
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChitosan-based glycated polyampholyte nanogels for copper-catalysed Fenton-like reaction†
Yeonjoo
Jung
,
Eunseo
Lee
,
So-Lee
Baek
and
Sang-Min
Lee
 *
*
Department of Chemistry, The Catholic University of Korea, Bucheon, Gyeonggi-do 14662, Korea. E-mail: smlee120@catholic.ac.kr
First published on 10th April 2024
Abstract
For several decades, there has been great interest in the application of polysaccharide nanogels as nanoscale platforms that integrate polymer scaffolds to confer distinctive biochemical properties. Notably, nanogels, utilizing metal coordination as an efficient cross-linking strategy to enhance structural integrity and responsiveness, have emerged as nanoscale catalysts, broadening their utility in controlled drug delivery, sensing, and biomedical applications. In this study, we present a facile method for preparing chemically modified nanogels based on chitosan, facilitated by Cu(II) coordination for a Fenton-like reaction. The chitosan scaffold undergoes modifications through ethylenediaminetetraacetate (EDTA) conjugation and non-enzymatic glycation, yielding water-soluble structures across a wide pH range. Cu(II) chelation facilitates coordination-mediated cross-linking, resulting in the formation of nanogels with multiple Cu(II)-chelated domains that resemble artificial enzymes. The resulting Cu(II)-containing nanostructures exhibit altered catalytic activity attributed to the distinctive chemical environment of self-folded polysaccharide scaffolds. Spectroscopic monitoring reveals different kinetic pathways in Cu(II)-catalysed Fenton-like reactions mediated by self-folded polysaccharide-based nanostructures containing Cu(II)-chelating active sites. These results demonstrate the potential of polysaccharide nanogels as advanced materials with versatile functionalities in catalytic applications.
Introduction
Enzyme catalysis plays a crucial role in biological processes governed by intricate molecular mechanisms. An important aspect of enzyme catalysis lies in the self-folding protein structure, which plays a pivotal role in forming the active site of an enzyme.1 Therefore, the three-dimensional conformation of enzymes is finely tuned, facilitating a specific reaction at the active site through the interaction with versatile substrate molecules at the binding pocket. Moreover, metal cofactors further contribute to the catalytic prowess of enzymes, serving as essential components that orchestrate precise chemical transformations.2,3 Inspired by these natural systems, there is growing interest in the development of self-assembled nanostructures as artificial enzyme-like catalysts with important implications for sustainable chemistry.4–6 However, challenges still persist in achieving artificial smart nanocatalysts that replicate the specific functionalities of natural enzymes.7Nanogels, emerging as versatile nanoscale platforms, exhibit unique characteristics through the integration of polymer scaffolds, which imparts desirable properties.8 Specifically, the utilization of polysaccharides in nanogel construction enhances biocompatibility and provides a means for cost-effective mass production for various applications.9,10 As a key feature in nanogel design, metal coordination facilitates cross-linking of the polymer matrix, contributing to the structural integrity and responsiveness of the nanogels.11 Furthermore, with metal coordination for the catalysis of specific reactions, nanogels can play an essential role as nanoscale catalysts.12,13 As such, combined with the self-folding capability of nanogels, the synergy of catalytic functions further extends their applicability in controlled drug delivery, sensing, and other biomedical applications, which establishes nanogels as promising candidates for advanced materials with multifaceted functionalities.14
Herein, we demonstrate the facile formation of chemically modified chitosan-based nanogel structures mediated by the coordination of Cu(II) as a catalyst for Fenton-like reaction. For Cu(II) chelation, ethylenediaminetetraacetate (EDTA) is employed on the chitosan (CS) backbone through amide ligation, and the remaining glucosamine (GlcN) units are further modified by non-enzymatic glycation to improve the water solubility of the chitosan scaffold over a wide pH range. The subsequent Cu(II) chelation provides facile coordination-mediated cross-linking of polymer scaffolds, allowing the formation of nanogel structures with multiple Cu(II)-chelated domains (Scheme 1), similar to artificial enzymes with metal cofactors for specific catalytic reactions. More importantly, the resulting Cu(II)-containing nanostructures exhibit catalytic activity significantly modified from that of free Cu(II) due to the unique chemical environment of the self-folding polysaccharide scaffolds. Based on spectroscopic monitoring, Cu(II)-catalysed Fenton-like reaction can proceed via a completely different kinetic pathway with these nanogel catalysts, which further demonstrates the potential of these nanogels as advanced materials with versatile functionalities in catalytic applications.
 | ||
| Scheme 1 Preparation of Cu(II)-chelated nanogels with polyampholytic Glc-ECS scaffolds. | ||
Results and discussion
Preparation of polyampholytic ECS and Glc-ECS
Glycated, EDTA-tethered chitosan (Glc-ECS) was obtained by the two-step modification of native CS via carbodiimide-mediated peptide coupling of EDTA at pH 4–5 followed by in situ glycation of glucosamine (GlcN) units in aqueous solution (Scheme S1 in the ESI†). Compared to native CS, which is soluble only in acidic solutions, EDTA-tethered CS (ECS) showed improved solubility at pH 7, allowing facile glycation under neutral conditions. The initial EDTA modification was verified by 1H NMR and FT-IR analyses. In the 1H NMR spectrum in D2O, a new proton peak 2′ at 3.8 ppm appeared in ECS due to the amide formation by EDTA conjugation (Fig. 1A).15 Additionally, the H signal at C2′ (peak 3 at 2.9 ppm) of GlcN units in ECS was shifted to higher fields (peak 3′ at 3.1 ppm) by amide formation, further confirming EDTA conjugation.16 In FT-IR analysis, the presence of carbonyl groups in EDTA was confirmed by a stretching band at 1730 cm−1 in the ECS at pH 2 (Fig. 1B), while both asymmetric and symmetric COO− stretching bands (1587 and 1408 cm−1, respectively) were observed under basic conditions, which also confirmed successful EDTA conjugation.17 | ||
| Fig. 1 (A) 1H NMR spectra of ECS (red) and native CS (blue) in D2O. Also shown is the 1H NMR spectrum of free EDTA for comparison (green). The peak “Ac” is due to the N-acetyl moieties at GlcN residues in CS. (B) FT-IR spectra of ECS prepared under acidic (blue) and basic conditions (red). For comparison, the FT-IR spectrum of native CS is also shown (green). (C) pH titration curves of ECS (the molar ratio of EDTA modification, ε = 0.2 and 0.5) and glycated CS with 0.1 M aqueous NaOH. For comparison, the titration curves of native CS and free EDTA are also shown. Each sample was measured at 1.0 mg mL−1. *In native CS, the pH change above ∼6.5 cannot be measured because of significant precipitation. | ||
After the EDTA modification, the ECS solution was titrated with aqueous NaOH (0.1 M), and the subsequent pH change was monitored to confirm the polyampholytic structure of ECS. Compared to native CS, which shows a single plateau at pH 5.0–6.5 due to the proton-buffering activity of GlcN units (pKa ∼6.5),17 the ECS with 0.2 equiv. EDTA modification (ε = 0.2, compared to the GlcN units of the CS scaffold) exhibited significantly improved solubility with an equivalence point (Veq) of 0.87 mL NaOH (Fig. 1C). ECS with higher EDTA modification (ε = 0.5) showed a Veq of 1.06 mL at an increased volume of NaOH due to the high fraction of EDTA moieties, while glycated CS without EDTA conjugation, for comparison, required only a small amount of NaOH (Veq = 0.48 mL) for neutralization due to the reduced portion of free GlcN units by glycation. As such, pH titration analysis suggested a quantitative modification of the ECS depending on the EDTA feeding dose, resulting in a polyampholyte structure of the ECS after EDTA conjugation.
To protect the reactive GlcN units of the CS scaffold, non-enzymatic glycation of the ECS was achieved with aqueous glucose by Maillard reaction, where reducing sugars can react with versatile amino-containing molecules such as amino acids and proteins. It has been suggested that during the reaction, several reaction intermediates are generated through Schiff base formation, which eventually exhibits characteristic fluorescence and UV-vis absorbance.18 In the early stages of the reaction, initial fluorescent intermediates are formed, typically with an absorbance of 294 nm, while the fluorescence attenuates with a development of brown colour as the final melanoidin compounds are formed through the Amadori rearrangement.19 As such, ECS was incubated at 60 °C for 24 h with an excess of glucose (∼10-fold molar ratio relative to GlcN units), and subsequent fluorescence emission was monitored in aqueous solution. As shown in Fig. 2A and Fig. S1,† fluorescence emission peaks at 390 and 415 nm developed with the corresponding fluorescence excitation observed at 340 nm (Fig. 2A, inset), indicating the progress of the Maillard reaction for the glycation of the ECS scaffold.20 Additionally, the absorption band at 294 nm also confirmed glycation in the ECS by Maillard reaction (Fig. 2B).19 After incubation for an additional 24 h, however, the colour of the solution changed to brown due to the formation of melanoidin, a late-stage advanced glycation end-product (Fig. 2B, inset).
 | ||
| Fig. 2 (A) Fluorescence emission spectra of Glc-ECS, depending on the feeding molar ratio of Glc to GlcN units in CS for glycation (λEx = 290 nm). Inset shows the corresponding fluorescence excitation spectra (λEm = 390 nm). (B) UV-vis absorption changes of Glc-ECS by glycation. Inset shows the Glc-dependent color change of Glc-ECS (5.0 mg mL−1) by melanoidin formation after 48 h. (C) pH-dependent zeta potential changes of ECS and glycated ECS. Isoelectric points (pI) of each sample are presented in the legend. (Inset) pH-dependent transmittance (%T) changes of aqueous ECS, Glc-ECS, and native CS measured at 600 nm by UV-vis spectroscopy. | ||
Given the zwitterionic nature of the polyampholytic ECS, pH-dependent charge conversion of the ECS was observed through zeta potential measurements in aqueous solution.21 The isoelectric point (pI) of ECS (ε = 0.2) was observed at pH 5.66, while ECS of ε = 0.5 showed a lower pI at 4.57 (Fig. 2C) consistent with a higher level of anionic EDTA modification. Glycation of the ECS led to further decrease of pI to 4.36, confirming that further neutralization of the cationic GlcN units in Glc-ECS was achieved by glycation. Although the ECS (ε = 0.2) solution became turbid under isoelectric conditions (Fig. 2C, inset), as expected by DLVO theory, subsequent glycation further increased the overall solubility of Glc-ECS, indicating that highly improved aqueous solubility was achieved for Glc-ECS even under isoelectric conditions.
Glc-ECS can be utilized as a polymer scaffold for further metal chelation via EDTA ligands.15,22 As such, an equimolar amount of aqueous Cu(II) relative to the GlcN units of Glc-ECS was added and agitated at room temperature for 48 h for chelation. To avoid significant precipitation due to interchain cross-linking, the chelation reaction was performed under slightly diluted conditions (∼1 mg mL−1).23 In contrast to the native CS in aqueous solution, which showed significant precipitation upon Cu(II) addition under the same conditions, Glc-ECS maintained excellent solubility in aqueous Cu(II) solution (Fig. 3A, inset). After the purification of unreacted Cu(II) ions by centrifugal filtration, the Glc-ECS/Cu complex exhibited characteristic absorption peaks at 260 and 720 nm, which were consistent with the Cu-to-acetate metal to ligand charge transfer (MLCT) and d–d transition of Cu(II), respectively (Fig. 3A).24,25 FT-IR analysis also confirmed the Cu(II) chelation to Glc-ECS by shifting the characteristic stretching bands of the carboxylate ligand in EDTA moieties. Upon Cu(II) chelation, the separation frequency (Δν) between the symmetric and asymmetric stretching bands of carboxylates in EDTA increased significantly from 180 to 230 cm−1 (Fig. 3B), which is similar to that of the previously reported metal complexes with carboxylate ligands.17
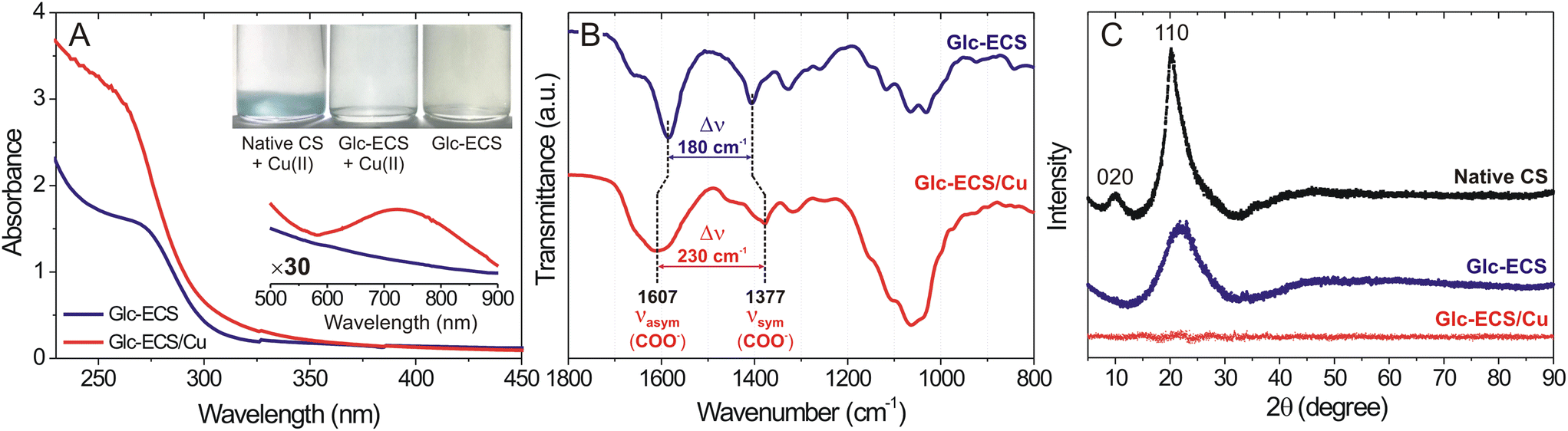 | ||
| Fig. 3 (A) UV-vis absorption spectra of Glc-ECS (blue) and Glc-ECS/Cu (red). Inset shows ×30-magnified spectra at 500–900 nm to verify the absorption peak of Cu(II) at 740 nm. The digital photographic image confirms no precipitation observed after Cu(II) chelation to Glc-ECS. (B) FT-IR spectra show the increase of Δν after Cu(II) chelation. (C) Powder X-ray diffraction (XRD) analysis of Glc-ECS and Glc-ECS/Cu. | ||
Cu(II) chelation was further confirmed through powder X-ray diffraction (XRD) analysis. Due to the crystal structure of native CS by hydrogen bonding, strong diffraction peaks by (110) and (020) reflections were observed at 20.1° and 9.9°, respectively (Fig. 3C).26 However, EDTA conjugation and subsequent glycation significantly reduced the peak intensity at 20.1°, resulting in a substantial increase in full width at half-maximum (FWHM) from 0.1 to 18.8 and the complete absence of 020 reflection, indicating that the crystallinity of Glc-ECS was reduced by chemical modifications.26,27 In particular, the diffraction peak from the 110 reflection completely disappeared after Cu(II) chelation due to the disruption of the crystal structure of Glc-ECS/Cu, as observed previously.26,27 Taken together, facile Cu(II) chelation for Glc-ECS was successfully achieved in aqueous solution.
Thermal degradation studies
Cu(II) chelation on the polymer scaffold significantly modified the thermal stability of Glc-ECS as observed by thermogravimetric analysis (TGA) and corresponding differential thermogravimetric (DTG) profiles. Compared to glycated CS, which exhibited a single degradation temperature (Td) at 256 °C (Fig. 4A) similar to native CS,28 Glc-ECS showed a two-step degradation process with an additional Td observed at 305 °C (Fig. 4B), presumably due to EDTA conjugation and possible cross-linking inside the Glc-ECS scaffold. After Cu(II) chelation, however, the second Td substantially decreased to 278 °C without a significant effect on the first Td (Fig. 4C). This decrease of the second Td clearly indicates that the thermal degradation was promoted by the known catalytic activity of Cu(II) for thermolysis at the chelating EDTA tethered on Glc-ECS.29 After complete degradation, a Cu(II) loading of approximately 17 wt% in Glc-ECS/Cu was estimated by comparing the final residues at 900 °C.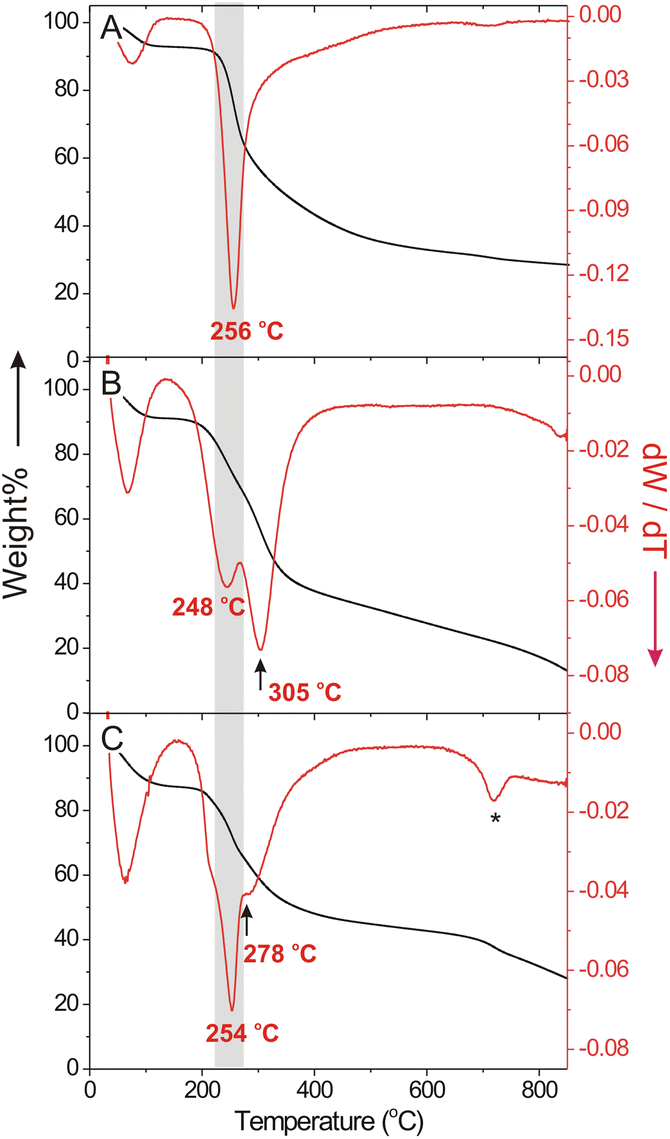 | ||
| Fig. 4 TGA thermogram and differential thermogravimetric (DTG) curves of (A) glycated CS, (B) Glc-ECS, and (C) Glc-ECS/Cu. Asterisk at 718 °C indicates the decomposition of residual sulfate ions from CuSO4. | ||
Preparation of Cu(II)-chelated nanoparticles and viscosity analyses
Given that metal coordination can be exploited as a facile cross-linking strategy for polymer-based nanogel formation,11 the hydrodynamic volume change due to Cu(II) chelation was monitored by dynamic light scattering (DLS). Compared to Cu(II)-free Glc-ECS, which exhibited a hydrodynamic diameter (DH) of 232 ± 73 nm in an intensity-based value, the DH of Glc-ECS/Cu decreased to 128 ± 45 nm after Cu(II) chelation and the polydispersity index (PdI) also decreased from 0.477 to 0.251 (Fig. 5A and Fig. S2†), indicating that compact nanogel particles with narrow dispersities were obtained by Cu(II) chelation. Transmission electron microscopy (TEM) observation also confirmed the DLS results (Fig. 5B and Fig. S3†), showing spherical particles of 83 ± 16 nm and multiple Cu(II)-chelated domains formed in Glc-ECS/Cu (Fig. S4†).30 Interestingly, XPS analysis revealed significant reduction of Cu(II) to Cu(I) in Glc-ECS/Cu (Fig. 5C and Fig. S5†). Strong satellite peaks in the Cu 2p spectrum are typically observed in the Cu(II) state, whereas Cu(I) complexes have been reported to display only subtle satellites.31,32 Hence, it is highly anticipated that the reducing Glc moieties in Glc-ECS can reduce Cu(II) during the chelation reaction. Consistent with the cationic metal binding to the anionic polymer scaffold,33 the zeta potential was significantly neutralized (ζ = −1.91 mV) after Cu(II) chelation (Fig. 5D), which further confirmed the successful formation of Glc-ECS/Cu nanogels. The resulting nanogels exhibited excellent colloidal stability in aqueous solution for 1 month, while moderate aggregation was observed after lyophilisation (Fig. S6†).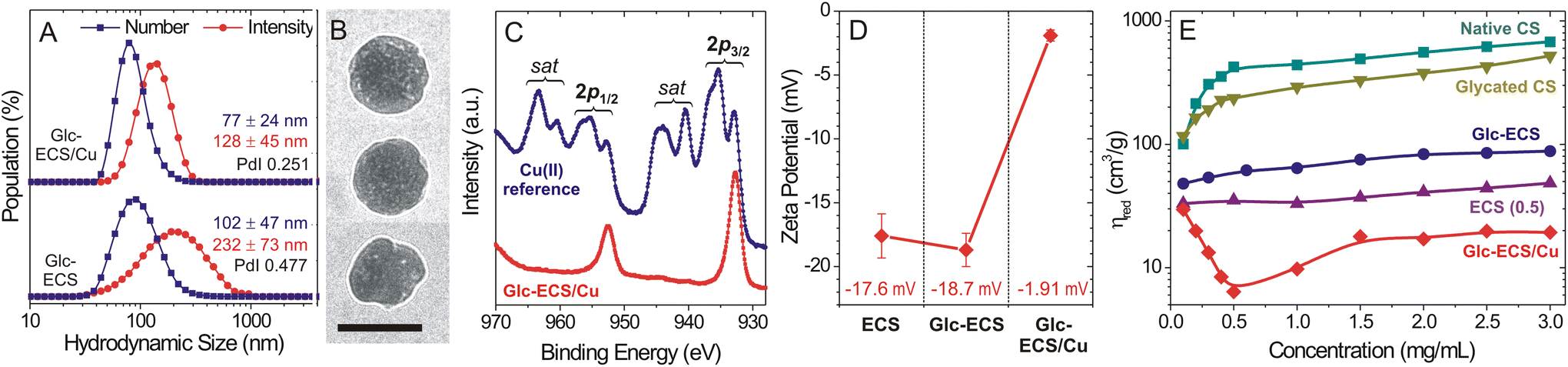 | ||
| Fig. 5 (A) Hydrodynamic size distribution of Glc-ECS before and after the Cu(II) chelation. (B) TEM image of Glc-ECS/Cu (scale bar = 100 nm). (C) XPS analysis of the Cu 2p region in Glc-ECS/Cu. CuSO4 was measured with Cu(II) as a reference and ‘sat’ indicates the satellite peaks in Cu(II). (D) Zeta potentials of ECS, Glc-ECS, and Glc-ECS/Cu measured at pH 7.0. (E) Viscosity analysis of CS derivatives measured in 20 mM acetate buffer (pH 4.0) containing 100 mM NaCl at room temperature. | ||
Chemical modification and Cu(II) chelation significantly changed the viscosity of the diluted sample solutions. When the reduced viscosity (ηred) of aqueous CS derivatives was estimated under acidic conditions (pH 4.0, 100 mM NaCl) at 25 °C (Fig. 5E), native CS showed a steady increase in ηred as a function of concentration according to the Huggins relationship, but a sharp decrease in ηred was observed under highly diluted conditions (<0.5 mg mL−1), which is a typical property of the semiflexible polyelectrolytes.34 Compared with native CS, glycated CS showed a slight decrease in viscosity in the same concentration range, while the viscosity of ECS decreased sharply under the same measurement conditions possibly due to partial intra-polymer cross-linking within the ECS domains.23,28 For both ECS and Glc-ECS, the sharp deviation from the Huggins relationship under diluted conditions mostly disappeared, despite a slight increase in the viscosity of Glc-ECS, which can be attributed to the enhanced entanglement of the polymer scaffold by glycation. More importantly, Cu(II) chelation induced a completely different rheological behaviour in Glc-ECS/Cu, showing a further reduction in viscosity at intermediate concentrations due to the formation of compact domains inside the nanogels consistent with DLS/TEM results. Under highly diluted conditions, however, Cu(II) ions can reversibly dissociate from the polymer scaffold (Fig. S7†), eventually resulting in the nanogels swelling and undergoing an increase in their viscosity close to that of Glc-ECS.17
Catalytic activity of Glc-ECS/Cu for Fenton-like reaction
To investigate the catalytic activity of Glc-ECS/Cu, we monitored the degradation rate of methylene blue (MB) as an organic substrate for Cu(II)-catalysed Fenton-like reaction. To this end, we used 40 μM MB and 0.6 mM Cu catalyst (Glc-ECS/Cu or free Cu(II) ions) with H2O2 (80 or 120 mM), which generated a final solution pH of ∼5.8, requiring no further pH adjustment for the dissolution of aqueous Cu(II). The temporal degradation of MB was then monitored by UV-vis absorption at 25 °C (Fig. 6A and D). As previously reported, pseudo-first-order kinetics were obtained with the free Cu(II) catalyst when the reaction progress was plotted by monitoring the absorbance of MB at 664 nm (Fig. 6B and C).35,36 Although the overall kinetics of Cu(II)-catalysed Fenton-like reactions are governed by a relatively slow Cu(II) reduction step, which generally exhibits second-order kinetics,37 apparent pseudo-first order kinetic results were observed in our study, presumably due to modifications from the optimal reaction conditions.38,39 | ||
| Fig. 6 UV-vis analysis of MB decomposed by (A–C) free Cu(II)- and (D–F) Glc-ECS/Cu-catalyzed Fenton-like reaction under the conditions of [Cu2+] = 0.6 mM and [MB] = 40 μM at 25 °C. (A and D) Time-dependent change of UV-vis absorption spectra of MB with 80 mM H2O2. (B) First-order fit of free Cu(II)-catalyzed reaction. (C) Second-order fit of free Cu(II)-catalyzed reaction deviates from linearity. (E) Second-order fit of Glc-ECS/Cu-catalyzed reaction. (F) Reaction kinetics at pH 7.0 and 10 mM H2O2. Inset shows the enhanced stability of Glc-ECS/Cu before and after the Fenton-like reaction at pH 7.0. | ||
In stark contrast, Glc-ECS/Cu showed significant deviation from the first-order fit (Fig. S8†), whereas second-order kinetics were apparently observed by linear regression (Fig. 6E). This different kinetics can be attributed to the local microenvironment of the Cu(II)-chelated catalytic site inside the self-folded polysaccharide scaffold of Glc-ECS/Cu, which is not present in free Cu(II) ions.24,40 Generally, EDTA is often used as a quencher in Fenton-like reactions38 due to its multivalent chelating effect, which can block all active sites of the Cu(II) catalyst and retard the reduction of Cu(II), eventually reducing the overall reaction rate as shown in Fig. 6B.38 Nevertheless, the observed catalytic activity for Glc-ECS/Cu confirms the presence of reactive sites on Cu(II) chelated to EDTA tethered to the polymer scaffold. Additionally, the Glc-mediated reduction of Cu(II) in Glc-ECS/Cu, as observed in Fig. 5C, can further facilitate the reactions, resulting in the altered kinetics from that of free Cu(II). Indeed, the addition of methanol as a radical scavenger substantially reduced the reaction rate of free Cu(II), showing a 33.1% reduced catalytic activity (Fig. 6B),41 whereas relatively small effects were observed with the addition of methanol in the Glc-ECS/Cu-catalysed reaction (Fig. 6E).
Finally, the degradation of MB was monitored at low H2O2 (10 mM) and pH 7, where the consumption of H2O2 can be accelerated by the enhanced kinetics of the Fenton-like reactions.35 Under these conditions, no significant precipitation was observed for Glc-ECS/Cu, in stark contrast to that with free Cu(II), which substantially precipitated out of the solution due to the formation of unstable Cu(I) intermediates during the reaction (Fig. 6F, inset). Interestingly, two-step reaction kinetics were observed, showing an initial rapid phase followed by a slow steady-state phase (Fig. 6F and Fig. S9†). These kinetic features can be possibly attributed to the burst generation of hydroxyl radicals in the initial stage, while the second stage can be characterized by a diffusion-controlled steady state to recover the consumed H2O2 around the catalyst after an induction period.42 These results demonstrate the potential capability of Glc-ECS/Cu as an artificial enzyme-like nanocatalyst for Fenton-like reactions.
Conclusions
We demonstrated the development of polysaccharide nanogels as an advanced nanoscale platform for catalytic applications. The incorporation of EDTA and subsequent glycation for the chemical modification of native CS result in improved water solubility over a wide pH range and enhance the versatility of nanogels by exploiting metal coordination. In particular, Cu(II) chelation has been proven to be an efficient cross-linking strategy for enhancing the structural integrity and catalytic activity for Fenton-like reaction. The resulting nanostructures exhibited multiple Cu(II)-chelated domains similar to artificial enzymes, and showed significantly modified catalytic activity attributed to the distinctive chemical environment of self-folded polysaccharide scaffolds. Given that these results can contribute towards broadening our understanding of the design and applications of polysaccharide nanogels, the demonstrated ability to tailor their properties through chemical modification and metal coordination for catalysis suggests the potential of these nanogels to serve as highly adaptable and efficient nanocatalysts in diverse biomedical and technological contexts. Further modification of various stimuli-responsive functional groups can make these nanogels smart catalytic systems with great potential.43,44Experimental section
Materials
Chitosan (low molecular weight, Mν = 70 kDa estimated, 75–85% deacetylation), N-(3-dimethylaminopropyl)-N′-ethyl-carbodiimide hydrochloride (EDC, commercial grade, powder), ethylenediamine-tetraacetic acid (EDTA, ACS reagent), and copper(II) sulfate pentahydrate (ACS reagent, ≥98.0%) were purchased from Aldrich Chemical Co. (Milwaukee, WI) and used without further purification. Regenerated Cellulose (RC) dialysis membranes were purchased from Spectrum Laboratories (Thermo Scientific, Waltham, MA). Deionized (DI) water was obtained from Human Power I+ Scholar-UV (Human Corporation, Seoul, Korea) (18.2 MΩ cm resistivity). Native CS was dissolved in pH 2 aqueous solution for complete dissolution.General methods and synthetic procedures
Instrumental analyses were carried out at the Cooperative Center for Research Facilities of the Catholic University of Korea. Fourier-transform nuclear magnetic resonance (NMR) spectroscopy was carried out on an AVANCE III 300 MHz spectrometer. The chemical shifts of 1H NMR spectra are reported in ppm against residual solvent resonance as the internal standard (DMSO-d6 = 2.5 ppm; D2O = 4.8 ppm). Measurements were performed after dissolving 10 ± 1 mg of sample in 0.7 mL deuterated solvent in a 5 mm NMR tube. DLS and zeta (ζ) potential measurements were carried out with a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Unless noted otherwise, all samples were dispersed in 10 mM phosphate buffer (pH 7.4) for measurements. The data reported represent an average of 10 measurements with five scans each. Thermogravimetric analysis (TGA) was carried out with an Auto Thermogravimetric Analyzer (TGA 2) (Mettler-Toledo, Columbus, USA) under a nitrogen (N2) atmosphere. The samples (5 mg) were heated from 30 to 850 °C at a constant heating rate of 10 °C min−1. XRD data were generated using a Miniflex 600 X-ray diffractometer (Rigaku, Austin, TX, USA) with Cu-Kα radiation at λ = 1.5405 Å. The samples were observed in the range 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ = 3–90° and at a rate of 10° min−1.
θ = 3–90° and at a rate of 10° min−1.
Ultraviolet-visible (UV-vis) absorption spectra were obtained using a Lambda 35 spectrophotometer (PerkinElmer, Waltham, MA). Fluorescence emission/excitation spectra were obtained on a FluoroMate FS-2 fluorometer (Scinco Co., Ltd. Seoul, Korea) at a scan speed of 100 nm min−1 (λEx = 290 nm, λEm = 390 nm, slit width = 2.5 nm). Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy measurements were carried out with a Bruker TENSOR II FTIR spectrometer equipped with a Miracle Single Reflection Horizontal ATR accessory (PIKE Technologies, Inc., Madison, WI). The solution viscosity was measured with an Ubbelohde viscometer (SI Analytics GmbH, Mainz, Germany). For the measurements, a dilute solution of each sample was prepared by dissolving the samples in 20 mM acetate buffered solution (pH 4.0, 100 mM NaCl), and the measurements were carried out at 25 °C.
Transmission Electron Microscopy (TEM) was carried out with a Talos L120C (FEI, Brno, Czech Republic) with a beam voltage of 120 kV and a slow-scan charge-coupled device at the National Instrumentation Center for Environmental Management in Seoul National University. The sample solution (20 μL) was placed on a copper grid (300 mesh) for 30 s, and the excess solution was blotted with filter paper. The grid was dried at room temperature for 24 h. The samples were observed without staining. High Resolution Scanning Electron Microscopy (HRSEM) observation was carried out with a HITACHI SU8220 (Hitachi, Tokyo, Japan) at Korea Basic Science Institute (KBSI, Metropolitan Seoul Center). X-ray photoelectron spectroscopy (XPS, ESCALAB 250) measurements were performed with an AXIS UltraDLD system (Kratos Analytical, Manchester, UK) using a monochromatic Al Kα X-ray source (15 keV, 150 W) under a base pressure of 3 × 10−9 Torr at Korea Basic Science Institute (KBSI, Busan Center). The samples were concentrated by centrifugal filtration (100k MWCO), drop-cast onto a Si wafer and dried overnight in the dark.
where η and ηs are the viscosity of the nanogel solution and solvent, respectively, which can be also derived from the ratio of the corresponding flow times in the viscometer. The specific viscosity (ηsp) of the nanogel solution was obtained from the following equation

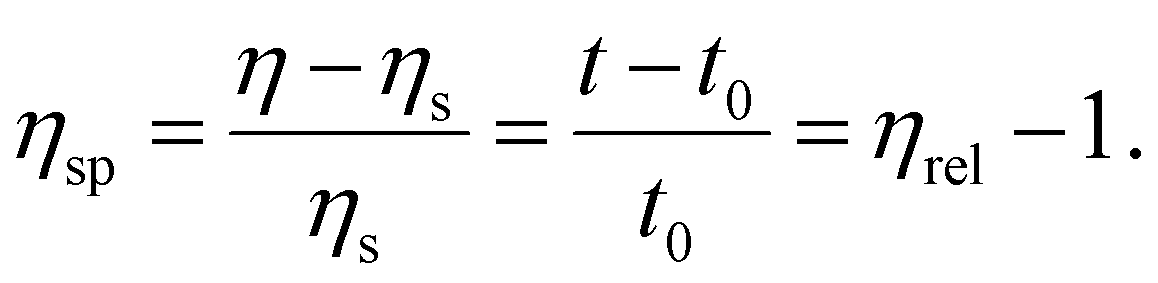
Then, the reduced viscosity (ηred) of the nanogel solution was obtained from the following equation

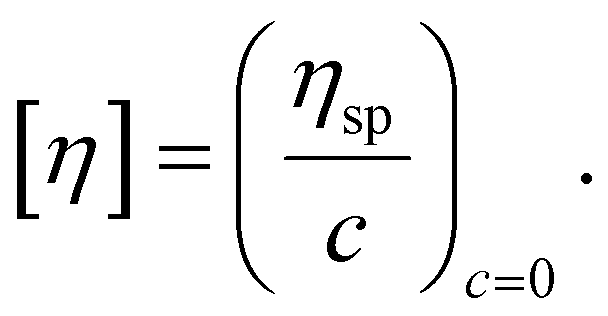
The intrinsic viscosity was employed to determine the Mν of native chitosan using the reported Mark–Houwink parameters (a = 0.78, K = 58.5 × 10−5 dL g−1).45
Author contributions
All authors have given approval to the final version of the manuscript. Credit: Yeonjoo Jung: investigation (lead); formal analysis; data curation (lead); and writing – review & editing; Eunseo Lee: investigation (supporting); data curation (supporting); validation; and writing – review & editing; So-Lee Baek: methodology and writing – review & editing; and Sang-Min Lee: conceptualization; writing – original draft; writing – review & editing; supervision; funding acquisition; and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government, MSIT (no. NRF-2022R1A2C2004768). Korea Basic Science Institute (KBSI) is acknowledged for SEM (Metropolitan Seoul Center) and XPS analysis (Busan Center).References
- S. S. Chaturvedi, D. Bím, C. Z. Christov and A. N. Alexandrova, Chem. Sci., 2023, 14, 10997–11011 RSC.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- C. Adamson and M. Kanai, Org. Biomol. Chem., 2021, 19, 37–45 RSC.
- J. Shi, Y. Wu, S. Zhang, Y. Tian, D. Yang and Z. Jiang, Chem. Soc. Rev., 2018, 47, 4295–4313 RSC.
- B. Chang, L. Zhang, S. Wu, Z. Sun and Z. Cheng, Chem. Soc. Rev., 2022, 51, 3688–3734 RSC.
- W. Xu, Y. Wu, W. Gu, D. Du, Y. Lin and C. Zhu, Chem. Soc. Rev., 2024, 53, 137–162 RSC.
- Y. Lyu and P. Scrimin, ACS Catal., 2021, 11, 11501–11509 CrossRef CAS.
- A. Scotti, M. F. Schulte, C. G. Lopez, J. J. Crassous, S. Bochenek and W. Richtering, Chem. Rev., 2022, 122, 11675–11700 CrossRef CAS PubMed.
- H. Wang, J. Qian and F. Ding, J. Mater. Chem. B, 2017, 5, 6986–7007 RSC.
- A. Plucinski, Z. Lyu and B. V. K. J. Schmidt, J. Mater. Chem. B, 2021, 9, 7030–7062 RSC.
- X. Zhang, Y. Tang, P. Wang, Y. Wang, T. Wu, T. Li, S. Huang, J. Zhang, H. Wang, S. Ma, L. Wang and W. Xu, New J. Chem., 2022, 46, 13838–13855 RSC.
- Z. Wei, H. Duan, G. Weng and J. He, J. Mater. Chem. C, 2020, 8, 15956–15980 RSC.
- Á. Molnár, Coord. Chem. Rev., 2019, 388, 126–171 CrossRef.
- X. Zhang, G. Li, G. Chen, D. Wu, Y. Wu and T. D. James, Adv. Funct. Mater., 2021, 31, 2106139–2106139 CrossRef CAS.
- S. Fujita and N. Sakairi, RSC Adv., 2016, 6, 10385–10392 RSC.
- X. Shen, L. Zhang, X. Jiang, Y. Hu and J. Guo, Angew. Chem., Int. Ed., 2007, 46, 7104–7107 CrossRef CAS PubMed.
- R. H. Kang, J. Y. Kwon, Y. Kim and S. M. Lee, Langmuir, 2017, 33, 9091–9099 CrossRef CAS PubMed.
- E. H. Ajandouz, V. Desseaux, S. Tazi and A. Puigserver, Food Chem., 2008, 107, 1244–1252 CrossRef CAS.
- Z. Jiang, L. Wang, W. Wu and Y. Wang, Food Chem., 2013, 141, 3837–3845 CrossRef CAS PubMed.
- X. Xia, T. Zhou, J. Yu, H. Cui, F. Zhang, K. Hayat, X. Zhang and C. T. Ho, J. Agric. Food Chem., 2023, 71, 8569–8579 CrossRef CAS PubMed.
- P. Xu, G. Bajaj, T. Shugg, W. G. Van Alstine and Y. Yeo, Biomacromolecules, 2010, 11, 2352–2358 CrossRef CAS PubMed.
- A. Shahzad, W. Miran, K. Rasool, M. Nawaz, J. Jang, S. R. Lim and D. S. Lee, RSC Adv., 2017, 7, 9764–9771 RSC.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS PubMed.
- S. Thanneeru, S. S. Duay, L. Jin, Y. Fu, A. M. Angeles-Boza and J. He, ACS Macro Lett., 2017, 6, 652–656 CrossRef CAS PubMed.
- T. Baran, I. Sargin, M. Kaya and A. Menteş, Carbohydr. Polym., 2016, 152, 181–188 CrossRef CAS PubMed.
- Y. Wang, A. Pitto-Barry, A. Habtemariam, I. Romero-Canelon, P. J. Sadler and N. P. E. Barry, Inorg. Chem. Front., 2016, 3, 1058–1064 RSC.
- S. L. Baek, Y. Kim, Y. Jang and S. M. Lee, ACS Macro Lett., 2022, 11, 1129–1135 CrossRef CAS PubMed.
- S. Dubinsky, G. S. Grader, G. E. Shter and M. S. Silverstein, Polym. Degrad. Stab., 2004, 86, 171–178 CrossRef CAS.
- J. A. Pomposo, A. J. Moreno, A. Arbe and J. Colmenero, ACS Omega, 2018, 3, 8648–8654 CrossRef CAS PubMed.
- T. M. Ivanova, K. I. Maslakov, A. A. Sidorov, M. A. Kiskin, R. V. Linko, S. V. Savilov, V. V. Lunin and I. L. Eremenko, J. Electron Spectrosc. Relat. Phenom., 2020, 238, 146878–146878 CrossRef CAS.
- K. Roy and C. S. Gopinath, Anal. Chem., 2014, 86, 3683–3687 CrossRef CAS PubMed.
- H.-W. Shin, H. Sohn, Y.-H. Jeong and S.-M. Lee, Langmuir, 2019, 35, 6421–6428 CrossRef CAS PubMed.
- H. Yang, Q. Zheng and R. Cheng, Colloids Surf., A, 2012, 407, 1–8 CrossRef CAS.
- H. J. Lee, H. Lee and C. Lee, Chem. Eng. J., 2014, 245, 258–264 CrossRef CAS.
- W. Yuan, C. Zhang, H. Wei, Q. Wang and K. Li, RSC Adv., 2017, 7, 22825–22835 RSC.
- A. N. Pham, G. Xing, C. J. Miller and T. D. Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS.
- X. Yuan, A. N. Pham, G. Xing, A. L. Rose and T. D. Waite, Environ. Sci. Technol., 2012, 46, 1527–1535 CrossRef CAS PubMed.
- J. F. Perez-Benito, J. Inorg. Biochem., 2004, 98, 430–438 CrossRef CAS PubMed.
- N. K. Pandey, H. B. Li, L. Chudal, B. Bui, E. Amador, M. B. Zhang, H. M. Yu, M. L. Chen, X. Luo and W. Chen, Mater. Today Phys., 2022, 22, 100587 CrossRef CAS.
- L. Wang, B. Li, D. D. Dionysiou, B. Chen, J. Yang and J. Li, Environ. Sci. Technol., 2022, 56, 3386–3396 CrossRef CAS.
- K. A. Johnson, FEBS Lett., 2013, 587, 2753–2766 CrossRef CAS PubMed.
- J. M. Park, M. H. Choi, E. Lee and S. M. Lee, ACS Appl. Polym. Mater., 2023, 5, 7411–7419 CrossRef CAS.
- N. Morimoto, S. Hirano, H. Takahashi, S. Loethen, D. H. Thompson and K. Akiyoshi, Biomacromolecules, 2013, 14, 56–63 CrossRef CAS PubMed.
- M. R. Kasaai, Carbohydr. Polym., 2007, 68, 477–488 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00055b |
| This journal is © The Royal Society of Chemistry 2024 |