
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-line-of-sight synthesis and characterization of a conformal submicron-thick cationic polymer deposited on 2D and 3D substrates†
Hunter O.
Ford
 a,
Brian L.
Chaloux
b,
Battogtokh
Jugdersuren
c,
Xiao
Liu
d,
Christopher A.
Klug
b,
Joel B.
Miller
e,
Xiaobing
Zuo
f,
Michael W.
Swift
g,
Michelle D.
Johannes
g,
Jeffrey W.
Long
b,
Debra R.
Rolison
*b and
Megan B.
Sassin
*b
a,
Brian L.
Chaloux
b,
Battogtokh
Jugdersuren
c,
Xiao
Liu
d,
Christopher A.
Klug
b,
Joel B.
Miller
e,
Xiaobing
Zuo
f,
Michael W.
Swift
g,
Michelle D.
Johannes
g,
Jeffrey W.
Long
b,
Debra R.
Rolison
*b and
Megan B.
Sassin
*b
aNRL–NRC Postdoctoral Associate in the Chemistry Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA
bChemistry Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA. E-mail: debra.rolison@nrl.navy.mil; megan.sassin@nrl.navy.mil
cJacobs Engineering Group, Hanover, MD 21076, USA
dAcoustics Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA
eEmeritus, Chemistry Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA
fX-ray Science Division, Argonne National Laboratory, Lemont, IL 60439, USA
gMaterials Science & Technology Division, U.S. Naval Research Laboratory, Washington, DC 20375, USA
First published on 19th February 2024
Abstract
The non-line-of-sight nature of initiated chemical vapor deposition (iCVD) makes it an amenable technique for depositing thin, conformal polymer films on three-dimensional (3D) high surface-area substrates. Adding to the versatility of iCVD, we process the deposited film in situ to impart additional functionality. Herein we describe fabricating submicron-thick positively charged polymer films with labile anions, a class of materials that brings ion-transport regulation to water desalination, gas separations, and energy-storage systems. Coatings of iCVD-deposited poly(4-dimethylaminomethylstyrene) (pDMAMS) on planar and 3D substrates are reacted with an alkylating/cross-linking reagent to yield quaternary ammonium-containing pDMAMS+. By optimizing deposition conditions, we demonstrate conformal coatings of pDMAMS and pDMAMS+ spanning a thickness range of 30–1000+ nm. Chemical and mechanical characterization confirms the successful quaternization of the iCVD-generated films, subsequent ion exchange of various counterions (Br−, HCO3−, and OH−), and enhanced modulus of the crosslinked pDMAMS+ polymer. The use of grazing-incidence X-ray scattering reveals the films are noncrystalline and contain ion clusters with anion-dependent geometry. By understanding the impact of deposition conditions and functionalization on subsequent physicochemical-mechanical properties, we lay the groundwork for using iCVD-derived pDMAMS+ films in targeted applications.
Introduction
The technique known as initiated chemical vapor deposition (iCVD) enables coating complex substrates with conformal, submicron-thick polymer layers of a wide variety of chemistry and function. Demonstrated applications in the literature include iCVD-generated coatings for biocidal medical device treatment, hydrophobic surface modifications, targeted-release drug delivery, and energy-storage applications.1–9Initiated CVD relies on free-radical polymerization, albeit one that is mediated through adsorption of monomer and activated initiator to the substrate surface.10 In a typical iCVD process, monomer and initiator are vaporized and brought into the deposition chamber under controlled flow and pressure. Within the deposition chamber, hot filaments decompose the initiator molecules in the gas phase into active radicals. Monomer and free-radical molecules adsorb to the cooler substrate surface where polymerization begins. The coating thickness and the degree of polymerization depend on the reactant concentrations in the vapor phase and the temperature of the substrate.10,11 This design enables iCVD generation of polymer films from any monomer chemistry that can be vaporized and free-radical initiated.
Initiated CVD is a non-line-of-sight technique because polymer film growth occurs by adsorption of monomer and radical to a substrate surface from the vapor phase. Any surface in contact with the vapor phase can be coated with polymer, which contrasts with line-of-sight techniques such as magnetron sputtering that require a direct path between the source and substrate. The non-line-of-sight nature of iCVD makes this technique particularly well suited to deposit thin polymer films within three-dimensional (3D) structures that are otherwise difficult to coat homogeneously and conformally. The ever-increasing complexity of 3D architected substrates, whether they are for medical devices, electrochemical electrodes and components, or sensors, warrants further development of new iCVD-derived polymer chemistries.
Ionomeric polymers, specifically positively charged ionomers, have applications in ion-transport regulation in desalination, gas separation, and fuel cell and battery systems.12–15 One route to an iCVD-derived positively charged polymer involves copolymerization of an amine-functionalized acrylate monomer with vinylbenzyl chloride, where both monomers polymerize and undergo amine quaternization on the substrate surface simultaneously.4,9 Here we report iCVD synthesis of an ionomeric positively charged polymer coating based on an isomerically pure polymer of poly(4-dimethylaminomethylstyrene) (pDMAMS). Post deposition, vapor-phase alkylation converts the iCVD-generated pDMAMS film to a positively charged anion-labile form (pDMAMS+). This route decouples the deposition and quaternization reactions, leading to enhanced process design flexibility. We characterize the submicron-thick pDMAMS and alkylated pDMAMS+ polymers in terms of morphology, chemical composition, and mechanical properties.
The counterbalancing anions we report are relevant for energy storage, but the ease of anion exchange without damage to the film conformality and integrity allows this approach to be tailored to any application requiring a submicron-thick polymer with mobile anions. Coupling the post-deposition introduction of fixed ionic charge and ease of ion exchange to our extensive characterization establishes the foundation for design and delivery of multifunctional polymers with wide-ranging impact.
Results and discussion
Generating ionomeric films
