
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical polymers in optoelectronic and spintronic applications
Hyunki
Yeo†
 a,
Suman
Debnath†
a,
Baiju P.
Krishnan†
a and
Bryan W.
Boudouris
*ab
a,
Suman
Debnath†
a,
Baiju P.
Krishnan†
a and
Bryan W.
Boudouris
*ab
aCharles D. Davidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: boudouris@purdue.edu
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
First published on 20th December 2023
Abstract
Radical polymers hold great potential as solid-state conducting materials due to their distinctive charge transport mechanism and intriguing optical properties resulting from their singly occupied molecular orbital energy levels. Furthermore, the paramagnetic nature of their open-shell structures broadens their applicability, allowing them to be magnetic field-active while also offering promising spin transport properties. These molecular design features position radical polymers as interesting materials for next-generation quantum information systems as well. In this review, we highlight the progress regarding several stable open-shell radical macromolecular architectures. We commence by examining their synthetic methods along with the mechanisms governing charge transport in such materials, followed by emphasizing their significant development of solid-state optoelectronic materials, and we conclude by discussing their emerging roles in spintronic applications.
1. Introduction
Free radicals are often considered highly reactive intermediates in organic chemical reactions. However, after Gomberg's discovery of the triphenylmethyl radical with a measurable lifetime,1 the development of various radicals has improved their stability through the control of electronic and steric factors.2 With these advances in the design and syntheses of various stable radical groups, there have also been many developments in radical-containing macromolecules.2 In recent decades, because of the scope of structural diversity of open-shell macromolecules, materials with novel properties have been designed and implemented across multiple application spaces.3–5Specifically, radical polymers have emerged as promising solid-state optoelectronic materials.6 These polymers pass charge at localized radical sites that can communicate with each other by electron exchange in both electrolyte-based and solid-state devices when these radical sites are in close proximity.7 Because open-shell macromolecules transport charge in the solid state, they are being considered as future candidates for modern optoelectronic technologies. Moreover, open-shell materials have low-lying excited states due to the presence of singly occupied molecular orbitals (SOMOs), leading to intriguing optical properties. Because of these promising properties, open-shell materials have been used as a supplement to conjugated macromolecules. However, deciphering the nature of charge transport processes in a class of macromolecules and an appropriate molecular design to place charge-active sites within a critical distance to enable better electronic communication could open new avenues in the development of organic open-shell based electronic materials.8,9
Moreover, open-shell radical small molecules and polymers exhibit magnetic properties resulting from the interaction of the spin moments of the unpaired electron, extending their range of applications.10–12 Interest in nanoelectronic devices that use quantum phenomena for their operation has increased in recent years.13–16 In particular, devices that use the spin of an electron are being actively explored because spins can be manipulated in a faster and more energy-efficient manner.15,17 As a result, spintronics are expected to have faster switching times and lower power consumption than conventional organic electronic devices based solely on electron transport. Moreover, the energy levels of spins are discrete, and the associated quantum states can be tuned and coherently manipulated by external electromagnetic fields.18,19 While spintronics based on radical polymers have been little explored, they have the potential to transform the field of spin-based electronics, optoelectronics, and diagnostics due to ease of fabrication, tunable molecular, magnetic, and electronic structures.10,12,20
This review is intended to provide a survey of the state-of-the-art in macromolecular materials bearing radical motifs and the opportunities presented by their electronic, magnetic, and spin properties (Fig. 1). We begin with a discussion of the various known types of open-shell radical molecules, the classes of macromolecular architecture involving open-shell molecules, their chemical synthetic strategies, and charge transport in these materials. Then, we focus on electronic applications of these functional materials, which are mainly focused on organic light-emitting devices (OLEDs). Next, we discuss applications for open-shell macromolecular materials in solid-state spintronic devices. We conclude with our own assessment of the current state of the art and opportunities in this growing field, as well as challenges that should be addressed for the further development of these exciting functional materials.
 | ||
| Fig. 1 Schematic representation of macromolecules with open-shell character. On the right side of the figure, future potential applications such as spin valves and organic magnets are shown. The top section of the left side shows how electronic transport in these materials occurs between the open-shell molecules and the charged state of the open-shell molecules. The blue spheres represent the open-shell molecules attached to the polymer backbone, and the red spheres represent the redox state of the open-shell molecules. Pictured on the bottom left is the application of these polymers in light-emitting devices. | ||
2. Molecular design and synthesis of open-shell micromoles
Various types of open-shell molecules, such as (a) carbon-centered radicals, (b) nitrogen-centered radicals, (c) oxygen-centered radicals, and (d) nitroxide radicals, are used in design of radical polymers (Fig. 2).3 Among them, nitroxide radicals are the most common radical species, including 2,2,6,6-tetramethylpiperidinyloxy (TEMPO), 2,2,5,5-tetramethyl-1-pyrrolidinyloxy (PROXYL), and N-tert-butyl-N-oxy-aminobenzene, which are incorporated into radical polymers and polyradicals due to their relatively high stability. Similarly, radical polymers based on oxygen-centered radical groups, such as the phenoxyl and galvinoxyl open-shell groups, have been reported. Although nitrogen-centered open-shell groups, such as tetrahydro-s-tetrazin-1-(2H)-yl (verdazyl), dithiadiazolyl, and Blatter radicals, have also been investigated in small molecules, polymers based on them have been reported rarely due to either poor stability or complicated synthetic pathway. Pure hydrocarbon ensembles are less stable. These structures usually use combinations of carbon-centered radicals covalently bonded to multiple aromatic rings; the simplest example is the triarylmethyl radical. | ||
| Fig. 2 The chemical structures of the various open-shell species based on the nitroxyl group, oxygen-, carbon-, and nitrogen- atoms that are reported in the literature. | ||
Based on the structure or connectivity of open-shell molecules in macromolecules, open-shell macromolecules are classified into the following three categories: (1) non-conjugated radical polymers, (2) conjugated radical polymers (CRPs), and (3) polyradicals. The following subsections provide an overview of the chemical strategies used in the syntheses of these materials.
2.1. Non-conjugated radical polymers syntheses
Macromolecules with non-conjugated backbones and stable open-shell units are an emerging class of organic electronic materials with many potential applications in solid-state electronics.21 The charge transport and redox activity of these polymers primarily depend on the radical side groups, while the polymer backbone primarily determines the thermal and mechanical properties of the radical polymers. Therefore, the electronic and mechanical properties of these materials can be controlled with a known chemical strategy that allows the incorporation of new radical groups and the different architecture of the backbone.The radical groups are incorporated into the polymer backbone by (a) direct polymerization of monomers bearing pendant open-shell groups, (b) polymerization of a protected closed-shell monomer (i.e., a radical precursor), which requires an additional post-polymerization modification step, and (c) the attachment of open-shell groups to pre-synthesized polymer structures. The following section details synthetic strategies for developing non-conjugated radical polymers.
Direct polymerization routes allow for the syntheses of radical polymers without further modification, and these include ionic (i.e., either cationic or anionic) polymerizations, metathesis polymerizations, and ring-opening polymerizations.3 Therefore, these routes are often preferred when they are synthetically possible. Ionic polymerization is considered one of the most useful methods for the syntheses of radical polymers because the ionic terminus of the propagating chain does not interfere with the radical groups. The first stable radical polymers with pendant nitroxide radicals were synthesized by carbanionic polymerization of 2,2,6,6-tetramethylpiperidinyloxymethacrylate (TMA) using phenylmagnesium bromide (Fig. 3a).22 However, this strategy was somewhat hindered due to partial crosslinking during polymerization. To overcome this challenge, Nishide and co-workers used a moderately nucleophilic anionic polymerization initiator, methyl methacrylate-capped diphenylhexyllithium (MMALi), which suppressed the side reaction between the nitroxide radical of the TEMPO unit and the carbanion of diphenylhexyllithium (Fig. 3a).23 In turn, this modified strategy gave poly(2,2,6,6-tetramethylpiperidinyloxymethacrylate) (PTMA) with well-controlled molecular weight, a narrow molecular weight distribution, high yield, and exactly 1.0 radical per monomer unit. Similarly, many researchers have used anionic or anionic coordinated ring opening polymerization (ACROP) to synthesize radical polymers. In one example, ACROP initiators (e.g., ZnEt2/H2O) were highly suitable for unsaturated proxy-containing epoxide ring syntheses, resulting in a poly(proxyethylene oxide) polymer with full radical content (Fig. 3b).24 In contrast, the cationic polymerization of nitroxide radical monomers has been less studied because the radicals were susceptible to degradation by cationic initiators. However, low-temperature cationic polymerization of 4-vinyloxyl-TEMPO using boron trifluoride etherate (BF3·OEt2) as a catalyst has been reported,25 but this resulted in a TEMPO-containing poly(vinyl ether) gel, presumably due to an inevitable side reaction with the nitroxide radical (Fig. 3c).
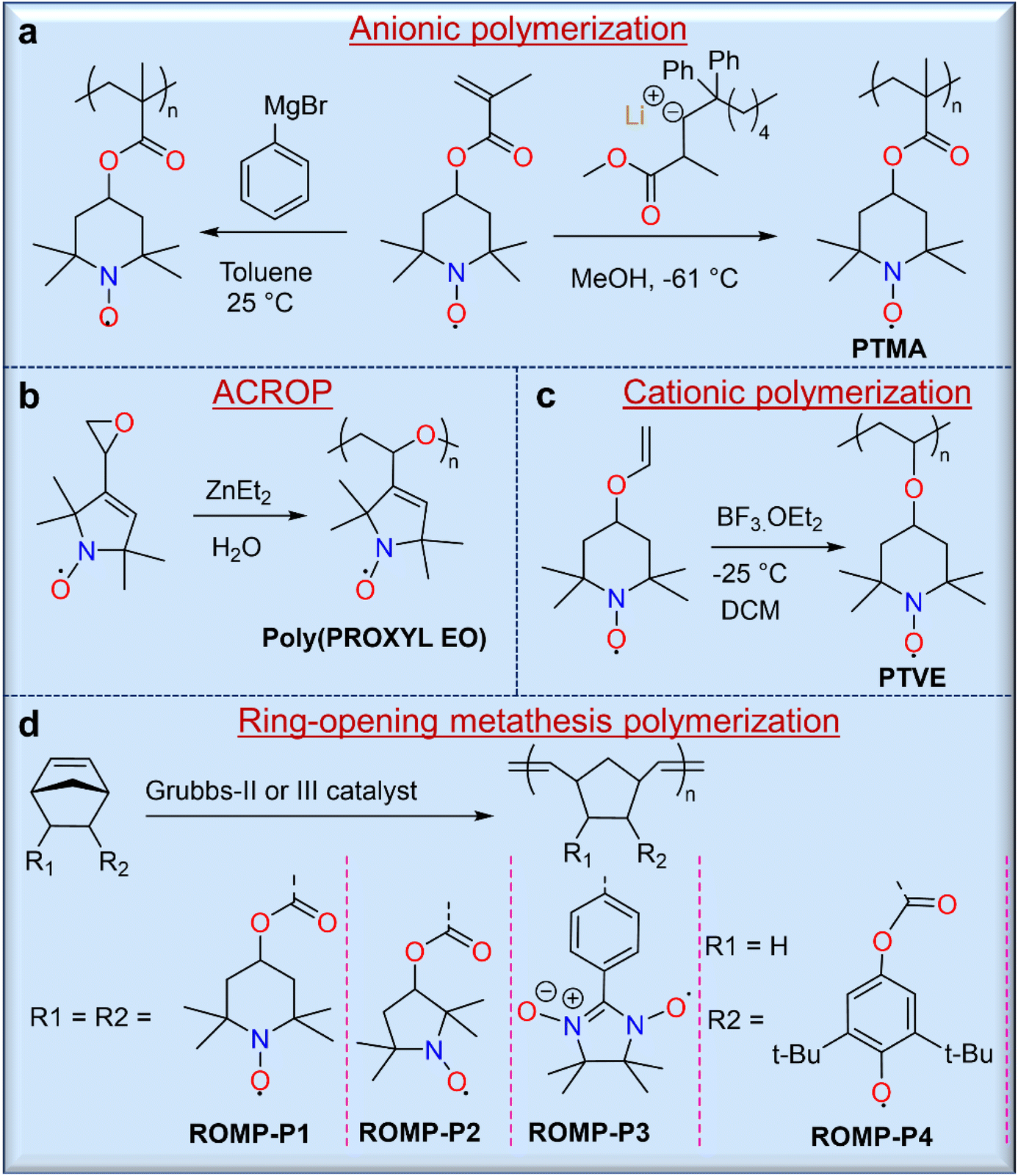 | ||
| Fig. 3 Non-conjugated radical polymers synthesized from open-shell monomers using (a) anionic, (b) anionic coordination ring opening (ACROP), (c) cationic, and (d) ring-opening metathesis polymerization techniques. | ||
In addition, the ring-opening metathesis polymerization (ROMP) of TEMPO-containing norbornene26 or 7-oxanorbornene27 monomers with second- or third-generation Grubbs catalysts has been carried out to obtain radical polymers without quenching the radical centers. For example, a series of TEMPO-containing norbornene monomers was polymerized by ROMP using the second-generation Grubbs catalyst in DCM solvent by Masuda and coworkers, yielding 2,3-endo, exo- and 2,3-endo, endo-polymers with very high molecular weights (Mn) of ∼185 kg mol−1 and 137 kg mol−1, respectively (Fig. 3d).26 It is noted that the charge storage performance of these two polymers is different, although their structures are similar. While the advantages of ROMP include mild polymerization conditions, the ability to achieve high molecular weights, and good reaction control of monomers with large radical groups, caution should be exercised when solution-based processing is desired for end-use applications, as endo and exo conformations can drastically affect solubility.
The strategy of polymerizing a protected closed-shell monomer (i.e., a radical precursor) using free radical polymerization (FRP) and living radical polymerization (LRP) techniques is as important as the direct polymerization strategy because of the relatively versatile and robust nature of these synthetic methods. In this strategy, the target radical polymers are prepared by oxidation or deprotection after polymerization of these precursor monomers. The first demonstration of this strategy was described by Okawara and co-workers in 1972 using azobisisobutylnitrile (AIBN)-initiated free polymerization of 2,2,6,6-tetramethylpiperidin-4-yl methacrylate (TMPM),28 which yielded a polymer precursor that was converted to a nitroxide-containing polyacrylate by oxidation with H2O2/Na2WO4 (Fig. 4a). Similarly, Nishide and co-workers used a silyl-protected nitroxylstyrene monomer that allowed direct free radical polymerization with AIBN to give a radical precursor polystyrene.29 Deprotection of the tert-butyldimethylsilyl (TBDMS) groups from the radical precursor polymers with TBAF, followed by mild oxidation with silver or manganese oxide, led to the final radical nitroxide polymers, poly(nitroxylstyrene) (Fig. 4a). Other radical polymers, including nitronylnitroxy,30 phenoxy (galvinoxyl),31 6-oxoverdazyl,32 and 1,1,3,3-tetramethylisoindolin-2-yloxyl (TMIO)33 radical polymers, were also synthesized by similar methods.
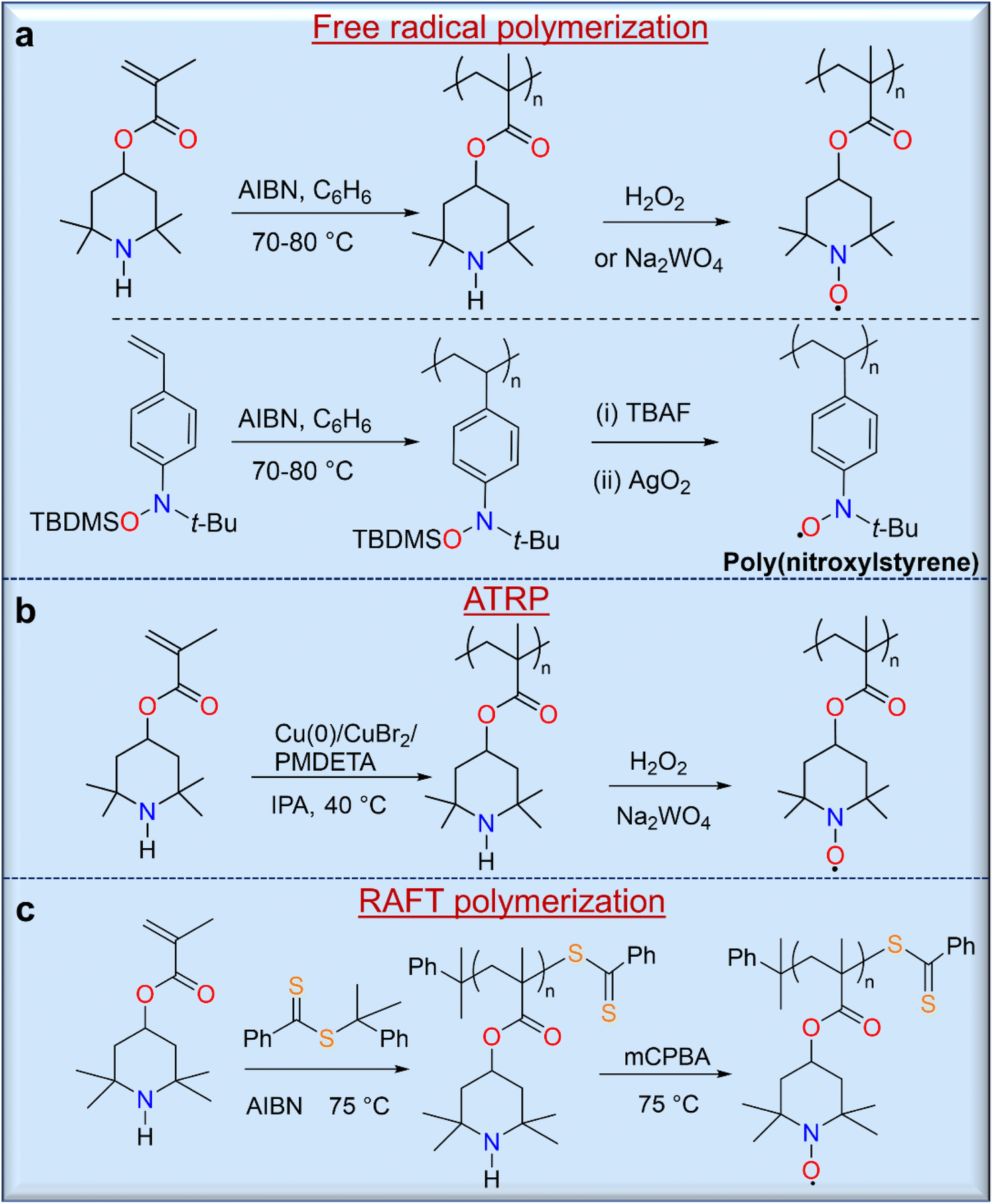 | ||
| Fig. 4 Non-conjugated radical polymers synthesized from open-shell precursor monomers through (a) free radical polymerization, (b) ATRP and (c) RAFT polymerization. In these examples, the precursor polymer obtained was converted to corresponding radical polymer by oxidation or deprotection after the polymerization reaction. | ||
The LRP of organic radical precursor monomers is attractive because of the large control over molecular weight and the controlled nature of polymerizations, and used these methods for the synthesis of various open shell macromolecules. Thus, Gohy and co-workers reported the atom transfer radical polymerization (ATRP) of TMPM using a catalyst system of Cu(0)/CuBr2/N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA),34 resulting in a radical polymer with a molecular weight of approximately 11 kg mol−1 and a narrow molecular weight distribution (Đ = 1.11) with a high conversion rate of 98% (Fig. 4b). Similarly, reversible addition–fragmentation chain transfer (RAFT) polymerization can be used for the synthesis of radical polymers such as well-defined PTMA (Fig. 4c).35 Although RAFT polymerization provides potential control over molecular weight, prolonged polymerization leads to an increase in dispersity due to intermolecular chain transfer and a loss of control by the RAFT end group, which can occur due to aminolysis of thiocarbonylthio compounds by the secondary amine monomer. In addition, the final oxidation of the precursor polymer can lead to an insoluble crosslinked product due to the presence of the terminal sulfur-containing component. On the other hand, removal of the RAFT-terminus with excess AIBN leads to a methyl-terminated PTMPM that can be easily oxidized to PTMA without crosslinking.7 RAFT polymerization of monomers containing free amines is a difficult task because these free amines cause aminolysis of thiocarbonylthio chain transfer agents, which stop polymerization and cleave the thiocarbonylthio end groups. Performing RAFT polymerizations at acidic pH would minimize this problem; however, the TMPM hydrochloride monomers only polymerized up to ∼60% polymer conversion.36 Protecting the radical moiety of hydroxy-TEMPO with a methyl37 or phenyl38 functionality is another solution to these problems. Thus, Blinco and co-workers have demonstrated the synthesis of PTMA by RAFT polymerization of methoxyamine-protected TEMPO methacrylate (MTMA) and subsequent cleavage of the temperature-stable methoxyamine functionality by oxidative treatment of PMTMA with meta-chloroperbenzoic acid.37 This polymerization shows a linear first-order behavior over the first 5 h up to a conversion of 7% with a dispersity of 1.13. The versatility of this method opens the possibility of controlled and facile synthesis of various PTMA polymer architectures with relatively low dispersity and highly targeted molecular weights.35
Post-polymerization modification, in which radical groups are added to pre-synthesized polymer structures, is another powerful strategy for preparing radical polymers. For example, the copper-catalyzed alkyne–azide cycloaddition (CuAAC) “click” reaction was used to install TEMPO groups on polythiophene by clicking 4-propargyl-TEMPO with azido-functionalized polythiophene.39 Similarly, siloxane-based radical polymers were synthesized by hydrosilylation of poly(methylhydrosiloxane) (PMHS) with 4-allyl-2,2,6,6-tetramethylpiperidine-N-oxyl ether in the presence of a platinum or rhodium catalyst.40 The post-modification method generally follows statistical incorporation because the radical content can be quantitatively measured.41 Although several non-conjugated radical polymers have been synthesized using these known chemical strategies, it is still a challenge to design a molecular structure that has great potential for the targeted applications, so a deep understanding of the structural properties is a key factor in developing high-performance devices using these materials.
2.2. Conjugated radical polymers
Open-shell molecules attached to conjugated macromolecules form a subclass of radical polymers called conjugated radical polymers (CRPs).42 Tuning the structure of the open-shell molecules and the conjugation length of the polymer would also provide the opportunity to control the optical and electrochemical properties of this class of materials. The syntheses of these polymers are often different from that of non-conjugated polymers. In the last decade, CRPs with different conjugated polymer backbones such as polyacetylene,43 polythiophene,44 polypyrrole,42 and poly(3,4-ethylenedioxylthiophene) (PEDOT)45 have been reported. CRPs with thiophene42- and pyrrole44,45-polymer backbones are usually synthesized by oxidation polymerization directly on an electrode surface (Fig. 5a). For instance, Armand and co-workers synthesized PEDOT with a stable nitroxide radical using an electropolymerization route (Fig. 5b).45 In addition to the syntheses of TEMPO polymers, oxidation polymerization was used for the synthesis of polythiophene with a verdazyl radical group46 and poly(3-phenoxyl-substituted thiophene).47 Taking advantage of the oxidation polymerization of dopamine, the auto-oxidation polymerization of a dopamine-functional TEMPO derivative on the substrate was also realized.48 Additionally, the copper-catalyzed alkyne–azide “click” reaction has been used as an alternative method for the preparation of a CRP with a polythiophene backbone, but polymers with a high content of radical moieties are not completely soluble in many solvents. The methods of Stille coupling and metal-catalyzed polymerization have been used to prepare CRPs. The synthesis of a CRP with polyacetylene backbone by Rh-catalyzed polymerization has been reported, but this produces polymers with high dispersity and even an insoluble fraction. Similarly, Stille coupling polymerization of 1,3-bisdiphenylene-2-phenylallyl (BDPA) analogues with thienyl distannanes gave BDPA-based polymers in 96% yield with a high molecular weight of 35 kg mol−1 (Đ = 2.70). These BDPA-based polymers were converted into corresponding radical polymers by deprotonation of the proton at the benzylic position of fluorene with excess potassium tert-butoxide and subsequent oxidation of the BDPA anions to radicals with potassium ferricyanide (Fig. 5c).49 | ||
| Fig. 5 Conjugated radical polymers with (a) polythiophene- and (b) PEDOT-backbones synthesized from corresponding open-shell- monomers by oxidative polymerization. (c) Synthesis of a BDPA-based radical polymer by Stille coupling. The obtained precursor polymer was further treated by proton abstraction with a base, followed by oxidation to give a BAPA radical polymer. | ||
2.3. Polyradicals
Because polyradicals are macromolecules composed of repeating open-shell units and do not have separate backbone and open-shell components, they often exhibit a high degree of radical delocalization. According to Rajca's classification, there are two main types of polyradicals (Fig. 6). Class I includes polyradicals with open-shell groups repeating along the polymer backbone, and class II includes polyradicals in which the open-shell groups are attached to the conjugated backbone.50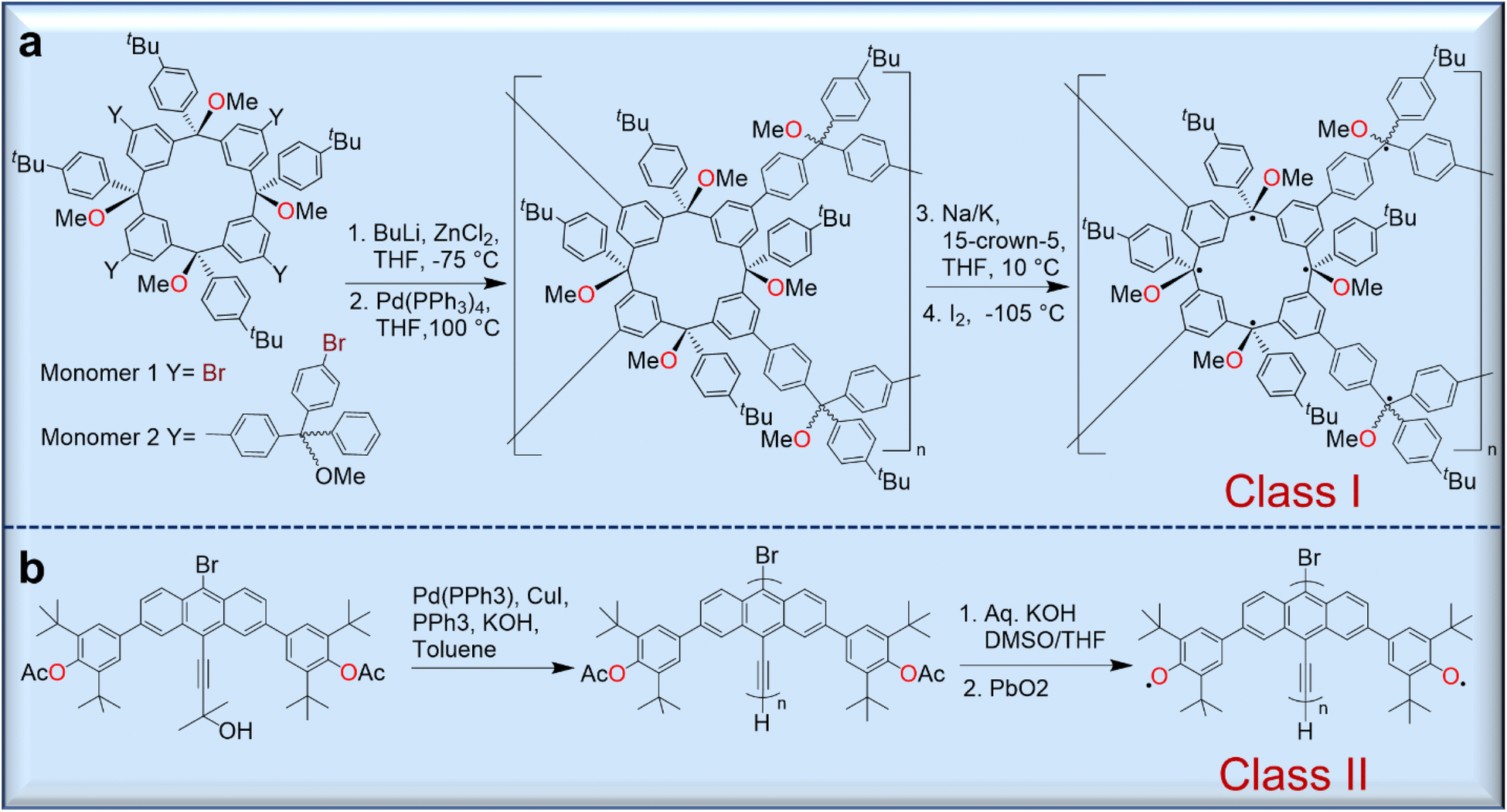 | ||
| Fig. 6 Syntheses of representative polyradicals of class I (a) and class II (b) according to Rajca's classification protocol. | ||
Although they exhibit a high-spin ground state and long-range magnetic ordering, only a few polyradicals are known due to the complicated synthesis strategies. However, the Rajca group has prepared numerous class I polyradicals with high spin content, including dendritic and star-branched structures based on the alternating connection of 1,3-phenylene-ferromagnetic coupling units (fCUs) and two arylmethyl spin sites.50 However, because defects are present in the dendrites that disrupt the ferromagnetic spin coupling in these polyradicals, the average values of S for these radicals are limited to S = 5 or less. To avoid these problems, dendritic–macrocyclic and macrocyclic–macrocyclic structures were designed using the spin cluster approach to maximize the number of spin sites.50 Moreover, elaboration of the spin-cluster approach leads to a polymacrocyclic highly interconnected π-conjugated polyradical network with multiple spin-coupling pathways through the alternating connectivity of two types of radical modules with ferromagnetic/ferrimagnetic arrangement of spins, which should allow large net S values.51–53 These polymer networks were synthesized by Pd-catalyzed Negishi coupling of two tetrafunctionalized macrocyclic monomers yielding a polymer further treated with metal (Na/K/15-crown-5) to give a corresponding carbopolyanion. These anions yielded the target polyradical after oxidation with iodine (Fig. 6a).51,52
Class II polyradicals, on the other hand, are less studied and are often based on phenoxyl-bearing conjugated polymers.54 For instance, the synthesis of poly(9,10-anthrylenethynylene)-based polyradicals bearing phenoxyls in the anthracene backbone was achieved through the polymerization of the corresponding bromoethynylanthracene monomer using a Pd(0) catalyst (Fig. 6b).54 Although there are some difficulties in their synthesis, the promising magnetic and electronic properties of these materials could be future functional molecules for organic magnets which will be discussed in the section of magnetic applications.
The synthetic development of radical polymers and polyradicals is steadily increasing and has accelerated greatly in the last two decades. With the help of modern chemistry, many advances have led to three types of open-shell macromolecules with different types of radicals. Elucidating the structure–function relationship within these macromolecules can lead to a deeper understanding of the transport mechanisms in these materials, which would be of great importance for future technological advances. In addition, by tuning of chemical structure of these materials, there is a possibility of altering the physical properties of these materials, which paves a way to control the performance of these materials in future applications. Furthermore, the development of precise molecular structures provides fundamental insights into the electronic structure and structure–property relationships of polyradical materials.
3. Charge transport in radical polymers
Because of the electronic properties of polymers bearing open-shell units, they can serve as charge conduction sites in potential electronic devices such as organic light-emitting devices (OLEDs), solar cells, and transistors. For a relatively long period of time, nitroxide-based non-conjugated radical polymers exhibited low electronic conductivity because of their insulating backbone components, which lowers the density of charge carrier sites. The solid-state electrical conductivity of non-conjugated radical polymers, such as PTMA, is in the range of 10−5 to 10−11 S cm−1.7,55 Therefore, various studies have been carried out to understand and explain the electronic transport mechanism in these materials.56Based on theoretical calculations of reorganization energies of different radical species, the calculated reorganization energies decrease with increasing delocalization of radicals.56 In this study, the highest reorganization energy was calculated for TEMPO, and the lowest for the galvinoxyl radical. Thus, maximal delocalization leads to significant electronic coupling, which results in high charge transfer in species with lower reorganization energy. Electron transport in radical polymers also depends on charge carrier density, and higher radical loadings are required for significant charge transport.57 In addition to these factors, polymer morphology and radical-to-radical distance are critical for efficient charge transport. Using atomistic molecular dynamic simulations of the radial distributions between radicals in the modeled amorphous PTMA structure, the effective electron transfer distance is ≤5.5 Å, which is responsible for most (>85%) of the charge transfer in the radical polymer.57
Furthermore, many polymer scientists have become interested in these materials over the past two decades because they exhibit fast redox kinetics, and numerous promising radical polymer conductors with different molecular architectures have been developed. For example, the chemical nature of pendant open-shell groups affects the conductivity of the polymer films by almost an order of magnitude.58 The conductivity of PTMA increases to a maximum value of (1.52 ± 0.3) × 10−5 S cm−1 when doped with ∼2.5% PTMA cation sites formed by oxidation of PTMA, suggesting that moderate doping of the radical polymers could improve their ability to conduct charges in the solid state compared with unmodified radical polymer materials.58 We reported that the conductivity of an ether-based organic radical polymer increases dramatically up to 0.20 S cm−1 over a distance of 600 nm or less, due to local organization of percolating radicals at or below this length scale by thermal annealing (Fig. 7a).21 This study suggests that this local organization leads to a high probability of radical-to-radical (i.e., site-to-site) charge hopping by increasing the local concentration of radicals and flexible ether bonds.59 We quantified the electrical conductivity in an organic crystal based on TEMPO molecules with an electrical conductivity of ∼0.03 S cm−1, which is one of the highest values for non-conjugated radical conductors over a length scale of 1 μm.60
 | ||
| Fig. 7 (a) Schematic representation of how organic radical polymer PTEO [poly(4-glycidyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl)] conduct electricity and how a flexible structure that helps to increase the polymer's conductivity at length scales of 600 nm or less. Adapted with permission from ref. 63. Copyright 2018 The American Association for the Advancement of Science. (b) Chemical structures of polythiophene conjugated radical polymers (CRPs) with alkyl spacers (n = 4, 6, 8) and control polymer P3BT. (c) Cyclic voltammograms (CVs) of CRPs and P3BT at 0.5 mV s−1. (d) OCP monitoring for 6 h. The electrodes were first charged by linear sweep voltammetry to 4.2 V and held at constant potential for 50 s (inset: illustration of CRPs before and after open-circuit potential decay). (e) Illustration of the internal electron-transfer process occurring for the CRPs. Adapted with permission from ref. 61. Copyright 2017 John Wiley and Sons. | ||
There were attempts made to improve the conductivity of these polymers by replacing the aliphatic backbone with a conjugated chain such as polythiophene.61 However, these CRPs exhibit low conductivities (i.e., in the range of 10−7–10−9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) S cm−1) due to internal electron transfer between the conjugated backbone and the radical group (Fig. 7b–e).61 However, the situation is different when radical pendant groups such as the galvinoxyl and phenoxyl radicals are incorporated into a polythiophene backbone, which increased the electrical conductivity of the polymer from 2.5 × 10−9 S cm−1 to 3.6 × 10−6 S cm−1 when the radical content increased to 0.93 radical/monomer unit because the radical groups facilitated interchain hole transfer relative to internal electron transfer.62
S cm−1) due to internal electron transfer between the conjugated backbone and the radical group (Fig. 7b–e).61 However, the situation is different when radical pendant groups such as the galvinoxyl and phenoxyl radicals are incorporated into a polythiophene backbone, which increased the electrical conductivity of the polymer from 2.5 × 10−9 S cm−1 to 3.6 × 10−6 S cm−1 when the radical content increased to 0.93 radical/monomer unit because the radical groups facilitated interchain hole transfer relative to internal electron transfer.62
The combination of synthetic efforts and theoretical understanding of electron transport in open-shell macromolecules has driven the successful development of electronic device applications.60,63 For example, efficient light-emitting diodes can be developed due to the doublet emission properties of radical-based species.64 In addition, organic spintronics is currently being explored due to the spin properties of these macromolecular systems, although these applications are still at infant stage.10–12 In the following sections, we focus on these emerging research areas with these electronic and magnetically active materials.
4. Electronic applications
Recently, open-shell macromolecules have shown promise in multiple organic electronic devices due to their mechanical robustness, versatile synthetic mythologies, and tunable electronic structures. Organic radicals and radical polymers have an extensive scope for various potential applications such as batteries, energy storage devices, memory devices, and OLEDs due to their special electronic and photophysical properties. Controlling their redox chemistry permits their use in batteries and energy storage devices. Due to these interesting chemical and physical properties, they have been utilized in a wide range of potential applications and have also discussed in detail these electrochemical applications in other comprehensive reviews.65–69 In the following section, we describe the significant role of OLEDs in organic electronics compared with the liquid-crystal displays (LCDs), electroluminescent processes, findings of emission mechanisms for organic emitters in radical based small molecules, and the structure-properties relationship of these open shell molecules in OLEDs.4.1. Applications of open-shell macromolecules in organic light-emitting diodes (OLEDs)
OLEDs play a significant role in organic electronics after first being introduced in 198770 due to their lower energy consumption, flexibility, light weight and device-fabrication compared with the LCDs.71 In most systems, electroluminescent processes occur through the 1:3 ratio of singlet and triplet excitons forms.72 Due to the Pauli exclusion principle, most triplet excitons are lost in traditional OLEDs.72 Thus, the internal quantum efficiency (IQE) of conventional closed-shell organic emitters cannot exceed 25%, and the external quantum efficiency (EQE) often does not exceed 5% in the case of traditional fluorescent OLEDs.73,74 To address this issue, spin–orbit coupling using heavy metals can significantly improve intersystem crossing and the phosphorescence decay rate, and thus, phosphorescent metal complexes expand the efficiencies of OLEDs.75 Alternatively, molecules with thermally activated delayed fluorescence (TADF) properties or triplet–triplet annihilation have achieved high IQEs in OLEDs (Fig. 8a and b).76–78 These triplet-excitons collecting approaches were all constructed on an even number of electrons-based closed-shell molecules. The doublet nature of the ground and excited states created open-shell molecules with an odd number of electrons emissive neutral radicals considered as probable ideal OLED emitters.64 In this effort, charge transfer becomes spin allowed, so the theoretical IQE of molecules that utilize radical-based materials as photoluminescence layers can reach 100%.
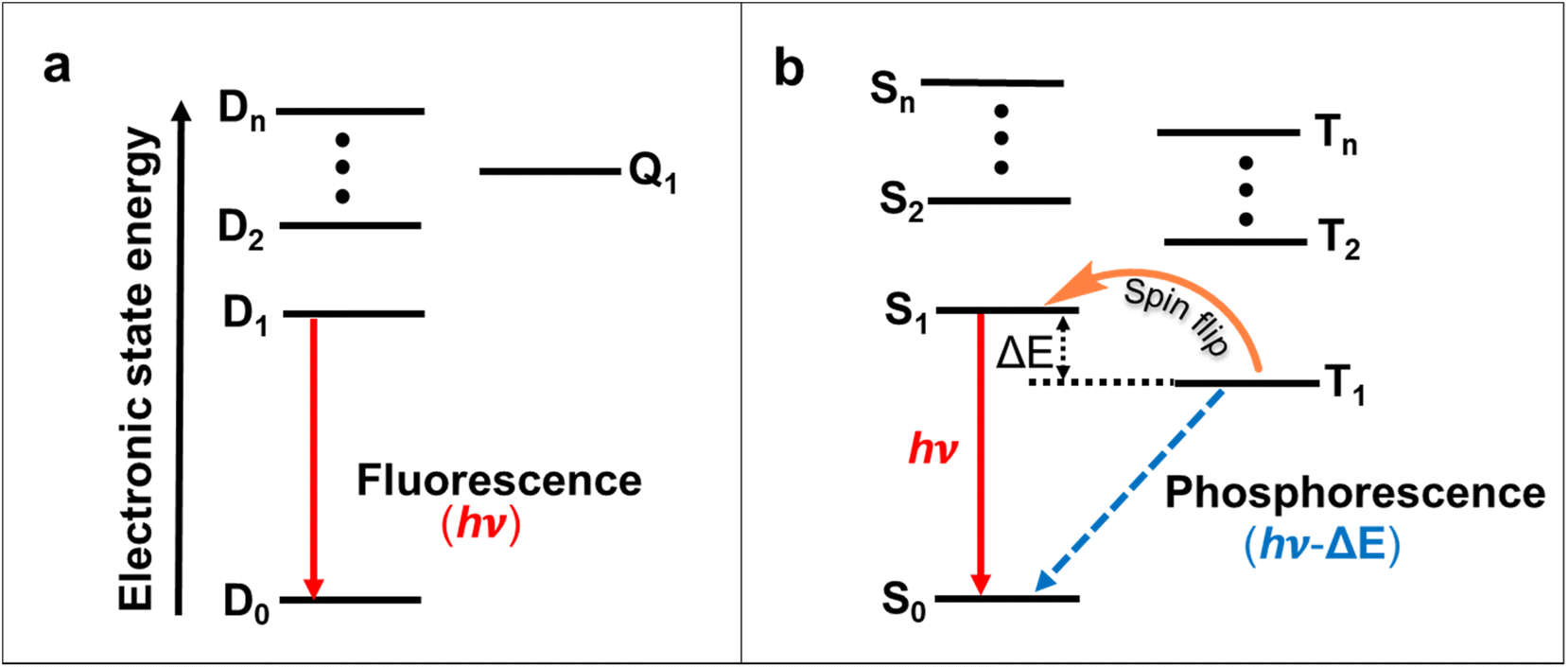 | ||
| Fig. 8 (a) Emission mechanisms for doublet- and singlet-/triplet-based organic emitters. Jablonski energy diagrams of the (a) doublet-quartet manifold, indicating doublet–doublet fluorescence. (b) Singlet–triplet manifold indicating singlet–singlet fluorescence, triplet–singlet phosphorescence, and spin flip processes, e.g., intersystem crossing (ISC, T1 → S1) in TADF; triplet–triplet annihilation (TTA, 2 T1 → S1 + S0). | ||
The doublet fluorescent in organic radicals is from materials that are mostly based on chlorinated triphenylmethyl radicals, including perchlorotriphenylmethyl (PTM),79–81 tris-2,4,6-trichlorophenylmethyl radical (TTM),82–84 and (3,5-dichloro-4-pyridyl) bis(2,4,6-trichlorophenyl) methyl (PyBTM)85,86 derivatives. To date, OLEDs based on organic radicals have mostly centered on red luminescent TTM derivatives. Li and co-workers introduced emissive conjugated radicals TTM-1Cz and TTM-2Cz in the OLEDs in 2015.87 The charge transfer from carbazole to the TTM center was the reason behind the emission of TTM-1Cz and TTM-2Cz, and the magneto-electroluminescence measurements confirmed the doublet nature of their excited states. TTM-1Cz-doped 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) showed a better EQE (4.3%) for OLEDs compared with the unfunctionalized TTM (EQE 2.8%) due to the intramolecular charge transfer between the carbazole donor's highest occupied molecular orbital (HOMO) and the TTM acceptor's singly-occupied molecular orbital (SOMO).88,89 Further, a sequence of new emission-neutral conjugated radicals was introduced by connecting electron-withdrawing benzimidazole groups on TTM radicals in OLED devices.90 The EQE of the radical-based OLEDs was improved to 10.6% by incorporation of the 1,5-diazarcarbazole modified TTM radical (TTM-DACz).91 Furthermore, the EQE was improved to 27% in the TTM-NCz system.92
Radical-containing polymers are promising for improving the stability of these OLED devices. The significant importance of high-efficiency OLEDs has progressed by appropriate host materials in polymer systems for application as an emitter. Li and coworkers introduced pendant carbazolyl group-based poly(meta-styrene) derivatives as a host for efficient solution-processed organic light-emitting diodes. For example, TTM-1Cz was attached to a polystyrene backbone to synthesize PS-CzTTM macromolecule (Fig. 9a), and this macromolecule displayed 24.4% and 37.6% photoluminescent quantum efficiency (PLQY) in solid state film and solution states, respectively.92,93 PS-CzTTM was incorporated as an emission layer in the OLEDs due to its photostability, reduced aggregation-caused quenching effect, and high photoluminescent quantum efficiency (PLQE). The half-life of emission intensity (1.6 × 104 s in cyclohexane solution) has improved up to 300 times compared with the TTM mono radical under same conditions.94 The devices were prepared with construction as ITO/PEDOT:PSS/PVK/emissive layer (EML)/B3PYMPM/LiF/Al to determine the potential of PS-CzTTM in OLEDs (Fig. 9b). Host–guest-based PS-CzTTM OLEDs (TPBi) showed deep-red emissions peaking at 685 nm with maximum EQEs of 3.0%, whereas the host-free OLEDs showed 0.9% EQEs (Fig. 9c). Besides the higher EQE, PS-CzTTM: TPBi OLED also improved brightness, charge balance, and reduced efficiency roll-off effect compared with the host-free OLED. The emission process was maintained and confirmed by electroluminescent (EL) spectra of host-free and host–guest devices at 12 V operating voltage (Fig. 9d). The PS-CzTTM-based OLEDS showed higher stability of operating current density (8.1 mA cm−2) and higher solution processability compared with the small radical-based OLEDs. Parallelly, Yang's group introduced super acid-catalyzed carbazole-conjugated backbones-based radical polymers with 1.8% EQE in electroluminescent devices to avoid the use of heavy metal catalysts in polymerization.95 In fact, the first photoluminescence studies of PTEO recently were reported.96 PTEO had high photoluminescence intensity in its aggregated state, which occurs below the glass transition temperature. This state is due to the intermolecular non-covalent interactions among the TEMPO units. Radical-based OLEDs have been developed only for a few years and only a few radicals have been studied; thus, there are challenges but abundant opportunities to improve their performances via smart molecular design and engineering.
