 66–82 CrossRef CAS PubMed.
- E. Fabersani, M. C. Abeijon-Mukdsi, R. Ross, R. Medina, S. González and P. Gauffin-Cano, Front. Immunol., 2017, 8, 1–15 Search PubMed.
- I. Doytchinova, Molecules, 2022, 27, 1496 CrossRef CAS PubMed.
- S. Galmés, B. Reynés, M. Palou, A. Palou-March and A. Palou, J. Agric. Food Chem., 2021, 69, 5281–5296 CrossRef PubMed.
- J. Wu, M. Dong, C. Rigatto, Y. Liu and F. Lin, NPJ Digit. Med., 2018, 1, 7 CrossRef PubMed.
- M. Filippi, T. Buchner, O. Yasa, S. Weirich and R. K. Katzschmann, Adv. Mater., 2022, 34, 2108427 CrossRef CAS PubMed.
- L. Gijzen, D. Marescotti, E. Raineri, A. Nicolas, H. L. Lanz, D. Guerrera, R. Van Vught, J. Joore, P. Vulto, M. C. Peitsch, J. Hoeng, G. Lo Sasso and D. Kurek, SLAS Technol., 2020, 25, 585–597 CrossRef CAS PubMed.
- N. Gjorevski, B. Avignon, R. Gérard, L. Cabon, A. B. Roth, M. Bscheider and A. Moisan, Lab Chip, 2020, 20, 3365–3374 RSC.
- M. Morelli, D. Kurek, C. P. Ng and K. Queiroz, Biomedicines, 2023, 11, 619 CrossRef CAS PubMed.
- D. Marrero, F. Pujol-Vila, D. Vera, G. Gabriel, X. Illa, A. Elizalde-Torrent, M. Alvarez and R. Villa, Biosens. Bioelectron., 2021, 181, 113156 CrossRef CAS PubMed.
- A. Valiei, J. Aminian-Dehkordi and M. R. K. Mofrad, APL Bioeng., 2023, 7, 011502 CrossRef CAS PubMed.
- Y. Guo, R. Luo, Y. Wang, P. Deng, T. Song, M. Zhang, P. Wang, X. Zhang, K. Cui, T. Tao, Z. Li, W. Chen, Y. Zheng and J. Qin, Sci. Bull., 2021, 66, 783–793 CrossRef CAS PubMed.
- M. J. Workman, J. P. Gleeson, E. J. Troisi, H. Q. Estrada, S. J. Kerns, C. D. Hinojosa, G. A. Hamilton, S. R. Targan, C. N. Svendsen and R. J. Barrett, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 669–677.e2 CrossRef PubMed.
- C. Beaurivage, E. Naumovska, Y. Chang, E. Elstak, A. Nicolas, H. Wouters, G. Van Moolenbroek, H. Lanz, S. Trietsch, J. Joore, P. Vulto, R. Janssen, K. Erdmann, J. Stallen and D. Kurek, Int. J. Mol. Sci., 2019, 20, 5661 CrossRef CAS PubMed.
- A. Apostolou, R. A. Panchakshari, A. Banerjee, D. V. Manatakis, M. D. Paraskevopoulou, R. Luc, G. Abu-Ali, A. Dimitriou, C. Lucchesi, G. Kulkarni, T. I. Maulana, M. Kasendra, J. S. Kerns, B. Bleck, L. Ewart, E. S. Manolakos, G. A. Hamilton, C. Giallourakis and K. Karalis, Cell. Mol. Gastroenterol. Hepatol., 2021, 12, 1719–1741 CrossRef CAS PubMed.
- M. S. Jeon, Y. Y. Choi, S. J. Mo, J. H. Ha, Y. S. Lee, H. U. Lee, S. D. Park, J.-J. Shim, J.-L. Lee and B. G. Chung, Nano Convergence, 2022, 9, 8 CrossRef CAS PubMed.
- S. Min, N. Than, Y. C. Shin, G. Hu, W. Shin, Y. M. Ambrosini and H. J. Kim, Sci. Rep., 2022, 12, 22641 CrossRef CAS PubMed.
- M. Maurer, M. S. Gresnigt, A. Last, T. Wollny, F. Berlinghof, R. Pospich, Z. Cseresnyes, A. Medyukhina, K. Graf, M. Gröger, M. Raasch, F. Siwczak, S. Nietzsche, I. D. Jacobsen, M. T. Figge, B. Hube, O. Huber and A. S. Mosig, Biomaterials, 2019, 220, 119396 CrossRef CAS PubMed.
- W. L. K. Chen, C. Edington, E. Suter, J. Yu, J. J. Velazquez, J. G. Velazquez, M. Shockley, E. M. Large, R. Venkataramanan, D. J. Hughes, C. L. Stokes, D. L. Trumper, R. L. Carrier, M. Cirit, L. G. Griffith and D. A. Lauffenburger, Biotechnol. Bioeng., 2017, 114, 2648–2659 CrossRef CAS PubMed.
- C. D. Edington, W. L. K. Chen, E. Geishecker, T. Kassis, L. R. Soenksen, B. M. Bhushan, D. Freake, J. Kirschner, C. Maass, N. Tsamandouras, J. Valdez, C. D. Cook, T. Parent, S. Snyder, J. Yu, E. Suter, M. Shockley, J. Velazquez, J. J. Velazquez, L. Stockdale, J. P. Papps, I. Lee, N. Vann, M. Gamboa, M. E. LaBarge, Z. Zhong, X. Wang, L. A. Boyer, D. A. Lauffenburger, R. L. Carrier, C. Communal, S. R. Tannenbaum, C. L. Stokes, D. J. Hughes, G. Rohatgi, D. L. Trumper, M. Cirit and L. G. Griffith, Sci. Rep., 2018, 8, 4530 CrossRef PubMed.
- Q. Ramadan, H. Jafarpoorchekab, C. Huang, P. Silacci, S. Carrara, G. Koklü, J. Ghaye, J. Ramsden, C. Ruffert, G. Vergeres and M. A. M. Gijs, Lab Chip, 2013, 13, 196–203 RSC.
- Q. Ramadan and L. Jing, Biomed. Microdevices, 2016, 18, 11 CrossRef PubMed.
- P. Shah, J. V. Fritz, E. Glaab, M. S. Desai, K. Greenhalgh, A. Frachet, M. Niegowska, M. Estes, C. Jäger, C. Seguin-Devaux, F. Zenhausern and P. Wilmes, Nat. Commun., 2016, 7, 11535 CrossRef CAS PubMed.
- W. Zhao, Y. Yao, T. Zhang, H. Lu, X. Zhang, L. Zhao, X. Chen, J. Zhu, G. Sui and W. Zhao, Front. Bioeng. Biotechnol., 2022, 10, 1035647 CrossRef PubMed.
- A. E. Cherwin, H. N. Templeton, A. T. Ehrlich, B. H. Patlin, C. S. Henry and S. A. Tobet, Lab Chip, 2023, 23, 4126–4133 RSC.
- H. J. Kim and D. E. Ingber, Integr. Biol., 2013, 5, 1130 CrossRef CAS PubMed.
- U. M. N. Cao, Y. Zhang, J. Chen, D. Sayson, S. Pillai and S. D. Tran, Int. J. Mol. Sci., 2023, 24, 3232 CrossRef CAS PubMed.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- A. Richardson, L. A. Schwerdtfeger, D. Eaton, I. Mclean, C. S. Henry and S. A. Tobet, Anal. Methods, 2020, 12, 297–303 RSC.
- M. Baydoun, A. Treizeibré, J. Follet, S. B. Vanneste, C. Creusy, L. Dercourt, B. Delaire, A. Mouray, E. Viscogliosi, G. Certad and V. Senez, Micromachines, 2020, 11(2), 150 CrossRef PubMed.
- H. Eslami Amirabadi, J. M. Donkers, E. Wierenga, B. Ingenhut, L. Pieters, L. Stevens, T. Donkers, J. Westerhout, R. Masereeuw, I. Bobeldijk-Pastorova, I. Nooijen and E. Van De Steeg, Lab Chip, 2022, 22, 326–342 RSC.
- Mimetas, OrganoPlate® 3-lane 40, https://www.mimetas.com/files/products/OrganoPlate%203-lane%2040/<?pdb_no 2023?>2023<?pdb END?>_Productflyer_3_lane_40_Specification%20sheet.pdf, (accessed August 2, 2023) Search PubMed.
- Mimetas, https://www.mimetas.com/en/home/ Search PubMed.
- T. J. Long, P. A. Cosgrove, R. T. Dunn, D. B. Stolz, H. Hamadeh, C. Afshari, H. McBride and L. G. Griffith, Drug Metab. Dispos., 2016, 44, 1940–1948 CrossRef CAS PubMed.
- P. Vulto, S. Podszun, P. Meyer, C. Hermann, A. Manz and G. A. Urban, Lab Chip, 2011, 11, 1596 RSC.
- P. Vulto, G. Medoro, L. Altomare, G. A. Urban, M. Tartagni, R. Guerrieri and N. Manaresi, J. Micromech. Microeng., 2006, 16, 1847–1853 CrossRef.
- P. Vulto, G. Dame, U. Maier, S. Makohliso, S. Podszun, P. Zahn and G. A. Urban, Lab Chip, 2010, 10, 610–616 RSC.
- S. J. Trietsch, E. Naumovska, D. Kurek, M. C. Setyawati, M. K. Vormann, K. J. Wilschut, H. L. Lanz, A. Nicolas, C. P. Ng, J. Joore, S. Kustermann, A. Roth, T. Hankemeier, A. Moisan and P. Vulto, Nat. Commun., 2017, 8, 262 CrossRef PubMed.
- N. Tsamandouras, W. L. K. Chen, C. D. Edington, C. L. Stokes, L. G. Griffith and M. Cirit, AAPS J., 2017, 19, 1499–1512 CrossRef CAS PubMed.
- V. De Gregorio, G. Imparato, F. Urciuolo, M. L. Tornesello, C. Annunziata, F. M. Buonaguro and P. A. Netti, Adv. Healthcare Mater., 2017, 6, 1601199 CrossRef PubMed.
- K. H. Benam, S. Dauth, B. Hassell, A. Herland, A. Jain, K.-J. Jang, K. Karalis, H. J. Kim, L. MacQueen, R. Mahmoodian, S. Musah, Y. Torisawa, A. D. Van Der Meer, R. Villenave, M. Yadid, K. K. Parker and D. E. Ingber, Annu. Rev. Pathol.: Mech. Dis., 2015, 10, 195–262 CrossRef CAS PubMed.
- Z. Zhao, X. Chen, A. M. Dowbaj, A. Sljukic, K. Bratlie, L. Lin, E. L. S. Fong, G. M. Balachander, Z. Chen, A. Soragni, M. Huch, Y. A. Zeng, Q. Wang and H. Yu, Nat. Rev. Methods Primer, 2022, 2, 94 CrossRef CAS PubMed.
- I. Russo, P. Zeppa, P. Iovino, C. Del Giorno, F. Zingone, C. Bucci, A. Puzziello and C. Ciacci, J. Immunol. Methods, 2016, 438, 1–10 CrossRef CAS PubMed.
- B. Martinez, L. A. Schwerdtfeger, A. Richardson, S. A. Tobet and C. S. Henry, Anal. Chem., 2022, 94, 9987–9992 CrossRef CAS PubMed.
- J. McCright, A. Sinha and K. Maisel, Cell. Mol. Bioeng., 2022, 15, 479–491 CrossRef CAS PubMed.
- M. Kasendra, A. Tovaglieri, A. Sontheimer-Phelps, S. Jalili-Firoozinezhad, A. Bein, A. Chalkiadaki, W. Scholl, C. Zhang, H. Rickner, C. A. Richmond, H. Li, D. T. Breault and D. E. Ingber, Sci. Rep., 2018, 8, 2871 CrossRef PubMed.
- A. Sontheimer-Phelps, D. B. Chou, A. Tovaglieri, T. C. Ferrante, T. Duckworth, C. Fadel, V. Frismantas, A. D. Sutherland, S. Jalili-Firoozinezhad, M. Kasendra, E. Stas, J. C. Weaver, C. A. Richmond, O. Levy, R. Prantil-Baun, D. T. Breault and D. E. Ingber, Cell. Mol. Gastroenterol. Hepatol., 2020, 9, 507–526 CrossRef PubMed.
- C. P. Miller, W. Shin, E. H. Ahn, H. J. Kim and D.-H. Kim, Trends Biotechnol., 2020, 38, 857–872 CrossRef CAS PubMed.
- A. Shanti, N. Hallfors, G. A. Petroianu, L. Planelles and C. Stefanini, Front. Pharmacol., 2021, 12, 711307 CrossRef CAS PubMed.
- S. S. Hinman, A. Massaro, Y. Wang, C. E. Sims, R. Kim and N. L. Allbritton, Adv. Biol., 2022, 6, 2200129 CrossRef CAS PubMed.
- C. Moysidou, A. M. Withers, A. J. Nisbet, D. R. G. Price, C. E. Bryant, C. Cantacessi and R. M. Owens, Adv. Biol., 2022, 2200015 CrossRef PubMed.
- G. Noel, N. W. Baetz, J. F. Staab, M. Donowitz, O. Kovbasnjuk, M. F. Pasetti and N. C. Zachos, Sci. Rep., 2017, 7, 45270 CrossRef CAS PubMed.
- R. T. Abraham and A. Weiss, Nat. Rev. Immunol., 2004, 4, 301–308 CrossRef CAS PubMed.
- B. Taciak, M. Białasek, A. Braniewska, Z. Sas, P. Sawicka, Ł. Kiraga, T. Rygiel and M. Król, PLoS One, 2018, 13, e0198943 CrossRef PubMed.
- M. Kubacz, A. Kusowska, M. Winiarska and M. Bobrowicz, Cancers, 2022, 15, 235 CrossRef PubMed.
- M. Ackermann, H. Kempf, M. Hetzel, C. Hesse, A. R. Hashtchin, K. Brinkert, J. W. Schott, K. Haake, M. P. Kühnel, S. Glage, C. Figueiredo, D. Jonigk, K. Sewald, A. Schambach, S. Wronski, T. Moritz, U. Martin, R. Zweigerdt, A. Munder and N. Lachmann, Nat. Commun., 2018, 9, 5088 CrossRef PubMed.
- J. Zeng, S. Y. Tang, L. L. Toh and S. Wang, Stem Cell Rep., 2017, 9, 1796–1812 CrossRef CAS PubMed.
- S. Suwanpitak, N. Promnakhon, R. Netsrithong and M. Wattanapanitch, in Induced Pluripotent Stem (iPS) Cells, ed. Nagy A. and Turksen K., Springer US, New York, NY, 2022, vol. 2454 Search PubMed.
- S. Iriguchi, Y. Yasui, Y. Kawai, S. Arima, M. Kunitomo, T. Sato, T. Ueda, A. Minagawa, Y. Mishima, N. Yanagawa, Y. Baba, Y. Miyake, K. Nakayama, M. Takiguchi, T. Shinohara, T. Nakatsura, M. Yasukawa, Y. Kassai, A. Hayashi and S. Kaneko, Nat. Commun., 2021, 12, 430 CrossRef CAS PubMed.
- M. Morsink, N. Willemen, J. Leijten, R. Bansal and S. Shin, Micromachines, 2020, 11, 849 CrossRef PubMed.
- L. Van Os, B. Engelhardt and O. T. Guenat, Front. Bioeng. Biotechnol., 2023, 11, 1191104 CrossRef PubMed.
- P. Moura Rosa, N. Gopalakrishnan, H. Ibrahim, M. Haug and Ø. Halaas, Lab Chip, 2016, 16, 3728–3740 RSC.
- G. Goyal, P. Prabhala, G. Mahajan, B. Bausk, T. Gilboa, L. Xie, Y. Zhai, R. Lazarovits, A. Mansour, M. S. Kim, A. Patil, D. Curran, J. M. Long, S. Sharma, A. Junaid, L. Cohen, T. C. Ferrante, O. Levy, R. Prantil-Baun, D. R. Walt and D. E. Ingber, Adv. Sci., 2022, 9, 2103241 CrossRef PubMed.
- L. M. Delong and A. E. Ross, Lab Chip, 2023, 23, 3034–3049 RSC.
- A. Mercado-Perez and A. Beyder, Nat. Rev. Gastroenterol. Hepatol., 2022, 19, 283–296 CrossRef PubMed.
- L. C. Delon, Z. Guo, A. Oszmiana, C.-C. Chien, R. Gibson, C. Prestidge and B. Thierry, Biomaterials, 2019, 225, 119521 CrossRef CAS PubMed.
- H. C. Lin, JAMA, 2004, 292, 852–858 CrossRef CAS PubMed.
- J. Bures, World J. Gastroenterol., 2010, 16, 2978 CrossRef CAS PubMed.
- R. Singhal and Y. M. Shah, J. Biol. Chem., 2020, 295, 10493–10505 CrossRef CAS PubMed.
- L. Zheng, C. J. Kelly and S. P. Colgan, Am. J. Physiol.-Cell Physiol., 2015, 309, C350–C360 CrossRef CAS PubMed.
- F. J. Gonzalez, C. Xie and C. Jiang, Nat. Rev. Endocrinol., 2019, 15, 21–32 CrossRef CAS PubMed.
- S. Jalili-Firoozinezhad, F. S. Gazzaniga, E. L. Calamari, D. M. Camacho, C. W. Fadel, A. Bein, B. Swenor, B. Nestor, M. J. Cronce, A. Tovaglieri, O. Levy, K. E. Gregory, D. T. Breault, J. M. S. Cabral, D. L. Kasper, R. Novak and D. E. Ingber, Nat. Biomed. Eng., 2019, 3, 520–531 CrossRef CAS PubMed.
- J. W. Hickey, Y. Dong, J. W. Chung, S. F. Salathe, H. C. Pruitt, X. Li, C. Chang, A. K. Fraser, C. A. Bessell, A. J. Ewald, S. Gerecht, H. Mao and J. P. Schneck, Adv. Mater., 2019, 31, 1807359 CrossRef PubMed.
- M. Saitakis, S. Dogniaux, C. Goudot, N. Bufi, S. Asnacios, M. Maurin, C. Randriamampita, A. Asnacios and C. Hivroz, eLife, 2017, 6, e23190 CrossRef PubMed.
- R. Sridharan, B. Cavanagh, A. R. Cameron, D. J. Kelly and F. J. O'Brien, Acta Biomater., 2019, 89, 47–59 CrossRef CAS PubMed.
- M. Friedemann, L. Kalbitzer, S. Franz, S. Moeller, M. Schnabelrauch, J.-C. Simon, T. Pompe and K. Franke, Adv. Healthcare Mater., 2017, 6, 1600967 CrossRef PubMed.
- A. K. Blakney, M. D. Swartzlander and S. J. Bryant, J. Biomed. Mater. Res., Part A, 2012, 100A, 1375–1386 CrossRef CAS PubMed.
- Y. Zeng, J. Yi, Z. Wan, K. Liu, P. Song, A. Chau, F. Wang, Z. Chang, W. Han, W. Zheng, Y.-H. Chen, C. Xiong and W. Liu, Eur. J. Immunol., 2015, 45, 1621–1634 CrossRef CAS PubMed.
- N. Yissachar, Y. Zhou, L. Ung, N. Y. Lai, J. F. Mohan, A. Ehrlicher, D. A. Weitz, D. L. Kasper, I. M. Chiu, D. Mathis and C. Benoist, Cell, 2017, 168, 1135–1148.e12 CrossRef CAS PubMed.
- M. Jang and H. N. Kim, BioChip J., 2023, 17, 133–146 CrossRef CAS.
- Y. Guo, X. Chen, P. Gong, G. Li, W. Yao and W. Yang, Int. J. Mol. Sci., 2023, 24, 4089 CrossRef CAS PubMed.
- Y. Wan and T. Zuo, Gastroenterol. Rep., 2022, 10, goac009 CrossRef PubMed.
- C. Villemin, A. Six, B. A. Neville, T. D. Lawley, M. J. Robinson and G. Bakdash, Trends Immunol., 2023, 44, 44–59 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |