
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thienoisoindigo-based recyclable conjugated polymers for organic electronics†
Naoya
Nozaki
a,
Azalea
Uva
 b,
Hidetoshi
Matsumoto
a,
Helen
Tran
*bcd and
Minoru
Ashizawa
*a
b,
Hidetoshi
Matsumoto
a,
Helen
Tran
*bcd and
Minoru
Ashizawa
*a
aDepartment of Materials Science and Engineering, Tokyo Institute of Technology, Meguro-ku, Tokyo, Japan. E-mail: ashizawa.m.aa@m.titech.ac.jp
bDepartment of Chemistry, University of Toronto, Toronto, Ontario, Canada. E-mail: tran@utoronto.ca
cDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Toronto, Ontario, Canada
dAcceleration Consortium, University of Toronto, Toronto, Canada
First published on 8th December 2023
Abstract
Imine-based semiconducting polymers based on thiophene-flanked diketopyrrolopyrrole (TDPP) are widely used to realize naturally disposable electronic devices. However, TDPP easily decomposes under mildly acidic conditions, limiting its potential for use in recyclable systems. Herein, we designed and synthesized two chemically recyclable thienoisoindigo (TII)-based polymers bearing an imine bond. These polymers were prepared from polycondensation reactions of the dialdehyde-functionalized monomer TII-(CHO)2 with p-phenylenediamine (PD) to produce p(TII-PD) and with 2,6-naphthalenediamine (2,6ND) to produce p(TII-2,6ND), respectively. Using ultraviolet-visible-near infrared spectroscopy, nuclear magnetic resonance, and mass spectroscopy, we examined the recyclability of both polymers. Under mildly acidic conditions, the imine-based polymers fully degrade into the original TII-(CHO)2 in as little as one day. Moreover, the recovered TII-(CHO)2 monomer is chemically stable for up to 6 months under acidic conditions, allowing us to isolate the monomer in high yield (>90%). Using the recovered TII-(CHO)2 monomer, we prepared recycled polymers, re-p(TII-PD) and re-p(TII-2,6ND). The recycled polymers displayed nearly the same electrical properties as the pristine polymers, with field-effect transistor mobilities in the order of 10−2–10−3 cm2 V−1 s−1. These results demonstrate the versatility of the TII-based monomer unit for developing fully recyclable semiconducting polymers.
Introduction
The chemical recycling of polymers into high quality monomers is a key step towards a circular economy.1–3 To achieve recyclability in polymeric systems, emphasis is placed on the molecular design of the polymers, where dynamic chemical motifs are utilized to enable controlled depolymerization into defined by-products that can be repolymerized. By this process, degradation by-products can be isolated, purified, and used once again for repolymerization. Examples of dynamic chemistries that allow for recyclability in polymeric systems include diketoenamines,4,5 acetals,6–8 and cyanurate9 motifs. These linkages have well-understood degradation mechanisms, allowing for the isolation of stable monomeric compounds that can be used to re-synthesize pristine polymers.The synthetic strategy for designing chemically recyclable polymers can be adopted to electron-conducting organic polymers, where conjugation along the backbone must be considered. Semiconducting polymers are a promising class of electron-conducting materials for next-generation electronic devices due to their advantageous properties that make them flexible,10–13 stretchable,14–16 biocompatible,17–19 or degradable20–22 for applications in organic photovoltaics,23,24 biological sensing,25–27 and bioelectronics.28–30 Significant efforts have gone into synthesizing new degradable semiconducting polymers for applications in transient electronics31–33 using cleavable moieties such as imines,34–36 imidazoles,37,38 alkynes39 or oxadiazoles.40 However, limited reports on the chemical recyclability of semiconducting polymers exist because it remains a challenge to design π-conjugated polymers that can degrade into stable by-products.
The imine bond is regarded as a highly desirable chemical linkage because it maintains π-conjugation along the polymer backbone while also providing a specific cleavage site. However, most degradable imine-based semiconducting polymers (i.e., poly(azomethine)s) yield degradation by-products that are not easily recoverable. This is attributed to the molecular design of the polymeric system, where often the by-products are unstable under the degradation conditions, contributing to the inability to recover intact monomers. For example, poly(azomethine)s based on thiophene-flanked diketopyrrolopyrrole (TDPP)41,42 are degradable upon hydrolysis of the imine bond, but the TDPP units are not recoverable because the diketopyrrolopyrrole (DPP) moiety undergoes a lactam ring hydrolysis in the presence of acid.17 This has prompted efforts to design monomers that are chemically stable under acidic conditions.43 For example, Collier and coworkers44 recently reported dihydropyrrolo[3,2-b]pyrrole-containing poly(azomethine)s, which produce stable by-products upon acid hydrolysis. Son and coworkers45 developed a π-conjugated polymeric system based on N1,N4-bis(thiophen-2-ylmethylene)benzene-1,4-diamine that degrades into a stable dialdehyde monomer upon acid hydrolysis, allowing for isolation and utilization of the monomer to prepare new polymers via imine condensation. Although both examples are positive steps towards developing recyclable semiconducting polymers, there is yet to be an example that illustrates closed-loop recycling of a π-conjugated monomer that can be recovered to prepare a pristine semiconducting polymer.
Building upon previously reported imine-based semiconducting polymers, we designed new chemically recyclable semiconducting polymers with stable degradation by-products that can be recovered for repolymerization. In this content, we focused on thienoisoindigo (TII),46,47 a thiophene-based analogue of isoindigo (IIG),48,49 as the recoverable monomer. Both TII and IIG monomers have been extensively used to synthesize semiconducting polymers for organic electronics,50–54 with a TII-based copolymer reported to have a hole-transporting field-effect transistor (FET) mobility of 10 cm2 V−1 s−1.55 In contrast to TDPP monomers, we found that TII monomers do not decompose in the presence of neat trifluoroacetic acid (TFA), a desirable characteristic for imine-based polymers that degrade under acidic conditions (Fig. 1). Building upon the chemical stability of TII under acidic conditions, we designed and synthesized two chemically recyclable TII-based semiconducting polymers, p(TII-PD) and p(TII-2,6ND), via an imine-based polycondensation reaction. We report the chemical, thermal, and photophysical properties of these polymers. Additionally, we investigate the charge carrier transport of each polymer through FET device fabrication and study their degradation lifetimes under acidic conditions. Upon acid hydrolysis, both polymers produce the original dialdehyde TII monomer (TII-(CHO)2), allowing us to recover the building block in high yields (>90%). Remarkably, the recycled polymers have comparable properties to the pristine polymers, implying that the recovered TII-(CHO)2 monomer is ideal for preparing high-quality recycled polymers. Regarding charge transport performance, the recycled polymers performed on par with pristine polymers, indicating no loss in electronic properties. This study demonstrates the versatility of the TII-based unit for constructing recyclable semiconducting polymers, contributes to our understanding of how by-products will form upon cleavage of a degradable polymer, and highlights the importance of designing degradable semiconducting polymers with chemically stable monomeric units. Broadly, this work further contributes to our understanding of designing degradable semiconducting polymers with known degradation by-products.
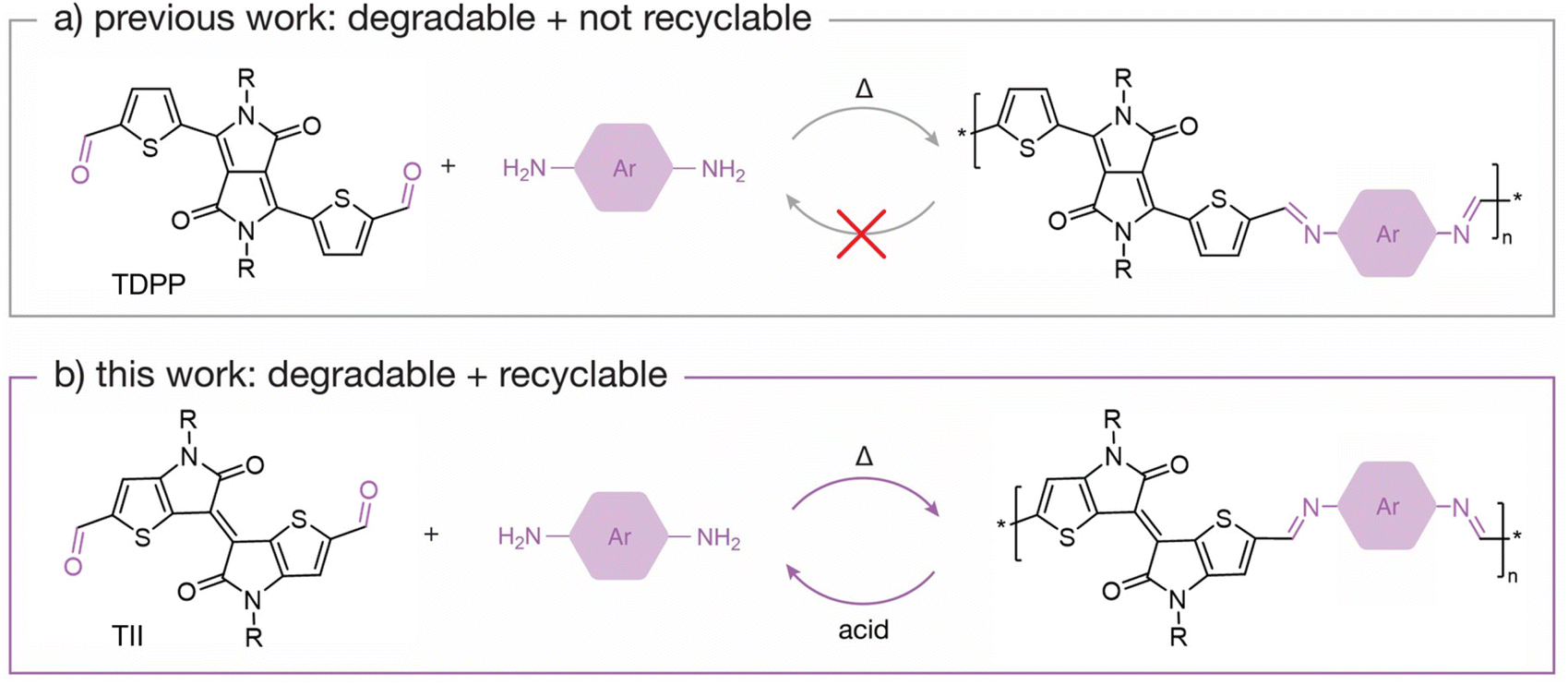 | ||
| Fig. 1 Schematic scheme of (a) non-recyclable TDPP-based polymers and (b) recyclable TII-based polymers. | ||
Results and discussion
Synthesis and polymer characterization
The synthesis of TII-(CHO)2 and two TII imine-based polymers is described in Fig. 2a, and the experimental details including nuclear magnetic resonance (NMR) data are provided in the ESI (Fig. S1–S5†). For incorporation of the TII unit into an imine-based polymer system, we selectively functionalized the α-positions of the thiophene rings on the TII core with dialdehydes, producing TII-(CHO)2 (Fig. 2a). The dialdehyde-modified monomer TII-(CHO)2 was prepared via deprotonation with lithium diisopropylamide (LDA) and subsequently quenched with dimethylformamide (DMF).17,42,50 To increase solubility for thin-film processability,36,56 TII-(CHO)2 was synthesized with branched alkyl side chains. We selected p-phenylenediamine (PD) and 2,6-naphthalenediamine (2,6ND) as the diamine compounds because PD is commercially available and 2,6ND is easily prepared. The polymers were synthesized via polycondensation reactions using a catalytic amount of p-toluenesulfonic acid (PTSA) and an excess amount of calcium chloride (CaCl2) as the drying agent (Fig. 2a). Considering the solubility of diamines in polycondensation reactions, we used toluene for p(TII-PD) and chlorobenzene for p(TII-2,6ND), respectively. The polymers were purified by precipitating the crude polymer solutions into methanol (MeOH), followed by Soxhlet extractions to remove impurities and low molecular weight oligomers. Owing to the slight and unclear 1H-NMR peak of the terminal formyl group, coming from low solubility of both polymers, molecular weight could not be obtained through end-group analysis.36,57 Therefore, the molecular weights and dispersities (Đ) of the polymers were determined by high-temperature gel permeation chromatography (HT-GPC) using 1,2,4-trichlorobenzene at 135 °C against polystyrene standards (Table 1, Fig. S6, and Table S1†), a method that is commonly used to determine the molecular weight of many acid-degradable polymers.34,37–41 The Mn values for p(TII-PD) and p(TII-2,6ND) were 22.3 kDa and 19.2 kDa, respectively, with dispersity values in the typical range for poly(azomethine)s synthesized via imine polycondensation reactions.58 Thermogravimetric analysis (TGA) results shown in Fig. S7a† and Table 1 indicate that these polymers exhibit reliable thermal stability with a thermal decomposition temperature (5% weight loss) above 330 °C, which is ideal for the organization of the polymer into thin film microstructures upon annealing. However, no noticeable thermal transitions were observed for both polymers (Fig. S7b and S7c†) in the differential scanning calorimetry (DSC) profiles.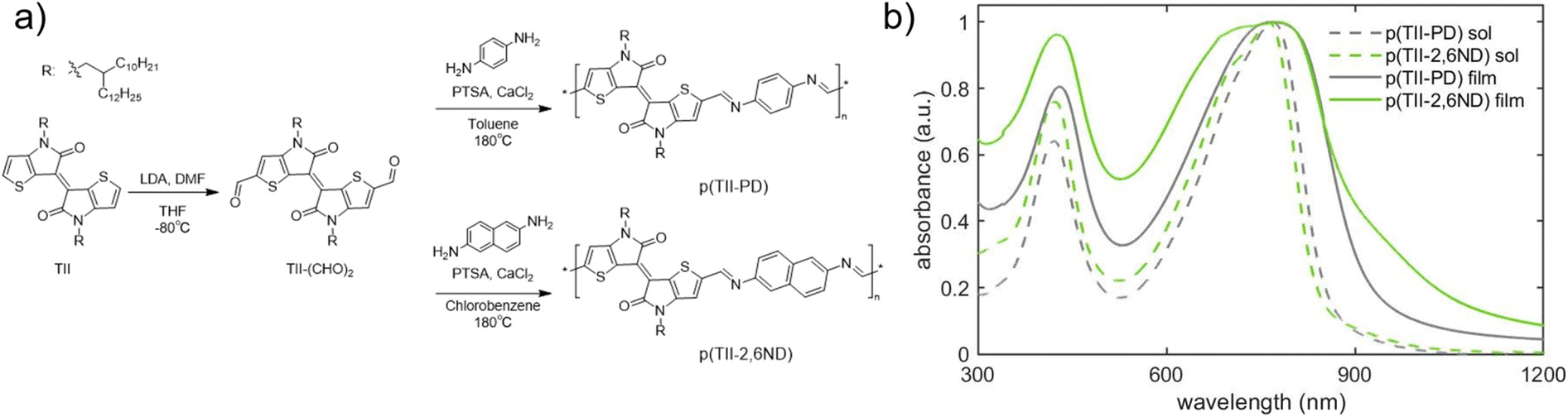 | ||
| Fig. 2 (a) Synthetic scheme of TII-(CHO)2 and TII polymers; (b) UV-vis-NIR spectra of polymer solutions (in chlorobenzene) and thin films. | ||
