
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow introduction of hydrolyzable moieties in POx influences particle formation – a library approach based on block copolymers comprising polyesters†
Natalie E.
Göppert
ab,
Antje
Vollrath
ab,
Leanne M.
Stafast
ab,
Steffi
Stumpf
ab,
Bianca
Schulze
ab,
Stephanie
Hoeppener
 ab,
Christine
Weber
ab and
Ulrich S.
Schubert
*ab
ab,
Christine
Weber
ab and
Ulrich S.
Schubert
*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
First published on 12th September 2023
Abstract
A library of twelve fully degradable, amphiphilic block copolymers based on degradable poly(2-alkyl-2-oxazoline) analogues (dPAOx) and polyesters was synthesized via strain-promoted azide–alkyne cycloaddition of azido terminated dPAOx and cyclooctyne-initiated poly(ε-caprolactone) or poly(L-lactic acid), respectively. Different amounts of glycine moieties (15%, 32% and 47%) were incorporated in the hydrophilic dPAOx through oxidation of poly(ethylene imine) and consecutive re-acylation using acetyl or propionyl chloride. The resulting block copolymers were characterized in detail by means of NMR spectroscopy, size exclusion chromatography and matrix-assisted laser desorption ionization mass spectrometry. The polymers’ degradability was confirmed by a stepwise hydrolysis of the polyesters under alkaline conditions, followed by cleavage of the dPAOx under acidic conditions. A high-throughput nanoprecipitation method using a liquid handling robot was applied to investigate the influence of the glycine moieties on the particle formulation and the stability in direct comparison to block copolymers comprising poly(2-ethyl-2-oxazoline)s (PEtOx). Stable particle dispersions were obtained for most formulations even without the utilization of surfactants. Electrophoretic light scattering revealed an increase of the zeta potential with increasing amount of glycine units in all cases, an effect which appeared more prominent for the degradable PEtOx based block copolymers compared to those comprising degradable poly(2-methyl-2-oxazoline). The new amphiphilic block copolymers seem promising for encapsulation of drugs into fully degradable carrier materials.
Introduction
Environmental pollution through (micro)plastics is a huge problem that has been realized in public, as has the switch to degradable packaging materials. However, the average person is much less aware of the everyday use of water-soluble polymers despite its presence in many products. Among synthetic polymers, poly(ethylene glycol) (PEG) is probably one of the most widely used materials in that context. Besides its use for industrial purposes, it forms part of, e.g., many cosmetic products and, hence, of our wastewater.1 As its removal is not trivial, the non-degradable PEG makes its way into the environment.Although much less abundant, polymers are also applied in the field of nanomedicine.2 The encapsulation of drugs in hydrophobic polymer nanocarriers to improve drug administration efficacy and drug loading is one example. Biodegradable hydrophobic polyesters, such as poly(ε-caprolactone) (PCL), poly(L-lactic acid) (PLA) or the copolymer poly(lactic-co-glycolic acid) (PLGA), are commonly used for this purpose.3 These polyesters can be degraded under various conditions, e.g. thermally or by hydrolysis. The latter can take place intracellularly, e.g., mediated by enzymes, or by pH-value.4,5 However, such formulations also contain hydrophilic polymers as building blocks to, e.g., enhance the blood circulation time of the nanocarriers.6,7 PEG is the gold standard in that respect.8 Its drawbacks, such as the occurrence of antibodies or allergic reactions, have led to an intense search for alternative materials within the scientific community.9,10 The hydrophilic poly(2-oxazoline)s (POx) are among the range of polymers that seem promising candidates for a potential replacement of PEG. Various examples of amphiphilic block copolymers comprising a hydrophilic POx and a hydrophobic polyester block were presented in literature.11–13 However, beside a long-term degradation via oxidation,14–16 PEG is considered non-degradable via hydrolysis. Hydrolysis of the POx amide moieties requires harsh conditions yielding linear poly(ethylene imine) (PEI). Thereby, the polymer backbone remains intact.
Recently we reported the development of a synthesis route towards degradable poly(2-alkyl-2-oxazoline) (dPAOx) analogues via a post-polymerization modification method to overcome this issue.17 Their degradability by hydrolysis was confirmed under acidic as well as enzymatic conditions. The synthetic method is based on the hydrolysis of poly(2-ethyl-2-oxazoline) (PEtOx), oxidation of the resulting linear PEI, and subsequent re-acylation (Scheme 1). Among a broad series of dPAOx, the poly(2-methyl-2-oxazoline) (dPMeOx) as well as poly(2-ethyl-2-oxazoline) analogues (dPEtOx) were hydrophilic.17 Although a method to obtain block copolymers with dPAOx segments was recently developed by the bio-orthogonal strain-promoted azide–alkyne cycloaddition (SPAAC),18 the properties of these materials remain mostly unexplored to date.
 | ||
| Scheme 1 Schematic representation of the synthesis route yielding fully degradable block copolymers comprising dPAOx and polyester segments as well as their PEtOx-b-PLA and PEtOx-b-PCL analogues. | ||
In general, degradable alternatives have to compete with their conventional analogues in terms of performance. In our case, the glycine moieties introduced in the oxidation step represent the main structural difference between POx and dPAOx (Scheme 1). The degree of oxidation (DO) of the oxidized PEI (oxPEI) represents hence a reasonable factor to be taken into account. Re-acylation using acetyl as well as propionyl chloride transformed each oxPEI into the respective dPMeOx or dPEtOx. The approach towards these degradable hydrophilic building blocks enabled a high comparability within the synthesized library since the building blocks were obtained with the same block length and the same DO for each dPEtOx/dPMeOx pair (Fig. 1). Moreover, block copolymers containing the PEtOx can be obtained from the same batch of starting material, enabling a direct comparison. PLA and PCL were chosen as hydrophobic building blocks to result in a library of twelve fully degradable block copolymers (Fig. 1).
 | ||
| Fig. 1 Generalized overview presenting the library approach towards fully degradable amphiphilic block copolymers. | ||
This set of polymer carrier materials resembles a starting point to exploit initially the properties of these systems albeit further variations of the polymer composition remain possible. We first focussed on the set of polymers presented in Fig. 1 since it enables potentially the formulation of drug carriers in aqueous dispersions.
In terms of formulation of particles, e.g., by nanoprecipitation, the parameters, such as, solvent/non-solvent ratio, polymer concentration, as well as processing parameters, such as, stirring speed or further purification steps have to be optimized as an essential step.19 For our studies we opted for a direct nanoprecipitation of the polymer library from tetrahydrofuran (THF) only. In this case, a direct comparison of the performance of the various fully backbone degradable block copolymers with their PEtOx-based counterparts is ensured. We hence focused on the variation of polymer concentration as well as the evaluation of repeatability of the formulation. Both parameters were tested in a high-throughput approach using a liquid handling robot.20
Experimental section
Materials
2-Ethyl-2-oxazoline (EtOx, 99+%) and triethylamine (NEt3, 99.7%) were obtained from Acros Organics. 2-Ethyl-2-oxazoline was dried over barium oxide and distilled under argon atmosphere. (1R,8S,9S)-Bicyclo[6.1.0]non-4-in-9-yl-methanol (BCN-OH), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%), benzoic acid (≥99.5%, ACS reagent), toluene (≥99.8%, anhydrous), and methyl tosylate (MeOTs, 98%) were obtained from Sigma Aldrich. MeOTs was dried over calcium hydride, distilled under reduced pressure and stored under argon atmosphere. Sodium azide (NaN3, 99%) was purchased from abcr. Hydrochloric acid (37%) was obtained from Fisher Chemicals. Aqueous hydrogen peroxide solution (30% w/w) was received from Carl Roth. Propionyl chloride (>98.0%) and ε-caprolactone (CL, >99%) were purchased from Tokyo Chemical Industry (TCI). CL was dried over calcium hydride, distilled under reduced pressure and stored under argon atmosphere. Acetyl chloride (≥90%) was obtained from Merck Schuchardt. Amberlite IRA-67 was obtained from Merck Supelco and washed serval times with deionized water before use. N,N-Dimethylformamide (DMF) and acetonitrile were dried in a solvent purification system (SPS, Pure solv EN, InnovativeTechnology). All other chemicals and solvents were purchased from commercial suppliers and used without further purification unless noted otherwise.Poly(L-lactic acid) was synthesized via 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (mTBD) catalyzed ring-opening polymerization from L-lactide (LA) initiated by BCN-OH as described in our recent publication.21
General methods and instrumentation
Proton (1H) nuclear magnetic resonance (NMR) spectra were obtained using a Bruker AC 300 MHz spectrometer. The measurements were performed at room temperature using either D2O, d4-methanol or deuterated chloroform as a solvent. Chemical shifts (δ) are given in parts per million (ppm) relative to the residual non-deuterated solvent resonance signal.Size exclusion chromatography (SEC) was measured on an Agilent 1200 series system equipped with a PSS degasser, a G1310A pump, a G1329A auto sampler, a Techlab oven at 40 °C, a G1362A refractive index detector (RID) and a PSS GRAM guard/30/1000 Å column (10 μm particle size). N,N-Dimethylacetamide (DMAc) with 0.21 wt% LiCl was used as an eluent at a flow rate of 1 mL min−1. Polystyrene (PS) standards (400 to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1) were used to calculate the molar masses.
000000 g mol−1) were used to calculate the molar masses.
Matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI TOF MS) was executed on a rapifleX MALDI-TOF/TOF system from Bruker Daltonics equipped with a smartbeam™ 3D laser (355 nm wavelength). The spectra were measured in the positive reflector mode. 2,5-Dihydroxybenzoic acid (DHB) or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) were used as matrices. Sodium trifluoroacetate (NaTFA) or sodium iodide (NaI) were added as doping salts. A baseline subtraction and an external calibration with poly(methyl methacrylate) (PMMA) 5000 or 10000 g mol−1 standards from Polymer Standards Service (PSS) were performed subsequent to the measurement.
Attenuated total reflection (ATR) infrared (IR) spectroscopy was performed on a Shimadzu IRAffinity-1 CE system equipped with a quest ATR diamond extended range X – single-reflection-ATR accessory with a diamond crystal.
The polymerizations of BCN-PCL and BCN-PLA were performed in a glovebox manufactured by MBRAUN equipped with an UNIlab inert gas purification system, a vacuum pump and high efficiency box filters HEPA H13.
The high-throughput formulation (HT formulation) was realized by assistance of a liquid handling robot (Cybio Felix, Analytik Jena GmbH) that was equipped with a choice head R 96/250 μL and programmed with the CyBio Composer software.
Dynamic light scattering (DLS) was measured in a 96 well plate using the DynaPro Platereader III (Wyatt Technology Europe GmbH) and the Software DYNAMICS®. For each sample three acquisitions with three seconds each were performed at 25 °C. The used parameters for the size calculations were set to dn/dc = 0.185 and the RI (at 830 nm) to 1.58. The auto-attenuation was on and the plates were closed with sealing tape (Sigma Aldrich) during the measurements.
For electrophoretic light scattering (ELS) measurements and additional DLS investigations, the Zetasizer (Malvern Panalytical Ltd) was used. To this end, 75 μL of the dispersion were further diluted with 900 μL of water and transferred into a zeta cell and measured thrice for 10 seconds at 25 °C for both methods.
For the scanning electron microscopy (SEM) imaging of the particles, samples were prepared by dropping 10 μL on a mica substrate and drying it for 3 hours in the fume hood. Subsequently, the samples were coated with a thin layer of platinum (4 nm) via sputter coating (CCU-010 HV, Safematic). SEM was measured utilizing a Sigma VP Field Emission Scanning Electron Microscope (Carl-Zeiss AG). The micrographs were acquired with the InLens detector at a 4 or 6 kV acceleration voltage.
:[I] of 40:1 and a monomer concentration of 4 M, MeOTs (2.269 mL, 2.800 g, 15.03 mmol), and EtOx (61 mL, 60 g, 0.61 mol, 41 equiv.) were dissolved in dry acetonitrile (87 mL) in a pre-dried flask under argon atmosphere. The reaction mixture was stirred at 82 °C for 7 hours. Subsequently, NaN3 (4.9 g, 0.075 mol, 5.0 equiv.) was added, and the reaction mixture stirred at 70 °C for an additional 14 hours. An aliquot was removed and analyzed by means of 1H NMR spectroscopy (conversion quantitative). The excess of NaN3 was removed by filtration. The reaction solution was diluted with chloroform (600 mL) and washed with sat. aq. sodium bicarbonate solution (1000 mL) and brine (3 × 1000 mL). Removing of the volatiles under reduced pressure yielded PEtOx-N3 as a colorless solid (yield: 52 g, 86%).
Monomer conversion: quantitative. DPtheor. = 41. Mn,theor. = 4100 g mol−1.
1H NMR spectroscopy (300 MHz, CDCl3): δ = 3.62–3.27 (br, 164H, CH2–CH2), 3.10–2.91 (br, 3H, CH3–N), 2.57–2.11 (br, 82H, CO–CH2–CH3), 1.40–0.82 ppm (br, 123H, CO–CH2–CH3); Mn,NMR = 4100 g mol−1; DPNMR = 41.
SEC (DMAc, 0.21 wt% LiCl, RI detection, PS cal.): Mn = 8500 g mol−1; Đ = 1.10.
ATR-IR: ν(N3) = 2103 cm−1.
MALDI TOF MS (DCTB + NaTFA): Mn = 4400 g mol−1; Đ = 1.05; [H3C(C5H9NO)nN3 + Na]+, [H(C5H9NO)nN3 + Na]+, [H3C(C5H9NO)nOH + Na]+, [H3C(C5H9NO)nN + Na]+, [H(C5H9NO)nOH + Na]+ and [H(C5H9NO)nN + Na]+ observed.
M n,theor. = 1900 g mol−1.
1H NMR spectroscopy (300 MHz, CD3OD): δ = 3.53–3.43 (br, CH3–CH3, EtOx unit), 2.78 (br, 160H, CH2–CH2, ethylene imine unit), 2.51–2.45 (br, CO–CH2–CH3, EtOx unit), 1.34–1.09 ppm (br, CO–CH2–CH3, EtOx unit); degree of hydrolysis (DH) = 99%.
ATR-IR: ν(N3) = 2099 cm−1.
General procedure for the synthesis of poly(ethylene imine-stat-glycine) with varying degree of oxidation (DO)
The oxidation of PEI was adapted from a literature procedure with various equivalents of hydrogen peroxide to result in different DO.24 The PEI-N3 was dissolved in MeOH (0.02 g mL−1). An aqueous hydrogen peroxide solution (30% w/w) was added dropwise, and the reaction solution was stirred at room temperature for 3 days. Afterwards the volatiles were removed under reduced pressure, the residues were dissolved in deionized water and freeze-dried. Details on the oxidation to yield different degrees of oxidation are provided in the ESI.†General procedure for the synthesis of poly(2-alkyl-2-oxazoline-stat-glycine)
The synthesis of the dPAOx-N3 was performed according to our recent publication.18 The respective oxPEI-N3 was pre-dried at 70 °C for 2 hours. Dry DMF (6 mL g−1) was added to dissolve the polymer under argon atmosphere. Triethylamine (4 equiv. per amino unit) was added and the reaction mixture was cooled in an ice bath. A solution of 3 equivalents of acyl chloride with respect to the amino moieties was added in dry DMF dropwise to the reaction mixture followed by stirring at room temperature for 20 hours. Subsequently, precipitated triethylammonium chloride was removed by filtration and volatiles were removed under reduced pressure. The residue was dissolved in deionized water and purified with Amberlite IRA 67 ion exchange resin. The resin was filtered off and water was removed under reduced pressure. The residue was dissolved in methanol and precipitated in cold diethyl ether (−80 °C) twice. Dissolving in deionized water and freeze drying yielded the dPAOx as light brown to brown solids.Details on the synthesis of the different dPAOx-N3 are provided in the ESI.†
:1:0.5, the reaction solution was prepared under nitrogen atmosphere in a glovebox using ε-caprolactone (3.87 g, 34 mmol), BCN-OH (26 mg, 0.173 mmol) and TBD (12 mg, 0.09 mmol) in 17.5 mL dry toluene. The reaction proceeded under stirring at room temperature and samples were taken every hour (1 to 13 hours) and quenched in order to determine the monomer conversion by 1H NMR spectroscopy as well as the molar mass distribution by means of SEC. The solution was quenched after 13.86 hours by addition of an excess of benzoic acid (17 mg, 0.13 mmol). After removal of an aliquot to determine the monomer conversion by 1H NMR spectroscopy (35%), toluene was removed under reduced pressure. The crude polymer was dissolved in chloroform, and precipitated in methanol (−22 °C). Subsequent to drying in vacuo overnight, a white powder was obtained (950 mg).
1H NMR (300 MHz, CDCl3): δ = 4.18 (d, 2H, CH2–CO–O, BCN), 4.08 (t, 2H, CO–CH2–CH2–CH2–CH2–CH2–O, εCL unit), 2.65–2.68 (br, 4H, 2 × CH2, BCN ring), 2.33 (t, 2H, CO–CH2–CH2–CH2–CH2–CH2–O, εCL unit), 1.62–1.72 (br, 4H, CO–CH2–CH2–CH2–CH2–CH2–O, εCL unit), 1.35–1.45 ppm (br, 2H, CO–CH2–CH2–CH2–CH2–CH2–O, εCL unit).
SEC (DMAc, 0.21 wt% LiCl, RI detection, PS calibration): Mn = 16100 g mol−1; Đ = 1.43.
MALDI TOF MS (DCTB + NaI): Mn = 7400 g mol−1; Đ = 1.02; [C10H13O(C6H10O2)nH + Na]+ and [HO(C6H10O2)nH + Na]+ observed.
General procedure for the synthesis of the poly(2-alkyl-2-oxazoline-stat-glycine)-b-polyester block copolymers
The azido-terminated PEtOx, dPMeOx or dPEtOx building block and the polyester building block were dissolved in dichloromethane or DMF (cpolymers = 0.016 mol L−1), depending on their solubility. Dichloromethane was used for block copolymers comprising dPEtOx and PEtOx, whereas DMF was used for block copolymers containing dPMeOx. The solutions were stirred for 20 hours at ambient temperature. Afterwards the solvent was removed under reduced pressure, the residue was dissolved in chloroform and precipitated in methanol. Experimental details regarding the different block copolymer syntheses are provided in the ESI.†Degradation studies
The degradation was performed in two steps. In the first step the PEtOx32%-b-PLA (50 mg, 4 μmol) was dissolved in 1 M NaOH (1.3 mL, 3 mmol L−1) and stirred overnight at room temperature to degrade the PLA. Subsequently, the mixture was neutralized with 1 M HCl and freeze-dried. The degradation products were analyzed without any further purification. To degrade the dPEtOx32% in the second step, the mixture resulting from the first step was dissolved in 6 M HCl (1.3 mL, 3 mmol L−1) and stirred overnight at 90 °C. Afterwards, the mixture was neutralized with 1 M NaOH and dried under reduced pressure. The degradation products were analyzed without any further purification.High-throughput formulation and characterization
High-throughput (HT) formulation was performed with a liquid handling robot that used disposable tips with a volume of maximum 250 μL for the liquid transfer. At first, the polymers were dissolved in THF with a polymer concentration of 10 mg mL−1 with help of a warm water bath (T = 40 °C) and short vortexing steps in between for 10 seconds. In total, 1 mL stock solutions of each polymer were prepared. Prior to further usage, all solutions were filtered through a 0.45 μm poly(tetrafluoroethylene) (PTFE) syringe filter (Applichrom GmbH). Subsequently, four stock solutions per plate were divided into three parts with Vmin = 300 μL each and filled into three wells in the last row (H1–H3, H4–H6, H7–9, H10–12) of a 96 well plate (Greiner Bio-One GmbH) (see Fig. 6). This setup resulted in four plates that were used for the formulation of the sixteen polymers in total. The liquid handling robot was subsequently programmed to perform a dilution series of the polymers from the initial stock solutions with the following concentrations: 10, 8, 6, 5, 4, 3, 2 and 1 mg mL−1. Additionally, two 96 well plates (polypropylene 392 μL per well, flat bottom clear, Greiner Bio-One GmbH) were filled with 200 μL of purified water per well (GenPure ultrapure water system, Thermo Scientific).For the subsequent formulation, 20 μL of all 96 polymer solutions were transferred in parallel by the robot into the water containing plate number 1. The solvent/non-solvent ratio was 1:10. The polymer solution was injected into the water phase and the dispersion was directly mixed by aspiration and release thrice. The plate was located on a bioshaker that allowed immediate shaking for improved mixing after preparation. After 1 minute of shaking, the plates with the dispersions were removed from the liquid handling robot and stored in the fume hood for 1 hour for solvent evaporation. The same step was repeated for plate number 2 to end up with six formulations (in total two plates, three wells per concentration). The final particle concentration was in the range of 0.09 to 0.91 mg mL−1 in water dependent on the initial polymer concentration applied. Afterwards, 10 μL of the dispersion were transferred with help of the robot into another 96 well plate that contained 100 μL of purified water for subsequent HT-DLS analysis. Additionally, for the samples prepared with the initial polymer concentration of 3 mg mL−1 the zeta potential was measured. Moreover, the dispersions prepared with the initial polymer concentration of 3 mg mL−1 were also subjected to SEM imaging in order to visualize the particles and analyze their morphology.
Results and discussion
Synthesis of the clickable dPMeOx-N3 and dPEtOx-N3
To enable the attachment of the polyester block via SPAAC, the initial step towards the dPAOx-b-polyester library was the synthesis of an azido-terminated PEtOx to be utilized as a starting material in the post-polymerization synthesis towards the clickable dPAOx.17 The step was straightforward as the living cationic oxazolinium species, occurring due to the cationic ring-opening polymerization (CROP) mechanism, can be readily terminated with nucleophiles such as azide. A degree of polymerization (DP) of 40 was targeted as it ensured that a sufficient amount of glycine repeating units is incorporated in each chain even at a low DO. At the same time, the identification of the end group is still feasible at the corresponding molar mass of Mn,theo = 4000 g mol−1. Hence, PEtOx-N3 was obtained according to a literature procedure with minor adjustments yielding a polymer with a narrow molar mass distribution (Đ = 1.10).25,26 The presence of the resulting ω-terminal azido end group was confirmed by ATR-IR spectroscopy (ESI Fig. 1–3†), revealing the characteristic azido vibration band at 2103 cm−1. Also, MALDI TOF mass spectrometry (ESI Fig. 5†) confirmed the attachment of the end group. dPAOx were obtained from the PEtOx-N3 starting material via a three step post polymerization route adapted from our recent publications.17,18 It comprises the hydrolysis of the amide groups using 6 M hydrochloric acid resulting in the azido-terminated poly(ethylene imine) (PEI-N3), the consecutive oxidation with hydrogen peroxide solution leading to the formation of randomly distributed degradable glycine moieties in the backbone (oxPEI-N3) and, finally, the re-acylation of the remaining amino functionalities to re-introduce N-acyl-ethylene imine functionalities (Scheme 1 and ESI Fig. 1–4†). In line with literature reports, the azido moiety remained intact in PEI-N3 as well as in oxPEI-N3 as verified by ATR-IR spectroscopy.18,27To investigate the influence of the amount of incorporated glycine moieties in the targeted dPAOx-containing block copolymers and the resulting nanoparticle properties, the DO of the oxPEI-N3 was varied (Table 1). Whereas a synthetic proof of concept has been demonstrated previously for a rather high DO of 50%,18 the dPAOx are likely to exhibit significantly different properties compared their non-degradable counterparts. We hence included oxPEI-N3 with a DO of 47%, 32% and 15% into the study presented here.
| Polymer | DOa [%] |
SEC, DMAc | ELS and DLS | |||
|---|---|---|---|---|---|---|
|
M
nb [g mol−1] |
Đb |
ζc [mV] |
D
hc [nm] |
PDIc | ||
| a Determined by 1H NMR spectroscopy of the oxPEI-N3. b Determined by SEC in DMAc (0.21 wt% LiCl, PS calibration, RI detection). c Zeta potential ζ and hydrodynamic diameter Dh determined by ELS or DLS, respectively, using the Malvern Zetasizer. Samples were prepared using initial polymer concentrations of 3 mg mL−1. Instable formulations resulting in particle aggregation and in high PDI values > 0.25 are highlighted with an asterisk (*). | ||||||
| PEtOx-N3 | 0 | 8500 | 1.10 | — | — | — |
| PEI-N3 | 0 | — | — | — | — | — |
| oxPEI15%-N3 | 15 | 2200 | 1.75 | — | — | — |
| oxPEI32%-N3 | 32 | 1500 | 1.64 | — | — | — |
| oxPEI47%-N3 | 47 | 1500 | 1.62 | — | — | — |
| dPEtOx15%-N3 | 15 | 4200 | 1.49 | — | — | — |
| dPMeOx15%-N3 | 15 | 4300 | 1.85 | — | — | — |
| dPEtOx32%-N3 | 32 | 2800 | 1.56 | — | — | — |
| dPMeOx32%-N3 | 32 | 3800 | 1.82 | — | — | — |
| dPEtOx47%-N3 | 47 | 2700 | 1.57 | — | — | — |
| dPMeOx47%-N3 | 47 | 2800 | 1.99 | — | — | — |
| BCN-PLA | — | 17300 |
1.15 | −30 | 181 | 0.12 |
| BCN-PCL | — | 16100 |
1.43 | −22 | 259 | 0.18 |
| PEtOx-b-PLA | 0 | 22000 |
1.05 | −23 | 70 | 0.36* |
| PEtOx-b-PCL | 0 | 20900 |
1.09 | −13 | 97 | 0.25* |
| dPEtOx15%-b-PLA | 15 | 23300 |
1.26 | +18 | 164 | 0.19 |
| dPEtOx15%-b-PCL | 15 | 20600 |
1.32 | +20 | 156 | 0.10 |
| dPMeOx15%-b-PLA | 15 | 21800 |
1.23 | −22 | 145 | 0.15 |
| dPMeOx15%-b-PCL | 15 | 21300 |
1.32 | −13 | 170 | 0.10 |
| dPEtOx32%-b-PLA | 32 | 21200 |
1.10 | +29 | 149 | 0.15 |
| dPEtOx32%-b-PCL | 32 | 18900 |
1.29 | +31 | 149 | 0.13 |
| dPMeOx32%-b-PLA | 32 | 23400 |
1.23 | −9 | * | — |
| dPMeOx32%-b-PCL | 32 | 21200 |
1.32 | +8 | 330 | 0.30* |
| dPEtOx47%-b-PLA | 47 | 19700 |
1.09 | +15 | 134 | 0.14 |
| dPEtOx47%-b-PCL | 47 | 18500 |
1.30 | +28 | 144 | 0.13 |
| dPMeOx47%-b-PLA | 47 | 21000 |
1.23 | −5 | 713 | 0.34* |
| dPMeOx47%-b-PCL | 47 | 22500 |
1.39 | +16 | 158 | 0.12 |
Moreover, a variation of the hydrophilic dPAOx building block was achieved by introducing N-acetyl ethylene imine and N-propionyl ethylene imine functionalities in the last step of the synthesis sequence. The resulting polymers resemble the repeating units of PMeOx as well as PEtOx with randomly distributed glycine repeating units. The increasing amount of glycine moieties within the dPAOx chains is clearly visible from the 1H NMR spectra (Fig. 2 for dPEtOx, ESI Fig. 6† for dPMeOx). The backbone signals broaden, and the signal assigned to the glycine amide proton increases in intensity. In addition, ATR-IR spectroscopy revealed the presence of the characteristic azido band at around 2100 cm−1 for all dPMeOx-N3 and dPEtOx-N3 (ESI Fig. 1–3†).
 | ||
| Fig. 2 Overlay of the 1H NMR spectra (300 MHz, D2O) of PEtOx-N3 and the dPEtOx-N3 with different degree of oxidation dPEtOx15%-N3, dPEtOx32%-N3 and dPEtOx47%-N3. The spectra are normalized according to the intensity of signal A. | ||
Synthesis of the amphiphilic block copolymers from dPMeOx-N3 and dPEtOx-N3
As representatives of commonly used degradable hydrophobic polymers, PLA and PCL were chosen as counterparts for the SPAAC to yield fully degradable polymers. The cyclooctyne moiety required for that purpose was introduced by utilizing BCN-OH as an initiator for the ring-opening polymerization (ROP) of LA and CL, respectively, resulting in BCN-PLA and BCN-PCL.18,28 In addition to 1H NMR spectroscopy, in particular MALDI TOF MS confirmed the presence of the BCN α-end group (ESI Fig. 7†).21A library of fourteen block copolymers was synthesized via SPAAC using the PEtOx-N3, the six dPAOx and the two polyester building blocks as presented in Fig. 1. In general, the dPAOx and PEtOx blocks were utilized in excess to account for polymer chains that might lack the azide end group. The excess was readily removed during purification of the block copolymers by precipitation in methanol.
The 1H NMR spectra of the block copolymers revealed the signals of both respective building blocks as exemplified for the block copolymers comprising polymers with a DO = 15%, namely dPMeOx15%-b-PLA, dPMeOx15%-b-PCL, dPEtOx15%-b-PLA and dPEtOx15%-b-PCL in Fig. 3. Similar overlays of the 1H NMR spectra of the block copolymers with higher DO can be found in the ESI (Fig. 8–10†).
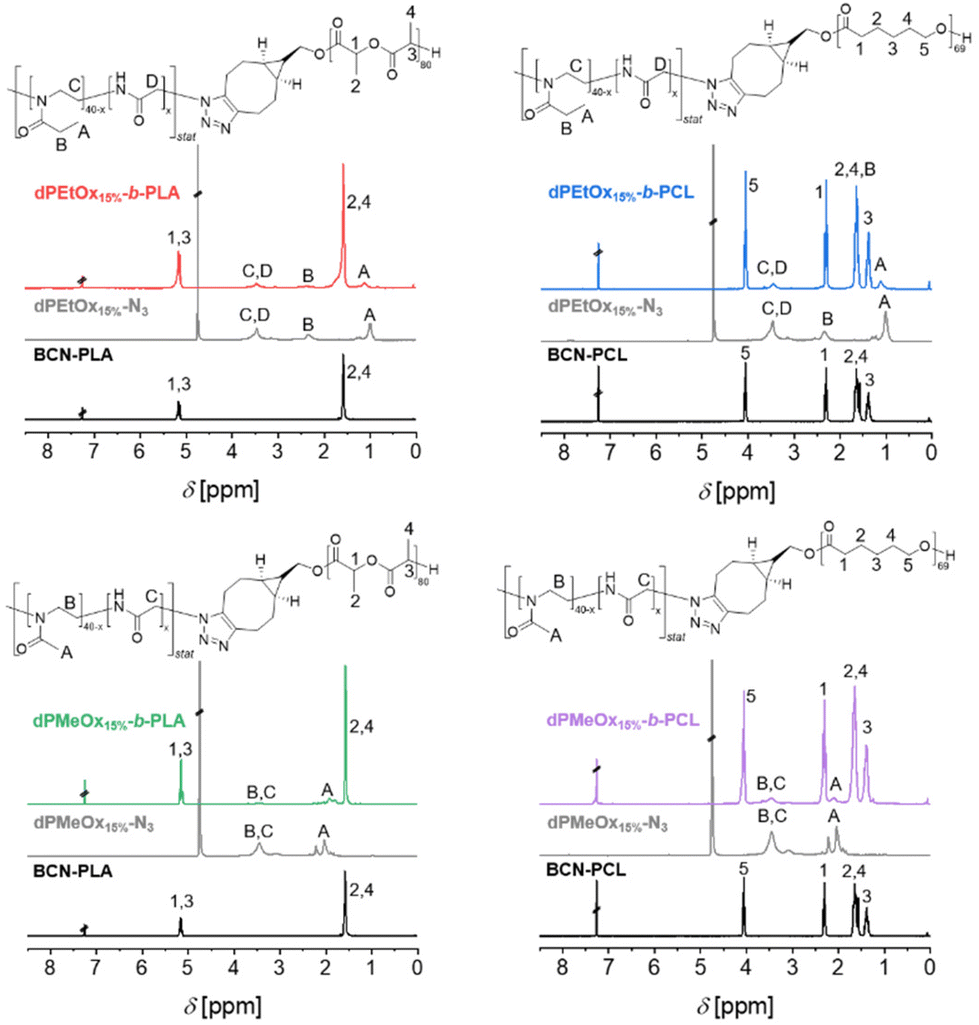 | ||
| Fig. 3 1H NMR spectra (300 MHz, D2O or CDCl3) of the polymers comprising dPAOx blocks with 15% DO dPMeOx15%-b-PLA, dPEtOx15%-b-PLA, dPMeOx15%-b-PCL, dPEtOx15%-b-PCL and the corresponding building blocks dPMeOx15%-N3, dPEtOx15%-N3, BCN-PLA and BCN-PCL, respectively, with assignment of the signals to the schematic representation of the structure. | ||
The successful SPAAC was also proven by the absence of the azido vibration band in the ATR-IR spectra of the block copolymers (ESI Fig. 11–14†) due to the transformation of the azido end group into a 5-membered heterocycle. Moreover, the ATR-IR spectra revealed two carbonyl vibration bands for the block copolymers corresponding to the ester moiety of PLA or PCL (1750 cm−1), and the amide moiety of PEtOx or dPAOx (1640 cm−1), respectively.
The SEC elugrams of the block copolymers revealed a clear shift towards lower elution volumes compared to those of the individual building blocks. The respective overlays are exemplified in Fig. 4 for polymers based on dPAOx with a DO of 15% (dPMeOx15%-b-PLA, dPMeOx15%-b-PCL, dPEtOx15%-b-PLA and dPEtOx15%-b-PCL). The corresponding elugrams of the other polymers can be found in the ESI (Fig. 15 and 16†). Molar mass distributions of block copolymers comprising dPAOx blocks tended to be broader compared to those of PEtOx-b-PLA and PEtOx-b-PCL. This was already observed in SEC analysis of the dPAOx building blocks and expected because dPAOx were obtained in three consecutive post-polymerization modification steps (ESI Fig. 4†). Combining the information from SEC and NMR spectroscopy confirmed the successful attachment of the two building blocks for all block copolymers.
 | ||
| Fig. 4 Overlay of the SEC elugrams (DMAc, RI detection) of polymers comprising dPAOx blocks with 15% DO dPMeOx15%-b-PLA, dPEtOx15%-b-PLA, dPMeOx15%-b-PCL, dPEtOx15%-b-PCL and the corresponding building blocks dPMeOx15%-N3, dPEtOx15%-N3, BCN-PLA and BCN-PCL, respectively. | ||
MALDI TOF MS analysis of the block copolymers is hampered due to high molar masses. In particular, the statistical distribution of the glycine units complicated ionization of the fully degradable block copolymers. Even though an isotopic pattern could not be resolved, the spectrum of PEtOx-b-PCL clearly revealed a signal at 12000 g mol−1 which corresponds to the theoretical molar mass of the block copolymer, additionally confirming the successful SPAAC (ESI Fig. 17†).
Degradation studies
Due to the introduction of the glycine units, the library comprising dPAOx is supposed to be fully backbone degradable. The block copolymers’ degradability was exemplarily confirmed by a stepwise hydrolysis of the building blocks in dPEtOx32%-b-PLA (Fig. 5). In line with the higher stability of the dPAOx’ amide functionalities, only the polyester block degraded under alkaline conditions using 1 M aqueous NaOH solution. Besides a shift of the SEC elugram towards the elution volume corresponding to the dPEtOx32%-N3 building block, the degradation was confirmed by the 1H NMR spectrum. Instead of signals assigned to the PLA block, lactic acid was detected. In contrast, the signals assigned to the dPEtOx block remained visible. A complete degradation was obtained in a second step using 6 M aqueous HCl solution at 90 °C. Detection of the 1H NMR signals of the degradation products, namely glycine, propionic acid and the amino ethyl-based degradation products, confirmed that. The latter resulted from the former N-acyl-ethylene imine repeating units after cleavage of the side chains. Furthermore, SEC analysis revealed the absence of polymers signals. The amphiphilic block copolymers were hence proven to be fully backbone degradable.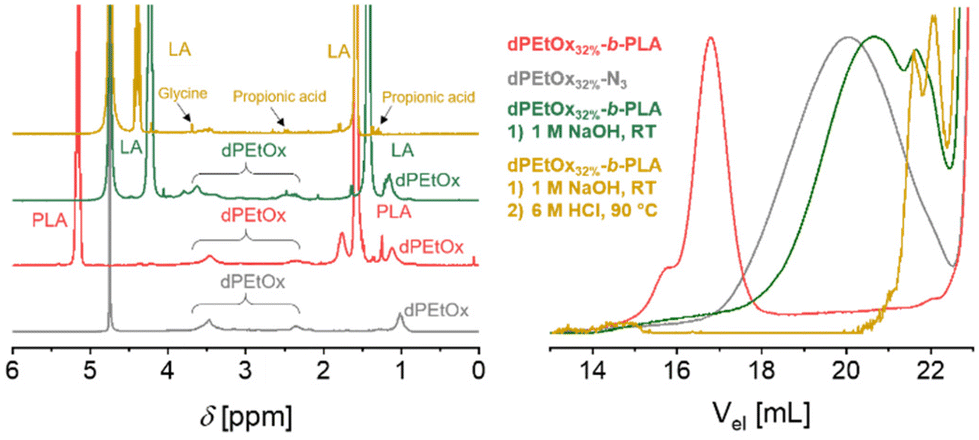 | ||
| Fig. 5 Characterization of the stepwise degradation of dPEtOx32%-b-PLA. Left: Overlay of the 1H NMR spectra of dPEtOx32%-N3, dPEtOx32%-b-PLA, dPEtOx32%-b-PLA after the treatment with 1 M NaOH at ambient temperature overnight and dPEtOx32%-b-PLA after the treatment with 6 M HCl at 90 °C overnight. Right: Overlay of the SEC elugrams (DMAc, RI detection). | ||
High-throughput formulation of particles
The synthesized polymers should be well suited as nanocarriers for drug delivery applications. However, their ability to form particles and the resulting properties need to be studied to reveal possible structural differences between the fully degradable dPAOx-based block copolymers and their PEtOx containing counterparts. Size, shape (morphology), and zeta potential are the most vital physicochemical properties of particles with intended use in pharmaceutical applications. These characteristics are strongly determined by the initial polymer properties, the formulation and purification method, but also by additives (e.g. drugs or surfactants) applied during the formulation.19 To evaluate structure–property relationships in view of the particle formulation performance, high-throughput (HT) nanoprecipitation was applied to all fully degradable copolymers as well as their partially degradable counterparts based on PEtOx (Fig. 6).20,29 The homopolymers PLA and PCL were used as additional control materials. | ||
| Fig. 6 Experimental layout of the particle formulation screening via HT-nanoprecipitation using a liquid handling robot for particle formulations in a 96 well plate format and subsequent size distribution analysis of the particles in 96 well plates using HT-DLS. | ||
The initial polymer concentration was varied to investigate how the size range of the particles changes with concentration since it is well-known that it has a direct influence on the size and stability of the particles.30 The formulations were performed without the addition of surfactants to enable a direct comparison of the block copolymers containing POx or dPAOx.
Following the HT-formulation, DLS analysis was performed in a HT fashion as well. The size and size distribution of the particles are provided via the Z-average values and polydispersity index (PDI), which were both calculated from the intensity-based distribution by cumulant analysis (ESI Fig. 18, 19 and ESI Tables 1–4†). Additionally, the standard deviation of the mean sizes was calculated by using the following equation: 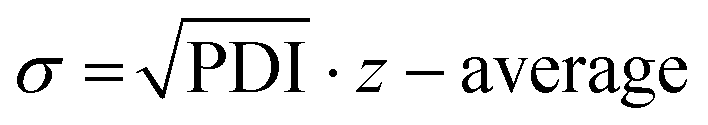 .31,32 The Z-average values are plotted with σ in Fig. 7.
.31,32 The Z-average values are plotted with σ in Fig. 7.
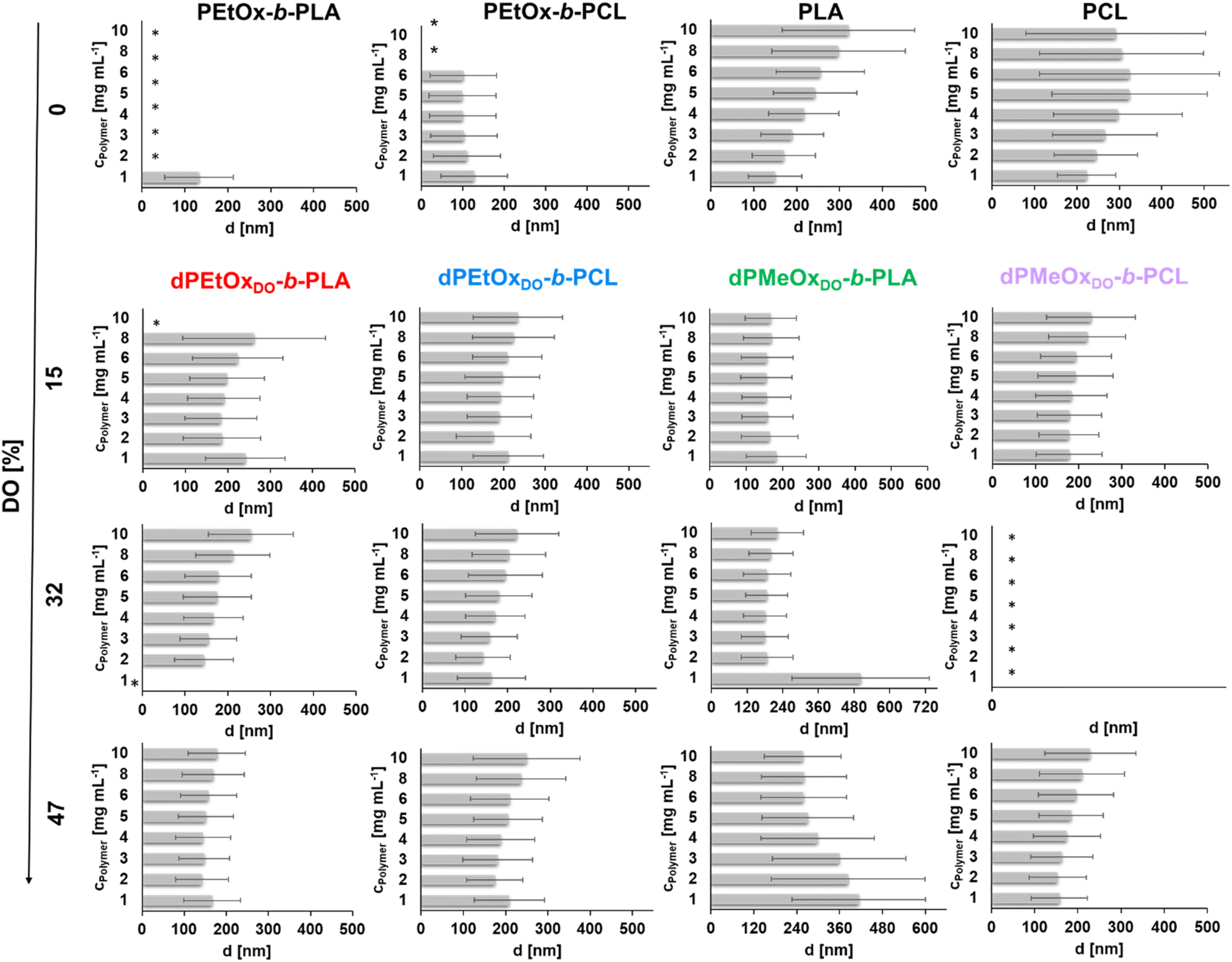 | ||
Fig. 7 The mean average particle sizes (Z-average value presented in grey bars) in dependency of the initial polymer concentration plotted with the standard deviation σ that was calculated by 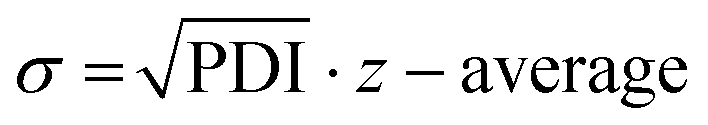 . *Indicates unstable formulations or formulations providing only insufficient size measurements resulting in unreliable DLS data. . *Indicates unstable formulations or formulations providing only insufficient size measurements resulting in unreliable DLS data. | ||
For the PLA and PCL homopolymers a slight trend of increasing mean particle size with increasing initial polymer concentration was observed until a concentration of 5 mg mL−1 (Fig. 7 and ESI Tables 1–4†). The trend continued until initial concentrations of 10 mg mL−1 for PLA. The overall width of the particle size distribution also increased with the polymer concentration.
The influence of the attachment of a PEtOx block was investigated next. The results are shown in Fig. 7 and ESI Fig. 18, 20, 21† as well as ESI Tables 1 and 2.† Surprisingly, the partially degradable PEtOx-b-PLA did not form stable and defined particles under the chosen conditions. A similar tendency was observed for PEtOx-b-PCL. Here, rather low mean average sizes below 133 nm but high PDI values between 0.31 and 0.45 were observed. In contrast, all fully backbone degradable block copolymers based on dPEtOx formed particles with mean sizes between 142 nm and 262 nm. A slight trend of increasing Z-average with increasing polymer concentration was found (ESI Fig. 18†). The PDI values remained below 0.23 with a few exceptions at very low or very high polymer concentrations.
Block copolymers containing dPMeOx segments with a DO of 15% formed stable dispersions, almost independent of the initial polymer concentration (Fig. 7 and ESI Fig. 19–21, ESI Tables 3, 4†). In contrast, the formulation of dPMeOx with a DO of 32% was only successful for dPMeOx32%-b-PLA, whereas strong aggregation was observed for dPMeOx32%-b-PCL. A further increase of the DO of the dPMeOx segments enabled reliable particle formulation for the dPMeOx47%-b-PCL, while resulting in broad particle size distributions for dPMeOx47%-b-PLA.
In summary, a “POxylation effect” on the average particle size was observed lowering the Z-average values in most cases.12 Presumably, the hydrophilic PEtOx as well as dPAOx segments tend to arrange themselves towards the surface of the particles in aqueous dispersion. Such altered surface properties would manifest in the respective zeta potentials. The zeta potential provides information about the surface charge and is one factor that influences the stability of the dispersions. High absolute zeta potential values (>30 mV) lead to increasing electrostatic repulsion of the particles between each other, which reduces the aggregation tendency of the particles and enhances their stability in dispersion.31,32
To investigate how the altered properties of the polymers influenced the zeta potential of the particles, ELS measurements were performed with the samples formulated from 3 mg mL−1 solutions as stable particles with similar size distributions were observed for this concentration in most cases (Fig. 8 and ESI Fig. 21†).
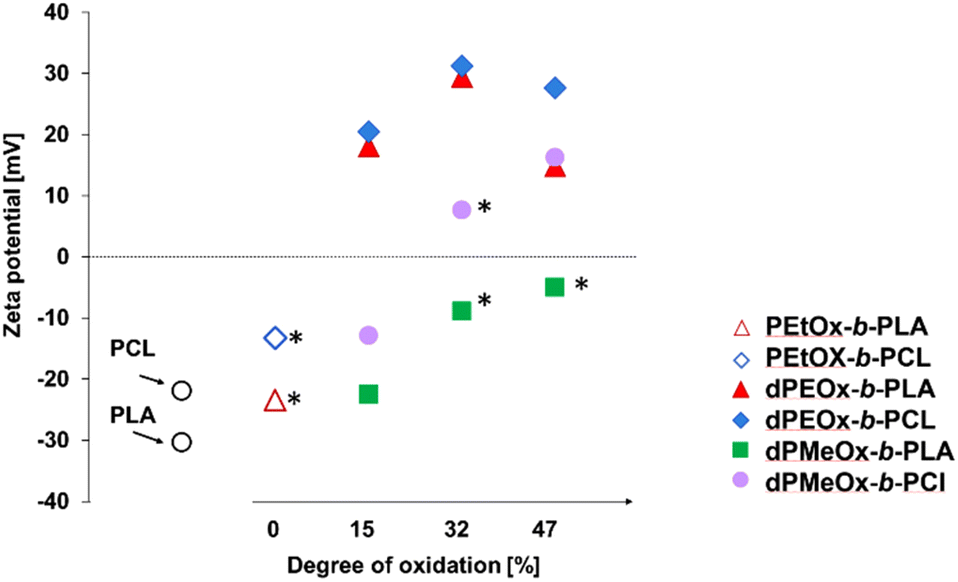 | ||
| Fig. 8 Zeta potential values of particles in dependency of the DO of the attached POx or dPAOx block determined by ELS measurements of the particles prepared with an initial polymer concentration of c = 3 mg mL−1. Instable formulations resulting in particle aggregation and in high PDI values > 0.3 are highlighted with an asterisk (*). | ||
For the bare polyester particles formed from PLA and PCL, highly negative zeta potentials of −30 mV and −22 mV, respectively, were measured (Fig. 8 and Table 1). The attachment of the PEtOx block decreased the absolute value, whereby the zeta potential remained negative. In contrast, the fully degradable dPEtOx-b-PLA and dPEtOx-b-PCL revealed positive zeta potentials between +15 mV and +31 mV for all DO values.
The zeta potential also increased for the dPMeOx-b-PLA samples with increasing DO but remained below zero. The same trend was observed for the dPMeOx-b-PCL samples. The zeta potential of samples with DO of 15% remained negative (−13 mV for dPMeOx15%-b-PCL), whereas the zeta potential switched to positive values for samples with higher DO (+8 mV for the dPMeOx32%-b-PCL and +16 mV for dPMeOx47%-b-PCL).
The influence of the DO on the zeta potential is very prominent: The amount of introduced glycine moieties in the hydrophilic block increased the zeta potential. The presence of non-acylated ethylene imine repeating units could explain the switch in general. However, the trend with increasing DO would be coincidental as each acylation was performed independently. POx feature exclusively hydrogen bond accepting sites, as has been well understood for decades.33,34 The incorporated glycine units in dPAOx result in the introduction of additional hydrogen bond donating moieties (ESI Fig. 22†). This might lead to a rearrangement of the hydration shell around the hydrophilic particle surface which influences the zeta potential.
The zeta potential affects significantly the success of the particle formulations as high absolute zeta potential values enhance the repulsion of the particles from each other and, thus, prevent them from aggregation. An insufficient repulsion and hence aggregation of the particles was observed for dPMeOx32%-b-PCL and dPMeOx47%-b-PLA samples where zeta potentials were found to be close to zero (<10 mV, Fig. 8). The unstable particle dispersions revealed broad size distributions, as well as high standard deviations between the different formulations as displayed in Fig. 9. In contrast, stable formulations were well repeatable in the utilized experimental HT-nanoprecipitation setup, as indicated by the low standard deviation values from six independent formulations conducted for each polymer.
 | ||
| Fig. 9 Z-average values of particles prepared from the fully degradable dPAOx-based block copolymers and their PEtOx containing counterparts as well as PLA and PCL homopolymers (highlighted via dotted line) in dependency of the initial polymer concentration used for the formulation. The standard deviation (SD) refers to the deviations for n = 6 individual formulations. No SD bars are shown if they fall into the symbol. | ||
The library approach as well as the repeatability studies were conducted in a high-throughput workflow utilizing a combination of a pipetting robot for HT-nanoprecipitation and HT-DLS data analysis. This workflow enabled fast and reproducible particle formulations. Such broad investigations were significantly simplified by the HT-nanoprecipitation workflow. However, more detailed investigations cannot be performed in a HT-fashion and, thus are accessible only for selected samples. Providing information about the particle morphology, SEM is one example for such a method. Hence, we limited SEM measurements to the particle suspensions prepared with 3 mg mL−1 (ESI Fig. 23 and 24†). The SEM investigations confirmed the formation of small particles in the size range of 100 to 300 nm for most polymers. No or aggregated particles were found for samples where already DLS indicated instabilities of the dispersion. Some particles were irregularly shaped and appeared smaller compared to the DLS results.
Conclusions
A library of twelve amphiphilic block copolymers based on the combination of hydrophilic dPAOx and hydrophobic polyesters was obtained from three starting materials, i.e. PEtOx, PLA and PCL. Different amounts of glycine moieties were introduced in the POx segments through a consecutive hydrolysis of PEtOx, oxidation and re-acylation yielding degradable PMeOx as well as PEtOx analogues. The fully degradable block copolymers comprising PLA as well as PCL blocks obtained via SPAAC were studied with respect to their ability to yield stable particle dispersions in a HT-formulation approach. An increased amount of introduced glycine moieties increased the zeta potential of the particles, which significantly influenced the overall stability of the formulations. While even low amounts of glycine moieties in PEtOx resulted in a switch of the zeta potential from negative to positive values, the effect was less prominent for formulations based on dPMeOx. Even without the use of surfactants, stable dispersions could be obtained from each dPAOx type, dependent on the combination with the polyester and the DO. The next logic steps will be to further optimize the formulation conditions as well as to utilize the obtained systems for drug delivery applications by the encapsulation of active pharmaceutical ingredients in the new fully degradable amphiphilic block copolymers.Author contributions
Natalie E. Göppert – synthesis of PEtOx-N3, all dPAOx-N3 and all block copolymers, characterization of all polymers, degradation studies, manuscript preparation. Antje Vollrath – particle formulation and characterization, manuscript preparation. Leanne M. Stafast – synthesis of BCN-PCL and BCN-PLA, correction of the manuscript. Steffi Stumpf – SEM measurements. Bianca Schulze – polymer synthesis support. Stephanie Hoeppener – correction of the manuscript. Christine Weber – development of concept, manuscript preparation, manuscript correction. Ulrich S. Schubert – correction of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the support of Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (project number 316213987, SFB 1278, projects A01, A04, A06 and Z01). The rapifleX mass spectrometer was funded by the Thüringer Aufbaubank (TAB, funding ID: 2016 IZN 0009). The SEM facilities of the Jena Center for Soft Matter (JCSM) were established with a grant from the DFG (project number 255916678). Furthermore, the authors acknowledge Nicole Fritz and Ursula Eberhardt for assistance with MALDI TOF MS measurements. Biorender was used to create graphical illustrations.References
- L. Pietrelli, S. Ferro, A. P. Reverberi and M. Vocciante, Chemosphere, 2021, 273, 129725 CrossRef CAS PubMed.
- B. Daglar, E. Ozgur, M. E. Corman, L. Uzun and G. B. Demirel, RSC Adv., 2014, 4, 48639–48659 RSC.
- K. E. Washington, R. N. Kularatne, V. Karmegam, M. C. Biewer and M. C. Stefan, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2017, 9, 1446 CrossRef.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- A. Larrañaga and E. Lizundia, Eur. Polym. J., 2019, 121, 109296 CrossRef.
- V. P. Torchilin and V. S. Trubetskoy, Adv. Drug Delivery Rev., 1995, 16, 141–155 CrossRef CAS.
- A. L. Klibanovl, K. Maruyamal, V. P. Torchilin and L. Huang, FEBS Lett., 1990, 268, 235–237 CrossRef.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS.
- M. D. McSweeney, Z. C. Versfeld, D. M. Carpenter and S. K. Lai, Clin. Transl. Sci., 2018, 11, 162–165 CrossRef.
- T.-L. Cheng, P.-Y. Wu, M.-F. Wu, J.-W. Chern and S. R. Roffler, Bioconjugate Chem., 1999, 10, 520–528 CrossRef CAS PubMed.
- S. C. Lee, Y. Chang, J.-S. Yoon, C. Kim, I. C. Kwon, Y.-H. Kim and S. Y. Jeong, Macromolecules, 1999, 39, 1847–1852 CrossRef.
- M. Dirauf, C. Grune, C. Weber, U. S. Schubert and D. Fischer, Eur. Polym. J., 2020, 134, 109801 CrossRef CAS.
- G. Le Fer, C. Le Cœur, J.-M. Guigner, C. Amiel and G. Volet, Polymer, 2019, 171, 149–160 CrossRef CAS.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS.
- B. Pidhatika, M. Rodenstein, Y. Chen, E. Rakhmatullina, A. Muhlebach, C. Acikgoz, M. Textor and R. Konradi, Biointerphases, 2012, 7, 1 CrossRef CAS.
- R. Konradi, C. Acikgoz and M. Textor, Macromol. Rapid Commun., 2012, 33, 1663–1676 CrossRef CAS PubMed.
- N. E. Göppert, M. Kleinsteuber, C. Weber and U. S. Schubert, Macromolecules, 2020, 53, 10837–10846 CrossRef.
- N. E. Göppert, M. Dirauf, C. Weber and U. S. Schubert, Polym. Chem., 2021, 12, 5426–5437 RSC.
- K. Y. Hernandez-Giottonini, R. J. Rodriguez-Cordova, C. A. Gutierrez-Valenzuela, O. Penunuri-Miranda, P. Zavala-Rivera, P. Guerrero-German and A. Lucero-Acuna, RSC Adv., 2020, 10, 4218–4231 RSC.
- I. Y. Perevyazko, J. T. Delaney, A. Vollrath, G. M. Pavlov, S. Schubert and U. S. Schubert, Soft Matter, 2011, 7, 5030–5035 RSC.
- L. M. Stafast, N. Engel, H. Görls, C. Weber and U. S. Schubert, Eur. Polym. J., 2023, 184, 111779 CrossRef CAS.
- K. Kempe, R. Hoogenboom, M. Jaeger and U. S. Schubert, Macromolecules, 2011, 44, 6424–6432 CrossRef CAS.
- L. Tauhardt, K. Kempe, K. Knop, E. Altuntaş, M. Jäger, S. Schubert, D. Fischer and U. S. Schubert, Macromol. Chem. Phys., 2011, 212, 1918–1924 CrossRef CAS.
- C. Englert, M. Hartlieb, P. Bellstedt, K. Kempe, C. Yang, S. K. Chu, X. Ke, J. M. García, R. J. Ono, M. Fevre, R. J. Wojtecki, U. S. Schubert, Y. Y. Yang and J. L. Hedrick, Macromolecules, 2015, 48, 7420–7427 CrossRef CAS.
- T. Rudolph, N. Kumar Allampally, G. Fernandez and F. H. Schacher, Chem. – Eur. J., 2014, 20, 13871–13875 CrossRef CAS PubMed.
- S. Osawa, T. Ishii, H. Takemoto, K. Osada and K. Kataoka, Eur. Polym. J., 2017, 88, 553–561 CrossRef CAS.
- E. Altuntas, K. Knop, L. Tauhardt, K. Kempe, A. C. Crecelius, M. Jager, M. D. Hager and U. S. Schubert, J. Mass Spectrom., 2012, 47, 105–114 CrossRef CAS PubMed.
- S. A. van den Berg, H. Zuilhof and T. Wennekes, Macromolecules, 2016, 49, 2054–2062 CrossRef CAS.
- J. Kimmig, T. Schuett, A. Vollrath, S. Zechel and U. S. Schubert, Adv. Sci., 2021, 8, 2102429 CrossRef CAS.
- C. J. Martínez Rivas, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari, S. A. Galindo Rodríguez, R. A. Roman, H. Fessi and A. Elaissari, Int. J. Pharm., 2017, 532, 66–81 CrossRef.
- C. N. Lunardi, A. J. Gomes, F. S. Rocha, J. De Tommaso and G. S. Patience, Can. J. Chem. Eng., 2021, 99, 627–639 CrossRef CAS.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS.
- F. P. Chen, A. E. Ames and L. D. Taylor, Macromolecules, 1990, 23, 4688–4695 CrossRef CAS.
- C. H. Chen, J. Wilson, W. Chent, R. M. Davis and J. S. Riffle, Polymer, 1994, 35, 3587–3591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00085k |
| This journal is © The Royal Society of Chemistry 2024 |