
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the thermal response of 3D-printed bilayer hydrogels via architectural control using binary ethanol–water solvent systems†
Francis
Klincewicz‡
 a,
Subhash
Kalidindi‡
b and
LaShanda T. J.
Korley
*ab
a,
Subhash
Kalidindi‡
b and
LaShanda T. J.
Korley
*ab
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: lkorley@udel.edu
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA
First published on 14th August 2024
Abstract
While stimuli-responsive materials can be prepared via many established procedures, digital light processing (DLP) 3D printing offers a simple and robust technique for the fabrication of hydrogels, including spatially-defined bilayer hydrogels. The use of synthesis solvent mixtures has recently gained attention as a facile alternative to more complicated chemical modifications to tune hydrogel morphology by exploiting solvent-monomer interactions and cononsolvency which, by extension, modulates stimuli-response time and magnitude. In this work, we utilized a binary solvent system consisting of ethanol and water to induce morphological changes within a thermally-responsive poly(N-isopropyl acrylamide) (pNIPAAm) hydrogel during polymerization. By varying the ratio of ethanol to water, we demonstrated that hydrogel properties, such as crosslink density, pore morphology, and thermal response, can be tuned and correlated. While mass expulsion was fastest in gels prepared in 100% ethanol, we found that gels prepared in 75%–25% ethanol–water and 50%–50% ethanol–water maintained mechanical integrity at high temperatures, allowing expulsion of water mass without large amounts of contraction. We utilized the experimental findings from the monolayer hydrogel studies and investigated the response of bilayer structures comprised of pNIPAAm hydrogel layer and a non-responsive poly(2-hydroxyethyl acrylate) (pHEA) hydrogel layer and applied a mathematical model to better understand the fundamental kinematics of these bilayer systems in response to temperature. We also demonstrated the utility of these bilayer hydrogels for use in soft robotics applications. Overall, this work highlights that modulation of binary solvent mixture ratios is a strategy that enables control of morphological and mechanical features of stimuli-responsive hydrogels via 3D printing.
1. Introduction
Natural systems, such as pine cones and vine tendrils, have the innate ability to display structural changes in response to environmental cues and external stimuli.1,2 In recent years, researchers have drawn inspiration from nature to design synthetic, stimuli-responsive soft materials.1,3,4 Specifically, pine cone scales possess bilayer structures comprised of an “active” responsive layer and a “passive” non-responsive layer. This geometry allows pine cone scales to curve inwards upon exposure to moisture due to a mismatch in response between the active and passive layers.1,5,6 This phenomenon of curvature in response to stimuli can be utilized for actuation and response in synthetic hydrogels by mimicking the bilayer geometry found in natural systems. Specifically, bilayer hydrogels containing thermally-responsive active layers have gained significant attention due to their application in soft robotics without the need for external electrical circuitry.7 Various studies have demonstrated that tuning the morphology and crosslink densities in active, thermally-responsive hydrogels via modulation of crosslinker content leads to better control over the degree of actuation.8 However, the tunability of the morphology of the active layer of bilayer hydrogels using solvent mixtures has not been well-studied, presenting opportunities to engineer materials for various applications.Poly(N-isopropylacrylamide) (pNIPAAm) is a common choice for the active layer in bilayer hydrogels. These hydrogels, which feature both hydrophilic and hydrophobic moieties, actuate due to a coil-to-globule transition at a lower critical solution temperature (LCST), at which pendant groups shift from favoring chain-solvent to intra-chain interactions, leading to the expulsion of water from the hydrogel and causing a volumetric transition.9,10 Linear homopolymer chains of pNIPAAm experience a sharp LCST around 32 °C; however, crosslinked, hydrated pNIPAAm networks can exhibit a broad transition temperature range.9 Multiple factors, including morphology,11 copolymerization of NIPAAm with other monomers such as acrylic acid (AA), furfuryl methacrylate (FMA), and 2-hydroxyethyl acrylate (HEA),10 and inclusion of additives,12 have been shown to impact the temperature and sharpness of this transition. While previous studies on manipulating the volume phase transition of pNIPAAm hydrogels for actuators have focused on copolymerization with other polymers,13–15 more recent investigations have examined the impacts of network architecture and morphology on the thermal response.16,17 This tunable thermal transition makes bilayer systems consisting of a pNIPAAm active layer and a non-responsive hydrogel passive layer ideal candidates for various applications, including soft robotics, temperature sensors, and healthcare devices.10,18 However, modulation of the morphology of the pNIPAAm-based active layer through bilayer fabrication processes has not been well-studied.
Traditionally, hydrogels are fabricated via thermal- or photo-initiated polymerization methods while their shape or size is defined using a poly(dimethylsiloxane) (PDMS) or poly(tetrafluoroethylene) (PTFE) mold.19 Some of the common limitations for fabrication in molds are the inability to rapidly create complex geometries and required use of multi-step photolithography processes with harsh chemicals.20,21 Digital light processing (DLP), three-dimensional (3D) printing, in which a photo-sensitive resin is sequentially polymerized to build up 3D geometries, can circumvent these challenges.22,23 This process offers several advantages, such as rapid yet inexpensive fabrication, reproducibility despite complex geometry, and avoidance of harsh chemicals, and presents an attractive alternative to reliably manufacture stimuli-responsive hydrogels.24 3D printing typically offers better interlayer bonding in bilayer gels due to the presence of active radicals during layer exchange that facilitate the formation of cross-layer covalent bonds.25 Additionally, many applications require complex architectures with multiple materials interfaced together, which is possible through exchanging resin vats25 or other sophisticated microfluidic methods.26 However, multi-material interfaces often are a locus of crack formation and material failure; efficient contraction without delamination requires sufficient interfacial strength.2,27,28
DLP 3D printing of pNIPAAm has been reported utilizing both custom-built26,29,30 and commercially available25 printers. However, DLP 3D printing of bilayer systems comprised of porous pNIPAAm hydrogels using a binary solvent system has not been well-explored. Compared to traditional polymerization methods, DLP requires greater amounts of photoinitiator and monomer to overcome oxygen inhibition and rapidly form a network that can support subsequent layers.31,32 In addition to synthetic challenges, common photoinitiators that absorb in the UV range of commercial 3D printers can reach saturation limits in pure water at lower concentrations than those required for easy printability.33 Consequently, previous reports of DLP 3D printing have primarily utilized ethanol as a solvent.25,26,29 However, many molecular and nanoscale additives of interest (e.g., nanoclays and cellulose nanocrystals) are more stable in dispersions of water than other polar solvents, such as ethanol.34,35 Therefore, for successful incorporation of these additives and a balance between photoinitiator solubility and dispersion stability, there is a need to understand the effect of the introduction of water as a cosolvent in printable hydrogel systems on the resulting polymer morphology and thermal response. Although several studies have investigated the use of binary solvent systems with water as a cosolvent, reliable manufacturing of these materials via DLP 3D printing is a facile pathway for further examination.
Solvents used in the preparation of polymer systems can have impacts beyond solubility and stability, influencing pore morphology and thermal behavior, and are often referred to as “synthesis solvents”.11,15,36,37 The crosslink density and pore morphology can be easily tuned by varying the concentration and type of synthesis solvent. De la Hoz Siegler et al. found that high solvent polarity decreases the molecular weight between crosslinks in pNIPAAm solutions by acting as a chain transfer agent.36 In the case of a binary cosolvent mixture of dimethyl sulfoxide (DMSO) and water, the solubility of pNIPAAm chains in the synthesis solvent was shown to cause large variations in the pore morphology by Alsaid et al.16 The hydrogels polymerized in either pure DMSO or water displayed closed, macroporous structures, while those polymerized in near-equal ratios of DMSO and water exhibited open pore structures with uneven texture, due to predicted competition between solvents causing phase separation, also known as cononsolvency.
Applying this synthesis solvent principle in a bilayer system, we also can explore the role of the solvent mixture on the polymerization of a similar, yet non-responsive, monomer, to investigate the relative effect of cononsolvency and chain transfer. HEA is a prime candidate, sharing similar molar mass and acrylate reactivity to NIPAAm, but without the large hydrophilicity imbalance that gives pNIPAAm its distinctive thermal response. In a similar system of a bilayer of pNIPAAm and hydroxyethyl acrylamide (HEAm) by Wang et al.,38 cononsolvency was utilized as a stimulus in addition to temperature, in which a mixture of ethanol and water caused the contraction of the active layer. Ethanol as a cosolvent to water has been shown to dramatically affect the swelling equilibrium and deswelling rate in a pNIPAAm hydrogel polymerized over a long period.11 Ethanol as a synthesis solvent for pHEA has been shown to yield hydrogels with larger pores compared to those polymerized from water.39 However, these insights only apply to traditionally-assembled hydrogels, and 3D printing utilizes significantly faster polymerization rates and impose spatial constraints that may result in differing morphological features. Therefore, it is vital to understand the impact of synthesis solvents on thermoresponsive hydrogel structure and properties fabricated via 3D printing.
In this work, pNIPAAm and pHEA hydrogels were characterized and subsequently interfaced to form a bilayer structure via DLP 3D printing utilizing binary mixtures of ethanol and water. Preferential curvature, resulting from the response of the pNIPAAm layer and non-response of the pHEA layer, was induced when these hydrogels were exposed to water above the LCST of pNIPAAm. Using this facile design, we systematically explored the impact of solvent content on the hydrogel structure, properties, and functionality. We investigated controlled actuation of the pHEA–pNIPAAm bilayer systems by tuning the synthesis solvent. Our focus encompassed pore morphology, mechanical properties, and thermal response, providing valuable insights into the balance of cononsolvency and chain transfer effects on these bio-inspired materials. This understanding serves as a foundation for advancing the development of stimuli-responsive materials for a variety of applications.
2. Methods
Materials
2-hydroxyethyl acrylate (HEA), 2,4,6-trimethylbenzoyldiphenyl phosphine oxide (TPO), and Rhodamine B were purchased from Sigma Aldrich. N,N′-methylenebis(acrylamide) (MBA) was purchased from Alfa Aesar. N-isopropylacrylamide (NIPAAm) was purchased from Tokyo Chemical Industry Co. 200-proof anhydrous ethanol was purchased from Decon Laboratories, Inc. 2-Isopropanol was purchased from Fisher Scientific. All chemicals were used as received without further purification. Deionized (DI) water was purified using a Milli-Q Academic (Millipore-Sigma).Resin preparation
To prepare resin formulations, first, ethanol and water were added to a vial in volumetric ratios of either 100–0, 75–25, or 50–50 (Table S1†). For NIPAAm-based resins, 18.4 g of NIPAAm monomer was added to 10 mL of ethanol or ethanol–water mixture to which MBA crosslinker was added at 1 mol% relative to the monomer (0.26 g mL−1 of solvent). Similarly, for HEA-based resins, 19.6 mL of HEA monomer was added to 10 mL of ethanol or ethanol–water mixture to which MBA crosslinker was added at 0.1 mol% relative to the monomer (0.03 g mL−1 of solvent). Finally, for both resins, TPO was added to the monomer solutions at a concentration of 0.06 g mL−1 of solvent.Hydrogel fabrication
To print the pHEA and pNIPAAm monolayers independently, either a HEA or NIPAAm resin solution (∼3 mL) was poured into the resin vat of the printer and sequentially irradiated with UV light for 16 seconds per 0.1 mm printed sublayer. To manufacture the pHEA:pNIPAAm bilayer hydrogels, printing was paused upon the formation of a desired number of HEA layers, which was facilitated by the removal of the print head from the vat during this time. The HEA resin was then poured out of the vat, followed by rinsing the vat with isopropanol and washing the print head with ethanol to remove excess unreacted resin. Then, NIPAAm resin was poured into the cleaned vat, and upon resuming printing, the layers of pNIPAAm were printed onto the existing pHEA layers to form pHEA:pNIPAAm bilayers. All prints were exposed in air to a 405 nm UV light (13 mW cm−2) in a UV chamber (SainSmart, Inc.) for 15 minutes to completely polymerize any unreacted initiator or monomer.
Hydrogels prepared with monomer NIPAAm or HEA are noted “NIPAAm X–Y” or “HEA X–Y”, where they are prepared with X vol% of ethanol and Y vol% of water comprising the solvent of the printing solution; for example, NIPAAm 50–50 resin was prepared using 50 vol% of ethanol and 50 vol% of water.
Characterization
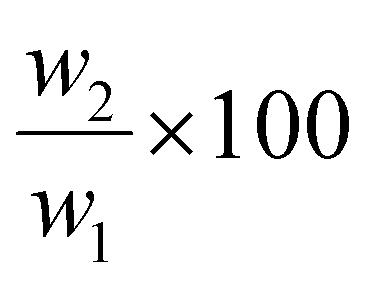 .
.
Dynamic mechanical analysis (DMA)
Responsiveness
To quantify the reversibility of the actuation of the bilayer hydrogels, rectangular prisms (7 mm × 20 mm × 2 mm) were printed and subjected to three heat–cool cycles between 60 °C and room temperature (∼23 °C) in a Petri dish. For further curvature analysis, the photographs of hydrogels were captured using a Google Pixel 7a phone camera at specific time intervals (0, 5, 15, and 30 minutes).
3. Results and discussion
Fabrication of controls and bilayers
Inspired by controlled actuation in pinecone scales (Fig. 1A), we fabricated stimuli-responsive, 3D printed monolayer and bilayer hydrogels and investigated the effect of processing conditions on the monolayer hydrogels and the subsequent impact of those conditions on the curvature of the bilayers.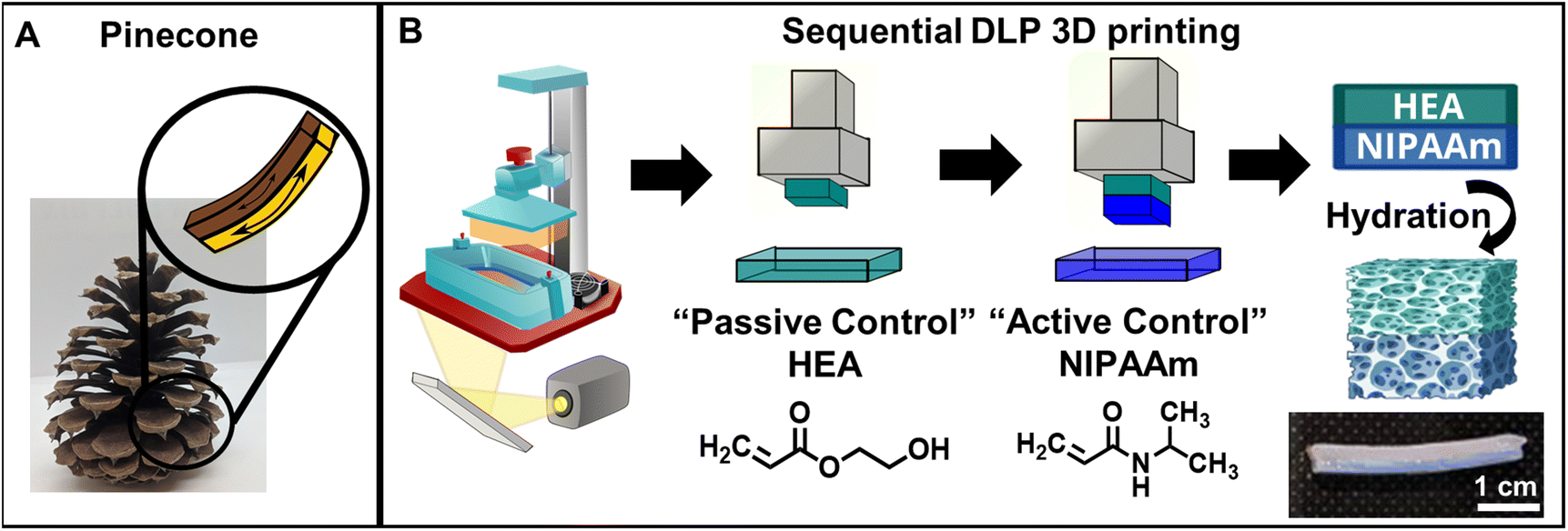 | ||
| Fig. 1 Fabrication of bilayer hydrogels using sequential DLP 3D printing. A. Natural inspiration from pinecone bilayer structure. B. Overall schematic for fabrication of monolayer and bilayer hydrogels using DLP printing. | ||
Linear pNIPAAm chains have been observed to have reduced solubility in mixtures of alcohol and water, with a maximum effect at a volumetric ratio of around 50% ethanol and 50% water, although they are soluble in both ethanol and in water.40,41 To investigate the effect that this cononsolvency may have on the resulting polymer structure during polymerization, two volumetric ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water ratios (50:50 v/v and 75:25 v/v) were selected to compare against an ethanol-only solution. To investigate the relative effect of cononsolvency versus solvent polarity, pHEA hydrogels also were printed. In this work, we varied the ethanol–water volume ratios in the monomer precursor solution containing NIPAAm or HEA, MBA (crosslinker), and TPO (photoinitiator) dissolved in a mixture of ethanol and water according to the ratios in Table S1.† Hydrogels prepared with X vol% of ethanol and Y vol% of water comprising the solvent of the printing solution are noted NIPAAm or HEA X–Y; for example, NIPAAm 50–50 resin was prepared using 50 vol% of ethanol and 50 vol% of water.
:water ratios (50:50 v/v and 75:25 v/v) were selected to compare against an ethanol-only solution. To investigate the relative effect of cononsolvency versus solvent polarity, pHEA hydrogels also were printed. In this work, we varied the ethanol–water volume ratios in the monomer precursor solution containing NIPAAm or HEA, MBA (crosslinker), and TPO (photoinitiator) dissolved in a mixture of ethanol and water according to the ratios in Table S1.† Hydrogels prepared with X vol% of ethanol and Y vol% of water comprising the solvent of the printing solution are noted NIPAAm or HEA X–Y; for example, NIPAAm 50–50 resin was prepared using 50 vol% of ethanol and 50 vol% of water.
Fig. 1B shows a schematic representation of the 3D printing-based fabrication of the monolayer and bilayer hydrogels containing pHEA and/or pNIPAAm using a commercial 3D DLP printer. First, monolayer prints (i.e., only NIPAAm or HEA resin) were printed and characterized. Gel contents of all printed networks were ∼90% as shown in Fig. S1,† suggesting that the mixture of solvents does not significantly affect the monomer incorporation into the network during polymerization. After hydration with DI water, all hydrogels of both polymers swelled to ∼1.5–1.6 times their original length, while pHEA absorbed slightly less mass by ratio (∼4 times its original mass compared to ∼4.5 times for pNIPAAm hydrogels) (Fig. S2†). Since the dimensional swelling is related to the crosslink density of the polymer networks,42 the crosslink density of the pHEA and pNIPAAm hydrogels are all similar despite the HEA printing solution containing one tenth the added bifunctional crosslinker (Table S1†). The additional crosslinking in the pHEA hydrogel can be attributed to an self-crosslinking mechanism reported previously for HEA, as well as potentially some diacrylate impurities in the monomer.43–45 To further investigate the crosslink density, the plateau moduli of as-printed (i.e., unhydrated) networks were found using DMA (Fig. S3†). As shown in Table 1 and Fig. S4,† the calculated crosslink densities of the pHEA networks and pNIPAAm networks exhibit the same trend, i.e., the 100–0 networks have a lower crosslink density compared to the 75–25 or 50–50 networks. The crosslink density of the pHEA networks increases from 53 mol m−3 in the HEA 100–0 and 50 mol m−3 in the HEA 75–25 to 62 mol m−3 in the HEA 50–50. The NIPAAm 100–0 also has a lower crosslink density at 38 mol m−3 compared to 47 and 63 mol m−3 in the NIPAAm 75–25 and NIPAAm 50–50, respectively. The solvent composition is expected to impact either the solubility of the network as it polymerizes through cononsolvency or the crosslink density due to the ability of the solvent to induce chain transfer.16,36 Since pHEA is typically unaffected by cononsolvency, any change in crosslink density can be attributed to chain transfer. Both pHEA and pNIPAAm show an increase in crosslink density, which is attributed to the solvent polarity during polymerization; the solubility of reaction mixture dictates free radical diffusion and chain transfer to solvent molecules.36 Achieving a range of crosslink density presents an avenue for swelling and actuation tunability for pNIPAAm networks via modulation of the synthesis solvent. It is important to note that, while the crosslink density can be varied using different crosslinker concentrations, the focus of this study is the utilization of synthesis solvent to modulate crosslink density and thereby impact the swelling and actuation tunability of pNIPAAm networks prepared via DLP 3D printing.
| Material | Ethanol–water ratio | Pore diameter (μm) | Crosslink density (mol m−3) | Diffusion coefficient (105 cm2 s−1) |
|---|---|---|---|---|
| NIPAAm | 100–0 | 9.3 ± 3.1 | 38 ± 24 | 0.55 |
| 75–25 | 11.1 ± 5.4 | 47 ± 6 | 2.2 | |
| 50–50 | 18.3 ± 7.7 | 63 ± 9 | 1.3 | |
| HEA | 100–0 | 2.3 ± 0.8 | 53 ± 4 | 1.3 |
| 75–25 | 1.3 ± 0.5 | 50 ± 4 | 1.8 | |
| 50–50 | 7.3 ± 2.3 | 62 ± 10 | 4.6 |
To fabricate bilayer hydrogels, ten sublayers of HEA monomer solution were printed, after which the vat was emptied and filled with NIPAAm monomer solution with the same ethanol–water solvent ratio to print the bilayer pNIPAAm–pHEA hydrogels as shown in Fig. 1B. After 3D printing, both the monolayer and bilayer gels were subjected to post-curing (405 nm UV light, 13 mW cm−2) for 15 min and finally immersed in DI water for at least 24 hours to allow for equilibrium swelling. The photograph in Fig. 1B shows one representative rectangular prism-shaped hydrated bilayer hydrogel, in which both the active and passive layer are 1 mm thick. Because multi-material printing also can lead to gradient structures,46,47 several imaging techniques were used to characterize the interface and distinguish if interdiffusion was occurring. To aid visualization of the two layers, pink Rhodamine B dye was added to the HEA resin before printing. A photographic image of the interface (Fig. S5A†) shows qualitatively that the pHEA and pNIPAAm layers are distinct, separate layers due to the lack of color gradient between the dyed pHEA region and the transparent pNIPAAm region. Darkfield microscopy (Fig. S5B†) as well as the corresponding fluorescence microscopy (Fig. S5C†) images of the hydrated bilayer hydrogel containing a rhodamine B dye-added pHEA layer and the neat pNIPAAm layer also shows a lack of fluorescence in the pNIPAAm layer. Further evidence of the two distinct pNIPAAm and pHEA layers in the bilayer can be found from an SEM image of a lyophilized hydrated bilayer in Fig. S5D,† which shows two distinct pore structures for the pHEA and pNIPAAm layers. During bilayer printing, the pHEA layer was rinsed briefly with ethanol while the NIPAAm resin was loaded into the printer. The lack of interdiffusion shows that this simple process successfully rinses excess monomers from the surface, preventing interdiffusion. At the same time, the lack of delamination upon lyophilization and handling demonstrates that the interlayer bond is sufficient to bear internal stress in subsequent experiments during which there will be a swelling mismatch upon stimulus.
It is important to note that monolayer hydrogels with thicknesses ranging between 0.1 and 0.5 mm can be reliably printed; however, these hydrogels were quite fragile and often fractured during handling. To circumvent these issues, 1 mm thick monolayer and 2 mm thick bilayers were fabricated to ensure sufficient mechanical integrity. This platform for reproducible 3D printing of bilayer hydrogels is shown in Fig. 1B.
Tuning of pore morphology using ethanol–water ratios
After printing and subjecting to post-curing (405 nm UV light, 13 mW cm−2), monolayer networks were swollen in DI water for at least 24 hours, forming porous hydrogels. Fig. 2A and B show photographs and the corresponding SEM images of pNIPAAm and pHEA monolayers, respectively, prepared using 100–0, 75–25, and 50–50 ethanol–water volumetric ratios. At all three ratios, the pNIPAAm hydrogels (Fig. 2A) were optically transparent, while the pHEA hydrogels (Fig. 2B) were opaque. We attribute the opacity to a higher solid fraction (Fig. S12†) in the pHEA hydrogels (55–80%) compared to the pNIPAAm hydrogels (25–30%). This opacity is likely due to the internal light scattering from dense network structures formed during polymerization. The hydroxyl groups in pHEA strongly hydrogen bond with both water and other hydroxyl groups, and can cause local aggregation during polymerization, which can lead to local dense networks.48,49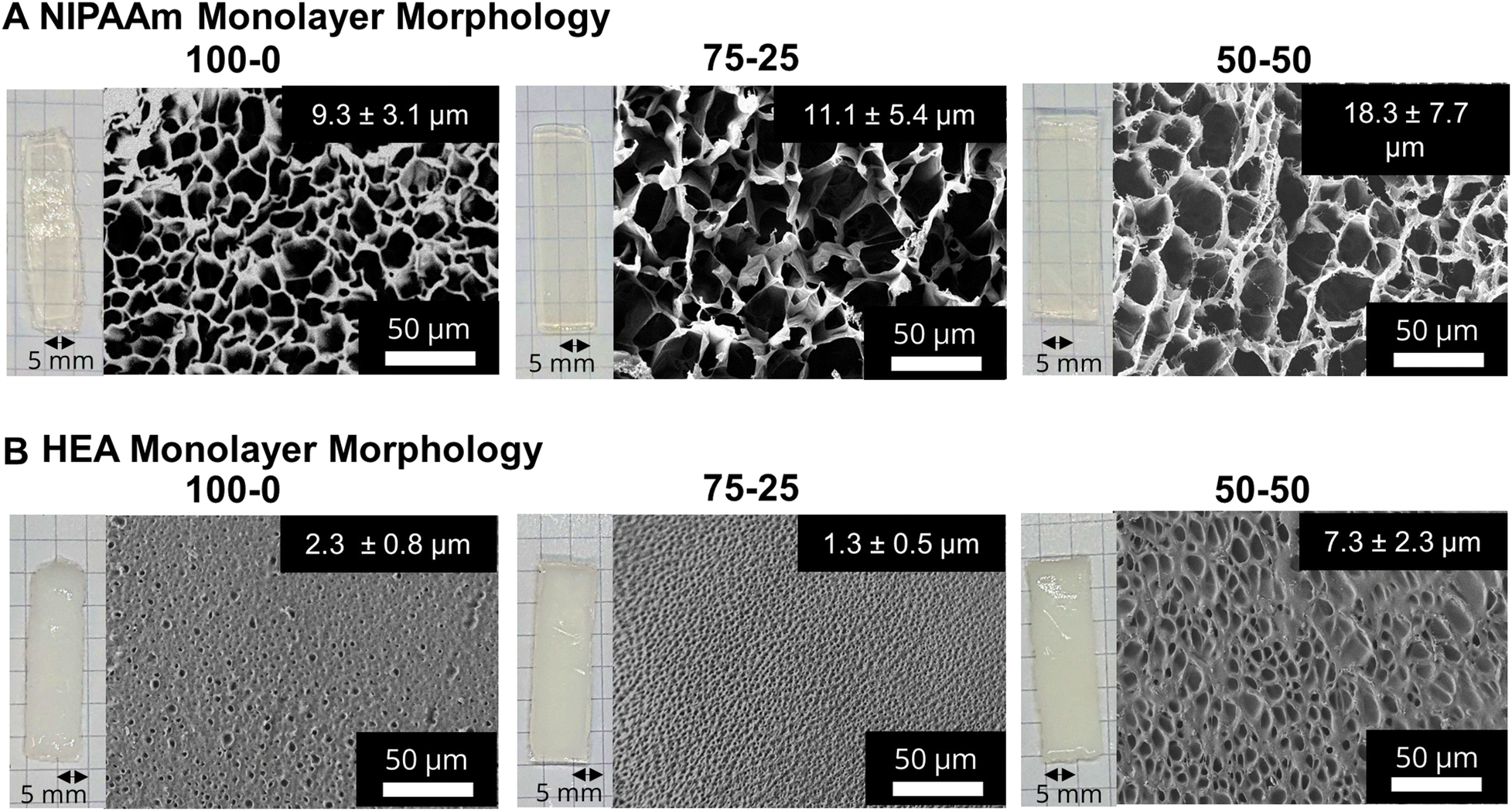 | ||
| Fig. 2 Hydrated pNIPAAm and pHEA hydrogel monolayer morphology and pore sizes. Photographs and corresponding SEM micrographs of A. pNIPAAm hydrogels prepared using varying ethanol–water volumetric ratios and B. pHEA hydrogels made using varying ethanol–water ratios (100–0 v/v, 75–25 v/v, or 50–50 v/v). Sample X–Y refers to a hydrogel printed from a solution containing X% ethanol and Y% water. Optical images were taken in front of graph paper with grid lines with 5 mm spacing. All micrographs were taken at 500× magnification. Pore sizes were calculated from image analysis of SEM images. | ||
SEM micrographs of the monolayer hydrogels, also shown in Fig. 2A, confirm the porous nature of the pNIPAAm hydrogels. The SEM micrographs consistently show the presence of thin borders around large pores in pNIPAAm hydrogels. By contrast, the SEM micrographs in Fig. 2B elucidate that the pHEA hydrogels do not display the same large pores, but rather microporous void spaces throughout the hydrogel. The pHEA pore structure, featuring a large solid fraction and low porosity, is indicative of a dense network formed during polymerization. Localized aggregation during polymerization of pHEA has been observed previously and attributed to hydrogen bonding between pendant hydroxyl groups and solvent (i.e., water or ethanol), as well as between other hydroxyl groups on the same chain.50–53 It is currently accepted that pHEA is not affected by cononsolvency, so the pore structure should remain unchanged by the ethanol–water ratio. However, other studies have shown that polymerization of HEA in either ethanol or in water independently may influence the porous nature of the pHEA network.39
Yet, cononsolvency during polymerization has been shown to lead to pore size variations in pNIPAAm hydrogels.11 The morphological differences between the NIPAAm 100–0 hydrogels and the NIPAAm 75–25 and 50–50 hydrogels align with the expected impact of cononsolvency. Qualitatively, the NIPAAm 75–25 and 50–50 hydrogels exhibited textured protrusions in the lyophilized gels that were not present in the NIPAAm 100–0 system. Nonuniformity in the pore walls can be attributed to portions of the network being unable to incorporate together seamlessly due to competitive solvation during polymerization.16 Measurement of pore size via image analysis yielded an increase in pore diameter with increasing water content, from 9.3 μm in the NIPAAm 100–0 hydrogel to 11.1 μm in the NIPAAm 75–25 hydrogel and 18.3 μm in the NIPAAm 50–50 hydrogel, as summarized in Table 1. Pore size can be correlated with crosslink density, as both are related to the mass uptake swelling of the network.11 Larger pores increase the water uptake while a higher crosslink density decreases water uptake via the Flory–Rehner relation.42 However, the water in the pores is free water, and does not interact greatly with the polymer network.19 We calculated the crosslink densities in Table 1 for the pNIPAAm networks before swelling, and thus conclude that water absorbed into the pores is independent of the crosslink density and dependent on the effect of the cononsolvency.
To connect these morphological differences to transport properties, we examined the elution of Rhodamine B dye out of these hydrogels. Briefly, the pNIPAAm and pHEA hydrogels were 3D printed and allowed to swell in DI water for 24 h, after which they were immersed into a 0.2 mM Rhodamine B dye solution for 48 h before being placed in clear DI water. For 72 h, 20 μL aliquots of the solution were drawn and the absorbance of the solution was examined using a UV-Vis spectrometer. The linear region of the concentrations of eluted dye are provided in Fig. S6,† and the calculated diffusion coefficients are summarized in Table 1. The NIPAAm 100–0 hydrogel was calculated to have a lower diffusion coefficient (0.55 × 10−5 cm2 s−1) than both the NIPAAm 75–25 and 50–50 hydrogels (2.2 × 10−5 and 1.3 × 10−5 cm2 s−1, respectively). The pHEA hydrogels showed a similar behavior with the HEA 100–0 hydrogel having a lower diffusion coefficient (1.3 × 10−5 cm2 s−1) than both the HEA 75–25 and 50–50 hydrogels (1.8 × 10−5 and 4.6 × 10−5 cm2 s−1, respectively). Solute diffusion is related to both the network mesh size and the interdiffusion of water through the pores of the hydrogel.19 With increasing pore size, the solute diffusion was expected to increase due to increased interdiffusion through free water. However, for the NIPAAm hydrogels, the diffusion through the polymer networks may be counteracted by the higher crosslink densities of the NIPAAm 75–25 and 50–50 hydrogels in Table 1. The diffusion of Rhodamine B in a pNIPAAm hydrogel may be affected by many factors, including adsorption within the network and homogeneity of the network.54 Inhomogeneous networks can be a result of cononsolvency during a polymerization due to the mix of coil and globule states of NIPAAm chains during polymerization.16 However, we observed the expected increase in diffusion coefficient corresponding to increased pore size, which suggests the interdiffusion through the free water dominates the effect of any inhomogeneity or increased crosslink density. Previous studies of pNIPAAm hydrogels have also shown an increase in diffusion coefficient with increased pore size.54 While it is expected that hydrogels with larger pore sizes will have faster diffusion, the calculated diffusion coefficient is also influenced by polymer–solute interactions In pHEA a higher amount of rhodamine B molecules diffuse into the network than in pNIPAAm, despite immersion in the same concentration of solution. In addition to diffusion through the pores, the nitrogen and oxygen in cationic rhodamine B interacts with the pHEA's hydroxyl group via hydrogen bonding and non-specific interactions leading to higher concentration of rhodamine B uptake. In pNIPAAm, there are minimal interactions between pNIPAAm's amide groups and rhodamine B molecules, so the pore tortuosity dominates the diffusion.55
Thermal response of monolayer hydrogels
The LCST of these pNIPAAm hydrogels marks a thermal transition of the chains within the crosslinked network from a coiled to globular state, with accompanying expulsion of water mass and contraction of the network volume as water is released. To characterize the effect of the morphological differences on the magnitude and sharpness of the thermal transition, we studied both the mass loss and spatial contraction of the hydrogels at a range of temperatures (30, 45 and 60 °C). To locate the temperature at the onset of the thermal transition, thermal profiles were investigated using DSC. As shown in Fig. S7,† endotherms with onset temperatures of 31.0, 32.4, and 31.9 °C were observed for the NIPAAm 100–0, 75–25, and 50–50 hydrogels respectively, which is consistent with the LCST (∼32 °C) of pNIPAAm reported in literature.10 The trend of the LCSTs (75–25 and 50–50 > 100–0) is consistent with the crosslink density values calculated in Table 1. This correlation highlights that an increase in crosslink density shifts the LCST of pNIPAAm hydrogels, and presents another way of fine-tuning the thermal response of these systems.56We examined the responsiveness of the monolayer hydrogels containing either pHEA or pNIPAAm to multiple temperatures (35, 45, and 60 °C) above the LCST as shown in Fig. 3. To explore the sharpness of the thermal transition, we measured the contraction of the active pNIPAAm and passive pHEA controls at three temperatures above the determined LCST of pNIPAAm hydrogels. Specifically, 35 °C is just above the LCST (32 °C), and 45 °C and 60 °C are significantly above the LCST without substantial evaporation from the heated water. The hydrogel mass and diameter were measured at various time points over a 90-minute period. The results at the 30-minute time point are reported in Fig. 3. At all temperatures and solvent contents, the pNIPAAm hydrogels expelled more mass and contracted more lengthwise than the pHEA hydrogels. Even with the cononsolvency effects, the pNIPAAm hydrogels do not lose their LCST thermal response, nor do the pHEA hydrogels exhibit significant actuation. It is noted that, at 60 °C, the pHEA hydrogels also show a small amount of contraction, which can possibly be attributed to syneresis or small amounts of aluminum from the resin vat creating supramolecular attractions to bring chains together as shown in literature.49 Expulsion of mass and contraction in length are shown as respectively 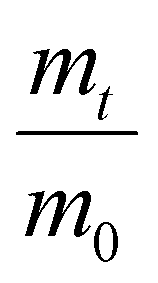 and
and 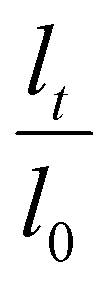 , where mt and lt is the mass and length at time t and m0 and l0 is the equilibrium hydrated mass or length. For example, if a hydrogel had expelled 25% of its mass, then the new mass would be 75% of the swollen hydrogel or a ratio of 0.75. The length change and mass expulsion ratios are comparable for the NIPAAm 75–25 and 50–50 hydrogels across all temperatures, within 0.05 for length changes and 0.03 for mass changes. However, at all three temperatures, the NIPAAm 100–0 hydrogel contracted in mass and length the most, with ratios ∼0.05–0.23 less in mass, 0.05–0.10 less for all lengths. At 35 °C, the NIPAAm 75–25 and 50–50 hydrogels showed minimal longitudinal contraction (0.98 and 0.95, respectively) while expelling mass (0.81 and 0.81, respectively). At 45 °C, the NIPAAm 75–25 hydrogel exhibited a larger scale contraction (0.81) while the NIPAAm 50–50 hydrogel displayed limited contraction (0.95), despite both demonstrating similar mass expulsions (0.84 and 0.83, respectively). Finally, at 60 °C, the NIPAAm 75–25 and 50–50 hydrogels showed a similar length contraction to the 35 and 45 °C values (0.87 and 0.91, respectively), but considerably more mass expulsion than the 35 and 45 °C values (0.57 and 0.61). The decrease of mass without changes in length, and by extension, volume, means there must be a change in density as well. A change in density implies that the hydrogel must be at least partially evacuated of water, despite the pores not collapsing. This observed pore integrity can aid reversibility over repeated actuation cycles, which will be explored in future studies. The increase in pore size of the NIPAAm 75–25 and 50–50 hydrogels were hypothesized to lead to faster and more pronounced actuation than the NIPAAm 100–0 hydrogel due to a reduced diffusion pathway required for water to travel from the interior of the hydrogel.57,58 However, the same trends are consistent over time as shown in Fig. S8.† The hydrogel mass and diameter were measured at various time points (0, 5, 15, 30, 60, and 90 minutes) over a 90-minute period. The results at the 30-minute time point are reported in Fig. 3. Furthermore, even after significant heating time, the hydrogels did not return to their as-printed masses, which indicated that despite chain collapse, there was still bound water in these networks. A small amount of entrapped water within the hydrogel is frequently observed in pNIPAAm hydrogels.11,56 To investigate the stability of the pore structure, SEM micrographs of lyophilized NIPAAm 100–0, NIPAAm 75–25, and NIPAAm 50–50 hydrogels after 30 minutes in a 60 °C water bath were obtained (Fig. S9†). The average pore size of the NIPAAm 100–0 hydrogel (7.8 ± 2.3 μm) (Fig. S9A†) is smaller compared to the unheated NIPAAm 100–0 hydrogel (9.8 ± 3.1 μm) in Fig. 2. A t-test (n = 100 measurements for each value) shows that the pore sizes are significantly different (p < 0.001). The smaller average pore size suggests that the overall hydrogel contracts due to pore shrinkage when exposed to water at 60 °C. By contrast, the exterior of the NIPAAm 75–25 and NIPAAm 50–50 hydrogels exhibited dense skin layers, with porous interiors (Fig. S9B and C†). A skin layer has been reported previously to occur when rapid transition occurs in the outer part of a hydrogel and prevents the transport of water from the interior of the hydrogel.59 Water must diffuse through this thick, hydrophobic boundary instead of through the pores of the hydrogel. Both NIPAAm 75–25 and 50–50 hydrogels expelled less mass than the NIPAAm 100–0 hydrogel, and mass loss without lateral contraction was observed in the NIPAAm 75–25 and 50–50 hydrogels at 35 °C, and again in the NIPAAm 50–50 hydrogel at 45 °C. Formation of a skin layer may explain the mitigated water mass expulsion in the NIPAAm 75–25 and 50–50 hydrogels due to the hindrance of diffusion from the interior. Furthermore, the decreased diameter change of the NIPAAm 75–25 and 50–50 hydrogels may be explained by this skin layer stabilizing the internal pore structure against collapse, even without the free water bound inside the pores. Therefore, we attribute the variation of thermal response of the monolayer hydrogels to the formation of a skin layer upon heating.
, where mt and lt is the mass and length at time t and m0 and l0 is the equilibrium hydrated mass or length. For example, if a hydrogel had expelled 25% of its mass, then the new mass would be 75% of the swollen hydrogel or a ratio of 0.75. The length change and mass expulsion ratios are comparable for the NIPAAm 75–25 and 50–50 hydrogels across all temperatures, within 0.05 for length changes and 0.03 for mass changes. However, at all three temperatures, the NIPAAm 100–0 hydrogel contracted in mass and length the most, with ratios ∼0.05–0.23 less in mass, 0.05–0.10 less for all lengths. At 35 °C, the NIPAAm 75–25 and 50–50 hydrogels showed minimal longitudinal contraction (0.98 and 0.95, respectively) while expelling mass (0.81 and 0.81, respectively). At 45 °C, the NIPAAm 75–25 hydrogel exhibited a larger scale contraction (0.81) while the NIPAAm 50–50 hydrogel displayed limited contraction (0.95), despite both demonstrating similar mass expulsions (0.84 and 0.83, respectively). Finally, at 60 °C, the NIPAAm 75–25 and 50–50 hydrogels showed a similar length contraction to the 35 and 45 °C values (0.87 and 0.91, respectively), but considerably more mass expulsion than the 35 and 45 °C values (0.57 and 0.61). The decrease of mass without changes in length, and by extension, volume, means there must be a change in density as well. A change in density implies that the hydrogel must be at least partially evacuated of water, despite the pores not collapsing. This observed pore integrity can aid reversibility over repeated actuation cycles, which will be explored in future studies. The increase in pore size of the NIPAAm 75–25 and 50–50 hydrogels were hypothesized to lead to faster and more pronounced actuation than the NIPAAm 100–0 hydrogel due to a reduced diffusion pathway required for water to travel from the interior of the hydrogel.57,58 However, the same trends are consistent over time as shown in Fig. S8.† The hydrogel mass and diameter were measured at various time points (0, 5, 15, 30, 60, and 90 minutes) over a 90-minute period. The results at the 30-minute time point are reported in Fig. 3. Furthermore, even after significant heating time, the hydrogels did not return to their as-printed masses, which indicated that despite chain collapse, there was still bound water in these networks. A small amount of entrapped water within the hydrogel is frequently observed in pNIPAAm hydrogels.11,56 To investigate the stability of the pore structure, SEM micrographs of lyophilized NIPAAm 100–0, NIPAAm 75–25, and NIPAAm 50–50 hydrogels after 30 minutes in a 60 °C water bath were obtained (Fig. S9†). The average pore size of the NIPAAm 100–0 hydrogel (7.8 ± 2.3 μm) (Fig. S9A†) is smaller compared to the unheated NIPAAm 100–0 hydrogel (9.8 ± 3.1 μm) in Fig. 2. A t-test (n = 100 measurements for each value) shows that the pore sizes are significantly different (p < 0.001). The smaller average pore size suggests that the overall hydrogel contracts due to pore shrinkage when exposed to water at 60 °C. By contrast, the exterior of the NIPAAm 75–25 and NIPAAm 50–50 hydrogels exhibited dense skin layers, with porous interiors (Fig. S9B and C†). A skin layer has been reported previously to occur when rapid transition occurs in the outer part of a hydrogel and prevents the transport of water from the interior of the hydrogel.59 Water must diffuse through this thick, hydrophobic boundary instead of through the pores of the hydrogel. Both NIPAAm 75–25 and 50–50 hydrogels expelled less mass than the NIPAAm 100–0 hydrogel, and mass loss without lateral contraction was observed in the NIPAAm 75–25 and 50–50 hydrogels at 35 °C, and again in the NIPAAm 50–50 hydrogel at 45 °C. Formation of a skin layer may explain the mitigated water mass expulsion in the NIPAAm 75–25 and 50–50 hydrogels due to the hindrance of diffusion from the interior. Furthermore, the decreased diameter change of the NIPAAm 75–25 and 50–50 hydrogels may be explained by this skin layer stabilizing the internal pore structure against collapse, even without the free water bound inside the pores. Therefore, we attribute the variation of thermal response of the monolayer hydrogels to the formation of a skin layer upon heating.
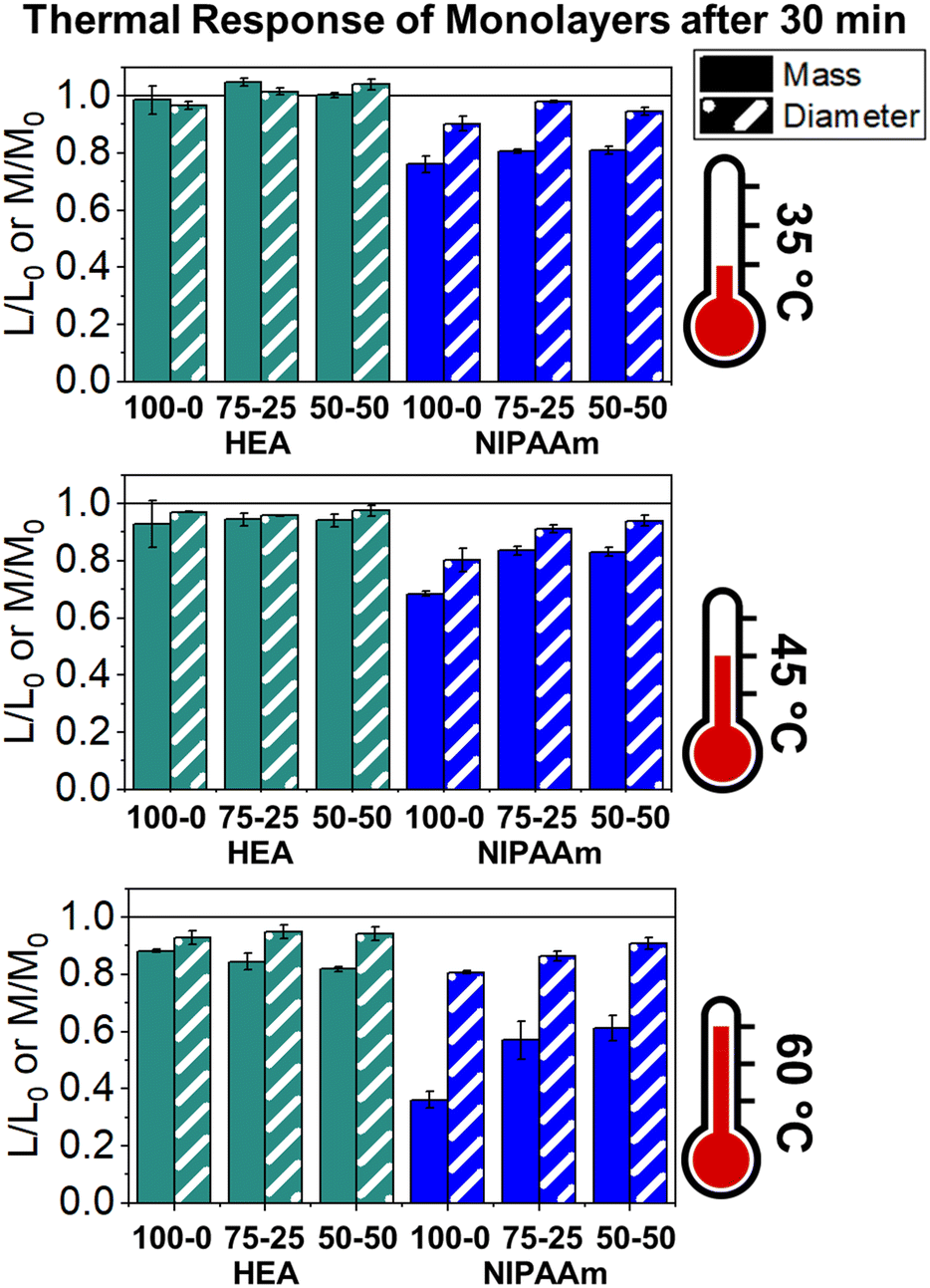 | ||
| Fig. 3 Thermal response of pNIPAAm and pHEA monolayer hydrogels at 35, 45, and 60 °C. Ratio of hydrogel diameter and mass after 30 minutes in a water bath at 35, 45, and 60 °C relative to the initial length or mass. | ||
To investigate the impact of the skin layer formation on the mechanical properties of the hydrogels, we conducted immersion DMA. A small sinusoidal strain was applied to hydrogels immersed in DI water at 60 °C, above the LCST, with the platen dynamically maintaining a constant axial force as the hydrogel contracted. As shown in Fig. S10A and B,† the water-immersed pNIPAAm hydrogels displayed a storage modulus of ∼0.2 MPa at room temperature. After the addition of 60 °C water, the storage moduli of the NIPAAm 100–0, NIPAAm 75–25, and NIPAAm 50–50 hydrogels rapidly increased to values of 0.62, 0.73, and 0.86 MPa, respectively, as shown in Fig. S10C.† After water expulsion, the NIPAAm 100–0 returns to its original value, likely due to pore collapse contracting the network. The NIPAAm 75–25 and NIPAAm 50–50 hydrogels retain the same modulus increase, likely due to the skin layer providing mechanical support to prevent collapse. The lack of mechanical property change in the NIPAAm 75–25 and 50–50 hydrogels suggests that despite water being expelled from the network, the network still shows the same amount of elastic activity. Unlike the NIPAAm 100–0 hydrogel, the NIPAAm 75–25 and NIPAAm 50–50 hydrogels form a skin layer upon heating, which may be creating a hydrostatic pressure within the porous structure beneath the skin layer.60 Despite interstitial water being expelled from between the chains, the pressurized hydrogels may maintain their mechanical integrity due to the intact pores. By contrast, the NIPAAm 100–0 hydrogel does not form a skin layer, leading to pore collapse upon heating above the LCST, which would be expected to lead to a stiffer gel due to a higher polymer volume fraction. However, the NIPAAm 100–0 hydrogel modulus displays the opposite trend, possibly due to the oscillatory strain allowing water to enter the collapsed hydrogel and surround the polymer network, thus inducing plasticization despite the thermodynamic phase separation. Overall, we can conclude that the larger pore dimensions seen in the NIPAAm 75–25 and 50–50 hydrogels do not translate to a faster or greater degree of actuation, due to the formation of a skin layer.
Thermal response of bilayers
To probe the effects of morphological changes on the curvature response, bilayers were fabricated by sequentially printing pHEA and pNIPAAm gels. As discussed above, micrographs in Fig. S5† demonstrated that the two materials had a strong bond without interdiffusion, suggesting either covalent bonds between layers or van der Waals interactions.25To demonstrate the versatility of the 3D printing technique, bilayers in the shape of a butterfly, a gripper, and a rectangular prism were printed and hydrated (Fig. 4A). Comparison of the time-dependent actuation at 60 °C of the rectangular prism bilayer gels fabricated from HEA and NIPAAm layers using three ethanol–water volume ratios (100–0, 75–25, and 50–50) is shown in Fig. 4B. Bilayers can be evaluated by their Gaussian curvature, which for a circular arc is equivalent to the reciprocal of the radius of curvature. Across all three bilayers, the actuation was minimal over the first five minutes, after which visible curvature began. The five-minute time delay also was observed in the progressive contraction of the diameters of the independent pNIPAAm monolayers as shown in Fig. S8B.† The curvature was quantified by the data points connected by solid lines in Fig. 4C. Although the 50–50 bilayer had a higher initial curvature than the 100–0 bilayer, the rate of curvature increase eventually slowed and plateaued. Simultaneously, the 100–0 bilayer steadily increased in curvature and resulted in a larger final curvature than either the 75–25 or 50–50 bilayer. This plateauing is possibly indicative of skin layer formation in the 75–25 and 50–50 bilayers preventing further expulsion, as also seen in the monolayers as similarly observed in other studies.59
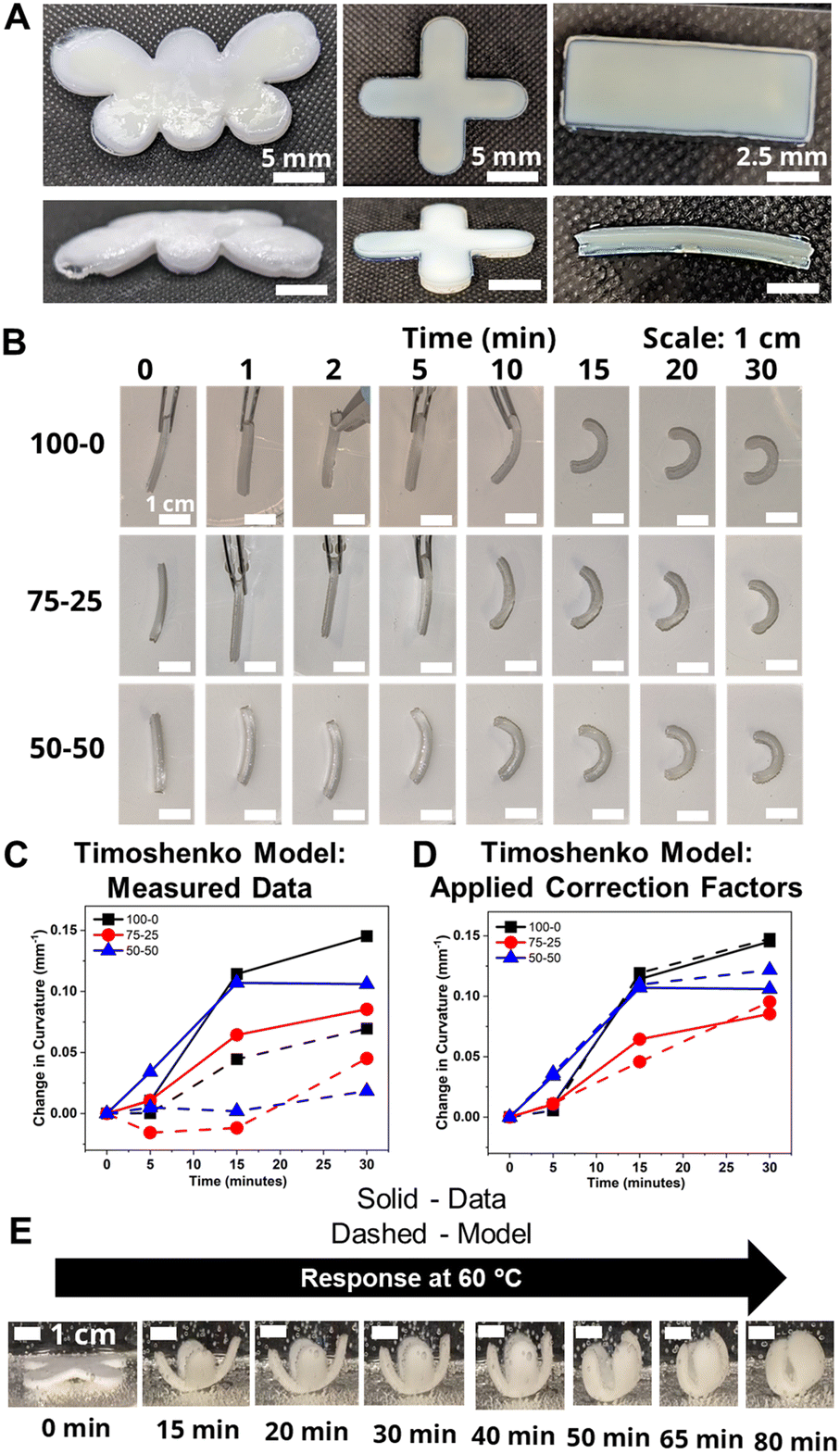 | ||
| Fig. 4 Bilayers with various geometries and their response to 60 °C water A. Photographs of butterfly-shaped, cross-shaped, and rectangular-shaped, hydrated bilayer hydrogels (left to right). B. Progressive curvature response at 60 °C in DI water of bilayer hydrogels printed from solutions with ethanol–water ratios of 100–0, 75–25, and 50–50, respectively. C. Comparison of measured curvatures (solid lines) of the bilayers in (B) to the Timoshenko kinematic model utilizing measured parameters of the hydrogels. D. Comparison of measured curvatures (solid lines) of the bilayers in (B) to the Timoshenko kinematic model utilizing modified parameters. E. Demonstration of actuation of a bilayer gripper printed from 100–0 printing solution. Scale bar in (B) and (E) is 1 cm. | ||
To investigate the relationship between the progressive response of the active pNIPAAm and pHEA hydrogels and contraction of the pNIPAAm–pHEA bilayer hydrogels, we compared the experimental curvature data to the predicted curvature of the kinematic model by Timoshenko (eqn (1)).61 The curvature of the hydrogels can be represented by the following kinematic model61
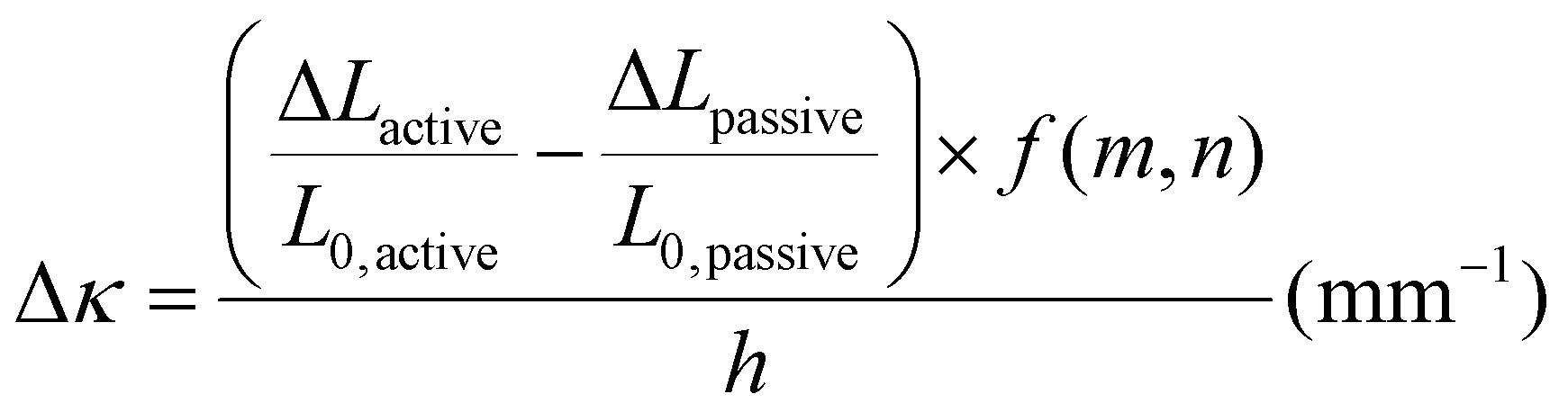 | (1) |
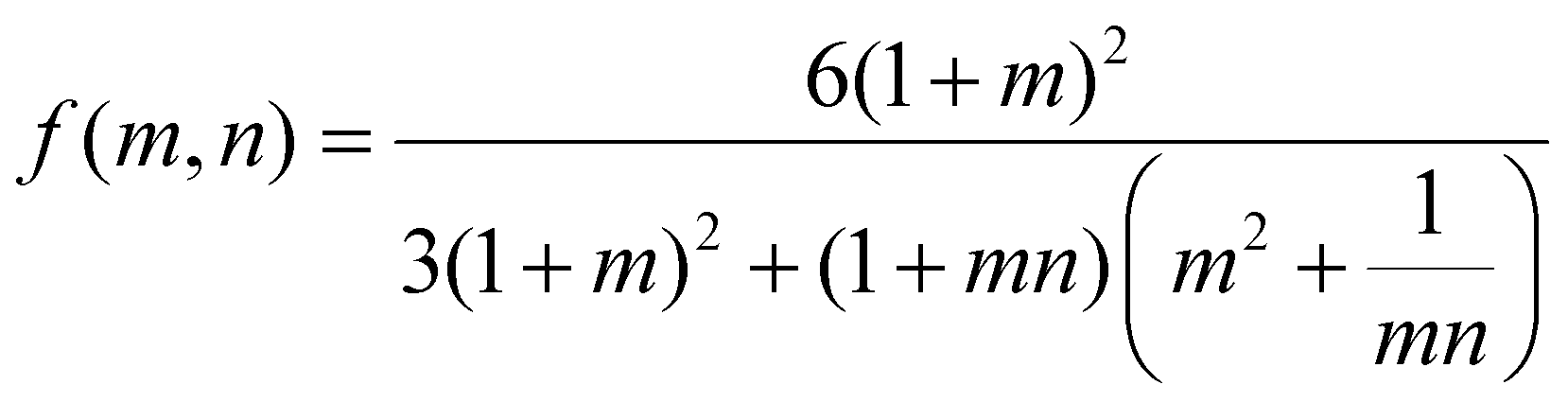 | (2) |
Experimentally-found values were utilized for the parameters of the model, and the resultant curvature was compared against to the experimental data in Fig. 4C. The storage moduli found via immersion DMA (Fig. S10†) were used as the modulus ratio parameter in the model, while the relative length changes were found from the cylindrical active and passive controls (Fig. S8†). The curvature of the bilayers imaged in Fig. 4B is shown as data points connected by solid lines in Fig. 4C, while the predictions from the model at those points are shown connected with dashed lines. A substantial difference between the prediction and the experimental data was observed across all three solvent ratios. Contraction of the pNIPAAm monolayers was observed to be isotropic between the thickness and diameter of the hydrogels, and similar isotropic behavior was observed in the active layer within the bilayers.62,63
To investigate possible reasons for differences between the experimental data and the model predictions, a sensitivity analysis of the effects of the input parameters to the predicted curvature was conducted. The input parameters are the ratio of the layer thicknesses, the ratio of their elastic moduli, the total thickness, and the relative lengthwise contraction of the two layers. The ratio of the two moduli did not have a major influence on the final curvature. Additionally, while we found that decreasing the bilayer thickness can increase the predicted curvature, modification of that parameter was unable to match the data behavior, even after accounting for continuous thickness contraction over time. The parameter with the most influence over the predicted curvature is the relative contraction of the two layers. This parameter was immediately promising; the surprising predicted negative curvature in the 75–25 system is due to the weak contraction of the NIPAAm 75–25 hydrogel at 60 °C, which is slightly less than the minimal actuation of the HEA 75–25 hydrogel at 60 °C. Hypothesizing that the pNIPAAm hydrogel contracts a greater amount when interfaced with pHEA than alone, correction factors were applied to the pNIPAAm monolayer contractions (i.e.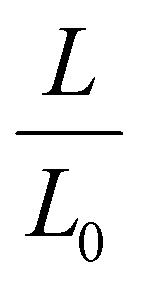 reported in Fig. S8†) input into eqn (1), resulting in a modified Timoshenko model (eqn (3)).
reported in Fig. S8†) input into eqn (1), resulting in a modified Timoshenko model (eqn (3)).
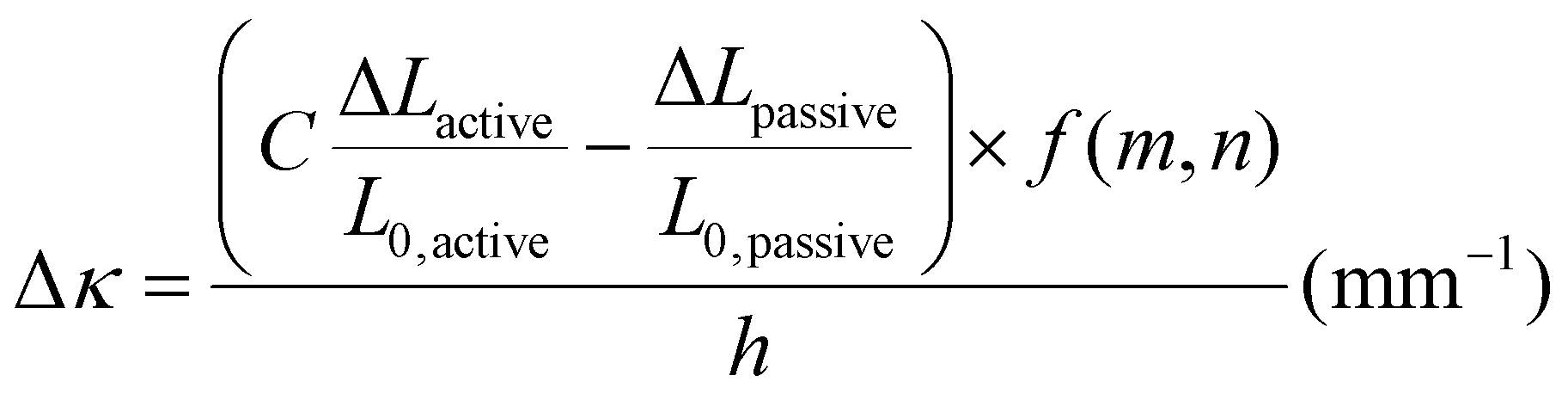 | (3) |
The length contraction ratio of the pNIPAAm monolayer was multiplied by the correction factor prior to inclusion into the Timoshenko model; for example, a correction factor of 0.95 means the monolayer contraction fit best with a 5% increase in mass expulsion. To fit the model, one correction factor was insufficient, as the 5 min data point always was significantly closer to the model than the 15 min or 30 min data point. By applying a correction factor to the 5 min data point and a second one for the others, a much closer fit was found. Briefly, the 5 min and 15-and-30 min correction factors were: 0.99 and 0.83, for the 100–0 bilayer; 0.95 and 0.89, for the 75–25 bilayer; and 0.94 and 0.79, for the 50–50 bilayer. The corrected model plots are shown in Fig. 4D. This modification of the active layer contraction behavior suggests that the interfacing of the active and passive layer dominates the thermal behavior of the active layer. One potential explanation is that the interface between the pNIPAAm and pHEA hydrogels allows for the egress of water, while the formation of a skin layer in the active pNIPAAm control may have prevented the flow of water out of the hydrogel network. Because the 5 min correction factor was less than the 15-to-30 min correction factor, there may be a time dependence to this observed phenomenon between those regimes. The skin layer has been shown to only form after the surface layers of water have been expelled,59,60 which matches the actuation behavior observed experimentally. Overall, the analysis shows that the contraction of thermally-responsive hydrogels in bilayers is dependent on multiple additional factors such as skin layer formation and additional water diffusion pathways from the interfaced passive layer. While models have demonstrated the effect of constrained deswelling on responsive hydrogels, this modified Timoshenko model provides a pathway for facile comparison of the constrained deswelling to known properties of the unconstrained hydrogel.62,63
The utility of the bilayer hydrogel system for soft robotics or sensors was demonstrated. We designed a soft gripper as a proof-of-concept model for an actuating device (Fig. 4E). The four arms of the cross-shaped, bilayer gripper all simultaneously curve, anchored at the base by the center of the cross. Engineering of bilayer hydrogel materials via functionalization with carboxylic groups or amine groups as well as addition of nanofillers into hydrogels will help expand applications in health care devices and sensors.64,65
To investigate the reversible thermal actuation, bilayer hydrogels (100–0, 75–25 and 50–50) were first immersed in 60 °C hot water for 30 minutes and subsequently immersed into room temperature (∼23 °C) water for 90 minutes, which was repeated for three cycles. The curvature of the bilayer hydrogels at each time point was measured using ImageJ from the captured photographs. During the three heat–cool cycles, the bilayer hydrogel consistently curved (∼0.15 mm−1) upon exposure to 60 °C water for 30 minutes and reversibly retained its initial curvature upon exposure to room temperature conditions. As a representative study, Fig. S11† shows the reversible thermal actuation of the 50–50 bilayer hydrogels for three heat–cool cycles. This study demonstrates the thermoreversible actuation of these responsive bilayer hydrogels.
4. Conclusions
In this work, we demonstrated the fabrication of bio-inspired, thermally-responsive hydrogel bilayers utilizing 3D printing and the tunability of hydrogel morphology using a binary ethanol–water solvent system to investigate their structure–property–functionality relationship. The pore size and crosslink density in thermally-responsive pNIPAAm was tuned by varying ethanol–water ratios in the precursor monomer solutions, while passive pHEA hydrogels were less affected by the modification of the precursor monomer solutions. We demonstrated that increasing the water content in the precursor monomer solutions results in increased pore size up to two-fold, primarily due to the role of the ethanol–water solvent mixture inducing a cononsolvency effect and acting as a chain transfer agent. Through a solute diffusion protocol, heating study, and DMA analysis, the varied pore morphology is correlated to transport behavior, thermal transitions, and crosslink density. Bilayer hydrogels prepared with varied ethanol–water solutions showed tunable actuation in response to temperature. Finally, a bilayer gripper showed promising potential towards soft robotics. Overall, this work highlights an opportunity for tunability of bilayer structures through modification of the additive manufacturing conditions rather than modification of the backbone chemistry of the starting materials. Furthermore, the complex nature of the bilayer curvature may be further understood through theoretical modeling.Author contributions
F. K. and L. T. J. K. conceived the idea. F. K. and S. K. performed experiments and analysis. F. K. and S. K. co-wrote the initial draft. L. T. J. K. supervised the work and obtained funding. All authors contributed to the writing and editing of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge Will Quintana for conducting preliminary experiments for this project. This work was supported by the National Science Foundation (NSF) PIRE: Bio-inspired Materials and Systems under grant number OISE 1844463. This material is based upon work supported by the University of Delaware Graduate College through the Unidel Distinguished Graduate Scholar Award. The authors acknowledge the use of facilities and instrumentation supported by NSF through the University of Delaware Materials Research Science and Engineering Center DMR 2011824. This work was supported as part of the Center for Plastics Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences at the University of Delaware under award DE-SC0021166. Access to the SEM was provided by the Keck Microscopy Center at the University of Delaware.References
- E. Reyssat and L. Mahadevan, J. R. Soc., Interface, 2009, 6, 951–957 CrossRef CAS PubMed.
- S. L. M. Alexander and L. T. J. Korley, Soft Matter, 2017, 13, 283–291 RSC.
- S. L. M. Alexander, L. E. Matolyak and L. T. J. Korley, Macromol. Mater. Eng., 2017, 302, 1700133 CrossRef.
- Q. Zhao, Y. Wang, H. Cui and X. Du, J. Mater. Chem. C, 2019, 7, 6493–6511 RSC.
- C. Dawson, J. F. V. Vincent and A. M. Rocca, Nature, 1997, 390, 668–668 CrossRef CAS.
- K. Song, E. Yeom, S. J. Seo, K. Kim, H. Kim, J. H. Lim and S. Joon Lee, Sci. Rep., 2015, 5, 9963 CrossRef CAS PubMed.
- S. Poppinga, C. Zollfrank, O. Prucker, J. Rühe, A. Menges, T. Cheng and T. Speck, Adv. Mater., 2018, 30, 1703653 CrossRef PubMed.
- S. Xiao, M. Zhang, X. He, L. Huang, Y. Zhang, B. Ren, M. Zhong, Y. Chang, J. Yang and J. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 21642–21653 CrossRef CAS PubMed.
- P. Chatterjee, A. Dai, H. Yu, H. Jiang and L. L. Dai, J. Appl. Polym. Sci., 2015, 132, 42776 CrossRef.
- M. M. Rana and H. De La Hoz Siegler, Polymers, 2021, 13, 3154 CrossRef CAS PubMed.
- C. S. Biswas, V. K. Patel, N. K. Vishwakarma, A. K. Mishra and B. Ray, J. Appl. Polym. Sci., 2011, 121, 2422–2429 CrossRef CAS.
- X. Li, M. Li, L. Tang, D. Shi, E. Lam and J. Bae, Mater. Chem. Front., 2023, 7, 5989–6034 RSC.
- J. C. Garbern, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2010, 11, 1833–1839 CrossRef CAS PubMed.
- V. Müller, R. Matthes, M. Wagner, M. Bros, P. Dreier and H. Frey, Polym. Chem., 2023, 14, 2599–2609 RSC.
- X. Zhang, L. Zhou, X. Zhang and H. Dai, J. Appl. Polym. Sci., 2010, 116, 1099–1105 CrossRef CAS.
- Y. Alsaid, S. Wu, D. Wu, Y. Du, L. Shi, R. Khodambashi, R. Rico, M. Hua, Y. Yan, Y. Zhao, D. Aukes and X. He, Adv. Mater., 2021, 33, 2008235 CrossRef CAS PubMed.
- D. M. Solis and A. Czekanski, Soft Matter, 2022, 18, 3422–3429 RSC.
- P. Imrie and J. Jin, Macromol. Mater. Eng., 2023, 2300272 Search PubMed.
- R. Foudazi, R. Zowada, I. Manas Zloczower and D. L. Feke, Langmuir, 2023, 39, 2092–2111 CrossRef CAS PubMed.
- J. Del Barrio and C. Sánchez Somolinos, Adv. Opt. Mater., 2019, 7, 1900598 CrossRef.
- I. Ertugrul, Micromachines, 2020, 11, 518 CrossRef PubMed.
- G. Zhu, Y. Hou, J. Xu and N. Zhao, Adv. Funct. Mater., 2021, 31, 2007173 CrossRef CAS.
- H. Ding, M. Dong, Q. Zheng and Z. L. Wu, Mol. Syst. Des. Eng., 2022, 7, 1017–1029 RSC.
- M. Champeau, D. A. Heinze, T. N. Viana, E. R. de Souza, A. C. Chinellato and S. Titotto, Adv. Funct. Mater., 2020, 30, 1910606 CrossRef CAS.
- J. Odent, S. Vanderstappen, A. Toncheva, E. Pichon, T. J. Wallin, K. Wang, R. F. Shepherd, P. Dubois and J. M. Raquez, J. Mater. Chem. A, 2019, 7, 15395–15403 RSC.
- D. Han, C. Yang, N. X. Fang and H. Lee, Addit. Manuf., 2019, 27, 606–615 CAS.
- S. L. M. Alexander, S. Ahmadmehrabi and L. T. J. Korley, Soft Matter, 2017, 13, 5589–5596 RSC.
- N. D. Wanasekara, L. E. Matolyak and L. T. J. Korley, ACS Appl. Mater. Interfaces, 2015, 7, 22970–22979 CrossRef CAS PubMed.
- D. Han, Z. Lu, S. A. Chester and H. Lee, Sci. Rep., 2018, 8, 1963 CrossRef PubMed.
- D. Han, C. Farino, C. Yang, T. Scott, D. Browe, W. Choi, J. W. Freeman and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 17512–17518 CrossRef CAS PubMed.
- H. Goodarzi Hosseinabadi, D. Nieto, A. Yousefinejad, H. Fattel, L. Ionov and A. K. Miri, Appl. Mater. Today, 2023, 30, 101721 CrossRef PubMed.
- Y. Bao, Macromol. Rapid Commun., 2022, 43, 2200202 CrossRef CAS PubMed.
- F. Puza and K. Lienkamp, Adv. Funct. Mater., 2022, 32, 2205345 CrossRef CAS.
- N. H. Tran, G. R. Dennis, A. S. Milev, G. S. K. Kannangara, P. Williams, M. A. Wilson and R. N. Lamb, J. Colloid Interface Sci., 2006, 297, 541–545 CrossRef CAS PubMed.
- Y. Kimura and K. Haraguchi, Langmuir, 2017, 33, 4758–4768 CrossRef CAS PubMed.
- M. M. Rana, A. Rajeev, G. Natale and H. De la Hoz Siegler, J. Mater. Res. Technol., 2021, 13, 769–786 CrossRef CAS.
- H. Tokuyama, N. Ishihara and S. Sakohara, Eur. Polym. J., 2007, 43, 4975–4982 CrossRef CAS.
- X. Wang, H. Huang, H. Liu, F. Rehfeldt, X. Wang and K. Zhang, Macromol. Chem. Phys., 2019, 220, 1800562 CrossRef.
- M. M. Pradas, G. Ribelles, A. S. Aroca, G. G. Ferrer, J. S. Antón and P. Pissis, Polymer, 2001, 42, 4667–4674 CrossRef.
- M. J. A. Hore, B. Hammouda, Y. Li and H. Cheng, Macromolecules, 2013, 46, 7894–7901 CrossRef CAS.
- H. G. Schild, M. Muthukumar and D. A. Tirrell, Macromolecules, 1991, 24, 948–952 CrossRef CAS.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- Y. Y. Liu, J. Lü and Y. H. Shao, Macromol. Biosci., 2006, 6, 452–458 CrossRef CAS PubMed.
- A. G. Andreopoulos, Biomaterials, 1989, 10, 101–104 CrossRef CAS PubMed.
- C. J. Thrasher, J. J. Schwartz and A. J. Boydston, ACS Appl. Mater. Interfaces, 2017, 9, 39708–39716 CrossRef CAS PubMed.
- Y. C. Huang, Q. P. Cheng, U. S. Jeng and S. H. Hsu, ACS Appl. Mater. Interfaces, 2023, 15, 5798–5810 CrossRef CAS PubMed.
- T. Yang, Y. Hu, C. Wang and B. P. Binks, ACS Appl. Mater. Interfaces, 2017, 9, 22950–22958 CrossRef CAS PubMed.
- H. Fan, D. Naohara, W. Li, X. Li and J. P. Gong, Polym. Chem., 2024, 15, 2104–2111 RSC.
- J. Zhou, D. Shi, T. Kaneko, W. Dong and M. Chen, Macromol. Rapid Commun., 2023, 2300488 Search PubMed.
- A. T. Gökçeören, B. F. Şenkal and C. Erbil, J. Polym. Res., 2014, 21, 370 CrossRef.
- I. Castilla Cortázar, A. Vidaurre, G. Gallego Ferrer, M. Monleón Pradas, J. L. Gómez Ribelles and J. M. Meseguer Dueñas, J. Non-Cryst. Solids, 2001, 287, 130–134 CrossRef.
- M. Přádný, M. Dušková-Smrčková, K. Dušek, O. Janoušková, Z. Sadakbayeva, M. Šlouf and J. Michálek, J. Polym. Res., 2014, 21, 579 CrossRef.
- C. G. Gomez, C. I. Alvarez Igarzabal and M. C. Strumia, Polymer, 2004, 45, 6189–6194 CrossRef CAS.
- F. Tsunomori and H. Ushiki, Phys. Lett. A, 1999, 258, 171–176 CrossRef CAS.
- X. Guo, Z. Liu, Z. Tong, N. Jiang and W. Chen, Environ. Technol., 2023, 44, 1691–1704 CrossRef CAS PubMed.
- X. Z. Zhang, Y. Y. Yang and T. S. Chung, Langmuir, 2002, 18, 2538–2542 CrossRef CAS.
- H. Warren, D. J. Shepherd, M. in het Panhuis, D. L. Officer and G. M. Spinks, Sens. Actuators, A, 2020, 301, 111784 CrossRef CAS.
- S. Pruksawan, Z. Lin, Y. L. Lee, H. L. Chee and F. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 46388–46399 CrossRef CAS PubMed.
- M. Li and J. Bae, Polym. Chem., 2020, 11, 2332–2338 RSC.
- Y. Kaneko, R. Yoshida, K. Sakai, Y. Sakurai and T. Okano, J. Membr. Sci., 1995, 101, 13–22 CrossRef CAS.
- S. Timoshenko, J. Opt. Soc. Am., 1925, 11, 233 CrossRef CAS.
- T. Morimoto and F. Ashida, Int. J. Solids Struct., 2015, 56–57, 20–28 CrossRef.
- J. Yoon, S. Cai, Z. Suo and R. C. Hayward, Soft Matter, 2010, 6, 6004 RSC.
- L. Hu, Q. Zhang, X. Li and M. J. Serpe, Mater. Horiz., 2019, 6, 1774–1793 RSC.
- Y. Ma, Y. Gao, L. Liu, X. Ren and G. Gao, Chem. Mater., 2020, 32, 8938–8946 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00032c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |