
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradation of a lithium cobalt oxide cathode under high voltage operation at an interface with an oxide solid electrolyte†
Kotaro
Ito
 a,
Kazuhisa
Tamura
b,
Keisuke
Shimizu
c,
Norifumi L.
Yamada
d,
Kenta
Watanabe
a,
Kota
Suzuki
c,
Ryoji
Kanno
c and
Masaaki
Hirayama
*ac
a,
Kazuhisa
Tamura
b,
Keisuke
Shimizu
c,
Norifumi L.
Yamada
d,
Kenta
Watanabe
a,
Kota
Suzuki
c,
Ryoji
Kanno
c and
Masaaki
Hirayama
*ac
aDepartment of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan. E-mail: hirayama@mac.titech.ac.jp
bMaterials Sciences Research Center, Japan Atomic Energy Agency, 1-1-1 Koto, Sayo, Hyogo 679-5148, Japan
cResearch Center for All-Solid-State Battery, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
dInstitute of Materials Structure Science, High Energy Accelerator Research Organization, 203-1 Shirakata, Tokai, Ibaraki 319-1106, Japan
First published on 3rd April 2024
Abstract
Lithium (de)intercalation of layered rocksalt-type cathodes in high-voltage regions is of great importance for achieving a high energy density in lithium batteries. The reversible capacity of LiCoO2 at high voltages is not well known because of oxidative side reactions with the electrolyte species. In this study, a model thin-film battery was fabricated using an epitaxially grown LiCoO2 cathode and an amorphous Li3PO4 solid electrolyte to suppress oxidative degradation. The film battery operated stably at high voltages, ranging up to 4.6 V, without severe side reactions of LiCoO2 and Li3PO4, resulting in a reversible capacity greater than 200 mA h g−1. However, the charge–discharge capacities of the battery decreased with cycling at 4.7 V. In situ synchrotron X-ray diffraction studies revealed an irreversible structural change in LiCoO2 at 3.0 V after charging at 4.7 V. Structural degradation occurred both in the bulk and surface regions of the LiCoO2 film, indicating intrinsic irreversibility of the crystal structure changes of highly delithiated LiCoO2, although the LiCoO2/electrolyte interface remained stable.
Introduction
Currently, technologies for large-scale energy storage and the electrification of power sources are required to achieve a sustainable society. Lithium-ion batteries (LIBs) are representative devices used in various mobile devices and automotive power sources, because they deliver high voltages and currents, and can be manufactured in a variety of shapes.1–3 However, it is necessary to develop rechargeable batteries with higher energy densities than those of conventional LIBs for large-scale energy storage and power sources. Energy densities of both cathodes and anodes must be increased for realizing a full battery with a high energy density. It is particularly necessary to increase the energy densities of cathodes, because the LiCoO2 (LCO) cathode has a lower theoretical capacity (137 mA h g−1 at 4.2 V vs. Li/Li+)4,5 than the graphite anode (372 mA h g−1) used in conventional LIBs.6 In fact, olivine-type structural materials, such as LiFePO4,7 Li-excess layered rock-salt materials, including Li2MnO3–LiMeO2 (Me: transition metal),8–10 and others, have been widely studied as cathode materials of LIBs for achieving a high energy density at a low cost.Although LCO is utilized as a cathode in conventional LIBs, its theoretical capacity is basically limited to 130–150 mA h g−1 by its upper limit of an operating potential of approximately 4.2 V.4 The maximum capacity of LCO is theoretically 274 mA h g−1 when Li ions are completely deintercalated from the crystal lattice,11 and realizing this capacity with an LCO cathode requires an operating potential higher than 4.2 V. However, LCO degrades at high potentials owing to the instability of its delithiated crystal structure. The structural degradation has been considered to proceed by oxygen release from the LCO lattice, which occurs simultaneously with side reactions involving oxidative decomposition of species in organic liquid electrolytes.12 This leads to the formation of a thermodynamically stable spinel phase (Co3O4 or/and LiCo2O4).13 The spinel phases lose oxygen atoms in further reactions to form the rock salt phase CoO.14 These structural degradations initiate at the interface and then extend to the bulk region.15 Thus, it is important to stabilize the LCO/organic electrolyte interfaces to increase the reversible capacity of LCO at high potentials. Measures, such as the formation of a cathode–electrolyte interface (CEI)16,17 and doping other metal ions into LCO,18,19 have been adopted to suppress side reactions. However, the degradation of LCO was not completely suppressed by these manipulations.
Solid oxide electrolytes are known to have a wide practical potential window (≥5 V) because solid-state batteries with high-potential cathodes or low-potential anodes operate stably.20,21 Although the upper limit of the thermodynamic potential window obtained by first-principles calculations is approximately 4 V vs. Li/Li+,22,23 they are considered to be kinetically stable with a slow decomposition reaction rate, or the electrochemical stability and ionic conductivity of the decomposition products are high.22 Moreover, the comprised elements of the solid electrolyte, except the carrier ions, are fixed at each position of the crystal lattice, and the transference numbers of the carrier ions are almost 1. Consequently, both oxygen release and elution of active materials are suppressed by using a solid electrolyte. Thus, the structural degradation of LCO can be suppressed at the interface with oxide electrolytes, leading to a superior reversible capacity compared to that at the LCO/liquid electrolyte interfaces. However, there is a lack of knowledge regarding the lithium (de)intercalation of LCO at high electrode potentials using solid oxide electrolytes. This might be due to the technical difficulty in fabricating bulk-type all-solid-state batteries and in characterizing the structure and electrochemical properties of the interfacial phenomena.
Thin-film batteries provide a simple reaction field suitable for analyzing electrochemical reactions during charge–discharge processes. Among thin-film electrodes, epitaxial-film electrodes offer several advantages for clarifying the interfacial reaction mechanism. First, they have very flat surfaces with a roughness of several nanometers, which provides a two-dimensional interface. Second, the film thickness can be controlled in the range of 10–100 nm, which enhances the electrochemical properties in the surface regions.24 Third, the nanosized film electrodes show a very small volumetric change during lithium (de)intercalation, allowing reaction analysis without mechanical effects such as particle cracking or changes in the conductive pathways. Fourth, flat electrode surfaces may enable surface structural changes to be detected using in situ surface scattering/spectroscopy techniques.25–30 We have recently clarified the operating mechanisms of Li2MnO3 using thin-film batteries with Li2MnO3 epitaxial films,31–33 which demonstrates the feasibility of using epitaxial film systems to investigate interfacial reactions in all-solid-state batteries.
In this study, thin-film batteries with LCO as the cathode and amorphous Li3PO4 (LPO) as the solid electrolyte were fabricated as model systems by exposing the (104) facets of LCO. The charge–discharge properties were characterized at different upper cutoff voltages to clarify the electrochemical stability at high voltages. The structural changes in the LCO film during the charge–discharge reactions were investigated using in situ synchrotron X-ray diffraction (SXRD). The study findings clarify the maximum upper cutoff voltage for stable lithium (de)intercalation of LCO, and the degradation mechanism of LCO at the electrochemical interface with the electrochemically stable solid electrolyte is discussed.
Experimental
Radio frequency (RF) magnetron sputtering (QAM-4ST, ULVAC) with a background pressure of 10−5 Pa or less was used to deposit SrRuO3 (SRO) as the current collector and LCO as the cathode on a single-crystalline SrTiO3(100) (STO(100)) substrate (crystal base). To prevent lithium deficiency in the film, an excess of the lithium target, Li1.4CoOx (Toshima Manufacturing Co., Ltd.), was used. The conditions for RF magnetron sputtering were as follows: a mixed gas of argon and oxygen at 0.75 Pa with argon and oxygen flow rates of 14 and 2 sccm, respectively, a substrate temperature of 600 °C, a 150 mm distance between the target and substrate, and an RF power of 110 W.The crystal structure of the LCO thin film grown was analyzed using both X-ray diffraction (XRD; ATX, Rigaku, Cu-Kα) and X-ray reflectometry (XRR; ATX, Rigaku, Cu-Kα). The surface morphology and bonding states were examined via atomic force microscopy (AFM; AFM5300E, Hitachi High-Tech Co.) and Raman spectroscopy (Lambda Vision, Micro-RAM300, 532 nm, 100 mW laser, 520 cm−1 calibration), respectively. Neutron reflectometry (NR) was performed using SOFIA (BL-16, Materials and Life Science Facility, Japan Proton Accelerator Research Complex, Tokai, Japan) and a time-of-flight reflectometer.34–36 NR spectra were collected in the single-flame mode at 0.3, 0.75, and 1.8° with a footprint of 15 × 15 mm2, and the individual data points were combined. Spectral fitting was performed using Motofit software.37 All the structural characterization techniques of the LCO films were conducted in air.
The amorphous LPO solid electrolyte and Li anode films were deposited on LCO/SRO/STO via RF magnetron sputtering and vacuum thermal evaporation, respectively. This resulted in a model thin-film battery with a Li/LPO/LCO/SRO/STO structure. After the fabrication, the thin film batteries were transferred to an Ar-filled glovebox using an air-tight transfer case. The charge–discharge characteristics of the model thin-film battery were evaluated under Ar using a potentiostat–galvanostat system (SP-300, Biologic) with constant current–constant voltage cycling. The cutoff voltage range was set to 4.2–4.7 V. The charge–discharge capacity per unit weight of LCO was calculated using the electrode area, density, and thickness obtained by XRR analysis.
To monitor the changes in the crystal structure of the LCO film in situ during charge–discharge reactions, SXRD experiments were performed at SPring-8 (Japan) using a κ-type diffractometer located at beamline BL22XU. The measurements were conducted under a low pressure (approximately below 50 Pa) continuously decompressed using a rotary pump that could not prevent lithium anode deactivation. The states of charge and discharge were controlled by charging and discharging with constant-current and constant-voltage modes using a potentiostat–galvanostat (SP-300, Biologic). A 15 keV undulator light source with a wavelength of λ = 0.82518 Å was employed as the excitation source. The measurements were conducted using a coordinate system comprising two components (H, K) parallel to the surface and a third component (L) perpendicular to the surface. This coordinate system is referred to as the reciprocal coordinate system (H, K, L). To study the surface structural changes, in-plane XRD patterns were collected at different incident angles below and above the critical angle, at which the penetration depth of the X-rays was limited to several nanometers.
Results & discussion
Fig. 1 shows the out-of-plane and in-plane XRD patterns and Raman spectrum of LCO prepared by RF magnetron sputtering on an SRO/STO(100) substrate. Diffraction peaks at 45.1° and 100.3° are observed in the patterns recorded along the out-of-plane [001] direction. These peaks were attributed to the 104 reflections of LCO with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (Fig. 1a). When measured along the [010] direction in the plane, the 01−4 and 02−8 diffraction peaks of LCO appeared at 45.1° and 100.3°, respectively (Fig. 1b). The ϕ scan of the 01−4 reflection (Fig. 1c) indicates a four-fold symmetry at intervals of 90°. In the in-plane measurements along the [110] direction, the reflection peak of LCO 1–20 was detected at 66.1° (Fig. 1d). The ϕ scan of the 1–20 reflection also suggested a four-fold symmetry at 90° intervals (Fig. 1e). The 1–20 reflection should exhibit a two-fold symmetry, because there is no equivalent plane other than the (1–20) plane itself. Therefore, the results of the ϕ scan on the 1–20 reflection indicate that the LCO(104) film is composed of domains rotated every 90° with respect to the [001] direction of the axis. According to these results, the obtained LCO thin film was oriented with LCO[104]//STO[100] in the out-of-plane direction and LCO[1−20]//STO[110] and LCO[1−20]//STO[−110] in the in-plane direction, indicating that LCO(104) was epitaxially grown along the [001] direction. The Raman spectrum of the LCO film shows only two distinct peaks at 488 and 596 cm−1, which can be attributed to the Eg (O–Co–O bending) and A1g (O–Co–O stretching) modes in LCO with a layered rock-salt structure (Fig. 1f).38 No peaks derived from impurities, such as Co3O4 (522 and 691 cm−1) and cubic LiCoO2 (doublet at ∼600 cm−1). Thus, a thin film of single-phase layered rock-salt LCO(104) was epitaxially deposited on the SRO/STO substrate by RF magnetron sputtering, without any impurities. In a previous study, LCO(104) was epitaxially deposited on STO(100) by pulsed laser deposition (PLD).39 Therefore, the epitaxial growth of LCO(104) on the STO(100) substrate is reasonable.
m (Fig. 1a). When measured along the [010] direction in the plane, the 01−4 and 02−8 diffraction peaks of LCO appeared at 45.1° and 100.3°, respectively (Fig. 1b). The ϕ scan of the 01−4 reflection (Fig. 1c) indicates a four-fold symmetry at intervals of 90°. In the in-plane measurements along the [110] direction, the reflection peak of LCO 1–20 was detected at 66.1° (Fig. 1d). The ϕ scan of the 1–20 reflection also suggested a four-fold symmetry at 90° intervals (Fig. 1e). The 1–20 reflection should exhibit a two-fold symmetry, because there is no equivalent plane other than the (1–20) plane itself. Therefore, the results of the ϕ scan on the 1–20 reflection indicate that the LCO(104) film is composed of domains rotated every 90° with respect to the [001] direction of the axis. According to these results, the obtained LCO thin film was oriented with LCO[104]//STO[100] in the out-of-plane direction and LCO[1−20]//STO[110] and LCO[1−20]//STO[−110] in the in-plane direction, indicating that LCO(104) was epitaxially grown along the [001] direction. The Raman spectrum of the LCO film shows only two distinct peaks at 488 and 596 cm−1, which can be attributed to the Eg (O–Co–O bending) and A1g (O–Co–O stretching) modes in LCO with a layered rock-salt structure (Fig. 1f).38 No peaks derived from impurities, such as Co3O4 (522 and 691 cm−1) and cubic LiCoO2 (doublet at ∼600 cm−1). Thus, a thin film of single-phase layered rock-salt LCO(104) was epitaxially deposited on the SRO/STO substrate by RF magnetron sputtering, without any impurities. In a previous study, LCO(104) was epitaxially deposited on STO(100) by pulsed laser deposition (PLD).39 Therefore, the epitaxial growth of LCO(104) on the STO(100) substrate is reasonable.
 | ||
| Fig. 1 (a–e) X-ray diffraction patterns for LiCoO2 deposited on a SrRuO3/SrTiO(100) substrate by RF magnetron sputtering: (a) 2θ/ω scan along the [001] direction of the SrTiO3(100) substrate; (b) 2θχ/φ scan along the [010] direction of the SrTiO3(100) substrate; (c) φ scan of the LiCoO2(01−4) reflection; (d) 2θχ/φ scan along the [110] direction of the SrTiO3(100) substrate; (e) φ scan of the LiCoO2(1−20) reflection. (f) Raman spectrum of the LiCoO2 film. | ||
To clarify the film structure in detail, the fabricated LCO thin film was further investigated using NR. Table 1 lists the structural parameters of the films extracted via fitting analysis. The neutron scattering length density (nSLD) of the surface layer is 1.33 × 10−6 Å−2, which is evidently lower than those of the other layers. The low density of the surface layer is due to the formation of impurity phases, such as Li2CO3 (3.49 × 10−6 Å−2), LiOH (0.06 × 10−6 Å−2), and Li2O (0.81 × 10−6 Å−2), upon exposing the sample to atmospheric conditions for NR measurements. The LCO film was fabricated with a Li-excess target material. Therefore, the film surface contained excess Li species, which were probably transformed into surface impurity phases when exposed to ambient conditions. Surface layer formation has been reported for air-exposed LiCoO2 films.26,30 The nSLD value of the LCO layer is 3.71 × 10−6 Å−2, which can be converted to a density of 4.9–5.0 g cm−3. This density is close to the theoretical mass density (dth = 5.1 g cm−3), suggesting that the obtained LCO film is of high purity. AFM measurements revealed the presence of voids among the square-shaped LCO islands grown on the SRO/STO substrate, as shown in Fig. 2. The slightly lower density of LCO compared to its theoretical density may be attributed to the presence of voids or a decrease in density caused by excess Li. The NR and AFM results also suggest that the LCO film had a relatively smooth surface.
| Layer | nSLD/10−6 Å−2 | Theoretical mass density dth/g cm−3 | Calculated mass density dcalc/g cm−3 | Thickness l/nm | Roughness σ/nm |
|---|---|---|---|---|---|
| Surface layer | 1.33 | — | — | 5.8 | 3.0 |
| LiCoO2 | 3.71 | 5.05 | 4.94 | 34.0 | 1.9 |
| SrRuO3 | 5.23 | 6.58 | 6.54 | 32.6 | 5.7 |
| SrTiO3 | 3.53 | 5.14 | 5.11 | — | 1.5 |
 | ||
| Fig. 2 AFM images of the LiCoO2 (LCO) film deposited on a SrRuO3/SrTiO3(100) substrate by RF magnetron sputtering, showing gaps between the LCO grains. Scan areas of the images are (a) 2 μm × 1 μm and (b) 500 nm × 250 nm. | ||
Fig. 3 shows the constant-current and constant-voltage charge–discharge curves and cycle properties of the Li/LPO/LCO/SRO/STO thin-film battery over the voltage range of 3.0 to 4.2 V at a constant current rate of 1 C (5.10 μA cm−2). The initial open-circuit voltage was 1.8 V. During the first charge–discharge cycle, the irreversible capacity was confirmed. When x in LixCoO2 is larger than 1, the capacity at <3.16 V appears.40 The results of NR in Table 1 and Fig. S1† suggest that the obtained LCO epitaxial film has excess Li derived from the Li-excess elemental composition (Li/Co > 1) of the target material used in RF magnetron sputtering. Therefore, the initial irreversible capacity is due to the excess Li present in the system. From the second cycle onward, the charge–discharge capacities were consistent with the theoretical capacity corresponding to LiCoO2 ↔ Li0.5CoO2 at ≤4.2 V and the coulombic efficiency remained at almost 100%. The voltage plateau of the battery was confirmed at approximately 3.9 V in the charge–discharge curves. This plateau region corresponds to Li+ (de)intercalation with the transition between two different hexagonal phases. In addition, two small peaks appeared at >4.0 V in the dQ/dV plot (Fig. S2†). These peaks are due to hexagonal → monoclinic and monoclinic → hexagonal phase transitions of LCO, respectively.41 The initial discharge capacity of the battery was 138.5 mA h g−1, and after 130 cycles, the discharge capacity decreased slightly to 137.2 mA h g−1, with the retention rate being greater than 99%. These results indicate that the charge–discharge processes proceed reversibly in the voltage range of 3.0 to 4.2 V, with full utilization of the capacity in the range of 3.0 to 4.2 V. It has been reported that the exposed area of the LCO(104) surface is inversely related to the resistance to Li-ion migration.42 Thus, the capacities corresponding to the theoretical value during cycling and high reversibility indicate the exposure of the LCO(104) surface to the LPO electrolyte at the LCO/LPO interface. These results suggest that the LCO epitaxial film fabricated by RF magnetron sputtering can be utilized as a model system for investigating the phase stability of LCO. Previously, thin-film batteries were fabricated using PLD43,44 and RF sputtering methods.45,46 However, most of these batteries did not achieve a capacity close to the theoretical capacity, and the unit of capacity was not standardized by weight. In this study, the model battery demonstrates the theoretical capacity per weight of LCO, also suggesting the suitability for investigating the degradation mechanism.
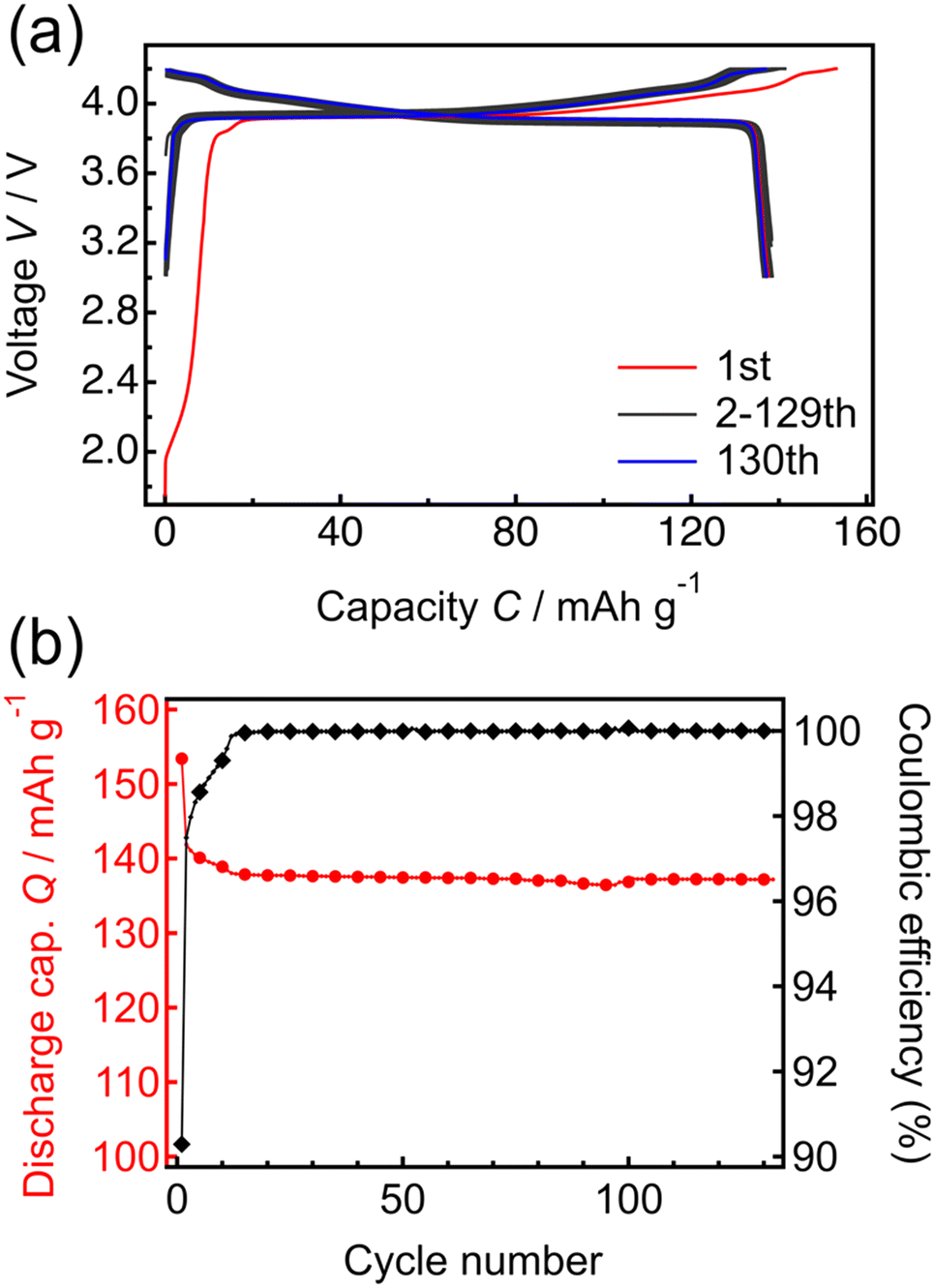 | ||
| Fig. 3 (a) Charge–discharge curves for the thin-film battery (battery structure: Li/Li3PO4/LiCoO2/SrRuO3/SrTiO3). (b) Cycle dependence of the specific discharge capacity and coulombic efficiency of the thin-film battery. | ||
To investigate the phase stability of LCO at high voltage, charge–discharge tests were continuously performed in the constant-current and constant-voltage modes by varying the cutoff voltage between 4.2 and 4.7 V (Fig. 3). Charge–discharge tests were performed at each cutoff voltage for 10 cycles. Charge–discharge tests at a cutoff voltage of 4.2 V were performed after the test at each higher cutoff voltage to compare the behavior at 4.2 V with the initial charge–discharge behavior at 4.2 V. The initial charge–discharge curves at 4.2 V (Fig. 4(a)) are almost the same as those in Fig. 3. When the cutoff voltage was increased from 4.2 to 4.5 V, the charge–discharge capacities increased with the increase in the cutoff voltage. The capacities showed high retention ratios. Moreover, the capacities measured at 4.2 V after the operation with each cutoff voltage hardly differed from the initially measured charge–discharge capacities at the same voltage (i.e., 4.2 V). After 10 cycles of charge–discharge at 4.5 V, the capacity retention rate was 99.3%. The coulombic efficiency almost remained at 100% during operation with cutoff voltages in the range of 4.2 to 4.5 V, indicating that the battery steadily operated without decomposition, elution, or other processes.
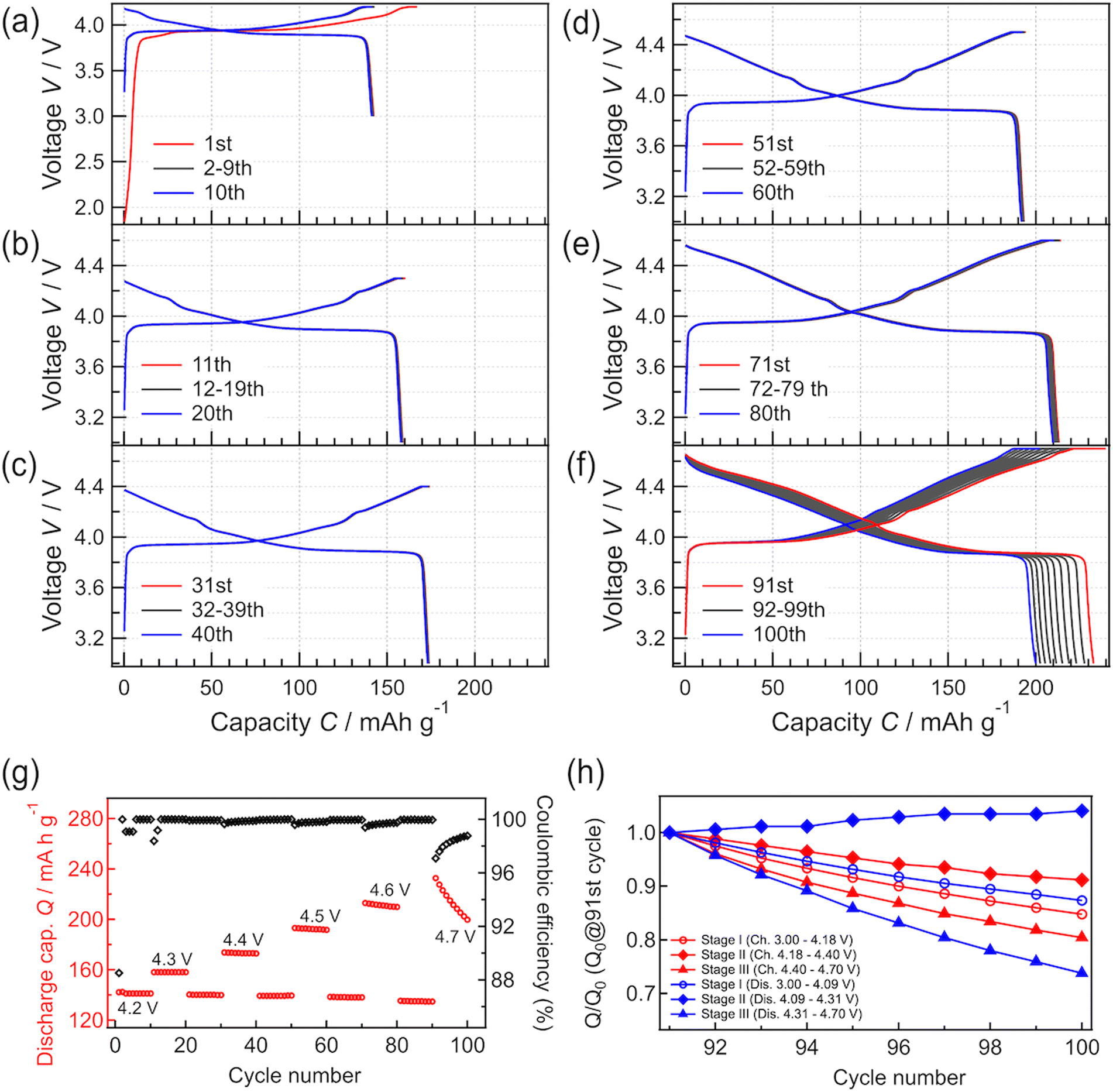 | ||
| Fig. 4 (a–f) Charge–discharge curves recorded at different cutoff voltages. (g) Discharge capacities obtained with different cutoff voltages (4.2, 4.3, 4.4, 4.5, 4.6, and 4.7 V). The discharge cutoff voltage was fixed at 3.0 V. (h) Charge–discharge capacity retention ratios at each voltage stage on 4.7 V charge/discharge. The process for fabricating (h) is summarized in Fig. S2.† | ||
When the cutoff voltage was increased to 4.6 V (Fig. 4(e)), the discharge capacity decreased slightly, resulting in a 98.5% retention ratio after 10 cycles. The capacities measured at the cutoff voltage of 4.2 V also decreased slightly after operation at 4.6 V. When the cutoff voltage was increased further to 4.7 V (Fig. 4(f)), the capacity decreased significantly, resulting in an 85.8% capacity retention ratio after 10 cycles. The coulombic efficiency increased with charge–discharge cycling. Based on the results in Fig. 4(a–f), the capacity change of the system can be divided into three stages: (I) a stable range at low voltages, including the main plateau region at 3.9 V, (II) a stable range at high voltages after the main plateau region, and (III) an unstable operation range at higher voltages. The changes in capacity observed at each stage are summarized in Fig. 4(h). The charge–discharge capacities in stages I and III showed relatively poor cycle retention compared to those in stage II. The charge–discharge reactions are limited at the end of charge and discharge, which might be due to an increase in the reaction resistance.
To investigate the degradation mechanism during the charge–discharge process at the upper cutoff voltage of 4.7 V, the changes in the crystal structure of LCO were investigated by in situ SXRD, as shown in Fig. 5. The SXRD data obtained in situ in different charge states of the battery are shown in Fig. 5(c) with open circles (no. 1–11). The battery exhibited an irreversible capacity derived from excess Li during the first charge, as shown in Fig. 3(a) and 4(a). After the first charging to 3.88 V (no. 2), which was just before the main plateau region, the 003 peak intensity increased slightly, relative to that observed in the as-fabricated battery (no. 1). This seems to be due to the electrochemical oxidation of impurities in the film battery. Owing to the lithium deintercalation at 4.2 V in the plateau region, the 003 peak shifted to lower angles (no. 3). The 003 peak shifted to higher angles at 4.6 V (no. 4) because of the phase transition from O3-type LCO with a cubic close-packed lattice to H1-3-type LCO. H1-3-type LCO has an intermediate structure consisting of O3-type LCO and O1-type LCO with a hexagonal close-packed lattice.11,47 This was confirmed from the oxidation peak observed between 4.5 and 4.6 V in the dQ/dV plot (Fig. 5(d)). After discharging to 3.0 V (no. 5), the 003 peak returned to its original position (before charging) without a significant change in the peak intensity. Moreover, the discharge capacity of 200 mA h g−1 is very close to those observed in Fig. 4(e). These results are consistent with the reversible lithium (de)intercalation of LCO under the charge–discharge with an upper cutoff voltage of 4.6 V. At the second cycle, the thin-film battery was charged to 4.7 V. Two diffraction peaks were observed at H = 0.503 and H = 0.534, which can be assigned to the 003 and 001 peaks of H1-3-type and O1-type LCO, respectively (no. 6). An oxidation peak observed between 4.6 and 4.7 V in the dQ/dV plot supported the phase transition from H1-3-type to O1-type LCO. These diffraction peaks disappeared after the subsequent discharging to 3.0 V (no. 7), and the 003 peak of lithiated O3-type LCO was observed at H = 0.480 after the first discharge (no. 5), indicating lithium intercalation in H1-3-type and O1-type LCO to generate O3-type LCO. However, the 003 peak was less intense than that during the first discharge. Moreover, the peak intensities observed after discharge gradually decreased in the subsequent cycles operated between 4.7 and 3.0 V (no. 7–11). The decrease in the peak intensity may correspond to the decrease in the discharge capacity (Fig. 5(e)). As no diffraction peaks derived from the other phases were observed in the XRD patterns, the O3-type structure was retained during cycling. Furthermore, the width at half maximum of the 003 peak hardly changed, indicating no significant change in the crystallinity of the LCO film or reaction distribution in the film. These results suggest that atomic arrangement changes, such as cation exchange (Li and Co) and oxygen defect formation in the O3-type structure, lead to a decrease in the amount of lithium showing (de)intercalation activity and/or an increase in the intercalation reaction resistance.
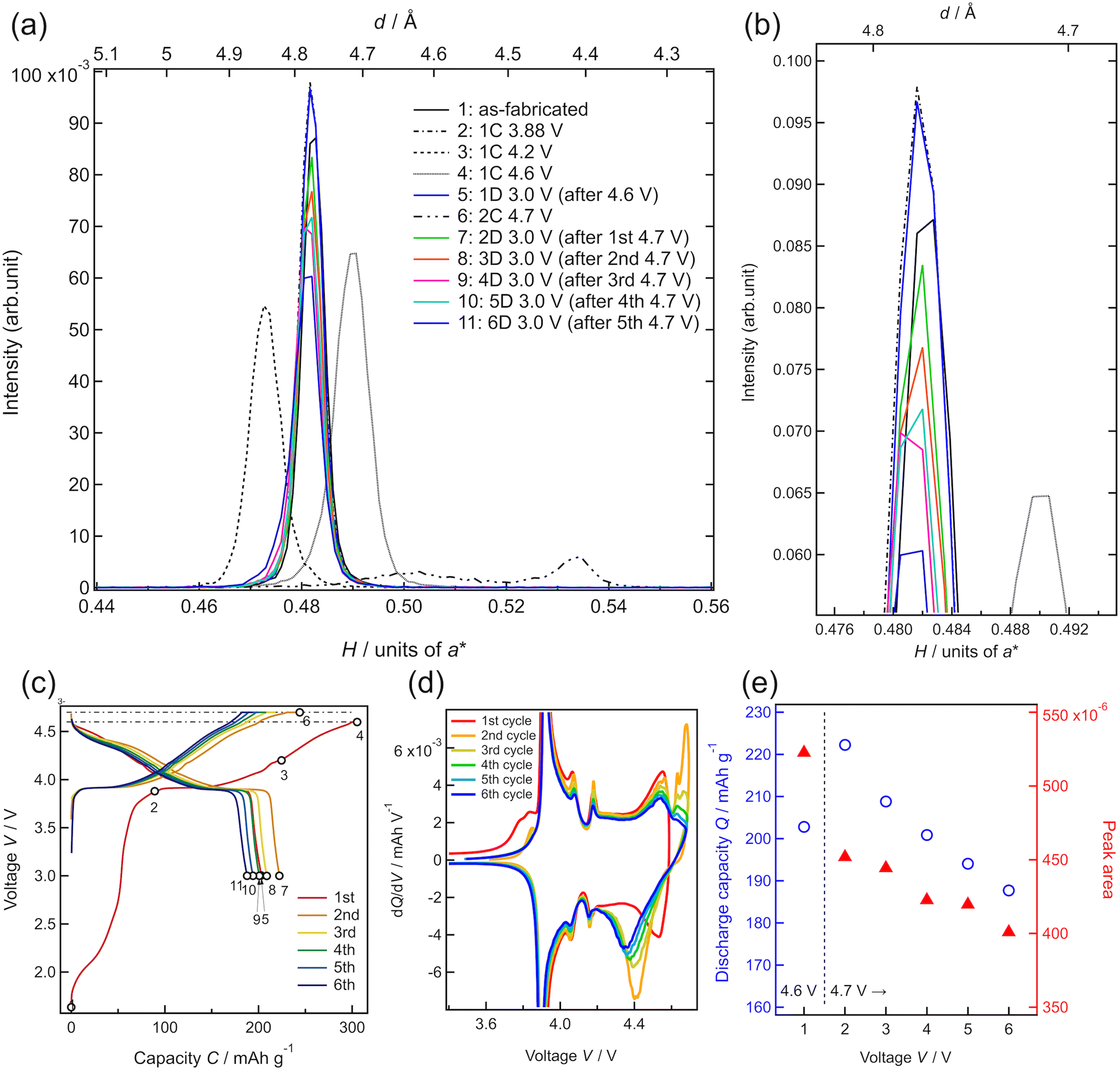 | ||
| Fig. 5 (a and b) Synchrotron X-ray diffraction patterns of a thin-film battery (Li/Li3PO4/LiCoO2/SrRuO3/SrTiO3) along the out-of-plane [111] direction of the SrTiO3(100) substrate. (c) Charge–discharge curves and (d) differential capacity (dQ/dV) plots obtained during in situ SXRD. The in situ SXRD measurement points are indicated using black open circles. (e) Discharge capacity retention ratio corresponding to charge–discharge at 4.7 V and LiCoO2(003) peak area change. | ||
To clarify whether the structural degradation proceeded at the surface and/or the whole region of the LCO film, in situ SXRD along the in-plane directions was conducted with different incident angles of the X-rays. Fig. 6 shows the in-plane SXRD patterns of the in-plane 10−8, 1−20, and 0−14 peaks of LCO collected before initial deintercalation (3.88 V, no. 2) and at 3.0 V after initial charging to 4.6 V (no. 5) and 4.7 V (no. 7). No significant changes in the peak position or intensity were observed for all diffraction peaks after charging to 4.6 V and discharging to 3.0 V, indicating the reversible structural changes at both surface and bulk regions of the LCO film. In contrast, all diffraction peaks of LCO showed a decrease in intensity at 3.0 V after the initial charging to 4.7 V from those at the initial state. These results are consistent with the irreversible structural changes observed in the out-of-plane 003 peak (Fig. 5(a)). Remarkably, there was no significant difference between the area ratios of the peaks for the surface and whole regions (Table S1†), indicating that irreversible structural changes occurred over the whole region of the 34 nm-thick LCO film.
 | ||
| Fig. 6 Synchrotron X-ray surface diffraction patterns (in-plane) of a thin-film battery (Li/Li3PO4/LiCoO2/SrRuO3/SrTiO3). (a and b) HH0 scans along the [110] direction of the SrTiO3(100) substrate; (c and d) H00 scans along the [100] direction of the SrTiO3(100) substrate. Reflections of the regions near the bulk (a and c) and interface (b and d) were obtained by adjusting the incident angle of synchrotron X-rays. | ||
In lithium-ion batteries, the structural degradation of LCO with oxygen release occurs at high voltages of above 4.2 V simultaneously with side reactions involving oxidative decomposition of species in organic liquid electrolytes,12,48 leading to an increase in the interfacial resistance.49 From our SXRD results, the phase transition between O3-type and H1-3-type LCO proceeds reversibly at the bulk and surface regions during the initial charging to 4.6 V and discharging to 3.0 V. The structural degradation of H1-3-type LCO, which originates from interfacial side reactions, may be suppressed by using a Li3PO4 solid electrolyte that is less susceptible to oxidation.50 This interpretation is consistent with the experimental findings that reversible lithium (de)intercalation of LCO can be realized at high voltages by modifying the interface between LCO and organic electrolytes with a stable oxide.51–56 In contrast, the LCO film showed an irreversible structural change in the whole region under operation with an upper cutoff voltage of 4.7 V. Although the phase transition between the H1-3 and O1-type LCO is accompanied by a large volumetric change, physical or mechanical degradation, such as particle cracking, is negligible for film electrodes with an extremely small thickness. Thus, the phase transition between the O3-type and O1-type LCO with the intermediate H1-3-type LCO is poorly reversible, even if the LCO/electrolyte interface remains stable. The phase transition between the O1 and O3 structures requires the gliding of the CoO2 layers, accompanied by a large change in the interlayer distances. This may cause irreversible structural changes, such as the exchange of Li and Co ions between the original octahedral positions, movement of cation species to tetrahedral positions, and oxygen release from the lattice, although it is difficult to directly observe the detailed atomic arrangements in thin-film electrodes. Recently, we reported that O3-type Li2MnO3 film cathodes irreversibly change to an O1-type structure during the initial charging to 4.8 V, and O1-type Li2MnO3 shows a reversible structural change without reverting to the O3-type structure during lithium (de)intercalation.32 This suggests that the O1 structure can be stabilized by modifying the atomic species and their arrangements. Our experimental results demonstrate that the reversible capacity of the LCO cathode can be extended to approximately 200 mA h g−1 by utilizing a solid–solid interface with excellent electrochemical stability. Further improvement in the intercalation capacity of layered rock-salt cathodes requires the development of a bulk material with high compatibility for O1-type structure-mediated phase transitions.
Conclusions
A thin-film battery with a Li/LPO/LCO/SRO/STO structure was fabricated with the epitaxial LCO(104) film. The thin-film batteries delivered steady lithium deintercalation and intercalation of LCO at high voltages ranging up to approximately 4.6 V without severe degradation. In contrast, the charge–discharge capacities decreased when charged at 4.7 V. During lithium deintercalation from 4.6 V to 4.7 V, the crystal structure of LCO changed from the H1-3 type to the O1 type with a large decrease in the interlayer distances, resulting in an irreversible structural change to the original O3 phase in the subsequent discharge process. The irreversible structural change occurred in the whole region of the 34 nm-thick LCO film. The structural degradation of LCO was not initiated by side reactions with the LPO electrolyte at the interface but was mainly associated with the poor reversibility of structural changes among the O3, H1-3, and O1 phases. All-solid-state batteries using oxide electrolytes with excellent electrochemical stability can be utilized up to the maximum reversible capacity, constrained by the inherent structural stability of the cathode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research on Innovative Areas [Grant No. 19H05793]. Neutron diffraction experiments were performed as projects approved by the Japan Proton Accelerator Research Complex (2019B0358). This work was partially supported by the SOLiD-EV project (JPNP18003) commissioned by the New Energy and Industrial Technology Development Organization (Japan). Synchrotron X-ray experiments were performed under the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2018A3635, 2018B3635, and 2022B3741). We would like to thank Mr. Subin Song for his kind assistance in Raman spectroscopy measurements.Notes and references
- B. E. Worku, S. Zheng and B. Wang, Int. J. Energy Res., 2022, 46, 14609–14626 CrossRef CAS.
- M. Gutsch and J. Leker, J. Energy Storage, 2022, 52(Part C), 105030 CrossRef.
- S. M. Abu, M. A. Hannan, M. S. Hossain Lipu, M. Mannan, P. J. Ker, M. J. Hossain and T. M. I. Mahlia, J. Cleaner Prod., 2023, 394, 136246 CrossRef CAS.
- K. Takada, N. Aotani, K. Iwamoto and S. Kondo, Solid State Ionics, 1995, 79, 284–287 CrossRef CAS.
- L. P. L. M. Rabou and A. Roskam, J. Power Sources, 1995, 54, 316–318 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CrossRef CAS.
- D. Di Lecce, R. Brescia, A. Scarpellini, M. Prato and J. Hassoun, ChemSusChem, 2016, 9, 223–230 CrossRef CAS PubMed.
- R. Wang, X. Li, L. Liu, J. Lee, D. H. Seo, S. H. Bo, A. Urban and G. Ceder, Electrochem. Commun., 2015, 60, 70–73 CrossRef CAS.
- K. Hikima, S. Taminato, Y. Hinuma, K. Shimizu, K. Suzuki, M. Hirayama, S. Yasuno, K. Tamura and R. Kanno, Batteries Supercaps, 2021, 4, 493–503 CrossRef CAS.
- H. Kim, S. Yoon, S. Koo, J. Lee, J. Kim, M. Cho and D. Kim, ACS Appl. Mater. Interfaces, 2022, 14, 9057–9065 CrossRef CAS PubMed.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114–1123 CrossRef CAS.
- S. P. Kühn, K. Edström, M. Winter and I. Cekic-Laskovic, Adv. Mater. Interfaces, 2022, 9, 2102078 CrossRef.
- H. Tan, S. Takeuchi, K. K. Bharathi, I. Takeuchi and L. A. Bendersky, ACS Appl. Mater. Interfaces, 2016, 8, 6727–6735 CrossRef CAS PubMed.
- W. Samarakoon, J. Hu, M. Song, M. Bowden, N. Lahiri, J. Liu, L. Wang, T. Droubay, K. Koirala, H. Zhou, Z. Feng, J. Tao and Y. Du, J. Phys. Chem. C, 2022, 126, 15882–15890 CrossRef CAS.
- C. Qin, Y. Jiang, P. Yan and M. Sui, J. Power Sources, 2020, 460, 228126 CrossRef CAS.
- Y. Yan, S. Weng, A. Fu, H. Zhang, J. Chen, Q. Zheng, B. Zhang, S. Zhou, H. Yan, C. W. Wang, Y. Tang, H. Luo, B. W. Mao, J. Zheng, X. Wang, Y. Qiao, Y. Yang and S. G. Sun, ACS Energy Lett., 2022, 7, 2677–2684 CrossRef CAS.
- A. Fu, J. Lin, Z. Zhang, C. Xu, Y. Zou, C. Liu, P. Yan, D. Y. Wu, Y. Yang and J. Zheng, ACS Energy Lett., 2022, 7, 1364–1373 CrossRef CAS.
- Z. Zhuang, J. Wang, K. Jia, G. Ji, J. Ma, Z. Han, Z. Piao, R. Gao, H. Ji, X. Zhong, G. Zhou and H. M. Cheng, Adv. Mater., 2023, 35, 1–14 Search PubMed.
- W. Kong, D. Zhou, D. Ning, W. Yang, D. Wong, J. Zhang, Q. Li, J. Yang, C. Schulz and X. Liu, J. Electrochem. Soc., 2021, 168, 030528 CrossRef CAS.
- A. Kim, S. Woo, M. Kang, H. Park and B. Kang, Front. Chem., 2020, 8, 1–13 CrossRef PubMed.
- M. Hallot, B. Caja-Munoz, C. Leviel, O. I. Lebedev, R. Retoux, J. Avila, P. Roussel, M. C. Asensio and C. Lethien, ACS Appl. Mater. Interfaces, 2021, 13, 15761–15773 CrossRef CAS PubMed.
- X. Cai, W. Bao, L. Zhao, Y. Zuo, H. Zhao, L. Su, Y. Zhang, H. Zhang and J. Xie, Adv. Sustainable Syst., 2023, 2300392, 4–9 Search PubMed.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- S. Taminato, M. Hirayama, K. Suzuki, N. L. Yamada, M. Yonemura, J. Y. Son and R. Kanno, Chem. Commun., 2015, 51, 1673–1676 RSC.
- M. Hirayama, H. Ido, K. Kim and W. Cho, J. Am. Chem. Soc., 2010, 132, 15268–15276 CrossRef CAS PubMed.
- H. Zhou, J. Izumi, S. Asano, K. Ito, K. Watanabe, K. Suzuki, F. Nemoto, N. L. Yamada, K. Aso, Y. Oshima, R. Kanno and M. Hirayama, Adv. Energy Mater., 2023, 2302402, 1–12 Search PubMed.
- J. Nakayama, H. Zhou, J. Izumi, K. Watanabe, K. Suzuki, F. Nemoto, N. L. Yamada, R. Kanno and M. Hirayama, Adv. Mater. Interfaces, 2024, 11, 1–9 Search PubMed.
- S. Taminato, M. Hirayama, K. Suzuki, K. Kim, Y. Zheng, K. Tamura, J. Mizuki and R. Kanno, J. Mater. Chem. A, 2014, 2, 17875–17882 RSC.
- S. Taminato, M. Hirayama, K. Suzuki, K. Kim, K. Tamura and R. Kanno, J. Phys. Chem. C, 2018, 122, 16607–16612 CrossRef CAS.
- M. Hirayama, N. Sonoyama, T. Abe, M. Minoura, M. Ito, D. Mori, A. Yamada, R. Kanno, T. Terashima, M. Takano, K. Tamura and J. Mizuki, J. Power Sources, 2007, 168, 493–500 CrossRef CAS.
- K. Hikima, K. Shimizu, H. Kiuchi, Y. Hinuma, K. Suzuki, M. Hirayama, E. Matsubara and R. Kanno, Commun. Chem., 2022, 5, 1–8 CrossRef PubMed.
- K. Hikima, Y. Hinuma, K. Shimizu, K. Suzuki, S. Taminato, M. Hirayama, T. Masuda, K. Tamura and R. Kanno, ACS Appl. Mater. Interfaces, 2021, 13, 7650–7663 CrossRef CAS PubMed.
- K. Hikima, K. Shimizu, H. Kiuchi, Y. Hinuma, K. Suzuki, M. Hirayama, I. Matsubara and R. Kanno, J. Am. Chem. Soc., 2022, 144, 236–247 CrossRef CAS PubMed.
- N. L. Yamada, N. Torikai, K. Mitamura, H. Sagehashi, S. Sato, H. Seto, T. Sugita, S. Goko, M. Furusaka, T. Oda, M. Hino, T. Fujiwara, H. Takahashi and A. Takahara, Eur. Phys. J. Plus, 2011, 126, 1–13 CrossRef.
- K. Mitamura, N. L. Yamada, H. Sagehashi, N. Torikai, H. Arita, M. Terada, M. Kobayashi, S. Sato, H. Seto, S. Goko, M. Furusaka, T. Oda, M. Hino, H. Jinnai and A. Takahara, Polym. J., 2013, 45, 100–108 CrossRef CAS.
- M. Yonemura, M. Hirayama, K. Suzuki, R. Kanno, N. Torikai and N. L. Yamada, J. Phys.: Conf. Ser., 2014, 502, 012054 CrossRef CAS.
- A. Nelson, J. Appl. Crystallogr., 2006, 39, 273–276 CrossRef CAS.
- J. I. Hata, M. Hirayama, K. Suzuki, N. Dupré, D. Guyomard and R. Kanno, Batteries Supercaps, 2019, 2, 454–463 CrossRef CAS.
- Z. Li, S. Yasui, S. Takeuchi, A. Creuziger, S. Maruyama, A. A. Herzing, I. Takeuchi and L. A. Bendersky, Thin Solid Films, 2016, 612, 472–482 CrossRef CAS PubMed.
- J. M. Rosolen, P. Ballirano, M. Berrettoni, F. Decker and M. Gregorkiewitz, Ionics, 1997, 3, 345–355 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS.
- S. Takeuchi, H. Tan, K. K. Bharathi, G. R. Stafford, J. Shin, S. Yasui, I. Takeuchi and L. A. Bendersky, ACS Appl. Mater. Interfaces, 2015, 7, 7901–7911 CrossRef CAS PubMed.
- K. Kawashima, T. Ohnishi and K. Takada, ACS Appl. Energy Mater., 2020, 3, 11803–11810 CrossRef CAS.
- S. Shiraki, T. Shirasawa, T. Suzuki, H. Kawasoko, R. Shimizu and T. Hitosugi, ACS Appl. Mater. Interfaces, 2018, 10, 41732–41737 CrossRef CAS PubMed.
- T. Ohnishi and K. Takada, ACS Omega, 2022, 7, 21199–21206 CrossRef CAS PubMed.
- S. Kobayashi, K. Nishio, M. Wilde, K. Fukutani, R. Shimizu and T. Hitosugi, J. Phys. Chem. C, 2023, 127, 4684–4688 CrossRef CAS.
- Z. Chen, Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1604 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- B. Wang, J. B. Bates, F. X. Hart, B. C. Sales, R. A. Zuhr and J. D. Robertson, J. Electrochem. Soc., 1996, 143, 3203–3213 CrossRef CAS.
- H. Kawasoko, S. Shiraki, T. Suzuki, R. Shimizu and T. Hitosugi, ACS Appl. Mater. Interfaces, 2018, 10, 27498–27502 CrossRef CAS PubMed.
- S. Verdier, L. El Ouatani, R. Dedryvère, F. Bonhomme, P. Biensan and D. Gonbeau, J. Electrochem. Soc., 2007, 154, 2007 CrossRef.
- J. Cho, Y. J. Kim and B. Park, Chem. Mater., 2000, 12, 3788–3791 CrossRef CAS.
- D. Takamatsu, S. Mori, Y. Orikasa, T. Nakatsutsumi, Y. Koyama, H. Tanida, H. Arai, Y. Uchimoto and Z. Ogumi, J. Electrochem. Soc., 2013, 160, A3054–A3060 CrossRef CAS.
- Z. Chen and J. R. Dahn, Electrochem. Solid-State Lett., 2002, 5, 1503202 Search PubMed.
- A. Yano, K. Hikima, J. Hata, K. Suzuki, M. Hirayama and R. Kanno, J. Electrochem. Soc., 2018, 165, A3221–A3229 CrossRef CAS.
- S. Taminato, M. Hirayama, K. Suzuki, K. Tamura, T. Minato, H. Arai, Y. Uchimoto, Z. Ogumi and R. Kanno, J. Power Sources, 2016, 307, 599–603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00251a |
| This journal is © The Royal Society of Chemistry 2024 |