
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSonication labile PEG-based hydrogel system for biological component suspension and subsequent degradation†
Meagan N.
Arguien
a,
Joshua T.
Kamps
b,
Sarah A.
Muth
c,
Marianela
Trujillo-Lemon
d and
Christopher N.
Bowman
 *ad
*ad
aMaterials Science & Engineering Program, University of Colorado, Boulder, Colorado 80309, USA. E-mail: Christopher.Bowman@colorado.edu
bDepartment of Chemistry, University of Colorado, Boulder, Colorado 80309, USA
cDepartment of Molecular, Cellular & Developmental Biology, University of Colorado, Boulder, Colorado 80309, USA
dDepartment of Chemical and Biological Engineering, University of Colorado, Boulder, Colorado 80309, USA
First published on 23rd September 2024
Abstract
This work synthesizes poly(ethylene glycol) (PEG) macromers that incorporate a phthalaldehyde moiety located at crosslink junctions. This location facilitates the severing of the polymer network when bond scission of the phthalaldehyde unit occurs with mechanical stimulation. As these networks degrade, the mechanical properties are analyzed to better understand how sonication driven degradation affects a polymer network – specifically looking at the degradation profile in hydrogel systems through the mass loss and storage modulus profiles. Comparison of hydrogels containing phthalaldehyde units with hydrogels that do not have the mechanophore pre- and post-mechanical stimulation provides evidence that the incorporation of the mechanophore at these crosslink junctions reduces the storage modulus by a factor of ten and results in greater than 90% decrease of the gel mass after 15 minutes of probe sonication, leading to breaking the network into soluble daughter fragments. The network degradation conditions of these hydrogels are shown to be compatible with biological component suspension and release for applications such as localized payload release.
Introduction
Degradable hydrogel systems have the potential to serve in a multitude of applications, ranging from drug delivery1–3 and tissue engineering to scaffolding in additive manufacturing applications2,4 due to their high water content, mechanical properties, and biocompatibility. These material properties are derived from crosslinked (either covalently or via noncovalent intermolecular attractions) hydrophilic polymers that are capable of retaining a significant amount of water in an aqueous environment.1,5,6The extensive use of hydrogels has resulted in an increase in methods for their controlled degradation to facilitate material removal at the end of the intended service life or the transition of a material's mechanical properties at various points throughout the application to better suit a changing or evolving environment.7–9 Through adaptations in the backbone of various hydrogel systems, triggerable degradability can be ‘built in’ to a network. Specifically, degradation can be triggered through various means including applications of thermal,10–12 chemical,7,13–16 mechanical,17,18 photo,5,14,19–27 and biological stimuli.28–33 For example, hydrolytic degradation of hydrogels has become a particularly favorable mechanism for biological applications. This means of degradation gained traction in the 1970s with the study of poly(lactones)34 that evolved into work in the early 2000s where poly(caprolactones) were utilized in the synthesis of drug delivery materials with biocompatible and tunable mechanical properties whose degradation kinetics could be varied through copolymerizations that provide a range of crystallinity and hydrophilicity of the polymer network.35–40 Notably, thiol–ene networks polymerized through a radical-mediated photochemistry result in step-growth networks that have been shown to be more homogeneous and also provide additional spatiotemporal control to the polymerization in comparison to non-photopolymerized step growth polymerizations such as the non-photo triggered Michael-type or the thermally initiated while still providing polymer degradability when functionalized appropriately.13,41,42 However, the lack of spatiotemporal control over the degradation process of hydrolytically degradable systems has given rise to numerous networks that degrade via photo triggers where both chain and step growth systems have been analyzed for applications ranging from drug delivery and cell scaffolding or encapsulation to hydrogel photoresists.5,20,21,26,27,43–45 Spatiotemporal control of polymer degradation is necessary in many applications where localized material property changes are desired. This approach has been applied in tissue engineering with the selective degradation of scaffolding or variation of material mechanical properties to direct cell growth30,31 and also with the release of a targeted payload at specific locations.10,32,46
As applications for hydrogels become increasingly complex, the need for environmentally sensitive degradation mechanisms grows accordingly. The scope of hydrogel degradation has benefited from the increasing interest in both degradable and chemically recyclable polymers. Among the less explored options for degradation is the integration of mechanophores in polymer networks to degrade through force driven bond cleavage. Mechanical degradation provides spatiotemporal control with the ability to target a location by focusing sound waves (such as ultrasonic) which are sustainable even in applications with optically opaque barriers where other degradation methods – such as photodegradation – have diminishing control. Ultrasound resolution is directly related to the wavelength used which is dictated by the speed of sound through a given medium and the frequency of the sound waves, resulting in wavelengths on the order of tens of millimeters for frequencies 1–30 MHz.47 Additionally, methods of mechanical degradation such as sonochemical degradation are particularly useful in stimuli sensitive biomedical contexts due to the compatibility of many biological environments with ultrasound.48 Mechanically degradable hydrogel systems that employ covalent mechanophores have been explored for use in force-reporting systems such as with spiropyran–merocyanine transitions.49 However, most of the mechanical dissolution methodologies for a hydrogel network have been achieved by cleavage of physical rather than covalent crosslinks. Phthalaldehydes offer a unique opportunity in network integration as they have been previously used in a poly-mechanophore system that was shown to degrade into monomer units upon sonication stimulation.17 Recently, an allyl-functionalized phthalaldehyde derivative was used in a poly(phthalaldehyde) copolymer as a crosslinker to generate a sonication-labile thiol–ene network,50 highlighting the potential of incorporating poly(phthalaldehyde)s into thermosets to confer mechanical material responses. These units, however, have yet to be incorporated into a hydrogel network successfully due to their hydrophobic character.
Here, a multi-arm poly(ethylene glycol)-based hydrogel is prepared with a phthalaldehyde unit built into the network near crosslink junctions to facilitate the degradation of a hydrogel system upon exposure to sonication. The sensitivity and stability of these networks is investigated, and payload release is demonstrated after mechanical stimulation of the hydrogel.
Experimental section
Materials
The PEG macromers, 4-arm 10K PEG-OH and 4-arm 10K PEG-SH, were purchased from JenKem Technology; 5-norbornene-2-carboxylic acid was purchased from TCI chemicals; phthalaldehyde, butylated hydroxytoluene (BHT), pyridine, sodium hydride, thionyl chloride, and lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) were purchased from Sigma-Aldrich.Norbornene acyl chloride synthesis (bicyclo[2.2.1]-hept-2-ene-5-carbonyl chloride, 5-norbornene carbonyl chloride)
The preparation of bicyclo[2.2.1]-hept-2-ene-5-carbonyl chloride was modified from a previously reported approach:51 5-norbornene-2-carboxylic acid (110.53 g, 0.8 mol) was mixed with thionyl chloride (190.35 g, 1.6 mol) in a 500 mL round-bottom flask, and then refluxed under dry nitrogen gas at 70 °C for 3 h (Scheme 1). Excess SOCl2 was evaporated under reduced pressure. The crude product was further purified by distillation to eliminate a colored impurity, yielding a transparent liquid (71% yield) (IR and NMR in ESI 1–3†).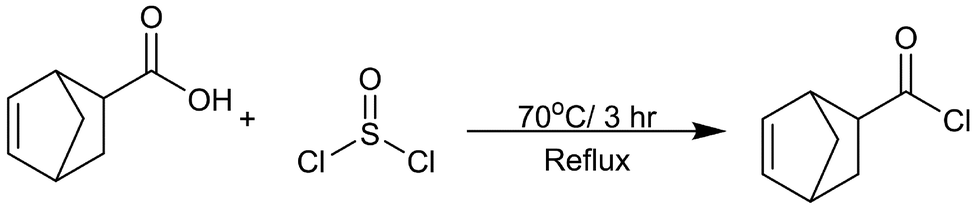 | ||
| Scheme 1 Synthesis for the norbornene acyl chloride that begins with the nucleophilic attack of the corresponding norbornene carboxylic acid by thionyl chloride at the carbonyl oxygen. This results in an intermediate that releases a chlorine anion which facilitates a second nucleophilic attack that results in the formation of the acyl chloride and release of sulfur dioxide and hydrogen chloride. | ||
PEG-p-norbornene macromer synthesis
4-arm PEG-OH (10K, 1 equiv., 0.2 mmol) was dissolved in dry dichloromethane (DCM) (30 mL) and added to a 250 mL flame dried round bottom flask with a magnetic stir bar. The reaction was cooled to 0 °C under inert conditions (N2). Sodium hydride (60% dispersion in mineral oil, 5 equiv., mmol) was added to the flask and stirred for 30 minutes at 0 °C. Phthalaldehyde (4 equiv., 3.2 mmol) dissolved in dry DCM (4 mL) was added and stirred for 1 hour at 0 °C. Pyridine (6 equiv., 4.8 mmol) and norbornene acyl chloride (5 equiv., 4 mmol) were added consecutively to the reaction flask and allowed to stir for 30 minutes and the reaction was allowed to warm up to room temperature (Scheme 2). The mixture was quenched in a dropwise fashion with methanol (5 mL). A rotary evaporator was used to concentrate the product until the mixture was approximately a 5 mL slurry. Methanol (140 mL) was added to the mixture. The mixture was cooled to −78 °C, centrifuged, and the solid precipitate was collected resulting in a white solid once vacuum dried. | ||
| Scheme 2 Synthesis of PEG-phthalaldehyde-Norbornene (PEG-p-Norb) macromer. Initial steps form the acetal connection between PEG core and the phthalaldehyde. The final step is a capping mechanism that provides a norbornene functional group via a substitution reaction. | ||
It is important to note that this reaction is highly variable, largely arising from being highly sensitive to water and the quality and purity of the reactants. It was found that canulating the reactants into the reaction vessel worked most aptly and best results were observed with fresh sodium hydride, lyophilized PEG-OH, and re-crystallized phthalaldehyde. Numerous synthetic repetitions of this macromer were performed, and variations were observed in the phthalaldehyde and norbornene functionalization as shown in ESI Fig. 4–7;† however, gelation and degradation were observed after each of multiple syntheses of the material if sufficient degrees of functionalization were met (supported by Flory-Stockmayer calculations as shown in the ESI†). All data presented within the main text in this paper corresponds to the NMRs shown in ESI Fig. 4 and 8.† The degree of functionalization for the degradable system presented in the body of this paper was determined to be 37% norbornene and 13% phthalaldehyde and the degree of functionalization for the control, non-degradable system was determined to be 48% norbornene.
Control PEG-norbornene macromer synthesis
A control macromer was synthesized following the afore mentioned synthesis with the exception of the phthalaldehyde addition.Hydrogel formation
The synthesized PEG-based macromers were solubilized in a LAP stock solution (0.1 wt% LAP in DI water) to form varied weight percent hydrogels (2–20 wt%). The norbornene systems were solubilized with equimolar amounts of a 4-arm 10K PEG-SH. The solution was pipetted between two glass slides equipped with spacers (100 μm–2 mm) treated with RainX (Scheme 3). The solution was exposed to 365 nm UV light (Acticure 4000 mercury-lamp with a 365 nm band gap filter) with an intensity of 20 mW cm−2 for 5 minutes to cure the hydrogels. The resultant hydrogels were placed into individual glass vials and swollen in DI water at room temperature under ambient conditions.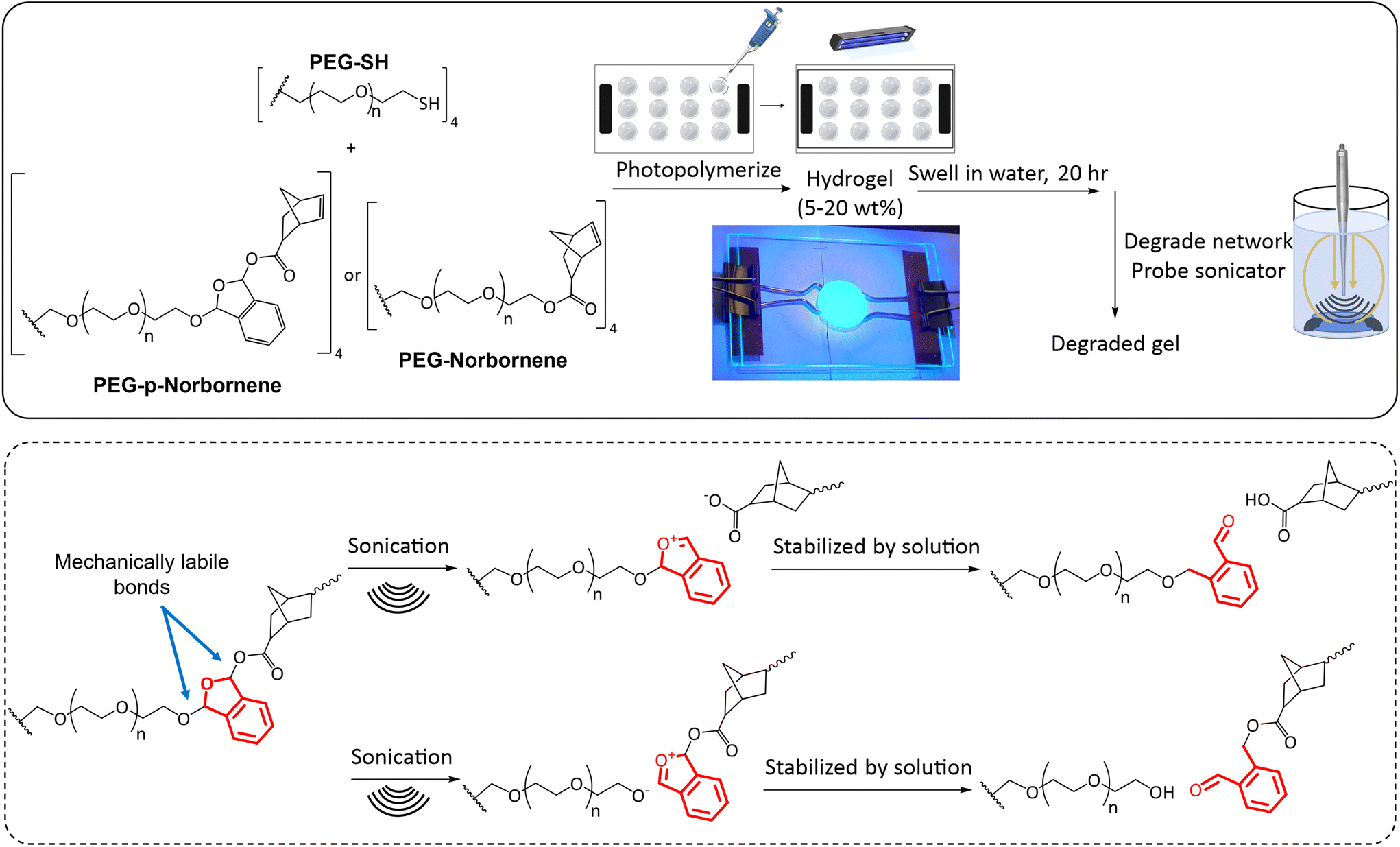 | ||
| Scheme 3 Chemical structures of PEG macromers and method for hydrogel synthesis. Equimolar amounts of thiol and norbornene functional macromers were added to a LAP solution and then photopolymerized. Polymer gels were then analyzed in swollen and degraded states. The proposed sites of bond breakage with mechanical stimulation are indicated with the phthalaldehyde unit shown in red. These bond breakage sites are hypothesized based on bond breakage locations within a poly(phthalaldehyde) system.53 Additional analysis would be needed to analyze the bond breakage location as NMR peaks of the degradation product end groups were convoluted due to the vastly greater number of repeat units in the PEG core, thus obscuring the signal from functional groups of the degradation products. | ||
Rheometry
Rheological measurements were obtained with Ares-G2 TA Instruments DH3 rheometer. Experiments were carried out at 25 °C, maintained by TA's Peltier plate temperature control, with an 8 mm quartz plate. Measurements were collected with 10% strain, a frequency of 10 rad s−1, and 0.105 N axial force. A 365 nm UV light source with an intensity of 20 mW cm−2 was used to irradiate the samples for photorheology with the light source being turned on after a 30 s baseline.Sonication degradation
A Fisher Scientific Sonic Dismembrator, Model 100 was used to degrade the hydrogels mechanically (operating frequency of 20 kHz). Hydrogels were allowed to swell completely in DI water and initial rheological data or mass was collected. Hydrogels were placed in 5 mL of DI water for sonication and the probe was placed ∼5 mm from the bottom of the vial. The vials were kept on ice during the sonication treatment to prevent heating of the hydrogels. Hydrogels were sonicated at 7 W for predetermined lengths of time. Subsequent rheological or mass data was then collected post-treatment (the sonication time points were chosen knowing the time scale at which the hydrogel networks degraded completely).Lyophilization
Lyophilization was used to obtain the dry weight of the hydrogels post sonication degradation for mass loss studies. Hydrogels were frozen in liquid nitrogen and lyophilized using a LABCONCO FreeZone FreezeDry System at −53 °C and 0.02 mbar for 24 hours.Eqn (1) was used to calculate mass loss of the hydrogels over time.
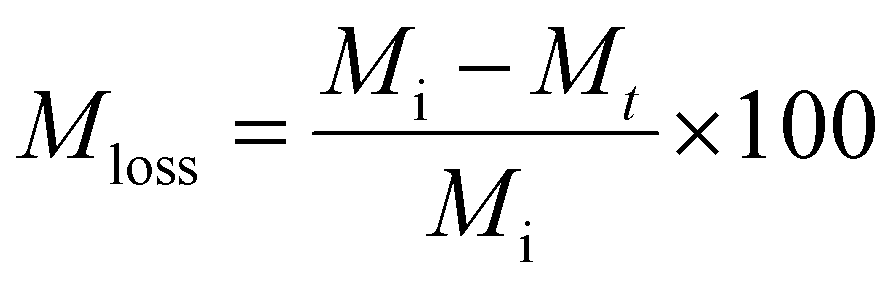 | (1) |
The initial “dry” mass of hydrogels was determined via the weight percent and volume of polymer precursor solution. Following the sonication degradation of the hydrogels, the supernatant was removed to remove soluble degradation products and the hydrogels were lyophilized again to obtain a final dry weight.
Heat degradation
Hydrogels were placed in glass vials with 5 mL of DI water and the caps were loosely twisted on. The oven was allowed to come to temperature (60 °C, 80 °C, or 100 °C) and the solutions were placed in the oven for predetermined lengths of time to determine the effect of elevated temperatures on the hydrogel network degradation.Blood assay
The following procedure was adapted from the Haeussler Group.52 Fresh human whole blood was obtained from Vitalant with ethylenediaminetetraacetic acid (EDTA) added as the anti-coagulant (1.8 mg mL−1). Whole blood was diluted in phosphate-buffered saline (PBS, 20 mL) and washed four times by centrifugation (10 min, 2000 rpm, 15 °C) to isolate the red blood cells (RBCs). PBS was added to mimic physiological conditions with 40% RBCs (vol/vol). This solution was then used in place of the DI water for hydrogel synthesis. The hydrogels created were 100 μm thick and contained 12 wt% macromer. To analyze the effects of each component on the RBCs, the various solutions (ESI Table 1†) were diluted to a final concentration of 2% RBCs (vol/vol) and three samples of each solution (100 μL) were pipetted into a 96-well round bottom plate. The assay plate was tapped to adequately mix all solutions of polymer and RBCs. This plate was then centrifuged to pellet out aggregated RBCs. The supernatant of each well was then transferred to a new plate. This new plate was put in a plate reader to assess hemolysis by measuring the absorbance of the solution at 570 nm. An attempt was made to redisperse the RBC pellets left in the original plate with a PBS solution to qualitatively determine the level of hemagglutination that occurred at each condition. A virgin 40% RBC (vol/vol) in PBS solution was used as a negative control for hemolysis and hemagglutination. Sonication of RBCs and PBS (5 W, level 5, 90 s) was used as a positive control for hemolysis. Blood exposed to oxygen for an extended period of time was used as a positive control for hemagglutination. Percent hemolysis was calculated according to the absorbance of the positive control (extended sonication of RBCs) and negative control (PBS and RBCs) using eqn (2). | (2) |
Results and discussion
Hydrogel polymerization
Initial data was collected on hydrogels of various composition to determine the ability of these macromer systems to polymerize, the effect the phthalaldehyde unit inclusion has on the storage modulus of the hydrogels, and to gather a baseline for samples prior to mechanical stimulation. Fig. 1 contains a representative polymerization plot for the photopolymerizations of PEG-p-Norb and PEG-Norb at 15 wt% macromer. 12 μl of this solution was pipetted onto the rheometer base plate and an 8 mm quartz head was lowered to 250 μm. After the initial 30 seconds to collect a baseline, a 365 nm UV light source (20 mW cm−2) was directed onto the solution to photopolymerize the material into gels. The average storage modulus of the phthalaldehyde containing systems was less than the non-phthalaldehyde systems. This difference may be due to the difference in norbornene functionalization of the control macromer versus the norbornene functionalization in the phthalaldehyde containing macromer. NMR integration suggests that approximately 48% of the control PEG is functionalized by a norbornene while in the phthalaldehyde containing material approximately 37% of the PEG is functionalized with a norbornene (ESI Fig. 4 and 8†). The incorporation of a phthalaldehyde unit via the method described here locates a mechanically labile bond near the crosslink junctions of the network once polymerized (Scheme 3). It is important to note that the precise configuration of each arm of the PEG macromers synthesized is not known with it being possible that each of the arms is capped with a phthalaldehyde, a norbornene, a phthalaldehyde and norbornene, or remain uncapped as alcohol functional groups. An attempt to determine if the phthalaldehyde units were capped with a norbornene was made by synthesizing a 5K monofunctional PEG-p-Norb (following the same procedure as noted for the 4-arm system). The monofunctional PEG-p-Norb was solubilized in water and 1 M HCL acid was added (until a pH of 3 was achieved) as a means of removing the phthalaldehyde via the hydrolytically degradable acetal linkage formed with successful phthalaldehyde incorporation into the macromer. NMRs of these starting and post-acid products were taken. Comparison of the pre- and post-acid wash functional group integrations of phthalaldehydes and norbornenes suggests that each time a phthalaldehyde unit was removed from the polymer chain, a norbornene was also removed (the norbornene integration decreased by approximately 49% and the phthalaldehyde integration decreased by approximately 56%) (ESI Fig. 9 and 10†). This behavior suggests that the majority of phthalaldehyde units that attach to the PEG chain are indeed capped by a norbornene, therefore adding in to the network during polymerization and facilitating the breaking of crosslinks when mechanically stimulated. Utilizing this key point, Flory-Stockmayer calculations for the gel point and ability of the network to degrade were performed. These calculations demonstrated that both gelation and degradation are possible with the functionalization of the PEG macromer that was achieved (calculations and assumptions are noted in the ESI†).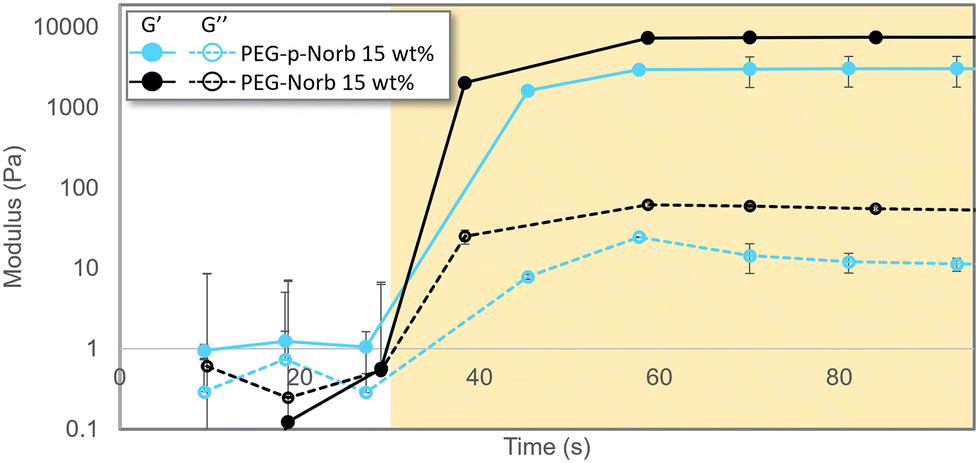 | ||
| Fig. 1 Photo-rheological data of gelation for norbornene systems at 15 wt%. The shaded region indicates when the sample was being irradiated by a UV source (0.1 wt% LAP, 365 nm, 20 mW cm−2, 250 μm thick). Systems reach their max storage modulus plateau within tens of seconds. For both compositions, phthalaldehyde containing systems have a decreased storage modulus. Error bars represent mean ± SD, n = 3. | ||
Gel in degradation conditions
The effect of temperature on the norbornene hydrogel systems was investigated to analyze how the material would respond to both the elevated temperature that occurs during sonication as well as temperature increases that might occur in applications of these materials. Gel samples were made with PEG-Norb as a comparative control to the PEG-p-Norb gels to allow determination of the effect of including a phthalaldehyde unit. The gels were made to be 15 wt% and 500 μm thick. Each gel was completely swollen post photocure and then heated for 15 minutes at the given temperature while being submersed in water. The effect of various temperatures was evaluated through rheological data of the storage modulus for the various conditions. As seen in Fig. 2a the norbornene control gel did not have a significant change in storage modulus upon heating. However, the PEG-p-Norb storage modulus increased approximately six-fold. This trend was further explored by heating the norbornene systems for extended periods of time at 100 °C. The storage modulus increased approximately five-fold in the system containing a phthalaldehyde unit (Fig. 2b). We hypothesize that this behavior is caused by a change in the hydrophobic nature of the gels during heating, thus causing a change in the degree of swelling that is achieved after heating. This trend is supported by the gels returning to their original storage moduli after having completely cooled as shown in the ESI (Fig. 11†).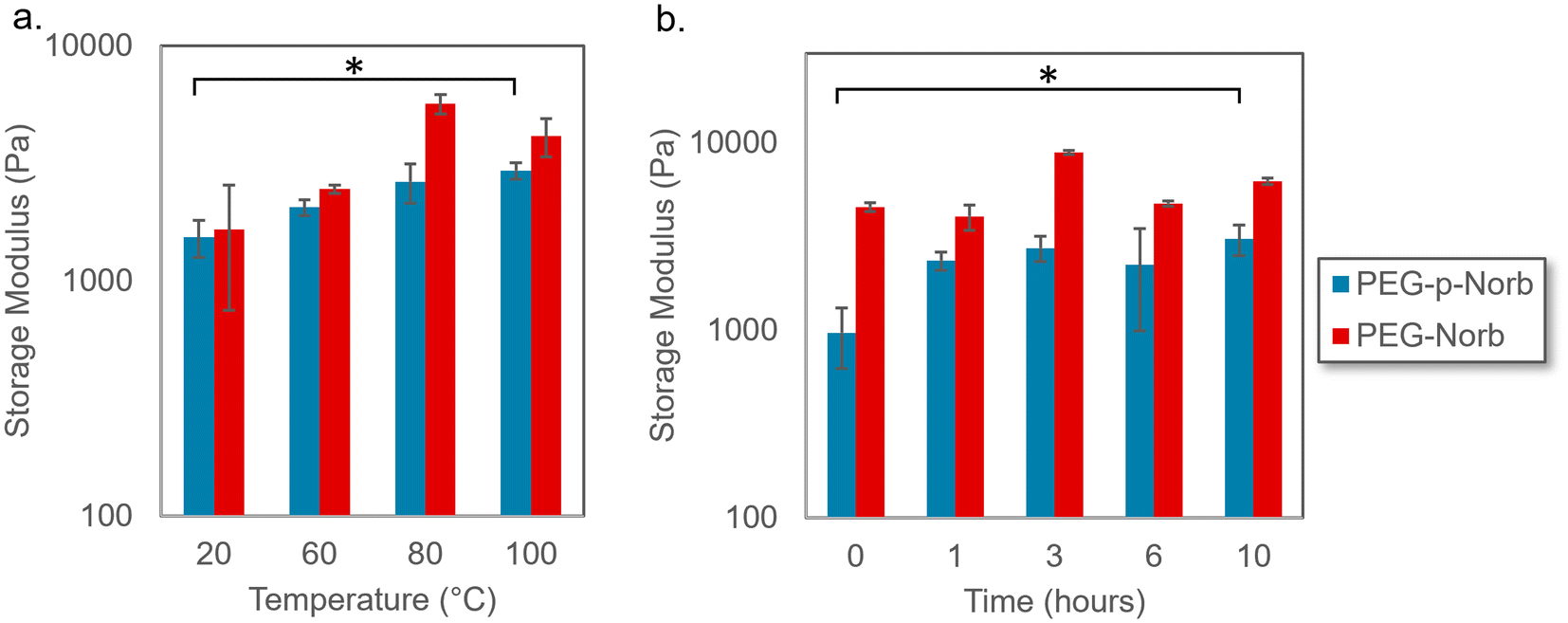 | ||
| Fig. 2 (a) Rheological data illustrating the effect on the storage modulus of heating the norbornene hydrogel systems for 15 minutes at various temperatures and (b) at 100 °C for extended periods of time. Minimal difference in storage modulus was seen for the PEG-norbornene systems in either test while the PEG-p-norbornene samples showed a significant increase in storage modulus when comparing the highest temperature or longest time point to the initial material. Error bars represent mean ± SD, n = 4. Significance tests were performed using two-tailed, heteroscedastic Student's t-test. * p < 0.01. | ||
Sonication degradation
Rheological and mass loss data over time were collected for the norbornene systems as the hydrogels were sonicated with a probe sonicator. The rheological data was collected from 15 wt% hydrogels that were 500 μm thick. The samples were sonicated at a power output of 7 W with a frequency of 20 Hz in 10 mL of DI water while the vials were kept on ice. The PEG-p-Norb gels showed significant loss in storage modulus as sonication time increased (Fig. 3a) while the control PEG-Norb gels displayed a minimal change in storage modulus. The storage modulus is correlated to the number of crosslinks in a polymer network suggesting that the sonication stimulus resulted in breakage of the polymer network of the phthalaldehyde containing materials.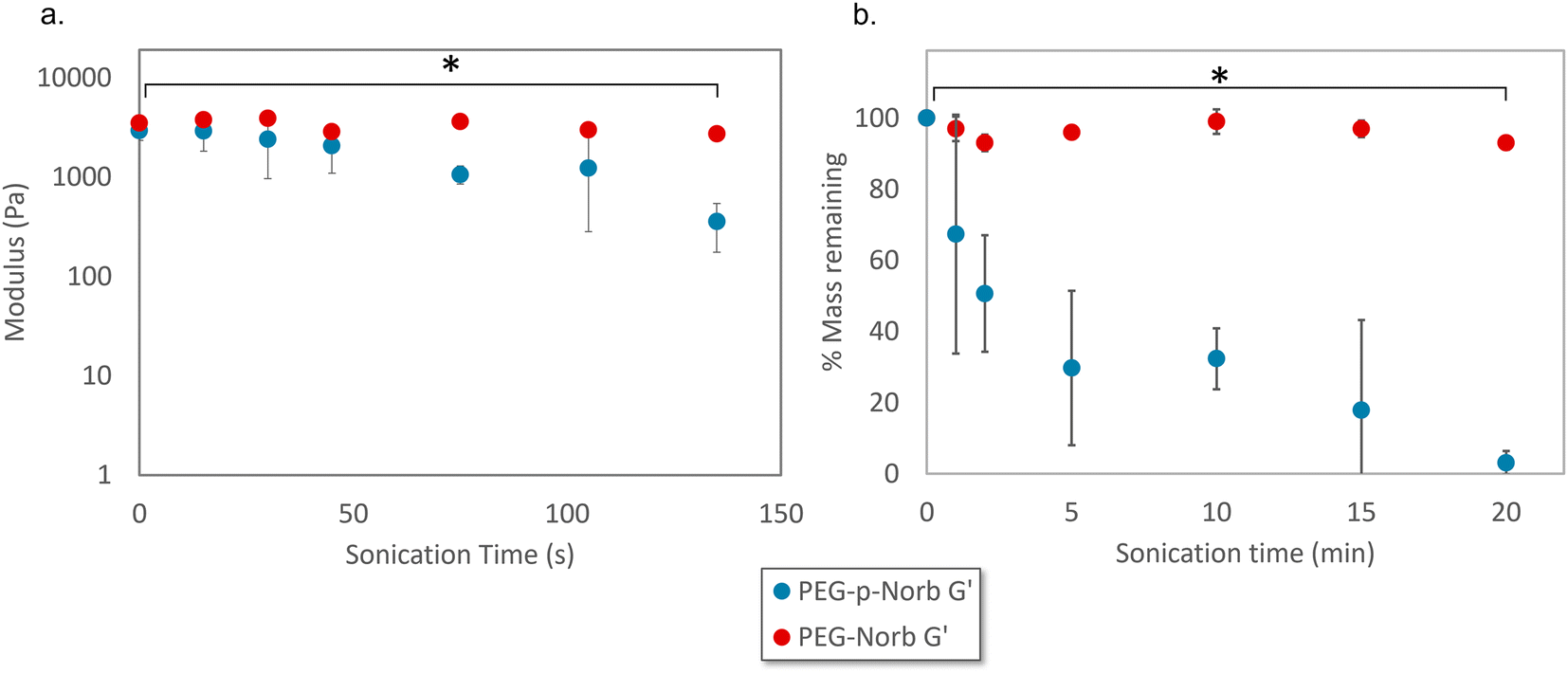 | ||
| Fig. 3 (a) Change in storage modulus and (b) mass loss of hydrogels when sonication is performed on a hydrogel system (power output of 7 W, 20 Hz, in 10 mL of DI water on ice). The phthalaldehyde containing system displayed a significant decrease in both storage modulus and mass with increased sonication time. Error bars represent mean ± one standard deviation, n = 3. p-Values were calculated with Welch's t-test. * p < 0.05. | ||
Mass loss data for these systems was taken from 15 wt% hydrogels that were 1 mm thick. The gels were sonicated at a power output of 7 W with a frequency of 20 Hz in 10 mL of DI water while on ice. The phthalaldehyde containing gels displayed significant and increasing mass loss as sonication time increased (Fig. 3b).
The combination of these two data sets indicates that the addition of the mechanophore into the network facilitates mechanical degradation. It is likely the hydrogels degrade through a combination of surface and bulk degradation as evidenced by the trends seen in Fig. 3 as well as the appearance of the gels during mechanical stimulation. GPC of the resultant soluble macromer fragments indicates that degradation of the hydrogel network produces fragments of similar molecular weight to the initial macromer used to form the network (ESI Fig. 12†).
Blood assay
The ability for these hydrogels to be used in optically dense biological conditions where photochemical degradation mechanisms may not be applicable was tested through the suspension and release of horse serum and red blood cells. Biocompatibility of the sonication degradation method for the PEG-p-Norb systems was initially tested with the suspension of horse serum in 100 μm thick 5 wt% PEG-p-Norb hydrogels photopolymerized with 365 nm UV light for 30 seconds at 20 mW cm−2. Equine serum was diluted in 1× PBS at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. The degradation for these gels was done with probe sonication at a power output of 5 W and a frequency of 20 Hz for 90 seconds on ice. The degradation of the hydrogel containing horse serum resulted in a lower absorbance at 350 nm and 550 nm in comparison to the denatured proteins, indicating minimal denaturation of the horse serum proteins under these degradation conditions (ESI Fig. 13†). The phthalaldehyde ring absorbs more strongly at 350 nm as compared to 550 nm which contributes to the greater absorbance measurement of both the control and degraded samples where there are phthalaldehyde units present in the degraded gel fragments. Comparatively, the protein absorbance at these two wavelengths also varies, but to a lesser degree.
:10. The degradation for these gels was done with probe sonication at a power output of 5 W and a frequency of 20 Hz for 90 seconds on ice. The degradation of the hydrogel containing horse serum resulted in a lower absorbance at 350 nm and 550 nm in comparison to the denatured proteins, indicating minimal denaturation of the horse serum proteins under these degradation conditions (ESI Fig. 13†). The phthalaldehyde ring absorbs more strongly at 350 nm as compared to 550 nm which contributes to the greater absorbance measurement of both the control and degraded samples where there are phthalaldehyde units present in the degraded gel fragments. Comparatively, the protein absorbance at these two wavelengths also varies, but to a lesser degree.
After seeing that the sonication conditions required for release of the suspended proteins resulted in minimal change in the absorbance of the solution at 350 nm and 550 nm, indicating minimal denaturation of the proteins, red blood cells (RBCs) were suspended in the hydrogels to test an optically dense condition. Percent hemolysis was measured to determine if RBCs could be encapsulated and released with sonication. 12 wt% macromer hydrogels of equimolar amounts of PEG-p-Norb and PEG-SH were used to make 100 μm thick gels with 40% (vol/vol) RBCs in PBS which were exposed to 365 nm UV light for 30 seconds at 20 mW cm−2 (0.1 wt% initiator). The gels were sonicated at a power output of 5 W with a frequency of 20 Hz for 30 seconds while the vials were kept on ice to ensure minimal effects from temperature changes during sonication. Samples were also prepared from the individual components of the hydrogels (ESI Table 1†) to evaluate the effects of the network components on the RBCs, thereby serving as standard control studies. A solution of PBS and RBCs (40% vol/vol) was used as the negative control for hemagglutination and hemolysis. The effect of the free radicals produced during the photopolymerization process was evaluated with a solution of RBCs and LAP (6 wt%) exposed to 365 nm UV light (5 mW cm−2, 30 s, 100 μm thick sample). A positive control of hemagglutination was measured from a solution of RBCs exposed to oxygen until coagulation occurred. The positive control for hemolysis was taken from a solution of RBCs probe sonicated at a power output of 5 W and a frequency of 20 Hz for 30 seconds on ice. 100 μl of each solution was pipetted into a 96-well round-bottom plate and the absorbance of hemoglobin was measured at 570 nm on a plate reader (hemolysis due to individual hydrogel components is included in ESI Fig. 14†). The results showed that the individual effects of the various hydrogel components resulted in no hemolysis of the RBCs (Fig. 4). Additionally, the RBCs encapsulated in a hydrogel network were less hemolyzed than those sonicated under similar conditions without the protective hydrogel presence. While each solution with an individual component did not show aggregation of any RBCs, the experimental conditions showed minimal aggregation in comparison to the positive controls. These results indicate that the hydrogel network may act as a protective shield to the RBCs, reducing the impact of the probe sonication on the cells and thereby minimizing hemolysis and hemagglutination during degradation. This outcome would prove useful for the suspension of various bio-components for drug delivery applications where the intended release location doesn't have a distinguishable pH, is unreachable by thermal means, and cannot be triggered photochemically due to optical density of the surrounding tissues.
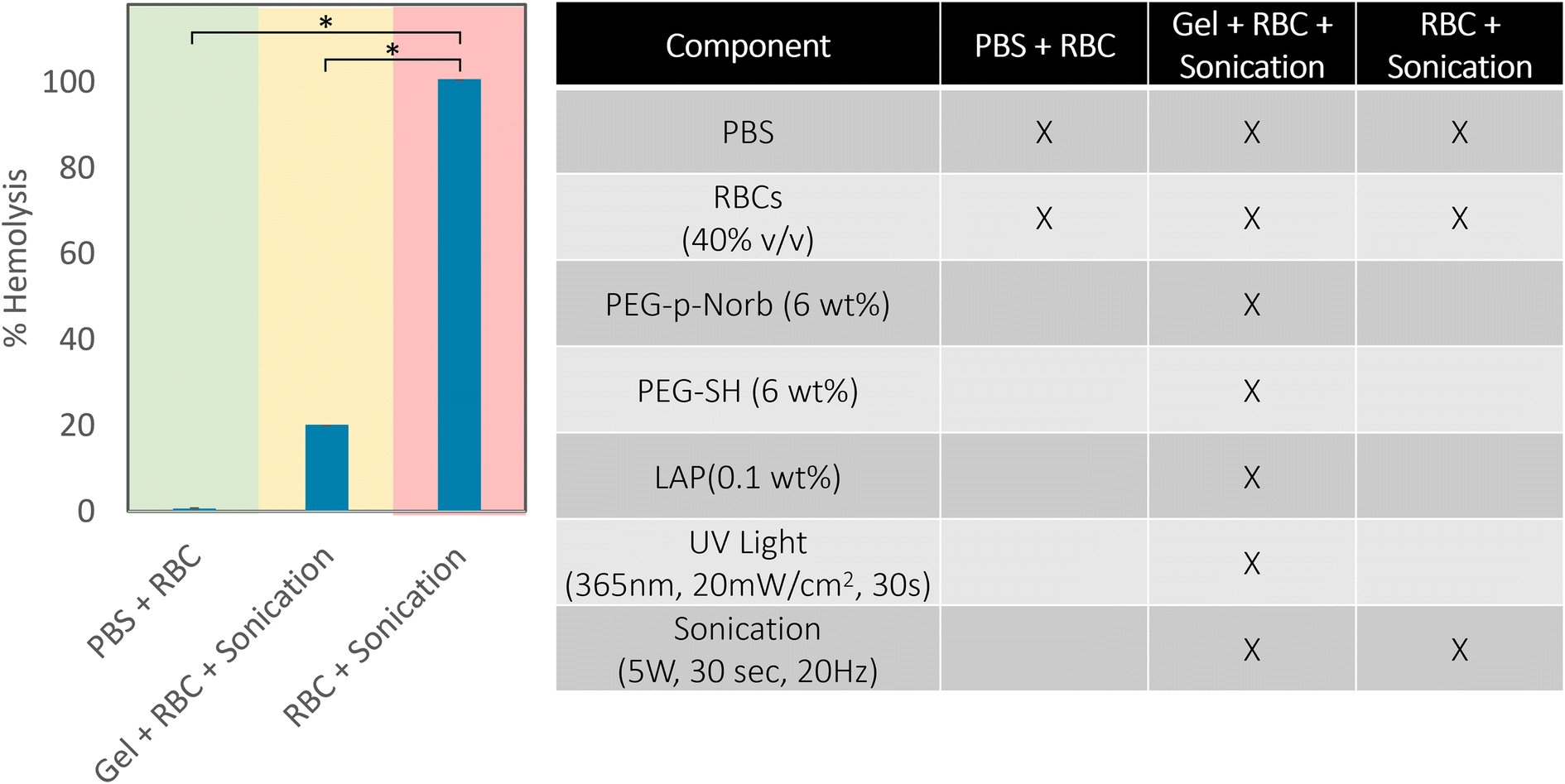 | ||
| Fig. 4 Percent hemolysis of red blood cells using a plate reader assay (measured at 570 nm) in the PEG-p-Norb hydrogel system.54 The conditions for these tests are shown in the adjoining table, the negative control is indicated by the green shade and the positive control is shaded red. The normal and extended sonication of hydrogels with suspended RBCs resulted in significantly less hemolysis of the RBCs compared to the positive controls. Error bars represent mean ± SD. p-Values were calculated with Welch's t-test. * p 0.05. | ||
Conclusions
This work demonstrates the inclusion of a mechanophore unit (phthalaldehyde) in a PEG-based hydrogel network that facilitates degradation of the material at biologically sustainable conditions. The degradation of these phthalaldehyde-containing networks results in water soluble products that can be washed away for applications requiring the removal of a scaffold and the degradation can be controlled through sonication energy dose. Additionally, the material used in this work was shown to provide a method for encapsulation of proteins or red blood cells that allows for release from the gel network without complete denaturation of the proteins or annihilation of the cells. This provides a means of creating mechanically degradable crosslinked polymers with similar chemical and mechanical characteristics to their non-mechanophore functionalized counterparts that could be used in applications ranging from cell scaffolds to drug delivery devices. Due to the ability of sonication waves to penetrate biological tissues more uniformly than heat or light, spatiotemporal control can be maintained in the degradation of these sonication labile networks. The incorporation of a mechanophore, specifically a phthalaldehyde, would be readily achieved in different polymer systems including other biopolymers for which it would be desirable to make them ultrasound degradable. The phthalaldehyde unit is shown here to be attached to a PEG macromer through the reaction of an alcohol with an aldehyde, forming an acetal linkage. This reaction and subsequent incorporation of the phthalaldehyde results in mechanical lability when the resulting macromer-phthalaldehyde unit is polymerized in such a way that the phthalaldehyde is located near crosslink junctions so that, upon activation, the mechanically labile bond results in network degradation.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors do not have any conflicts of interest to declare.Acknowledgements
This work was financially supported by DARPA (W911NF-19-2-0024). Additional thanks to Dr Benjamin Fairbanks for his input with respect to editing of the manuscript.References
- S. W. Kim, Y. H. Bae and T. Okano, Hydrogels: Swelling, Drug Loading, and Release, Pharm. Res., 1992, 9, 283–290 CrossRef CAS.
- E. Caló and V. V. Khutoryanskiy, Biomedical applications of hydrogels: A review of patents and commercial products, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- S. Khan, A. Ullah, K. Ullah and N. Rehman, Insight into hydrogels, Des. Monomers Polym., 2016, 19, 456–478 CrossRef CAS.
- R. Edri, I. Gal, N. Noor, T. Harel, S. Fleischer, N. Adadi, O. Green, D. Shabat, L. Heller, A. Shapira, I. Gat-Viks, D. Peer and T. Dvir, Personalized Hydrogels for Engineering Diverse Fully Autologous Tissue Implants, Adv. Mater., 2019, 31, 1803895 CrossRef.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- A. Borzacchiello and L. Ambrosio, in Hydrogels: Biological Properties and Applications, ed. R. Barbucci, Springer Milan, Milano, 2009, pp. 9–20 Search PubMed.
- K. Y. Lee, K. H. Bouhadir and D. J. Mooney, Controlled degradation of hydrogels using multi-functional cross-linking molecules, Biomaterials, 2004, 25, 2461–2466 CrossRef CAS PubMed.
- P. Szymczyk-Ziółkowska, M. B. Łabowska, J. Detyna, I. Michalak and P. Gruber, A review of fabrication polymer scaffolds for biomedical applications using additive manufacturing techniques, Biocybern. Biomed. Eng., 2020, 40, 624–638 CrossRef.
- B. Hosseinzadeh and M. Ahmadi, Degradable Hydrogels: Design Mechanisms and Versatile Applications, Mater. Today Sustain., 2023, 100468 Search PubMed.
- J. Hu, Y. Chen, Y. Li, Z. Zhou and Y. Cheng, A thermo-degradable hydrogel with light-tunable degradation and drug release, Biomaterials, 2017, 112, 133–140 CrossRef CAS.
- Y.-Y. Jo, K.-G. Lee, J. C. Bragg, C.-C. Lin and H. Kweon, Structural and thermal characteristics of photocrosslinked silk fibroin - PEG hydrogel, Int. J. Ind. Entomol., 2016, 32, 35–40 Search PubMed.
- G. I. Sandakov, L. P. Smirnov, A. I. Sosikov, K. T. Summanen and N. N. Volkova, in Physics of Polymer Networks, Steinkopff, Darmstadt, 1992, vol. 90, pp. 235–240 Search PubMed.
- H. Shih and C.-C. Lin, Cross-Linking and Degradation of Step-Growth Hydrogels Formed by Thiol–Ene Photoclick Chemistry, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- D. Klinger and K. Landfester, Dual Stimuli-Responsive Poly(2-hydroxyethyl methacrylate-co-methacrylic acid) Microgels Based on Photo-Cleavable Cross-Linkers: pH-Dependent Swelling and Light-Induced Degradation, Macromolecules, 2011, 44, 9758–9772 CrossRef CAS.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Fréchet, A Novel Strategy for Encapsulation and Release of Proteins: Hydrogels and Microgels with Acid-Labile Acetal Cross-Linkers, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef CAS PubMed.
- C. Mo, R. Luo and Y. Chen, Advances in the Stimuli-Responsive Injectable Hydrogel for Controlled Release of Drugs, Macromol. Rapid Commun., 2022, 43, 2200007 CrossRef CAS.
- G. I. Peterson and A. J. Boydston, Kinetic Analysis of Mechanochemical Chain Scission of Linear Poly(phthalaldehyde), Macromol. Rapid Commun., 2014, 35, 1611–1614 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova, C.-C. Chang and S. L. Craig, Enhanced polymer mechanical degradation through mechanochemically unveiled lactonization, Nat. Commun., 2020, 11, 4987 CrossRef CAS PubMed.
- L. Wang, M. Neumann, T. Fu, W. Li, X. Cheng and B.-L. Su, Porous and responsive hydrogels for cell therapy, Curr. Opin. Colloid Interface Sci., 2018, 38, 135–157 CrossRef CAS.
- E. R. Ruskowitz and C. A. DeForest, Photoresponsive biomaterials for targeted drug delivery and 4D cell culture, Nat. Rev. Mater., 2018, 3, 1–17 CrossRef.
- D. R. Griffin and A. M. Kasko, Photodegradable Macromers and Hydrogels for Live Cell Encapsulation and Release, J. Am. Chem. Soc., 2012, 134, 13103–13107 CrossRef CAS PubMed.
- I. I. Eyenga, W. W. Focke, L. C. Prinsloo and A. T. Tolmay, Photodegradation: a solution for the shopping bag “visual pollution” problem?, Macromol. Symp., 2002, 178, 139–152 CrossRef CAS.
- V. X. Truong, K. M. Tsang, G. P. Simon, R. L. Boyd, R. A. Evans, H. Thissen and J. S. Forsythe, Photodegradable Gelatin-Based Hydrogels Prepared by Bioorthogonal Click Chemistry for Cell Encapsulation and Release, Biomacromolecules, 2015, 16, 2246–2253 CrossRef CAS.
- C. S. Ki, H. Shih and C.-C. Lin, Facile preparation of photodegradable hydrogels by photopolymerization, Polymer, 2013, 54, 2115–2122 CrossRef CAS.
- J. Jiang, M. Warner, O. Phillips, A. Engler and P. A. Kohl, Tunable transient and mechanical properties of photodegradable Poly(phthalaldehyde), Polymer, 2019, 176, 206–212 CrossRef CAS.
- M. Villiou, J. I. Paez and A. del Campo, Photodegradable Hydrogels for Cell Encapsulation and Tissue Adhesion, ACS Appl. Mater. Interfaces, 2020, 12, 37862–37872 CrossRef CAS.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Mechanical Properties and Degradation of Chain and Step-Polymerized Photodegradable Hydrogels, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- G. H. Bhuvaneswari, in Recycling of Polyurethane Foams, ed. S. Thomas, A. V. Rane, K. Kanny, A. VK and M. G.Thomas, William Andrew Publishing, 2018, pp. 29–44 Search PubMed.
- M. M. Sadat Ebrahimi and H. Schönherr, Enzyme-Sensing Chitosan Hydrogels, Langmuir, 2014, 30, 7842–7850 CrossRef CAS PubMed.
- W. Zhao, X. Jin, Y. Cong, Y. Liu and J. Fu, Degradable natural polymer hydrogels for articular cartilage tissue engineering, J. Chem. Technol. Biotechnol., 2013, 88, 327–339 CrossRef CAS.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Designing degradable hydrogels for orthogonal control of cell microenvironments, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- J. Li and D. J. Mooney, Designing hydrogels for controlled drug delivery, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- A. M. Hawkins, T. A. Milbrandt, D. A. Puleo and J. Z. Hilt, Synthesis and analysis of degradation, mechanical and toxicity properties of poly(β-amino ester) degradable hydrogels, Acta Biomater., 2011, 7, 1956–1964 CrossRef CAS.
- A. Schindler, R. Jeffcoat, G. L. Kimmel, C. G. Pitt, M. E. Wall and R. Zweidinger, in Contemporary Topics in Polymer Science, ed. E. M. Pearce and J. R. Schaefgen, Springer US, Boston, MA, 1977, vol. 2, pp. 251–289 Search PubMed.
- H. Chen, J. Bei and S. Wang, Hydrolytic degradation of polyester–polyether block copolymer based on polycaprolactone/poly(ethylene glycol)/polylactide, Polym. Adv. Technol., 2000, 11, 180–184 CrossRef CAS.
- A. K. S. Chandel, C. U. Kumar and S. K. Jewrajka, Effect of Polyethylene Glycol on Properties and Drug Encapsulation–Release Performance of Biodegradable/Cytocompatible Agarose–Polyethylene Glycol–Polycaprolactone Amphiphilic Co-Network Gels, ACS Appl. Mater. Interfaces, 2016, 8, 3182–3192 CrossRef CAS.
- H. Bramfeldt, P. Sarazin and P. Vermette, Characterization, degradation, and mechanical strength of poly(D,L-lactide-co-ε-caprolactone)-poly(ethylene glycol)-poly(D,L-lactide-co-ε-caprolactone), J. Biomed. Mater. Res., Part A, 2007, 83, 503–511 CrossRef PubMed.
- A. O. Helminen, H. Korhonen and J. V. Seppälä, Cross-Linked Poly(ε-caprolactone/D,L-lactide) Copolymers with Elastic Properties, Macromol. Chem. Phys., 2002, 203, 2630–2639 CrossRef CAS.
- D. K. Han and J. A. Hubbell, Synthesis of Polymer Network Scaffolds from l-Lactide and Poly(ethylene glycol) and Their Interaction with Cells, Macromolecules, 1997, 30, 6077–6083 CrossRef CAS.
- M. P. Hiljanen-Vainio, P. A. Orava and J. V. Seppälä, Properties of ε-caprolactone/DL-lactide (ε-CL/DL-LA) copolymers with a minor ε-CL content, J. Biomed. Mater. Res., 1997, 34, 39–46 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS PubMed.
- Q. Xu, L. Guo, S. A, Y. Gao, D. Zhou, U. Greiser, J. Creagh-Flynn, H. Zhang, Y. Dong, L. Cutlar, F. Wang, W. Liu, W. Wang and W. Wang, Injectable hyperbranched poly(β-amino ester) hydrogels with on-demand degradation profiles to match wound healing processes, Chem. Sci., 2018, 9, 2179–2187 RSC.
- M. A. Azagarsamy, D. D. McKinnon, D. L. Alge and K. S. Anseth, Coumarin-Based Photodegradable Hydrogel: Design, Synthesis, Gelation, and Degradation Kinetics, ACS Macro Lett., 2014, 3, 515–519 CrossRef CAS PubMed.
- D. Y. Wong, D. R. Griffin, J. Reed and A. M. Kasko, Photodegradable Hydrogels to Generate Positive and Negative Features over Multiple Length Scales, Macromolecules, 2010, 43, 2824–2831 CrossRef CAS.
- L. Ren and Y. T. Lim, Degradation-Regulatable Architectured Implantable Macroporous Scaffold for the Spatiotemporal Modulation of Immunosuppressive Microenvironment and Enhanced Combination Cancer Immunotherapy, Adv. Funct. Mater., 2018, 28, 1804490 CrossRef.
- G. Burke, Z. Cao, D. M. Devine and I. Major, Preparation of Biodegradable Polyethylene Glycol Dimethacrylate Hydrogels via Thiol-ene Chemistry, Polymers, 2019, 11, 1339 CrossRef PubMed.
- T. M. Robinson, in Physics for Medical Imaging Applications, Springer, 2007, pp. 101–110 Search PubMed.
- G. Kim, V. M. Lau, A. J. Halmes, M. L. Oelze, J. S. Moore and K. C. Li, High-intensity focused ultrasound-induced mechanochemical transduction in synthetic elastomers, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10214–10222 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Force-induced activation of covalent bonds in mechanoresponsive polymeric materials, Nature, 2009, 459, 68–72 CrossRef CAS.
- S. Soars, J. Kamps, B. Fairbanks and C. Bowman, Stimuli-Responsive Depolymerization of Poly(Phthalaldehyde) Copolymers and Networks, Macromol. Chem. Phys., 2021, 222, 2100111 CrossRef CAS.
- S. Biswas, K. D. Belfield, R. K. Das, S. Ghosh and A. F. Hebard, Block Copolymer-Mediated Formation of Superparamagnetic Nanocomposites, Chem. Mater., 2009, 21, 5644–5653 CrossRef CAS.
- K. E. S. Locock, T. D. Michl, J. D. P. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Guanylated Polymethacrylates: A Class of Potent Antimicrobial Polymers with Low Hemolytic Activity, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed.
- A. M. DiLauro, J. S. Robbins and S. T. Phillips, Reproducible and Scalable Synthesis of End-Cap-Functionalized Depolymerizable Poly(phthalaldehydes), Macromolecules, 2013, 46, 2963–2968 CrossRef CAS.
- H.-C. Mahler, W. Friess, U. Grauschopf and S. Kiese, Protein aggregation: Pathways, induction factors and analysis, J. Pharm. Sci., 2009, 98, 2909–2934 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00161c |
| This journal is © The Royal Society of Chemistry 2024 |