We adapt iCVD protocols previously reported by Gleason and co-workers for generating poly(dimethylaminomethylstyrene)2,10,11 but synthesize isomerically pure 4-DMAMS as a monomer rather than using the commercially available ortho–para mixture. The NMR spectrum of 4-DMAMS monomer (Fig. S1†) verifies molecular identity and purity. The isomerically pure polymer (pDMAMS) allows for more direct experimental and computational comparison between iCVD-derived and bulk-synthesized polymers.16Fig. 1 illustrates the deposition and functionalization processing steps. Films are deposited, annealed above the glass-transition temperature (Tg: 82 °C; Fig. S2†), and subsequently reacted with a vapor-phase alkylating agent (1-chloro-3-bromopropane) to transform the amine pendent group to quaternary ammonium functionality (pDMAMS+). The labile, charge-balancing counterions incorporated during alkylation (Cl−/Br−) are readily exchanged for other anions of interest (Br−, HCO3−, and OH−); the anion-exchanged forms are designated as pDMAMS+(Br−), pDMAMS+(HCO3−), and pDMAMS+(OH−). We characterize the morphology, structure, and composition of the pDMAMS-based films as a function of iCVD deposition conditions, processing steps, and incorporated anion. | ||
| Fig. 1 Synthetic and processing protocol to generate anion-conducting pDMAMS+ polymer via iCVD polymerization of 4-dimethylaminomethylstyrene (DMAMS) using tert-butyl peroxide (TBPO) initiator. | ||
Evaluation of deposition conditions on pDMAMS film conformality
We find that substrate temperature impacts the uniformity and smoothness of pDMAMS coatings. Films deposited at a substrate temperature of 40 °C are optically textured (Fig. 2A), indicating islanding effects during polymer deposition. Raising substrate temperature to 50 or 60 °C generates more uniform films (Fig. 2B and C). Nanoscale morphology measured using atomic force microscopy quantitatively tracks these trends with a root-mean-square (RMS) roughness of 8.9 nm at 40 °C (Fig. 2D) but comparably low RMS values at 50 °C and 60 °C (0.58 and 0.38 nm, respectively; Fig. 2E and F). We set the substrate temperature at 60 °C for all iCVD-generated films in this report. The smooth morphology (RMS roughness ≤ 1 nm) is retained after annealing, quaternization, and ion exchange (Fig. 3). | ||
| Fig. 2 Optical images (top row) and atomic force micrographs (bottom row) of pDMAMS films deposited at different iCVD substrate temperatures on Au-coated glass microscope slides (A and D) 40 °C; (B and E) 50 °C; (C and F) 60 °C. | ||
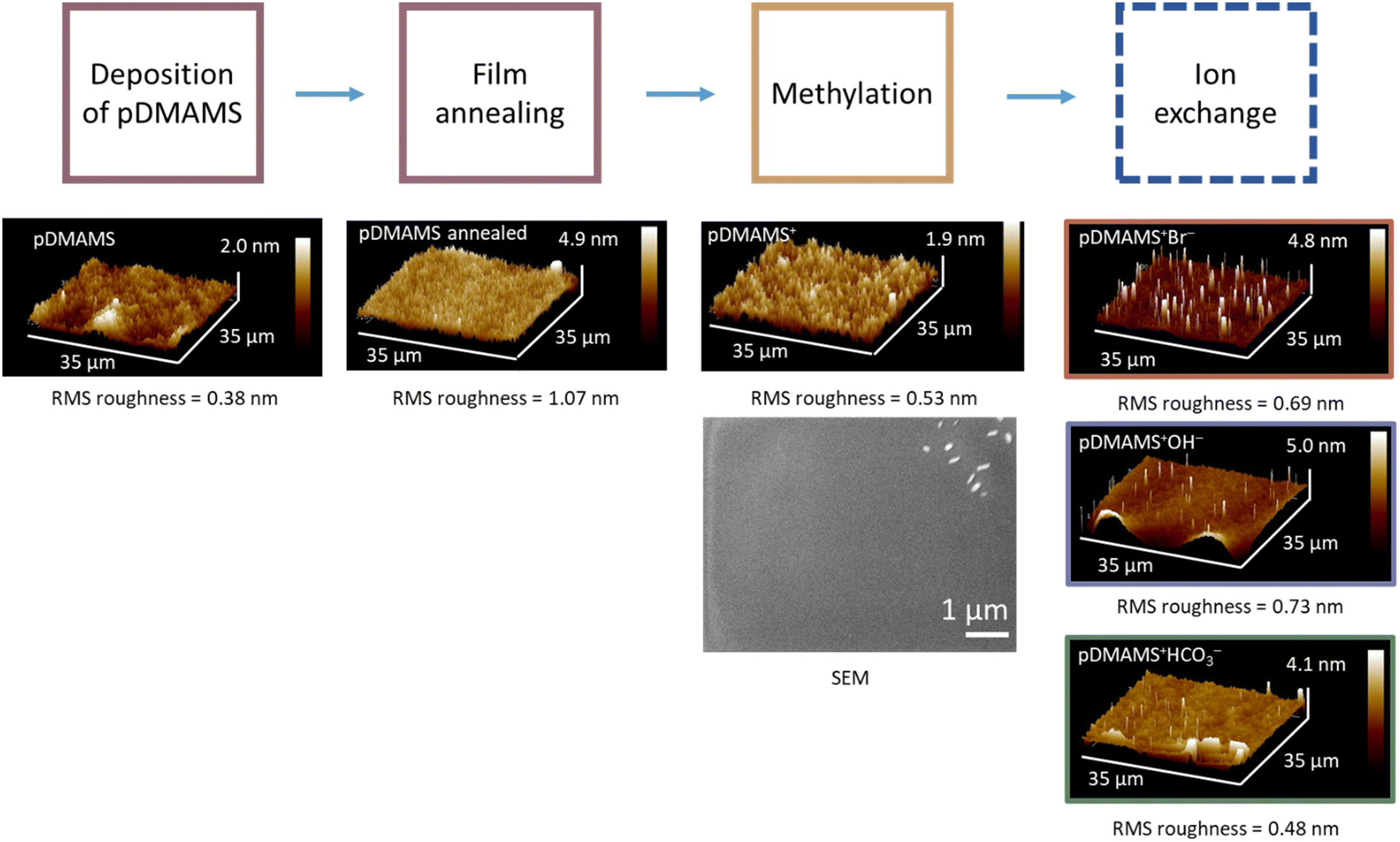 | ||
| Fig. 3 Atomic force and scanning electron microscopic analyses of pDMAMS films as a function of processing step. The scanning electron micrograph includes contamination particles to show image is in focus. | ||
Scanning electron microscopy (SEM) confirms the conformality of pDMAMS deposited on 2D (Au-coated glass) and 3D (zinc metal sponge) substrates (Fig. 4). The Zn metal sponge is selected as a model 3D substrate owing to its interconnected pore network and relevance in alkaline batteries where anions are the active ionic charge carrier. On 2D Au-coated glass substrates (Fig. 4A), the SEM-derived thickness (358 nm) is comparable to the profilometry-measured thickness (359 ± 10 nm) for the given deposition time (Fig. 5). Low-magnification SEM imaging of a cross-sectioned millimeter-thick Zn sponge reveals a uniform coating of pDMAMS within the interior surfaces (Fig. 4B). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping corroborates the efficacy of iCVD for non-line-of-sight coating in a complex 3D morphology, showing co-location of the C signal from the pDMAMS with the Zn and O maps. At higher magnification, the EDS maps of a cleaved Zn particle confirm that carbon from the polymer is preferentially located at the edges of the cleaved Zn particle (Fig. 4C).
 | ||
| Fig. 4 Scanning electron micrographs of cross-sections of iCVD-derived pDMAMS coated on (A) planar Au-coated glass substrate; (B) 3D Zn sponge electrode; and (C) cleaved Zn particle within 3D Zn sponge. | ||
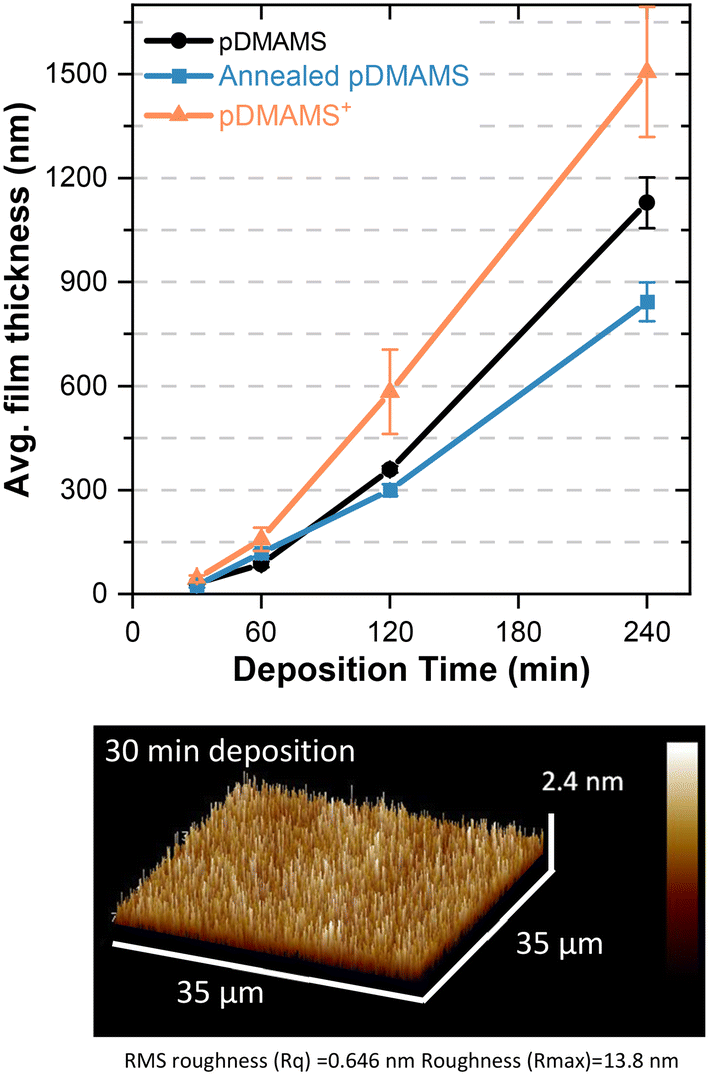 | ||
| Fig. 5 Top: Film thickness as a function of deposition time and processing step. Films are deposited on Au-coated glass slides. Bottom: Atomic force micrograph of 31 nm thick pDMAMS film obtained after 30 min of iCVD. | ||
Film thickness vs. time of deposition
An advantage of iCVD is facile control of the thickness of the deposited film; deposition time determines the thickness of the resultant polymer. Control of film thickness is important, such as in battery systems where the thinnest coating possible that can withstand the physico-electrochemical processes at the electrodes improves energy density by decreasing the mass and volume of inactive material. The growth of pDMAMS films is nonlinear (Fig. 5), with deposition rates slow in the beginning and faster at later times. This result is consistent with previous reports on pDMAMS thickness growth via iCVD.2 Even at the shortest time, 30 min, corresponding to a thickness of 31 nm, the film morphology remains conformal and pinhole free according to AFM. At the longest deposition time (4 h), the film thickness exceeds 1000 nm.Annealing and alkylation impact the film thickness. Generally, thickness decreases after annealing, which may result from a densification of the film stemming from cross-linking and/or the removal of small molecules (e.g., absorbed water, volatile unreacted organics). After alkylation, which introduces charge into the film, film thickness increases from introduction of additional mass and cross-linking of polymer chains, the latter of which prevents relaxation of the polymer into higher density chain-packing conformations. Charge overlap minimization/electrostatic chain repulsion between quaternized backbone pendant nitrogen atoms may also result in less dense chain packing.17
The standard deposition time for the majority of experiments in this study is 120 min, corresponding to an as-deposited film thickness of 359 ± 10 nm, an annealed film of 300 ± 17 nm, and an alkylated film of 570 ± 106 nm. The thickness of the ion-exchanged forms pDMAMS+(Br−) and pDMAMS+(HCO3−) are 580 ± 59 nm and 502 ± 83 nm, respectively, indicating the ion-exchange process does not markedly alter post-alkylation film thickness.
Physicochemical characterization of iCVD-derived pDMAMS and derivatives
For a primary assessment of chemical structure as a function of processing step, we analyze iCVD-derived pDMAMS films using solid-state 13C nuclear magnetic resonance spectroscopy (13C NMR) and attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR). To obtain sufficient 13C NMR signal for submicron-thick films of pDMAMS, we iCVD-coat conformal pDMAMS on another 3D substrate, high surface-area silica fiber paper (Fig. S3†). The 13C NMR and ATR-FTIR spectra of iCVD-derived pDMAMS are nearly identical to those of solution-polymerized bulk pDMAMS (Fig. 6A and B).10 Thus, solution-polymerized pDMAMS can serve as a surrogate for iCVD-derived pDMAMS for rapid screening of general materials properties18 or when large quantities of material are required. For reference, the estimated diffusive molecular weight (MD) of solution-polymerized bulk pDMAMS is 350 kDa (Fig. S4†). Additional characterization of bulk pDMAMS (1H and 13C NMR, ATR-FTIR) are found in Fig. S5 and S6.†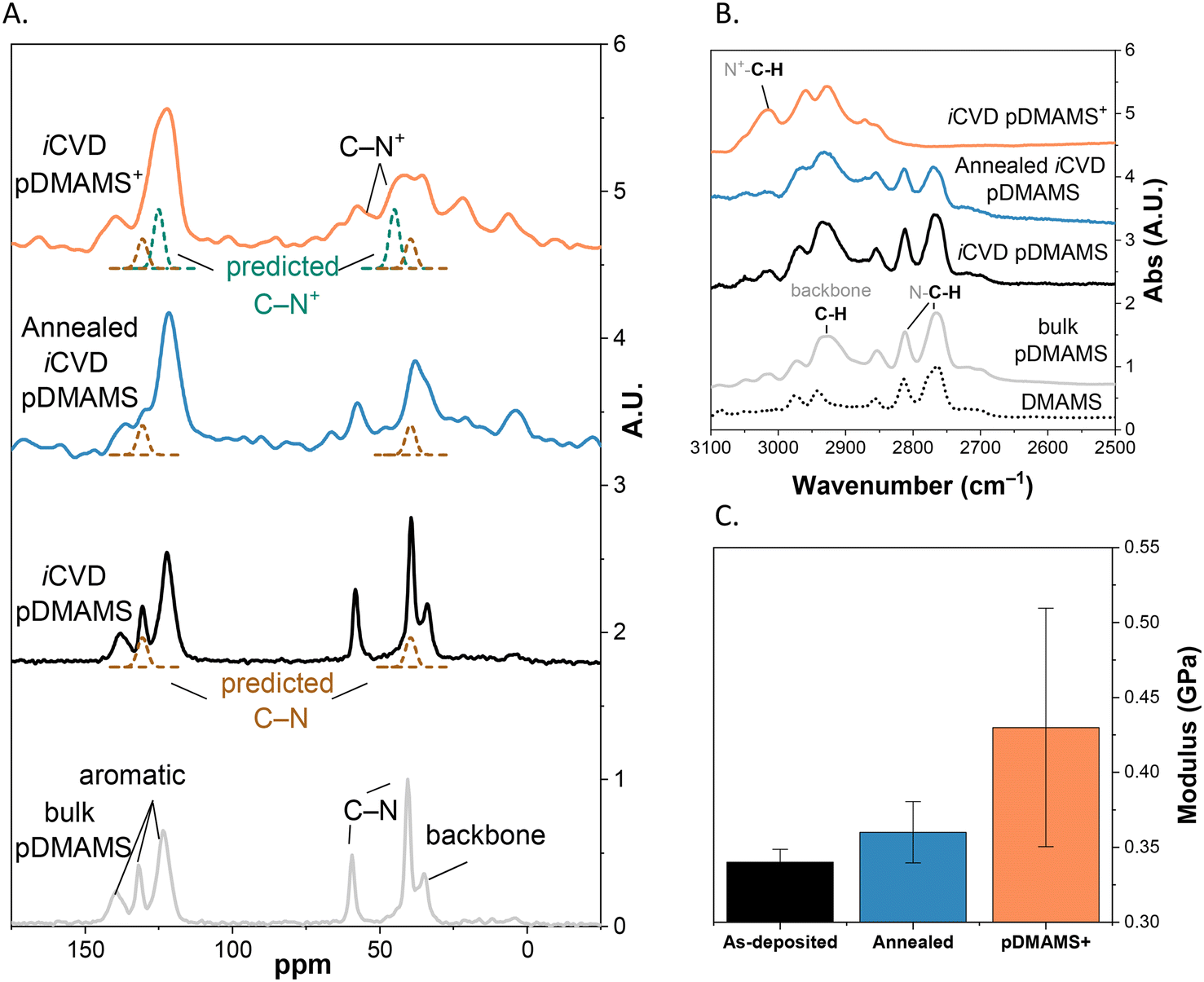 | ||
| Fig. 6 Chemical and mechanical characterization of iCVD-generated and solution-polymerized pDMAMS as a function of processing step to generate pDMAMS+ (A) 13C solid-state NMR; (B) ATR-FTIR (peaks were assigned by referencing the National Institute of Standards and Technology (NIST) spectral database for polystyrene, trimethylamine, and tetramethylammonium chloride); and (C) shear moduli. | ||
After initial deposition, iCVD-derived pDMAMS films are thermally annealed at 100 °C under vacuum. Solid-state 13C NMR shows that peaks generally broaden after annealing, indicating a greater variety in local physical and chemical environments (Fig. 6A). The peaks associated with amine-coupled C–H modes in ATR-FTIR (2770, 2810 cm−1) also diminish, which we attribute to cross-linking through the pendant nitrogen functionality (Fig. 6B), coincident with a decrease in film thickness (Fig. 5). Annealing-induced cross-linking increases the shear modulus of the pDMAMS film from 0.34 to 0.36 GPa (Fig. 6C). For context, the shear modulus of amorphous polystyrene is ∼1.2 GPa.19 Our density functional theory (DFT) calculations further support this interpretation of cross-linked densification after annealing, finding that linking two DMAMS units at their amine centers (and liberating trimethylamine) is energetically favorable by 0.17 eV, an energy barrier surmountable at the annealing temperature (Fig. S7†).
Alkylation of pDMAMS with 1-chloro-3-bromopropane to generate pDMAMS+ leads to the expected peak shift in the 13C NMR spectra associated with converting C–N to C–N+ bonds (Fig. 6A) and an increase in ATR-FTIR intensity at 3020 cm−1, which occurs in conjunction with disappearance of the amine-coupled C–H modes at 2770 cm−1 and 2810 cm−1 (Fig. 6B). The additional cross-links introduced upon alkylation further increase the shear modulus by 26% (Fig. 6C). These mechanical changes in the post-alkylated polymer correlate to an increase in the local structural order compared to native pDMAMS, indicated by a decrease in internal friction as a function of temperature (Fig. S8A†). We discuss polymer structural arrangement measured using X-ray scattering in a later section.
X-ray photoelectron spectroscopy (XPS) further confirms the conversion of pDMAMS to pDMAMS+ and the successful exchange of anions within the film. These chemical state data also allow us to estimate the film cross-linking density and ion-exchange capacity (IEC). For pDMAMS, the C1s region is dominated by a primary peak at 284.5 eV (typical for alkyl and related carbon speciation), while the N1s region reveals a pair of peaks at 399 eV and 400 eV that are attributed to C–N amine functionality (Fig. 7A and B).20 After alkylation to generate pDMAMS+, a secondary peak appears in both the C 1s and N 1s spectra at 286 eV and 402 eV, respectively. The appearance of these new peaks, concomitant with a decrease in the intensity of the C–N tertiary amine peak, show successful conversion of ∼65% of the pendant amines to quaternary C–N+ functionality.21 This degree of alkylation translates to an ion-exchange capacity (IEC) of ∼2.7 meq g−1 for pDMAMS+ (see eqn (S1) in ESI†), a moderately high value with respect to IEC values reported in the literature.22
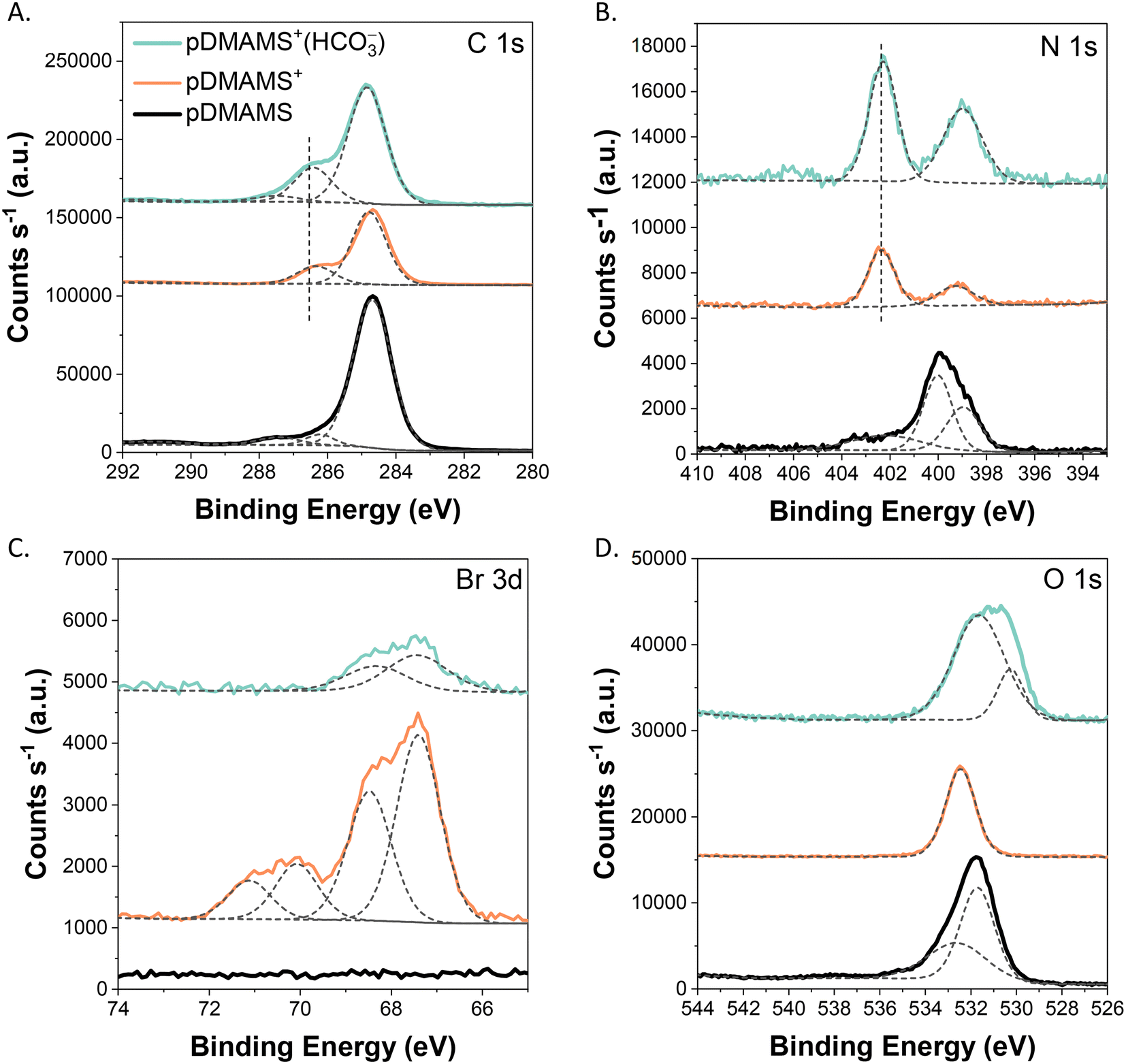 | ||
| Fig. 7 X-ray photoelectron spectra for pDMAMS, pDMAMS+, and pDMAMS+(HCO3−): (A) C 1s; (B) N 1s; (C) Br 3d; and (D) O 1s. | ||
Gentle ion-beam milling through the thickness of the pDMAMS+ film coupled with collection of spectra after specified milling intervals reveals that the boundary N+/N ratio persists into the first 90 nm of the ∼507 nm thick pDMAMS+ film, indicating that alkylation is not solely surface-sited (Fig. S9A†). After solution-based exchange to generate pDMAMS+(HCO3−), the N+/N ratio decreases slightly to 54%, and stays consistent through the ion-milled depth of the film. We attribute the decrease in quaternary nitrogen upon exchange to HCO3− to swelling-induced mechanical elimination of the quaternary groups, hinting at a design shortcoming when the charged moieties participate in the cross-link.
Quantification of the photoelectron intensity assigned to the halide chemical state in pDMAMS+ is used to estimate cross-linking density. As expected, pDMAMS+ contains both bromide and chloride species (Fig. 7C and S9F†) that originate from the organohalogens in the alkylating agent (1-chloro-3-bromopropane) and then serve as charge-balancing species for the ammonium functionality. The two broad Br 3d peaks are consistent with the spin–orbit couples (3d5/2/3d3/2) of bromide (67 eV, 68 eV) and unreacted alkyl bromine (70 eV, 71 eV). The presence of organobromine arises when the chloro end of the alkylating agent reacts with a tertiary amine but the Br-terminated end remains unreacted.23,24 A similar ionic–organic blend is observed in the Cl 1s spectra (Fig. S9F†). To test the hypothesis that pDMAMS+ contains some portion of unreacted alkyl bromine/chlorine, we treat pDMAMS+ with a solution of ethanolic trimethylamine, which reacts any residual alkyl halogen to generate Br−, and find decreased intensity of the organobromine peaks at 70 eV and 71 eV (Fig. S10†). From the relative atom ratios for N+/N and Br−/Br, and given that 65% of the nitrogen in pDMAMS+ exists as N+, we calculate a cross-linking density of 49%, indicating a high degree of cross-linking (see eqn (S2) in ESI†).
The XPS analyses of the pDMAMS+ films after ion exchange confirm successful exchange of Cl−/Br− for other anions of interest. The anion-exchange protocol requires immersing pDMAMS+ films in a solution containing the desired anion (e.g., OH− or HCO3−). The pDMAMS+ film is exchanged to OH− using a solution-based protocol and then exposed to CO2 in ambient air to generate HCO3−. The Br− peak in the Br 1s spectra decreases, translating to 90% exchange of Br− for HCO3−. The additional peak observed in the O 1s spectra at 530 eV, attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from inclusion of HCO3−, also corroborates successful ion exchange (Fig. 7D).20 The depth-profile XPS results (Fig. S9E†) reveal that pDMAMS+(HCO3−) has significantly higher (and stable) oxygen content relative to the halogenated counterpart. The O content of the pristine pDMAMS film is attributed to incorporation of initiated tert-butyl peroxide radicals in the polymer at either the chain initiation or termination steps and to unterminated radical chains reacting with molecular oxygen upon removing the film from the iCVD deposition chamber.2
O from inclusion of HCO3−, also corroborates successful ion exchange (Fig. 7D).20 The depth-profile XPS results (Fig. S9E†) reveal that pDMAMS+(HCO3−) has significantly higher (and stable) oxygen content relative to the halogenated counterpart. The O content of the pristine pDMAMS film is attributed to incorporation of initiated tert-butyl peroxide radicals in the polymer at either the chain initiation or termination steps and to unterminated radical chains reacting with molecular oxygen upon removing the film from the iCVD deposition chamber.2
Nanoscale morphology of pDMAMS and pDMAMS+via grazing incidence small- and wide-angle X-ray scattering (GISAXS/GIWAXS)
The techniques of GISAXS/GIWAXS are well suited for probing the nanoscale structure of thin films, especially for amorphous soft materials where structure can have a dramatic impact on ion transport.25Fig. 8 displays the 2D GISAXS patterns and 1D horizontal GISAXS/GIWAXS line-cuts for select ambient relative humidity (RH)-equilibrated pDMAMS films; the 2D GIWAXS patterns and 1D vertical GISAXS line-cuts can be found in Fig. S11.† The 2D GISAXS patterns (Fig. 8A) indicate no strong periodic ordering of scattering domains within the films in either the XY (parallel to substrate) or Z (perpendicular to substrate) planes, although such ordering is sometimes observed for dry bulk polymers of similar chemistry.26–28 Thus, any scattering domains are spatially distributed randomly in these films. In the GIWAXS portion of the 1D horizontal linecuts, the presence of an amorphous halo at q = 1.2–1.4 Å−1 (W1, d = 0.5 nm given that d = 2π/q) indicates the various pDMAMS films are noncrystalline (Fig. 8B).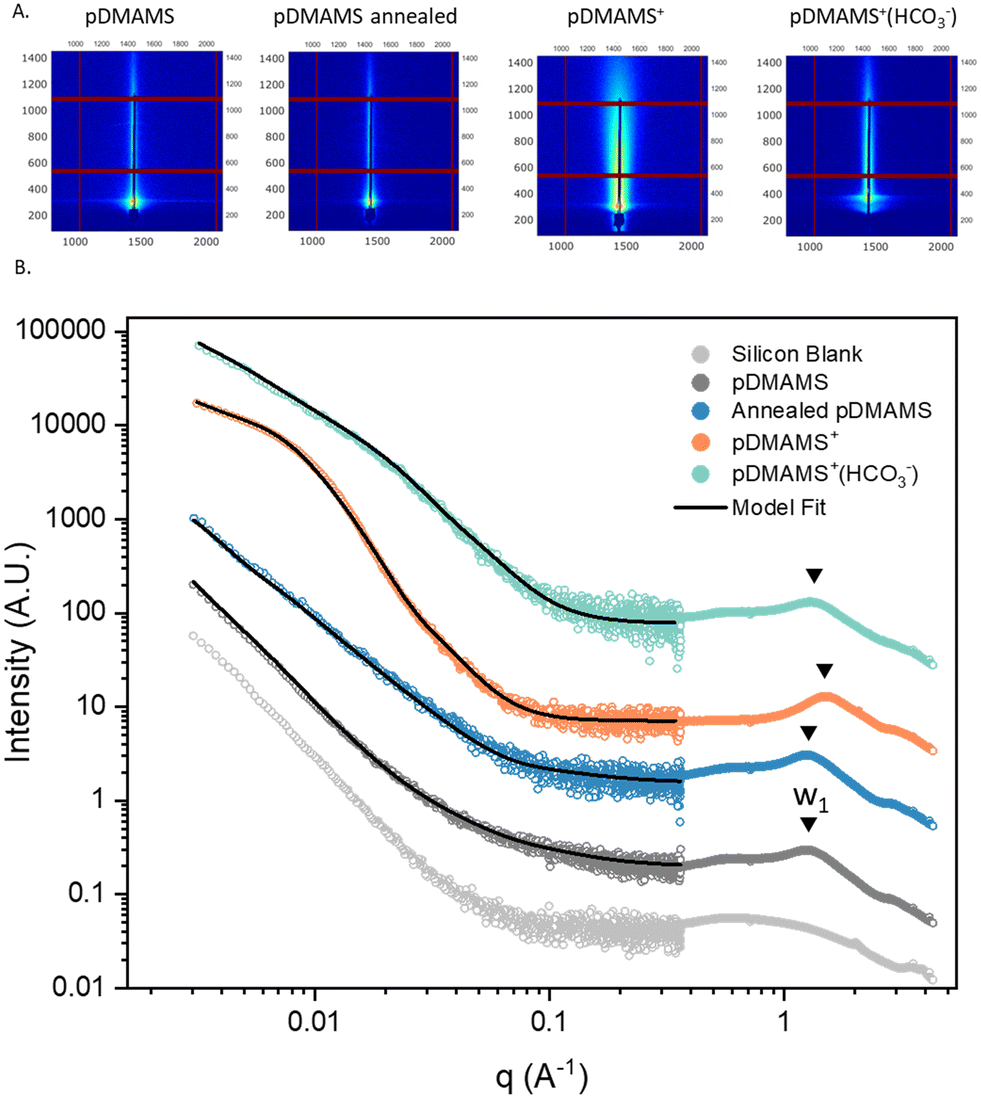 | ||
| Fig. 8 (A) Experimental 2D GISAXS spectra and (B) experimental and GP-modeled 1D horizontal line cut GISAXS/GIWAXS spectra of the series of pDMAMS films on silicon wafers, equilibrated at ambient conditions. | ||
Modeling the data using the multi-level unified function (eqn (S3A and B)†) provides some geometric description of the scattering domains. This analysis yields radius of gyration (Rg), power factor P relating to the shape, and prefactors G and B relating to the scattering domain volume, surface area, and contrast. Table S2† contains the model parameters obtained by fitting the data in Fig. 8. Each spectrum is fit with a two-level model, with level 1 representing larger structures (low q) and level 2 representing smaller structures (high q).
The annealed pDMAMS spectra can be fit with Rg2 set to 1.1 nm, the radius of gyration calculated for a pDMAMS chain in our recent work,16 however as-deposited pDMAMS can only be fit using a larger Rg2 value. The decrease in Rg1 and Rg2 upon annealing pDMAMS is consistent with our hypothesis of chain rearrangement-induced densification. Introducing charge to the polymer significantly alters the GISAXS spectra, where a clear knee is present in the spectra for pDMAMS+ and pDMAMS+(HCO3−). For pDMAMS+, Rg2 is 10.5 nm and the associated power P2 is 3.76, meaning the primary scattering structure in this system is larger than that of uncharged pDMAMS and has a sharp scattering length density contrast with the surrounding medium. We attribute this feature to an ionic cluster containing high-contrast halogen anions. A secondary structure with Rg1 = 48.1 nm and P1 = 2.59 is taken to be a semi-compact arrangement of the primary structures into a longer-range ionic domain. Upon exchanging the halides for HCO3−, Rg1 increases to 99.0 nm and P1 decreases to 1.69, while Rg2 and P2 both decrease to 7.3 nm and 3.2, respectively. Fig. S12† shows the GISAXS experimental and modeled spectra replotted with denotations for Rg values in q space. We interpret these results as the expansion and decreased density of the larger secondary structure due to the softer and lower scattering length density of the HCO3− anion. The modeled scattering data indicate that the ionic domains in pDMAMS+(HCO3−) are less aggregated than in pDMAMS+.
The GISAXS/GIWAXS results confirm that introducing charge into the polymer impacts the nanoscale morphology of the film. The different structural arrangement of ionic groups within the polymers should impact ion and mass transport through the material, an effect that we explore in detail elsewhere.29
Conclusions
Post-deposition modification of an iCVD-derived pDMAMS film with alkylating agent 1-bromo-3-chloropropane yields a positively charged ionomer with labile anions, a material with applications in diverse fields in which mass- and ion-transport regulation is desirable. Successful alkylation and subsequent ion exchange to prepare pDMAMS+(anion−) are confirmed using NMR, ATR-FTIR, and XPS. Through optimization of the deposition conditions, the polymer films are demonstrated to be both conformal on 2D and complex irregular 3D substrates and smooth at the nanoscale. Morphological differences induced by alkylation and the identity of the charge-balancing anion are assessed using GISAXS/GIWAXS, where the polymers are found to be noncrystalline and to contain ion-rich domains of varying shape. The demonstration and extensive characterization of a conformal submicron-thick anion-conducting polymer provide a foundation for the design and synthesis of related materials using iCVD. The ability to non-line-of-sight deposit this class of materials on complex substrates with facile control of thickness will advance a variety of application spaces, including medical devices, desalination, and energy storage.Experimental
Synthesis of 4-dimethylaminomethylstyrene monomer (4-DMAMS)
Aqueous dimethylamine (Thermo Fisher, 40% wt/wt aqueous solution, 250 mL of a 7.9 M solution, 1.98 mol), 53.99 g of K2CO3 (0.391 mol), and a magnetic stir bar were added to a 500 mL round-bottom flask and chilled to 0 °C in an ice bath. An addition funnel was attached to the flask to which 58.17 g of 1-(chloromethyl)-4-vinylbenzene (Oakwood Products, 98%, 0.381 mol) was added after filtration through a plug of neutral alumina to remove the t-butylcatechol inhibitor. Neat 1-(chloromethyl)-4-vinylbenzene was added dropwise to the stirring aqueous dimethylamine solution under N2 flow, forming a pale-yellow emulsion, which was stirred for 16 h and allowed to warm slowly to room temperature. Volatiles (e.g., excess dimethylamine) were subsequently removed under vacuum at ambient temperature causing the emulsion to separate into a colorless aqueous lower layer and yellow organic upper layer. The organic layer was separated, then the aqueous layer was washed several times with ethyl acetate. The multiple organic fractions were combined and washed several times with deionized water, dried over magnesium sulfate, and concentrated in vacuo. The crude yellow oil was vacuum distilled at 100 °C then filtered through neutral alumina to remove colored impurities (e.g., trace inhibitor that had carried over during distillation). The isomerically pure 4-dimethylaminomethylstyrene (4-DMAMS) was isolated as a colorless oil (49.98 g, 81% yield). 1H NMR (CDCl3, 400.13 MHz, Fig. S1A†): δ 7.40 (d, 2H, J = 15.3 Hz), 7.30 (d, 2H, J = 15.3 Hz), 6.74 (dd, 1H, Jtrans = 17.6 Hz, Jcis = 10.8 Hz), 5.77 (d, 1H, J = 17.6 Hz), 5.25 (d, 1H, J = 10.8 Hz), 3.44 (s, 2H), 2.27 (s, 6H). 13C[1H] NMR (CDCl3, 100.61 MHz, Fig. S1B†): δ 138.64, 136.68, 136.47, 129.26, 126.13, 113.42, 64.13, 45.37. IR (ATR, Fig. S6†): ν (cm−1) = 3085 (w), 2975 (m), 2940 (m), 2855 (w), 2815 (s), 2765 (s), 1805 (w), 1700 (vw), 1630 (s), 1610 (w), 1565 (w), 1510 (s), 1465 (s), 1455 (s), 1440 (s), 1405 (s), 1360 (s), 1320 (w), 1285 (w), 1255 (s), 1200 (w), 1175 (s), 1145 (s), 1110 (m), 1095 (m), 1030 (vs), 1015 (s), 990 (vs), 945 (w), 900 (vs), 860 (vs), 845 (m), 820 (vs), 765 (w), 725 (s), 715 (w), 640 (w), 625 (vw), 570 (m).The 4-DMAMS was stabilized with CuCl2 (forming a dark red solution) for long-term storage and for use in solution-based polymerization and initiated chemical vapor deposition.
Synthesis of bulk solution-polymerized pDMAMS
2,2′-Azobis(isobutyronitrile) (AIBN, Aldrich, 98%), 10.62 mg (64.7 μmol) was dissolved in 2.2050 g of unstabilized 4-DMAMS (13.67 mmol). The solution was subsequently deoxygenated by freeze–pump–thaw cycling and flame sealed under vacuum in a thick-walled Pyrex tube. The tube was immersed in a boiling water bath (100 °C) for 120 min then removed from the bath, cracked open, and the resulting hazy, viscous liquid was dissolved in 10 mL CHCl3. This solution was subsequently precipitated dropwise into ice cold CH3CN (Fisher Chemical, 99.5%), yielding a tacky white solid. After isolating the solid and drying it under vacuum at ambient temperature, 1.371 g of a brittle, off-white (CHCl3 soluble) foam was obtained (62% monomer conversion). The glass-transition temperature (Tg) of pDMAMS was measured to be 82 °C using differential scanning calorimetry (DSC, TA Instruments Q100 Series Fig. S2†) performed in aluminum pans under N2 at a heating/cooling rate of 20 K min−1. The experimentally measured Tg is consistent with the computationally calculated Tg of 83.4 °C.161H NMR (CDCl3, 400.13 MHz, Fig. S5A†): δ 6.9–6.8 (br, 2H), 6.4–6.3 (br, 2H), 3.29 (s, 2H), 2.15 (br, 6H), 1.7 (br, 1H), 1.3–1.2 (br, 2H). 13C[1H] NMR (CDCl3, 100.61 MHz, Fig. S5B†): δ 144 (br), 135.5, 128.5, 127.3, 64.0, 45.2, 44 (br), 40.1. IR (ATR, Fig. S6†): ν (cm−1) = 3380 (br, w), 3085 (vw), 3045 (w), 3015 (w), 2970 (m), 2930 (s), 2855 (m), 2810 (m), 2765 (s), 1795 (vw), 1700 (w), 1650 (w), 1610 (w), 1575 (vw), 1510 (s), 1455 (s), 1420 (m), 1360 (s), 1310 (w), 1255 (s), 1210 (vw), 1175 (m), 1145 (m), 1095 (m), 1030 (s), 1015 (vs), 980 (m), 940 (m), 855 (vs), 810 (vs), 725 (m), 710 (m), 545 (s).Poly(4-dimethylaminomethylstyrene) (pDMAMS) is soluble in a wide variety of polar and nonpolar solvents, including: CHCl3, ethyl acetate, hexanes, and methanol. Acetonitrile was the only solvent identified as suitable for precipitation of the polymer from solution.
Poly(4-dimethylaminomethylstyrene) films were prepared by creating a 10% w/v stock solution of polymer in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (by volume) mixture of diethyl ether, chloroform, and xylenes. The viscous solution was pipetted onto glass cover slips and dried under flowing air at room temperature. To create thicker films, additional aliquots of solution were added to previously deposited films after drying and before heat treating. Films and substrates were vacuum annealed at 50 °C for 16 h to remove residual solvent.
:1:1 (by volume) mixture of diethyl ether, chloroform, and xylenes. The viscous solution was pipetted onto glass cover slips and dried under flowing air at room temperature. To create thicker films, additional aliquots of solution were added to previously deposited films after drying and before heat treating. Films and substrates were vacuum annealed at 50 °C for 16 h to remove residual solvent.
Analysis of pDMAMS MW by 1H DOSY (diffusion ordered spectroscopy) NMR
As an alternative to gel-permeation chromatography, diffusion-ordered NMR spectroscopy (1H DOSY) was used to estimate the average molecular weight of bulk pDMAMS synthesized by free-radical polymerization. A sequence of 32 bipolar pulsed-field gradient (PFG) 1H NMR spectra with applied z-gradients ranging from g = 0.96–38.5 G cm−1 were acquired in randomized order on a Bruker AVANCE NEO NMR spectrometer using the “Oneshot45” pulse sequence outlined by Botana and colleagues.30 A gradient length (δ) of 2 ms, delay time (Δ) of 1 s, acquisition time of 1 s, and recycle delay of 9 s were used for these acquisitions. The series of 1D 1H spectra was subsequently inverse Laplace transformed in the z-gradient strength (F1) domain using the Stejskal–Tanner relationship with processing routines built into Bruker TopSpin 4.0.6 software package, resulting in a diffusion-ordered, pseudo-2D spectrum plotting average diffusion coefficient (D, m2 s−1) vs.1H chemical shift (Fig. S4†).
with processing routines built into Bruker TopSpin 4.0.6 software package, resulting in a diffusion-ordered, pseudo-2D spectrum plotting average diffusion coefficient (D, m2 s−1) vs.1H chemical shift (Fig. S4†).
The average diffusion coefficient of the polymer (DpDMAMS) = 3.0 × 10−7 cm2 s−1 under these conditions; the corresponding diffusion coefficient of the monomer (DDMAMS) = 1.5 × 10−5 cm2 s−1. Using the Stokes–Einstein relationship  as a means to relate diffusion coefficient to molecular size vis-à-vis radius of gyration (rg ∝ √ number of repeat units), the radius of gyration of pDMAMS is ∼47× the radius of gyration of DMAMS monomer, equating to a degree of polymerization (DP) ≈ 2200 (diffusion average molecular weight MD ≈ 350 kDa). In theory, the number average molecular weight (MN) should be ∼11 kDa based on DP ≈ 65 assuming 62% monomer conversion, quantitative initiation from AIBN, and no chain transfer to monomer (creating more polymer chains than expected). Polydispersity (Đ = MW/MN) cannot be measured directly by DOSY NMR but is likely to be high due to MD (observed) ≫ MN (theoretical). Since the half-life of AIBN is <10 min at 100 °C, a high polydispersity likely results from significant chain branching rather than slow initiation.
as a means to relate diffusion coefficient to molecular size vis-à-vis radius of gyration (rg ∝ √ number of repeat units), the radius of gyration of pDMAMS is ∼47× the radius of gyration of DMAMS monomer, equating to a degree of polymerization (DP) ≈ 2200 (diffusion average molecular weight MD ≈ 350 kDa). In theory, the number average molecular weight (MN) should be ∼11 kDa based on DP ≈ 65 assuming 62% monomer conversion, quantitative initiation from AIBN, and no chain transfer to monomer (creating more polymer chains than expected). Polydispersity (Đ = MW/MN) cannot be measured directly by DOSY NMR but is likely to be high due to MD (observed) ≫ MN (theoretical). Since the half-life of AIBN is <10 min at 100 °C, a high polydispersity likely results from significant chain branching rather than slow initiation.
iCVD of pDMAMS
Substrates were loaded into the center of the iCVD chamber (GVD ilab coating system) which was then evacuated to 50 mTorr. Fresh 4-dimethylaminomethylstyrene (DMAMS) monomer and tert-butyl peroxide (TBPO, Sigma-Aldrich synthesis grade) initiator were loaded into the monomer and initiator jars, respectively. Unless otherwise noted, the monomer jar and line were heated to 70 °C and 80 °C, respectively, the initiator jar and line were heated to 25 °C and 50 °C, respectively, the substrate stage temperature was maintained at 60 °C with a recirculating chiller, and the chamber and viewport temperatures were maintained at 80 °C and 60 °C, respectively. The impact of stage temperature on film morphology is shown in Fig. 2. Initiator flow rate was set to 1.2 sccm with the use of a nitrogen-calibrated mass-flow controller (MKS instruments 1479A11CR1BM-SPCAL), while monomer flow rate was regulated with the use of a needle valve. The operating chamber pressure was maintained at 210 mTorr using an automated butterfly valve and the tungsten filaments were set to 35 V (1.93 A, 220 °C). The amount of monomer and initiator added to the jars was such that the pressure in the chamber would not fall below 190 mTorr during the deposition. For 120 min of deposition, 2 mL of DMAMS was used. After the allotted deposition time, the chamber was evacuated for 30 min with the stage maintained at 60 °C to remove unreacted components.Films of pDMAMS were deposited onto various substrates. For FTIR, profilometry thickness measurements, X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), and scanning electron microscopy (SEM) pDMAMS was deposited onto 25 mm × 35 mm Au-coated glass slides. These substrates were prepared by cleaning glass microscope slides (VWR) in a KOH bath (water, ethanol, potassium hydroxide), drying, and then sputter coating with ∼50 nm of gold using a Denton Vacuum Desk IV. For NMR, silica-fiber filter paper (Pallflex TISSUQUARTZ-2500QAT-UP) was heat treated in a muffle furnace at 500 °C overnight in air, then coated with pDMAMS via iCVD for 240 min (120 min on each side). For demonstration of deposition within a complex 3D substrate, 3D zinc sponges were prepared according to a recent patent.31
Post-deposition processing of pDMAMS films
As-deposited pDMAMS films were annealed under dynamic vacuum at 100 °C for 24 h, removed from the oven while hot, and allowed to cool under ambient laboratory conditions. Annealed films were quaternized by placing them on a glass shelf in a ∼1 L glass vacuum desiccator containing 2 mL of 1-bromo-3-chloropropane (Alfa Aesar, 99%) in a glass Petri dish at the bottom of the desiccator. The desiccator was vacuum sealed, then placed into an 80 °C oven. After 24 h, the desiccator was removed from the oven and recovered films placed under dynamic vacuum for 24 h at 80 °C to remove any unreacted material and yield the pDMAMS+ form. The pDMAMS+(Br−) form was obtained by placing pDMAMS+ in an ion-exchange bath of 0.1 M potassium bromide (KBr, Fisher Scientific Certified Reagent) in deionized water for 1 h, followed by three rinses with DI water. Because direct exchange using aqueous KOH delaminated the films, the pDMAMS+(OH−) form was obtained by placing the film in 0.1 M tetramethylammonium hydroxide (Sigma, min. 97%) in isopropanol (Fisher, ACS+) for 1 h under a dry nitrogen atmosphere, followed by three rinses with isopropanol. The pDMAMS+(HCO3−) form was obtained by exposing pDMAMS+(OH−) to CO2 in ambient atmosphere.Characterization
Author contributions
Hunter O. Ford: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review and editing. Brian L. Chaloux: conceptualization, data curation, formal analysis, investigation, methodology, validation, writing – review and editing. Battogtokh Jugdersuren: data curation, formal analysis, investigation. Xiao Liu: data curation, formal analysis, investigation. Christopher A. Klug: data curation, formal analysis, investigation, writing – review and editing. Joel B. Miller: data curation, formal analysis, investigation. Xiaobing Zuo: data curation, formal analysis, investigation. Michael W. Swift: formal analysis, investigation, writing – review and editing. Michelle D. Johannes: formal analysis, writing – review and editing. Jeffrey W. Long: conceptualization, resources, validation, writing – review and editing. Debra R. Rolison: conceptualization, validation, formal analysis, funding acquisition, writing – review and editing. Megan B. Sassin: conceptualization, validation, formal analysis, funding acquisition, writing – review and editing.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This research was supported by the U.S. Office of Naval Research. H. O. F. is an NRL–National Research Council postdoctoral associate. We also acknowledge the use of resources at the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science user facility at Argonne National Laboratory and is based on research supported by the U.S. DOE Office of Science-Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the ESI† NE2020 project, grant agreement no 654000.References
- M. H. Wang, X. X. Wang, P. Moni, A. D. Liu, D. H. Kim, W. J. Jo, H. Sojoudi and K. K. Gleason, CVD Polymers for Devices and Device Fabrication, Adv. Mater., 2017, 29, 1604606 CrossRef PubMed.
- T. P. Martin, S. E. Kooi, S. H. Chang, K. L. Sedransk and K. K. Gleason, Initiated Chemical Vapor Deposition of Antimicrobial Polymer Coatings, Biomaterials, 2007, 28, 909–915 CrossRef CAS PubMed.
- G. Choi, G. M. Jeong, M. S. Oh, M. Joo, S. G. Im, K. J. Jeong and E. Lee, Robust Thin Film Surface with a Selective Antibacterial Property Enabled via a Cross-Linked Ionic Polymer Coating for Infection-Resistant Medical Applications, ACS Biomater. Sci. Eng., 2018, 4, 2614–2622 CrossRef CAS PubMed.
- E. Çitak, H. Testici, M. Gürsoy, E. Sevgili, H. T. Daği, B. Özturk and M. Karaman, Vapor Deposition of Quaternary Ammonium Methacrylate Polymers with High Antimicrobial Activity: Synthetic Route, Toxicity Assessment, and Durability Analysis, J. Vac. Sci. Technol., A, 2020, 38, 043203 CrossRef.
- P. Moni, A. Al-Obeidi and K. K. Gleason, Vapor Deposition Routes to Conformal Polymer Thin Films, Beilstein J. Nanotechnol., 2017, 8, 723–735 CrossRef CAS PubMed.
- N. Chen, B. Reeja-Jayan, J. Lau, P. Moni, A. D. Liu, B. Dunn and K. K. Gleason, Nanoscale, Conformal Polysiloxane Thin Film Electrolytes for Three-Dimensional Battery Architectures, Mater. Horiz., 2015, 2, 309–314 RSC.
- Y. Yoo, B. G. Kim, K. Pak, S. J. Han, H. S. Song, J. W. Choi and S. G. Im, Initiated Chemical Vapor Deposition (iCVD) of Highly Cross-Linked Polymer Films for Advanced Lithium-Ion Battery Separators, ACS Appl. Mater. Interfaces, 2015, 7, 18849–18855 CrossRef CAS PubMed.
- S. H. Baxamusa, S. G. Im and K. K. Gleason, Initiated and Oxidative Chemical Vapor Deposition: A Scalable Method For Conformal and Functional Polymer Films on Real Substrates, Phys. Chem. Chem. Phys., 2009, 11, 5227–5240 RSC.
- M. Joo, J. Shin, J. Kim, J. B. You, Y. Yoo, M. J. Kwak, M. S. Oh and S. G. Im, One-Step Synthesis of Cross-Linked Ionic Polymer Thin Films in Vapor Phase and Its Application to an Oil/Water Separation Membrane, J. Am. Chem. Soc., 2017, 139, 2329–2337 CrossRef CAS PubMed.
- T. P. Martin and K. K. Gleason, Combinatorial Initiated CVD for Polymeric Thin Films, Chem. Vap. Deposition, 2006, 12, 685–691 CrossRef CAS.
- G. Ozaydin-Ince and K. K. Gleason, Transition Between Kinetic and Mass Transfer Regimes in the Initiated Chemical Vapor Deposition from Ethylene Glycol Diacrylate, J. Vac. Sci. Technol., A, 2009, 27, 1135–1143 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Anion-Exchange Membranes in Electrochemical Energy Systems, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- A. Alabi, A. AlHajaj, L. Cseri, G. Szekely, P. Budd and L. D. Zou, Review of Nanomaterials-Assisted Ion Exchange Membranes For Electromembrane Desalination, npj Clean Water, 2018, 1, 10 CrossRef CAS.
- C. M. Gabardo, C. P. O'Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Continuous Carbon Dioxide Electroreduction to Concentrated Multi-Carbon Products Using a Membrane Electrode Assembly, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- M. Groß, N. Röttgen, S. Volz and C. Cremers, Oxygen and Carbon Dioxide Separation at Cathodes of Anion Exchange Membrane Cells, 2022 Meet. Abstr. Electrochem. Soc., MA2022-02, 1627.
- Y. C. Kim, B. L. Chaloux, D. R. Rolison, M. D. Johannes and M. B. Sassin, Molecular Dynamics Study of Hydroxide Ion Diffusion in Polymer Electrolytes, Electrochem. Commun., 2022, 140, 107334 CrossRef CAS.
- B. Q. Kim, J. Chae, J. Q. Kim, K. Kim and S. Q. Choi, Interfacial Shear Rheology of Perfluorosulfonic Acid Ionomer Monolayers at the Air/Water Interface, J. Rheol., 2019, 63, 947–959 CrossRef CAS.
- H. O. Ford, B. L. Chaloux, M. W. Swift, C. A. Klug, Y. Kim, J. W. Long, M. D. Johannes, D. R. Rolison and M. B. Sassin, 3D All Solid-State Ag–Zn Batteries: A Non-Strategic Chemistry to Meet DoD Energy and Power Needs, Proc. 49th Power Sources Conf., 2023, in the press.
- J. E. Mark, Polymer Data Handbook, Oxford University Press, 2009 Search PubMed.
- G. Beamson and D. Briggs, High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database, Wiley, Chichester, 1992 Search PubMed.
- R. K. Blundell and P. Licence, Quaternary Ammonium and Phosphonium Based Ionic Liquids: A Comparison of Common Anions, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC.
- S. Maurya, S. H. Shin, Y. Kim and S. H. Moon, A Review on Recent Developments of Anion Exchange Membranes for Fuel Cells and Redox Flow Batteries, RSC Adv., 2015, 5, 37206–37230 RSC.
- K. Oai, Y. Inoue, A. Nakao, K. Fukazawa and K. Ishihara, Antibacterial Effect of Nanometer-Size Grafted Layer of Quaternary Ammonium Polymer on Poly(Ether Ether Ketone) Substrate, J. Appl. Polym. Sci., 2020, 137, e49088 CrossRef.
- L. Smykalla, P. Shukrynau, M. Korb, H. Lang and M. Hietschold, Surface-Confined 2D Polymerization of a Brominated Copper-Tetraphenylporphyrin on Au(111), Nanoscale, 2015, 7, 4234–4241 RSC.
- M. A. Hickner, Water-Mediated Transport in Ion-Containing Polymers, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 9–20 CrossRef CAS.
- H. S. Dang and P. A. Jannasch, Comparative Study of Anion-Exchange Membranes Tethered with Different Hetero-Cycloaliphatic Quaternary Ammonium Hydroxides, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC.
- E. A. Weiber and P. Jannasch, Ion Distribution in Quaternary-Ammonium-Functionalized Aromatic Polymers: Effects on the Ionic Clustering and Conductivity of Anion-Exchange Membranes, ChemSusChem, 2014, 7, 2621–2630 CrossRef CAS PubMed.
- M. Ozawa, T. Kimura, K. Otsuji, R. Akiyama, J. Miyake, M. Uchida, J. Inukai and K. Miyatake, Structurally Well-Defined Anion-Exchange Membranes Containing Perfluoroalkyl and Ammonium-Functionalized Fluorenyl Groups, ACS Omega, 2018, 3, 16143–16149 CrossRef CAS PubMed.
- H. O. Ford, B. L. Chaloux, Y. Kim, J. W. Long, D. R. Rolison and M. B. Sassin, Submicron-Thick Single-Anion Conducting Polymer Electrolytes, RSC Appl. Interfaces, 2024 10.1039/d3lf00257h.
- A. Botana, J. A. Aguilar, M. Nilsson and G. A. Morris, J-Modulation Effects in DOSY Experiments and Their Suppression: The Oneshot45 Experiment, J. Magn. Reson., 2011, 208, 270–278 CrossRef CAS PubMed.
- R. H. DeBlock, J. F. Parker, J. W. Long, D. R. Rolison and C. N. Chervin, Fabrication and Fusion of Zinc Particles in Porous Electrodes, US Pat., 11710818B1, 2023 Search PubMed.
- D. Banfi and L. Patiny, https://www.nmrdb.org: Resurrecting and Processing NMR Spectra On-Line, Chimia, 2008, 62(4), 280–281 CrossRef CAS.
- A. M. Castillo, L. Patiny and J. Wist, Fast and Accurate Algorithm for the Simulation of NMR Spectra of Large Spin Systems, J. Magn. Reson., 2011, 209, 123–130 CrossRef CAS PubMed.
- C. Steinbeck, S. Krause and S. Kuhn, NMRShiftDB Constructing a Free Chemical Information System with Open-Source Components, J. Chem. Inf. Comput. Sci., 2003, 43, 1733–1739 CrossRef CAS PubMed.
- Y. W. Pei, D. J. H. Cant, R. Havelund, M. Stewart, K. Mingard, M. P. Seah, C. Minelli and A. G. Shard, Argon Cluster Sputtering Reveals Internal Chemical Distribution in Submicron Polymeric Particles, J. Phys. Chem. C, 2020, 124, 23752–23763 CrossRef CAS.
- P. J. Cumpson, J. F. Portoles and N. Sano, Material Dependence of Argon Cluster Ion Sputter Yield in Polymers: Method and Measurements of Relative Sputter Yields for 19 Polymers, J. Vac. Sci. Technol., A, 2013, 31, 020605 CrossRef.
- P. J. Cumpson, J. F. Portoles and N. Sano, Observations on X-Ray Enhanced Sputter Rates in Argon Cluster Ion Sputter Depth Profiling of Polymers, Surf. Interface Anal., 2013, 45, 601–604 CrossRef CAS.
- Y. J. Hofstetter and Y. Vaynzof, Quantifying the Damage Induced by X-Ray Photoelectron Spectroscopy Depth Profiling of Organic Conjugated Polymers, ACS Appl. Polym. Mater., 2019, 1, 1372–1381 CrossRef CAS.
- C. M. Goodwin, Z. E. Voras, X. Tong and T. P. Beebe, Jr., Soft Ion Sputtering of PAni Studied by XPS, AFM, TOF-SIMS, and STS, Coatings, 2020, 10, 967 CrossRef CAS.
- X. Liu, M. R. Abernathy, T. H. Metcalf, B. Jugdersuren, J. C. Culbertson, M. Molina-Ruiz and F. Hellman, Comparing Amorphous Silicon Prepared by Electron-Beam Evaporation and Sputtering Toward Eliminating Atomic Tunneling States, J. Alloys Compd., 2021, 855, 157431 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient Iterative Schemes for ab initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H–Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. Jónsson, G. Mills and K. Jacobsen, Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions, Classical and Quantum Dynamics, in Condensed Phase Simulations, ed. B. Berne, G. Ciccoti and D. F. Coker, World Scientific Publishing, Hackensack, NJ, 1998, pp. 385–404 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00256j |
| This journal is © The Royal Society of Chemistry 2024 |