Enhancing charge transport in isoindigo-based donor–acceptor copolymers by combining ionic doping with polar alkoxy side chains†
Received
19th March 2024
, Accepted 30th April 2024
First published on 1st May 2024
Abstract
Side chains of polymers play a crucial role in manipulating polymer interchain interactions, especially polar side chains that promote strong molecular stacking and facilitate ionic diffusion. Here, we first synthesize a series of isoindigo-based donor–acceptor copolymers with varying ratios of linear alkoxy and branched alkyl side chains (PII2TC8C10, PII2TPEO25, PII2TPEO40, PII2TPEO63). Among this group of polymers, PII2TPEO25 with partial alkoxy chains exhibits a high field-effect mobility of 1.98 ± 0.05 × 10−1 cm2 V−1 s−1, comparable to PII2TC8C10 with all branched alkyl side chains. After doping with an ionic additive, i.e., tetrabutylammonium perchlorate (TBAP), the hole mobility of PII2TPEO25 was significantly increased up to 0.37 cm2 V−1 s−1, which is almost twice the mobility of its counterpart without the ionic additive. The increase in charge mobility with the addition of TBAP is mainly attributed to the fact that the presence of the ionic species inhibits the torsion of the isoindigo-based copolymer backbone, which is induced by the alkoxy side chains. Polymers with alkoxy side chains have smaller torsion angles and larger crystal sizes than polymers without alkoxy side chains. This study demonstrates that the binding of polar alkoxy side chains to ionic additives is important for charge transport in donor–acceptor copolymers.
Introduction
Donor–acceptor conjugated polymer materials offer opportunities for the development of next-generation organic electronics, including field-effect transistors (FETs),1–12 solar cells,13–17 and memory devices.18–21 Solution-processable conjugated polymers generally consist of two parts: a π-conjugated backbone and flexible solubilizing side chains. The structures of the backbone and side chains of conjugated polymers directly affect their molecular crystallization and energy levels. Therefore, the molecular design of the side chain and backbone structures is crucial for electronic properties and device performance.
Many backbone structures have been demonstrated over the past few years.22–26 For example, isoindigo (ID) and diketopyrrolopyrrole (DPP) molecules have been shown to be strong electron-accepting units for donor–acceptor polymers, exhibiting high performance in solar cells and FETs.27–33 In addition, side chains are often incorporated into conjugated polymer chains to enhance their solubility in common organic solvents, to tune their intermolecular interaction, and to adjust their molecular stacking in the solid state.34,35 Recently, side-chain engineering in donor–acceptor copolymers has attracted more attention. It has been shown that several strategies for side-chain structural design can induce tighter π–π stacking and manipulate the morphology of conjugated polymers, including distant branching positions of alkyl chains36 and the use of siloxane-terminated solubilizing side chains,37 semifluorinated alkyl chains,38 and polar ethylene glycol chains.39 As mentioned above, side-chain engineering is becoming increasingly important for improving the semiconducting properties of conjugated polymers.
Recently, ion doping in FETs has been reported and studied by several research groups. For example, electrochemical transistors (OECTs) are mainly composed of an electrolyte and a conjugated polymer. Ions in the electrolyte diffuse into the polymer, thereby modulating the transport of carriers and ions in the channel during device operation.40–43 Several research groups have used conventional three-electrode electrochemical cells to measure the effect of different properties on poly(3-hexylthiophene) (P3HT)-based OECTs.44 Guo et al. investigated the effect of charge injection on OECTs using side-chain-free conjugated polymers.45
In addition, other groups have delved into controlling the threshold voltage, on/off current ratio, and charge mobility in FETs through n-type and p-type dopants.46–52 Such doping induces more charge carriers in organic semiconductors, thereby improving electronic properties.46,47,50,53 Conjugated polymers such as polycarbazoles, polyfluorenes, and polythiophenes have been investigated by many research groups with various dopants including FeCl3, NOPF6, and F4TCNQ.54,55 Recent studies have found that doping with ionic additives improves the charge transport ability of polymer semiconductors and provide nonvolatile memory properties that further demonstrate biological synaptic behavior.56,57 Previously, we investigated ion-associated transistors doped with tetrabutylammonium perchlorate (TBAP) in DPP-based conjugated polymers. These devices utilize anions to transfer electrons into the conjugated polymer, thereby reducing the torsional angle of the polymer backbone. This phenomenon leads to current enhancement and has the potential for long-term memory.58 Anions can transfer electrons into the conjugated polymer or reduce the torsion angle of the polymer backbone. Luo et al. used an ionic dopant, namely tetramethylammonium iodide (TMAI), to enhance the charge mobility of conjugated polymers.59 Heeney et al. found that a conjugated polymer doped with tetrabutylammonium fluoride (TBAF) showed a desirable current response that was independent of the gate voltage.60 Although doping methods can significantly improve the charge mobility of polymer FETs, the relationship between side chains and ionic dopants for charge transport in conjugated polymers remains poorly understood.
In this work, we investigate the effect of polar alkoxy chains on ion-doped conjugated polymers to understand how the side chain manipulates the ion-doping effect. Polar alkoxy chains were chosen as the side chains for the study due to the following reasons: (i) polar alkoxyl chains have a strong ion transport capacity and (ii) polar alkoxyl chains have strong dipole–dipole interactions. The chemical structure of the alkoxy chains is identical to that of poly(ethylene oxide) (PEO), which is the most promising polymer electrolyte for ion transport in solid-state lithium-ion batteries. Therefore, the addition of alkoxy chains promotes the distribution of ions in the polymer matrix. In addition, the polar alkoxy group induces dipole–dipole interactions between the side chains, leading to tight molecular stacking and thus improving device performance. To investigate the effect of alkoxy chains on ionic doping in conjugated polymers, we synthesized a series of isoindigo-bithiophene polymers with different ratios of alkoxy chains and branched alkyl side chains (Fig. 1). We systematically investigated the thermal, optical, and electrochemical properties of these polymers. In addition, the morphology and molecular stacking structure of the polymer films were characterized using atomic force microscopy (AFM) and grazing-incidence X-ray diffraction (GIXD). Our results showed that the addition of the ionic additive TBAP to the alkoxy side chains enhanced the charge mobility of the conjugated polymers. This study provides insight into the molecular design principles of polar side chains and the doping effects of polymer FETs.
