
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePatterned organic layers on gold surfaces prepared by electro-grafting of photolabile-protected aryl diazonium salts†
Max
Taras
a,
Jean-Francois
Bergamini
a,
Paula A.
Brooksby
b,
Philippe
Hapiot
 a,
Corinne
Lagrost
ac and
Yann R.
Leroux
*a
a,
Corinne
Lagrost
ac and
Yann R.
Leroux
*a
aUniv Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France. E-mail: yann.leroux@univ-rennes.fr
bSchool of Physical and Chemical Sciences and MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Canterbury, Christchurch 8041, New Zealand
cUniv Rennes, CNRS, UAR ScanMAT 2025, F-35000 Rennes, France
First published on 21st March 2024
Abstract
The electroreduction of aryl diazonium salts is a powerful technique for functionalizing many surfaces. To widen the use of aryl diazonium salts in many more applications, new strategies allowing their use with classical photolithography techniques are essential. Herein, we describe the synthesis and properties of an aryl diazonium salt protected by a photolabile group. The photolabile protecting groups allow the facile patterning of an organic layer on gold substrates by photolithography upon exposure to soft UV light (365 nm). The resulting patterned films are characterized using optical microscopy and scanning electrochemical microscopy (SECM).
Introduction
The electroreduction of aryl diazonium salts1 is now a common technique used to functionalize various materials including conductors, semiconductors and insulating surfaces.2,3 It has the advantage to produce robust interfaces in a short time frame (second to minutes).4 The reduction of an aryl diazonium salt leads to the formation of aryl radicals that are highly reactive intermediates. These intermediates react with the substrate, but also attack the already attached aryl groups, leading to multilayered disordered organic films.5 For many applications, control of the film structure at the monolayer scale is an essential pre-requisite. Hence, for almost a decade, some novel strategies to deposit only monolayer films via the reduction of aryl diazonium salts have been proposed,6–10 including our groups.11–14 In general, the patterning of surfaces is very important for the fabrication of devices as biosensors15 or in electronics.16 For these purposes, there are many strategies to pattern monolayers deposited on gold17–19 or silicon20,21 surfaces and even, to a less extent, on carbon materials.22Patterning with aryl diazonium salts is relatively new, and there is only a limited number of publications describing patterning of organic films grafted by aryl diazonium salt reduction. Previously, we reported on the use of atomic force microscopy (AFM) combined with aryl diazonium salt grafting to obtain patterned organic films deposited onto flat carbon materials.23 Here, an AFM probe tip was used to intentionally remove a section of the electro-grafted organic layer, leaving exposed carbon that was functionalized with a second modifier. More recently, Zambelli and coworkers used microfluidics in association with AFM (FluidFM) for a similar purpose.24 Here, they created patterns with diverse shapes and topologies by confining the aryl diazonium ion solution in the microchannel of FluidFM probes. Other scanning probe microscopies have been used to pattern surfaces in combination with aryl diazonium ion reduction, notably scanning electrochemical microscopy (SECM). For instance, to avoid unwanted spontaneous grafting, Cougnon et al.25 described the in situ production of aryl diazonium ions through the electrochemical reduction of nitrophenyl to aniline derivatives (in the presence of sodium nitrate) under the SECM tip. If a sufficiently negative potential is applied to the substrate, it allows the controlled deposition of organic films just under the SECM tip. Other techniques, generally adapted from methods developed for other modifiers, have been successfully applied to pattern substrates with aryl diazonium ion reduction. We can cite inkjet printing,26 microcontact printing (μCP)27 or nanosphere lithography28 as remarkable examples of methods used successfully to pattern surfaces with aryl diazonium ion reduction.
Photolithography29 is one of the most popular techniques in industries and laboratories to pattern surfaces with a high degree of precision. Even though this was developed decades ago, it continues to be improved. Unfortunately, while reduction of aryl diazonium ions can be triggered by light in the presence of photosensitizers,30 due to their instability, they can spontaneously graft surfaces31 which is a major drawback considering photolithography applications. One possibility is to use iodonium salts instead of aryl diazonium salts. They are much more stable in regards with their low reduction potentials, and direct photopatterning using deep-UV light (254 nm) or with the help of photosensitizers in solution has been reported.32,33 Regarding other technologies, as self-assembled monolayers (SAMs) of alkanethiols on coinage metals34 or silanization at oxide surfaces,35 many examples in the literature can be found where photolithography has been successfully used to pattern monolayer films.36–54 Diverse photolabile functional groups have been described in the literature for this purpose as diazoketo51 or p-methoxyphenacyl derivatives,52 but o-nitrobenzyl derivatives36–50 are by far the most used. Their synthesis is well-known and largely described in the literature.
Also, they can be cleaved easily with soft UV light at 365 nm. They have been used for various purposes ranging from hydrophobic/hydrophilic switching37 to protein immobilization and patterning.41,46,48
In this study, as a proof of concept, we present the synthesis and characterization of a new aryl diazonium salt bearing a photolabile protecting group (Scheme 1). We have chosen o-nitrobenzyl derivatives as photolabile groups to protect 4-hydroxybenzenediazonium salts. As the maximum absorption of o-nitrobenzyl is in the UV-B region, we introduce a 3,4-methylenedioxy substituent, as others,55 to shift the maximum absorption to the UV-A region. After exposure to soft UV light (365 nm), o-nitrobenzyl derivative protecting groups are cleaved and organic films constituted of phenol are obtained (Scheme 1). Patterning of the deposited organic layer is performed using classical photolithography techniques and imaged by scanning electrochemical microscopy (SECM) and optical microscopy.
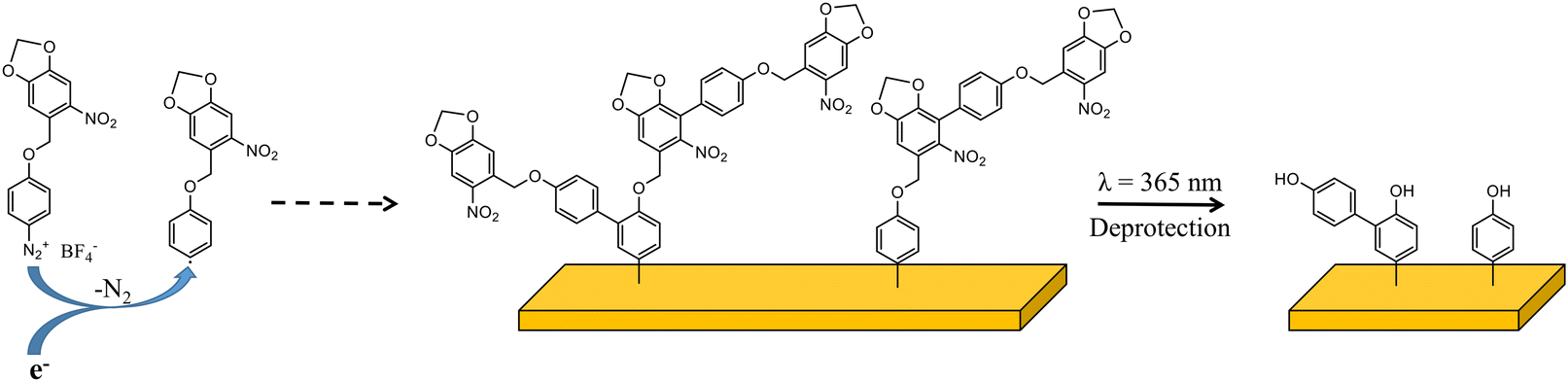 | ||
| Scheme 1 Schematic representation of aryl diazonium 1 and of the grafting process and the resulting organic deposit after deprotection. | ||
Results and discussion
The new aryl diazonium salt 1 (Fig. 1) possessing a photolabile protecting group was synthesized in 4 steps. First, 3,4-methylenedioxy-6-nitro-benzyl bromide was synthesized as previously described55 then used in the esterification of Boc-protected aminophenol (see the Experimental section). Selective deprotection of the Boc group was achieved using TFA under dark conditions. Finally, diazotization of the obtained aniline derivatives was performed in acetonitrile using NOBF4 as a diazotization agent. Fig. 1 shows the electrochemical reduction of this new aryl diazonium salt on a gold electrode. During the first scan (Fig. 1, black curve), we clearly observe two irreversible reduction peaks centered at 0.25 V and −0.1 V vs. SCE with no associated oxidation peaks. This behaviour is ascribed to the one-electron transfer from the electrode material to the aryl diazonium salt followed56 or concerted3 with the cleavage of the N2 group leading to the corresponding aryl radical. On gold electrodes, it has been demonstrated that the observation of multiple peaks during the electroreduction of aryl diazonium salts is due to the reduction of the molecules on different gold facets.57 Successive potential scans do not show additional electroactivity in this potential range and is characteristic of an insulating organic film deposited onto the electrode surface blocking further electroreduction of the salt.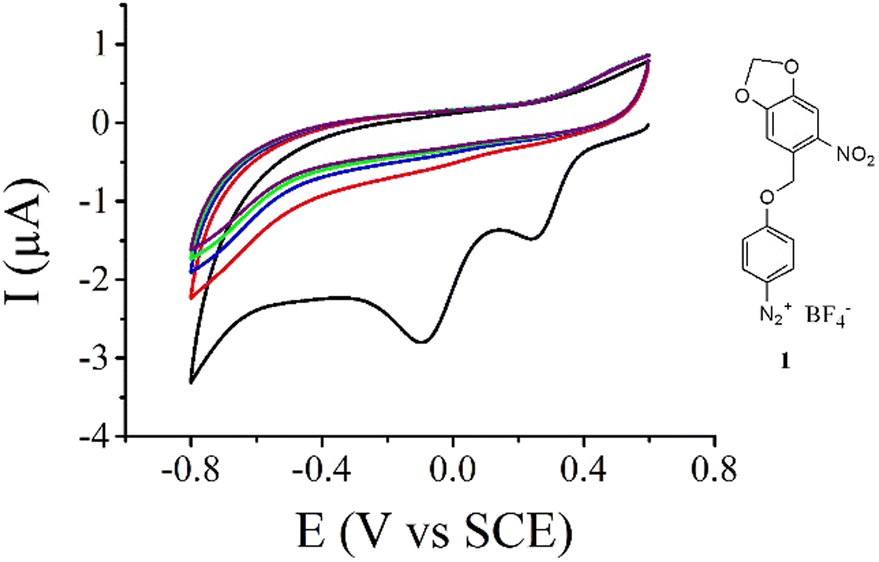 | ||
| Fig. 1 Five consecutive cyclic voltammograms of 10 mM aryl diazonium 1 in an acetonitrile solution containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte on a gold electrode. Scan rate = 50 mV s−1. First scan (black curve), second scan (red curve), third scan (blue curve), fourth scan (green curve) and fifth scan (wine curve) are presented. | ||
The electrochemical properties of the gold electrode surface were monitored using dopamine as a redox probe in solution. On the bare gold electrode (Fig. 2, black curve), we observed the expected reversible electrochemical signal of dopamine oxidation at half-wave potential E1/2 = 0.480 V vs. SCE with a peak-to-peak separation ΔE = 42 mV. After the electroreduction of 1 (Fig. 2, blue curve) as presented in Fig. 1, the cyclic voltammogram of dopamine oxidation becomes irreversible with a peak-to peak separation ΔE = 259 mV and a decrease of the peak current intensity. This is consistent with the deposition of a thin insulating organic film onto the electrode surface, with a layer thickness thin enough to still allow electron transfer through tunnelling.11,58
