
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering Pt–CeO2 interfaces for reverse water-gas shift (RWGS) reaction†
Kauê G. G.
dos Santos
a,
Alisson S.
Thill
 a,
Livia P.
Matte
a,
Gustavo Z.
Girotto
a,
Mateus V.
Costa
a,
Denise R.
Bohn
b,
Fernanda
Poletto
b and
Fabiano
Bernardi
*a
a,
Livia P.
Matte
a,
Gustavo Z.
Girotto
a,
Mateus V.
Costa
a,
Denise R.
Bohn
b,
Fernanda
Poletto
b and
Fabiano
Bernardi
*a
aPrograma de Pós-Graduação em Física, Instituto de Física, Universidade Federal do Rio Grande do Sul, Brazil. E-mail: bernardi@if.ufrgs.br
bPrograma de Pós-Graduação em Química, Instituto de Química, Universidade Federal do Rio Grande do Sul, Brazil
First published on 25th April 2024
Abstract
Nowadays, Pt–CeO2 interfaces are very popular in many applications. In particular, this system is widely used in catalysis for the reverse water gas-shift (RWGS) reaction aiming to stop the dangerous advancement of the global warming effect. Nevertheless, some complex atomic events occurring at this interface are still unclear. In this work, superhydrophobic Pt–CeO2 nanoparticles were used in the RWGS reaction aiming to shift the equilibrium of the RWGS reaction towards the formation of CO. It was demonstrated that this sample presents a highly reducible CeO2 surface and an easy tunability of the O vacancy population, which is the main active site of metal oxides in catalysis. Consequently, the Pt–CeO2 superhydrophobic sample presents improved performance towards CO formation in the RWGS reaction. During the RWGS reaction, the Pt nanoparticles suffer from the strong metal–support interaction (SMSI) effect that may hinder the catalytically active sites but, even so, the superhydrophobic Pt–CeO2 nanoparticles are active in the RWGS reaction. It opens new frontiers in the engineering of active superhydrophobic Pt–CeO2 interfaces with tunable O vacancy population.
Introduction
Carbon dioxide (CO2) is currently depicted as the most influential greenhouse gas responsible for global warming, primarily due to its high concentration in the atmosphere. The consequences of global warming include an increase in infectious diseases, a rise in sea level, and an increase in heat waves and other natural catastrophes like hurricanes.1–3 Considering this, in the last few decades, a great deal of research has been conducted to either capture or dissociate the CO2 molecule.4,5 A promising approach for dissociating the CO2 molecule is through the reverse water gas-shift (RWGS) reaction. The CO produced can be captured and utilized as a raw material for a variety of processes, such as the synthesis of polymers, hydrocarbons, and other valuable compounds like methanol, as well as a feedstock for renewable energy sources. However, the current catalysts employed in the RWGS reaction, such as Pt/CeO2, still operate at elevated temperatures, typically around 600 °C, and exhibit limited reactivity.6–9 Consequently, it restricts the use of the RWGS reaction against the global warming effect, thus impairing the advancement towards a healthy world.The best way to design improved catalysts for the RWGS reaction is to make a detailed description of the atomic events occurring during the reaction. The catalytic reaction depends on different phenomena like sintering, coking, poisoning, etc. In particular, the occurrence of the strong metal–support interaction (SMSI) effect,10 a famous metal/metal oxide interface effect, is closely related to the performance of the catalyst. The geometrical factor of the SMSI effect corresponds to the formation of a capping layer coming from the support and covering the metallic nanoparticles during the reduction treatment. Consequently, a strong change in the catalytically active sites available occurs at the surface of the nanoparticles, thus improving or impairing the catalytic activity.11,12 Given the above, it is crucial to determine the interface effects occurring during the RWGS reaction and their influence on the catalytic reaction.
Pt/CeO2 nanoparticles are relatively good catalysts for the RWGS reaction. In particular, CeO2 can easily alternate between its totally reduced (Ce(III), Ce2O3) and fully oxidized (Ce(IV), CeO2) states repeatedly.13 Oxygen vacancies work as adsorption sites for CO2 molecules during the RWGS reaction, so CeO2 is an interesting support for the reaction.14 An interesting idea is the use of hydrophobic supports for the reversible RWGS reaction. It allows shifting the RWGS equilibrium towards the formation of CO products, thus increasing the dissociation efficiency of the CO2 molecule. However, it is hard to find studies reporting the hydrophobicity property of metal oxide supports used in the RWGS reaction. In this work, to enhance the reactivity of Pt–CeO2 nanoparticles towards CO formation in the RWGS reaction at low temperatures, superhydrophobic CeO2 nanoparticles were synthesized and used together with Pt nanoparticles.
Experimental
Superhydrophobic CeO2 nanoparticles were synthesized using the precipitation method. Firstly, Ce(OH)3 nanoparticles were synthesized in a lyotropic liquid-crystalline medium composed of phytantriol and water, using Ce(NO3)3·6H2O as a precursor. In a round-bottom flask, 2.40 g of phytantriol and 0.24 g of the precursor Ce(NO3)3·6H2O were added. This mixture was treated with NaOH and subsequently with H2O2, as previously described,15 to obtain CeO2 nanoparticles. After this treatment, the mixture was dissolved in 10 ml of absolute ethanol and kept under gentle magnetic stirring for 8 h. After this period, the mixture was allowed to stand for 12 h. Subsequently, centrifugation was carried out at 3500 rpm for 15 min. The precipitated powder was separated by decantation, washed with a fresh aliquot of 8 ml of absolute ethanol, and centrifuged under the same previously described conditions. This process was repeated three times. After ethanol extractions, the precipitate was washed with 8 ml of Milli-Q water and centrifuged for 15 min at 3500 rpm. The remaining precipitate was kept in a desiccator under reduced pressure for 12 h for drying. The obtained powder exhibited a characteristic yellowish color. This sample will be hereafter named CeO2 Hyd. For comparison purposes, high surface area CeO2 nanoparticles were also synthesized in accordance with previous work,16 hereafter named CeO2 high S. In this case, 2 g of (NH4)2Ce(NO3)6 dissolved in 6 ml of distilled water was added dropwise to a saturated aqueous solution (14 mL) of ammonium carbonate under magnetic stirring. The pH value of the mixture was maintained at 9 by adding excess (NH4)2CO3. After 30 min, the resultant solution was filtered and a 2 ml aliquot of the sample was mixed with 2 ml of ethanol under magnetic stirring at 70 °C for 16 h. Nanoparticles that precipitated after this period were washed 3 times with water, centrifuged and left in a drying oven at 90 °C for 48 h. A commercial standard of CeO2 (Sigma-Aldrich code 211575) was also used and it is named CeO2 Std in this work.A commercial aqueous solution (Sigma Aldrich code 773875) containing dispersed Pt nanoparticles was mixed with the different CeO2 samples used in this work. In separate crucibles, 1 ml of the Pt aqueous solution and 19.5 mg of each CeO2 sample were added to obtain 8 wt% Pt–CeO2 nanoparticles. The crucibles were placed in an ultrasonic bath for 5 minutes to achieve a homogeneous mixture between Pt and CeO2. The mixture was dried under vacuum at room temperature in a desiccator for 24 hours. After this, 1 ml of absolute ethanol was added to each crucible, and the mixture was again homogenized by ultrasonic treatment and dried in a desiccator for 3 hours more. This process was repeated three times. It resulted in a powder consisting of Pt nanoparticles mixed with CeO2 nanoparticles.