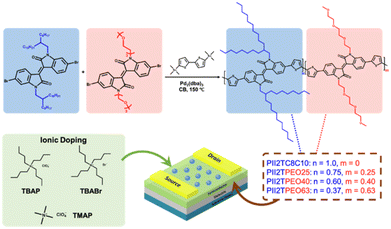 |
| | Fig. 1 The synthetic route of the studied isoindigo-based polymers, chemical structures of the ionic dopants, and the field-effect transistor device structure. | |
Experimental section
Materials
All the anhydrous solvents were purchased from Sigma-Aldrich (USA). Thiophene monomers, MgSO4, 2,3-dibromo-thiophene, Pd(PPh3)4, 6-bromoisatin, 6-bromooxindole, potassium carbonate, 1,1,3,3,5,5,5-heptamethyltrisiloxane, tri(o-tolyl) phosphine, and tris(dibenzylideneacetone)-dipalladium(0) were purchased from Sigma-Aldrich (USA), Alfa Aesar. (USA) and TCI (Japan) and used without further purification. The reagent for surface treatment, octadecyltrimethoxylsilane (95%) (OTS), was purchased from Gelest (USA).
N-Alkylation of 6,6′-dibromoisoindigo with alkoxy chains and branched alkyl side chain monomers
The detailed synthetic route is shown in Scheme S1.† 6,6′-Dibromoisoindigo (3 mmol) and potassium carbonate (12 mmol) were dissolved and mixed in dimethylformaldehyde (DMF) (50 mL). 1-(2-(2-Methoxyethoxy)-2-bromoethane) (alkoxy monomer) or 9-(bromomethyl)nonadecane (branched alkyl side chain monomer) (8 mmol) was added through a septum under an inert atmosphere at room temperature. The mixture was stirred overnight at 110–125 °C under nitrogen. The precipitates were collected by vacuum filtration and washed successively with dichloromethane and sodium chloride solution. The red solids were further purified by chromatography, eluting with dichloromethane and hexane (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 7:3). The combined fractions were concentrated to dryness to yield red powders.
:v = 7:3). The combined fractions were concentrated to dryness to yield red powders.
Polymerization of isoindigo-based polymers
The general method for synthesizing isoindigo-based conjugated polymers was as follows. 2-(Trimethylstannyl)-5-(5-(trimethylstannyl)thiophen-2-yl)thiophene monomer, tri-(otolyl)phosphine (16 mol% of 2-(trimethylstannyl)-5-(5-(trimethylstannyl)thiophen-2-yl)thiophene monomer), and tris(dibenzylideneacetone) dipalladium(0) (2 mol% with respect to 2-(trimethylstannyl)-5-(5-(trimethylstannyl) thiophen-2-yl)thiophene monomer) were dissolved in a microwave vessel using 5 ml of chlorobenzene under nitrogen. The vessel was microwaved at 180 °C for two hours. After end-capping with 2-(tributylstannyl)-thiophene and 2-bromothiophene at 200 °C for 5 min under microwave, the mixture was cooled and poured in methanol to form a crude polymer. The crude polymer was purified by Soxhlet extraction with methanol, acetone, and hexane, respectively. Finally, the powder was vacuum-dried at 40 °C. The characterization details are shown in the ESI.†
OFET fabrication and characterization
A bottom gate/top-contact transistor was employed. 300 nm-thick SiO2/Si acts as a gate dielectric/gate electrode. Octadecyltrimethoxylsilane (OTS) was modified on the SiO2 layer by spin-coating a 3 mM solution of OTS (in anhydrous toluene) at a spin rate of 3000 rpm. The OTS-treated wafers were then reacted in ammonia vapor at room temperature overnight. Finally, the OTS-treated wafers were rinsed using toluene and blown dry with nitrogen. All conjugated polymers were dissolved in dichlorobenzene (5 mg mL−1) at 110 °C overnight. The polymer solutions were coated onto the OTS-modified Si substrates by a solution shearing process. The gap between the top blade and the bottom substrate was around 100 μm. The conjugated polymer solution was sequentially placed on the substrate at a rate of 15 μL cm−2, and the shearing top blade was moving at a rate of 0.2–0.4 mm s−1. The sheared films were annealed at 170 °C under nitrogen for one hour. The top contact source/drain electrodes were defined using 80 nm thick gold. The channel length (L) and width (W) were 50 and 1000 μm, respectively. Electrical measurements were performed using a Keithley2634B under ambient conditions. The charge carrier mobility (μ) was calculated based on the following saturation-regime equation:
| IDS = μWCox(VG − VTH)2/2L |
where Cox is the capacitance per unit area = 10 nF cm−2 and VTH is the threshold voltage.
Results and discussion
Characterization of polymers
The chemical structures of the IID monomers with alkoxy chains were confirmed by 1H NMR (Fig. S1†). The chemical structures of the synthesized polymers were also confirmed by 1H NMR and elemental analysis, which are summarized in the ESI.† The average molecular weights (Mn) of PII2TC8C10, PII2TPEO25, and PII2TPEO40 were 30.8, 21.0, and 17.6 kg mol−1, respectively, and the corresponding polydispersity indices (PDIs), estimated from GPC, were 2.39, 2.00, and 2.10, respectively. It should be noted that due to the limited solubility, Mn and PDI are not available for PII2TPEO63 because it contains a high proportion of alkoxy side chains. In order to investigate the thermal stability of the polymers, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed, as shown in Fig. S2.† The thermal decomposition temperatures (Td, 5% weight loss) of PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63 were 389, 374, 379, and 381 °C, respectively, which indicates that the studied polymers have good thermal stability. On the other hand, no significant thermal transition was observed in the DSC traces, which is similar to the isoindigo-based polymers reported in the literature.
Optical properties
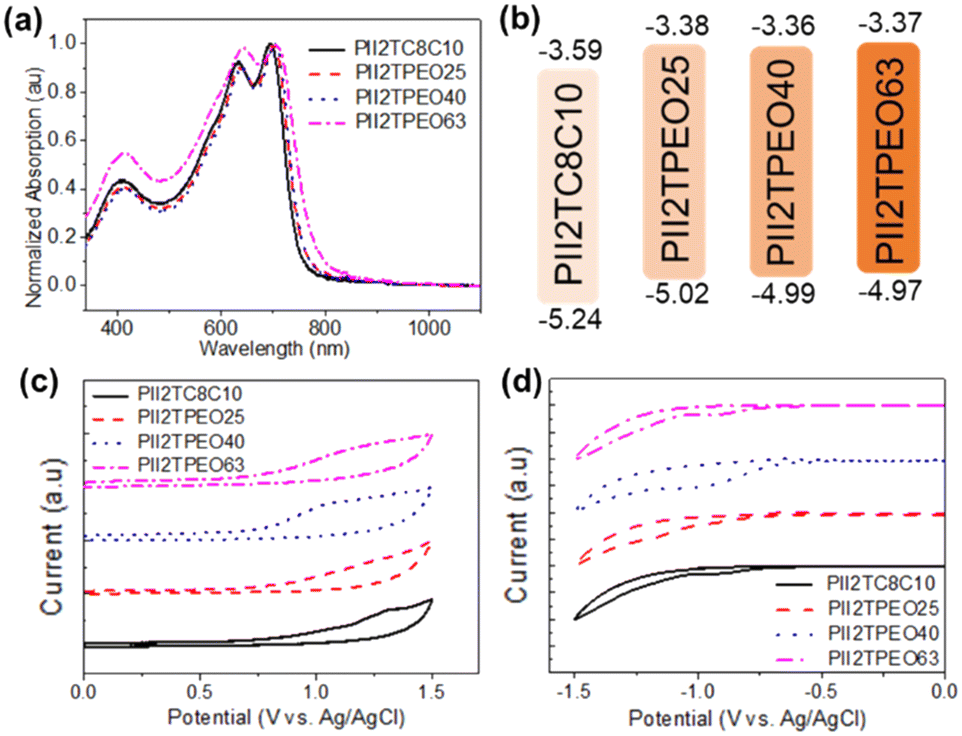
Optical characterization was carried out by UV-vis spectroscopy and electrochemical cyclic voltammetry. The absorption wavelength (λmaxfilm) and optical band gap (Egopt) of the polymer films are summarized in Table 1. The UV-vis spectra of the films are shown in Fig. 2a. As a reference polymer, PII2TC8C10 shows typical absorption bands of isoindigo-based donor–acceptor copolymers, with three characteristic bands at 408, 632, and 694 nm, respectively. It was observed that all absorption bands were red-shifted if there were more alkoxy side chains in the polymer. Specifically, PII2TPEO63, which has a higher percentage of alkoxy side chains, displays a red-shifted absorption band at 706 nm. This suggests that the presence of alkoxy side chains induces interactions between the polymer chains in the thin film. Based on the UV-visible data, their Egopt values were estimated from the onset of thin-film UV-vis absorption using the empirical formula Egopt = 1240/λonset, and the corresponding values for PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63 are 1.65, 1.64, 1.63, and 1.60 eV, respectively. Interestingly, among these polymers, PII2TPEO63 has the smallest band gap attributed to the large number of oxygen atoms in its alkoxy side chain. These oxygen atoms create additional dipole–dipole interactions with neighboring alkoxy side chains, thereby promoting a denser molecular packing between the polymer chain. It is worth noting that although PII2TPEO63 has a narrower band gap, its low solubility poses a challenge for device fabrication and results in poorer film quality.
Table 1 Optical characteristics of the studied isoindigo-bithiophene polymers
|
|
λ
max
film (nm) |
E
g
opt,
(eV) |
E
red
onset/LUMO (V/eV) |
E
OX
onset/HOMO (V/eV) |
LUMOb |
|
E
g = 1240/λ (nm).
LUMO = HOMO − Egopt.
|
| PII2TC8C10 |
408, 632, 694 |
1.65 |
−0.65/−3.54 |
1.04/−5.24 |
−3.59 |
| PII2TPEO25 |
413, 637, 700 |
1.64 |
−0.69/−3.50 |
0.83/−5.02 |
−3.38 |
| PII2TPEO40 |
414, 639, 702 |
1.63 |
−0.70/−3.49 |
0.80/−4.99 |
−3.36 |
| PII2TPEO63 |
413, 643, 706 |
1.60 |
−0.73/−3.46 |
0.78/−4.97 |
−3.37 |
 |
| | Fig. 2 (a) UV-vis spectra and (b) band gaps of the studied polymer films. CV curves for (c) oxidation and (d) reduction of the studied isoindigo-based polymers. | |
Electrochemical property
The electrochemical behaviors of the studied polymers were measured by cyclic voltammetry (CV), as shown in Fig. 2c and d. The energy levels of the polymers were estimated using the CV data; the highest occupied molecular orbital (HOMO) energy level was calculated from the following equation: HOMO = −[Eoxonset − Eferrocene1/2 + 4.8] eV, and the lowest unoccupied molecular orbital (LUMO) energy level was given by the difference between the HOMO energy level and the optical band gap. Their details are summarized in Table 1 and Fig. 2b. The HOMO energy levels of PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63 are −5.24, −5.02, −4.99, and −4.97 eV, respectively, while their LUMO energy level values are −3.59, −3.38, −3.36 and −3.37 eV, respectively. It is noteworthy that the incorporation of alkoxy side chains tends to upshift the energy levels of the HOMO and LUMO, which suggests the strong electron-donating ability of alkoxy side chains. On the other hand, the presence of alkoxy side chains enhances interchain interactions and contributes to this change. Stronger interchain interactions help to improve the molecular packing of the polymers. We also investigated the energy levels of these polymers after ionic doping, as shown in Fig. S3.† After doping with TBAP, the HOMO energy level values of PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63 were found to be −5.13, −5.04, −5.06, and −4.89 eV, respectively, while their LUMO energy level values were −3.50, −3.42, −3.46, and −3.38 eV, respectively. The TBAP-doped conjugated polymers show a similar pattern.
FET characteristics
In order to evaluate the charge transport properties of the polymer films, we employed the FET technique, and Table 2 summarizes the electrical characteristics of the polymer films sheared on OTS-modified SiO2/Si substrates. As shown in Fig. 3a, the isoindigo-based polymers exhibit typical p-type transfer characteristics, and we calculated the saturation-regime mobility using the slope of the drain current curve (Id)−0.5 as a function of the gate voltage (VG). In general, the higher the drain current, the higher the field-effect mobility. As summarized in Table 2, the charge carrier mobilities of the studied FETs are 1.91 × 10−1, 1.98 × 10−1, 8.36 × 10–2, and 1.03 × 10−2 cm2 V−1 s−1, corresponding to PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63, which have a relatively high on/off current ratio of about 105. It can be seen that PII2T devices with a higher proportion of alkoxy chains have lower mobility. One of the main reasons for the lower mobility is that the increase in alkoxy chains reduces the solubility of the polymer in solution during the film fabrication, resulting in a relatively lower mobility for the PII2TPEO63 film than that for the other polymer films.
Table 2 Summary of FET performance of isoindigo-based polymers without and with dopants
| Polymer |
Dopant |
Doping ratio (wt%) |
Mobilityavg (cm2 V−1 s−1) |
On/off ratio |
V
th
avg (V) |
| PII2TC8C10 |
— |
— |
(1.91 ± 0.09) × 10−1 |
∼105 |
−1.32 ± 2.62 |
| PII2TPEO25 |
— |
— |
(1.98 ± 0.50) × 10−1 |
∼104 |
−18.49 ± 8.36 |
| PII2TPEO40 |
— |
— |
(8.36 ± 0.84) × 10−2 |
∼105 |
−6.48 ± 2.44 |
| PII2TPEO63 |
— |
— |
(1.03 ± 0.36) × 10−2 |
∼104 |
−13.01 ± 4.6 |
| PII2TPEO25 |
TBAP |
0.5 |
(2.76 ± 0.25) × 10−1 |
∼105 |
−28.51 ± 2.83 |
| PII2TPEO25 |
TBAP |
1 |
(3.70 ± 0.34) × 10−1 |
∼106 |
−18.09 ± 3.14 |
| PII2TPEO25 |
TBAP |
3 |
(2.27 ± 0.11) × 10−1 |
∼106 |
−12.48 ± 0.92 |
| PII2TPEO25 |
TBAP |
5 |
(2.07 ± 0.20) × 10−1 |
∼106 |
−12.92 ± 2.99 |
| PII2TPEO25 |
TMAP |
1 |
(2.67 ± 0.20) × 10−1 |
∼105 |
−30.07 ± 3.59 |
| PII2TPEO25 |
TBABr |
1 |
(1.91 ± 0.18) × 10−1 |
∼105 |
−18.23 ± 8.52 |
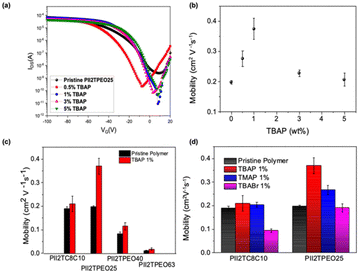 |
| | Fig. 3 FET characteristics of the studied polymers under different conditions: (a) transfer curves of the studied polymer films; (b) PII2TPEO25 doped with different ratios of TBAP; (c) different ratios of alkoxy side-chain conjugated polymers doped with 1% of TBAP; (d) PII2TC8C10 and PII2TPEO25 doped with different ionic additives. | |
To investigate the relationship between ionic species and solid-state conjugated polymers, the polymer films were doped with three different alkyl ammonium salts: namely, TBAP, tetramethylammonium perchlorate (TMAP), and tetrabutylammonium bromide (TBABr). Table 2 and Fig. 3 summarize the device performance under various ion doping conditions. Similar results were observed in the DPP-based polymers, as shown in Fig. S4.† Since PII2TPEO25 has a high mobility in the undoped state, it was initially chosen to study the effect of dopant concentration (TBAP) on its mobility. TBAP was studied for different doping ratios (0.5%, 1%, 3%, and 5% w/w), as shown in Fig. 3a. The hole mobility of the doped PII2TPEO25 device is simplified, as shown in Fig. 3b. It was found that the optimal doping concentration for the PII2TPEO25 device was 1 wt%. Its average hole mobility increases to 0.37 cm2 V−1 s−1, which is almost twice that of the undoped FET (0.19 cm2 V−1 s−1). Similar mobility enhancements were observed in the FETs of other polymers, as depicted in Fig. 3c. The output characteristics of the isoindigo-based transistors without ionic doping and the PII2TPEO25 device doped with different amounts of TBAP are shown in Fig. S5 and S6,† respectively.
Next, we examine dopants with different chemical structures to compare the effects of cations and anions. TBAP and TMAP have the same perchlorate anion (ClO4−) but different alkyl ammonium groups. In contrast, TBAP and TBABr have the same cation but different anions. As illustrated in Fig. 3d, for the PII2TC8C10- and PII2TPEO25-based FETs, the hole mobility of both TBAP- and TMAP-doped FETs increased, while the performance of the TBABr-doped FET decreased significantly in the PII2TC8C10 device and slightly decreased in the PII2TPEO25 device. The performance degradation is attributed to the fact that bromide ions (Br−), a strong reducing agent, react electrochemically with the polymer side chains during doping, and the binding of electrons to holes reduces the hole mobility. In contrast to bromide ions, the stability of the perchlorate anion is such that it does not react electrochemically with the polymer chains, thus avoiding degradation of the charge transport behavior. On the other hand, the length of the dopant chains between TBAP and TMAP also affects the stacking characteristics of the alkoxyl side chains. We noticed that the longer the dopant chain length, the tighter the molecular stacking. Interestingly, unlike the PII2TPEO series, PII2TC8C10 containing 1 wt% TBAP did not show a significant increase in charge carrier mobility, even with various ionic dopants. This clearly highlights the importance of alkoxy chains for improving charge transport in isoindigo-based polymers. Comparing the mobility of PII2TC8C10 and PII2TPEO25 doped with different dopants, TBAP consistently outperforms TMAP and TBABr. This suggests that TBAP is the best choice regardless of the presence or absence of alkoxy or alkyl side chains in the polymer. We finally analyzed the electron paramagnetic resonance (EPR) spectra to probe the role of the dopant (TBAP) in PII2TPEO25, as shown in Fig. S7.† We observed a paramagnetic radical signal in the undoped polymer, which was attributed to the presence of alkoxyl groups. Subsequently, 1 wt% TBAP doping enhanced the EPR intensity, indicating that the ions were effectively doped into the conjugated polymer.60 Finally, we compared the best performing device with previously reported ion-doped polymer transistors and summarized the results in Table 3, which helps to understand the significance of polar side-chain modification for ion-doped polymer transistors.
Table 3 Summary of previously reported ion-doped polymer transistors
| Ref. |
Polymer |
Dopant |
Mobilityavg (cm2 V−1 s−1) |
On/off ratio |
V
th
avg (V) |
|
46
|
PC61BM |
FPI |
8.70 × 10−2 |
∼107 |
21 |
|
47
|
P3HT |
F4-TCNQ |
1.43 × 10−2 |
∼104 |
5.1 |
|
48
|
TIPS-pentacene |
o-MeO-DMBI |
3.2 |
∼106 |
N/A |
|
52
|
PDPPFu-BTI |
TBAI |
8.4 × 10−3 |
∼104 |
29.2 |
|
52
|
PDPPSe-BTI |
TBAI |
1.54 × 10−1 |
∼105 |
41.5 |
| This work |
PII2TPEO25 |
TBAP |
3.70 × 10−1 |
∼106 |
−18.09 |
Molecular simulations
In the presence of an ionic additive (ClO4−), we analyzed the molecular structure of polymers with alkoxy side chains using the Gaussian 09 package program. Two variants of the isoindigo-bithiophene dimers were investigated: one with a methyl group (representing PII2TC8C10, as shown in Fig. S8†) and the other with an alkoxy side chain (representing a polymer based on PII2TPEO, as illustrated in Fig. 4). This approach facilitates the calculations by substituting long branched alkyl chains with methyl groups and alkoxy side chains with mono alkoxy groups. The torsion angles (θ1, θ2, and θ3) in the polymer chains were investigated. It was found that the torsional angle θ1, which represents the angle of the side chains before and after doping with an ionic additive, increased from 104° to 110° for the branched alkyl side chains, while it slightly increased from 85° to 87° for the alkoxyl side chain PII2T, which is attributed to the difference in the structure of the side chains. It was observed that the linear alkoxy chains exhibited less steric hindrance than the branched alkyl chains, thus allowing ion diffusion without largely affecting polymer conformation. The torsion angles θ2 and θ3 of both polymer backbones were significantly reduced by ClO4− doping, which is in agreement with previous literature findings.61,62 It is noteworthy that the change in the torsion angle θ3 of the bithiophene of PII2TC8C10 after doping with the ionic additive is negligible, suggesting that the coplanarity in the ion-doped polymers is enhanced. Furthermore, the coplanarity of the linear alkoxy chains is better than that of the branched alkyl chains (Table 4).
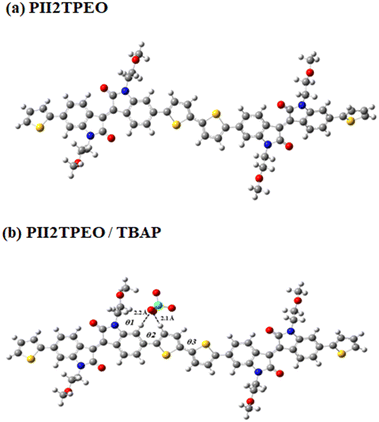 |
| | Fig. 4 Optimized structural geometries of (a) PII2TPEO and (b) PII2TPEO/ClO4− with mono alkoxy groups. | |
Table 4 Torsional angles of undoped and ClO4−-doped isoindigo-based polymers
| Polymer |
θ
1
|
θ
2
|
θ
3
|
| PII2TC8C10 |
−103.78 |
21.25 |
−15.00 |
| PII2TC8C10/ClO4− |
−110.09 |
10.11 |
−16.59 |
| PII2TPEO |
85.22 |
21.33 |
−15.24 |
| PII2TPEO/ClO4− |
86.89 |
9.47 |
1.5 |
Morphological characterization
The surface morphology and molecular stacking structures of the isoindigo-based donor–acceptor polymer films were investigated to analyze the relationship between side-chain effects and crystallinity within the polymer chains. Fig. 5 shows the tapping mode AFM phase images of the four polymer films. The average surface roughness (Ra) values of pristine PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO60 were 0.59 ± 0.1, 0.47 ± 0.22, 0.23 ± 0.07, and 2.81 ± 0.94 nm, respectively. Morphologically, worm-like structures are observed in PII2TC8C10 and PII2TPEO25, while PII2TPEO63 forms large aggregates due to its limited solubility. After doping with TBAP, small particles appear on the surface of TBAP-doped PII2TPEO25, resulting in an increase in surface roughness (Rms) from 0.51 nm to 2.09 nm (Fig. S9 in the ESI†). Although the formation of small aggregates is observed in the doped polymer films, this does not appear to result in a significant decrease in charge mobility, as shown by the results of FETs.
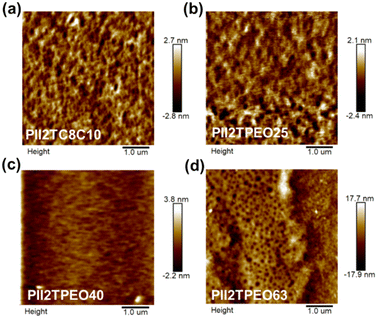 |
| | Fig. 5 AFM height images of the isoindigo-based polymer films of (a) PII2TC8C10, (b) PII2TPEO25, (c) PII2TPEO40, and (d) PII2TPEO63, respectively. | |
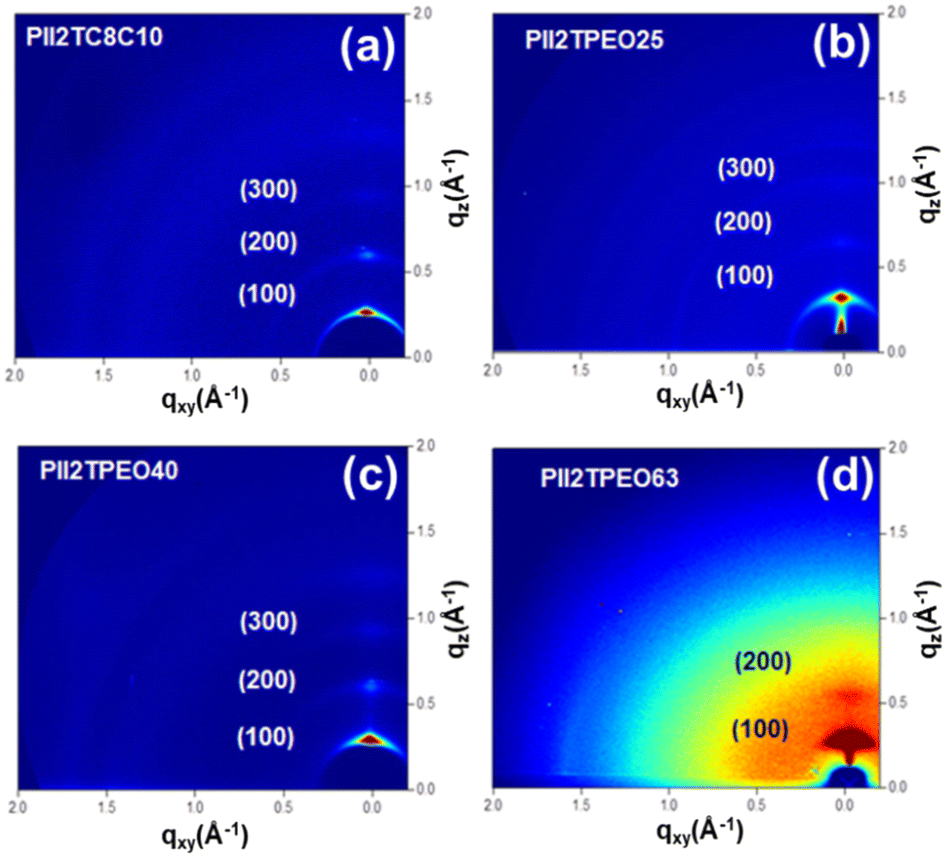
To investigate the crystalline structures of the PII2TPEO series further, we utilized the technique grazing-incidence X-ray diffraction (GIXD). The corresponding two-dimensional (2D) patterns are shown in Fig. 6, and 1D curves are shown in Fig. S10,† and the relevant crystallographic parameters are summarized in Table 5. As shown in Table 5, the isoindigo-based polymers show well-defined diffraction signals in the out-of-plane direction, with the main (100) diffraction peak corresponding to the lamellar d-spacing. The lamellar d-spacings for PII2TC8C10, PII2TPEO25, PII2TPEO40, and PII2TPEO63 are 19.71, 19.27, 18.35, and 16.57 Å, respectively. The corresponding π–π stacking distances are 3.75 to 3.80 Å. The lamellar spacing decreases significantly with the increase in the proportion of alkoxy chains, which is due to their shorter chain lengths. Moreover, the reduced steric hindrance of the linear chains may promote the interdigitation between the polymer chains, thus further shortening the lamellar spacing. PII2TC8C10, PII2TPEO25, and PII2TPEO40 exhibited stronger diffraction intensities and higher-order out-of-plane signals, suggesting that their self-assembly was more organized. After doping TBAP in PII2TPEO25, there was no significant difference in the lamellar d-spacing between the doped and pristine polymers. However, even with only 1 wt% of TBAP doping, the crystal size increased dramatically from 13.8 nm to 15.2 nm; conversely, the higher the doping ratio, the smaller the crystal size. The results suggest that the addition of a small amount of ionic dopant can induce larger crystal sizes, and finding the optimal crystal size will depend on the amount added, which will help to improve the mobility of FETs.
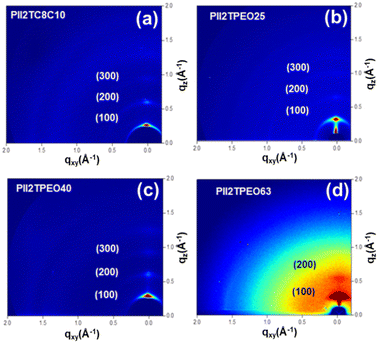 |
| | Fig. 6 2D GIXD patterns of (a) PII2TC8C10, (b) PII2TPEO25, (c) PII2TPEO40, and (d) PII2TPEO63, respectively. | |
Table 5 Crystalline structure parameters of pristine and ion-doped isoindigo-based polymers
| Polymer |
TBAP doping ratio (%) |
Lamellar spacing (Å) |
π–π spacing (Å) |
L
c (Å) |
| PII2TC8C10 |
0 |
19.71 |
3.80 |
139.11 |
| PII2TPEO25 |
0 |
19.27 |
3.75 |
138.31 |
| PII2TPEO40 |
0 |
18.35 |
N/A |
110.27 |
| PII2TPEO63 |
0 |
16.57 |
N/A |
96.39 |
| PII2TPEO25 |
0.5 |
18.68 |
3.68 |
146.30 |
| PII2TPEO25 |
1 |
18.14 |
3.67 |
152.76 |
| PII2TPEO25 |
5 |
19.35 |
3.71 |
143.04 |
Conclusions
Ion-doped thiophene-isoindigo donor–acceptor conjugated polymers with alkoxy side chains were successfully prepared. Notably, among these polymers, the mobility of PII2TPEO25 was as high as 0.19 cm2 V−1 s−1. Moreover, the addition of TBAP significantly enhanced the charge transport ability of the conjugated polymers with alkoxy side chains, and when doped with 1 wt% of TBAP, the crystallite size increased from 13.8 nm to 15.2 nm, and their mobility increased to 0.34 cm2 V−1 s−1 with an on/off current ratio of over 105. Gaussian calculations indicate that the increase in mobility is attributed to the reduction of the polymer backbone torsional angle after doping with the ionic additive TBAP. These findings emphasize the potential of incorporating alkoxy side chains in donor–acceptor copolymers to amplify the effect of ionic dopants. This study presents a new concept for the design of high-mobility polymer FETs through a synergistic combination of side-chain engineering and ionic doping.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
W. Y. Lee acknowledges the financial support from the Ministry of Science and Technology in Taiwan (110-2221-E-027-008-MY3 and 112-2221-E-027-012-MY3) and the Powerchip Semiconductor Manufacturing Corporation (112A0347). C.-C. C acknowledges financial supports from the National Science and Technology Council (NSTC) in Taiwan (111-2923-E-002-006-MY3, 112-2124-M-002-015, 112-2628-E-002-031, and 112-2223-E-002-008-MY4) and from the Top University Project of National Taiwan University (113NTUS05).
References
- A. L. Briseno, F. S. Kim, A. Babel, Y. Xia and S. A. Jenekhe, J. Mater. Chem., 2011, 21, 16461–16466 RSC.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS PubMed.
- Y.-J. Hwang, N. M. Murari and S. A. Jenekhe, Polym. Chem., 2013, 4, 3187–3195 RSC.
- G. Kim, A. R. Han, H. R. Lee, J. Lee, J. H. Oh and C. Yang, Chem. Commun., 2014, 50, 2180–2183 RSC.
- H.-W. Lin, W.-Y. Lee and W.-C. Chen, J. Mater. Chem., 2012, 22, 2120–2128 RSC.
- K. Mahmood, Z.-P. Liu, C. Li, Z. Lu, T. Fang, X. Liu, J. Zhou, T. Lei, J. Pei and Z. Bo, Polym. Chem., 2013, 4, 3563–3574 RSC.
- H.-C. Wu, S. J. Benight, A. Chortos, W.-Y. Lee, J. Mei, J. W. F. To, C. Lu, M. He, J. B. H. Tok, W.-C. Chen and Z. Bao, Chem. Mater., 2014, 26, 4544–4551 CrossRef CAS.
- Y. Yao, H. Dong and W. Hu, Polym. Chem., 2013, 4, 5197–5205 RSC.
- H. Yu, K. H. Park, I. Song, M.-J. Kim, Y.-H. Kim and J. H. Oh, J. Mater. Chem. C, 2015, 3, 11697–11704 RSC.
- H.-J. Yun, B. Lee Gi, S. Chung Dae, Y.-H. Kim and S.-K. Kwon, Adv. Mater., 2014, 26, 6612–6616 CrossRef CAS PubMed.
- X. Zhao, Y. Zhao, Q. Ge, K. Butrouna, Y. Diao, K. R. Graham and J. Mei, Macromolecules, 2016, 49, 2601–2608 CrossRef CAS.
- C. Y. Huang, X. F. Liao, K. Gao, L. J. Zuo, F. Lin, X. L. Shi, C. Z. Li, H. B. Liu, X. S. Li, F. Liu, Y. W. Chen, H. Z. Chen and A. K. Y. Jen, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS.
- J.-Y. Chen, H.-C. Wu, Y.-C. Chiu and W.-C. Chen, Adv. Energy Mater., 2014, 4, 1301665 CrossRef.
- L. Liu, J. Song, H. Lu, H. Wang and Z. Bo, Polym. Chem., 2016, 7, 319–329 RSC.
- S. Subramaniyan, T. Earmme, N. M. Murari and S. A. Jenekhe, Polym. Chem., 2014, 5, 5707–5715 RSC.
- J.-S. Wu, S.-W. Cheng, Y.-J. Cheng and C.-S. Hsu, Chem. Soc. Rev., 2015, 44, 1113–1154 RSC.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- Y.-H. Chou, H.-C. Chang, C.-L. Liu and W.-C. Chen, Polym. Chem., 2015, 6, 341–352 RSC.
- Q.-D. Ling, D.-J. Liaw, C. Zhu, D. S.-H. Chan, E.-T. Kang and K.-G. Neoh, Prog. Polym. Sci., 2008, 33, 917–978 CrossRef CAS.
- J.-T. Wang, S. Takashima, H.-C. Wu, Y.-C. Chiu, Y. Chen, T. Isono, T. Kakuchi, T. Satoh and W.-C. Chen, Adv. Funct. Mater., 2016, 26, 2695–2705 CrossRef CAS.
- H.-C. Wu, A.-D. Yu, W.-Y. Lee, C.-L. Liu and W.-C. Chen, Chem. Commun., 2012, 48, 9135–9137 RSC.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS.
- C. Thompson Barry and M. J. Fréchet Jean, Angew. Chem., Int. Ed., 2007, 47, 58–77 CrossRef PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- P. Sonar, H. S. Tan, S. Y. Sun, Y. M. Lam and A. Dodabalapur, Polym. Chem., 2013, 4, 1983–1994 RSC.
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed.
- Q. Liu, S. E. Bottle and P. Sonar, Adv. Mater., 2020, 32, 1903882 CrossRef CAS PubMed.
- D. D. Pei, Z. L. Wang, Z. X. Peng, J. D. Zhang, Y. F. Deng, Y. Han, L. Ye and Y. H. Geng, Macromolecules, 2020, 53, 4490–4500 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- X. Y. Wei, W. F. Zhang and G. Yu, Adv. Funct. Mater., 2021, 31, 2010979 CrossRef CAS.
- Q. Liu, K. Kanahashi, K. Matsuki, S. Manzhos, K. Feron, S. E. Bottle, K. Tanaka, T. Nanseki, T. Takenobu, H. Tanaka and P. Sonar, Adv. Electron. Mater., 2020, 6, 1901414 CrossRef CAS.
- J. Lee, S. A. Park, S. U. Ryu, D. Chung, T. Park and S. Y. Son, J. Mater. Chem. A, 2020, 8, 21455–21473 RSC.
- Y. Yamaguchi, Y. Kojiguchi, S. Kawata, T. Mori, K. Okamoto, M. Tsutsui, T. Koganezawa, H. Katagiri and T. Yasuda, Chem. Mater., 2020, 32, 5350–5360 CrossRef CAS.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- H.-G. Jeong, B. Lim, D. Khim, M. Han, J. Lee, J. Kim, J.-M. Yun, K. Cho, J.-W. Park and D.-Y. Kim, Adv. Mater., 2013, 25, 6416–6422 CrossRef CAS PubMed.
- C. Kanimozhi, N. Yaacobi-Gross, K. W. Chou, A. Amassian, T. D. Anthopoulos and S. Patil, J. Am. Chem. Soc., 2012, 134, 16532–16535 CrossRef CAS PubMed.
- R. Bhunia, E. K. Boahen, D. J. Kim, H. Oh, Z. Y. Kong and D. H. Kim, J. Mater. Chem. C, 2023, 11, 7485–7509 RSC.
- D. Rawlings, E. M. Thomas, R. A. Segalman and M. L. Chabinyc, Chem. Mater., 2019, 31, 8820–8829 CrossRef CAS.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- E. Zeglio and O. Inganäs, Adv. Mater., 2018, 30, 1800941 CrossRef PubMed.
- L. Q. Flagg, R. Giridharagopal, J. J. Guo and D. S. Ginger, Chem. Mater., 2018, 30, 5380–5389 CrossRef CAS.
- J. J. Guo, L. Q. Flagg, D. K. Tran, S. E. Chen, R. P. Li, N. B. Kolhe, R. Giridharagopal, S. A. Jenekhe, L. J. Richter and D. S. Ginger, J. Am. Chem. Soc., 2023, 145, 1866–1876 CrossRef CAS PubMed.
- C.-Z. Li, C.-C. Chueh, H.-L. Yip, F. Ding, X. Li and K. Y. Jen Alex, Adv. Mater., 2013, 25, 2457–2461 CrossRef CAS PubMed.
- L. Ma, W. H. Lee, Y. D. Park, J. S. Kim, H. S. Lee and K. Cho, Appl. Phys. Lett., 2008, 92, 063310 CrossRef.
- D. Naab Benjamin, S. Himmelberger, Y. Diao, K. Vandewal, P. Wei, B. Lussem, A. Salleo and Z. Bao, Adv. Mater., 2013, 25, 4663–4667 CrossRef CAS PubMed.
- D. Naab Benjamin, S. Zhang, K. Vandewal, A. Salleo, S. Barlow, R. Marder Seth and Z. Bao, Adv. Mater., 2014, 26, 4268–4272 CrossRef CAS PubMed.
- P. Wei, T. Menke, B. D. Naab, K. Leo, M. Riede and Z. Bao, J. Am. Chem. Soc., 2012, 134, 3999–4002 CrossRef CAS PubMed.
- Y. Zhang, H. Zhou, J. Seifter, L. Ying, A. Mikhailovsky, J. Heeger Alan, C. Bazan Guillermo and T.-Q. Nguyen, Adv. Mater., 2013, 25, 7038–7044 CrossRef CAS PubMed.
- Q. Liu, W. E. He, Y. Q. Shi, S. Otep, W. L. Tan, S. Manzhos, C. R. McNeill, X. G. Guo, P. Sonar, T. Michinobu and A. K. K. Kyaw, Chem. Mater., 2022, 34, 3140–3151 CrossRef CAS.
- Y. Nogami, H. Kaneko, T. Ishiguro, A. Takahashi, J. Tsukamoto and N. Hosoito, Solid State Commun., 1990, 76, 583–586 CrossRef CAS.
- M. Glaudell Anne, E. Cochran Justin, N. Patel Shrayesh and L. Chabinyc Michael, Adv. Energy Mater., 2014, 5, 1401072 CrossRef.
- K.-H. Yim, L. Whiting Gregory, E. Murphy Craig, J. M. Halls Jonathan, H. Burroughes Jeremy, H. Friend Richard and J.-S. Kim, Adv. Mater., 2008, 20, 3319–3324 CrossRef CAS.
- R. Bhunia, J. S. Kim, H. Kweon, D. J. Kim and D. Kim, Mater. Chem. Phys., 2022, 287, 126227 CrossRef CAS.
- D. H. Ho, D. G. Roe, Y. Y. Choi, S. Kim, Y. J. Choi, D. Kim, S. B. Jo and J. H. Cho, Sci. Adv., 2022, 8, eabn1838 CrossRef CAS.
- T. F. Yu, H. Y. Chen, M. Y. Liao, H. C. Tien, T. T. Chang, C. C. Chueh and W. Y. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 33968–33978 CrossRef CAS PubMed.
- H. Luo, C. Yu, Z. Liu, G. Zhang, H. Geng, Y. Yi, K. Broch, Y. Hu, A. Sadhanala, L. Jiang, P. Qi, Z. Cai, H. Sirringhaus and D. Zhang, Sci. Adv., 2016, 2, e1600076 CrossRef PubMed.
- Y. Han, Z. Fei, Y.-H. Lin, J. Martin, F. Tuna, T. D. Anthopoulos and M. Heeney, npj Flexible Electron., 2018, 2, 11 CrossRef.
- C. L. Donley, J. Zaumseil, J. W. Andreasen, M. M. Nielsen, H. Sirringhaus, R. H. Friend and J. S. Kim, J. Am. Chem. Soc., 2005, 127, 12890–12899 CrossRef CAS PubMed.
- H. W. Luo, C. M. Yu, Z. T. Liu, G. X. Zhang, H. Geng, Y. P. Yi, K. Broch, Y. Y. Hu, A. Sadhanala, L. Jiang, P. L. Qi, Z. X. Cai, H. Sirringhaus and D. Q. Zhang, Sci. Adv., 2016, 2, e1600076 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chu-Chen
Chueh
a,
Chu-Chen
Chueh