
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SlipO2Chip – single-cell respiration under tuneable environments†
Yuan
Cui
 a,
Milena De Albuquerque
Moreira
b,
Kristen E.
Whalen
c,
Laurent
Barbe
d,
Qian
Shi
d,
Klaus
Koren
e,
Maria
Tenje
d and
Lars
Behrendt
*a
a,
Milena De Albuquerque
Moreira
b,
Kristen E.
Whalen
c,
Laurent
Barbe
d,
Qian
Shi
d,
Klaus
Koren
e,
Maria
Tenje
d and
Lars
Behrendt
*a
aDepartment of Organismal Biology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden. E-mail: lars.behrendt@scilifelab.uu.se
bMyfab Uppsala, Uppsala University, Sweden
cBiology Department, Haverford College, USA
dDepartment of Materials Science and Engineering, Science for Life Laboratory, Uppsala University, Sweden
eDepartment of Biology, Aarhus University, Aarhus, Denmark
First published on 18th September 2024
Abstract
In disciplines like toxicology and pharmacology, oxygen (O2) respiration is a universal metric for evaluating the effects of chemicals across various model systems, including mammalian and microalgal cells. However, for these cells the common practice is to segregate populations into control and exposure groups, which assumes direct equivalence in their responses and does not take into account heterogeneity among individual cells. This lack of resolution impedes our ability to precisely investigate differences among experimental groups with small or limited sample sizes. To overcome this barrier, we introduce SlipO2Chip, an innovative glass microfluidic platform for precisely quantifying single-cell O2 respiration in the coordinated absence and presence of chemical solutes. SlipO2Chip comprises a wet-etched fused silica channel plate on the top and a dry-etched borosilicate microwell plate at the bottom. The microwells are coated with Pt(II) meso-tetra(pentafluorophenyl)porphine (PtTFPP), an O2 sensing optode material and an O2-independent reference dye. A custom 3D-printed holder facilitates the controlled horizontal movement (‘slipping’) of the channel plate over the microwell plate, thereby establishing or disrupting the fluid path over microwells. Collectively, these design elements enable the immobilization of single-cells in microwells, their exposure to controlled fluid flows, the coordinated opening and closing of microwells and repeated measurements of single-cell O2 respiration. Uniquely, by sequentially executing opening and closing it becomes possible to measure single-cell respiration prior to and after exposure to chemical solutes. In a proof-of-concept application, we utilized SlipO2Chip to measure the impact of increasing exposures of the marine bacterial signal 2-heptyl-4-quinolone (HHQ) on the dark respiration of the diatom Ditylum brightwellii at single-cell resolution. Results revealed a concentration-dependent decrease in per-cell O2 dark respiration, with a maximum reduction of 40.2% observed at HHQ concentrations exceeding 35.5 μM, and a half-maximal effective concentration (EC50) of 5.8 μM, consistent with that obtained via conventional bulk respiration methods. The ability of SlipO2Chip to sequentially assess the effects of chemical substances on single-cell O2 metabolism is advantageous for research where sample volumes are limited, such as clinical biopsies, studies involving rare microbial isolates, and toxicological studies aiming to address exposure effects while accounting for cell-to-cell variability.
Introduction
Exposure to chemicals can introduce cellular toxicity,1 a phenomenon investigated extensively in toxicology through various metabolic proxies such as oxygen (O2) respiration, changes in gene expression, or alterations in growth across biological entities. Traditional toxicological testing often involves segregating populations of organism into ‘controls’ and ‘exposed’ groups, and statistically comparing their responses to incremental chemical concentrations to obtain concentration–response relationships. This presumes uniform behavior across populations and potentially disregards the variability in responses among individuals, a problem particularly notable in unicellular organisms. While conventional approaches thus offer cost-efficiency and broad acceptance, they inherently obscure heterogeneous responses to chemical perturbations. The implication of this is particularly acute in medical microbiology, where, for instance, subsets of bacterial cells known as ‘persisters’ exhibit distinct resilience to antibiotic regimes,2 and individual cancer stem cells may drive the relapse of disease progression within predominantly healthy cellular environments.3 Similarly, in microbial ecology, chemically resistant cells may aid populations in tolerating and adapting to chemical perturbations,4,5 and microalgae can exhibit varying susceptibilities to infochemicals released by bacteria, with significant implications for their interaction with viruses and their role in marine ecosystems.6,7 These studies emphasize the significance of understanding the impact of cellular heterogeneity in the context of chemical perturbations, ideally on a per-cell basis.Microfluidic techniques provide the ability to precisely manipulate low fluid volumes (10−9 to 10−18 L)8 and have been used to expose tissues and individual cells to chemical solutes and other environmental stimuli.9,10 Microfluidic devices are often constructed from materials like silicone-based polymer, borosilicate and fused silica glass, or plastic and offer unparalleled control over microenvironments while minimizing sample volumes. In the realm of toxicology, microfluidics has been used to, for instance, measure toxic and inflammatory responses of lung tissues to silica nanoparticles,11 or to monitor the dynamics of cytotoxicity within effector cell populations against target cells.12 Microfluidic devices are also increasingly coupled to optical and electrical readouts for continuous measurements of single-cell isotope uptake,13 drugs in saliva,14 and glyphosate in drinking water.15 Significantly, the integration of optical sensors – ‘optodes’ – into microfluidics has emerged as a powerful technique to non-invasively measure O2 dynamics in and around biological systems. Optodes utilize the O2-dependent luminescence quenching16,17 and are characterized by their broad biocompatibility18 as well as resistance to photobleaching.19 When entrapped in a polymer matrix, optode materials can be coated onto optical fibers,20,21 particles18,22,23 and planar surfaces.24–26 This versatility has allowed researchers to integrate optodes and other optical sensors into microfluidic devices for monitoring the spatial distribution of O2 in liver organ model27 and human cell cultures28,29 and to determine the heterogeneity in O2 consumption among single-cells.30–33 Compared to other O2 sensing approaches often integrated into microfluidics for single-cell level O2 measurement, such as microelectrodes,34 optodes do not consume O2.35,36 This characteristic is particularly crucial for micro-volume measurements. Additionally, optodes offer several advantages, including easy preparation, no need for wire connections or specialized electrochemical equipment, and other benefits inherent to optical measurement techniques. Despite these advances, to our knowledge, there is currently no microfluidic approach capable of measuring single-cell respiration on a per-cell basis before and after exposure to chemical perturbations. This capability is essential for determining the effect of single-cell heterogeneity towards chemical treatments in both clinical and toxicological settings.
In this study, we introduce SlipO2Chip, a microfluidic device capable of measuring single-cell respiration rates before and after coordinated exposure to chemicals. SlipO2Chip draws inspiration from the original SlipChip technology,37 a glass microfluidic device developed to mix fluids within microwell arrays through coordinated slipping motions. In addition to the original design, SlipO2Chip integrates O2-sensitive optode materials and O2-insensitive reference dye into microwells to ratiometrically monitor O2 respiration among single-cells.38 Moreover, by orchestrating the opening and closing of microwells, single-cell respiration can be measured in the absence and presence of chemicals. In this study, we used SlipO2Chip to examine the effect of the bacterial signal 2-heptyl-4-quinolone (HHQ) on the respiration rates of individual diatom cells.39 We chose diatoms as a model system because diatoms are primarily unicellular and relatively fast growing, making them ideal candidates for testing the capabilities of SlipO2Chip. Additionally, diatoms are among the main primary producers in the ocean40,41 and their productivity and bloom formation are influenced, at least in part, by the chemical exchange occurring at the interface surrounding individual cells.42 This interface, known as the ‘phycosphere’, is the environment where bacteria release infochemical signals,43 including HHQ for the purpose of quorum sensing44,45 as well as secondary metabolites that directly affect the growth, metabolic activity, and bloom formation of microalgae.39,42,46–48 Secondary metabolites and infochemicals thus hold the potential to affect the health and productivity of diatoms as well as other phytoplankton, thereby mediating the primary productivity of aquatic ecosystems. Despite this crucial link, our understanding of their effects on individual diatom cells remains limited.
By combining microfabrication, O2 sensing and ecotoxicology, we present initial proof-of-concept data on the impact of HHQ on the respiration rates of individual diatom cells. While this application focuses on cells relevant to microbial ecology and ecosystems, we highlight the broader versatility of SlipO2Chip for exploring O2 metabolism across various cell types, extending its relevance to medical and toxicological investigations. We propose that the capacity of SlipO2Chip to analyze minute sample volumes could facilitate the detailed examination of individual cells from heterogeneous populations from rare patient samples, offering a unique opportunity to assess the cellular effects of specific drug treatments in a controlled manner. Thus, our innovative approach not only enhances our understanding of ecologically relevant cellular responses to chemical change but also supports clinically relevant investigations on targeted drug treatments and toxicity assessments.
Experimental
Cell culture
Cultures of the diatom Ditylum brightwellii were purchased from the Bigelow culture collection (accession number CCMP 3370). The size of Ditylum brightwellii, ranged between 50–120 μm in length and 10–40 μm in width, as determined by a standard brightfield microscope. Cells were cultivated in L1 media and grown at a temperature of 22 °C while being subjected to a 14/10 h light/dark cycle. L1 media was prepared by dissolving 35 g of sea salt (Instant Ocean®, US) in 1 L of deionized (DI) water and by adding 1 mL of 75 g L−1 sodium nitrate (NaNO3) solution, 5 g L−1 sodium dihydrogen phosphate monohydrate (NaH2PO4·H2O) solution, 30 g L−1 sodium metasilicate nonahydrate (Na2SiO3·9H2O) solution, trace element stock solution, respectively, and 0.5 mL of vitamin stock solution. The components of the vitamin and trace element stock solution are provided in Tables S1 and S2.† All stock solutions were supplied in the L1 media kit (NCMA at Bigelow Laboratory, US). To ensure cultures were in mid-exponential growth for experiments, diatom cells were inoculated into fresh L1 media at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 (volume/volume) dilution five days prior to experiments.
:6 (volume/volume) dilution five days prior to experiments.
Device components and operation
SlipO2Chip is comprised of three main components: a bottom microwell plate decorated with O2-sensitive optodes for detecting respiration, a top channel plate for introducing fluids and a holder to aid in the assembly and slipping of the channel plate against the microwell plate. Details on parts of the 3D printed holder and assembly of SlipO2Chip are provided in Fig. 1A, B and S1.† Before assembling the microwell plate, PDMS (0.25 mm thickness, SuperClear silicone sheet, BISCO) and PET films (0.05 mm thickness, Silflu 1R88001, Siliconature S.P.A., US) were precisely cut to match the shape and size of the channel plate using a desktop vinyl cutter (CAMM-1 GS-24, Roland DGA Co., US). The elastic PDMS film was first affixed to the channel plate to enhance liquid-tightness. Subsequently, the gas-impermeable PET film was layered on top of the PDMS to ensure gas-tightness and facilitate smooth sliding. This combined PDMS–PET layering not only promotes seamless operation but also effectively seals each microwell, minimizing cross-talk between them, as demonstrated in the sealing test (refer to Fig. 1C, S2† and the dedicated section below for more details). The microwell plate was secured in place by sandwiching it between two dedicated holder parts labeled ‘holder top’ and ‘holder bottom’ (Fig. S1†). Following the secure attachment of the microwell plate into the holder, the channel plate + PDMS–PET film was carefully placed onto the microwell plate and affixed using six pairs of neodymium magnets, two pairs positioned in the middle and four pairs positioned at the corners of the top channel plate (block magnet 10 × 3 × 2 mm and 20 × 4 × 3 mm, Supermagnete DE). The precise slipping movements, essential for opening and closing microwells at the microscale, were facilitated by a 3D-printed holder and two commercial micrometer screws (high precision micrometer head, Thorlabs Inc., US), each offering a resolution of ±1 μm.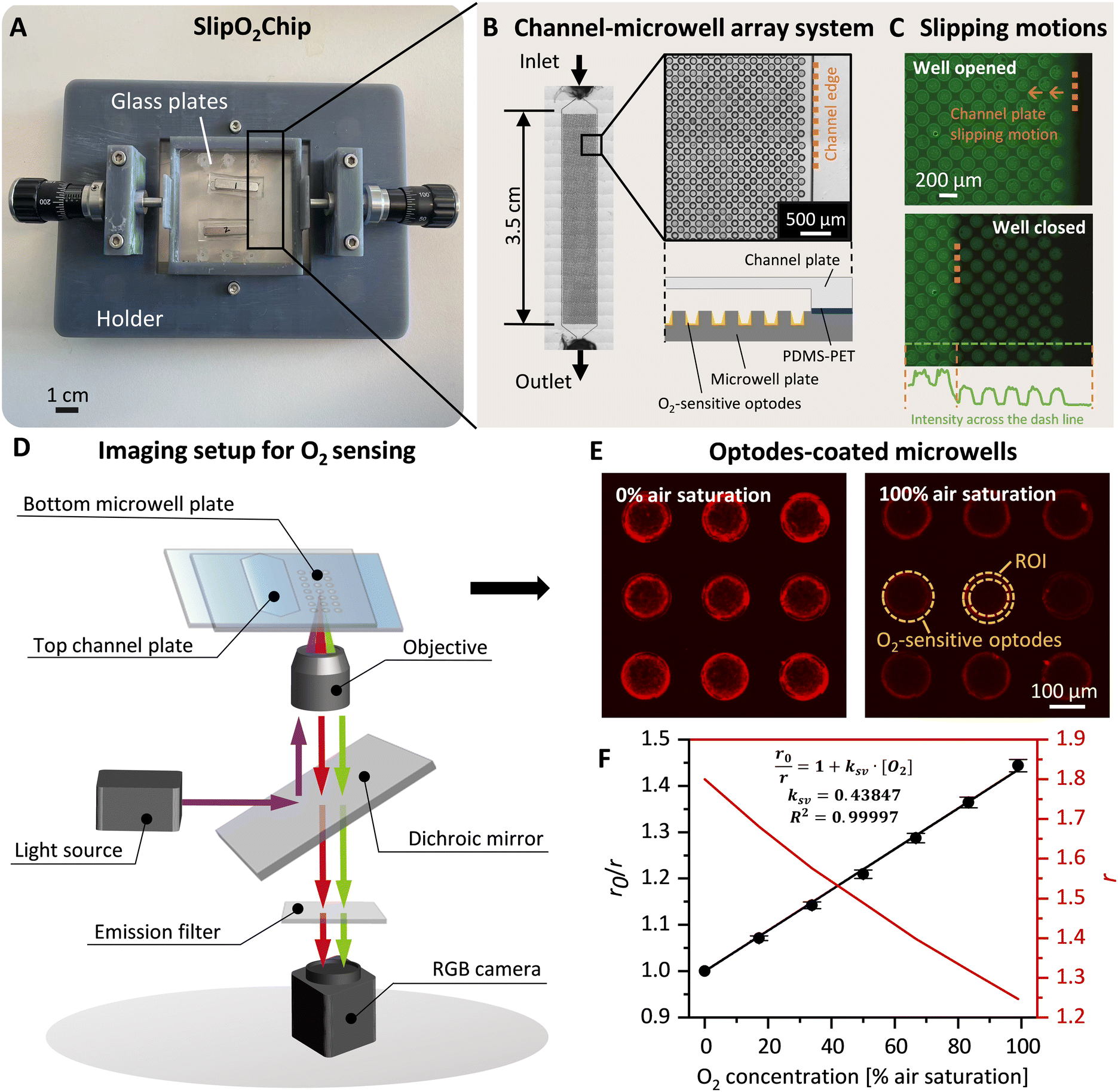 | ||
| Fig. 1 SlipO2Chip, a glass microfluidic device for repeated single-cell O2 respiration measurements under tuneable chemical microenvironments. (A) Image of the fully assembled SlipO2Chip. SlipO2Chip consists of two glass plates – a channel plate on the top and a microwell plate at the bottom, affixed together with neodymium magnets and secured within a 3D printed holder to facilitate controlled slipping of the top channel plate over the bottom microwell plate. An explosion diagram of the assembly of the frame and SlipO2Chip components is available in Fig. S1.† (B) Close-up of the bottom microwell array within SlipO2Chip and the fluid access resulting from assembling the top channel plate and the microwell plate. Each channel has dedicated inlet and outlet ports accessible through the bottom microwell plate. (C) Fluorescein was introduced into the fully assembled SlipO2Chip to visualize the controlled slipping motion used to close microwells and test the efficacy of the liquid seal during manipulations. Note the retained fluorescein solution in the individual microwells upon closure. The fluorescein intensity across the microwell array is provided and sharp peak edges and zero intensity in areas outside the closed microwells indicate the liquid-tightness of the sealed microwells (D) a schematic depiction of the microscopy imaging setup used to conduct ratiometric O2 measurements. UV light (395/25 nm) is provided via a SpectraX light source which excites the O2-sensitive optode material Pt(II) meso-tetra(pentafluorophenyl)porphine (PtTFPP) as well as a co-embedded reference dye. The resulting luminescence emission of PtTFPP (red) and reference dye (green) is captured by an RGB camera. During image-processing the RGB image is split into red and green channels in order to obtain ratiometric measurements of O2 concentrations. (E) Fluorescence image of deposited PtTFPP within microwells. Red rings signify the deposition of PtTFPP and reference dye primarily at the edges of the microwell. Here shown is the red luminescence intensity of the optode material at 0% and 100% air saturation, respectively. The area where the optode material was coated and the region of interest (ROI) which was segmented for subsequent O2 concentration measurements are both labeled. (F) Calibration of red/green ratiometric images across seven O2 concentrations and its description via the Stern–Volmer relationship (see eqn (1)). Shown are the Stern–Volmer constant (kSV), and the goodness of linear fit (R2), used to derive O2 concentrations from RGB images. | ||
Glass etching
The bottom microwell plate was built from a borosilicate glass wafer (borosilicate 4 inch, thickness = 1000 ± 30 μm, MicroChemicals GmbH, DE). The wafer was first subjected to plasma cleaning and subsequently 7 μm of aluminium (Al) was deposited via a sputtering process (CS 730 S, Von Ardenne GmbH, DE) to serve as a future etch mask. After sputtering, the wafer was oven-dried at 200 °C for 5 min to eliminate residual moisture and enhance the adhesion of the photoresist. To fabricate the metal mask, a 5 μm layer of photoresist AZ-10XT (AZ® 10XT, Merck KGaA, DE) was spin-coated onto the Al layer. The photoresist was soft baked at 110 °C for 2 min and subjected to four cycles of UV light (365 nm) for 30 s each, utilizing a mask aligner (MA6/BA6, Süss MicroTec GmbH, DE). Following UV exposure, the photoresist layer was developed (AZ 400K developer, Merck KGaA, DE) for 4 min and subsequently hard baked at 120 °C for 7 min. The Al layer was then etched using inductively coupled plasma – reactive ion etching (ICP–RIE) (SLR, PlasmaTherm LLC, US), yielding an Al mask comprising the desired microwell patterns. The AZ-10XT photoresist was removed using acetone (Acetone, VWR chemicals, US) and isopropanol (IPA) (2-propanol, VWR chemicals, US). The glass wafer, now patterned with the Al mask, was then processed in a reactive ion etching (RIE) machine (110 S/DE, Tegal Co., US) to etch the microwells to a depth of 50 μm. Following this process, the Al mask was removed using an Al etchant at 40 °C, prepared from phosphoric acid (H3PO4 85%, Sunchem AB, SE), acetic acid (CH3COOH 99%, Sunchem AB, SE) and nitric acid (HNO3 69% VLSI selectipur, BASF, DE) in a volumetric ratio of 29:5:1. To finalize the process, the glass wafer was immersed in buffered oxide etch (BHF, 7-1, VWR, US) for 30 min to smooth the dry-etched surface, followed by thorough rinsing in deionized water. The entire dry etching protocol is depicted in Fig. S3.†
The top channel plate was fabricated from a fused silica glass wafer (Fused silica JGS2 wafer 4 inch, thickness = 350 ± 25 μm, MicroChemicals GmbH, DE), via a wet etching process. Initial preparation steps included plasma cleaning of the glass wafer, followed by the deposition of a 1 μm thick molybdenum (Mo) layer using a sputter (CS 730 S, Von Ardenne GmbH, DE) to act as an etch mask. After this deposition, the wafer underwent a hydrophobic treatment within a vapor prime furnace (Star 2000, Imtec Inc., US), followed by the application of a 1 μm thick layer of photoresist (Microposit™ S1813™ G2 photoresist, Dow Inc., US) via spin-coating. The wafer was then soft baked at 110 °C for 2 min and subjected to UV exposure for 6 s in the mask aligner. The exposed photoresist was developed for 45 s (Microposit™ 351 developer, Dow Inc., US) and hard baked at 120 °C for 5 min. The channel pattern was then transferred onto the Mo layer through etching in a prepared Mo etchant, comprising water, HNO3, and hydrochloric acid (HCl 36%, BASF, DE) mixed in a volumetric ratio of 30:57:13, for 3 min. To protect the backside of the wafer from this etching process, this side of the wafer was taped with acid-resistant tape (SPV224R PVC Surface Protection tape, Nitto Denko Co., JP). The channels were then etched into the glass substrate using 50% hydrofluoric acid (HF 50%, VWR, US) at a etch rate of 1.5 μm min−1 for 37 min. After etching, the photoresist was removed through consecutive acetone and IPA baths, followed by a Mo etchant bath to eliminate the metal mask. The entire wet etching protocol is available in Fig. S3.†
After the etching process, wafer samples were sectioned using a dicing saw (DAD 361, Disco Co., JP). Furthermore, 2 mm diameter access holes were introduced on the bottom microwell plate by a laser cutter (AIO G+ 532 nm 5 W, Östling Märksystem AB, SE). The dimension and surface topography of etched structures were evaluated using a profilometer (Dektak 150 Stylus profiler, Bruker Nano Inc., US). Surface roughness was quantified in terms of the arithmetical mean roughness value (Ra), defined as the average of deviations from the mean line across the assessment length.
Optodes decoration and calibration
To functionalize the dry-etched microwells, the O2-sensitive optodes were deposited at the bottom of each microwell utilizing a film applicator tool. This procedure started with the preparation of an optode + reference dye solution with the following proportions: 1.5 mg PtTFPP (Frontier Scientific Inc., US), 1.5 mg MY yellow dye (Macrolex Fluorescent Yellow 10GN, Lanxess GmbH, DE), 1 g of polystyrene (Sigma-Aldrich Inc., US), and 2 g of silicone rubber49 (ELASTOSIL® E4, Wacker Chemie AG, DE) in 10 mL toluene (Sigma-Aldrich Inc., US). Subsequently, 80 μL of this mixture was pipetted onto the microwell plate and spread evenly across via a 50 μm applicator (4-sided bar 60, 50–200 μm, BYK-Gardner Gmbh, DE, Fig. S4†). By evaporating the toluene solvent, this resulted in the formation of an optode sensor film that adhered to the entire microwell plate. Excess sensor film outside the microwells was carefully removed using a scalpel. This ensured that only microwells were decorated with optode chemistries. Notably, due to edge effects the majority of the optode material was immobilized onto the sides of microwells. This ‘ring’ of optode chemistry was 3 ± 1 μm thick. The optode material in the central part of the microwell was measured to be 1 ± 1 μm thick. The thickness was characterized by a profilometer (Dektak 150 Stylus profiler, Bruker Nano Inc., US).Calibration entailed exciting optode-coated microwells with UV light (395/25 nm) for a duration of 50 ms across various O2 concentrations. Subsequently, the resulting optode-luminescence was captured using an RGB camera (DFK 33UX264 USB 3.0 color industrial camera, Image Source, DE). This setup, was integrated into a fully automated inverted Nikon Ti2-E microscope equipped with a perfect focus system (Nikon Inc., JPN) and a UV light source (SpectraX Light Engine, Lumencor Inc., US).
To process the obtained RGB images, the pixel-wise ratiometric signals (r) of the red emission (attributable to the O2-sensitive optodes) and the green emission (emanating from the O2-insensitive reference MY dye) at different O2 concentrations were extracted via Fiji Image Processing50 and subsequently analyzed through the application of the Stern–Volmer relationship:
 | (1) |
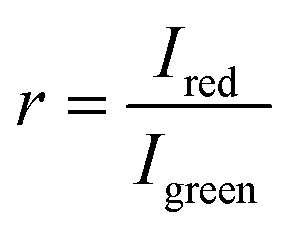 , signifies the ratio of luminescence intensities between the red and green channels in the presence of O2, being a function of O2 concentration [O2] (% air saturation); r0 denotes ratio between red and green luminescence intensities in the absence of O2; kSV represents the Stern–Volmer constant. The Stern–Volmer constants derived from each calibration set were used to transform the experimentally obtained luminescence signals into O2 concentrations.
, signifies the ratio of luminescence intensities between the red and green channels in the presence of O2, being a function of O2 concentration [O2] (% air saturation); r0 denotes ratio between red and green luminescence intensities in the absence of O2; kSV represents the Stern–Volmer constant. The Stern–Volmer constants derived from each calibration set were used to transform the experimentally obtained luminescence signals into O2 concentrations.
By conducting a 7-point calibration experiment in the gas phase (Fig. S5†), we determined that the performance of O2 optodes obeys a linear fit (Fig. 1E). Therefore, we performed simpler 2-point calibrations in the liquid phase for most calibrations.
Validation of liquid-tightness and gas-tightness
To validate the liquid tightness of the SlipO2Chip, fluorescein was introduced using a low-pressure pump system (OB1 MK3+ low-pressure pump, Elvesys Co, France) at 30 mbar pressure. This was carried out within the fully assembled device, with microwells left open. The pressure was lowered to 10 mbar stepwise over the course of 1 min and the top channel plate was subsequently slipped horizontally to close microwells. The ability of SlipO2Chip to effectively isolate liquid flow was confirmed by observing minimal presence of fluorescein solution outside the microwells. All fluorescein imaging was conducted using an RGB camera under blue light excitation (440/20 nm) and FITC specific emission (525 nm).To validate the gas-tightness of SlipO2Chip, the bottom microwell plate was positioned inside a customized chamber designed for precise control over gas composition. The plate was partitioned such that half was covered by a PDMS film (0.25 mm thickness) and the other half by a PDMS–PET film (0.25 + 0.05 mm thickness), with the PET side facing the microwells. Initially, the microwell plate was exposed to compressed air (CA) for a duration of 10 min. Subsequently, 100% nitrogen gas (N2) was introduced to the chamber. During this stepwise gas exposure, O2 concentrations within the microwells were monitored every 2 min using the automated microscopy equipped with an RGB camera, documenting the changes over a 60 min period to assess the effectiveness of the films in maintaining gas integrity.
Repeated single-cell O2 respiration experiments
Experiments began with cell loading, followed by iterative opening and closing of the SlipO2Chip to measure respiration, with and without chemical solutes. For cell loading, a 100 μL aliquot of 1:2 diluted mid-exponentially growing culture was manually pipetted into SlipO2Chip while microwells were open. Single-cells were isolated and captured in individual microwells. This step was repeated until the occupancy across the microwell array exceeded 20%.
To facilitate the introduction of L1 media into SlipO2Chip, the device inlet was connected to the low-pressure pump system. First, a pressure of 30 mbar was set to remove cells outside of microwells and subsequently the pressure was manually reduced over 1 min to a final pressure of 10 mbar. This procedure helped retain cells within microwells while effectively washing away excess cells outside of the microwells. Once a stable fluid flow was established, the system underwent a 10 min equilibration period. Subsequently, the channel plate was slipped horizontally to close microwells. To quantify the O2 respiration of cells isolated within microwells, all external light sources were deactivated. Subsequently, the microwells were exposed to a 50 ms long pulse of UV light (395/25 nm, power 35%) every 2 min over a duration of 16 min. The O2-dependent luminescence from the optode material was recorded using an RGB camera as described before. In each experiment, we utilized the ‘large image scanning mode’ within NIS Elements (AR 5.10.00, Nikon Inc., JPN) to image a total of 275 microwells. This approach generated RGB time-lapse sequences that captured changes in O2 signals among individual microwells. The O2 concentration within each microwell was calculated from the obtained ratiometric values r via the Stern–Volmer equation, utilizing the kSV constant obtained from the prior calibration step. Finally, single-cell O2 respiration rates were calculated using a linear fit to the O2 consumption over the entire 16 min observation period.
After the first O2 respiration measurement, microwells were re-opened by slipping the channel plate and by introducing fully oxygenated L1 media into the channel. Cells in microwells were left for 10 min under continuous fluid flow (10 mbar pump pressure). Following this incubation, microwells were closed again to conduct a second O2 respiration measurement on the exact same cells. This step was repeated for a third time. In combination, this experimental protocol assessed the effect of three repeated slipping motions on single-cell O2 respiration rates.
Single-cell O2 respiration under HHQ exposure
To explore the impact of the bacterial secondary metabolite HHQ on the O2 respiration of diatoms, microwells were filled with individual cells following previously described protocols. Following cell loading, the microwells containing cells were sealed and baseline respiration rates were obtained. These baseline respiration rates were obtained under standard L1 media supplemented with DMSO as a carrier control. Following these baseline measurements, the microwells were opened and the media in the channel replaced with L1 media augmented with a specific concentration of HHQ (0, 5, 10, 20 or 100 μM) and left to incubate for 10 min before closing microwells again. This process was iterated in five separate experiments to acquire per-cell respiration rates first under L1 media + carrier control, and subsequently under L1 media + different concentrations of HHQ. Switching between L1 media + DMSO and L1 media + HHQ was achieved through a manual three-way switching valve located upstream of the channel inlet. Experimental data, both before and after exposure to the different HHQ concentrations, was used in a concentration–response model and used to determine EC50 values via the following relationships: | (2) |
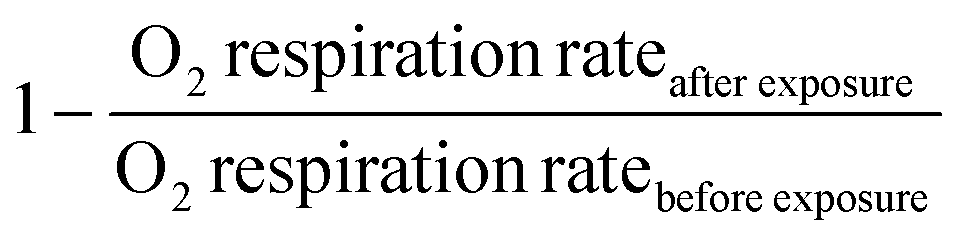 . Emin represents the minimal reduction in O2 respiration after HHQ exposure, while Emax represents the maximal reduction in O2 respiration after HHQ exposure. The term [HHQ] refers to the concentration of administered HHQ. EC50 is identified as the half-maximal effective concentration of HHQ concentration producing 50% of the maximal effect. The p value is the hill slope and quantifies the steepness of the concentration–response curve.
. Emin represents the minimal reduction in O2 respiration after HHQ exposure, while Emax represents the maximal reduction in O2 respiration after HHQ exposure. The term [HHQ] refers to the concentration of administered HHQ. EC50 is identified as the half-maximal effective concentration of HHQ concentration producing 50% of the maximal effect. The p value is the hill slope and quantifies the steepness of the concentration–response curve.
Bulk O2 respiration measurements
To benchmark the precision and accuracy of single-cell respiration measurements under HHQ exposures, the O2 respiration of the same diatom species was measured in commercial O2 sensing vials (OXVIAL4, PyroScience GmbH, DE). For each experiment, this entailed preparing a pair of 15 mL Falcon tubes, each filled with 5 mL of cell suspension at a density of 85–350 cells per mL. To one of the tubes, HHQ stock solution was added to achieve final concentrations of 0, 2.5, 5, 10, or 50 μM, while an equivalent volume of DMSO was added to the other tube as a carrier control. Both tubes were gently shaken for 10 min, and their contents were carefully transferred into two O2 sensing vials, ensuring no air was trapped in the headspace. A small stirring bar was added to each vial and the vials were placed on a magnetic stirrer for constant mixing during the measurement. The O2 concentration within each vial was monitored using a fiber-optic cable connected to a commercial 4-channel O2 sensing system (FireSting, Pyroscience GmbH, DE). This facilitated measurements of O2 consumption for the entire diatom population. Subsequent to the experiments, the cell numbers in each vial were manually counted using a Neubauer counting chamber and the resulting numbers used to calculate the per-cell respiration rates.Results and discussion
SlipO2Chip allows for the precise quantification of O2 respiration rates in single-cells under the coordinated absence and presence of user-controlled chemical solutes. SlipO2Chip integrates a movable channel plate on the top and a fixed microwell plate at the bottom. Embedded within the channel plate are three wet-etched fluidic channels, each 3.5 cm in length, 8 mm in width, and 70 μm in depth (Fig. 1A and B). These channels are strategically positioned to overlay a microwell array consisting of 25 × 155 microwells (total n = 3875) when moved horizontally into the ‘open’ position. The microwells have a diameter of 140 μm, a depth of 50 μm, and a pitch (center-to-center spacing) of 240 μm. The bottom of microwells have a roughness (Ra) of 200–1000 nm, and when coated with O2-sensitive optode chemistries, enable for measuring single-cell respiration rates when fully closed by horizontally moving the top plate (Fig. 2A). By utilizing strong neodymium magnets and a PDMS–PET film, SlipO2Chip reliably isolates individual microwells from both fluid flow (Fig. 1C) and atmospheric O2 (Fig. S2†). Fig. 3 illustrates the stepwise operation of SlipO2Chip, showcasing how the horizontal motion of the channel plate enables the opening and closing of microwells and how this facilitates cell loading, the introduction of chemical solutes, and measurements of single-cell respiration. Cell loading typically resulted in 20–60% of microwells being occupied by cells, with 80–90% of filled microwells containing single-cells, 10–20% containing doublets, while triplet occupation was rarely observed.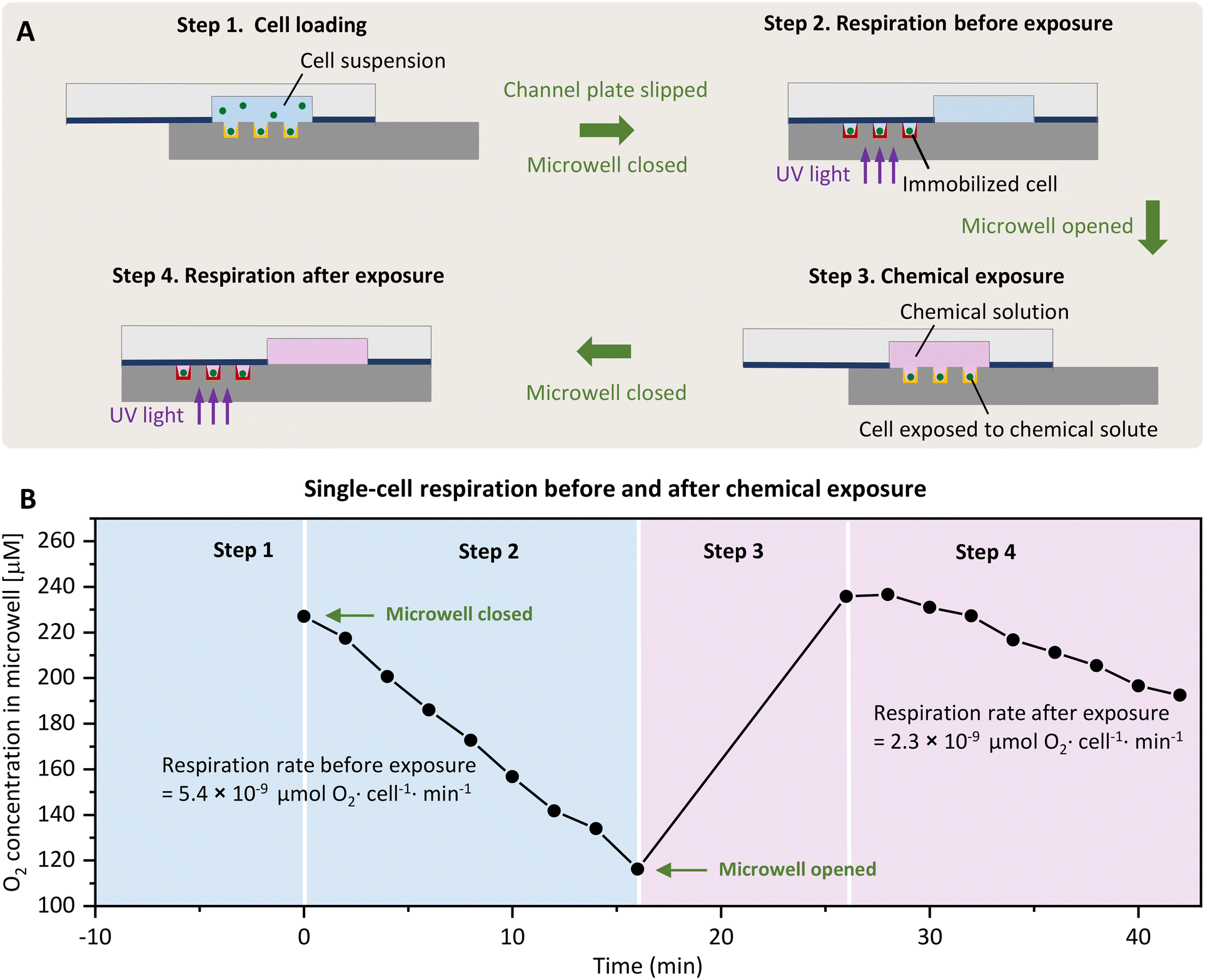 | ||
| Fig. 2 Schematic illustrating the coordinated opening and closing of SlipO2Chip for conducting single-cell O2 respiration measurements before and after chemical exposure. (A) Cross section of SlipO2Chip with the top channel plate and the bottom microwell plate. Please note the thin PDMS–PET film (in blue) and the deposited PtTFPP and reference dye (in yellow at the bottom of microwells). In step 1, to load cells, a diluted suspension of cells is introduced into the main channel, where gravity leads to their deposition into microwells. Cells located outside of microwells are removed by connecting the inlet to a low-pressure pump and flushing them out with media. In step 2, cells are isolated within closed microwells by horizontally sliding the top channel plate, facilitating the measurement of single-cell O2 respiration through deposited optode materials. In step 3, the top channel plate is slipped in the opposite direction to open microwells, facilitating the subsequent introduction of liquids such as fully oxygenated media or chemical solutions. The selection of these liquids is managed by a Y-shaped valve positioned outside of SlipO2Chip. In step 4, closing microwells for a second time allows for a repeated respiration measurement of the same cell, with the option of conducting this measurement under exposure to previously introduced chemicals, see step 3. (B) Exemplary data on O2 respiration of a single diatom cell before and after exposure to the chemical 2-heptyl-4-quinolone (HHQ) as outlined by steps 1–4. O2 respiration measurements were taken every 2 min over a span of 16 min. Rates of single-cell O2 respiration before and after introduction of HHQ were determined through linear fits of O2 consumption curves. | ||
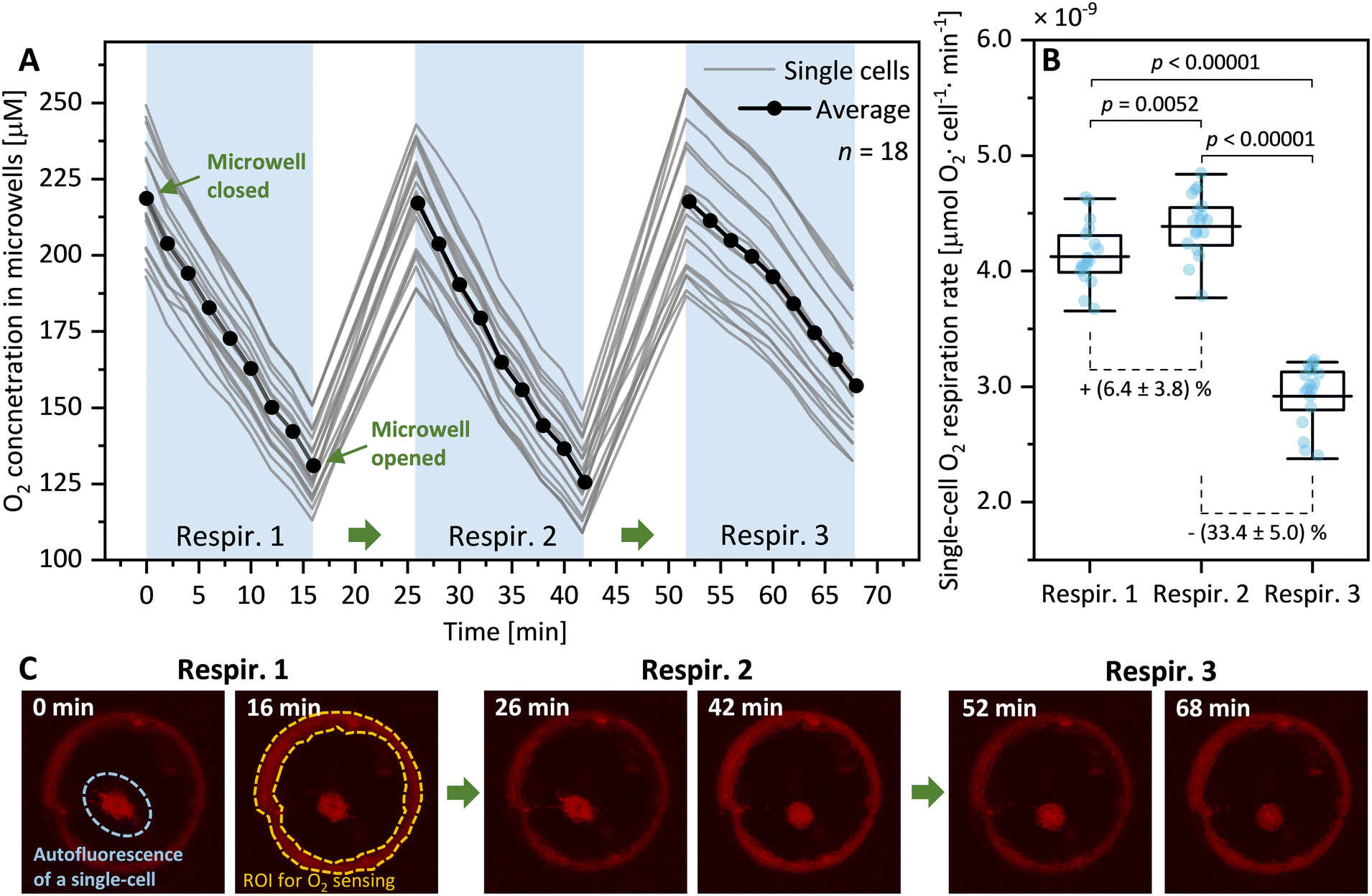 | ||
| Fig. 3 Opening and closing SlipO2Chip twice has negligible effects on diatom respiration. (A) Time resolved O2 concentrations measured in 18 microwells containing a single-cell of the diatom Ditylum brightwellii. Curves depict dark O2 consumption for individual cells upon repeated opening and closing of microwells. Repeated respiration measurements are labeled as ‘Respir. 1’, ‘Respir. 2’ and ‘Respir. 3’, respectively. Following each respiration measurements, cells were supplied with fully oxygenated media. (B) Box and whisker plot of the pairwise comparison of single-cell O2 respiration rates obtained from three sequential opening and closing cycles. Boxes depict the mean and whiskers quartiles, and bars showing 10% outliers of the O2 respiration rate data. Respiration rates were calculated from data presented in (A) and statistically analyzed in a pairwise fashion using a Mann–Whitney U test. The resulting p-values between groups are indicated above the box plot and average percent changes compared to neighboring groups are indicated below box plots. (C) Microscopy images of a representative microwell featuring a solitary diatom cell with embedded optodes and reference dye (highlighted by a red ring). The images depict the red luminescence intensity at the beginning and end of each opening and closing cycle, providing a visual representation of the changes in O2 concentration resulting from single-cell respiration within the microwells. The natural autofluorescence of the immobilized diatom cell is visible. This signal was not included in image processing as it lies outside the region of interest (ROI) used. | ||
Opening and closing SlipO2Chip has negligible effects on single-cell respiration
To assess whether the process of mechanically opening and closing microwells would cause stress to cells within microwells we conducted a simple test. This involved subjecting individual diatom cells (Ditylum brightwellii, n = 18) to three consecutive opening and closing cycles while measuring their respiration rate three times (Fig. 3A). This test revealed that the second opening and closing cycle resulted in a significant increase in respiration rates (by 6.4 ± 3.7%, p = 0.0052, Mann–Whitney test) compared to the first opening and closing cycle. Further, the third opening and closing cycle resulted in a significant reduction of respiration rates (by 33.4 ± 5.0%, p < 0.00001) compared to the second cycle. The results of this straightforward experiment suggest that opening and closing microwells twice has minimal impact on O2 respiration rates in single diatom cells. However, conducting a third cycle leads to notably significant changes in respiration rates. This emphasizes the importance of experimentally evaluating the effect of repeated slipping motions before conducting qualitative measurements of O2 respiration in other cell types. We did not investigate the specific nature of this effect, but we speculate that it could stem from mechanical stress arising from repeated slipping motions, potentially damaging the fragile silica shell of diatoms, or a combination of factors such as nutrient depletion or waste accumulation in the microwells. For this study, we have found that two consecutive opening and closing cycles are not only sufficient but also minimally impact diatom cells, effectively measuring the effects of chemical exposure both before and after treatment. Consequently, we have standardized this dual-cycle protocol for all subsequent experiments to ensure consistent and reliable data collection.HHQ affects single-cell diatom respiration in a concentration-dependent manner
To evaluate the efficacy of SlipO2Chip in orchestrating the exposure of single-cells to chemical solutes and measuring their impact on individual cell respiration, we designed an experiment using the bacterial infochemical HHQ. This molecule is a recognized quorum sensing signal produced by bacteria and plays a crucial role in their interactions with host phytoplankton. To assess the impact of HHQ on diatom respiration, we first exposed individual Ditylum brightwellii cells within SlipO2Chip to L1 media + carrier control. These baseline measurements revealed respiration rates ranging between 3.2 × 10−9 and 8.1 × 10−9 μmol O2 per cell per min (Fig. 4A, ‘before exposure’). While the range of these rates is comparable to respiration measurements conducted in bulk, both by us and by others on other diatom species (Table 1), they also underscore the significant cell-to-cell variability that is often overlooked in traditional bulk measurements.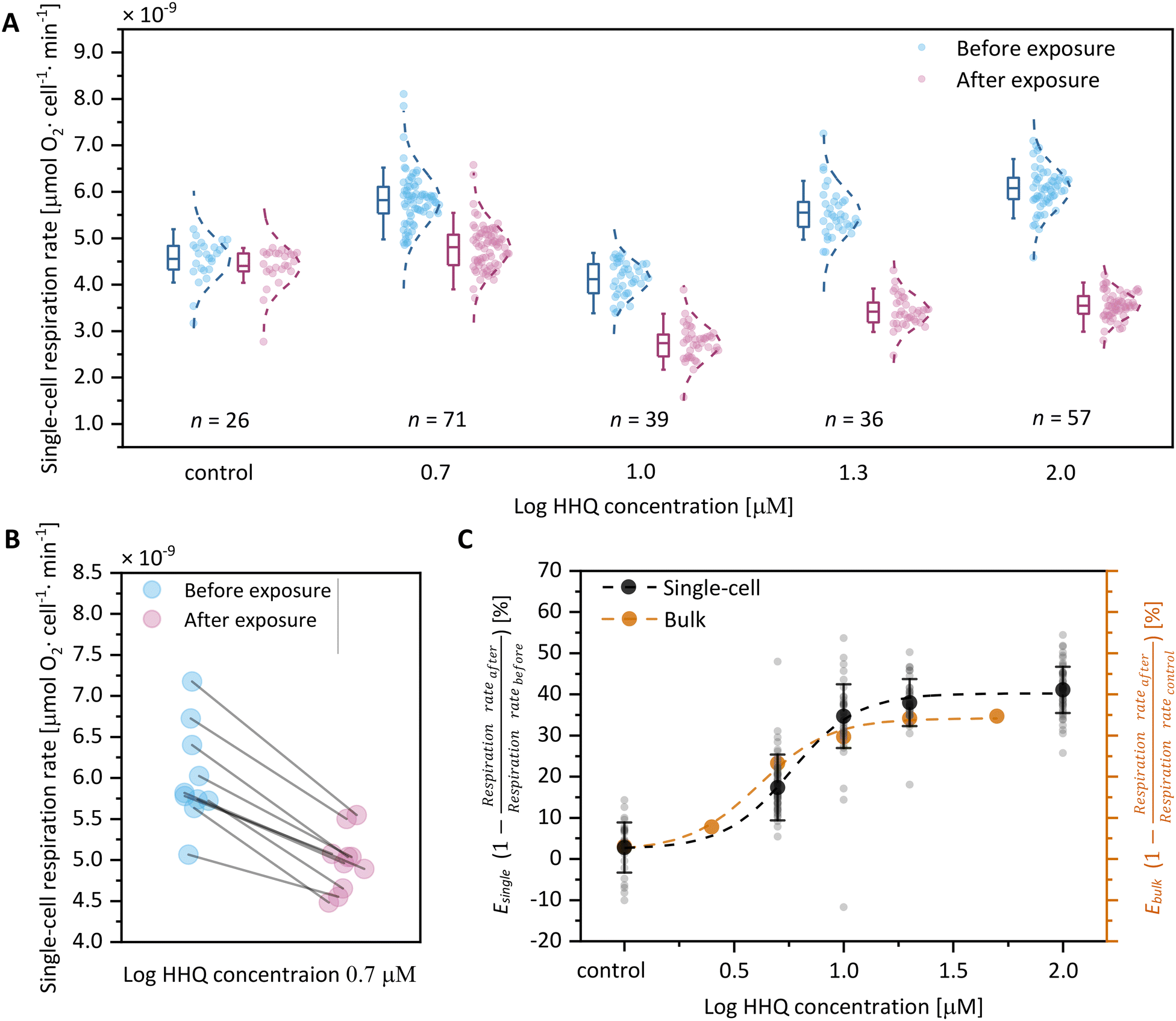 | ||
| Fig. 4 Increasing concentrations of HHQ reduce the O2 respiration of single diatom cells. (A) Assessment of the single-cell O2 respiration of the diatom Ditylum brightwellii before and after exposure to HHQ (0, 5, 10, 20, 100 μM) as measured by SlipO2Chip. In each HHQ exposure group, 26 to 71 single-cells were measured (exact numbers displayed below). The ‘after exposure’ measurement in the control group was included to account for any potential changes in baseline respiration rates caused by the slipping operation. Boxes depict the mean and whiskers quartiles, and bars showing 10% outliers of the O2 respiration rate data. Adjacent to each box, the normal distribution of the data is shown. (B) The O2 respiration rates of 10 diatom single-cells before and after HHQ exposure. These cells were randomly selected from the 5 μM exposure group. (C) Concentration–response relationship between the reduction of respiration rates (E) versus HHQ concentration. The reduction in respiration among investigated single cells was quantified by comparing respiration rates before and after exposure to HHQ. Here shown is this change for both single cells (individual black dots) and bulk measurements (brown dots). For single cells, the mean is shown as solid black dots and standard deviation is represented by error bars. Bulk measurements were only conducted once, thus no standard deviation is provided. | ||
| Diatom species | Equivalent spherical diameter (ESD) [μm] | Temperature [°C] | Methods | Respiration rate [μmol O2˙ per cell per min] |
|---|---|---|---|---|
| Ditylum brightwellii (this paper) | 20–100 | 20 | Optode-coated microwells (SlipO2Chip) | 3.2 × 10−9–8.1 × 10−9 |
| Ditylum brightwellii (this paper) | 20–100 | 20 | Fibre-optic O2 sensor (PyroScience) | 1.3 × 10−9–6.6 × 10−9 |
| Ditylum brightwellii 51 | 44.9 | 18 | Clark-type electrode (Triox EO200; RZ90 stirrer) | 4.5 × 10−6 |
| Thalassiosira punctigera 52 | 29.6 | 20 | Clark-type O2 electrode (Oxygraph+, Hansatech) | 1.8 × 10−8 |
| Thalassiosira weissflogii 52 | 15.1 | 20 | Clark-type O2 electrode (Oxygraph+, Hansatech) | 1.7 × 10−9 |
| Thalassiosira pseudonana 52 | 7.2 | 20 | Clark-type O2 electrode (Oxygraph+, Hansatech) | −3.3 × 10−8 |
| Phaeodactylum tricornutum 53 | 5.1 | 18 | Membrane inlet mass spectrometry (MIMS) | 1.8 × 10−10 |
After these baseline measurements, we introduced L1 media augmented with one of five concentrations of HHQ (0, 5, 10, 20 or 100 μM) into the main channel of SlipO2Chip and exposed cells for 10 min. Following this exposure period, microwells were closed and the respiration rates of each cell were measured again. We note that for each concentration of HHQ this involved a different set of immobilized diatom cells, since we can only execute two consecutive opening/closing cycles to ensure reliable results. Although different cells were used, the results of this experiment revealed a consistent decrease in respiration rates with increasing HHQ concentrations (Fig. 4A). Using single-cell measurements on baseline respiration (L1 media + carrier control) and post-exposure respiration (L1 + HHQ at varying concentrations), we calculated E values, representing the ratio of the reduction of respiration rate after chemical exposure and before exposure. E thus provides a straightforward measure of the inhibitory effect of HHQ on respiration of single diatoms by comparing it to its individual baseline respiration. Through this comparison, we observed a reduction in respiration rates of 2.7 ± 6.1% (0 μM HHQ, n = 26), 17.3 ± 8.0% (5 μM HHQ, n = 39), 34.7 ± 7.7% (10 μM HHQ, n = 71), 38.0 ± 5.7% (20 μM HHQ, n = 36), and 41.1 ± 5.6% (100 μM HHQ, n = 57) following exposure to HHQ. In order to further explore the effect of HHQ on diatom respiration, we fitted a classical concentration–response model on per-cell values of E (Fig. 4C, Table 2). This revealed a maximal reduction of single-cell respiration (Emax, single-cell) by 40.2 ± 1.4% compared to cells not exposed to HHQ at a concentration of 35.5 μM. Using the same concentration–response relationship, we also determined the half-maximal effective concentration (EC50, single-cell) for single-cells to be 5.8 ± 0.5 μM. These EC50 values, obtained from separate populations of individual cells before and after HHQ exposure, closely align with previously reported EC50 values for HHQ in diatom species such as C. closterium, P. tricornutum, and A. minutissimum, ranging from 1.4 μM to 4.9 μM.39
| Concentration-response model | E min | E max | EC 50 [μM] | p | R 2 |
|---|---|---|---|---|---|
| Single-cell | 2.4 ± 1.9% | 40.2 ± 1.4% | 5.8 ± 0.5 | 2.9 ± 0.8 | 0.99 |
| Bulk | 1.9 ± 2.2% | 34.2 ± 1.3% | 4.1 ± 0.4 | 2.7 ± 0.6 | 0.98 |