Optical and electrochemical properties
The optical energy gaps in solution and thin films along with EHOMO and ELUMO levels estimated by cyclic voltammetry (CV) and the absorption maxima for each polymer are summarized in Table 2. First, we examined the UV-vis-NIR absorption spectra of p(TII-PD) and p(TII-2,6ND) in chlorobenzene solutions and their thin films (Fig. 2b). In solution, p(TII-PD) and p(TII-2,6ND) showed similar absorption profiles, including a weak absorption band ranging from 300 nm to 500 nm and a strong energy absorption band ranging from 500 nm to 900 nm continuously. The red-shifted absorption profiles in thin films can be ascribed to the planarized polymer backbone in the solid state, leading to enhanced intermolecular interactions. The optical energy gaps of p(TII-PD) and p(TII-2,6ND) thin films determined from the onset of absorption were found to be 1.32 eV and 1.31 eV, respectively (Table 2). In the CV curves, both polymers exhibited an irreversible oxidation step, and a small irreversible reduction step was observed (Fig. S8†). Hence, the EHOMO values of both polymers were estimated from the onset of the oxidation step in the CV curves, and ELUMO values were calculated using the difference between the EHOMO and the optical energy gaps for each polymer thin film. From the oxidation onset potentials referenced to the Fc/Fc+ couple, EHOMO and ELUMO were estimated to be −5.21 eV and −3.89 eV for p(TII-PD) and −5.22 eV and −3.91 eV for p(TII-2,6ND), respectively (Table 2). Compared with previously reported TII copolymers,46,48 these results indicate that the imine-bond inserted between TII and aromatic units lowers both HOMO and LUMO levels.| Solution | Film | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| λ solmax (nm) | λ solonset (nm) |
E
solg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (eV) a (eV) |
λ with TFAmax (nm) | λ filmmax (nm) | λ filmonset (nm) |
E
filmgb (eV) |
E
HOMOc (eV) |
E
LUMOd (eV) |
|
| a E solg was calculated from λsolonset. b E filmg was calculated from λfilmonset. c E HOMO was determined using the onset curve of CV. d E LUMO was calculated using ELUMO = EHOMO + Efilmg. | |||||||||
| p(TII-PD) | 770 | 865 | 1.43 | 880 | 770 | 938 | 1.32 | −5.21 | −3.89 |
| p(TII-2,6ND) | 762 | 841 | 1.47 | 915 | 779 | 947 | 1.31 | −5.22 | −3.91 |
Degradation and recyclability studies
To quantitatively study the degradation of p(TII-PD) and p(TII-2,6ND) (Fig. 3a), we used UV-vis-NIR absorption spectroscopy. At first, we monitored the time-dependent UV-vis-NIR absorption profiles during polymer depolymerization in chlorobenzene at room temperature (Fig. 3c–e). Both TII-based polymers exhibited nearly the same degradation behaviour, regardless of the benzene and naphthalene diamine monomers, with the polymers undergoing complete depolymerization in one day. In the experiment, 250 μL of 1 M TFA(aq) was added to each polymer solution (0.5 mg of polymers in 50 mL of chlorobenzene). As the polymer degraded, the absorption peaks of p(TII-PD) and p(TII-2,6ND) solutions rapidly red-shifted after the addition of TFA, and these red-shifted peaks gradually decreased over time (approximately 4 hours until the peaks completely diminished). It is interesting to note the largely red-shifted peak profiles of both polymers by ca. 100–150 nm after the immediate addition of TFA. These red-shifted behaviours are hypothesized to be from the protonation of the nitrogen atom in the imine bond. This hypothesis is further supported by other protonated poly(azomethine)s in the literature,59–62 which undergo a structural polymer backbone alternation from an aromatic to quinoidal structure upon acidification (Fig. 3f and g).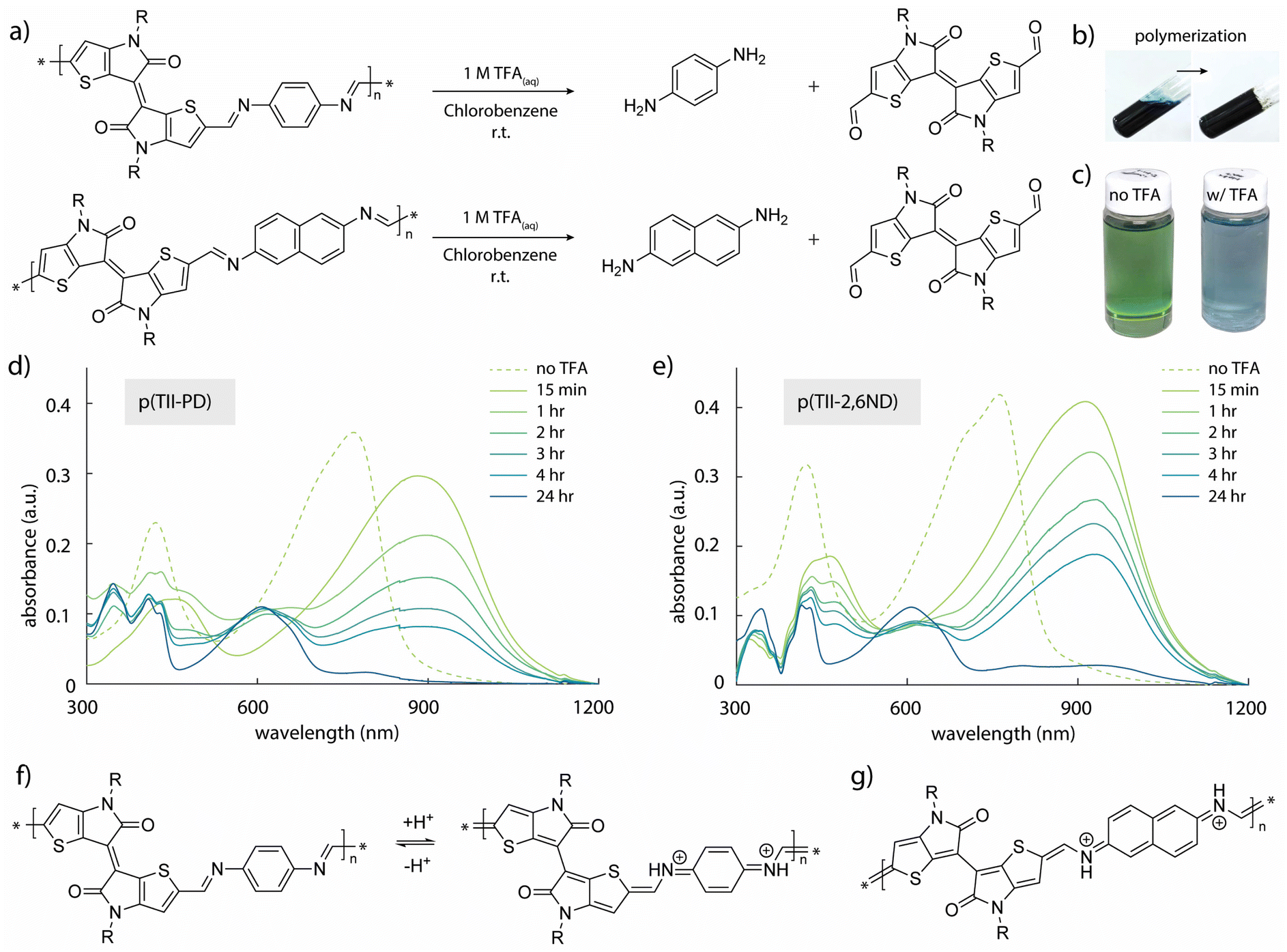 | ||
| Fig. 3 Degradation studies: (a) synthetic scheme of polymer degradation; (b) pictures taken before and after polymerization of p(TII-PD); (c) pictures taken before and after depolymerization of p(TII-PD) with TFA solution; UV-vis-NIR spectra during polymer degradation of (d) p(TII-PD) and (e) p(TII-2,6ND); the protonated aromatic-quinoidal transition for (f) p(TII-PD) and (g) p(TII-2,6ND), leading to red-shifted UV-vis-NIR spectra. | ||
After one day, the blue-shifted absorption containing three main peaks corresponding to the absorption profiles of the TII unit appeared, indicating that the polymer chains were completely degraded, and the TII-(CHO)2 monomer was recovered (Fig. 4a and b). Notably, the solution used for the degradation studies displayed the same absorption profiles even after 6 months (Fig. 4a). This indicates that TII-(CHO)2 is chemically stable under acidic hydrolysis conditions, originating from the unique chemical character of the TII unit.46,52 We hypothesize that the TII framework adopts a planar and rigid structure due to intramolecular sulfur–oxygen short contacts between the annulated thiophene and the lactam core, contributing to its overall chemical stability in the presence of acid. To recover a large quantity of the TII-(CHO)2 monomer, we increased the scale of polymer degradation to ca. 50 mg. p(TII-PD) and p(TII-2,6ND) were dissolved in chloroform and 5 mL of 1 M TFA(aq) was added. After 2 days of stirring at 45 °C to accelerate imine hydrolysis, the crude mixture was poured into water and extracted with dichloromethane. The TII-(CHO)2 extracted from the organic layer was purified using silica gel chromatography, and we retrieved the pure TII-(CHO)2 from p(TII-PD) in 95% yield and p(TII-2,6ND) in 91% yield, respectively. The recovered TII-(CHO)2 was fully characterized via1H-NMR and ESI-MS (Fig. 4c–e and S9†). Diamine monomers, PD and 2,6ND, were also isolated from the aqueous layer by NaOH treatment followed by extraction with dichloromethane, and finally both were characterized via1H-NMR (Fig. S10†) without further purification. These results indicate that the TII-(CHO)2 is a promising monomer unit for polymer recyclability in organic electronics.
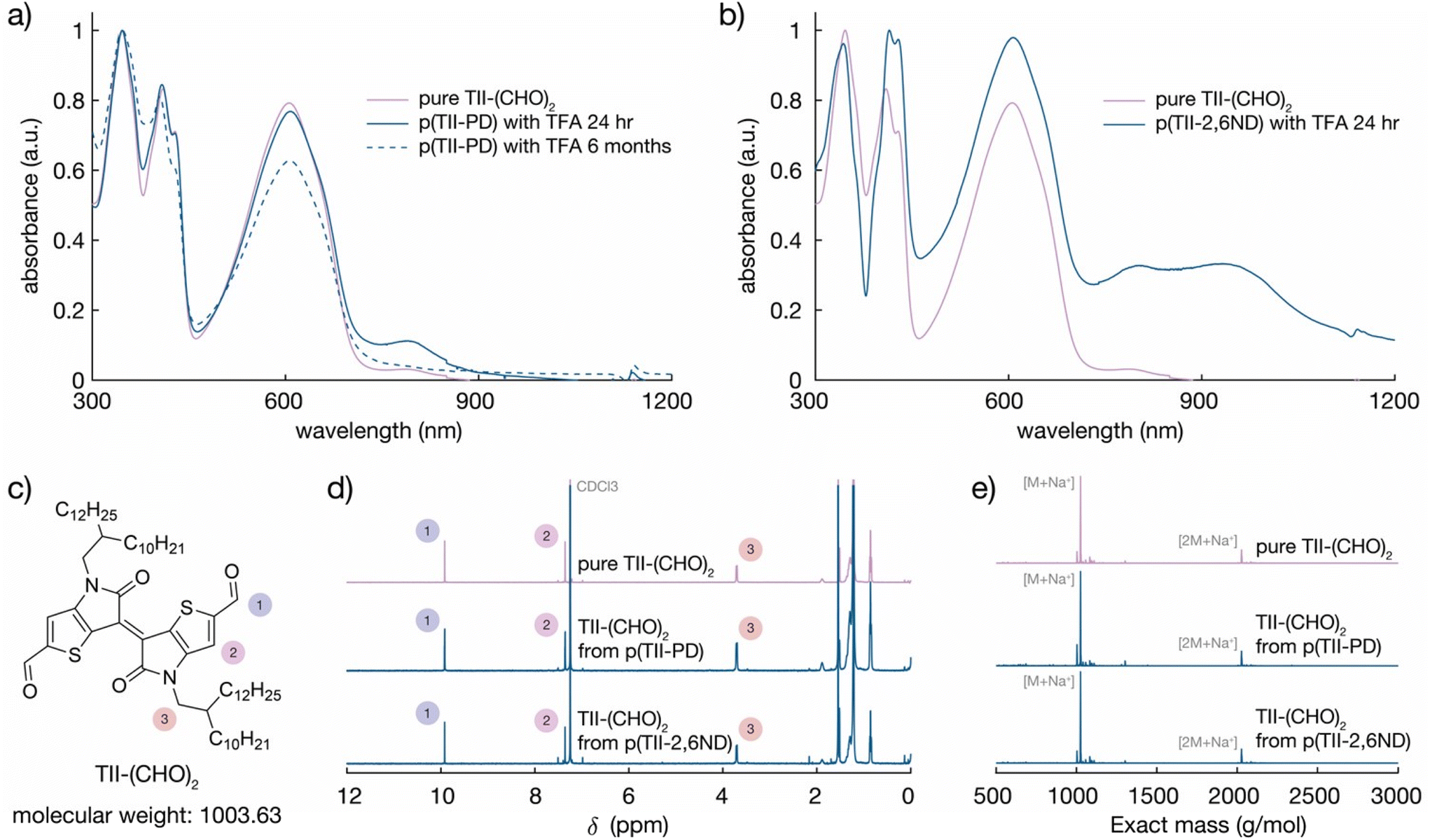 | ||
| Fig. 4 Comparison of the pure and recycled TII-(CHO)2: UV-vis-NIR absorption spectra of decomposed (a) p(TII-PD) and (b) p(TII-2,6ND) solutions after adding TFA solution, (c) the chemical structure and exact mass of TII-(CHO)2, (d) 1H-NMR assignment of recycled TII-(CHO)2 from p(TII-PD) and p(TII-2,6ND), and (e) ESI-HRMS spectra of recycled TII-(CHO)2 from p(TII-PD) and p(TII-2,6ND). | ||
Following the recovery of TII-(CHO)2, we synthesized recycled polymers, re-p(TII-PD) and re-p(TII-2,6ND), using the recovered TII-(CHO)2 monomer (Fig. 3b) under the same polymerization conditions as for the pristine polymers. The Mn values of the recycled polymers were 28.8 kDa and 21.6 kDa with dispersities of 6.9 and 4.0, respectively (Fig. S6 and Table S1†). We attribute the differences in Mn values of the recycled polymers compared to those of the pristine polymers to batch-to-batch variations. From the 1H-NMR results, both recycled polymers show the same spectra as the pristine polymers (Fig. S11†).
Charge carrier transport properties
To evaluate the charge carrier transport properties of the pristine and recycled polymers, we fabricated FET devices with a bottom-gate, top-contact configuration with Au source and drain electrodes. The active layer was spin-coated on n-doped Si substrates treated with octadecyltrimethoxysilane (OTMS) on the surface (300 nm SiO2 layer),63 and the as-spun films were heat-annealed at 250 °C for 15 minutes in a glovebox. Finally, Au source/drain electrodes were deposited onto the active layer under vacuum, with 50 μm channel length and 1000 μm channel width. All measurements were conducted under vacuum, and the extracted FET parameters are summarized in Table 3, wherein we estimated the mobilities from the saturation regions. Thin films of freshly prepared p(TII-PD) and p(TII-2,6ND) exhibited typical p-channel behaviours, as anticipated from the HOMO and LUMO levels calculated from the redox and the optical properties (Fig. 5a, b, S8, S12,† and Table 2). The average hole mobilities of both pristine polymers are in the order of 10−3 cm2 V−1 s−1, whereas thin films of p(TII-2,6ND) exhibited better FET performance with the maximum mobility of 0.016 cm2 V−1 s−1 than those of p(TII-PD). Additionally, we evaluated charge carrier transport properties of the thin films of the recycled polymers, re-p(TII-PD) and re-p(TII-2,6ND) (Fig. 5c, d, and S13†), and the extracted data are summarized in Table 3. Both recycled polymers showed typical p-channel gate moderation with mobilities in the order of 10−2–10−3 cm2 V−1 s−1, similar to the pristine polymers, confirming the chemical recyclability of TII-based polymers without a loss in performance. Differences in performance between the pristine and recycled polymers are attributed to the increase in molecular weight of recycled polymers compared to pristine polymers (Fig. S6 and Table S1†). We note that there is room for improving FET mobilities by changing the molecular design to adopt these materials for various applications. | ||
| Fig. 5 Typical transfer curves of pristine polymers (a) pristine p(TII-PD) and (b) pristine p(TII-2,6ND) and recycled polymers (c) re-p(TII-PD) and (d) re-p(TII-2,6ND). AFM height images of thin films of (e) p(TII-PD) and (f) p(TII-2,6ND) with 500 nm scale bar. | ||
|
μ
h
avea (10−3 cm2 V−1 s−1) |
μ maxh (10−3 cm2 V−1 s−1) | V th (V) | I on/Ioff | |
|---|---|---|---|---|
| a μ h ave was calculated from at least 10 devices. | ||||
| Pristine p(TII-PD) | 4.4 ± 0.7 | 5.8 | −4.5 ± 4.7 | 106 |
| Pristine p(TII-2,6ND) | 6.9 ± 4.5 | 16.2 | −18.9 ± 7.9 | 106 |
| Recycled re-p(TII-PD) | 9.1 ± 3.7 | 18.3 | −11.4 ± 4.6 | 106 |
| Recycled re-p(TII-2,6ND) | 14.1 ± 3.0 | 19.6 | −1.9 ± 7.0 | 104 |
Thin film morphology
To examine the interchain packing and orientations of the polymer thin films annealed at 250 °C, grazing incidence wide-angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM) measurements were performed (Fig. 5e, f, and S14†) and the results are summarized in Table 4. According to GIWAXS patterns (Fig. S14†), thin films of both p(TII-PD) and p(TII-2,6ND) adopt similar microstructures, wherein both thin films display an edge-on and face-on mixed bimodal orientation with intense (100) lamellar scattering up to (200) order diffraction in both out-of-plane and in-plane directions. The corresponding out-of-plane and in-plane lamellar distances were 27.94 Å and 26.48 Å for p(TII-PD) and 25.47 Å and 25.06 Å for p(TII-2,6ND), respectively. Both polymers displayed out-of-plane (010) diffraction peaks corresponding to π–π stacking distances of approximately 3.5 Å, which further supports the presence of bimodal polymer orientations. To gain further insight for examining thin film microstructures, we estimated the crystal coherence length (CCL) from out-of-plane (010) and in-plane (100) peaks.50–52 The larger CCL values of p(TII-PD) than those of p(TII-2,6ND) indicate that densely packed intermolecular aggregates are formed in thin films, which is desirable for charge carrier transport. However, the resulting FET characterizations do not agree with this trend; p(TII-2,6ND) showed slightly improved charge carrier mobility compared to p(TII-PD).| Out-of-plane | In-plane | |||||
|---|---|---|---|---|---|---|
| Lamellar spacinga (Å) | CCLb (Å) | π–π stacking c(Å) | CCLd (Å) | Lamellar spacinge (Å) | CCLd (Å) | |
| a Estimated from the primal (200) peak. b It was impossible to calculate due to the X-ray leakage. c π–π stacking was estimated from the (010) peak. d The CCL was calculated using the Scherrer equation. e Estimated from the primal (100) peak. | ||||||
| p(TII-PD) | 27.94 | N/A | 3.56 | 37.2 | 26.48 | 308.1 |
| p(TII-2,6ND) | 25.47 | N/A | 3.54 | 18.6 | 25.06 | 67.5 |
To study the origin of different FET performances, we examined the thin film morphology by atomic force microscopy (AFM). We note that the AFM images represent the top of the thin films and are a proxy for the morphology in the channel, as AFM of the bottom interface was difficult to obtain without surface damage due to repetitive transfers. We observed quite different morphologies in the AFM height images. Thin films of p(TII-PD) showed continuous nanofibril networks with many void-like structures, whereas thin films of p(TII-2,6ND) showed larger bundle-like aggregates connecting each domain (Fig. 5e and f). RMS roughness was 5.2 nm for p(TII-PD) and 7.5 nm for p(TII-2,6ND), and these results support the bundle-like large crystalline morphology in p(TII-2,6ND). When considering lamellar distances, p(TII-2,6ND) exhibited shorter distances by around 2 Å in both out-of-plane and in-plane directions than p(TII-PD), implying closer intermolecular interactions. This compact interchain packing would be associated with different thin film morphologies when observed by AFM. We hypothesize that the large bundle-like assembly made of p(TII-2,6ND) would contribute to the higher FET performance observed than that of p(TII-PD).
Conclusions
Incorporation of acid-stable monomeric units allows for recyclability in imine-based semiconducting polymers. In this study, we synthesized and characterized the chemical recyclability of two TII-based copolymers bearing imine bonds, p(TII-PD) and p(TII-2,6ND). Both polymers were synthesized by a polycondensation reaction using TII-(CHO)2 with PD and 2,6ND monomers, respectively. The two polymers exhibited similar optical and redox properties, and incorporation of the imine bond along the polymer backbone was found to lower both HOMO and LUMO levels. In the degradation studies, both polymers showed good degradability through rapid proton-doping before imine bond cleavage. The TII-(CHO)2 monomer showed high chemical stability under acidic conditions, allowing for isolation of the monomer in over 90% yield from depolymerized products. The recovered monomer can subsequently be used to prepare the recycled polymers of p(TII-PD) and p(TII-2,6ND). Remarkably, the electrical properties of the recycled polymers showed FET mobilities comparable to those of the pristine ones. Overall, we demonstrated the closed-loop chemical recycling of two semiconducting polymers based on TII monomers. The results obtained from this study suggest that the TII building block is a promising unit for constructing chemically recyclable semi-conducting polymers for organic electronics. We hope that the design parameters discussed herein will guide future design of degradable semiconducting polymers, with careful consideration placed on ensuring that the by-products are stable and ideally non-toxic at the quantities used in future applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by JSPS KAKENHI Grant Numbers JP 18H01730 (for H. M.) and JP 22K05047 (for M. A.), the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN2021-03554) (for H. T.), the Canadian Foundation for Innovation (CFI-JELF-41743) (for H. T.) and in part to funding provided to the University of Toronto’s Acceleration Consortium from the Canada First Research Excellence Fund (for H. T.). A. U. thanks the NSERC for her CGS-D graduate scholarship. The synchrotron radiation experiments were performed at the BL40B2 beamline in SPring-8 with the approval of JASRI (Proposal No. 2021B1106 and No. 2022B1474). The authors thank Dr Kimito Koizumi (Open Facility Center, Tokyo Institute of Technology) in the Materials Analysis Division for ESI-HRMS spectra, and Dr Hiroyasu Masunaga and Dr Noboru Ohta (Japan Synchrotron Radiation Research Institute: JASRI) for assistance in the GIWAXS experiments.References
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef PubMed.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- J. Demarteau, A. R. Epstein, P. R. Christensen, M. Abubekerov, H. Wang, S. J. Teat, T. J. Seguin, C. W. Chan, C. D. Scown, T. P. Russell, J. D. Keasling, K. A. Persson and B. A. Helms, Sci. Adv., 2022, 8, 8823 CrossRef PubMed.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2022, 13, 6484–6491 RSC.
- X. Y. Oh, Y. Ge and A. Goto, Chem. Sci., 2021, 12, 13546–13556 RSC.
- B. A. Abel, R. L. Snyder and G. W. Coates, Science, 2021, 373, 783–789 CrossRef CAS.
- Z. Lei, H. Chen, C. Luo, Y. Rong, Y. Hu, Y. Jin, R. Long, K. Yu and W. Zhang, Nat. Chem., 2022, 14, 1399–1404 CrossRef CAS.
- P. Wang, J. Yang, Y. Zhang, W. Hu and H. Dong, ACS Mater. Lett., 2022, 4, 1112–1123 CrossRef CAS.
- Y. Lee, H. Zhou and T. W. Lee, J. Mater. Chem. C, 2018, 6, 3538–3550 RSC.
- L. Lan, J. Chen, Y. Wang, P. Li, Y. Yu, G. Zhu, Z. Li, T. Lei, W. Yue and I. McCulloch, Chem. Mater., 2022, 34, 1666–1676 CrossRef CAS.
- Z. Zhang, M. Liao, H. Lou, Y. Hu, X. Sun, H. Peng, Z. Zhang, M. Liao, H. Lou, Y. Hu, X. Sun and H. Peng, Adv. Mater., 2018, 30, 1704261 CrossRef.
- M. Ashizawa, Y. Zheng, H. Tran and Z. Bao, Prog. Polym. Sci., 2020, 100, 101181 CrossRef CAS.
- J. Xu, S. Wang, G. J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y. H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B. H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 6320 CrossRef PubMed.
- J. Onorato, V. Pakhnyuk and C. K. Luscombe, Polym. J., 2017, 49, 41–60 CrossRef CAS.
- T. Lei, M. Guan, J. Liu, H. C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T. C. Huang, L. Shao, K. T. Cheng, J. B. H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107–5112 CrossRef CAS.
- J. A. Chiong, H. Tran, Y. Lin, Y. Zheng, Z. Bao, J. A. Chiong, Y. Zheng, H. Tran, Y. Lin and Z. Bao, Adv. Sci., 2021, 8, 2101233 CrossRef CAS.
- J. Tropp and J. Rivnay, J. Mater. Chem. C, 2021, 9, 13543–13556 RSC.
- J. A. Chiong, Y. Zheng, S. Zhang, G. Ma, Y. Wu, G. Ngaruka, Y. Lin, X. Gu and Z. Bao, J. Am. Chem. Soc., 2022, 144, 3717–3726 CrossRef CAS.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef CAS.
- A. Uva, A. Lin, J. Babi and H. Tran, J. Chem. Technol. Biotechnol., 2022, 97, 801–809 CrossRef CAS.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743–768 CrossRef CAS.
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M.-G. Kim, A. Facchetti, X. Guo, S. Shi, P. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, X. Guo, Y. Chen, A. Facchetti and M. Kim, Adv. Mater., 2019, 31, 1905161 CrossRef CAS.
- J. Borges-González, C. J. Kousseff and C. B. Nielsen, J. Mater. Chem. C, 2019, 7, 1111–1130 RSC.
- Y. Dai, S. Dai, N. Li, Y. Li, M. Moser, J. Strzalka, A. Prominski, Y. Liu, Q. Zhang, S. Li, H. Hu, W. Liu, S. Chatterji, P. Cheng, B. Tian, I. McCulloch, J. Xu and S. Wang, Adv. Mater., 2022, 34, 2201178 CrossRef CAS.
- K. Mahesh, S. Karpagam and K. Pandian, Top. Curr. Chem., 2019, 377, 1–39 CrossRef CAS.
- H. R. Lee, Y. Won and J. H. Oh, J. Polym. Sci., 2022, 60, 348–376 CrossRef CAS.
- I. B. Dimov, M. Moser, G. G. Malliaras and I. McCulloch, Chem. Rev., 2022, 122, 4356–4396 CrossRef PubMed.
- S. G. Higgins, A. Lo Fiego, I. Patrick, A. Creamer, M. M. Stevens, S. G. Higgins, A. Lo Fiego, I. Patrick, A. Creamer and M. M. Stevens, Adv. Mater. Technol., 2020, 5, 2000384 CrossRef.
- E. W. C. Chan, X. Sun and J. Travas-Sejdic, Macromolecules, 2023, 56, 3755–3773 CrossRef.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef.
- C. Li, C. Guo, V. Fitzpatrick, A. Ibrahim, M. J. Zwierstra, P. Hanna, A. Lechtig, A. Nazarian, S. J. Lin and D. L. Kaplan, Nat. Rev. Mater., 2020, 5, 61–81 CrossRef.
- G. Garbay, L. Giraud, S. M. Gali, G. Hadziioannou, E. Grau, S. Grelier, E. Cloutet, H. Cramail and C. Brochon, ACS Omega, 2020, 5, 5176–5181 CrossRef PubMed.
- T. R. Martin, L. Rynearson, M. Kuller, J. Quinn, C. Wang, B. Lucht, N. R. Neale, T. R. Martin, M. Kuller, N. R. Neale, L. Rynearson, B. Lucht, J. Quinn and C. Wang, Adv. Energy Mater., 2023, 13, 2203921 CrossRef CAS.
- A. Uva, A. Lin and H. Tran, J. Am. Chem. Soc., 2023, 145, 3606–3614 CrossRef CAS.
- H. Huang, W. Xie, Q. Wan, L. Mao, D. Hu, H. Sun, X. Zhang, Y. Wei, H. Huang, W. Xie, L. Mao, D. Hu, H. Sun, Y. Wei, Q. Wan and X. Zhang, Adv. Sci., 2022, 9, 2104101 CrossRef CAS.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef.
- S. Tian, Q. Yue, C. Liu, M. Li, M. Yin, Y. Gao, F. Meng, B. Z. Tang and L. Luo, J. Am. Chem. Soc., 2021, 143, 10054–10058 CrossRef CAS PubMed.
- B. R. Varju and D. S. Seferos, Polym. Chem., 2022, 13, 6386–6392 RSC.
- H. Tran, S. Nikzad, J. A. Chiong, N. J. Schuster, A. E. Peña-Alcántara, V. R. Feig, Y.-Q. Q. Zheng and Z. Bao, Chem. Mater., 2021, 33, 7465–7474 CrossRef CAS.
- H. Tran, V. R. Feig, K. Liu, H. C. Wu, R. Chen, J. Xu, K. Deisseroth and Z. Bao, ACS Cent. Sci., 2019, 5, 1884–1891 CrossRef CAS.
- X. Q. Xu, Z. Wang, R. Li, Y. He and Y. Wang, Chem. – Eur. J., 2018, 24, 9769–9772 CrossRef CAS PubMed.
- K. A. Bartlett, A. Charland-Martin, J. Lawton, A. L. Tomlinson and G. S. Collier, Macromol. Rapid Commun., 2023, 2300220 CrossRef PubMed.
- H. Jin, K. Kim, S. Park, J. H. Rhee, H. Ahn, D. J. Kim, K. Kim, J. H. Noh, T. S. Kim, E. Y. Shin and H. J. Son, Adv. Funct. Mater., 2023, 2304930 CrossRef CAS.
- A. Velusamy, C. H. Yu, S. N. Afraj, C. C. Lin, W. Y. Lo, C. J. Yeh, Y. W. Wu, H. C. Hsieh, J. Chen, G. H. Lee, S. H. Tung, C. L. Liu, M. C. Chen and A. Facchetti, Adv. Sci., 2021, 8, 2002930 CrossRef CAS.
- T. Hasegawa, M. Ashizawa and H. Matsumoto, RSC Adv., 2015, 5, 61035–61043 RSC.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- M. Ashizawa, N. Masuda, T. Higashino, T. Kadoya, T. Kawamoto, H. Matsumoto and T. Mori, Org. Electron., 2016, 35, 95–100 CrossRef CAS.
- T. Hasegawa, M. Ashizawa, J. Hiyoshi, S. Kawauchi, J. Mei, Z. Bao and H. Matsumoto, Polym. Chem., 2016, 7, 1181–1190 RSC.
- P. Wang, C. Xu, X. Zhang, Y. Shi, C. Wang, Y. Han, Y. Deng and Y. Geng, Macromol. Rapid Commun., 2023, 2300245 CrossRef.
- D. Yoo, T. Hasegawa, M. Ashizawa, T. Kawamoto, H. Masunaga, T. Hikima, H. Matsumoto and T. Mori, J. Mater. Chem. C, 2017, 5, 2509–2512 RSC.
- X. Wei, W. Zhang and G. Yu, Adv. Funct. Mater., 2021, 31, 2010979 CrossRef CAS.
- R. S. Ashraf, A. J. Kronemeijer, D. I. James, H. Sirringhaus and I. McCulloch, Chem. Commun., 2012, 48, 3939–3941 RSC.
- G. Kim, S. J. Kang, G. K. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- H. Murakami, K. Kobayashi, K. Suzuki, T. Yasuda, T. Kanbara and J. Kuwabara, Macromolecules, 2021, 54, 11281–11288 CrossRef CAS.
- L. Giraud, S. Grelier, E. Grau, L. Garel, G. Hadziioannou, B. Kauffmann, É. Cloutet, H. Cramail and C. Brochon, Molecules, 2022, 27, 4138 CrossRef CAS.
- J. G
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) siorowski, E. D. Głowacki, B. Hajduk, M. Siwy, M. Chwastek-Ogierman, J. Weszka, H. Neugebauer and N. S. Sariciftci, J. Phys. Chem. C, 2013, 117, 2584–2589 CrossRef.
siorowski, E. D. Głowacki, B. Hajduk, M. Siwy, M. Chwastek-Ogierman, J. Weszka, H. Neugebauer and N. S. Sariciftci, J. Phys. Chem. C, 2013, 117, 2584–2589 CrossRef. - P. Nitschke, B. Jarząbek, A. E. Bejan and M. D. Damaceanu, J. Phys. Chem. B, 2021, 125, 8588–8600 CrossRef CAS PubMed.
- B. Hu, X. Zhu, X. Chen, L. Pan, S. Peng, Y. Wu, J. Shang, G. Liu, Q. Yan and R. W. Li, J. Am. Chem. Soc., 2012, 134, 17408–17411 CrossRef CAS.
- N. Sung, Y. Hsu, A. Lin, A. Uva, H. Huang and H. Tran, Macromolecules, 2023, 56, 8947–8955 CrossRef.
- Y. Ito, A. A. Virkar, S. Mannsfeld, H. O. Joon, M. Toney, J. Locklin and Z. Bao, J. Am. Chem. Soc., 2009, 131, 9396–9404 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure, NMR, TGA, DSC, CV, FET output profiles, GIWAXS, and AFM data. See DOI: https://doi.org/10.1039/d3lp00209h |
| This journal is © The Royal Society of Chemistry 2024 |