
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn organohydrogel with tunable fluorescence and shape-memory property for advanced anti-counterfeiting†
Yu
Sun
ab,
Hui
Shang
ab,
Xiaoxia
Le
 *ab and
Tao
Chen
*abc
*ab and
Tao
Chen
*abc
aKey Laboratory of Marine Materials and Related Technologies, Zhejiang Key Laboratory of Marine Materials and Protective Technologies, Ningbo Institute of Material Technology and Engineering, Chinese Academy of Sciences, Ningbo 315201, China. E-mail: lexiaoxia@nimte.ac.cn; tao.chen@nimte.ac.cn
bSchool of Chemical Sciences, University of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, China
cCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China
First published on 17th February 2024
Abstract
Counterfeiting is a significant threat in the intricate realm of global commerce, casting shadows over industries, economies, and unsuspecting consumers. Fluorescent anti-counterfeiting labels have been widely used in the past, but their level of security is still relatively inadequate. Therefore, the ongoing research is aimed at improving security through the encapsulation of information within predefined geometric structures. Herein, fluorescent organohydrogels with a hydrophilic polymer network of poly(N,N-dimethylacrylamide-acrylic acid) (P(DMA-AAc)), containing blue fluorescent monomers (PyMA), and a hydrophobic polymer network, polystearyl methylacrylate (PSMA), are fabricated by two-step interpenetrating polymerization. Upon treatment with Fe3+, the blue fluorescence of organohydrogels is quenched owing to the intramolecular charge transfer (ICT) effect, which can be reinstated by adding H+. Furthermore, coupled with the shape memory function induced by the crystallization of PSMA, the organohydrogels enable the concealment of encoded fluorescent information in specific three-dimensional shapes. This work presents innovative possibilities for designing and constructing advanced anti-counterfeiting systems.
1. Introduction
In a time of globalization and technological progress, counterfeiting has become a significant obstacle with far-reaching consequences for various industries, economies, and public welfare.1–3 The consequences of counterfeiting go beyond economic considerations and include risks to public safety, damage to brand reputation, and the loss of consumer trust. Numerous materials and techniques have been developed to combat counterfeiting and have demonstrated effectiveness in reducing counterfeiting threats, including holograms,4–6 watermarks,7–9 Quick Response (QR) codes,10–12 Radio-Frequency Identification (RFID),13–15 and fluorescent patterns.16–18 Among these technologies, fluorescent materials have risen as a novel frontier in the realm of anti-counterfeiting owing to their polychromatic attributes and wavelength-dependent characteristics, enhancing their utility in information security applications. However, the security of numerous fluorescent information encryption strategies remains deficient, as they can be decoded with singular ultraviolet light. Hence, advancing the substrate materials of fluorescent information in additional dimensions can further enhance information security.Currently, efforts to address the issue of singular ultraviolet decoding of fluorescent anti-counterfeiting labels mainly focus on the construction of dual encryption systems, which can be achieved by controlling the fluorescence color for dynamic anti-counterfeiting and utilizing deformable substrate materials to conceal the loaded information. As the former is relatively difficult,19–21 modulation of substrate materials with fluorescent patterns to realize dual encryption by adding the spatial dimension becomes an effective strategy. Intelligent gel materials exhibit outstanding stimulus-responsiveness, stability, and modifiability, and are easy to integrate with fluorescence emitters.22,23 Simultaneously, their flexibility extends the possibilities of encryption in spatial and geometric shapes.24 Yang et al.25 prepared a gradient cross-linked fluorescent hydrogel responsive to temperature changes, serving as an information carrier readable under specific UV light irradiation, while self-deforming to reveal information as the temperature increases. Tang and coworkers26 designed a bilayer hydrogel incorporating synergistic deformation and fluorescence color change through electrostatic interactions and dynamic covalent bonding, enabling ionoprinting of fluorescent information. The encoded 2D bilayer hydrogel, capable of generating pre-designed 3D shapes to hide information, could only be read after the shapes were sequentially restored in the absence of background fluorescence. In our previous research,27 we introduced a fluorescent hydrogel featuring simultaneous Fe3+-responsive fluorescence quenching, borax-triggered shape memory, and inherent self-healing properties, concealing data loaded with Fe3+ ink within a complex 3D hydrogel origami structure to significantly enhance information security compared to its conventional 2D counterparts. However, the fluorescent hydrogel labels prepared in these studies were constrained by aqueous environments, posing a significant limitation to their further development.
This work synthesized the organohydrogels with both hydrophilic and hydrophobic networks, possessing reversible fluorescence switching and shape memory properties and exhibiting the capability to function in a desiccated state. The hydrophilic polymer network was synthesized through the copolymerization of N,N-dimethylacrylamide, acrylic acid, and 1-pyrenylmethyl acrylate (PyMA, blue fluorescence). Concurrently, the hydrophobic polymer network was derived from the polymerization of long-chain stearate methacrylate (SMA). Under the ultraviolet irradiation of 365 nm, PyMA emits blue fluorescence, which can be quenched by Fe3+ ions. This is due to the coordination of carboxylic acid groups on the hydrophilic polymer chains upon treatment with Fe3+ ions, which facilitates electron transfer between Fe3+ ions (electron acceptors) and PyMA fluorescent monomers (electron donors) via the ICT effect. Upon the additional introduction of H+, the ICT process28 between Fe3+ and PyMA would be terminated, thereby enabling the restoration of fluorescence color (Scheme 1a). Therefore, various fluorescent information can be loaded on the surface of the organohydrogels by ionoprinting and erased by treating with H+. Additionally, the inherent low-temperature-induced crystallinity of PSMA endowed shape memory capabilities to the organohydrogels (Scheme 1b), establishing a foundation for the spatial dimension encryption of fluorescent labels. Subsequently, the organohydrogels with desired patterns were deformed into a specific configuration to complete the information encryption process. The hidden information could be decoded by successive steps of shape recovery and UV irradiation (Scheme 1c). Furthermore, the loaded information could be reversibly erased, allowing for multiple repetitions of the writing–encryption–decryption process. The prepared organohydrogels improved the information storage capacity and increased the security level of information encryption, which hold promising prospects in the advanced anti-counterfeiting field.
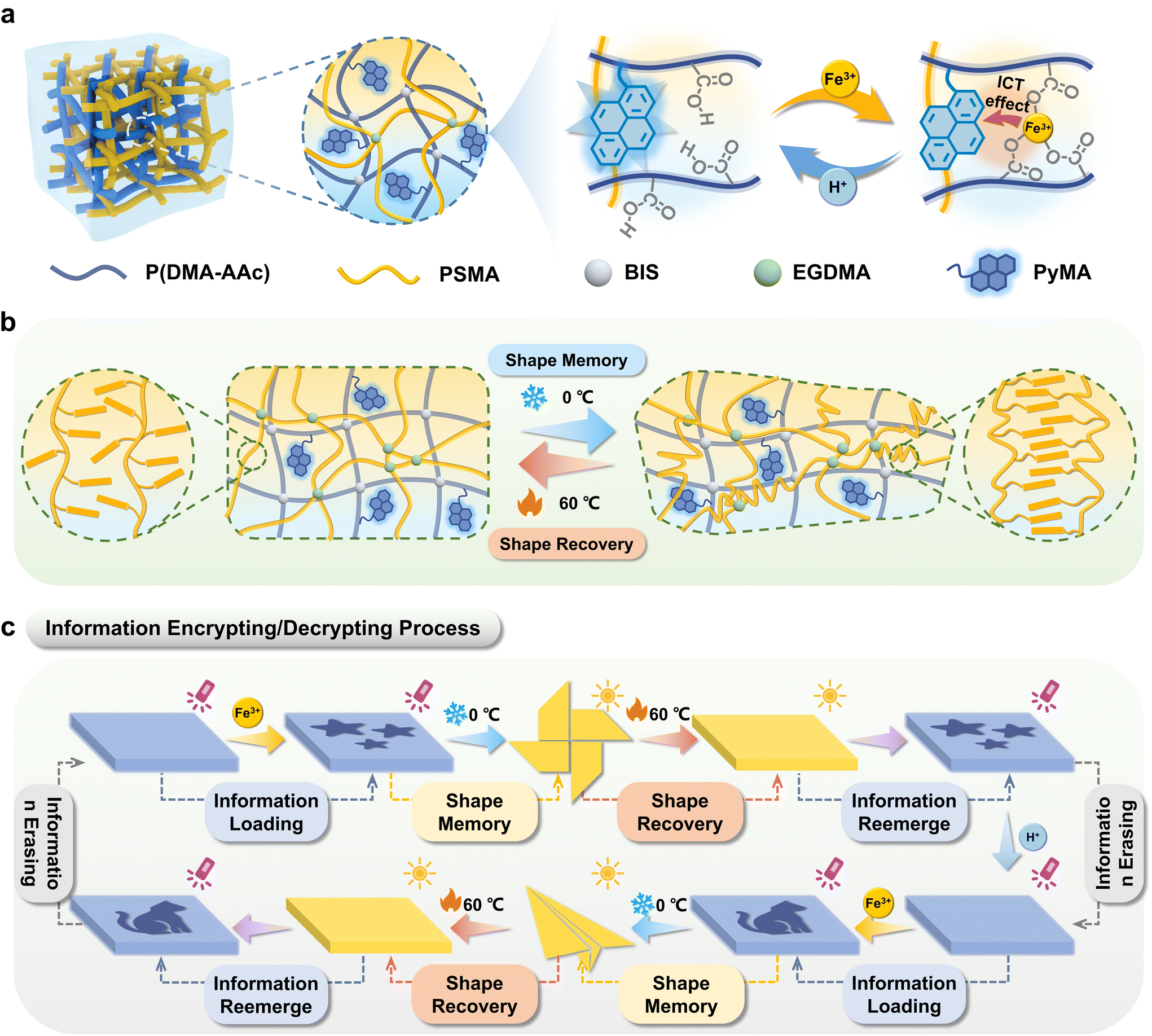 | ||
| Scheme 1 Schematic illustration of the fluorescent organohydrogels P(DMA-AAc-PyMA)/PSMA for advanced encryption and decryption processes. (a) The fluorescent organohydrogels were composed of a hydrophilic polymer network and a hydrophobic network. Due to the ICT effect, Fe3+ leads to the quenching of the blue fluorescence, and H+ reversibly restores the fluorescence colors. (b) Schematic diagram of the shape memory/recovery process of the organohydrogels P(DMA-AAc-PyMA)/PSMA. (c) Schematic diagram of the process of information encoding, encryption, and decryption. | ||
2. Experimental section
2.1 Materials
N,N-Dimethylacrylamide (DMA), N,N,N′′,N′-tetramethylethylenediamine (TEMED), N,N′′-methylene bis(acrylamide) (BIS), 2,2-diethoxyacetophenone (DEAP), ethylene dimethacrylate (EGDMA), 2-hydroxyethyl methacrylate (HEMA), 1-pyrenylbutyric acid and FeCl3·6H2O were purchased from Aladdin. Stearyl methylacrylate (SMA) was commercially provided by J&K Chemical Co., Ltd. Acrylic acid (AAc), HCl and DMSO were bought from Sinopharm Chemical Reagent Co., Ltd. The fluorescent monomer 1-pyrenylmethyl acrylate (PyMA) was synthesized according to our previous report. All reagents were used without any treatment or purification.2.2 Preparation of the P(DMA-AAc-PyMA) hydrogel
First, 0.216 g AAc and 2.673 g DMA were dissolved in 2.01 mL deionized water. Second, 10 mg PyMA and 3 mg BIS were dispersed in 5 mL DMSO. After mixing the above two solutions, 70 mg APS and 46 μL TEMED were gradually added. After rapid shaking and mixing, the mixed solution was transferred to a homemade reaction cell. After polymerization in an oven at 60 °C for 5 h, the hydrogels were placed in a large amount of acetone to remove deionized water and DMSO.2.3 Preparation of the P(DMA-AAc-PyMA)/PSMA organohydrogel
The naturally air-dried P(DMA-AAc-PyMA) hydrogels were immersed in oily precursor solutions consisting of 20 mL SMA, 10 mL ethanol, 100 μL DEAP, and 150 μL EGDMA and stored at 35 °C for 12 h in the dark. The fully soaked swollen hydrogels were then sandwiched between two pieces of quartz glass and polymerized under UV light for 2 h (365 nm, 50 W). Furthermore, the organohydrogels were soaked in ethanol in order to remove unreacted monomers and then placed in air to dry.2.4 Multi-level encryption and decryption of information
The prepared P(DMA-AAc-PyMA)/PSMA organohydrogels were ionoprinted by bringing them into contact with filter paper soaked with Fe3+ (0.1 M, 15 min) to load the corresponding information. To hide the loaded information, special shapes were fixed onto the organohydrogels and stored in a 4 °C freezer for 10 minutes. The original shape was restored by heating the organohydrogels to 60 °C, and the information could be revealed under UV light. In addition, the organohydrogels stained with Fe3+ can be brought into contact with filter paper soaked with H+ (0.1 M, 15 min) to erase the loaded information and further rewrite with Fe3+.2.5 Characterization
1H NMR spectra were obtained from a Bruker Avance III 400 MHz spectrometer. Rheological characterization was carried out on a stress-controlled rheometer (TA-dhr2) with a parallel plate (25 mm) in frequency sweep mode (from 0.1 to 100 rad s−1) at a constant shear strain of 1%. UV-Vis absorption and transmittance spectra were measured on a UV-Vis spectrophotometer (TU-1810, Purkinje General Instrument Co. Ltd). ATR-FTIR spectra were recorded on a Thermo Scientific Nicolet 6700 FT-IR spectrometer. Raman spectra were recorded on a confocal micro-Raman spectrometer (Renishaw inVia Reflex, Renishaw Instrument Co. Ltd). The digital photos of the hydrogels were taken under a UV lamp (ZF-5, 5 W, 365 nm). All fluorescence photographs were taken using the same UV lamp. Fluorescence intensity was recorded with a HPRIBA FL3-111 fluorescence spectrometer. The excitation wavelength was 365 nm. SEM images were taken using a scanning electron microscope (S-4800, Hitachi).3. Results and discussion
3.1 Fabrication of the P(DMA-AAc-PyMA)/PSMA organohydrogel
The synthesis of the fluorescent monomer (PyMA) involved the condensation of HEMA and 1-pyrenylbutyric acid (Fig. S1†), building upon established methodologies from previous work.29 The 1H NMR spectrum demonstrated the synthesis of the target fluorescent monomer, PyMA (Fig. S2†). The fluorescent organohydrogels were prepared by a two-step interpenetrating method (Fig. S3†). Specifically, the hydrogels, P(DMA-AAc-PyMA), were formed through copolymerization of the fluorescent PyMA monomer with acrylic acid (AAc) and N,N-dimethylacrylamide (DMA) in a mixed solvent system of water and dimethyl sulfoxide (DMSO), employing a free radical polymerization process with N,N′′-methylene bis(acrylamide) (BIS) as the crosslinker, ammonium persulfate (APS) as the initiator, and N,N,N′′,N′-tetramethylenediamine (TEMED) as the accelerator. Furthermore, acetone immersion was employed to remove excess water and DMSO. Following natural air-drying, the dried hydrogels were thoroughly immersed in an oil gel precursor solution with SMA as the monomer, ethylene dimethacrylate (EGDMA) as the crosslinker, and 2,2-diethoxyacetophenone (DEAP) as the photosensitizer. Subsequently, the hydrogels saturated with the precursor were subjected to polymerization under UV irradiation to obtain the hydrophobic polymer network, resulting in the formation of the organohydrogels (Fig. 1a). The P(DMA-AAc-PyMA)/PSMA organohydrogels for follow-up research were achieved by purging unreacted monomers with ethanol and subsequently air-drying at room temperature. Additionally, to fully coordinate the carboxyl group of acrylic acid, the P(DMA-AAc-PyMA)/PSMA organohydrogel samples for the subsequent characterization were soaked in FeCl3 solution (0.1 M) for 24 hours and naturally air-dried at room temperature.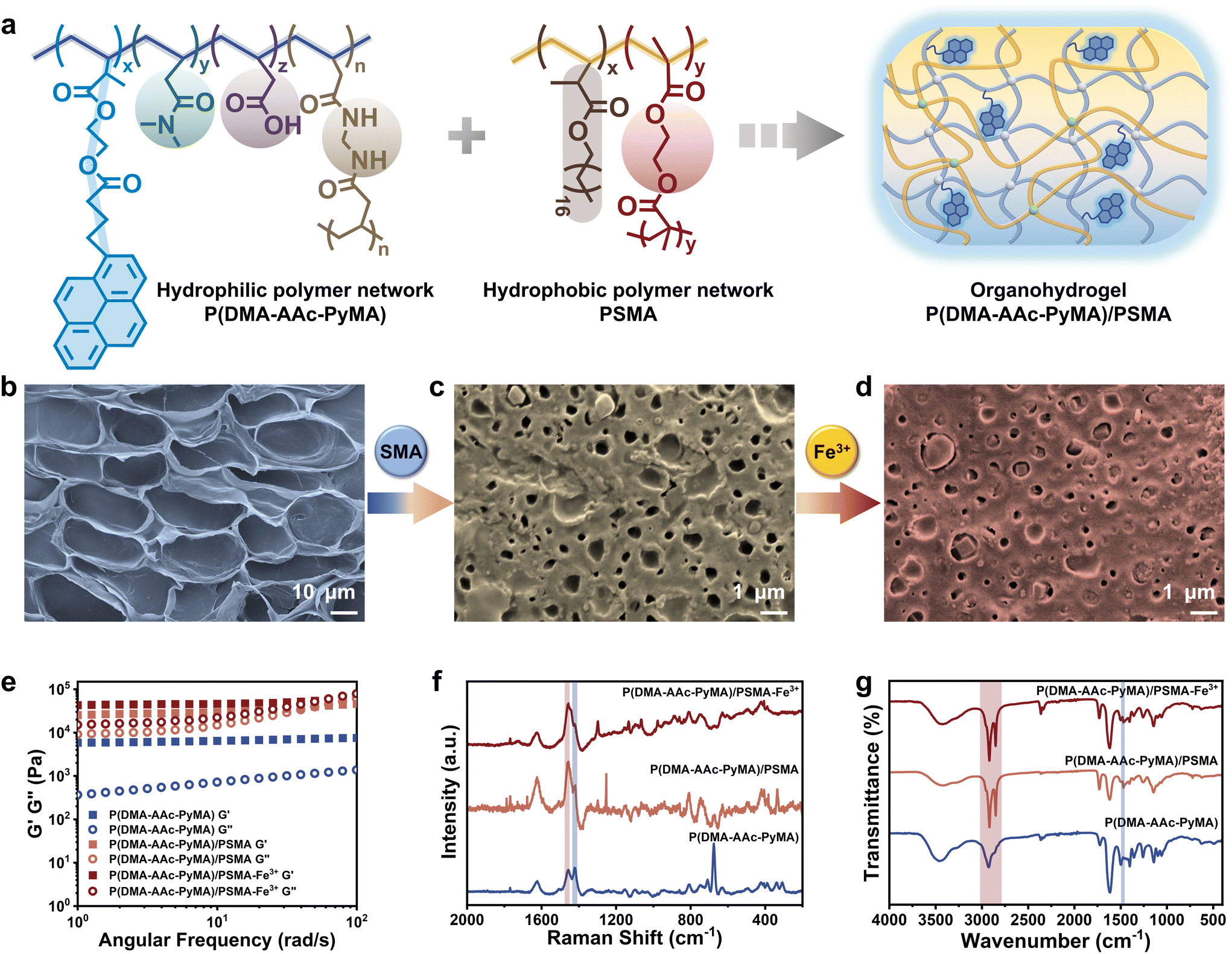 | ||
| Fig. 1 Characterization of P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels before and after treatment with Fe3+. (a) Schematic illustration of the internal molecular composition of the fluorescent organohydrogels. SEM images of (b) the P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels (c) before and (d) after treatment with Fe3+. (e) Storage modulus (G′) and loss modulus (G′′), (f) Raman spectra and (g) FT-IR spectra of P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels before and after treatment with Fe3+. | ||
3.2 Characterization of the P(DMA-AAc-PyMA)/PSMA organohydrogel
Compared to the hydrophilic P(DMA-AAc-PyMA) hydrogels (d = 22.13 ± 3.14 μm), the size of the porous structure of the P(DMA-AAc-PyMA)/PSMA organohydrogels showed an order of magnitude decrease (d = 0.61 ± 0.11 μm) as observed through a scanning electron microscope (SEM). Furthermore, the porous structure of the organohydrogels treated with Fe3+ immersion became denser (d = 0.14 ± 0.03 μm) (Fig. 1b–d and Fig. S4†) due to the coordination interaction between AAc and Fe3+, leading to an increase in the crosslinking density and a decrease in pore size. The progressive densification of the porous structure from hydrogels to organohydrogels was further verified by a stepwise increase in the dynamic mechanical properties of energy storage modulus (G′) and loss modulus (G′′) (Fig. 1e). When a second layer of the hydrophobic polymer network was introduced, both G′ and G′′ of the organohydrogels were enhanced by orders of magnitude, compared to those of P(DMA-AAc-PyMA) hydrogels. It was also observed that both G′ and G′′ were slightly increased when the organohydrogels were treated with Fe3+. The gradual reduction in transparency observed in both hydrophilic P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels before and after Fe3+ treatment (Fig. S5†) also served as evidence for densification.To further validate the changes in the chemical composition of P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels with or without Fe3+ treatment, the Raman spectra were measured and analyzed (Fig. 1f). The presence of a distinct single peak at 1455.06 cm−1 suggested the carboxyl bonding composition of ester molecules, proving that the PSMA hydrophobic network was introduced into the hydrogel's molecular network. Following Fe3+ treatment, the disappearance of the peak at 1418.79 cm−1 (associated with C–O stretching and –OH deformation) was a result of the coordination between Fe3+ and the –COOH groups. Similarly, P(DMA-AAc-PyMA) hydrogels and P(DMA-AAc-PyMA)/PSMA organohydrogels with or without Fe3+ treatment were analyzed by attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR). As shown in Fig. 1g, all samples showed an absorption peak at 1731.76 cm−1, caused by the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, and a characteristic peak at 2931.27 cm−1 generated by the antisymmetric stretching vibration of –CH2–. However, the intensity of this peak is significantly higher in the organohydrogels due to the increase in CO groups after the introduction of the PSMA polymer chain. At the same time, a new absorption peak appeared at 2850.27 cm−1, indicating an increase in the introduction of –CH2–, which indirectly proved the introduction of the hydrophobic polymer network of PSMA into organohydrogels. The elimination of the –OH bending peak at 1463.70 cm−1 subsequent to Fe3+ treatment underscored the coordination of Fe3+ with –COOH groups.
O groups, and a characteristic peak at 2931.27 cm−1 generated by the antisymmetric stretching vibration of –CH2–. However, the intensity of this peak is significantly higher in the organohydrogels due to the increase in CO groups after the introduction of the PSMA polymer chain. At the same time, a new absorption peak appeared at 2850.27 cm−1, indicating an increase in the introduction of –CH2–, which indirectly proved the introduction of the hydrophobic polymer network of PSMA into organohydrogels. The elimination of the –OH bending peak at 1463.70 cm−1 subsequent to Fe3+ treatment underscored the coordination of Fe3+ with –COOH groups.
3.3 Reversible fluorescence switching process of the prepared organohydrogel
Due to the copolymerization of PyMA on hydrophilic polymer network chains, the organohydrogels emitted blue fluorescence under 365 nm. The initial P(DMA-AAc-PyMA)/PSMA organohydrogels exhibited blue fluorescence. However, upon treatment with strongly oxidative Fe3+ ions, coordination occurred between Fe3+ ions and carboxylic acid groups on the hydrophilic polymer chains (Fig. 2a), where Fe3+ ions became excellent electron acceptors due to their electron-deficient nature, leading to ICT between Fe3+ and fluorescence monomers PyMA (electron donors). This ICT phenomenon resulted in the rapid quenching of fluorescence from the PyMA as depicted in Fig. S6.† As shown in Fig. 2b, the photoluminescence (PL) mapping spectrum demonstrated the singular blue-emitting center of P(DMA-AAc-PyMA)/PSMA organohydrogels. Comparatively, Fe3+ treatment led to the disappearance of the blue emission center.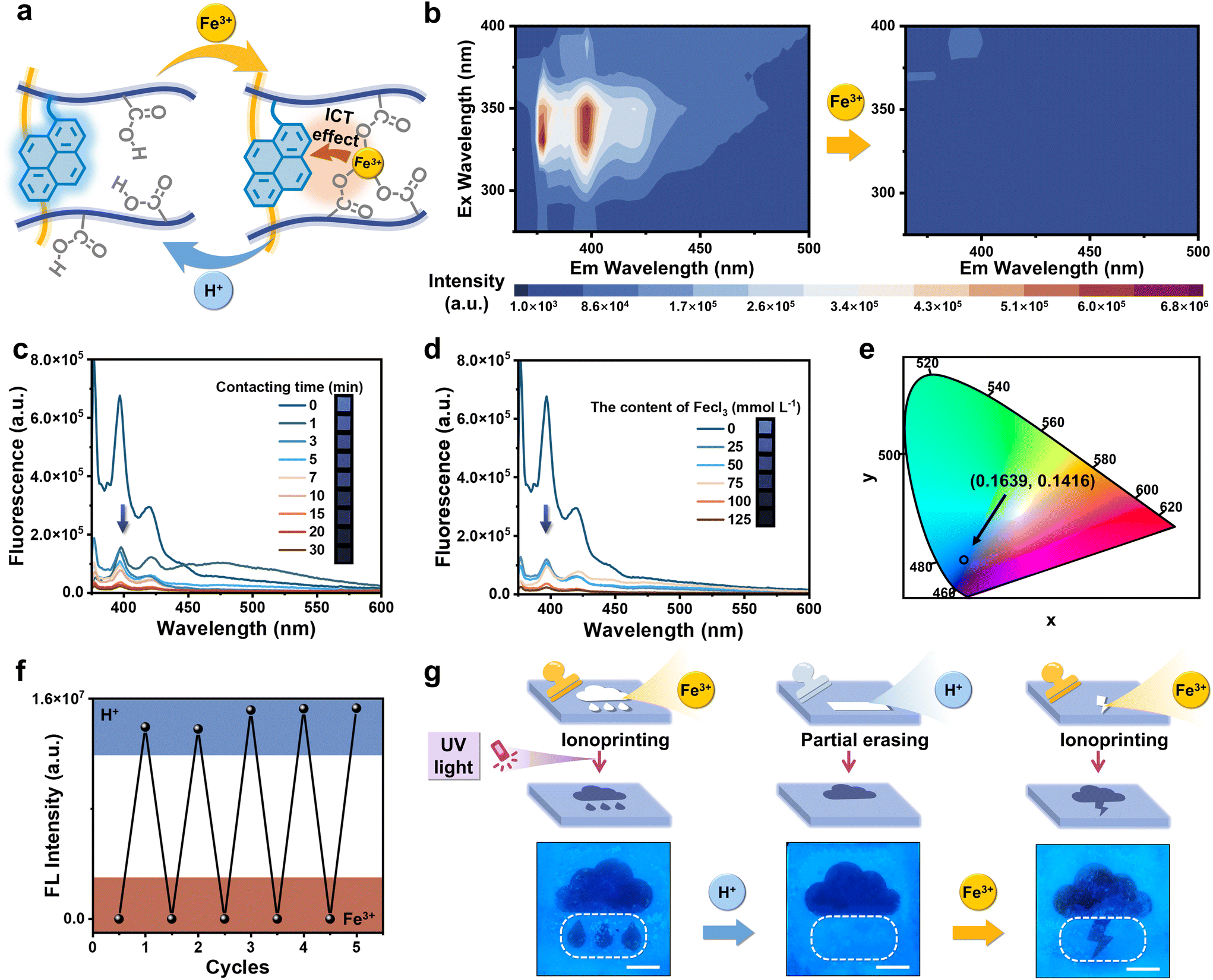 | ||
| Fig. 2 Reversible fluorescence switching process. (a) Schematic illustration of the reversible fluorescence switching mechanism. Due to the ICT effect, fluorescence was quenched when adding Fe3+, and subsequent treatment with H+ led to the restoration of fluorescence. (b) PL mapping spectra of P(DMA-AAc-PyMA)/PSMA organohydrogels before and after Fe3+ (0.1 M) treatment (24 h). (c) Fluorescence spectra of the organohydrogels after various times of Fe3+ (0.1 M) treatment. (d) Fluorescence spectra of the organohydrogels immersed in solutions (1 min) containing different Fe3+ contents. (e) The fluorescence color of P(DMA-AAc-PyMA)/PSMA organohydrogels under 365 nm UV irradiation on the CIE (1931) chromaticity diagram. (f) The cyclic changes in the fluorescence intensity of organohydrogels following successive treatments with Fe3+ (0.1 M, 24 h) and H+ (0.1 M, 24 h). (g) Schematic illustration and digital photos of the reversible fluorescence switching patterns were generated through the application of ionoprinting methodologies (scale bar: 1 cm). | ||
Furthermore, the degree of fluorescence quenching of the organohydrogels was also affected by the concentration of Fe3+ in solution as well as the soaking time. As shown in the fluorescence spectra of P(DMA-AAc-PyMA)/PSMA organohydrogels (Fig. 2c), the fluorescence intensity at 380 nm decreased with increasing time (0 to 30 min) of immersion in Fe3+ solution (0.1 M). With increasing immersion duration, there was not only a noticeable quenching of the fluorescence intensity but also a transformation of the organohydrogels. The organohydrogels displayed anisotropic swelling as the soaking time of one side of the organohydrogels in Fe3+ solution went from 0 min to 10 min (Fig. S7†), which is derived from chelation of Fe3+ within AAc, resulting in temporary cross-linking, and diminished the extent of swelling on one side. Relatively, when the immersion time was kept at 1 min, there was distinct weakening in the blue fluorescence with increasing Fe3+ content in the solution (0–125 mmol L−1) (Fig. 2d). Coordinates of the organohydrogels without Fe3+ treatment was (0.1639, 0.1416) in the Commission Internationale de L′ Eclairage (CIE), but the organohydrogels with Fe3+ treatment did not exhibit them (Fig. 2e). It is important to emphasize that the organohydrogels displayed excellent cycling stability through the ICT effect of Fe3+ and the uncoordinated action of H+, as seen in the fluorescence spectra (Fig. 2f). In light of the aforementioned remarkable reversible fluorescence quenching characteristics inherent in the organohydrogels, a sequential process encompassing information inscription, obliteration, and reinscription was achieved. A pattern of quenched fluorescence can be formed on the surface of an otherwise blue fluorescent organohydrogel by Fe3+ ion ionoprinting, and immediately thereafter, the introduction of H+ ions can cause the quenched portion of the fluorescence to reappear. Notably, H+ ions can be localized and brought into contact with the pattern of fluorescence quenching to achieve local recovery of local fluorescence. As illustrated in Fig. 2g, the initial introduction of the “raining” pattern was accomplished through Fe3+ (0.1 M, 15 min) ionoprinting. Subsequently, the “water drop” motif underwent erasure upon treatment with H+ (0.1 M, 15 min), and in a subsequent step, the “lightning” pattern was re-ionoprinted and loaded once more through Fe3+ application.
3.4 Advanced encryption and decryption based on the P(DMA-AAc-PyMA)/PSMA organohydrogel
The constitution of organohydrogels involved a hydrophobic PSMA polymer network, which exhibits the capability to form microcrystalline domains under defined temperature conditions (Fig. S8†).30 This inherent attribute imparted a shape memory function to the organohydrogels, enabling them to fold at high temperature (60 °C) and maintain a specific configuration at low temperature (0 °C) and subsequently recover the original shape when exposed to high temperatures again. Consequently, the integration of the reversible fluorescence switching phenomenon and shape memory characteristics in organohydrogels presented innovative avenues for implementing advanced information encryption and decryption strategies.As depicted in Fig. 3a, information encoding within the organohydrogels was initiated through the utilization of Fe3+ (0.1 M, 15 min) ionoprinting, and its reversible erasure was facilitated by the introduction of H+ (0.1 M, 15 min). Additionally, the written information underwent concealment via a shape memory process at lower temperatures (0 °C). The revelation of this information was contingent upon the restoration of the original shape at high temperatures (60 °C), permitting decryption when the organohydrogels were subjected to UV light at a wavelength of 365 nm. To exemplify the proof-of-concept for advanced encryption and decryption, organohydrogels with a precisely rendered Quick Response Code (QR I) can undergo encryption by fixing the shape of the pinwheel at 0 °C (Fig. 3b). Following a 15-minute exposure in an oven (60 °C), the organohydrogel reverted to a flat shape. Illumination under UV light (365 nm) enabled the reacquisition of QR I information, such as “UCAS”, accessible through smartphone scanning. The original information was erased through H+ processing, thereby preparing the organohydrogels for the introduction of novel information. Subsequent encryption involved the concealment of “wrong information” within the shape of a butterfly, allowing for decryption and erasure under analogous conditions. Furthermore, QR II, coded as “NIMTE”, was hidden within the geometric configuration of an airplane, and the information achieved encryption and decryption cycles facilitated by the shape memory and recovery process.
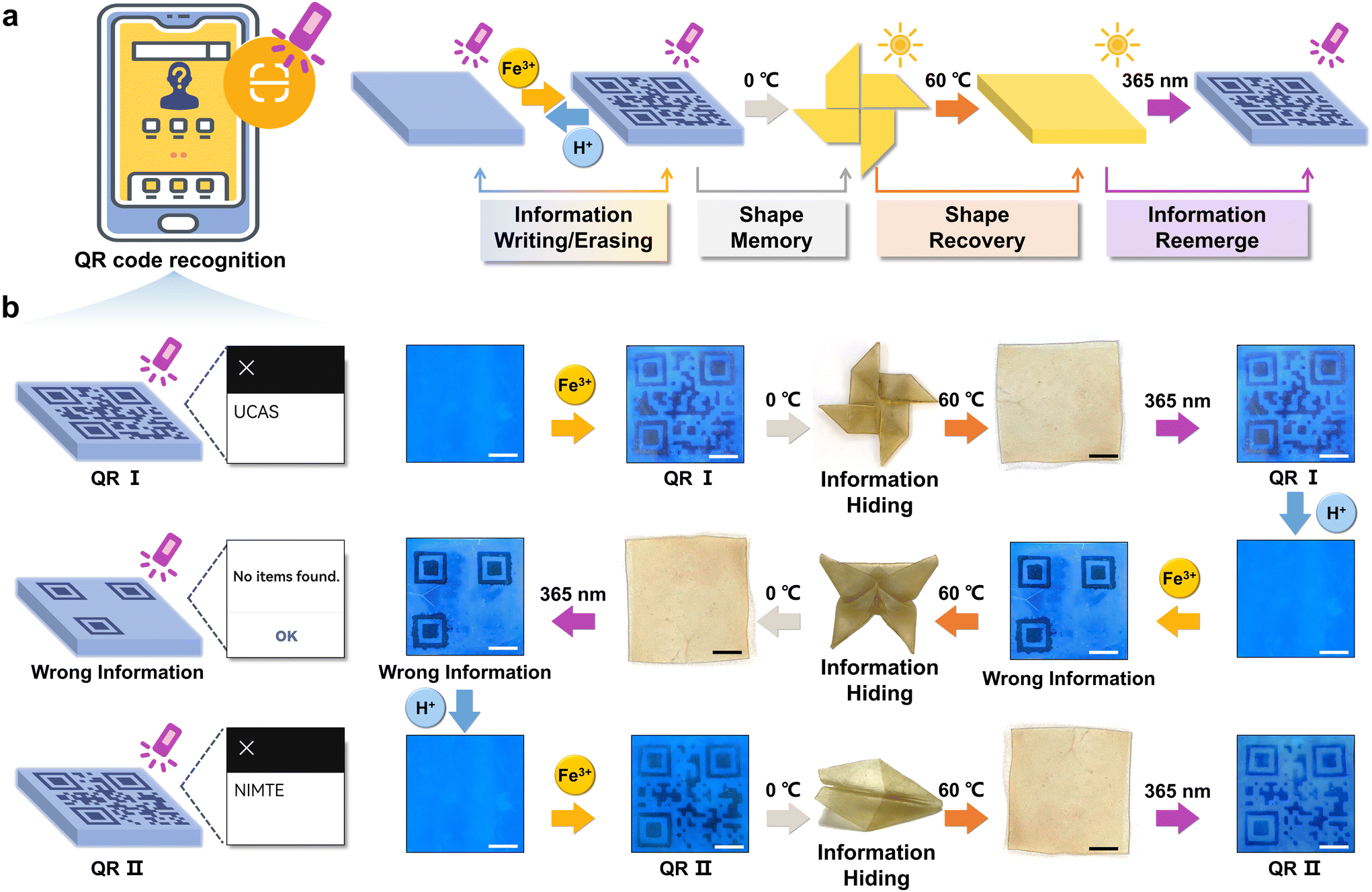 | ||
| Fig. 3 Information encoding, encryption, and decryption processes based on the fluorescent organohydrogels with reversible fluorescence switching changes and shape memory properties. (a) Schematic illustration of information encoding, encryption, and decryption processes. (b) QR codes are precisely ionoprinted onto the organohydrogels in the presence of Fe3+, and then the encoded information is further encrypted by a shape memory process. The hidden information could be obtained again under 365 nm UV light after shape recovery. H+ treatment could erase the ionoprinted information for recoding new patterns on the organohydrogel (scale bar: 1 cm). | ||
4. Conclusions
In this study, we reported an organohydrogel featuring dual hydrophilic and hydrophobic polymer networks, with the incorporation of the fluorescent monomers PyMA into the hydrophilic network. Hydroxy groups attached on the polymeric chains can coordinate with strongly oxidizing Fe3+, inducing the intramolecular charge transfer phenomenon which results in the fluorescence quenching of the organohydrogel. This quenching phenomenon can further be eliminated through treatment with H+. Additionally, the inherent crystalline properties of the hydrophobic polymer network contributed to the shape memory characteristics of the organohydrogel. The integration of reversible fluorescence switching with shape memory behavior facilitated advanced information encryption and decryption within the organohydrogels. In conclusion, our investigation proposes a novel approach for the design and fabrication of intelligent materials capable of advanced information storage. This innovation holds promise for enhancing information security and exhibits broad applicability in the realm of anti-counterfeiting.Author contributions
Yu Sun: conceptualization, methodology, investigation, visualization and writing (original draft); Hui Shang: conceptualization, investigation, validation and writing (review & editing); Xiaoxia Le: supervision and writing (review & editing); Tao Chen: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFB3204300), the National Natural Science Foundation of China (52103246), the Zhejiang Provincial Natural Science Foundation of China (LQ22E030015), the Natural Science Foundation of Ningbo (2023J408, 20221JCGY010301), the Ningbo International Cooperation Project (2023H019), and the Sino-German Mobility Program (M-0424).References
- R. Arppe and T. Sørensen, Nat. Rev. Chem., 2017, 1, 0031 CrossRef CAS.
- Y. Sun, X. Le, S. Zhou and T. Chen, Adv. Mater., 2022, 34, 2201262 CrossRef CAS PubMed.
- Y. Shen, X. Le, Y. Wu and T. Chen, Chem. Soc. Rev., 2024, 53, 606–623 RSC.
- D. Wen, F. Yue, G. Li, G. Zheng, K. Chan, S. Chen, M. Chen, K. Li, P. Wong, K. Cheah, E. Pun, S. Zhang and X. Chen, Nat. Commun., 2015, 6, 8241 CrossRef PubMed.
- X. Li, L. Chen, Y. Li, X. Zhang, M. Pu, Z. Zhao, X. Ma, Y. Wang, M. Hong and X. Luo, Sci. Adv., 2016, 2, e1601102 CrossRef PubMed.
- K. Lim, H. Liu, Y. Liu and J. Yang, Nat. Commun., 2019, 10, 25 CrossRef CAS PubMed.
- I. Cox, J. Kilian, F. Leighton and T. Shamoon, IEEE Trans. Image Process., 1997, 6, 1673–1687 CAS.
- F. Hartung and M. Kutter, Proc. IEEE, 1999, 87, 1079–1107 CrossRef.
- H. Hu, H. Zhong, C. Chen and Q. Chen, J. Mater. Chem. C, 2014, 2, 3695 RSC.
- S. Han, H. Bae, J. Kim, S. Shin, S. Choi, S. Lee, S. Kwon and W. Park, Adv. Mater., 2012, 24, 5924–5929 CrossRef CAS PubMed.
- M. You, M. Lin, S. Wang, X. Wang, G. Zhang, Y. Hong, Y. Dong, G. Jin and F. Xu, Nanoscale, 2016, 8, 10096–10104 RSC.
- M. Li, W. Yao, J. Liu, Q. Tian, L. Liu, J. Ding, Q. Xue, Q. Lu and W. Wu, J. Mater. Chem. C, 2017, 5, 6512–6520 RSC.
- R. Singh, E. Singh and H. Nalwa, RSC Adv., 2017, 7, 48597–48630 RSC.
- Y. Duroc and S. Tedjini, C. R. Phys., 2018, 19, 64–71 CAS.
- S. Terranova, F. Costa, G. Manara and S. Genovesi, Sensors, 2020, 20, 4740 CrossRef CAS PubMed.
- T. Ma, T. Li, L. Zhou, X. Ma, J. Yin and X. Jiang, Nat. Commun., 2020, 11, 1811 CrossRef CAS PubMed.
- C. Zhu, T. Bai, H. Wang, J. Ling, F. Huang, W. Hong, Q. Zheng and Z. Wu, Adv. Mater., 2021, 33, 2102023 CrossRef CAS PubMed.
- Q. Wang, B. Lin, M. Chen, C. Zhao, H. Tian and D. Qu, Nat. Commun., 2022, 13, 4185 CrossRef CAS PubMed.
- L. Ding and X. Wang, J. Am. Chem. Soc., 2020, 142, 13558–13564 CrossRef CAS PubMed.
- Z. Li, X. Ji, H. Xie and B. Tang, Adv. Mater., 2021, 33, 2100021 CrossRef CAS PubMed.
- K. Zhang, X. Zhou, S. Li, L. Zhao, W. Hu, A. Cai, Y. Zeng, Q. Wang, M. Wu, G. Li, J. Liu, H. Ji, Y. Qin and L. Wu, Adv. Mater., 2023, 35, 2305472 CrossRef CAS PubMed.
- Q. Ge, Z. Chen, J. Cheng, B. Zhang, Y. Zhang, H. Li, X. He, C. Yuan, J. Liu, S. Magdassi and S. Qu, Sci. Adv., 2021, 7, eaba4261 CrossRef CAS PubMed.
- X. Le, H. Shang, H. Yan, J. Zhang, W. Lu, M. Liu, L. Wang, G. Lu, Q. Xue and T. Chen, Angew. Chem., Int. Ed., 2021, 60, 3640–3646 CrossRef CAS PubMed.
- B. Wu, H. Lu, X. Le, W. Lu, J. Zhang, P. Théato and T. Chen, Chem. Sci., 2021, 12, 6472–6487 RSC.
- X. Zuo, S. Wang, Y. Zhou, C. Wu, A. Huang, T. Wang and Y. Yang, Chem. Eng. J., 2022, 447, 137492 CrossRef CAS.
- C. Yang, H. Xiao, L. Tang, Z. Luo, Y. Luo, N. Zhou, E. Liang, G. Wang and J. Tang, Mater. Horiz., 2023, 10, 2496–2505 RSC.
- Y. Zhang, X. Le, Y. Jian, W. Lu, J. Zhang and T. Chen, Adv. Funct. Mater., 2019, 29, 1905514 CrossRef CAS.
- S. Panja, N. Dwivedi and S. Saha, RSC Adv., 2016, 6, 105786–105794 RSC.
- X. Le, W. Lu, J. He, M. Serpe, J. Zhang and T. Chen, Sci. China Mater., 2019, 62, 831–839 CrossRef CAS.
- H. Shang, X. Le, Y. Sun, F. Shan, S. Wu, Y. Zheng, D. Li, D. Guo, Q. Liu and T. Chen, Adv. Opt. Mater., 2022, 10, 2200608 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00003j |
| This journal is © The Royal Society of Chemistry 2024 |