The wettability of the CeO2 samples was determined using contact angle measurements. For the measurements, about 10 mg of the nanoparticle powder was used to produce a homogeneous thin membrane supported on PVDF (polyvinylidene difluoride). Aiming to produce these membranes, the nanoparticle powder was sieved using a nylon mesh of 10, 25, or 40 μm size. Then, 10 mg of the nanoparticle powder was dispersed in isopropyl alcohol utilizing an ultrasound device, resulting in a homogeneous solution. A clean membrane was fixed between a glass funnel and a glass tube using a grapple. The solution was dropped inside the glass tube and, with the help of a vacuum pump attached to the glass funnel, passed through the membrane forming a thin layer of the nanoparticle powder on the membrane. The membrane was fixed on the sample holder for the contact angle measurements. A drop of 0.2 μl, 0.3 μl, or 0.5 μl of Milli-Q water was dropped over the membrane. For each drop volume, the procedure was repeated at least 10 times for all the different grain sizes (10, 25 and 40 μm). The contact angle value was determined through the Surftens software. The final contact angle was the average value obtained from the different measurements.
Transmission Electron Microscopy (TEM) images were obtained for the Pt and CeO2 nanoparticles in a JEOL JEM 1200 EXll microscope with an operating voltage of 80 kV at CMM BR-Sul-UFRGS. A drop of the aqueous solution containing the nanoparticles was dropped over a carbon-coated copper grid. The grid was left to dry before the TEM measurements. The size of the nanoparticles was manually obtained using ImageJ software. For each nanoparticle, two width measurements in perpendicular directions were taken and averaged to determine the nanoparticle diameter. High-Resolution TEM (HRTEM) images were obtained for the Pt–CeO2 nanoparticles using a Tecnai G2-20 – FEI SuperTwin microscope with an operating voltage of 200 kV at CM-UFMG. The samples were prepared with the same procedure used for the TEM measurements.
Small Angle X-ray Scattering (SAXS) measurements were conducted in the aqueous solution containing the Pt nanoparticles on the Nano-inXider (Xenocs) equipment at CNANO-UFRGS. A Cu Kα X-ray source with a wavelength of 1.54 Å and a Dectris Pilatus 3 detector with a semitransparent beamstop were used in the measurements. The aqueous solution containing the Pt nanoparticles was inserted in a borosilicate tube with a 2 mm external diameter, 80 mm length and 0.01 mm thickness. The measurements were performed in transmission mode in the 2θ range from 0.00° to 5.25° with a 60 s exposure time for each scan. The final pattern resulted from an average of 60 scans.
The Foxtrot software was used to convert symmetric 2D SAXS data into 1D data and to obtain the average between multiple scans.17 SAXS patterns were analyzed using the SASfit software.18 Raw SAXS data were subtracted by the background function
| I(q) = c0 + c1q + c4q−α | (1) |
Rutherford Backscattering Spectrometry (RBS) measurements were performed to determine the Pt amount deposited on each CeO2 sample. The RBS measurements were conducted at a TANDETRON 3 MV accelerator at the Laboratório de Implantação Iônica-UFRGS with a He beam of E0 = 2 MeV. The scattered ions were detected at θ = 165°. The measurements were conducted in a pressure of 10−6 mbar and with an ion current of around 10 nA. The amount of Pt supported on each sample was quantified using the following equation:
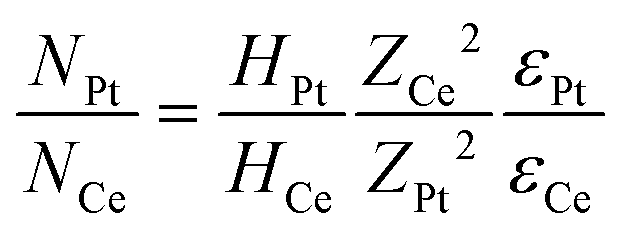 | (2) |
X-ray Photoelectron Spectroscopy (XPS) measurements were performed in the as-prepared Pt–CeO2 nanoparticles at the Laboratório Nacional de Nanotecnologia (LNNano – CNPEM) with a Thermo Scientific K-α X-ray photoelectron spectrometer using an Al Kα X-ray source (hv = 1486.6 eV) operating at 12 kV and with a 45° emission angle of the photoelectrons with respect to the incident X-rays. For the measurements, a thin layer of powder from each sample was dispersed on a carbon tape, attached to the sample holder, and introduced in an ultrahigh vacuum chamber (1 × 10−9 mbar). The measurements were performed in the survey, Pt 4f, Ce 3d, C 1s, and O 1s electronic regions of the samples. The measurements were conducted with an energy step of 0.33 and 0.05 eV, and a dwell time of 0.5 s, for the survey and high-resolution regions, respectively. The analyzer's energy calibration was performed using a standard Au foil (Au 4f7/2 peak at 84.0 eV). The C 1s peak position of adventitious carbon at 284.5 eV was also considered to verify possible charging effects.
The XPS data were analyzed using the XPSPeak software. In order to fit the high-resolution XPS spectra, a symmetric Gaussian–Lorentzian function with a 55% Lorentzian contribution was employed, as determined from analyzing the isolated and highest energy peak in the Ce 3d region at 916.7 eV. A Shirley-type background was used in all the cases. The FWHM values and the relative binding energy of a given component remained fixed across different measurements of different samples.
The Ce 3d XPS region was adjusted with 10 distinct components, where four of these represent the Ce(III) oxidation state and are named v0, v′, u0, and u′ and the other six components represent the Ce(IV) oxidation state and are named v, v′′, v′′, u, u′′ and u′′′, as described by Schierbaum.20 The Ce 3d XPS spectra of the samples were fitted simultaneously, constraining the FWHM and relative energy position of different spectra to be the same. The u0/v0 and u′/v′ intensity ratios were constrained to 1.5. The surface Ce(III) fraction (μsurf) was determined from this fitting using the XPS intensities as shown in eqn (3)
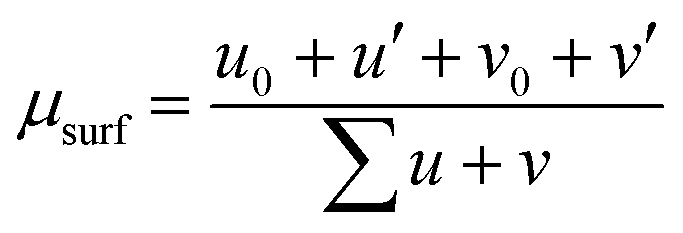 | (3) |
Fourier-Transform Infrared Spectroscopy (FTIR) measurements were performed at the Laboratório de Materiais Poliméricos (LAPOL-UFRGS) using the Spectrum 1000 Perkin Elmer equipment. The FTIR measurements were performed with 4 cm−1 resolution and the result is from the average of 32 scans. Sample preparation for FTIR measurements was performed by mixing the sample powder with KBr.
In situ XAS measurements were conducted at Laboratório Nacional de Luz Síncrotron (LNLS). The nanoparticle powder was mixed with boron nitride (BN) and compacted to make a homogeneous 5 mm diameter pellet before introduction into the tubular furnace existing at the beamline. The quantity of nanoparticles and BN depends on the edge probed and is described below. In all the cases, the samples were initially exposed to a reducing atmosphere with 100 ml min−1 5% H2 + 95% He flux. Then the samples were heated to 400 °C at a 10 °C min−1 rate under this atmosphere. At 400 °C, the samples were kept in the reducing atmosphere for 30 minutes. After this, a mixture of 75 ml min−1 20% CO2 + 80% He and 300 ml min−1 5% H2 + 95% He was introduced, initiating the RWGS reaction at a temperature of 400 °C for 1 hour. After the RWGS reaction, the samples were cooled to room temperature under 200 ml min−1 N2.
In situ time-resolved XANES (X-ray Absorption Near Edge Structure) measurements at the Ce L3 edge (5723 eV) were performed in transmission mode at the DXAS beamline at LNLS.21 For the measurements, pellets containing 8 mg of Pt–CeO2 nanoparticle powder mixed with 40 mg of BN were inserted into the tubular furnace. The XANES spectra were collected every 18 s during the full reduction treatment and RWGS reaction. The measurements made use of a curved Si(111) crystal (dispersive polychromator) and a CCD detector. The time resolution of the measurements was 100 ms. The in situ time-resolved XANES measurements were performed simultaneously with time-resolved mass spectrometry measurements to correlate the CeO2 oxidation state with the sample's reactivity.
The reduction and reactivity of the nanoparticles toward CO2 dissociation were investigated by time-resolved mass spectrometry measurements during the in situ time-resolved XANES measurements described. The reaction products were analyzed using a Pfeiffer Vacuum OmniStar mass spectrometer coupled to the tubular furnace. To remove possible signal fluctuations coming from the flux variations on the inserted gas, the signal from CO (m/q = 28) was normalized by the He (m/q = 2) signal detected, where m/q denotes the mass/charge ratio since He is an inert gas and it is not interacting with the sample and the H2/He and CO2/He ratios are fixed. The reactivity towards the formation of CO was determined through the CO/CO2 signal ratio normalized by the Pt–CeO2 mass of the sample.
The in situ time-resolved XANES spectra were fitted using a linear combination of arctangent and Lorentzian functions, as described by Takahashi et al.22 For this fitting, XANES spectra of Ce(IV) and Ce(III) standards were fitted with two and one Lorentzian function, respectively. It also used an arctangent function with the same parameters for both standards. In this way, it was possible to obtain the parameters (width and position) of the arctangent and Lorentzian functions for the Ce(IV) and Ce(III) standards that were then used as fixed parameters for the fitting of the in situ XANES spectra of the samples. Only the height of each Lorentzian function was varied in the fitting procedure of each XANES spectrum of the samples. The Ce(III) fraction (μ) was calculated by using eqn (4)
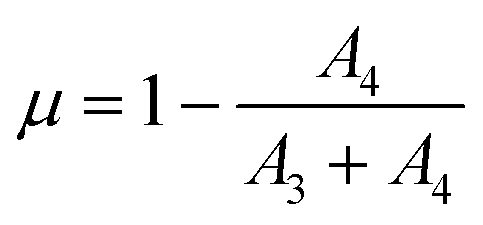 | (4) |
In situ Extended X-ray Absorption Fine Structure (EXAFS) measurements were performed at the Pt L3 edge (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV) in transmission mode at the XAFS2 beamline at LNLS23 to investigate the local atomic order around the Pt atoms during the thermal treatment. For the measurements, pellets containing 15 mg of Pt–CeO2 nanoparticle powder mixed with 40 mg of BN were inserted into the tubular furnace. In this beamline, the radiation emitted by a bending magnet was first collimated vertically by a Rh-coated cylindrical mirror, then the beam was monochromatized by a Si(111) double crystal monochromator and focused in the sample by a second Rh-coated mirror. The in situ EXAFS spectra were collected at the end of each gaseous treatment to ensure the achievement of a steady state of the sample (checked by short in situ XANES measurements). The measurements were performed using three ionization chambers filled with Ar. Standard Pt foil was used to calibrate the Si(111) monochromator. The spectra were acquired in the range of 11610–12025 eV with 0.5 and 1 eV per steps and 2 s per point and 3 s per point for the XANES and EXAFS regions, respectively. Two to four scans were collected to improve the signal-to-noise ratio.
564 eV) in transmission mode at the XAFS2 beamline at LNLS23 to investigate the local atomic order around the Pt atoms during the thermal treatment. For the measurements, pellets containing 15 mg of Pt–CeO2 nanoparticle powder mixed with 40 mg of BN were inserted into the tubular furnace. In this beamline, the radiation emitted by a bending magnet was first collimated vertically by a Rh-coated cylindrical mirror, then the beam was monochromatized by a Si(111) double crystal monochromator and focused in the sample by a second Rh-coated mirror. The in situ EXAFS spectra were collected at the end of each gaseous treatment to ensure the achievement of a steady state of the sample (checked by short in situ XANES measurements). The measurements were performed using three ionization chambers filled with Ar. Standard Pt foil was used to calibrate the Si(111) monochromator. The spectra were acquired in the range of 11610–12025 eV with 0.5 and 1 eV per steps and 2 s per point and 3 s per point for the XANES and EXAFS regions, respectively. Two to four scans were collected to improve the signal-to-noise ratio.
The EXAFS spectra were analyzed in accordance with the standard procedure of data reduction using IFEFFIT.24,25 The k2-weighted EXAFS signal k2χ(k) was extracted and then Fourier transformed using a Kaiser–Bessel window with a k range of 8.0 Å−1. The phase shift and amplitudes were obtained with the FEFF6 code26 by using metallic Pt, PtO2, and H2PtOH6 clusters of 6 Å radius. Single scattering events were considered in the fitting procedure of the coordination shell. The S02 value was fixed at 0.7 as obtained from the fitting of the Pt standard. The R-factor obtained from the analysis was always lower than 0.03, which demonstrates the excellent agreement between the proposed model and the experimental result.
Results and discussion
A typical TEM image of the Pt nanoparticles is shown in Fig. 1a, where it is possible to observe small nanoparticles with a mean diameter of 2.4 ± 0.4 nm and an almost circular projection in the plane. The SAXS pattern of the Pt nanoparticles is depicted in Fig. 1b. The big wavelength indicates the presence of small nanoparticles. The fitting result gives a lognormal size distribution of spherical particles of 3.0 ± 0.6 nm in diameter, in agreement with the TEM result. Fig. S1 of the ESI† shows the TEM images of the CeO2 nanoparticles synthesized, which present a mean diameter of 1.2 ± 0.2 nm and a surface area of 9.2 m2 g−1 (CeO2 Hyd), a mean diameter of 3 ± 2 nm and a surface area of 201 m2 g−1 (CeO2 high S) and a mean diameter of 26 ± 24 nm and a surface area of 38 m2 g−1 (CeO2 Std). Therefore, the Pt–CeO2 nanoparticles should consist of a mixture of Pt and CeO2 nanoparticles of similar sizes. A typical HRTEM image of the Pt–CeO2 Hyd sample is shown in Fig. 1c, where the presence of a uniform distribution of Pt–CeO2 interfaces constituted of individual Pt and CeO2 nanoparticles is observed. Furthermore, the contact angle measurement shows an average value of 157° for the CeO2 Hyd sample (Fig. S2 of the ESI†), which demonstrates a superhydrophobic behavior of the synthesized CeO2 sample. In the literature, it is well known that CeO2 presents a hydrophilic nature27 but it may become hydrophobic in nature after air exposure.28 Despite that, it is rare to find superhydrophobic CeO2 surfaces. The CeO2 Std sample measured in this work is highly hydrophilic since it adsorbs the water drop. It shows that the synthesized CeO2 Hyd sample greatly presents this unique property of superhydrophobicity, which may be highly useful for the RWGS reaction. The RBS spectra Fig. 1d show the presence of Pt, Ce, C, O, and Cl in all the samples. The Cl contamination comes probably from the Pt salt used in the synthesis process. In all the samples, the Pt amount was around 8 wt%, as expected.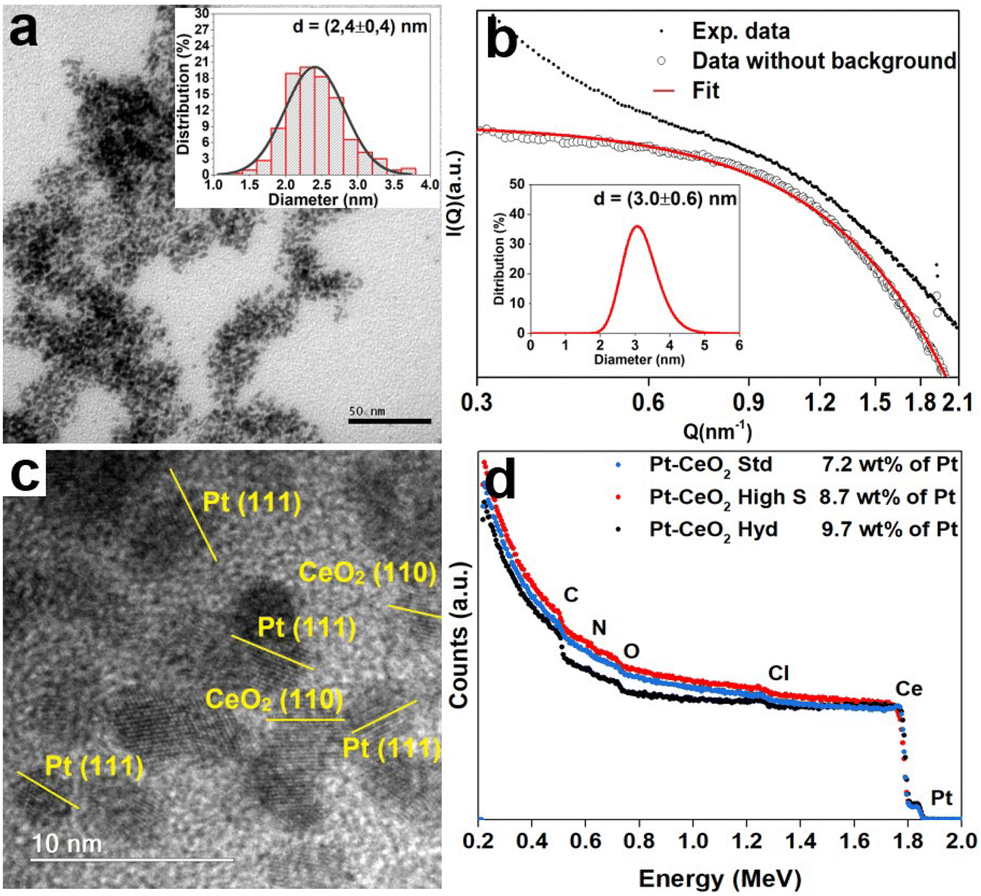 | ||
| Fig. 1 a) Typical TEM image of the commercial Pt nanoparticles and the histogram of size distribution (inset). b) SAXS pattern of the commercial Pt nanoparticles and the respective fitting result with the size distribution (inset). c) Typical HRTEM image of the Pt–CeO2 Hyd sample with the Pt and CeO2 nanoparticles identified. d) Normalized RBS spectra of the Pt–CeO2 Hyd, Pt–CeO2 high S and Pt–CeO2 Std samples in black, red and blue points, respectively. | ||
The XPS measurements at the Ce 3d electronic level for the as-prepared Pt–CeO2 samples are displayed in Fig. 2. The component labeled v′′′ at 916.7 eV serves as a direct indicator of the sample's oxidation state. A greater relative intensity in this component is correlated with a higher oxidation state. The Pt–CeO2 Hyd sample presents a small intensity for this component, indicating a highly reduced sample. The surface Ce(III) fractions obtained from the fitting for the as-prepared Pt–CeO2 nanoparticles were 0.77, 0.33, and 0.25 for the Pt–CeO2 Hyd, Pt–CeO2 high S, and Pt–CeO2 Std samples, respectively. The CeO2 nanoparticles synthesized in this work exhibited a high surface Ce(III) fraction in comparison to the literature,29–31 which is typically around 0.1. The high surface Ce(III) fraction values, especially for the Pt–CeO2 Hyd sample, give a large O vacancy population, which are the most active sites of metal oxides for catalysis.32 It is another promising result for using the synthesized nanoparticles in the RWGS reaction.
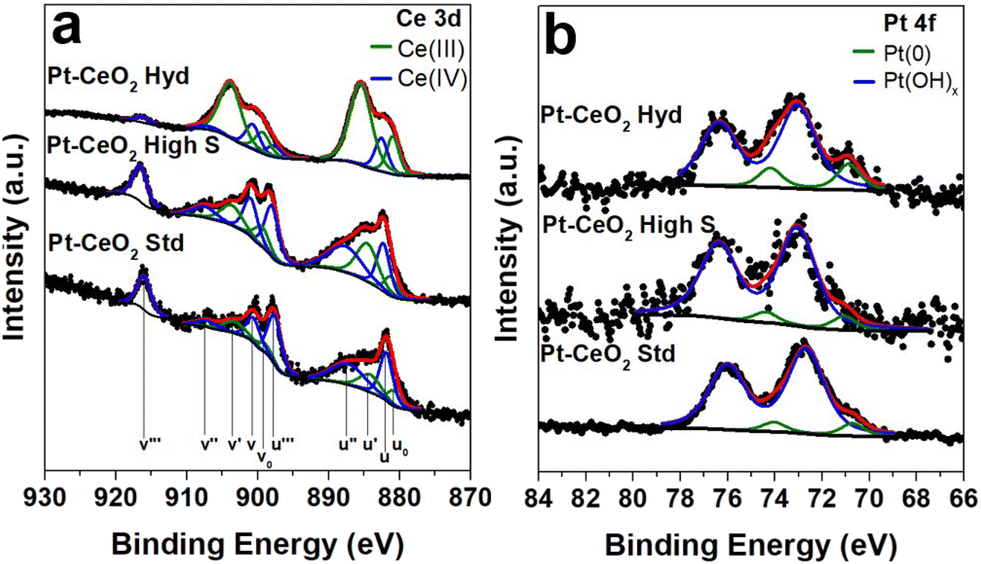 | ||
| Fig. 2 High resolution XPS spectra at a) Ce 3d and b) Pt 4f XPS regions of the as-prepared Pt–CeO2 nanoparticles. | ||
In the Pt 4f region, a consistent chemical environment around Pt atoms was observed across all the samples. A small metallic Pt component at 70.9 eV33 was detected, followed by the main contribution from Pt(OH)x at 73.0 eV33 coming from the surfactant present at the Pt nanoparticle surface. However, the posterior activation treatment, typically employed in catalysis, should be able to remove it and leave the Pt nanoparticles ready for the catalytic reaction.
The chemical components at Pt–CeO2 interfaces were studied by C 1s XPS measurements (Fig. S3 of the ESI†). In addition to the adventitious carbon, the presence of CO adsorbed at the CeO2 surface is observed in all the samples coming from air exposure. Interestingly, only the Pt–CeO2 Std sample presents CO bonding at the Pt surface. It may be associated with the low porosity of CeO2 in the standard sample, consequently, Pt nanoparticles are more exposed to the atmosphere and subjected to the formation of CO bonding at the surface. The CH3OH component appears due to the ethanol used during the synthesis process of CeO2. The phytantriol component in the C 1s XPS spectrum is expected at 282.7 eV due to the presence of highly reduced C atoms in the structure,34 but it is not observed in the Pt–CeO2 Hyd sample. FTIR measurements (Fig. S4 of the ESI†) were also conducted and the spectra are consistent with the findings in the XPS measurements. Moreover, it shows the absence of the characteristic bands of phytantriol.35
Mass spectrometry measurements were performed during the RWGS reaction. Fig. 3 shows the reactivity of the nanoparticles towards the formation of CO. The zero on the x-axis represents the beginning of the RWGS reaction at 400 °C. The reactivity of Pt–CeO2 Hyd, Pt–CeO2 high S, and Pt–CeO2 Std nanoparticles was determined as approximately 10%, 8%, and 3%, respectively. The Pt–CeO2 Hyd sample exhibits a much higher reactivity than the Pt–CeO2 Std sample and higher reactivity than the Pt–CeO2 high S sample, thus showing that the use of superhydrophobic nanoparticles results in increased reactivity for the RWGS reaction. In fact, the experiment was repeated for the CeO2 Hyd sample (without Pt) to investigate the effect of superhydrophobicity on the CO and H2O product formation. At 400 °C, the RWGS reaction started and a temperature-dependent study was conducted, heating the sample to 500 °C during the RWGS reaction. An increase of the reactivity with the temperature is expected, and a constant H2O/CO ratio due to the stoichiometry of the reaction. However, there is a strong drop to half of the value of the H2O/CO ratio with the increase of the temperature (Fig. S5a of the ESI†). It occurs because H2O molecules are bounded at the CeO2 surface due to the loss of the superhydrophobicity property of CeO2. Consequently, more CO molecules are detected at the mass spectrometer than H2O molecules and the H2O/CO ratio drops. Moreover, the reactivity starts to decrease as well (Fig. S5b of the ESI†), which shows that the loss of superhydrophobicity property impairs the reactivity towards CO production. It means that, indeed, the superhydrophobicity property helps on the reactivity towards CO formation. It is interesting to note that the Pt–CeO2 Hyd sample presents a smaller surface area than the Pt–CeO2 high S sample (9.2 m2 g−1 against 201 m2 g−1) but, even so, the superhydrophobicity property is enough to improve the reactivity of the nanoparticles. The reactivity normalized by the surface area gives a more realistic comparison between the samples since it allows evaluation of the performance of the active sites on each surface, despite the different amounts of active sites. This value for the Pt–CeO2 Hyd sample is almost 30 times bigger than the Pt–CeO2 high S one, which demonstrates the great advancement achieved in the present work. Furthermore, the reactivity towards CO formation is stable at least during 1 h of RWGS reaction.
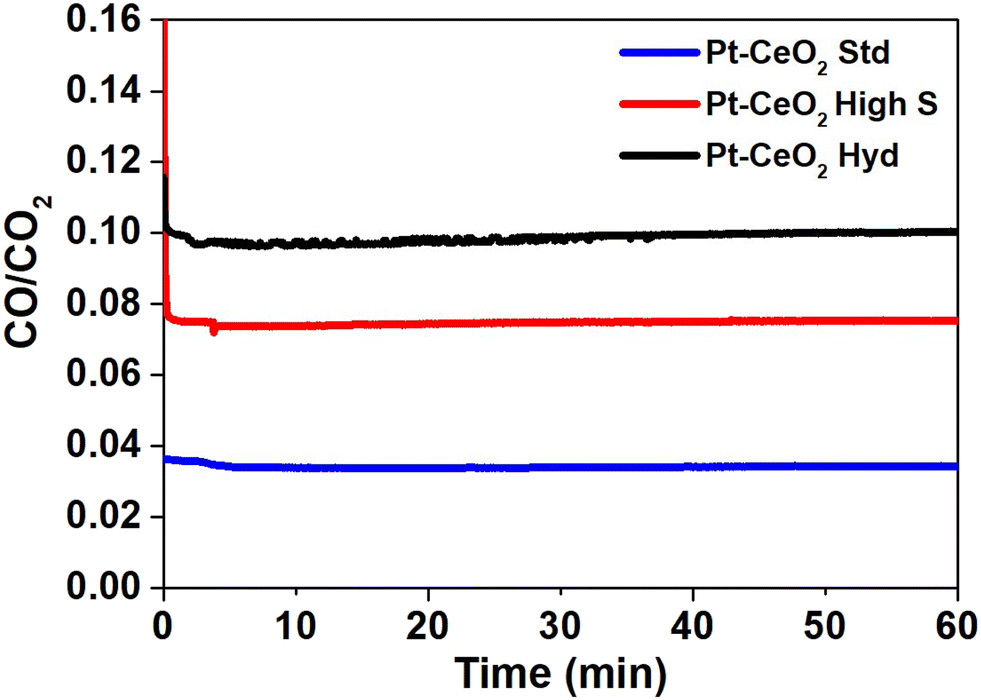 | ||
| Fig. 3 Normalized signal of the CO/CO2 ratio as a function of time during the RWGS reaction. | ||
Fig. S6 of the ESI† shows the H2O/CO ratio as a function of time obtained from mass spectrometry measurements during the RWGS reaction to identify whether the superhydrophobic property of the Pt–CeO2 Hyd sample is lost during the reaction at 400 °C. The result presents an almost constant H2O/CO ratio during the RWGS reaction, which shows that the superhydrophobic property was not lost during 1 h of RWGS reaction at 400 °C. The small increase observed (close to 3%) in the Pt–CeO2 Hyd sample occurs in the Pt–CeO2 high S sample (hydrophilic) as well and it is attributed to H2O molecules trapped at the connection walls of the mass spectrometer system. Thus, at least during 1 h of RWGS reaction at 400 °C, there is no evidence of the Pt–CeO2 Hyd sample losing the superhydrophobicity property. However, the increase of temperature induces the loss of the superhydrophobicity property, as shown in Fig. S5 of the ESI.†
Fig. 4 shows typical in situ time-resolved XANES spectra at the Ce L3 edge of the Pt–CeO2 samples. The spectra are shown every 5 min during the heating stage of the thermal treatment since this is the range of bigger changes observed. Fig. S7 of the ESI† shows the in situ time resolved XANES spectra at 400 °C, where no significant changes are observed. It can be observed that initially, all the samples exhibit a doublet, characteristic of the Ce(IV) oxidation state, although with visually different intensity ratios, which should imply different initial oxidation states. After starting the reduction treatment of the samples in the H2 atmosphere, a clear qualitative change from the Ce(IV) to Ce(III) oxidation state is observed in the Pt–CeO2 Hyd sample, with the appearance of a singlet, characteristic of the Ce(III) oxidation state, and a slight decrease in the height of the second peak in the Pt–CeO2 high S sample. The Pt–CeO2 Std sample does not show significant changes in the oxidation state during the reduction treatment. The Ce(III) fraction as a function of time, obtained from the analysis of the in situ XANES spectra of the three samples, is shown in Fig. 4d. Fig. S8 of the ESI† shows a typical fit result from the analysis conducted. During the heating process, all the samples showed a continuous increase in the Ce(III) fraction, except for the Pt–CeO2 Hyd sample, which stabilizes its oxidation state before reaching 400 °C. In a previous study,16 highly reducible CeO2 nanoparticles were synthesized. The nanoparticles were reduced at temperatures as low as 110 °C when exposed to a CO atmosphere. In particular, CeO2 high S nanoparticles presented a reduction temperature (defined as the inflection point of the curve of the Ce(III) fraction as a function of time) of 188 °C. In the present study, CeO2 high S nanoparticles presented a reduction temperature of 202 °C under H2 atmosphere, which makes sense since CO presents a higher reducing power than the H2 molecule in the CeO2 to Ce2O3 reduction reaction. The Ce(III) fraction values found are much higher than typical values from the literature for the Pt/CeO2 system after reduction treatment.29–31 In particular, Pastor-Pérez et al. achieved 37% of Ce(III) after reduction treatment at almost the same temperature used here (350 °C).31 In the present study, the Pt–CeO2 high S sample presents a higher Ce(III) fraction of 48%, and Pt–CeO2 Hyd is even more reduced with 98% of Ce(III) fraction. It is interesting to note that Pt/CeO2 nanoparticles should present the electronic effect of SMSI where a charge transfer from Pt to CeO2 is observed, thus helping with CeO2 reduction.30 For Pt–CeO2 Hyd nanoparticles, a much lower reduction temperature of 152 °C is found. It does not occur due to the charge transfer from Pt nanoparticles to CeO2 since the same experiment was repeated for CeO2 Hyd nanoparticles without Pt nanoparticles and almost the same reduction temperature was found (not shown here). In fact, H2-TPR measurements were conducted with the monitoring of the H2O signal using a mass spectrometer during sample reduction (Fig. S9 of the ESI†). The results point to Pt reduction from Pt(OH)x to Pt(0) starting at low temperatures of around 50 °C for all the samples. The Pt nanoparticles are fully reduced at around 175 °C. The reduction process of CeO2 starts at different temperatures. Pt–CeO2 Hyd starts to reduce earlier, followed by Pt–CeO2 high S and then the Pt–CeO2 Std sample, in accordance with the time evolution of the Ce(III) fraction (Fig. 4). It is consistent with a Pt–CeO2 interaction through charge transfer from Pt to CeO2, as previously reported,30 but the different extent on CeO2 reduction comes from the surface properties of the nanoparticles synthesized. This result shows the existence of easily reduced CeO2 nanoparticles, thus presenting plenty of oxygen vacancies, which are great active sites of metal oxides in catalysis (not restricted to the RWGS reaction).
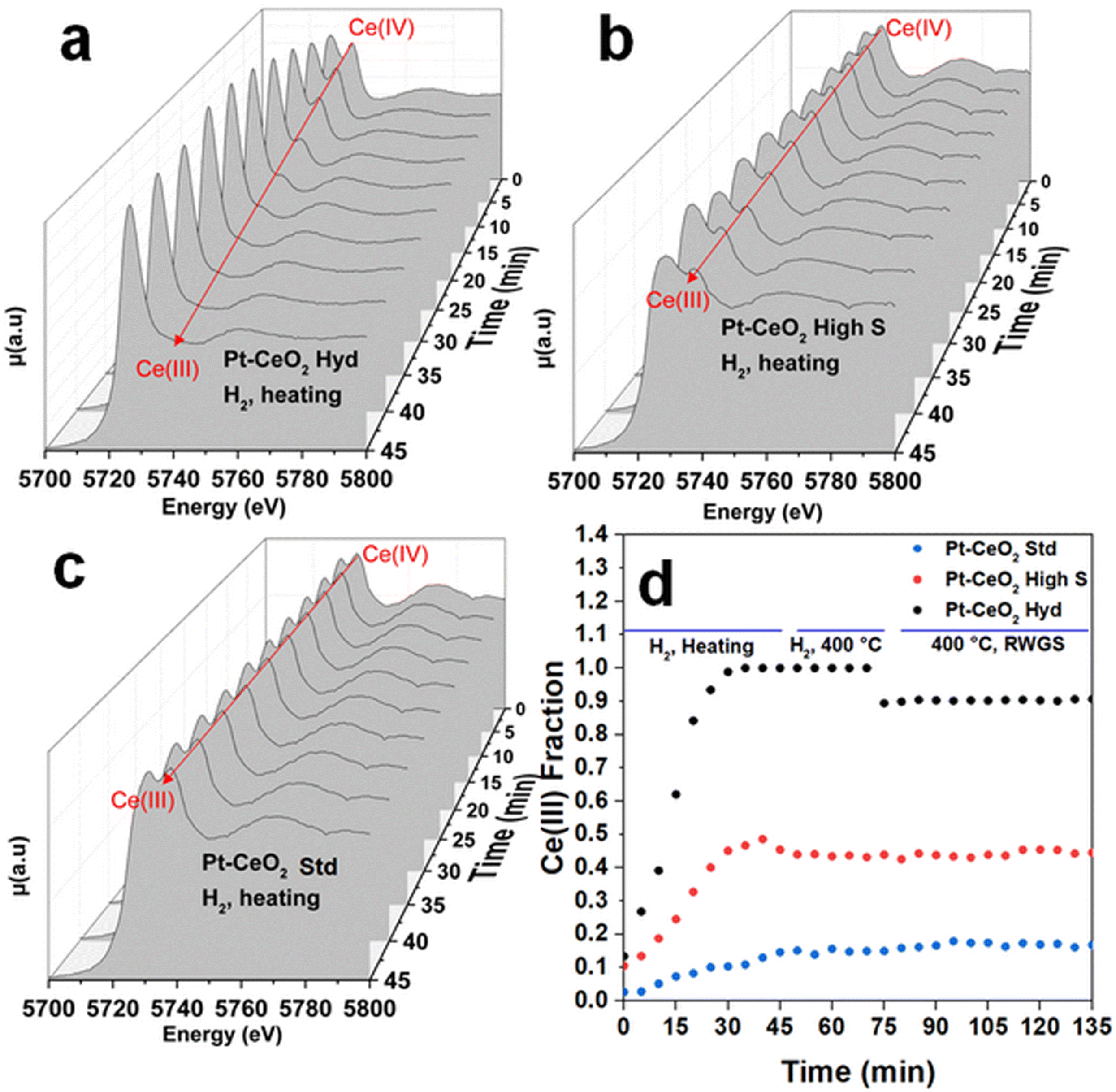 | ||
| Fig. 4 Time evolution of the in situ XANES spectra at the Ce L3 edge during heating treatment to 400 °C under H2 atmosphere for a) Pt–CeO2 Hyd, b) Pt–CeO2 high S and c) Pt–CeO2 Std samples. d) Time evolution of the Ce(III) fraction in the Pt–CeO2 samples during reduction and RWGS reaction. | ||
At 400 °C, the Ce(III) fraction becomes constant and stable in all the samples. During the RWGS reaction, the Ce(III) fraction remains the same in the Pt–CeO2 high S and Pt–CeO2 Std samples, while it slightly decreases by 10% in the Pt–CeO2 Hyd sample. Besides a small reduction, it occurs only at the beginning of the RWGS reaction and the Ce(III) fraction remains the same during 1 h of RWGS reaction. It indicates that the CO2 molecules are readily adsorbed at CeO2, then slightly oxidizing it, but it stabilizes quickly for the remaining time of reaction. It is observed that there is a direct relation between the reactivity towards CO formation and the O vacancy population in the CeO2 nanoparticles. The higher the O vacancy population, the higher the reactivity towards CO formation. In the literature it is possible to find studies reporting on the activation of CO2 in the RWGS reaction through the interaction between Pt and CeO2,36 frustrated Lewis pairs37 or O vacancies at the CeO2 surface.38 The slight oxidation of CeO2 in the Pt–CeO2 Hyd sample after CO2 insertion and the relation between reactivity towards CO formation and Ce(III) fraction indicates that the activation of CO2 occurs at the O vacancies of the CeO2 surface in this case. Then, in addition to the superhydrophobicity of the Pt–CeO2 Hyd sample, its great reducibility makes it highly interesting for catalytic applications. Considering this, the Pt–CeO2 Hyd sample is highly promising for future applications due to the high reactivity towards CO formation at a relatively low temperature and easy tunability (through the low temperature used) of the Ce oxidation state.
Fig. S10 of the ESI† shows the normalized in situ XAS spectra at the Pt L3 edge of the as-prepared Pt–CeO2 samples, during reduction treatment and RWGS reaction. It can be observed that initially, all spectra present high intensity right after the absorption edge, which is linked to a non-metallic state of Pt. This is in accordance with the XPS measurements at the Pt 4f electronic region. During the reduction treatment, the spectra of all the samples clearly changed to a metallic state, as expected. It is very similar to the findings reported in the literature.29 There is no further significant qualitative alteration observed during the RWGS reaction. The fitting of the XANES spectra with a linear combination of Pt and PtO2 standard spectra resulted in 100% Pt during both reduction and RWGS reactions.
Fig. 5 illustrates the k2-weighted EXAFS oscillations and the respective FT from in situ EXAFS measurements at the Pt L3 edge. The EXAFS oscillations of the as-prepared samples show a distinct pattern compared to that presented in the Pt foil. After reduction, the EXAFS oscillations are typical of metallic Pt but they are damped due to the high temperature of analysis. A comparison between the Fourier transform of the as-prepared Pt–CeO2 samples and the standard metallic Pt foil clearly reveals that the as-prepared Pt nanoparticles are not fully reduced since there is an important contribution at short distances from a Pt–OH scattering path. This statement is supported by the XPS measurements at the Pt 4f region, where a small metallic component was observed, followed by a huge contribution from hydroxyl groups bound to the Pt atoms. During reduction treatment, there is an increase in the intensity around 2.2 Å and 2.0 Å (not phase corrected), which are associated with Pt–Pt and Pt–O scattering paths, respectively, together with the disappearance of the Pt–(OH)x scattering. This indicates that the Pt atoms are reduced due to the thermal treatment applied, but a Pt–O component is still present.
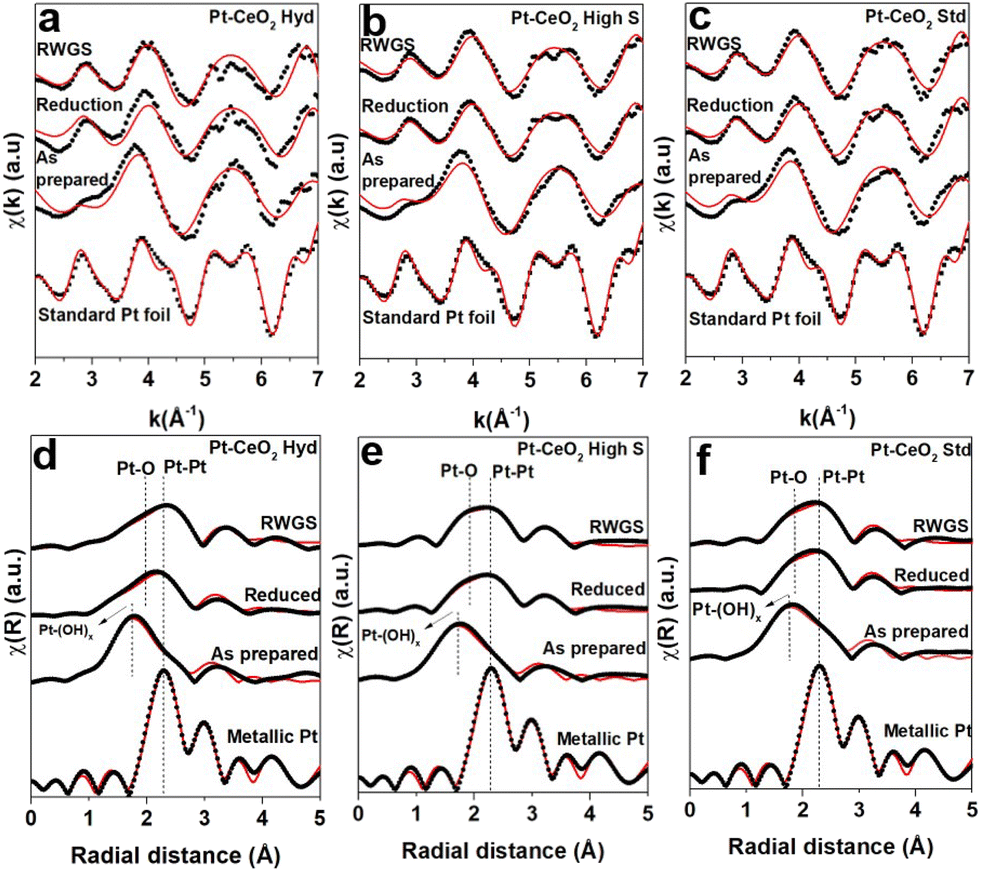 | ||
| Fig. 5 EXAFS oscillations from in situ EXAFS measurements at the Pt L3 edge of the as-prepared Pt–CeO2 samples, during reduction treatment and RWGS reaction (a–c) and the respective Fourier transform (d–f). The best fitting found is represented by the red line. | ||
Table S1 of the ESI† presents the parameters found during the fitting process described for the three samples. The Pt–Pt coordination number of the as-prepared nanoparticles points to a rough estimation of the size of a few nm,39 in agreement with the TEM and SAXS results. The Pt–(OH)x contribution is small when compared to the Pt–Pt one, which is consistent with the Pt–(OH)x bonding present at the surface of the nanoparticles. An increase in the coordination number associated with the Pt–Pt scattering path was also observed during reduction and RWGS reactions at 400 °C, which can be attributed to the sintering of Pt nanoparticles due to the elevated temperature.
Surprisingly, the Pt–O scattering path has an important contribution during the reduction treatment. The oxidation of Pt cannot be attributed to the gaseous atmosphere since only H2 and He were interacting with the sample during reduction treatment. At this temperature and atmosphere, it is expected that Pt reduces easily and becomes fully metallic.29 However, it is well known from the literature that Pt–CeO2 nanoparticles exhibit the geometrical factor of the SMSI effect under a reducing atmosphere at elevated temperatures.40,41 The formation of the capping layer covering the metallic nanoparticles is driven by the minimization of the surface energy.42 Metal nanoparticles typically present surface energy much higher than metal oxides. Despite the reduced size of the CeO2 nanoparticles as compared to typical systems in the literature, this relation still should hold due to the small size of the Pt nanoparticles as well and the typical variation of the surface energy of CeO2 with size in this size range.43 Then the presence of CeO2 nanoparticles smaller than Pt nanoparticles (Pt–CeO2 Hyd sample) should not avoid the geometrical factor of the SMSI effect whether there is indeed a Pt–CeO2 interface, as demonstrated in Fig. 1c. This effect can be responsible for the appearance of a strong Pt–O–Ce bond,44 which can explain the appearance of the Pt–O component. The geometrical factor of the SMSI effect may lead to a decrease or increase in the catalytic activity. Catalyst deactivation occurs due to the loss of catalytically active sites caused by the encapsulation of the metallic nanoparticles.4,12,40 On the other hand, in some cases the encapsulating layer may create new catalytically active sites at the surface11 that improve the catalytic activity. During the RWGS reaction, no new scattering path emerged. However, a slight increase in the coordination number associated with the Pt–O scattering path was observed. This increase is more evident in the Pt–CeO2 Hyd and Pt–CeO2 high S nanoparticles. The increase follows the same trend observed for the reactivity of the samples, indicating that it may come from byproducts of the RWGS reaction, especially related to the reduction of the CO2 molecules. It indicates that the gas is interacting with the Pt nanoparticles, even if the SMSI effect is present. It shows that the SMSI effect did not deactivate the samples, which can be explained by the penetration of the gas molecules through the encapsulating layer and interaction with the metallic surface,45 or by the presence of a partially encapsulated metallic nanoparticle. Finally, the Pt–CeO2 Hyd nanoparticles exhibit the best reactivity, even suffering from the SMSI effect, which may be attributed to the superhydrophobicity of CeO2 nanoparticles that shifts the reaction towards the formation of CO and its high reducibility.
Conclusions
This work demonstrated that superhydrophobic Pt–CeO2 interfaces are highly promising for the RWGS reaction with improved results even in comparison to high surface area Pt–CeO2 nanoparticles. The scenario is even more exciting because the superhydrophobic Pt–CeO2 nanoparticles are highly reducible and their O vacancy population is easily tunable through the temperature used in the reduction treatment. Besides the indication of the SMSI effect, these nanoparticles are still active toward CO formation in the RWGS reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by FAPERGS (Project Numbers 19/2551-0000703-5 and 19/2551-0001752-9), CNPq (Project numbers 19/2551-0000703-5, 19/2551-0001752-9 and 23/2551-0000177-2). K. G. G. dos S., A. S. T., L. P. M., G. Z. G., M. V. C. and F. B. thank the CNPq for the research grant. The authors also thank CNANO-UFRGS, CMM BR-Sul-UFRGS, LII-UFRGS, CEOMAT-UFRGS, LAPOL-UFRGS, LNLS, LNNano, and CM-UFMG for the use of the infrastructure.References
- I. Kurane, Osong Public Health Res. Perspect., 2010, 1, 4–9 CrossRef PubMed.
- P. Roy, S. C. Pal, R. Chakrabortty, I. Chowdhuri, A. Saha and M. Shit, J. Environ. Manage., 2023, 330, 117–187 CrossRef PubMed.
- M. Liu, G. A. Vecchi, J. A. Smith and T. R. Knutson, npj Clim. Atmos. Sci., 2019, 2, 1 CrossRef.
- W. T. Figueiredo, C. Escudero, V. Pérez-Dieste, C. A. Ospina and F. Bernardi, J. Phys. Chem. C, 2020, 124, 16868–16878 CrossRef CAS.
- D. H. Kim, S. W. Han, H. S. Yoon and Y. D. Kim, J. Ind. Eng. Chem., 2019, 23, 67–71 CrossRef.
- A. Goguet, S. Shekhtman, R. Burch, C. Hardacre, F. Meunier and G. Yablonsky, J. Catal., 2006, 237, 102–110 CrossRef CAS.
- C.-S. Chen, W.-H. Cheng and S.-S. Lin, Appl. Catal., A, 2003, 238, 55–67 CrossRef CAS.
- M. Ginés, A. Marchi and C. Apesteguía, Appl. Catal., A, 1997, 154, 155–171 CrossRef.
- W. Luhui, Z. Shaoxing and L. Yuan, J. Rare Earths, 2008, 26, 66–70 CrossRef.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- M. Ren, K. Qian and W. Huang, Chem. – Eur. J., 2019, 25, 15978–15982 CrossRef CAS PubMed.
- L. Wang, H. Liu, Y. Liu, Y. Chen and S. Yang, J. Rare Earths, 2013, 31, 969–974 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CrossRef CAS.
- F. Cao, Y. Xiao, Z. Zhang, J. Li, Z. Xia, X. Hu, Y. Ma and Y. Qu, J. Catal., 2022, 414, 25–32 CrossRef CAS.
- D. R. Bohn, F. O. Lobato, A. S. Thill, L. Steffens, M. Raabe, B. Donida, C. R. Vargas, D. J. Moura, F. Bernardi and F. Poletto, J. Mater. Chem. B, 2018, 6, 4920–4928 RSC.
- G. B. D. Mea, L. P. Matte, A. S. Thill, F. O. Lobato, E. V. Benvenutti, L. T. Arenas, A. Jürgensen, R. Hergenröder, F. Poletto and F. Bernardi, Appl. Surf. Sci., 2017, 422, 1102–1112 CrossRef.
- G. David and J. Perez, J. Appl. Crystallogr., 2009, 42, 892–900 CrossRef CAS.
- I. Brebler, J. Kohlbrecher and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 1587–1598 CrossRef PubMed.
- J. F. Ziegler, M. Ziegler and J. Biersack, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1818–1823 CrossRef CAS.
- K.-D. Schierbaum, Surf. Sci., 1998, 399, 29–38 CrossRef CAS.
- J. C. Cezar, N. M. Souza-Neto, C. Piamonteze, E. Tamura, F. Garcia, E. J. Carvalho, R. T. Neueschwander, A. Y. Ramos, H. C. N. Tolentino, A. Caneiro, M. J. M.-L. N. E. Massa, J. A. Alonso and J.-P. Itié, J. Synchrotron Radiat., 2010, 17, 93–102 CrossRef CAS PubMed.
- Y. Takahashi, H. Sakami and M. Nomura, Anal. Chim. Acta, 2002, 468, 345–354 CrossRef CAS.
- S. J. A. Figueroa, J. C. Mauricio, J. Murari, D. B. Beniz, J. R. Piton, H. H. Slepicka, M. F. de Sousa, A. M. Espíndola and A. P. S. Levinsky, J. Phys.: Conf. Ser., 2016, 712, 012022 CrossRef.
- D. C. Koningsberger and R. Prins, X-ray absorption: Principles, applications, techniques of EXAFS, SEXAFS and XANES, John Wiley and Sons, 1st edn, 1987 Search PubMed.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 4 CrossRef PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS PubMed.
- G. Azimi, R. Dhiman, H. Kwon, A. T. Paxson and K. K. Varanasi, Nat. Mater., 2013, 12, 315–320 CrossRef CAS PubMed.
- R. Lundy, C. Byrne, J. Bogan, K. Nolan, M. N. Collins, E. Dalton and R. Enright, ACS Appl. Mater. Interfaces, 2017, 9, 13751–13760 CrossRef CAS PubMed.
- S. Alayoglu, K. An, G. Melaet, S. Chen, F. Bernardi, L. W. Wang, A. E. Lindeman, N. Musselwhite, J. Guo, Z. Liu, M. A. Marcus and G. A. Somorjai, J. Phys. Chem. C, 2013, 117, 26608–26616 CrossRef CAS.
- A. S. Thill, A. S. Kilian and F. Bernardi, J. Phys. Chem. C, 2017, 121, 25323–25332 CrossRef CAS.
- L. Pastor-Pérez, V. Belda-Alcázar, C. Marini, M. M. Pastor-Blas, A. Sepúlveda-Escribano and E. V. Ramos-Fernandez, Appl. Catal., B, 2018, 225, 121–127 CrossRef.
- C. T. Campbell, J. Sci., 2005, 309, 713–714 CAS.
- M. Peuckert, Electrochim. Acta, 1984, 29, 1315–1320 CrossRef CAS.
- A. S. Thill, Development of highly promising NiO nanofoams for hydrogen storage application, PhD Thesis, UFRGS, 2023 Search PubMed.
- A. Misiûnas, Z. Talaikytë, G. Niaura and V. Razumas, Biologija, 2004, 4, 26–29 Search PubMed.
- F. Zhang, R. A. Gutiérrez, P. G. Lustemberg, Z. Liu, N. Rui, T. Wu, P. J. Ramírez, W. Xu, H. Idriss, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, ACS Catal., 2021, 11, 1613–1623 CrossRef CAS PubMed.
- W. Li, J. Gan, Y. Liu, Y. Zou, S. Zhang and Y. Qu, Angew. Chem., 2023, 135, 56–61 Search PubMed.
- A. Goguet, S. Shekhtman, R. Burch, C. Hardacre, F. Meunier and G. Yablonsky, J. Catal., 2006, 237, 102–110 CrossRef CAS.
- F. Rasera, A. S. Thill, L. P. Matte, G. Z. Girotto, H. V. Casara, G. B. D. Mea, N. M. Balzaretti, F. Poletto, C. Brito and F. Bernardi, ACS Appl. Nano Mater., 2023, 6, 6435–6443 CrossRef CAS.
- J. Z. Chen, A. Talpade, G. A. Canning, P. R. Probus, F. H. Ribeiro, A. K. Datye and J. T. Miller, J. Sci., 2020, 371, 4–710 Search PubMed.
- M. Abid and V. Paul-Boncour, Appl. Catal., A, 2006, 297, 48–59 CrossRef CAS.
- Q. Fu and T. Wagner, Surf. Sci. Spectra, 2007, 62, 431–498 CAS.
- V. Perebeinos, S.-W. Chan and F. Zhang, Solid State Commun., 2002, 123, 295–297 CrossRef CAS.
- E. A. Derevyannikova, T. Y. Kardash, A. I. Stadnichenko, O. A. Stonkus, E. M. Slavinskaya, V. A. Svetlichnyi and A. I. Boronin, J. Phys. Chem. C, 2019, 123, 1320–1334 CrossRef CAS.
- F. Bernardi, M. E. Grass, Y. P. Hong, R. Chang, N. Jabeen, C. Zhang, B. W. Eichhorn, B. Seo, S. Alayoglu, Z. Hussain, S. H. Joo and Z. Liu, Catal. Today, 2016, 260, 95–99 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00064a |
| This journal is © The Royal Society of Chemistry 2024 |