To further validate the accuracy of our single-cell concentration–response relationship, we conducted additional measurements of Ditylum brightwellii under increasing HHQ concentrations using standard commercial respiration vials (see Experimental section). This revealed that, in bulk, respiration is maximally reduced by approximately 34.2 ± 1.3% (Emax, bulk) at an HHQ concentration of 28.2 μM, compared to the control. Additionally, we determined the half-maximal effective concentration in bulk to be 4.1 ± 0.4 μM, which closely aligns with the earlier EC50 values of 5.8 ± 0.5 μM obtained for single-cells. Collectively, these findings demonstrate that HHQ has a stronger inhibitory effect on diatom respiration in single-cell measurements, with an Emax, single-cell of 40.2 ± 1.4%, compared to bulk measurements, which show an Emax, bulk of 34.2 ± 1.3%. This discrepancy may stem from undetected cell-to-cell variabilities in bulk measurements, potentially leading to the emergence of chemically tolerant subpopulations that are only discernible through single-cell analyses. While we did not validate this assertion, our findings underscore the significance of single-cell techniques, like SlipO2Chip, in evaluating these effects with a level of resolution typically absent in (eco)toxicological investigations. Additionally, single-cell measurements conducted also revealed a higher response efficiency (psingle-cell = 2.9 ± 0.8 vs. pbulk = 2.7 ± 0.6), indicating a greater effectiveness of HHQ at the single-cell level. We speculate that this effect could stem from the absence of cell-to-cell interactions and competition for resources within microwells, characterized by uniform exposure to surrounding media and limited chemical interchange between neighboring microwells. This stands in contrast to bulk environments, where cells encounter each other stochastically through mixing and collectively deplete nutrient levels. While bulk measurements thus offer rapid insights into collective effects of chemical exposure, single-cell analyses provide greater precision in measuring individual cell responses to chemical challenges.
Conclusions
Here, we introduce SlipO2Chip, a novel device designed for conducting single-cell respiration measurements in the coordinated presence and absence of chemical solutes. Similar to the original SlipChip, it consists of a microwell plate and moveable channel plate. However, in addition SlipO2Chip features an array of individual microwells coated with an O2 sensing optode material. By utilizing a 3D-printed frame and micrometer screws, the top channel plate can be slipped over the bottom microwell plate, allowing for the introduction of cells into microwells, their exposure to controlled fluid flows, and measurement of single-cell O2 respiration. In an initial proof-of-concept study, we applied SlipO2Chip to individual diatom cells, crucial contributors to planetary climate and biogeochemistry. Our experiments showed that the respiration rate of cells was only minimally affected by repeated slipping motions, at least when these motions were conducted only twice. Additionally, we demonstrated a concentration-dependent reduction in the respiration rate of individual diatom cells with increasing concentrations of HHQ, a quorum-sensing signal released by marine bacteria with demonstrated impacts on the physiology of marine diatoms. Having the ability to interrogate physiological responses of individual microalgal cells to bacterial infochemicals will open exciting avenues for understanding how phycosphere-based interactions contribute to global processes in modern oceans.We foresee that the ability of SlipO2Chip to measure respiration rates, a vital indicator of metabolism, both before and after chemical exposure within the same cell, will find broad applications in research fields dealing with scarce sample volumes. This includes areas like clinical settings and medical diagnostics, where precise and sensitive measurements on a few cells are indispensable. Furthermore, SlipO2Chip provides (eco)toxicologists and microbiologists with a refined tool to explore how cell heterogeneity influences the overall sensitivity of populations to chemicals and how this relates to the emergence of tolerant subpopulations. However, these potential applications of SlipO2Chip will necessitate further technical and conceptual refinements. Potential enhancements for the system encompass the development of a 2D O2 diffusion model, conceptualizing each microwell as a distinct O2 sink and source within an x–y coordinate framework. Additionally, tailoring microwells in various sizes and shapes could accommodate a broader spectrum of cell types, significantly reducing cross-talk. Integrating additional sensors to continuously monitor environmental parameters such as temperature and pH would further refine the precision and utility of this technology. Nevertheless, even in its current state, SlipO2Chip comprises a valuable tool for answering biological questions related to ecology, (eco)toxicology and microbiology at the single-cell level. As this technology advances, it may pave the way for novel inquiries into cellular metabolism and single-cell plasticity.
Data availability
All the data supporting this article are available upon reasonable request to the corresponding authors.Author contributions
Yuan Cui: methodology, data curation, formal analysis, writing – original draft; Milena De Albuquerque Moreira: methodology; Kristen E. Whalen: resources, conceptualization, writing – review & editing; Laurent Barbe: methodology; Qian Shi: methodology; Klaus Koren: supervision, conceptualization, methodology, writing – review & editing; Maria Tenje: supervision, conceptualization, methodology, funding acquisition, writing – review & editing; Lars Behrendt: conceptualization, funding acquisition, supervision, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Behrendt was supported by grants from the Independent Research Fund Denmark (DFF-1323-00747 & DFF-1325-00069), the Swedish Research Council (2019-04401), the Novo-Nordisk Foundation (NNF22OC0079370) and the Science for Life Laboratory. Tenje acknowledges financial support for the Knut and Alice Wallenberg foundation (WAF 2016.0112). Whalen acknowledges financial support from the National Science Foundation (IOS 2041748) and the Charles E. Kaufman Foundation Integrated Research and Education Grant (KA2021-121932). We acknowledge Myfab Uppsala for providing facilities and experimental support. Myfab is funded by the Swedish Research Council (2019-00207) as a national research infrastructure. We also acknowledge the Sinergia team (Oliver Müller, Dieter Baumgartner, Ermes Botte, Johannes Keegstra, Clara Martinez-Pérez, Carrara Francesco, Kang Soo Lee, Roman Stocker, Arti Ahluwalia) for input and discussions.Notes and references
- M. Whelan and M. Andersen, Toxicity pathways – from concepts to application in chemical safety assessment, 2013 Search PubMed.
- K. Lewis, Annu. Rev. Microbiol., 2010, 64, 357–372 CrossRef CAS PubMed.
- C. E. Meacham and S. J. Morrison, Nature, 2013, 501, 328–337 CrossRef CAS PubMed.
- S. C. Booth, M. L. Workentine, J. Wen, R. Shaykhutdinov, H. J. Vogel, H. Ceri, R. J. Turner and A. M. Weljie, J. Proteome Res., 2011, 10, 3190–3199 CrossRef CAS PubMed.
- B. S. Griffiths and L. Philippot, FEMS Microbiol. Rev., 2013, 37, 112–129 CrossRef CAS PubMed.
- N. Joffe, C. Kuhlisch, G. Schleyer, N. S. Ahlers, A. Shemi and A. Vardi, ISME J., 2024, 18, wrae038 CrossRef PubMed.
- E. Harvey, H. Yang, E. Castiblanco, M. Coolahan, G. Dallmeyer-Drennen, N. Fukuda, E. Greene, M. Gonsalves, S. Smith and K. Whalen, Aquat. Microb. Ecol., 2023, 89, 75–86 CrossRef.
- C. K. Dixit, A. K. Kaushik and A. Kaushik, Microfluidics for biologists, Springer, 2016 Search PubMed.
- G. Velve-Casquillas, M. Le Berre, M. Piel and P. T. Tran, Nano Today, 2010, 5, 28–47 CrossRef CAS PubMed.
- C. M. Leung, P. de Haan, K. Ronaldson-Bouchard, G.-A. Kim, J. Ko, H. S. Rho, Z. Chen, P. Habibovic, N. L. Jeon, S. Takayama, M. L. Shuler, G. Vunjak-Novakovic, O. Frey, E. Verpoorte and Y.-C. Toh, Nat. Rev. Methods Primers, 2022, 2, 33 CrossRef CAS.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- N. Subedi, L. C. Van Eyndhoven, A. M. Hokke, L. Houben, M. C. Van Turnhout, C. V. C. Bouten, K. Eyer and J. Tel, Sci. Rep., 2021, 11, 17084 CrossRef CAS PubMed.
- N. Musat, R. Foster, T. Vagner, B. Adam and M. M. M. Kuypers, FEMS Microbiol. Rev., 2012, 36, 486–511 CrossRef CAS PubMed.
- C. Andreou, M. R. Hoonejani, M. R. Barmi, M. Moskovits and C. D. Meinhart, ACS Nano, 2013, 7, 7157–7164 CrossRef CAS PubMed.
- G. Emonds-Alt, C. Malherbe, A. Kasemiire, H. T. Avohou, P. Hubert, E. Ziemons, J.-C. M. Monbaliu and G. Eppe, Talanta, 2022, 249, 123640 CrossRef CAS PubMed.
- W. R. Gruber, I. Klimant and O. S. Wolfbeis, Advances in Fluorescence Sensing Technology, ed. J. R. Lakowicz and R. B. Thompson, 1993, vol. 1885, pp. 448–457 Search PubMed.
- A. Mills, Platinum Met. Rev., 1997, 41, 115–127 CrossRef CAS.
- M. Moßhammer, K. E. Brodersen, M. Kühl and K. Koren, Microchim. Acta, 2019, 186, 126 CrossRef PubMed.
- D. Eastwood and M. Gouterman, J. Mol. Spectrosc., 1970, 35, 359–375 CrossRef CAS.
- M. Kühl, Methods Enzymol., 2005, 397, 166–199 Search PubMed.
- I. Klimant, V. Meyer and M. Kühl, Limnol. Oceanogr., 1995, 40, 1159–1165 CrossRef CAS.
- K. Koren, K. E. Brodersen, S. L. Jakobsen and M. Kühl, Environ. Sci. Technol., 2015, 49, 2286–2292 CrossRef CAS PubMed.
- S. Ahmerkamp, F. M. Jalaluddin, Y. Cui, D. R. Brumley, C. O. Pacherres, J. S. Berg, R. Stocker, M. M. M. Kuypers, K. Koren and L. Behrendt, Cells Rep. Methods, 2022, 2, 100216 CrossRef CAS PubMed.
- I. Klimant and O. S. Wolfbeis, Anal. Chem., 1995, 67, 3160–3166 CrossRef CAS.
- R. Glud, N. Ramsing, J. Gundersen and I. Klimant, Mar. Ecol.: Prog. Ser., 1996, 140, 217–226 CrossRef.
- Y. Amao, K. Asai and I. Okura, J. Porphyrins Phthalocyanines, 2000, 4, 292–299 CrossRef CAS.
- F. T. Lee-Montiel, S. M. George, A. H. Gough, A. D. Sharma, J. Wu, R. DeBiasio, L. A. Vernetti and D. L. Taylor, Exp. Biol. Med., 2017, 242, 1617–1632 CrossRef CAS PubMed.
- B. Ungerböck, V. Charwat, P. Ertl and T. Mayr, Lab Chip, 2013, 13, 1593 RSC.
- C. Grün, J. Pfeifer, G. Liebsch and E. Gottwald, Front. Bioeng. Biotechnol., 2023, 11, 1111316 CrossRef PubMed.
- T. W. Molter, S. C. McQuaide, M. T. Suchorolski, T. J. Strovas, L. W. Burgess, D. R. Meldrum and M. E. Lidstrom, Sens. Actuators, B, 2009, 135, 678–686 CrossRef CAS PubMed.
- J. R. Etzkorn, W.-C. Wu, Z. Tian, P. Kim, S.-H. Jang, D. R. Meldrum, A. K.-Y. Jen and B. A. Parviz, J. Micromech. Microeng., 2010, 20, 95017 CrossRef.
- P. Hai, T. Imai, S. Xu, R. Zhang, R. L. Aft, J. Zou and L. V. Wang, Nat. Biomed. Eng., 2019, 3, 381–391 CrossRef CAS PubMed.
- S. Kannan, P.-L. Ko, H.-M. Wu and Y.-C. Tung, Biomicrofluidics, 2023, 17, 5 CrossRef PubMed.
- C. S. Santos, A. J. Kowaltowski and M. Bertotti, Sci. Rep., 2017, 7, 11428 CrossRef PubMed.
- T. Yoshihara, Y. Hirakawa, M. Hosaka, M. Nangaku and S. Tobita, J. Photochem. Photobiol., C, 2017, 30, 71–95 CrossRef CAS.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286 RSC.
- K. Koren, S. L. Jakobsen and M. Kühl, Sens. Actuators, B, 2016, 237, 1095–1101 CrossRef CAS.
- L. Dow, F. Stock, A. Peltekis, D. Szamosvári, M. Prothiwa, A. Lapointe, T. Böttcher, B. Bailleul, W. Vyverman, P. G. Kroth and B. Lepetit, ChemBioChem, 2020, 21, 1206–1216 CrossRef CAS PubMed.
- K. Roberts, E. Granum, R. C. Leegood and J. A. Raven, Photosynth. Res., 2007, 93, 79–88 CrossRef CAS PubMed.
- S. Basu and K. Mackey, Sustainability, 2018, 10, 869 CrossRef.
- C. Kuhlisch, A. Shemi, N. Barak-Gavish, D. Schatz and A. Vardi, Nat. Rev. Microbiol., 2024, 22, 138–154 CrossRef CAS PubMed.
- J. R. Seymour, S. A. Amin, J. B. Raina and R. Stocker, Nat. Microbiol., 2017, 2, 1–12 Search PubMed.
- J.-F. Dubern and S. P. Diggle, Mol. BioSyst., 2008, 4, 882 RSC.
- N. Meyer, A. Bigalke, A. Kaulfuß and G. Pohnert, FEMS Microbiol. Rev., 2017, 41, 880–899 CrossRef CAS PubMed.
- F. J. Reen, M. J. Mooij, L. J. Holcombe, C. M. McSweeney, G. P. McGlacken, J. P. Morrissey and F. O'Gara, FEMS Microbiol. Ecol., 2011, 77, 413–428 CrossRef CAS PubMed.
- E. L. Harvey, R. W. Deering, D. C. Rowley, A. El Gamal, M. Schorn, B. S. Moore, M. D. Johnson, T. J. Mincer and K. E. Whalen, Front. Microbiol., 2016, 7, 59 Search PubMed.
- K. E. Whalen, J. W. Becker, A. M. Schrecengost, Y. Gao, N. Giannetti and E. L. Harvey, Microbiome, 2019, 7, 93 CrossRef PubMed.
- J. Ehgartner, P. Sulzer, T. Burger, A. Kasjanow, D. Bouwes, U. Krühne, I. Klimant and T. Mayr, Sens. Actuators, B, 2016, 228, 748–757 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- J. W. Rijstenbil, J. A. Wijnholds and J. J. Sinke, Mar. Biol., 1989, 101, 131–141 CrossRef CAS.
- J. Fan, F. Li, S. Hu, K. Gao and J. Xu, Limnol. Oceanogr., 2023, 68, 2512–2528 CrossRef.
- M. Li and J. N. Young, Photosynth. Res., 2023, 156, 205–215 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00420e |
| This journal is © The Royal Society of Chemistry 2024 |