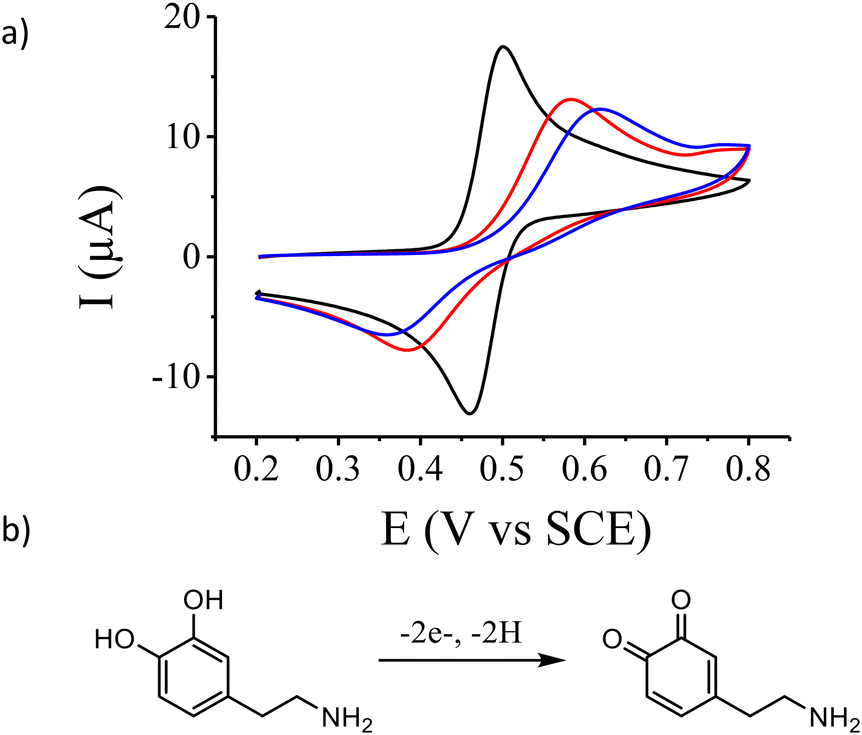 | ||
| Fig. 2 a) Cyclic voltammograms of 1 mM dopamine aqueous solution (+0.1 M H2SO4) on a bare gold electrode (black curve) and on a gold electrode functionalized with aryl diazonium 1 before (blue curve) and after (red curve) photo-deprotection at λ = 365 nm for 5 min (power: 40 mW cm−2). Scan rate = 100 mV s−1. b) Scheme of dopamine oxidation. | ||
In order to estimate the organic layer thickness, we performed ellipsometry and AFM scratching experiments.59 While ellipsometry is an optical and non-destructive technique, AFM scratching intentionally damages the organic film with the AFM tip removing a section of the film back to the gold substrate. Then, an AFM image is acquired and a line profile across the scratch is recorded to estimate the layer thickness. All estimated layer thicknesses are presented in Table 1. They are equal to 2.3 ± 0.6 nm and 2.1 ± 0.3 nm by ellipsometry and AFM experiments, respectively. Both values are close to each other and reinforce the conclusions based on the electrochemical data. In addition, as the length of the deposited molecule can be estimated to 1.3 nm, the electroreduction process leads to the deposition of a multilayer organic film on the gold surface although the orientation of the film is unknown, for example, perpendicular or tilted.
| Protected film | Deprotected film | |
|---|---|---|
| AFM | 2.1 ± 0.3 nm | 1.2 ± 0.3 nm |
| Ellipsometry | 2.3 ± 0.6 nm | 1.2 ± 0.1 nm |
Deprotection of the o-nitrobenzyl protecting group was achieved by exposing the functionalized gold surface to soft UV light (λ = 365 nm) for 5 minutes and should lead to an organic film composed of only phenol end groups. The electrochemical properties of the deprotected surface were investigated using dopamine as previously demonstrated. Compared to the cyclic voltammogram of dopamine on the modified surface before deprotection, we observed an increase of the peak current intensity and a decrease of the peak-to-peak separation equal to ΔE = 200 mV (Fig. 2, red curve). This is indicative of a faster charge transfer at the modified surface due to a decrease of the film thickness as expected after deprotection. This is also confirmed by ellipsometry and AFM scratching experiments on the deprotected surface which give an average film thickness of 1.2 nm (Table 1).
The estimated length of a phenol molecule is 0.6 nm, and hence the gold surface after deprotection is composed of a multilayer phenol film. To further evidence the efficiency of the deprotection process, the hydrophilic/hydrophobic properties of the organic film before and after photo-deprotection were investigated by contact angle measurements. As shown in Fig. 3, the static contact angle on the protected film is equal to 64° ± 5° and decreases to 44° ± 2° after photo-deprotection, as expected for a phenol multilayer film exhibiting a higher hydrophilic nature.
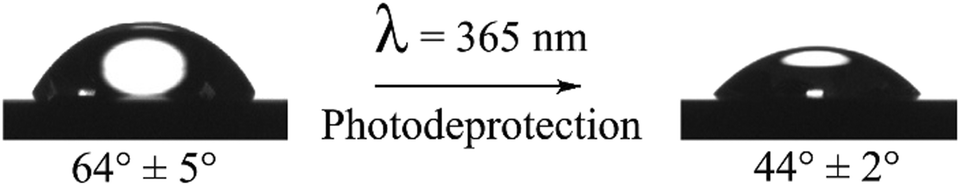 | ||
| Fig. 3 Images and contact angle measurement values of a 2 μL drop of pure water on the gold functionalized surface before and after photo-deprotection at λ = 365 nm for 5 min (power: 40 mW cm−2). | ||
XPS experiments were also performed on the functionalized gold surface before and after photo-deprotection. The survey spectra showed photoelectron peaks corresponding to elemental species C, O and Au as main components (Fig. S1†). The contribution due to the N 1s photoelectron is weakly visible before photodeprotection (1–1.5 at%) but still significant, and almost invisible after the deprotection step (Fig. S1†). Fig. 4 displays the XPS core level spectra in the N 1s region for gold samples modified with 1 before and after irradiation. Two peaks were clearly identified for the sample before irradiation. The peak at the highest binding energy at ca. 406 eV is assigned to the NO2 group.60,61 The second component located at 399.9 eV is due to the amino group,60,61 which could originate from contamination by atmospheric nitrogen as already reported and/or from the reduction of the nitro group under the X-ray beam during the XPS experiment.61,62 Such a phenomenon is very likely in our experiments since a large number of scans and an important dwell time was required for obtaining the N 1s signal with a good signal-to-noise ratio because of its weak contribution. When the modified gold sample undergoes 5 min irradiation at λ = 365 nm, the XPS signal exhibits a very different feature, with the disappearance of the component at 406 eV and a very small contribution at 399.9 eV. The latter could be due to contamination or to the presence of residual amino groups, produced upon the reduction of the nitro groups before the photodeprotection step. Interestingly, we also observed a decrease in intensity for C and O species while the contribution of the gold signal is increasing after irradiation (Fig. S2 and Table S1†). These observations clearly indicate the departure of the o-nitrobenzyl moiety upon 5 min irradiation.
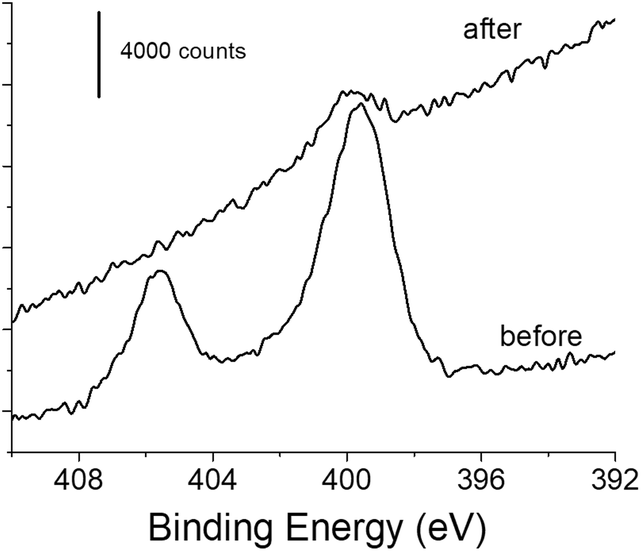 | ||
| Fig. 4 High resolution XPS core level spectra of the N 1s region of the functionalized gold surface before and after photo-deprotection at λ = 365 nm for 5 min (power: 40 mW cm−2). | ||
Then, we have gone ahead to undertake the photopatterning of the organic layer using a UV-LED masking system as a proof of concept for demonstrating the usefulness and versatility of the strategy. We employed a classical chrome mask consisting of lines of 25 μm width and 25 μm spacing. To reveal the photopatterning, we employed two different methods. First, we used scanning electrochemical microscopy (SECM) in feedback mode and under unbiased conditions to image the patterned organic layer. In the feedback mode, SECM provides a chemical view of the surface from the solution side. It uses the interaction of an electrogenerated probe (redox mediator) that diffuses from a tip electrode to the sample. The image contrast is provided by the differences between the charge transfer properties of the redox mediator toward different areas of the modified surface. As previously demonstrated by voltammetry (Fig. 2), the electron transfer properties of dopamine are sensitive to the variation of film thicknesses before and after deprotection, making it a very good probe to monitor the occurrence of the photodeprotection, even at a localized scale. Hence, we used dopamine as a redox mediator in SECM to image the patterned organic layer where the SECM tip scanned the surface at a fixed distance. As shown in Fig. 5a, we were able to observe the pattern constituted of lines of 25 μm width and 25 μm gap. The green lines correspond to the photo-deprotected area, with a higher current, and the blue lines represent the protected film. Taking advantages of the different hydrophilic/hydrophobic properties of both areas, optical images of the patterned organic film under a wet atmosphere were also recorded (Fig. 5b). As expected, we can easily observe the same patterns as those observed by SECM, demonstrating the efficiency of the method to produce ultrathin patterned organic films. Furthermore, as the electroreduction of aryl diazonium salts can be performed on a wide range of substrates, either conductors, semi-conductors or insulators, this method can be easily transferred to other metals or even to carbon materials, those latter being particularly difficult to pattern.
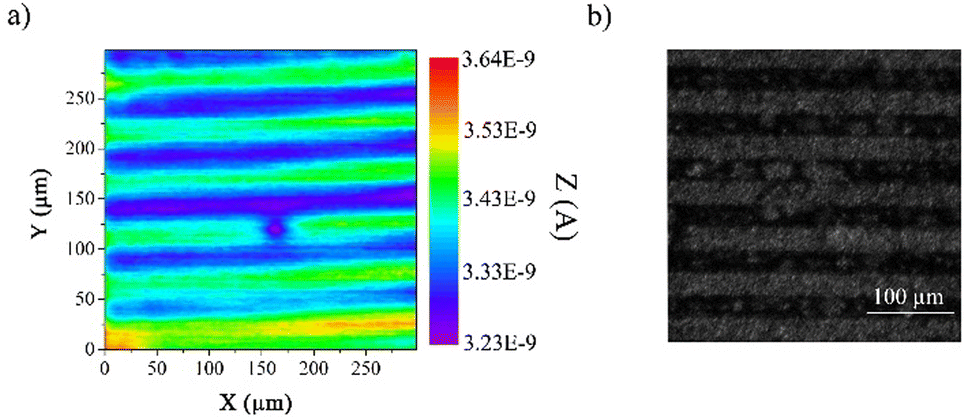 | ||
| Fig. 5 (a) Scanning electrochemical microscopy (SECM) image of the patterned functionalized gold surface using dopamine (1 mM) as a redox mediator in 0.1 M H2SO4 aqueous solution. The tip was a 5 μm radius disk Pt microelectrode and was maintained at 8 μm from the substrate. (b) Photograph of the patterned organic film via optical microscopy. | ||
Conclusions
In this work, we describe the synthesis of a new aryl diazonium salt bearing a photolabile group. Reductive electrochemical grafting is classically performed, leading to a protected multilayered film whose terminal group could be removed upon exposure to UV light. Subsequent photopatterning of such a modified surface could be easily achieved as demonstrated by SECM and optical imaging experiments. Because of the high versatility of the diazonium grafting procedure, the strategy paves the way toward the patterning of a wide range of materials through a very simple method using UV-A light that preserves the quality of the grafted moieties.Experimental
Chemicals and reagents
Commercially available reagents were used as received without further purification. N-Boc-4-aminophenol, dopamine, tetrabutylammonium hexafluorophosphate (NBuPF6) and a gold coated silicon wafer were purchased from Sigma-Aldrich. 3,4-Methylenedioxy-6-nitro-benzyl bromide was synthesized as previously described.55Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether (1:1) as an eluent in 69% yield. δH (300 MHz, CD3CN) 7.65 (s, 1H), 7.31 (d, J = 9.2 Hz, 2H), 7.26 (s, 1H), 6.93 (d, J = 9.3 Hz, 2H), 6.15 (s, 2H), 5.35 (d, J = 0.8 Hz, 2H), 1.47 (s, 9H).
:petroleum ether (1:1) as an eluent in 69% yield. δH (300 MHz, CD3CN) 7.65 (s, 1H), 7.31 (d, J = 9.2 Hz, 2H), 7.26 (s, 1H), 6.93 (d, J = 9.3 Hz, 2H), 6.15 (s, 2H), 5.35 (d, J = 0.8 Hz, 2H), 1.47 (s, 9H).
Electrochemical setup and procedures
All electrochemical measurements were performed with an Autolab PGSTAT 101 (Metrohm) and a conventional three-electrode system, comprising the modified substrate as the working electrode, platinum foil as the auxiliary electrode, and the (KCl saturated) SCE electrode (Metrohm) as the reference. The gold electrodes were purchased from CH Instruments, INC. (TX) as 2 mm-diameter disks. The electrodes were electrochemically polished by cyclic voltammetry with a 0.1 M sulfuric acid solution (H2SO4) from −0.3 V to 1.2 V for at least 50 cycles, rinsed with water and acetone and dried. The gold coated silicon wafers (Sigma-Aldrich) were rectangles of 12 × 18 mm2.Scanning Electrochemical Microscopy (SECM)
Scanning electrochemical microscopy (SECM) experiments were carried out with a 920D Scanning Electrochemical Microscope (CH Instruments, Inc.) equipped with an adjustable stage for tilt angle correction. All experiments were performed with a platinum ultra-micro-electrode (UME) (5 μm diameter) from CH Instruments as the working electrode, a platinum wire as the counter electrode and a silver wire coated with AgCl as the reference electrode. Probe approach curves and SECM images were obtained in 0.1 M sulfuric acid solution containing 1 mM of dopamine as the redox mediator. The applied potential at the tip was chosen at the diffusion plateau of the mediator. Prior to any experiment, UME parameters (a and RG) were determined independently from approach curves on an insulator sample (glass). Tilt correction was performed by recording three approach curves, one determining the (0, 0) position and the two others at 500 μm in the x (500, 0) and y (0, 500) directions. For SECM imaging, positioning of the UME at a fixed distance was performed by recording an approach curve at the (0, 0) position. Mira software, developed by Pr. Gunther Wittstock, was used to analyze and fit the experimental curve and extrapolate the distance from the substrate.63Patterning
Organic layer patterning was carried out with a UV-KUB 2 (Kloe SA). The sample was covered with a mask wielding a 5 × 5 mm2 square with bands of 25 μm width. The covered substrate was irradiated at 365 nm for 5 minutes then rinsed with acetone and dried with argon gas flow.Atomic Force Microscopy (AFM)
All atomic force microscopy (AFM) experiments were carried out with an NT-MDT Ntegra microscope in tapping mode. An ACL (AppNano Inc.) probe was employed for imaging. Experiments were performed to estimate the thickness of the organic layers by the AFM scratching procedure.59 The term AFM scratching is used here to describe the intentional damage to a modified layer on a relatively hard substrate. If the applied force is sufficient to disrupt the organic layer but not to damage the substrate, it is possible to remove a square trench in the deposit layer only. On the modified gold surface, a 0.5 × 0.5 μm2 scratch was made by moving the tip into contact mode with a set-point frequency at around 100 kHz. Note that careful examination confirmed that scratching of the underlying gold substrate did not occur. The images shown were recorded in tapping mode after scratching, and representative line profiles through each scratch are shown. In all experiments, deposited layers appear as continuous films with no obvious film segregation.Spectroscopic ellipsometry measurements
All spectroscopic ellipsometry experiments were carried out with an Alpha-SE (J.A. Woollam Co., Inc.). All samples were rinsed with acetone and dried with argon gas flow before measurement. To perform the thickness measurement, a model of the substrate was created for each sample. These models were used to measure the film permittivity parameters n and k with the Cauchy dispersion law:| n(λ, T) = (n1(T))/λ2 + (n2(T))/λ4 |
| k(λ, T) = (k1(T))/λ2 + (k2(T))/λ4 |
Contact angle measurements
Static contact angle measurements were conducted with an easy drop goniometer DSA25 (Krüss). The static contact angles of 2 μL of Milli-Q water drops were measured on 5 different spots. The contact angles were determined using tangent 1 and tangent 2 circle methods. Initially, a static contact angle of 83.6° ± 1.2° was determined for the bare gold substrate.X-ray photoelectron spectroscopy (XPS)
XPS data were collected with a NEXSA G2 (ThermoFischer Scientific) spectrometer using an Al Kα X-ray source working at 1486.6 eV and using a spot size of 200 μm2. Survey spectra (0–1000 eV) were acquired with an analyzer pass energy of 200 eV (1 eV per step); high resolution spectra used a pass energy of 50 eV (0.1 eV per step). Binding energies were referenced to the Au 4f7/2 peak at 84 eV. The peak areas were normalized by the manufacturer-supplied sensitivity factors (SC1s = 1, SN1s = 1.68, SAu4f = 20.73, SO1s = 2.88). For each sample, XPS survey spectra were recorded on three different locations, and we did not observe any difference.Author contributions
Max Taras: investigation, validation, and visualization. Jean-Francois Bergamini: investigation and visualization. Paula A. Brooksby: investigation and writing – review & editing. Philippe Hapiot: conceptualization, funding acquisition, and writing – review & editing. Corinne Lagrost: investigation, funding acquisition, supervision, and writing – review & editing. Yann Leroux: conceptualization, funding acquisition, writing – original draft, writing – review & editing, visualization, supervision, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to warmly thank Alison J. Downard for our past collaborations and her kind advice concerning this work. M. T. thanks the French Ministry of Research for financial support. The XPS measurements have been performed on an ASPHERYX platform (ScanMAT, UAR 2025 University of Rennes-CNRS; CPER MAT &Trans 2021-2027).Notes and references
- M. Delamar, R. Hitmi, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 5883–5884 CrossRef CAS.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- J. M. Hicks, Z. Y. Wong, D. J. Scurr, N. Silman, S. K. Jackson, P. M. Mendes, J. W. Aylott and F. J. Rawson, Langmuir, 2017, 33, 4924–4933 CrossRef CAS PubMed.
- J. K. Kariuki and M. T. McDermott, Langmuir, 2001, 17, 5947–5951 CrossRef CAS.
- L. T. Nielsen, K. H. Vase, M. Dong, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, J. Am. Chem. Soc., 2007, 129, 1888–1889 CrossRef CAS PubMed.
- C. Combellas, F. Kanoufi, J. Pinson and F. I. Podvorica, J. Am. Chem. Soc., 2008, 130, 8576–8577 CrossRef CAS PubMed.
- O. Fontaine, J. Ghilane, P. Martin, J.-C. Lacroix and H. Randriamahazaka, Langmuir, 2010, 26, 18542–18549 CrossRef CAS.
- A. Mattiuzzi, I. Jabin, C. Mangeney, C. Roux, O. Reinaud, L. Santos, J.-F. Bergamini, P. Hapiot and C. Lagrost, Nat. Commun., 2012, 3, 1130 CrossRef.
- T. Menanteau, E. Levillain and T. Breton, Chem. Mater., 2013, 25, 2905–2909 CrossRef CAS.
- Y. R. Leroux, H. Fei, J.-M. Noël, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef CAS PubMed.
- Y. R. Leroux and P. Hapiot, Chem. Mater., 2013, 25, 489–495 CrossRef CAS.
- L. Lee, H. Ma, P. A. Brooksby, S. A. Brown, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2014, 30, 7104–7111 CrossRef CAS.
- L. Lee, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2015, 31, 5071–5077 CrossRef CAS PubMed.
- P. Jonkheijm, D. Weinrich, H. Schröder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618–9647 CrossRef CAS.
- C. Zhou, G. Nagy and A. V. Walker, J. Am. Chem. Soc., 2005, 127, 12160–12161 CrossRef CAS PubMed.
- J. T. Koepsel and W. L. Murphy, ChemBioChem, 2012, 13, 1717–1724 CrossRef CAS PubMed.
- L. Yan, W. T. S. Huck and G. M. Whitesides, Journal of Macromolecular Science, Part C, 2004, 44, 175–206 CrossRef.
- J. L. Wilbur, A. Kumar, E. Kim and G. M. Whitesides, Adv. Mater., 1994, 6, 600–604 CrossRef CAS.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282–6304 CrossRef CAS.
- N. Herzer, S. Hoeppener and U. S. Schubert, Chem. Commun., 2010, 46, 5634–5652 RSC.
- M. M. Chehimi, Aryl Diazonium Salts: New Coupling Agents in Polymer and Surface Science, 2012, Print ISBN: 9783527329984, Online ISBN: 9783527650446, DOI:10.1002/9783527650446.
- P. A. Brooksby and A. J. Downard, Langmuir, 2005, 21, 1672–1675 CrossRef CAS PubMed.
- L. Hirt, R. R. Grüter, T. Berthelot, R. Cornut, J. Vörös and T. Zambelli, RSC Adv., 2015, 5, 84517–84522 RSC.
- C. Cougnon, F. Gohier, D. Bélanger and J. Mauzeroll, Angew. Chem., Int. Ed., 2009, 48, 4006–4008 CrossRef CAS.
- A. Garcia, N. Hanifi, B. Jousselme, P. Jégou, S. Palacin, P. Viel and T. Berthelot, Adv. Funct. Mater., 2013, 23, 3668–3674 CrossRef CAS.
- D. J. Garrett, J. Lehr, G. M. Miskelly and A. J. Downard, J. Am. Chem. Soc., 2007, 129, 15456–15457 CrossRef CAS PubMed.
- V.-Q. Nguyen, D. Schaming, D. L. Tran and J.-C. Lacroix, ChemElectroChem, 2016, 3, 2264–2269 CrossRef CAS.
- K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer and S. Mahajan, MRS Bull., 2004, 29, 512 CrossRef.
- M. Bouriga, M. M. Chehimi, C. Combellas, P. Decorse, F. Kanoufi, A. Deronzier and J. Pinson, Chem. Mater., 2013, 25, 90–97 CrossRef CAS.
- A. Adenier, N. Barré, E. Cabet-Deliry, A. Chaussé, S. Griveau, F. Mercier, J. Pinson and C. Vautrin-Ul, Surf. Sci., 2006, 600, 4801–4812 CrossRef CAS.
- J. Médard, C. Combellas, F. Kanoufi, J. Pinson, J. Chauvin and A. Deronzier, J. Phys. Chem. C, 2018, 122, 19722–19730 CrossRef.
- O. Guselnikova, E. Miliutina, R. Elashnikov, V. Burtsev, M. M. Chehimi, V. Svorcik, M. Yusubov, O. Lyutakov and P. Postnikov, Prog. Org. Coat., 2019, 136, 105211 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS PubMed.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS PubMed.
- K. Critchley, J. P. Jeyadevan, H. Fukushima, M. Ishida, T. Shimoda, R. J. Bushby and S. D. Evans, Langmuir, 2005, 21, 4554–4561 CrossRef CAS.
- K. Critchley, R. Ducker, J. P. Bramble, L. Zhang, R. J. Bushby, G. J. Leggett and S. D. Evans, J. Exp. Nanosci., 2007, 2, 279–290 CrossRef CAS.
- P. Prompinit, A. S. Achalkumar, X. Han, R. J. Bushby, C. Wälti and S. D. Evans, J. Phys. Chem. C, 2009, 113, 21642–21647 CrossRef CAS.
- C. Daengngam, S. B. Thorpe, X. Guo, S. V. Stoianov, W. L. Santos, J. R. Morris and H. D. Robinson, J. Phys. Chem. C, 2013, 117, 14165–14175 CrossRef CAS.
- S. Zhou, K. J. Metcalf, P. Bugga, J. Grant and M. Mrksich, ACS Appl. Mater. Interfaces, 2018, 10, 40452–40459 CrossRef CAS.
- B. A. Magill, X. Guo, C. L. Peck, R. L. Reyes, E. M. See, W. L. Santos and H. D. Robinson, Photochem. Photobiol. Sci., 2019, 18, 30–44 CrossRef CAS PubMed.
- T. Vossmeyer, S. Jia, E. DeIonno, M. R. Diehl, S. H. Kim, X. Peng, A. P. Alivisatos and J. R. Heath, J. Appl. Phys., 1998, 84, 3664–3670 CrossRef CAS.
- M. Nakagawa and K. Ichimura, Colloids Surf., A, 2002, 204, 1–7 CrossRef CAS.
- A. del Campo, D. Boos, H. W. Spiess and U. Jonas, Angew. Chem., Int. Ed., 2005, 44, 4707–4712 CrossRef CAS.
- J. M. Alonso, A. Reichel, J. Piehler and A. del Campo, Langmuir, 2008, 24, 448–457 CrossRef CAS PubMed.
- P. Stegmaier, J. M. Alonso and A. d. Campo, Langmuir, 2008, 24, 11872–11879 CrossRef CAS PubMed.
- M. Álvarez, J. M. Alonso, O. Filevich, M. Bhagawati, R. Etchenique, J. Piehler and A. del Campo, Langmuir, 2011, 27, 2789–2795 CrossRef PubMed.
- T. Konishi and K. Yamaguchi, Bull. Chem. Soc. Jpn., 2016, 89, 424–429 CrossRef CAS.
- T. Konishi and K. Yamaguchi, Chem. Lett., 2016, 45, 469–471 CrossRef CAS.
- R. Ganesan, H.-J. Lee and J.-B. Kim, Langmuir, 2009, 25, 8888–8893 CrossRef CAS PubMed.
- M. Millaruelo, L. M. Eng, M. Mertig, B. Pilch, U. Oertel, J. Opitz, B. Sieczkowska, F. Simon and B. Voit, Langmuir, 2006, 22, 9446–9452 CrossRef CAS PubMed.
- B. Leuschel, A. Gwiazda, W. Heni, F. Diot, S.-Y. Yu, C. Bidaud, L. Vonna, A. Ponche, H. Haidara and O. Soppera, Sci. Rep., 2018, 8, 10444 CrossRef PubMed.
- T. Griesser, J. Adams, J. Wappel, W. Kern, G. J. Leggett and G. Trimmel, Langmuir, 2008, 24, 12420–12425 CrossRef CAS.
- L. F. Tietze, M. Müller, S.-C. Duefert, K. Schmuck and I. Schuberth, Chem. – Eur. J., 2013, 19, 1726–1731 CrossRef CAS PubMed.
- L. Pichereau, L. Fillaud, N. Kostopoulos, E. Maisonhaute, T. Cauchy, M. Allain, J.-M. Noël, C. Gautier and T. Breton, J. Phys. Chem. Lett., 2022, 13, 11866–11871 CrossRef CAS.
- A. Benedetto, M. Balog, P. Viel, F. Le Derf, M. Sallé and S. Palacin, Electrochim. Acta, 2008, 53, 7117–7122 CrossRef CAS.
- P. Chen and R. L. McCreery, Anal. Chem., 1996, 68, 3958–3965 CrossRef CAS.
- F. Anariba, S. H. DuVall and R. L. McCreery, Anal. Chem., 2003, 75, 3837–3844 CrossRef CAS PubMed.
- A. Adenier, E. Cabet-Deliry, A. Chaussé, S. Griveau, F. Mercier, J. Pinson and C. Vautrin-Ul, Chem. Mater., 2005, 17, 491–501 CrossRef CAS.
- K. Roodenko, M. Gensch, J. Rappich, K. Hinrichs, N. Esser and R. Hunger, J. Phys. Chem. B, 2007, 111, 7541–7549 CrossRef CAS PubMed.
- P. Mendes, M. Belloni, M. Ashworth, C. Hardy, K. Nikitin, D. Fitzmaurice, K. Critchley, S. Evans and J. Preece, ChemPhysChem, 2003, 4, 884–889 CrossRef CAS.
- G. Wittstock, T. Asmus and T. Wilhelm, Fresenius' J. Anal. Chem., 2000, 367, 346–351 CrossRef CAS PubMed.
- Y. Aceta, J.-F. Bergamini, C. Lagrost, P. Hapiot and Y. R. Leroux, Langmuir, 2018, 34, 2410–2419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, XPS and AFM experiments. See DOI: https://doi.org/10.1039/d3lf00208j |
| This journal is © The Royal Society of Chemistry 2024 |