 | ||
| Fig. 9 (a) The chemical structure of PS-CzTTM. (b) Schematic diagram of the device structure of PS-CzTTM-based OLED. (c) EQE of host-free and host–guest OLEDs versus current density. (d) EL spectra of both host–guest and host-free OLEDs at 12 V operating voltage. Adapted with permission from ref. 93. Copyright 2023 American Chemical Society. | ||
5. Spintronic applications
Two distinct, yet interconnected, modes of transport, namely spin transport and charge transport, form the foundation of contemporary technological landscape of polymer-based electronics.97–99 These phenomena dominantly influence the migration of crucial physical and chemical entities within (radical) organic materials, thus paving the way for innovative electronic, magnetic, and informational technologies.100 Establishing the difference between spin transport, which involves the manipulation of intrinsic angular momentum, and charge transport, which centers around the movement of electric charge, is pivotal for harnessing their unique properties and engineering novel functionalities.65 Evaluating these distinct transport mechanisms and leveraging the principles of polymer science in practical applications are vital steps toward advancing diverse fields, ranging from semiconductor electronics to spintronics and quantum computing.10,101 In the following section, we discuss recent findings regarding magnetism and spin transport in radical polymers, focusing on their individual characteristics, underlying principles, and emerging applications.5.1. Magnetism in radical polymers
Although spintronic applications of radical polymers have recently been reported, the magnetism of radical-containing macromolecules have been studied more than 30 years.50,52,102 Since the first discovery of the triphenylmethyl stable radical, initial studies were focused on discovering the fundamental magnetic properties and evaluating the potential of these radical-containing macromolecules in various applications.103 Early investigations centered around understanding the electronic structure and magnetic interactions within these materials, aiming to unveil the origin of their magnetic behavior.In general, organic molecules are diamagnetic because of the large energy gap between the singlet ground state and the nearest triplet state (ΔEST). However, this scenario can be different in radical polymers, because the ΔEST is smaller and the thermal energy available at room temperature is sufficient for spin excitation. Moreover, the physical and chemical properties are tunable through synthetic modifications such that these materials are appropriate for magnetic applications in dynamic polarization techniques, organic magnets, and spintronics, as compared to inorganic counterparts. For instance, in small molecule and polyradical studies, smaller electron–proton coupling constants, J values, where ΔEST = 2J, were observed when methoxy groups were substituted to chloride groups in between tris(2,6-dimethoxyphenyl)methyl and perchlorotriphenylmethyl radical magnetism studies. To develop this correlation between functionalization and magnetism into polymers, the Azoulay group demonstrated two different donor–acceptor polymers with high and low spin, by simply tuning the functional group of monomers.104 This demonstration has successfully proved that the spin density in radicals is directly influenced by surrounding functional groups. Furthermore, Rajca's group proved that quantum spin numbers, S, can be modulated simply by linking polyradicals with either an antiferromagnetic coupling unit (aCU) or ferromagnetic coupling unit (fCU). This work highlights the synthesis of a class of high-spin materials with up to up to S = 5000 (Fig. 10).51 As the values of S are related to the number of ferromagnetically coupled unpaired electrons, materials with high S are considered as one of the possible candidates for organic polymer magnets. This class of macromolecules with paramagnetic nature, compared to diamagnetic closed-shell conjugated polymers, has brought the attention of radical polymers as a suitable material for potential spintronic applications. To date, researchers are still delving into the influence of chemical substitutions, molecular arrangements, and external stimuli on the magnetic properties of radical polymers. These foundational studies provide crucial insights into the relationship between the molecular architecture of radical-containing macromolecules and their magnetism, paving the way for subsequent advancements in the field.
 | ||
| Fig. 10 (a) Development of high-spin polyarylmethyls (Ar = 4-tert-butylphenyl) (b) magnetic field (H) dependence of the magnetization (M) of OMP-1 at T = 3.5 K. The solid line corresponds to the least-squares plot using linear combination of Langevin and Brillouin functions corresponding to average S = 5400. Adapted with permission from ref. 102. Copyright 2005 Elsevier. (c) Plot of χT versus T in a static field in presence of 0.5, 5 and 50 Oe. Adapted with permission from ref. 51. Copyright 2001, The American Association for the Advancement of Science. | ||
5.2. Spin relaxation length and time
Spin relaxation time (τs) can be expressed as the following equation. | (1) |
Also, the spin relaxation length can be expressed as ls = kτs0.5, where the k constant value differs within the magnetism of material.105,106 Here, τ↑↓ stands for the average time for an up-spin flip to a down-spin and τ↓↑ for the reverse. Furthermore, τs is a crucial parameter for spintronic devices because it sets the time and length scale for the loss of spin polarization. Spin relaxation in solids can be explained by two mechanisms, spin–orbit coupling (SOC) and hyperfine interactions.107 In general, organic materials, trend to show small but non-negligible SOC and hyperfine interactions, which leads to the comparably longer relaxation time to that of inorganic materials (Fig. 11a).108 SOC is an effect that describes the interaction between the electron's spin and its motion while it travels along the orbital within an electric field. While a non-zero spin particle travels along the electric field with velocity, it generates a field with a magnetic component, which interacts with the electric field. SOC is a relativistic effect that grows with the atomic number Z, typically in a scale of Z4.109 Because organic materials have lower Z numbers than inorganic metals, the SOC is usually small. The hyperfine interaction, which is also an important factor to determine spin relaxation, describes the interactions in between the nuclear spins adjacent to the electrons. By electron-nuclear flip, the hyperfine interaction directly influences τs, while dephasing in an order of Z−0.5 due to fluctuating nuclear spins occurs.110 In organic materials the hyperfine interaction is weak mainly originating from relatively small isotopes such as 1H, 13C and 14N, that once again, leads to a conclusion of longer τs and ls in organic materials. As spin polarization can be maintained longer in both time and length scale, materials with longer spin relaxation time are suitable materials to manufacture spin valve devices.111 In the context of electron paramagnetic resonance (EPR) spectroscopy investigations, it is commonly observed that inorganic materials typically exhibit spin relaxation times on the order of ns. In contrast, organic radicals have the capacity to significantly prolong this relaxation time, extending it to the order of μs. Recent discoveries from the Sirringhaus group have performed delaying the spin relaxation even up to 200 μs scale in a doped thiophene oligomer, once again proving organic radicals as suitable materials for spintronic applications (Fig. 11b).112
 | ||
| Fig. 11 (a) Illustration of spin relaxation in inorganic and organic materials, representing SOC and hyperfine interaction as barriers to diphase the spin polarity throughout the medium. (b) Spin lattice relaxation time versus Δg for all measured molecules. Error bars represent 95% confidence intervals. Dashed line shows the expected proportionality for relaxation via SOC fields. (c) Spin coherence time versus Δg for all measured molecules. Error bars show the 95% confidence intervals. Adapted with permission from ref. 112. Copyright 2017 Springer Nature. | ||
Organic spintronics has caught the attention of researchers over the past two decades as a promising research field where organic materials are applied to control or respond to a spin-polarized signal. Spintronic devices are potentially computationally faster and less power-intensive than traditional electronic devices, because the relevant energy scale for spin dynamics is smaller than that for manipulating charges (Fig. 12a).113,114 Notably, inorganic materials prove challenging for spintronics applications due to their short spin-relaxation time and length.115 This can be attributed to their pronounced larger spin–orbital coupling and hyperfine interaction, which collectively hinder their potential in this field (Fig. 12b). Therefore, conductive and paramagnetic radical polymers arise as a suitable candidate for spintronic devices because of their long spin relaxation time. For instance, the distance between ferromagnetic electrodes in spin valve devices should be designed in a smaller dimension than ls, to maintain the spin polarization within the device. Therefore, organic materials, especially organic radical polymers with long spin relaxation time are suitable materials for facile manufacturing spin valve devices along with several additional advances to inorganic materials, such as cost-efficiency, tunable mechanical and chemical properties, low-weight, etc.116,117 However, there are remaining issues to solve such as relatively low conductivity compared to metals that lead to conductivity mismatch problems, which the pioneers in this field are consistently making attempts to improve.
 | ||
| Fig. 12 Compared to (a) traditional electronics where interplays with the energy states of electrons, (b) spintronics operate in a much lower energy scale, due to spin splitting as two states, m = +1/2 and −1/2, when an external magnetic field is applied. The split spin states can fine split into further states due to SOC and hyperfine interaction. | ||
5.3. Tunnel magnetoresistance (TMR)
Organic radicals as a magneto-responsive material, have prompted extensive research involving both experimental and computational procedures. The initial results were demonstrated by measuring the magnetoresistance (MR) of junctions of single molecules, either in a monomer or oligomer form of organic radicals. In general, MR can be expressed as the following equation, | (2) |
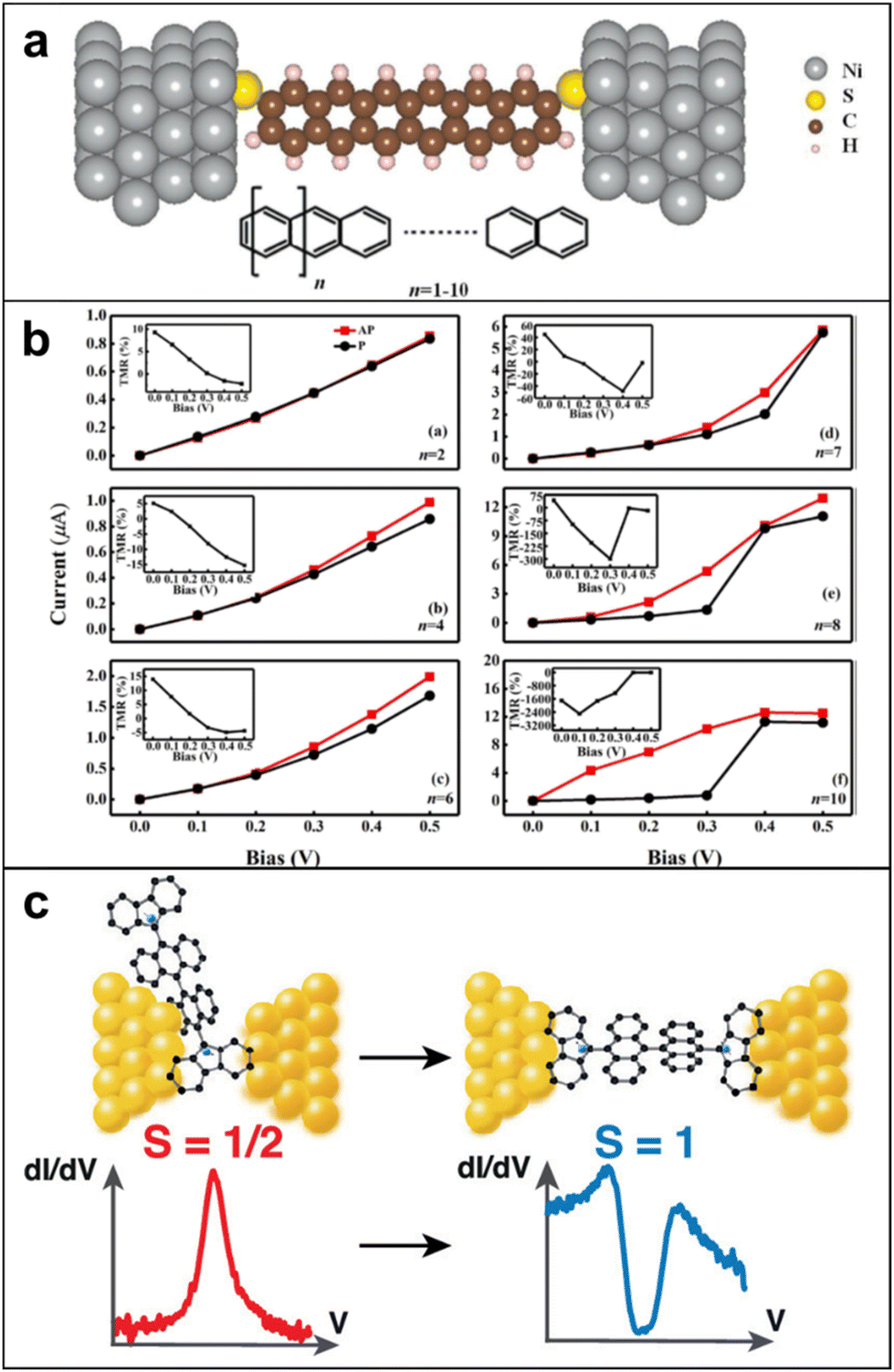 | ||
| Fig. 13 (a) The schematic structure for Ni/polyacene/Ni molecular junctions. (b) Current–voltage characteristics in parallel and antiparallel configurations with different molecular lengths (n = 2, 4, 6, 7, 8, 10, while the inserts are the corresponding TMR). TMR values show up to negative 2500% when n = 10 and bias is 0.1 V. Adapted with permission from ref. 122. Copyright 2021 The author(s). Published by Elsevier B.V. (c) The magnetic fingerprints depend on the exact configuration of the molecule in the junction, supported by measurements on a radical molecule with the same backbone but with one free spin, in which only Kondo anomalies are observed. Adapted with permission from ref. 123. Copyright 2022 The author(s). Published by American Chemical Society. | ||
5.4. Giant magnetoresistance (GMR)
Spin valves incorporating radical polymers as spinterface layers were engineered to quantify giant magnetoresistance (GMR) values with the aspiration to pave the way for future practical applications.124–126 In this type of measurement, either doped conjugated polymers or donor–acceptor systems were chosen due to their relatively higher conductivity compared to non-conjugated radical polymers, to avoid conductivity mismatch problems and enhance their stability in ambient conditions while the spinterface remains paramagnetic. In fact, researchers have designed and demonstrated several low-bandgap donor–acceptor polymers that perform up to a spin relaxation time of ∼1 μs.127 By applying a similar class of donor–acceptor polymer in spin valves Yu's group showed promising results for radical polymers to be a suitable material for spinterfaces that imply GMR effects.128 In this study, naphthalenediimide (NDI)-based conjugated polymer PNVT-CN-8 containing 2,3-bis(thiophen-2-yl)acrylonitrile units as a nonferromagnetic interlayer was studied as a spinterface material. They determined that a negative MR does not result from negative polarization at spin injection, but from the spin transport inside the donor–acceptor polymer itself, widening the capability of MR that radical polymers can perform from positive to even negative responses (Fig. 14a–f). On the whole, however, spin valve studies employing radical polymers are not well explored yet, with a low MR value (∼1%) compared to those of inorganic materials (∼100%). However, considering the benefits of organic materials to metal and possible applications such as flexible and transparent spintronics, we believe this field is an interesting direction to study. In light of the numerous benefits organic materials bring to the table, the exploration of spin valve studies involving radical polymers holds promise and invites further exploration in this captivating field of research.129,130 | ||
| Fig. 14 (a) Molecular structures of the conjugated polymer PNVT-CN-8. (b) Device structure of the spin valves based on PNVT-CN-8. (c) Current–voltage characteristics of the spin valves based on PNVT-CN-8 measured at different temperatures. (d) MR loops of all metal devices and the spin valves based on PNVT-CN-8 measured at 100 K. (e) Current dependence of the MR ratios for the spin valves based on PNVT-CN-8 measured at different temperatures. (f) MR loops of the spin valves based on PNVT-CN-8 measured under different currents at 300 K. Adapted with permission from ref. 128. Copyright 2020 Wiley-VCH GmbH. | ||
6. Conclusions and outlook
Numerous efforts in macromolecular design using radical polymers have enabled researchers to influence various solid-state optoelectronic and spintronic applications. Consequently, open-shell macromolecules have gained a solid foundation and significant presence in these realms. However, many foundational and applied principles still stymie the practical implementation of these promising materials in practical devices. Therefore, now is an opportune moment to expand the radical polymer community with the intention of reshaping the fundamental aspects of material systems.While vast prospects lie ahead for radical polymers, certain areas deserve immediate attention. These opportunities might be best achieved through the involvement of researchers with diverse and complementary skill sets currently not present in the radical polymers field. Specifically, the range of chemical structures with open-shell characteristics integrated into polymer frameworks has been fairly limited, even though new systems such as doner-acceptor polymers are being developed. For instance, the focus on nitroxide radicals in linking molecular design to end-use properties has been dominant. While there are valid reasons for this emphasis, it is unclear if nitroxide-containing macromolecules truly represent the broader class of materials. Furthermore, the position of nitroxide radicals in terms of ultimate end-use performance remains uncertain for many applications. Thus, there is an urgent call for chemists, physicists, and engineers to embrace the challenge of open-shell macromolecules to drive substantial breakthroughs in limits of macromolecular design (Fig. 15a).
 | ||
| Fig. 15 (a) Radical polymers exhibit multiple characteristics. First, charge transfer occurs between radical units, making them suitable for optoelectronic applications. Second, spin–spin interactions contribute to their efficacy in spintronic domains. These dual features offer benefits in both optoelectronic (upper, green) and spintronic (lower, green) applications. This is accompanied by open opportunities for conventional use cases (middle, red). (b) Radical polymers have the potential as materials that can merge the three areas of electronics, spintronics, and photonics. | ||
Similarly, the characterization of radical-containing polymers in solid-state applications has been hindered by their typically amorphous nature. This has impeded efforts to establish connections between macromolecular design and nanoscale attributes. Overcoming these issues requires advanced characterization techniques and building upon previous successes in this field. One way to overcome this issue would be computational modeling, especially recent advances that consider mass and electron transport, that can play a crucial role in refining our understanding of transport in radical polymers and establishing robust structure–function design principles. New developments in machine learning offer the potential to expedite the computational exploration of diverse radical-containing polymer chemistries at mesoscopic scales. Another way to address out the connections between design and nanoscale properties would be constructing materials with orientated morphology, such as organic radical single crystals (or semicrystalline radical polymers) and organic–inorganic hybrid materials. Crystalline materials will facilitate these studies in focusing on radical–radical interactions, because distance and morphology of radicals are known as the key factors of charge and spin transport.
Finally, we highlight the emerging realm of diverse spintronic investigations, such as chiral induced spin selectivity (CISS). CISS materials introduce the intriguing prospect of manipulating spin characteristics without the need for external magnetic fields or exchange interactions with magnetic elements.131,132 These materials, owing to their coiled chiral structure, function as spin and optical filters, preserving time-reversal symmetry and enabling localized electronic spin control.133,134 This capability opens doors to innovative spintronic device designs crucial for optical-quantum information science.135 Notably, CISS materials have found recent application in OLED technology, including a new generation of circular polarized luminescence (CPL) devices incorporating magnetic fields, giving rise to a new division of spintronics so-called ‘opto-spintronics’.136–140 Considering this development and the remarkable performances exhibited by radical polymers in both optoelectronics and spintronics domains, synergies between these areas hold substantial promise. This concept of merging disciplines can eventually expand to combinations of two or more of these areas of electronic, magnetic, and photonic applications (Fig. 15b).
In conclusion, the need for various research communities to dive into open-shell macromolecules is evident, and it is hoped that this endeavor provides the necessary context and references to inspire growth in this field. This approach aims to unlock the complete potential of radical polymers (and organic radicals in a wider scope), in terms of groundbreaking science and societal impact.
Author contributions
H. Y., S. D., and B. P. K equally contributed to writing the article. B. W. B. developed the concept of the manuscript and prepared the first version. All authors contributed to the discussion on the contents of the final version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This effort was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award DE-SC0021967 (the work of B. P. K.), by the National Science Foundation (NSF) under Award DMR-2321618 (the work of H. Y. and B. W. B.), and by the Air Force Office of Scientific Research (AFOSR) under Award FA9550-23-1-0240 (the work of S. D.). We gratefully thank the DOE, NSF, and AFOSR for their generous support.References
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC.
- S. Wang, A. D. Easley and J. L. Lutkenhaus, ACS Macro Lett., 2020, 9, 358–370 CrossRef CAS PubMed.
- K.-A. Hansen and J. P. Blinco, Polym. Chem., 2018, 9, 1479–1516 RSC.
- A. J. Wingate and B. W. Boudouris, J. Polym. Sci., Part A-1: Polym. Chem., 2016, 54, 1875–1894 CrossRef CAS.
- L. Rostro, A. G. Baradwaj and B. W. Boudouris, ACS Appl. Mater. Interfaces, 2013, 5, 9896–9901 CrossRef CAS PubMed.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- Y. Xie, K. Zhang, Y. Yamauchi, K. Oyaizu and Z. Jia, Mater. Horiz., 2021, 8, 803–829 RSC.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- M. A. Sabuj, C. Saha, M. M. Huda and N. Rai, Mol. Syst. Des. Eng., 2023, 8, 874–886 RSC.
- T. Y. Gopalakrishna, W. Zeng, X. Lu and J. Wu, Chem. Commun., 2018, 54, 2186–2199 RSC.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- T. Sugawara, H. Komatsu and K. Suzuki, Chem. Soc. Rev., 2011, 40, 3105–3118 RSC.
- R. Gaudenzi, E. Burzurí, D. Reta, I. D. P. R. Moreira, S. T. Bromley, C. Rovira, J. Veciana and H. S. J. van der Zant, Nano Lett., 2016, 16, 2066–2071 CrossRef CAS PubMed.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. van der Zant, C. Rovira, E. Burzurí and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294, 1488–1495 CrossRef CAS PubMed.
- S. Sanvito, Nat. Mater., 2007, 6, 803–804 CrossRef CAS PubMed.
- P. Murto and H. Bronstein, J. Mater. Chem. C, 2022, 10, 7368–7403 RSC.
- E. C. Lee, Y. C. Choi, W. Y. Kim, N. J. Singh, S. Lee, J. H. Shim and K. S. Kim, Chem. – Eur. J., 2010, 16, 12141–12146 CrossRef CAS PubMed.
- Y. Joo, V. Agarkar, S. H. Sung, B. M. Savoie and B. W. Boudouris, Science, 2018, 359, 1391–1395 CrossRef CAS PubMed.
- O. H. Griffith, J. F. W. Keana, S. Rottschaefer and T. A. Warlick, J. Am. Chem. Soc., 1967, 89, 5072–5072 CrossRef CAS.
- T. Sukegawa, H. Omata, I. Masuko, K. Oyaizu and H. Nishide, ACS Macro Lett., 2014, 3, 240–243 CrossRef CAS PubMed.
- K. Oyaizu, T. Suga, K. Yoshimura and H. Nishide, Macromolecules, 2008, 41, 6646–6652 CrossRef CAS.
- M. Suguro, S. Iwasa, Y. Kusachi, Y. Morioka and K. Nakahara, Macromol. Rapid Commun., 2007, 28, 1929–1933 CrossRef CAS.
- T. Katsumata, M. Satoh, J. Wada, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Rapid Commun., 2006, 27, 1206–1211 CrossRef CAS.
- J. Qu, T. Katsumata, M. Satoh, J. Wada and T. Masuda, Polymer, 2009, 50, 391–396 CrossRef CAS.
- T. Kurosaki, K. W. Lee and M. Okawara, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 3295–3310 CrossRef CAS.
- T. Suga, Y.-J. Pu, S. Kasatori and H. Nishide, Macromolecules, 2007, 40, 3167–3173 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751–754 CrossRef CAS PubMed.
- Y. Yonekuta, K. Susuki, K. Oyaizu and K. Honda, J. Am. Chem. Soc., 2007, 129, 14128–14129 CrossRef CAS PubMed.
- J. T. Price, J. A. Paquette, C. S. Harrison, R. Bauld, G. Fanchini and J. B. Gilroy, Polym. Chem., 2014, 5, 5223–5226 RSC.
- K.-A. Hansen, J. Nerkar, K. Thomas, S. E. Bottle, A. P. O'Mullane, P. C. Talbot and J. P. Blinco, ACS Appl. Mater. Interfaces, 2018, 10, 7982–7988 CrossRef CAS PubMed.
- O. Bertrand, B. Ernould, F. Boujioui, A. Vlad and J.-F. Gohy, Polym. Chem., 2015, 6, 6067–6072 RSC.
- X. Zhuang, C. Xiao, K. Oyaizu, N. Chikushi, X. Chen and H. Nishide, J. Polym. Sci., Part A-1: Polym. Chem., 2010, 48, 5404–5410 CrossRef CAS.
- T. Janoschka, A. Teichler, A. Krieg, M. D. Hager and U. S. Schubert, J. Polym. Sci., Part A-1: Polym. Chem., 2012, 50, 1394–1407 CrossRef CAS.
- K.-A. Hansen, L. C. Chambers, M. Eing, C. Barner-Kowollik, K. E. Fairfull-Smith and J. P. Blinco, ChemSusChem, 2020, 13, 2386–2393 CrossRef CAS PubMed.
- M. Aqil, A. Aqil, F. Ouhib, A. El Idrissi, C. Detrembleur and C. Jérôme, RSC Adv., 2015, 5, 85035–85038 RSC.
- T. K. Kunz and M. O. Wolf, Polym. Chem., 2011, 2, 640–644 RSC.
- M. Suguro, A. Mori, S. Iwasa, K. Nakahara and K. Nakano, Macromol. Chem. Phys., 2009, 210, 1402–1407 CrossRef CAS.
- T. Chi, S. Akkiraju, Z. Liang, Y. Tan, H. J. Kim, X. Zhao, B. M. Savoie and B. W. Boudouris, Polym. Chem., 2021, 12, 1448–1457 RSC.
- L. Xu, F. Yang, C. Su, L. Ji and C. Zhang, Electrochim. Acta, 2014, 130, 148–155 CrossRef CAS.
- J. Qu, T. Katsumata, M. Satoh, J. Wada and T. Masuda, Macromolecules, 2007, 40, 3136–3144 CrossRef CAS.
- M. Aydın, B. Esat, Ç. Kılıç, M. E. Köse, A. Ata and F. Yılmaz, Eur. Polym. J., 2011, 47, 2283–2294 CrossRef.
- N. Casado, G. Hernández, A. Veloso, S. Devaraj, D. Mecerreyes and M. Armand, ACS Macro. Lett., 2016, 5, 59–64 CrossRef CAS PubMed.
- S. Almubayedh and M. H. Chahma, New J. Chem., 2015, 39, 7738–7741 RSC.
- M. Miyasaka, T. Yamazaki, E. Tsuchida and H. Nishide, Polyhedron, 2001, 20, 1157–1162 CrossRef CAS.
- H. Woehlk, J. Steinkoenig, C. Lang, L. Michalek, V. Trouillet, P. Krolla, A. S. Goldmann, L. Barner, J. P. Blinco, C. Barner-Kowollik and K. E. Fairfull-Smith, Langmuir, 2018, 34, 3264–3274 CrossRef CAS PubMed.
- P. Wang, S. Lin, Z. Lin, M. D. Peeks, T. Van Voorhis and T. M. Swager, J. Am. Chem. Soc., 2018, 140, 10881–10889 CrossRef CAS PubMed.
- A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, Science, 2001, 294, 1503–1505 CrossRef CAS PubMed.
- A. Rajca, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 1999, 121, 6308–6309 CrossRef CAS.
- A. Rajca, J. Wongsriratanakul and S. Rajca, J. Am. Chem. Soc., 2004, 126, 6608–6626 CrossRef CAS PubMed.
- T. Kaneko, T. Makino, H. Miyaji, M. Teraguchi, T. Aoki, M. Miyasaka and H. Nishide, J. Am. Chem. Soc., 2003, 125, 3554–3557 CrossRef CAS PubMed.
- Y. Zhang, A. Park, A. Cintora, S. R. McMillan, N. J. Harmon, A. Moehle, M. E. Flatté, G. D. Fuchs and C. K. Ober, J. Mater. Chem. C, 2018, 6, 111–118 RSC.
- Y. Tan, N. C. Casetti, B. W. Boudouris and B. M. Savoie, J. Am. Chem. Soc., 2021, 143, 11994–12002 CrossRef CAS PubMed.
- D. C. Bobela, B. K. Hughes, W. A. Braunecker, T. W. Kemper, R. E. Larsen and T. Gennett, J. Phys. Chem. Lett., 2015, 6, 1414–1419 CrossRef CAS PubMed.
- L. Rostro, S. H. Wong and B. W. Boudouris, Macromolecules, 2014, 47, 3713–3719 CrossRef CAS.
- I. Yu, D. Jeon, B. Boudouris and Y. Joo, Macromolecules, 2020, 53, 4435–4441 CrossRef CAS.
- Z. Liang, S.-N. Hsu, Y. Tan, H. Tahir, H. J. Kim, K. Liu, J. F. Stoehr, M. Zeller, L. Dou, B. M. Savoie and B. W. Boudouris, Cell Rep. Phys. Sci., 2023, 4, 101409 CrossRef CAS.
- F. Li, D. N. Gore, S. Wang and J. L. Lutkenhaus, Angew. Chem., Int. Ed., 2017, 56, 9856–9859 CrossRef CAS PubMed.
- M. Tanaka, K. Hatta, T. Edura, K. Tsutsui, Y. Wada and H. Nishide, Polym. Adv. Technol., 2007, 18, 925–931 CrossRef CAS.
- J. Lutkenhaus, Science, 2018, 359, 1334–1335 CrossRef CAS PubMed.
- A. Obolda, M. Zhang and F. Li, Chin. Chem. Lett., 2016, 27, 1345–1349 CrossRef CAS.
- Y. Tan, S.-N. Hsu, H. Tahir, L. Dou, B. M. Savoie and B. W. Boudouris, J. Am. Chem. Soc., 2022, 144, 626–647 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
- S. Ezugwu, J. A. Paquette, V. Yadav, J. B. Gilroy and G. Fanchini, Adv. Electron. Mater., 2016, 2, 1600253 CrossRef.
- L. Ji, J. Shi, J. Wei, T. Yu and W. Huang, Adv. Mater., 2020, 32, 1908015 CrossRef CAS PubMed.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 CrossRef CAS.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908–912 CrossRef CAS PubMed.
- G. Cheng, K. T. Chan, W.-P. To and C.-M. Che, Adv. Mater., 2014, 26, 2540–2546 CrossRef CAS PubMed.
- S.-Y. Kim and J.-J. Kim, Org. Electron., 2010, 11, 1010–1015 CrossRef CAS.
- G. Gu, D. Z. Garbuzov, P. E. Burrows, S. Venkatesh, S. R. Forrest and M. E. Thompson, Opt. Lett., 1997, 22, 396–398 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- A. Obolda, Q. Peng, C. He, T. Zhang, J. Ren, H. Ma, Z. Shuai and F. Li, Adv. Mater., 2016, 28, 4740–4746 CrossRef CAS PubMed.
- C.-J. Chiang, A. Kimyonok, M. K. Etherington, G. C. Griffiths, V. Jankus, F. Turksoy and A. P. Monkman, Adv. Funct. Mater., 2013, 23, 739–746 CrossRef CAS.
- A. Heckmann, C. Lambert, M. Goebel and R. Wortmann, Angew. Chem., Int. Ed., 2004, 43, 5851–5856 CrossRef CAS PubMed.
- A. Heckmann, S. Dümmler, J. Pauli, M. Margraf, J. Köhler, D. Stich, C. Lambert, I. Fischer and U. Resch-Genger, J. Phys. Chem. C, 2009, 113, 20958–20966 CrossRef CAS.
- C.-H. Liu, E. Hamzehpoor, Y. Sakai-Otsuka, T. Jadhav and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 23030–23034 CrossRef CAS PubMed.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- S. Castellanos, V. Gaidelis, V. Jankauskas, J. V. Grazulevicius, E. Brillas, F. López-Calahorra, L. Juliá and D. Velasco, Chem. Commun., 2010, 46, 5130–5132 RSC.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed.
- T. Kusamoto, S. Kimura, Y. Ogino, C. Ohde and H. Nishihara, Chem. – Eur. J., 2016, 22, 17725–17733 CrossRef CAS PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 CrossRef CAS PubMed.
- E. Neier, R. A. Ugarte, N. Rady, S. Venkatesan, T. W. Hudnall and A. Zakhidov, Org. Electron., 2017, 44, 126–131 CrossRef CAS.
- Y. Gao, W. Xu, H. Ma, A. Obolda, W. Yan, S. Dong, M. Zhang and F. Li, Chem. Mater., 2017, 29, 6733–6739 CrossRef CAS.
- Z. Cui, S. Ye, L. Wang, H. Guo, A. Obolda, S. Dong, Y. Chen, X. Ai, A. Abdurahman, M. Zhang, L. Wang and F. Li, J. Phys. Chem. Lett., 2018, 9, 6644–6648 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- Q. Gu, A. Abdurahman, R. H. Friend and F. Li, J. Phys. Chem. Lett., 2020, 11, 5638–5642 CrossRef CAS PubMed.
- A. Abdurahman, Q. Peng, O. Ablikim, X. Ai and F. Li, Mater. Horiz., 2019, 6, 1265–1270 RSC.
- K. An, G. Xie, S. Gong, Z. Chen, X. Zhou, F. Ni and C. Yang, Sci. China: Chem., 2020, 63, 1214–1220 CrossRef CAS.
- Z. Wang, X. Zou, Y. Xie, H. Zhang, L. Hu, C. C. S. Chan, R. Zhang, J. Guo, R. T. K. Kwok, J. W. Y. Lam, I. D. Williams, Z. Zeng, K. S. Wong, C. D. Sherrill, R. Ye and B. Z. Tang, Mater. Horiz., 2022, 9, 2564–2571 RSC.
- L. Guo, Y. Qin, X. Gu, X. Zhu, Q. Zhou and X. Sun, Front. Chem., 2019, 7, 428 CrossRef CAS PubMed.
- P. Zilske, D. Graulich, M. Dunz and M. Meinert, Appl. Phys. Lett., 2017, 110, 192402 CrossRef.
- S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294, 1488–1495 CrossRef CAS PubMed.
- A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS.
- W. J. M. Naber, S. Faez and W. G. van der Wiel, J. Phys. D: Appl. Phys., 2007, 40, R205 CrossRef CAS.
- A. Rajca, Adv. Phys. Org. Chem, ed. J. P. Richard, Academic Press, 2005, vol. 40, pp. 153–199 Search PubMed.
- S. Rajca, A. Rajca, J. Wongsriratanakul, P. Butler and S.-M. Choi, J. Am. Chem. Soc., 2004, 126, 6972–6986 CrossRef CAS PubMed.
- K. S. Mayer, D. J. Adams, N. Eedugurala, M. M. Lockart, P. Mahalingavelar, L. Huang, L. A. Galuska, E. R. King, X. Gu, M. K. Bowman and J. D. Azoulay, Cell Rep. Phys. Sci., 2021, 2, 100467 CrossRef CAS.
- A. Fert and H. Jaffrès, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 184420 CrossRef.
- R. Mattana, J. M. George, H. Jaffrès, F. Nguyen Van Dau, A. Fert, B. Lépine, A. Guivarc'h and G. Jézéquel, Phys. Rev. Lett., 2003, 90, 166601 CrossRef CAS PubMed.
- A. Fert, Thin Solid Films, 2008, 517, 2–5 CrossRef CAS.
- V. I. Krinichnyi, H. K. Roth and M. Schrödner, Appl. Magn. Reson., 2002, 23, 1–17 CrossRef CAS.
- A. V. Khaetskii, D. Loss and L. Glazman, Phys. Rev. Lett., 2002, 88, 186802 CrossRef PubMed.
- I. A. Merkulov, A. L. Efros and M. Rosen, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 205309 CrossRef.
- S. Sanvito and A. R. Rocha, J. Comput. Theor. Nanosci., 2006, 3, 624–642 CrossRef CAS.
- S. Schott, E. R. McNellis, C. B. Nielsen, H.-Y. Chen, S. Watanabe, H. Tanaka, I. McCulloch, K. Takimiya, J. Sinova and H. Sirringhaus, Nat. Commun., 2017, 8, 15200 CrossRef PubMed.
- A. J. Drew, J. Hoppler, L. Schulz, F. L. Pratt, P. Desai, P. Shakya, T. Kreouzis, W. P. Gillin, A. Suter, N. A. Morley, V. K. Malik, A. Dubroka, K. W. Kim, H. Bouyanfif, F. Bourqui, C. Bernhard, R. Scheuermann, G. J. Nieuwenhuys, T. Prokscha and E. Morenzoni, Nat. Mater., 2009, 8, 109–114 CrossRef CAS PubMed.
- P. A. Bobbert, W. Wagemans, F. W. A. van Oost, B. Koopmans and M. Wohlgenannt, Phys. Rev. Lett., 2009, 102, 156604 CrossRef CAS PubMed.
- M. Cinchetti, K. Heimer, J.-P. Wüstenberg, O. Andreyev, M. Bauer, S. Lach, C. Ziegler, Y. Gao and M. Aeschlimann, Nat. Mater., 2009, 8, 115–119 CrossRef CAS PubMed.
- S. Pramanik, C. G. Stefanita, S. Patibandla, S. Bandyopadhyay, K. Garre, N. Harth and M. Cahay, Nat. Nanotechnol., 2007, 2, 216–219 CrossRef CAS PubMed.
- L. E. Hueso, J. M. Pruneda, V. Ferrari, G. Burnell, J. P. Valdés-Herrera, B. D. Simons, P. B. Littlewood, E. Artacho, A. Fert and N. D. Mathur, Nature, 2007, 445, 410–413 CrossRef CAS PubMed.
- K. Tagami and M. Tsukada, J. Phys. Chem. B, 2004, 108, 6441–6444 CrossRef CAS PubMed.
- Y. Matsuura and I. Taniguchi, Org. Electron., 2019, 69, 114–119 CrossRef CAS.
- M. S. Zöllner, R. Nasri, H. Zhang and C. Herrmann, J. Phys. Chem. C, 2021, 125, 1208–1220 CrossRef.
- R. Hayakawa, M. A. Karimi, J. Wolf, T. Huhn, M. S. Zöllner, C. Herrmann and E. Scheer, Nano Lett., 2016, 16, 4960–4967 CrossRef CAS PubMed.
- J. Qi, Y. Miao, Y. Cui, S. Qiu, J. Zhao, G. Zhang, J. Ren, C. Wang and G. Hu, Results Phys., 2021, 27, 104510 CrossRef.
- T. Y. Baum, S. Fernández, D. Peña and H. S. J. van der Zant, Nano Lett., 2022, 22, 8086–8092 CrossRef CAS PubMed.
- M. N. Baibich, J. M. Broto, A. Fert, F. N. Van Dau, F. Petroff, P. Etienne, G. Creuzet, A. Friederich and J. Chazelas, Phys. Rev. Lett., 1988, 61, 2472–2475 CrossRef CAS PubMed.
- D. Yu, S. Ding, J. Li, W. Mi, Y. Tian and W. Hu, J. Mater. Chem. C, 2022, 10, 2608–2615 RSC.
- E. Y. Tsymbal and D. G. Pettifor, Solid State Phys, ed. H. Ehrenreich and F. Spaepen, Academic Press, 2001, vol. 56, pp. 113–237 Search PubMed.
- M. E. Steelman, D. J. Adams, K. S. Mayer, P. Mahalingavelar, C.-T. Liu, N. Eedugurala, M. Lockart, Y. Wang, X. Gu, M. K. Bowman and J. D. Azoulay, Adv. Mater., 2022, 34, 2206161 CrossRef CAS PubMed.
- N. Zheng, X. Wang, Y. Zheng, D. Li, Z. Lin, W. Zhang, K.-J. Jin and G. Yu, Adv. Mater. Interfaces, 2020, 7, 2000868 CrossRef CAS.
- H. Gu, J. Guo, X. Zhang, Q. He, Y. Huang, H. A. Colorado, N. Haldolaarachchige, H. Xin, D. P. Young, S. Wei and Z. Guo, J. Phys. Chem. C, 2013, 117, 6426–6436 CrossRef CAS.
- Z. H. Xiong, D. Wu, Z. V. Vardeny and J. Shi, Nature, 2004, 427, 821–824 CrossRef CAS PubMed.
- T. P. Fay, J. Phys. Chem. Lett., 2021, 12, 1407–1412 CrossRef CAS PubMed.
- J. Luo and P. J. Hore, New J. Phys., 2021, 23, 043032 CrossRef CAS.
- A. C. Aragonès, D. Aravena, J. M. Ugalde, E. Medina, R. Gutierrez, E. Ruiz, V. Mujica and I. Díez-Pérez, Isr. J. Chem., 2022, 62, e202200090 CrossRef.
- A. M. Garcia, G. Martínez and A. Ruiz-Carretero, Front. Chem., 2021, 9, 722727 CrossRef CAS PubMed.
- A. Chiesa, A. Privitera, E. Macaluso, M. Mannini, R. Bittl, R. Naaman, M. R. Wasielewski, R. Sessoli and S. Carretta, Adv. Mater., 2023, 35, 2300472 CrossRef CAS PubMed.
- J. F. Sierra, J. Fabian, R. K. Kawakami, S. Roche and S. O. Valenzuela, Nat. Nanotechnol., 2021, 16, 856–868 CrossRef CAS PubMed.
- H. Zhong, B. Zhao and J. Deng, Adv. Opt. Mater., 2023, 11, 2202787 CrossRef CAS.
- S. Alwan and Y. Dubi, J. Am. Chem. Soc., 2021, 143, 14235–14241 CrossRef CAS PubMed.
- T. S. Metzger, H. Batchu, A. Kumar, D. A. Fedotov, N. Goren, D. K. Bhowmick, I. Shioukhi, S. Yochelis, I. Schapiro, R. Naaman, O. Gidron and Y. Paltiel, J. Am. Chem. Soc., 2023, 145, 3972–3977 CrossRef CAS PubMed.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |