
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free thiol-Michael addition for hydrogen bond-rich poly(dimethyl siloxane) networks with improved electromechanical properties and self-healing capabilities†
Pavle
Ramah
,
Liyun
Yu
,
Anders Egede
Daugaard
 and
Anne Ladegaard
Skov
*
and
Anne Ladegaard
Skov
*
The Danish Polymer Centre, Department of Chemical and Biochemical Engineering, Danish Technical University, 2800 Kongens Lyngby, Denmark. E-mail: al@kt.dtu.dk
First published on 15th July 2024
Abstract
Thiol-maleimide conjugation is a well-established coupling method in biochemistry but with little reported use within silicone materials. A facile synthetic route of functionalised poly(dimethyl siloxane) (PDMS) to a prepolymer species capable of non-metal-catalysed cross-linking via thiol-Michael addition is presented. Two systems are compared: maleimide (MI) terminated PDMS and its precursor, maleamic acid amide (MA) terminated PDMS. Despite the traditional view of maleamic acid amide functionalities as being of inferior value to their maleimide counterparts, we argue for their potential. The increased relative concentration of polar groups in a robust MA network matrix can be exploited for better electrical properties for self-healable dielectric elastomer actuators, as demonstrated by the MA networks with their quadruple hydrogen bonding centres, self-healing capabilities, increased polarity, good electrical breakdown strength, and increased dielectric permittivity over both commercial and MI PDMS networks.
Introduction
Poly(dimethyl siloxane) (PDMS) is a polymer known for its flexibility due to its low glass transition temperature (<−110 °C),1 and it finds use in many novel applications, such as soft robotics and dielectrics.2,3 PDMS is also a chemically inert, thermally stable, and commercially available material with a high level of hydrophobicity. For specific applications, a more hydrophilic nature is desirable. The non-polar character of PDMS is usually tuned by combining it with fillers and additives, chemically modifying it through side-chain polar groups, and applying surface activation methods.4 Such alterations are common in soft wearables and dielectric elastomers, where improved electrical properties benefit higher actuation and lower voltage demands.3 Metal-free elastomers enable curing or solidification under mild conditions.5 This enhances their potential applications, improves recyclability, and eliminates the negative impact of metals on the increased conductivity of the resulting elastomers. A low concentration (ppm) of a conductive metal catalyst may be beneficial for the electrical properties of the dielectric elastomers due to the charge trapping; however, at higher concentrations, the conductivity is increased and will negatively affect the electrical breakdown strength.6,7 While catalyst-free cross-linking is a frequent topic for silicone elastomers, their self-healing capabilities have been discussed rarely.8–11Hydrogen bonds between a proton donor and a proton acceptor can promote supramolecular organisation within a given polymer network and is one of the primary pathways towards self-healing materials.12 Being able to self-organise without external stimuli is essential for the ability to recover from damage. Due to their dynamic nature, responsiveness to external stimuli, and tunable strength, hydrogen bonds are commonly used to include self-healing ability in elastomers.13 The association constant of hydrogen bonds can vary greatly but approaches those of covalent bonds for multiple hydrogen bonding domains.14
Dielectric elastomers, amongst other products, can benefit from self-healing properties. During operation, dielectric elastomers can experience breakdown due to high applied voltages, reflected through topological failure. If this breakdown pinhole can be repaired through local reorganisation, it would improve performance and extend operational life.15 A low glass transition temperature in combination with physical or chemical bonding has previously been highlighted as a prerequisite for ambient self-healing.16 However, using strong hydrogen bonding domains in PDMS elastomers frequently requires a more elaborate synthesis procedure.17–19 Additionally, constraints in mobility and limitations in the crack closure process of a highly cross-linked network frequently require special conditions to achieve self-healing.20
As an alternative to these more elaborate synthetic pathways, we here investigate the use of Michael addition reactions as a simple approach toward self-healing dynamic networks. Michael additions are highly effective coupling reactions between a Michael donor (a nucleophile) and a Michael acceptor (an unsaturated carbonyl with an electron-withdrawing group). The reaction can be considered a click chemistry reaction, known for its rapid kinetics, selectivity, and minimal catalyst requirements.21 Maleimide (MI) moieties make strong Michael acceptors due to their high reactivity, facilitated by the five-atom N-ring's ring strain and its vinyl bond's electron-deficient character.21–23 The MI's controlled conversion into different functionalities under facile conditions is widely utilised.24–27 However, the Michael addition reaction between thiol and vinyl moieties has rarely been explored within silicones, and most commonly only in combination with some other chemistry, e.g. Diels Alder.28–30 Research has tended to focus on the radical thiol–ene reaction.31–34 This is primarily due to the thiol-Michael addition reaction's high dependence of the on reaction conditions, susceptible for low molecular weight compounds.35
In this work, a commercially available aminopropyl-terminated PDMS (APDMS) is converted in a two-step process into (i) α, ω – maleamic acid amide terminated PDMS (MA) through a ring-opening reaction of maleic anhydride with the terminal amine group to a carboxylate derivative, and (ii) α, ω – maleimide terminated PDMS (MI) through the ring-closure of MA PDMS. APDMS is commercially available, relatively cheap, and the aminopropyl has significant reactivity.36–38 The reaction conditions were based on similar materials reported, being primarily non-PDMS systems.21,28,39,40 The synthesised prepolymers were then cross-linked via thiol-Michael addition. Triethyl amine, a weak volatile base (b.p. 77 °C), was used as a catalyst, making the cross-linking metal-free and insensitive to O2.
While the MI functionality has found some use within PDMS synthesis, the MA functionality is mostly considered a MI precursor, due to the lower reactivity of its vinyl bond and its instability.41–43 This work shows both that (a) thiol-Michael addition is a robust method for synthesising highly cross-linked PDMS networks with unique properties, and that (b) MA-containing cross-linked networks have increased intermolecular interactions, due to having quadruple H bonding centres, which lead to emerging intrinsic self-healing, distinct mechanical properties, and improved dielectric permittivity.
Materials and methods
Materials
All chemicals were reagent grade and were used without additional purification. Maleic anhydride (MAnh), acetic anhydride (AcAnh), 2-methyl tetrahydrofuran (2mTHF), pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), and triethyl amine (TEA) were purchased from Merck Life Science. Aminopropyl terminated poly(dimethyl siloxane) (APDMS) was purchased from abcr GmbH (AB109371, 5000 g mol−1, 0.6–0.7% amine content, and AB153374, 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 0.11–0.12% amine content).
000 g mol−1, 0.11–0.12% amine content).
Methods
Tensile self-healing tests were performed as follows: a cut was made through the dogbone neck, with 0.05 mm left to help with subsequent alignment during healing. The sample was not mechanically pressed together in any way and was simply left to self-heal under room temperature for 24 h before being mechanically tested.
Both the MA5 and MI5 followed the same clean-up procedure: a reactive mixture with the prepolymer species was poured into a liquid–liquid extractor. It was washed with a saturated NaCl aqueous solution (3 × 50 mL), then with a saturated NaHCO3 solution (1 × 50 mL), and finally with deionised water (3 × 50 mL). The product was dried over MgSO4 and filtered through filter paper. Finally, the product was isolated as an oil after rotary evaporatation to dryness.
1H NMR (A5PDMS, 80 MHz): 2.67 (t, 4H, –NHCH2CH2CH2–), 1.49 (m, 4H, –NHCH2CH2CH2–), 0.52 (m, 4H, –NHCH2CH2CH2–), 0.07. Mn (A5PDMS): 4530 Da.
ATR-IR (A5PDMS): 2962, 2905, 1983, 1412, 1257, 1010, 863, 789, 700, 661.
The final product of MA5 was isolated as a pale yellow viscous liquid with a yield of 88%.
1H NMR (MA5, 80 MHz): 6.32 (d, 4H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 3.33 (q, 4H, –NHCH2CH2CH2–), 1.53 (m, 4H, –NHCH2CH2CH2–), 0.60 (m, 4H, –NHCH2CH2CH2–), 0.07 (s, 6H, –Si(CH3)2–). Mn (MA5): 4860 Da. Mn (MA25): 22980 Da.
CH–), 3.33 (q, 4H, –NHCH2CH2CH2–), 1.53 (m, 4H, –NHCH2CH2CH2–), 0.60 (m, 4H, –NHCH2CH2CH2–), 0.07 (s, 6H, –Si(CH3)2–). Mn (MA5): 4860 Da. Mn (MA25): 22980 Da.
ATR-IR (MA5): 2962, 2905, 1709, 1630, 1572, 1411, 1257, 1075, 1008, 862, 786, 700, 635.
In accordance with the general procedure, the longer chain MA25 was synthesised from amine-terminated PDMS (A25PDMS, 25000 g mol−1), in a yield of 80%.
The final product of MI was isolated as a deep brown viscous liquid with a yield of 85%.
1H NMR (MI5, 80 MHz): 6.65 (s, 4H, –CHCH–), 3.51 (t, 4H, –NCH2CH2CH2–), 1.60 (m, –NCH2CH2CH2–), 0.65 (m, 4H, –NCH2CH2CH2–), 0.07 (s, 6H, –Si(CH3)2–). Mn (MI5): 4860 Da. Mn (MI25): 22980 Da.
ATR-IR (MI5): 2962, 2905, 1800, 1714, 1691, 1409, 1257, 1077, 1008, 864, 786, 696, 661.
Analogously, the longer chain MI25 was synthesised from amine-terminated PDMS (A25PDMS, 22530 g mol−1), in a yield of 83%.
ATR-IR (MA5N): 2962, 2574, 1734, 1411, 1352, 1257, 1008, 864, 785, 697.
ATR-IR (MI5N): 2962, 1739, 1710, 1411, 1352, 1257, 1008, 864, 785, 697.
Results and discussion
A simple synthetic process was developed for functionalising an amine-terminated commercial PDMS with maleimides as shown in Scheme 1. It consisted of a two-step reaction – first, aminopropyl-terminated PDMS was reacted with maleic anhydride, under slight excess, to a maleamic acid amide-terminated PDMS (MA), which was either isolated and purified or continued reacting in the second step. In the second step, MA PDMS was reacted with acetic anhydride and triethyl amine to yield maleimide-terminated PDMS (MI). Both prepolymers, MA and MI, were consistently synthesised to high yields (>80%) from two different molar mass PDMSs (5 and 25 kDa). The total four resulting prepolymers were then cross-linked into networks using pentaerythritol tetrakis(3-mercaptopropionate) (PETMP), a four-functional Michael donor, under equimolar conditions, as shown in Scheme 1.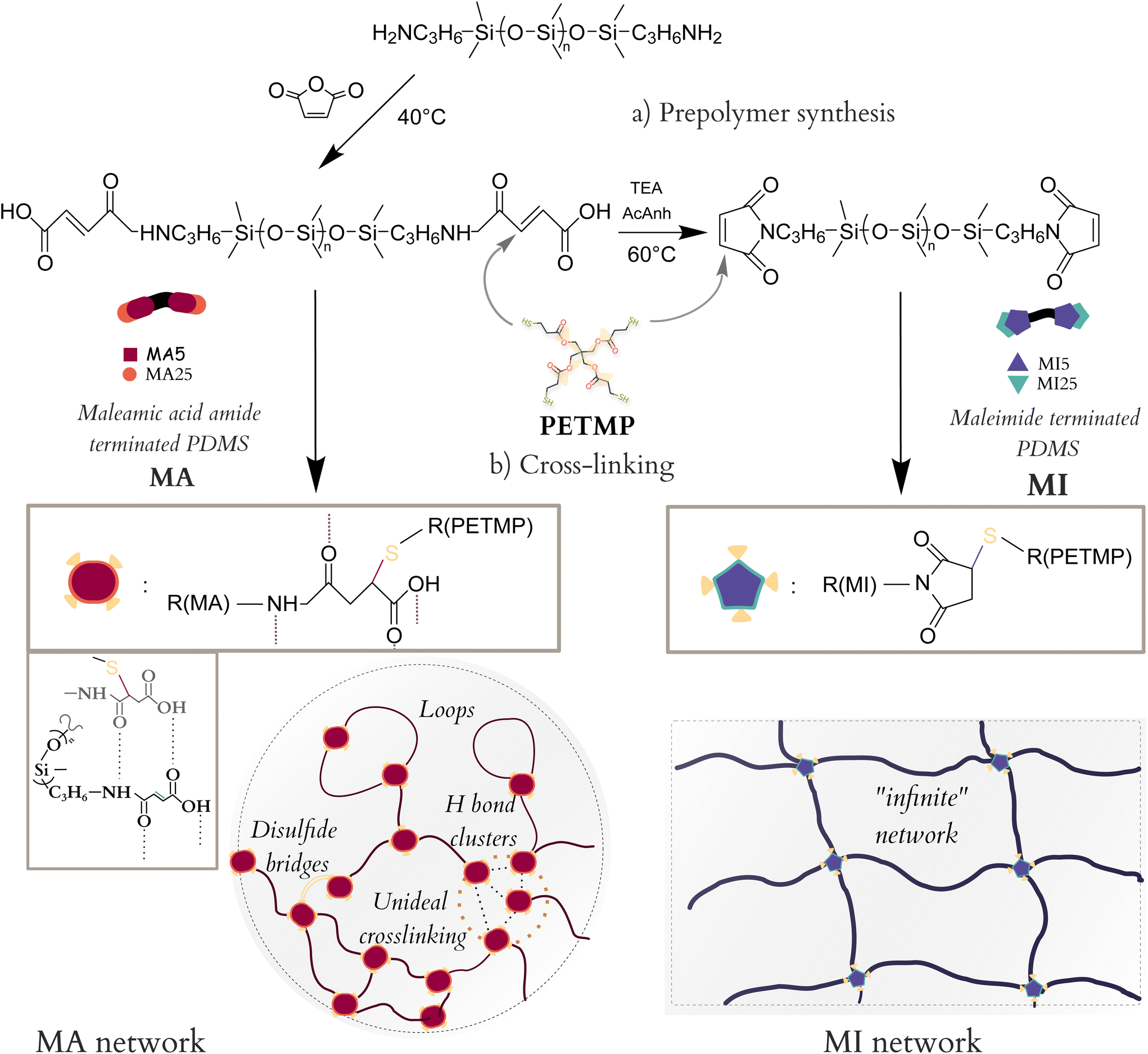 | ||
| Scheme 1 Synthesis of prepolymer species and their cross-linking via thiol-Michael addition into networks. Illustration of H bond interactions and intermolecular organisation of MA system (bottom left) and a traditional MI network (bottom right). | ||
From the ATR-IR spectra, the two products of the two synthetic steps are readily identifiable (Fig. 1a). The CO stretch is a characteristic peak for both the MA and MI systems. It is frequently found in the general range of 1650–1820 cm−1, depending on the functionalities (Fig. 1a).44 For the MA5, it appears as two peaks, at 1709 cm−1 and 1630 cm−1 showing the conjugated urethane linkage as well as the conjugated free carboxylic acid, which in combination with the broad N–H bending at 1572 cm−1 confirms the open structure. Meanwhile, MI, being an unsaturated cyclic imide, has three carbonyl stretches, at 1800, 1714 and 1691 cm−1,45 respectively. A5PDMS, in contrast, has no signal in this range, confirming the functionalisation of MA and MI.
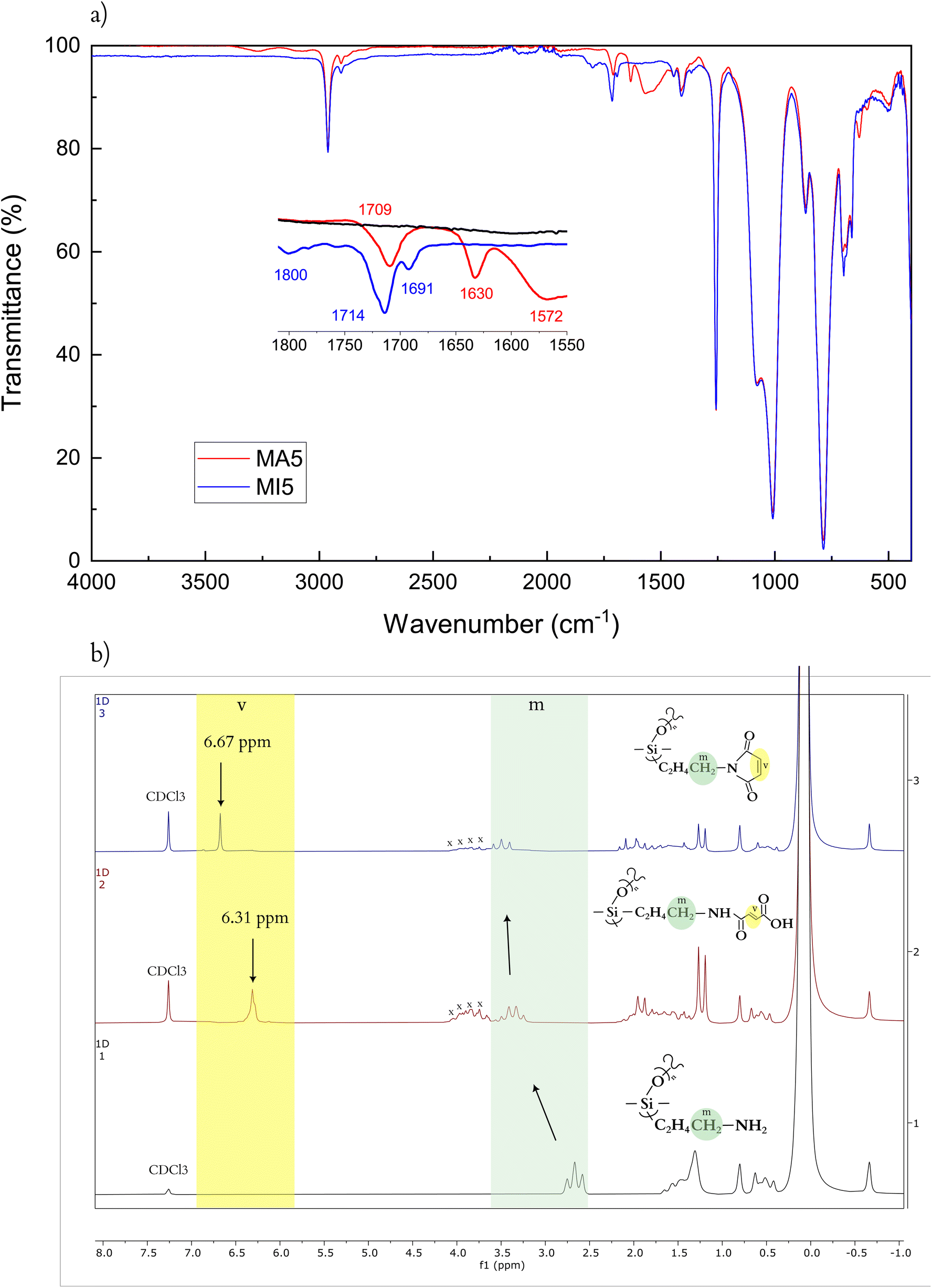 | ||
| Fig. 1 (a) ATR-IR spectra of A5PDMS (black), MA5 (red), and MI5 (blue) prepolymers. (b) 1H NMR of prepolymers A5PDMS (black), MA5 (red), and MI5 (blue). The position of –CHCH– is marked with “v”, the position of the terminal –CH2– of the aminopropyl group is marked with “m”, and peaks corresponding to solvent residue of 2mTHF with “x” (Fig. S1†). | ||
The synthesis of the prepolymers can be further investigated by 1H HMR (Fig. 1b), through the appearance of peaks attributed to the vinyl protons of MA5 and MI5, respectively. This peak appears with the conversion of A5PDMS to MA5 at 6.31 ppm, and shifts with the synthesis of MI5 to 6.67 ppm. Additionally, the functionalisation can be followed by the position and character of the protons associated with the terminal –CH2– of the aminopropyl group. The peak for A5PDMS is a triplet (2.58–2.75 ppm) that shifts to a characteristic 1:3:3:1 quartet upon the conversion to MA5 (3.22–3.47 ppm). This corresponds to the now secondary amine of the telechelic maleamic acid amide. Finally, in MI5, this peak is expressed as a triplet again (3.33–3.65 ppm) due to the loss of a proton upon imide formation.
Both MA and MI form a homogenous semi-transparent film at the end of the cross-linking according to the general procedure as previously described. However, their reactivities greatly differ. The MI5/25 networks regularly cure within an hour of solvent, even at room temperature. Meanwhile, the MA5/25 networks require post-curing at 75 °C overnight to ensure a high degree of network-forming reactions (Fig. S2†). This agrees well with the steric hindrance and lower reactivity of the vinyl bond of the MA system compared to that of the MI system.
All of the cross-linked networks have a significant gel fraction (>85%), comparable to classical PDMS networks, and do not dissolve in the presence of the solvent. Thus, cross-linking is considered successful and efficient (Table 1).
| Prepolymer | M n, g mol−1 | Yield, % | Cross-linked network | Gel. Frac., % |
|---|---|---|---|---|
| MA5 | 4860 | 81.6 | MA5 | 91.4 |
| MA25 | 22980 |
80.8 | MA25 | 90.3 |
| MI5 | 4860 | 85.3 | MI5 | 86.2 |
| MI25 | 22980 |
83.7 | MI25 | 88.1 |
ATR-IR spectra of the cross-linked network confirm the reaction of MA5 and MI5 prepolymers. Since the vinyl bond overlaps with the PDMS “fingerprint” region, cross-linking was confirmed by following the CO peaks. A shift of the CO peaks to higher wavelengths as a consequence of the consumption of the vinyl bond is observed for both systems after the cross-linking process – from 1709 to 1734 cm−1 for the MA5 network, and from 1691 and 1714 to 1710 and 1739 cm−1, respectively, for MI5. Additionally, the MA5 network spectrum shows a weak peak corresponding to the S–H stretch (2550 cm−1), suggesting excess PETMP in the system (Fig. 2a). However, a high level of gel fraction (90%) for MA5 systems indicates almost full integration of MA chains. MA chains possibly loop around PETMP, leading to one chain occupying two cross-linking thiol groups of the same PETMP molecule, as previously illustrated in Scheme 1b. Finally, the FTIR spectra of both the cross-linked MI and MA networks remain stable over a month (Fig. S3†). This stability is noteworthy, especially in the case of the MA5 network, considering the typical sensitivity of hydrogen-bond-rich systems to moisture.46
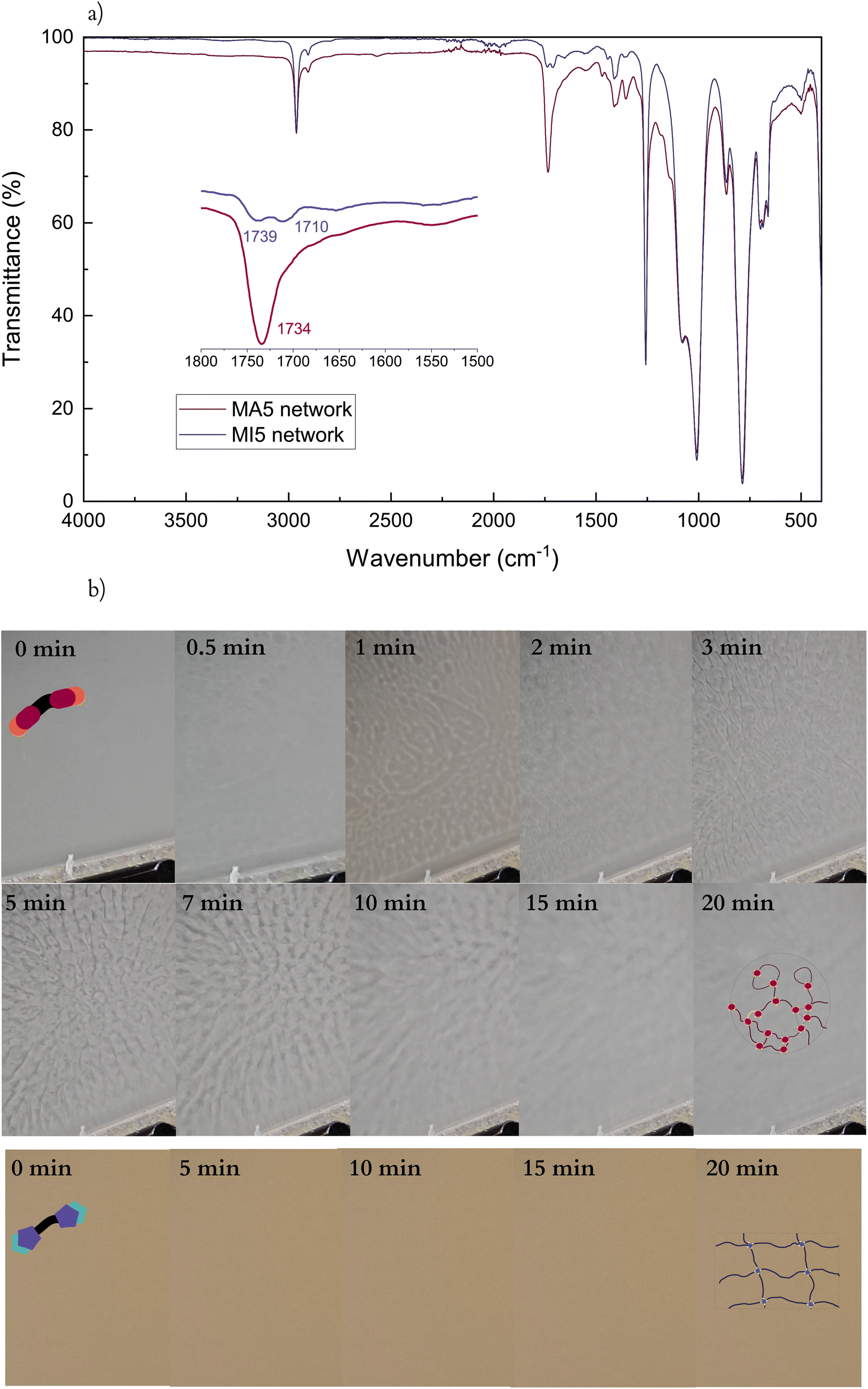 | ||
| Fig. 2 (a) ATR-IR spectra of MA5 (dark red) and MI5 (dark blue) cross-linked networks. (b) Images taken during the cross-linking reaction of both systems. A phase separation is evident in the MA5 network (upper) whereas the MI5 network (lower) does not undergo any visual change. | ||
During cross-linking, the MA networks transition through a visible phase separation within minutes of the start of the cross-linking process (Fig. 2b), unlike the MI networks. This difference in behaviour cannot be described by different amounts of solvents as both systems contain equal amounts. The ends of the MA prepolymers group in non-covalent clusters due to strong intermolecular interactions between them.47 These clusters begin reacting and the growing branched polymers separate from the monomer and solvent mixture. After 20 minutes of cross-linking, the reactive mixture has grown almost fully uniform and opaque, suggesting the bridging of these otherwise branched structures. Upon complete solvent evaporation, the film turns semi-transparent.
DSC spectra of the prepolymers and the corresponding cross-linked networks are given in Fig. S4.† The MI and MA short-chained prepolymers and networks do not show a characteristic cold crystallisation peak observed for all long-chained samples. This suggests that the increased relative polar group concentration and their intermolecular interactions inhibit crystallisation, since a similar reference system based on vinyl–hydride bonding shows clear crystallisation (Fig. S4a†).
Rheological properties of networks
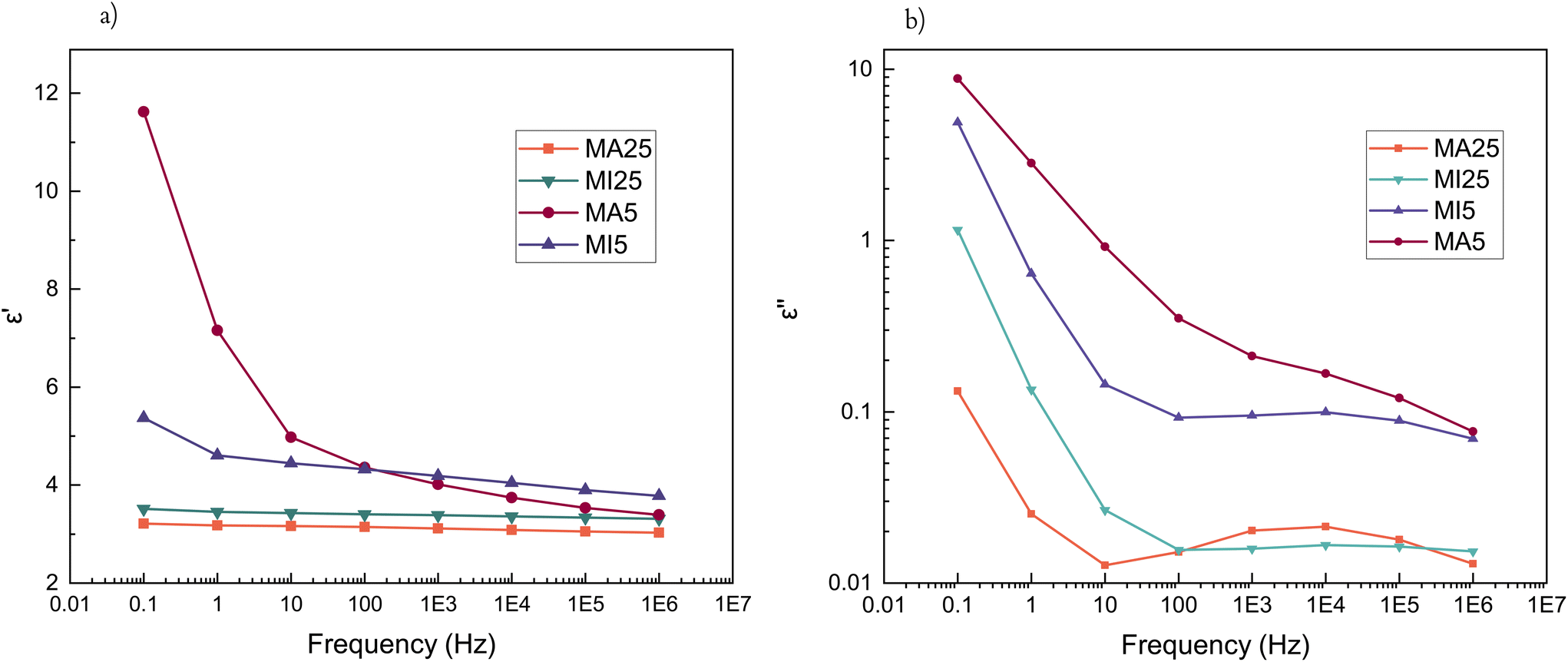
Rheological measurements were performed to investigate the hydrogen bonding capability of the two types of networks. The MA moiety possesses quadruple H bonding centres (hydroxyl and secondary amine as donors and two carbonyl oxygens as acceptors), compared to only acceptor dual H bonding centres (belonging to the carbonyl O atoms) of the MI systems (Scheme 1b).All cross-linked networks are found to be rubber-like (G′ > G′′) as expected (Fig. 3a and b).
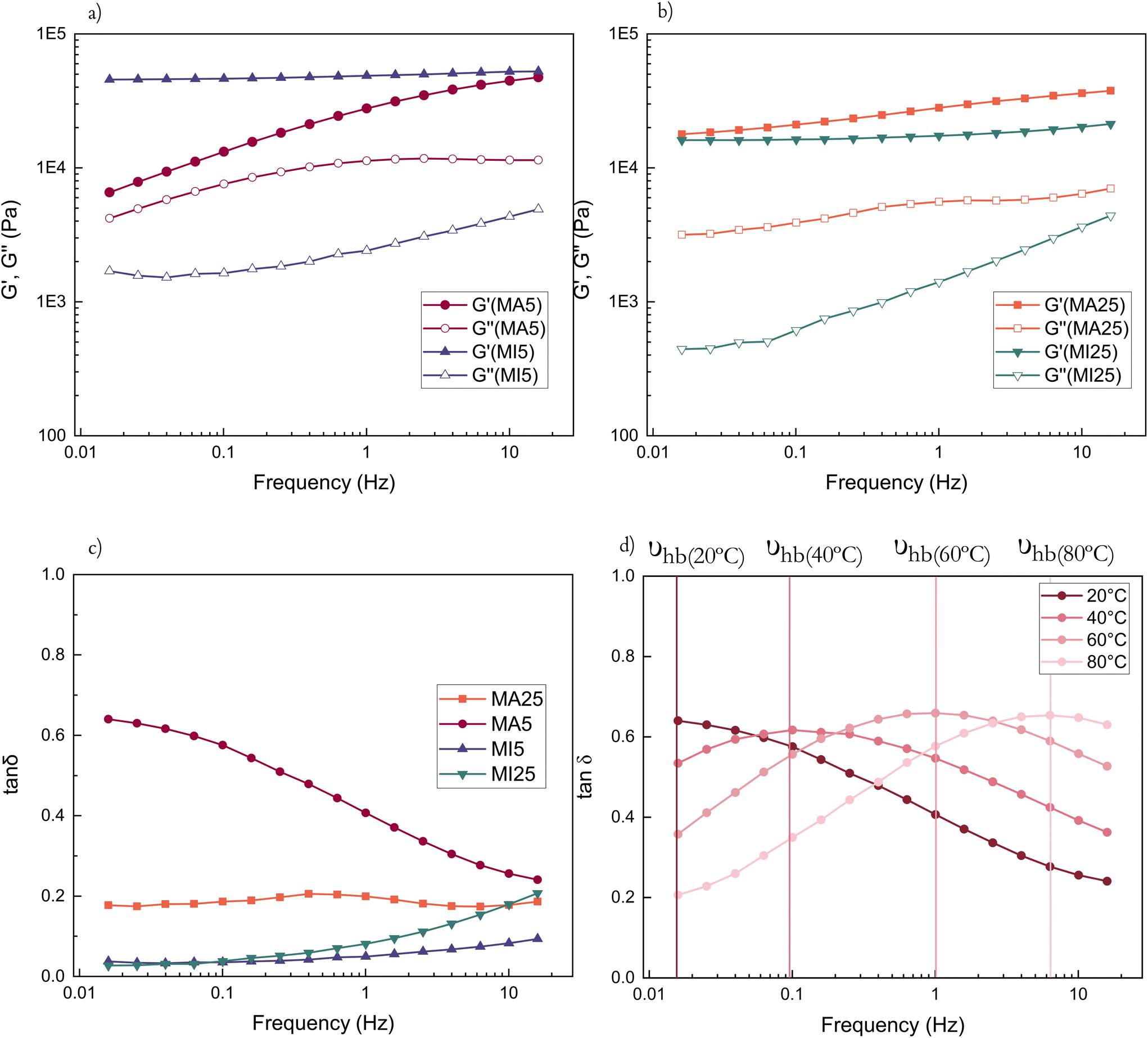 | ||
| Fig. 3 (a) Viscoelastic moduli of MA5 vs. MI5, (b) moduli of MA25 vs. MI25, (c) loss factors of all the samples, (d) the rheological loss factor of MA5 over four different temperatures with marked υhb corresponding to the peak position. | ||
For shorter chain networks, with a larger contribution of the MA moieties, hydrogen bonds function as a damping mechanism. This is evidenced through the MA5 network's energy dissipative response at lower frequencies (tanδ ≈ 0.65 (0.015 Hz)), where hydrogen bonds begin to break and reform, before their contribution to energy dissipation diminishes at higher frequencies as they become unable to respond quickly enough to the imposed strain (Fig. 3c). The maximum in the loss factor is outside the investigated frequency range in room temperature rheological experiments, but becomes apparent at increased temperatures (Fig. 3d). The peak of the loss factor occurs at a frequency at which the relaxation time of the hydrogen bonding-influenced chains matches the imposed frequency (marked υhb).48,49 With an increase in temperature, the chains have more kinetic energy and hydrogen bonds begin to break and reassemble rapidly. Hence their relaxation happens faster (Fig. 3d). In contrast, there are no damping peaks for the MI networks.
Generally, the MI networks have low tanδ values (<0.2) over the entire frequency range (Fig. S5a and S5b†). For the long-chain MA system, the rheological loss factor is essentially a constant value (Fig. S5c†). The maximum storage modulus of MA networks decreases with an increase in temperature, contrary to the MI networks – this is likely due to hydrogen bonding dominating the properties and their dissipative nature overcoming the entropic contribution at higher temperatures (Fig. S6†). Complex viscosities of the cross-linked samples can be found in ESI† and show shear-thinning behaviour, with viscosity decreasing with increasing frequency (Fig. S7†). This suggests that the terminal viscosity plateau appears for frequencies lower than the measured range. Interestingly, the MA5 network, due to increased relative concentration of hydrogen bonding domains, has a distinct behaviour with a smaller slope as a consequence of the wider, looser network possessing strong intermolecular organisation.
Non-linear mechanical properties of networks
Non-linear stress–strain measurements are found to complement the conclusions from linear viscoelastic measurements in contrasting the amide and imide functionalities. Table 2 summarizes common mechanical properties.| Sample | Y, MPa | σ ts, MPa | ε f, % |
|---|---|---|---|
| Young's modulus | Tensile strength | Strain at break | |
| MA5 | 0.824 ± 0.124 | 0.620 ± 0.022 | 134 ± 11.9 |
| MA25 | 0.255 ± 0.033 | 0.544 ± 0.045 | 454 ± 56.4 |
| MI5 | 0.957 ± 0.148 | 0.379 ± 0.148 | 46.6 ± 12.0 |
| MI25 | 0.423 ± 0.074 | 0.282 ± 0.003 | 112 ± 2.13 |
With respect to tensile test properties, the MI5 network shows an entirely linear response of the stress versus engineering strain, while MA5 has a noticeably more rubber-like behaviour with strain softening setting in after the linear regime (Fig. 4a). Considering that the two short-chain networks have comparable Young's moduli (0.824 and 0.967, respectively) and gel fractions (91.4% and 86.2%), this gives a clear indication of the contribution from hydrogen bonding in the MA networks. This type of intermolecular interaction between the functionalised ends of MA prepolymers, inherently situated close to the cross-linker, can be viewed as an additional local strengthening factor preventing failure. In particular, the MA5 network has both increased strain-at-break and tensile strength compared to its MI5 counterpart. Additionally, due to evidence of phase separation at the beginning of the cross-linking process and the lower reactivity giving sufficient time for reorganisation of the terminal polar clusters, the intermolecular structures caused by the hydrogen bonding between MA moieties might be contorting into loops, threads, and other similar entanglements. This would lead to multiple large branched structures held together by strong hydrogen bonding, unlike the conventional covalent “infinite network” of the MI network. These network imperfections can have a synergistic effect akin to a reversible cross-link.50,51
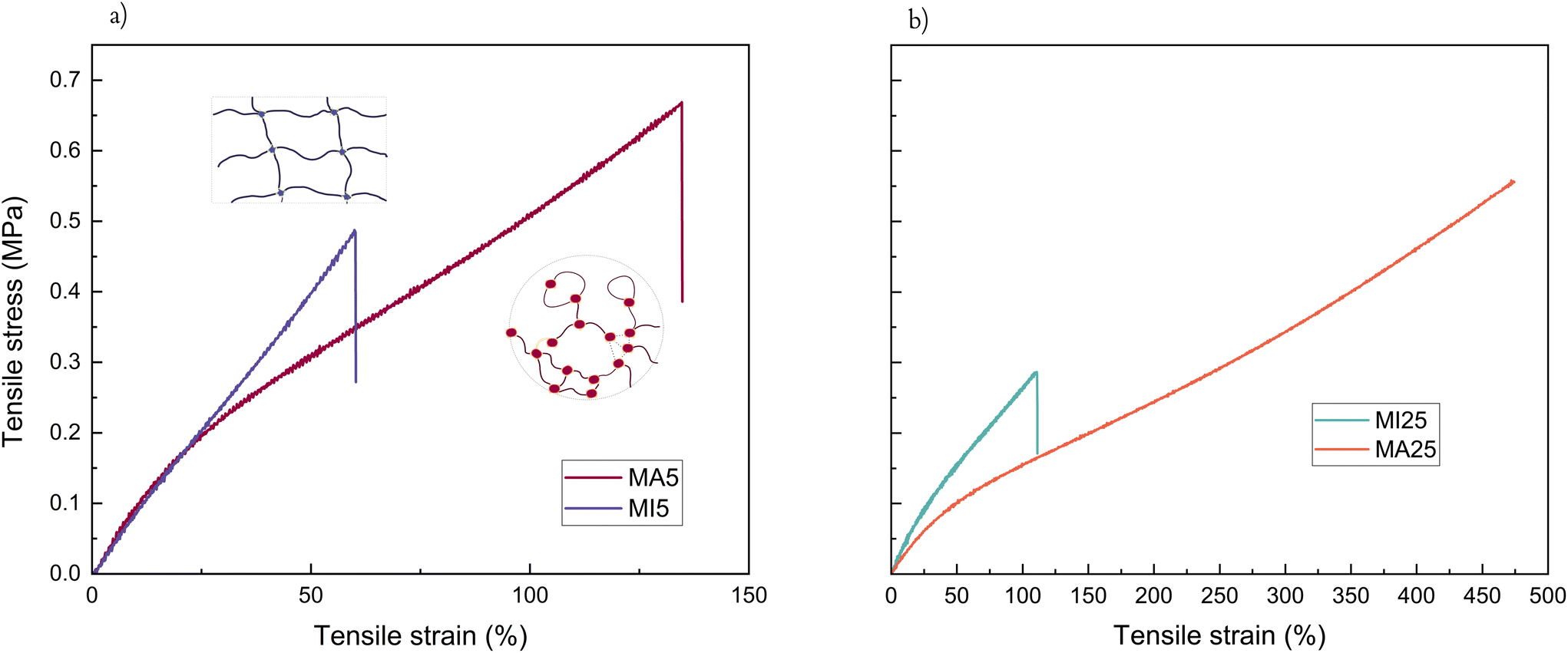 | ||
| Fig. 4 Tensile test of the four network samples where (a) short-chain networks (MA5 v MI5, illustration inset of the two hypothesized network structures), and (b) long-chain networks (MA25 v MI25). | ||
Similarly, the ultimate properties of the long-chain MA networks are improved due to the contribution of a larger level of chain association compared to the more brittle, hydrogen bond deficient MI networks (Fig. 4b).
Electrical properties of the networks
The electrical properties of PDMS elastomers play a vital role in their potential application as dielectric elastomers. The incorporation of a polar cross-linker and the polar terminal MA and MI moieties should improve dielectric performance, in particular MA, due to its secondary amine and hydroxyl group.All of the synthesised networks have a higher dielectric permittivity at room temperature than the commonly used commercial PDMS elastomer (Sylgard™ 184, εr = 2.73, as stated by the producer's specification sheet) (Fig. 5a). Ionic polarisation is typically observed at lower frequencies (<1 MHz), where there is sufficient times for charged entities to respond to the changing electric field. As expected, MA5 has a significantly larger contribution of ionic polarisation than its MI5 counterpart, which results in a significant improvement in its relative dielectric permittivity at lower frequencies. The increased εr is stronger for short-chain networks due to the relative increase in the concentration of polar groups.
 | ||
| Fig. 5 (a) Relative dielectric permittivity and (b) the dielectric loss of prepared elastomers. | ||
Ion polarisation and mobilisation in an electric field at lower frequencies lead to an increase in dielectric loss at room temperature (Fig. 5b). In the case of the MI networks, there are fewer polarisable centres, and their orientation is strained due to the spatial factor of the larger maleimide group. This is particularly notable for short-chain networks, where the concentration of polar terminal groups is larger. Consequently, the dielectric loss of MI5 network is smaller and starts to increase at lower frequencies than that of the MA5 counterpart.
The influence of temperature on the dielectric properties can further emphasise the distinct difference between the MA and MI networks. The short-chain MA5 network has a stronger temperature dependence than MI5 (Fig. 6). The MA5 network shows a strong temperature dependency on the dielectric permittivity for frequencies below 1000 Hz. The MI5 network has temperature-independent properties in the frequency range of approximately 102–106 Hz, whereas at lower frequencies, there is a slight increase in the dielectric permittivity with decreased frequency. However, high dielectric losses make further optimisation a requirement (Fig. 5b and S8†). The dielectric permittivity's dependence on temperature for the long-chain networks and an overview of the permittivity changes over a broader temperature range are given in Fig. S9 and S10,† respectively.
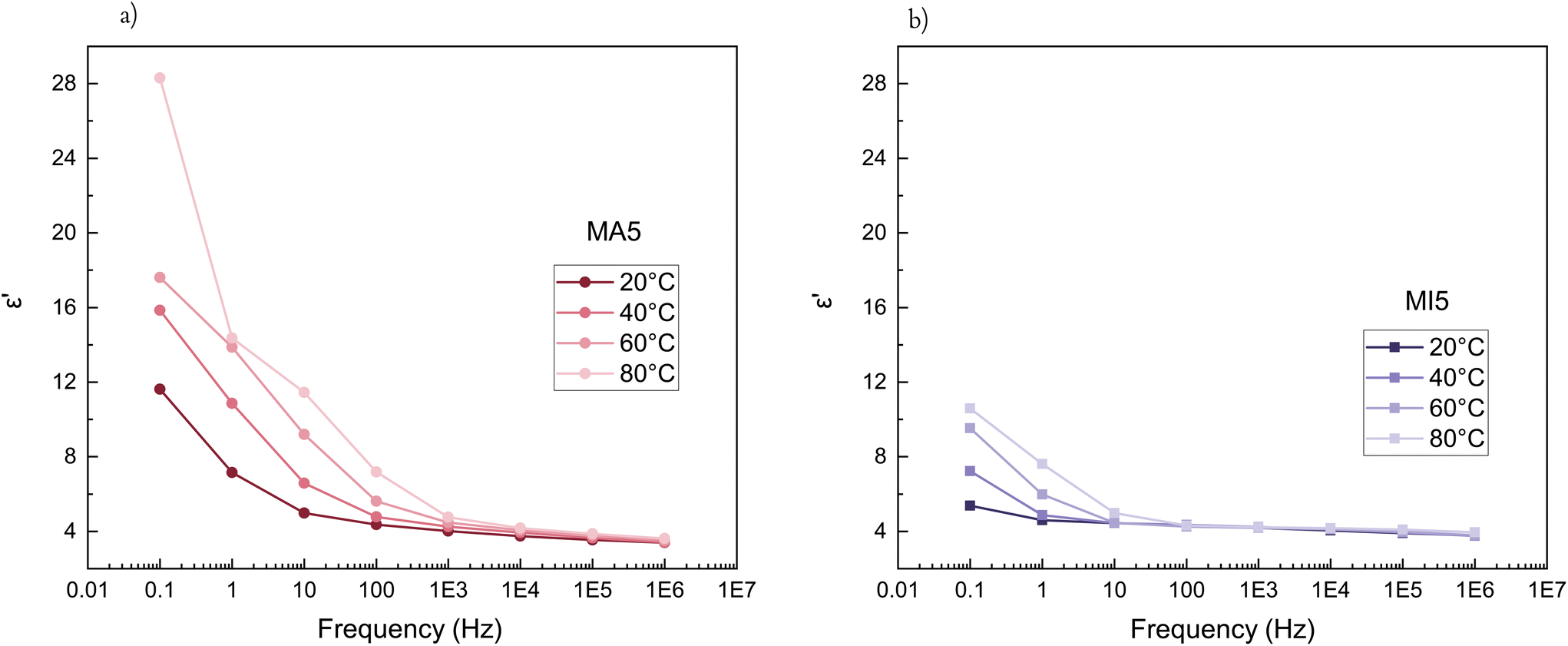 | ||
| Fig. 6 Relative dielectric permittivity of (a) MA5, and (b) MI5 at different temperatures. | ||
The hydrophilicity of the MA and MI networks is investigated. Both the short-chain MA and MI networks show significantly increased hydrophilicity (81° for MA5, 91° for MI5) compared to the literature data on PDMS (113.5°),52 while the longer-chain networks have intermediate values (99° for MA25, 95° for MI25) (Fig. S11†). MI and MA short-chain networks can then be considered hydrophilic or wettable PDMS, which increases their application potential.
Furthermore, both MI and MA networks show promise as dielectric elastomer actuators due to their high electric breakdown strength (Fig. S12,†Table 3), compared to commercial PDMS films (e.g. Sylgard™ 184, EBD = 24 kV mm−1, as stated by the producer's specification sheet). Commercial films commonly use a significant volume fraction of fillers, hence the performance of MA and MI PDMS can be further improved by careful selection of additives and further optimisation of the cross-linking process to improve homogeneity.
| Sample | E BD, kV mm−1 |
|---|---|
| MA5 | 65 ± 16 |
| MA25 | 64 ± 9 |
| MI5 | 77 ± 27 |
| MI25 | 93 ± 16 |
Investigation of self-healing of MA networks
Self-healing is not a property frequently associated with tightly cross-linked covalent networks. Due to constrained molecular mobility, the point of fracture cannot be circumvented through intermolecular diffusion. Normally, self-healing can be achieved by instead designing a dynamic covalent bonding network, where the reversibility of the chemical linkages depending on external conditions results in self-healing. Another approach is to design a thermoplastic elastomer with strong intermolecular interactions. Remarkably, both short-chain and long-chain MA networks have the ability to self-heal, whereas MI networks do not.In terms of self-healing efficiency, the MA networks show high recovery from an imposed cut (Fig. 7). Self-healing efficiency, as defined in eqn (1), for both MA5 and MA25 systems equaled 85%.
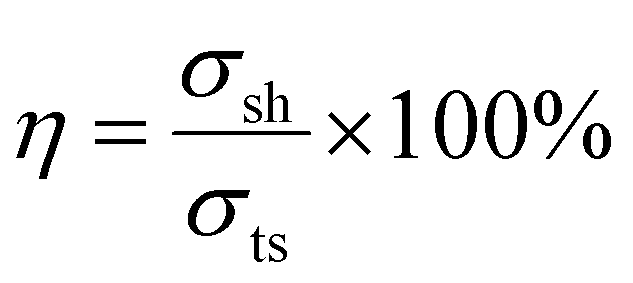 | (1) |
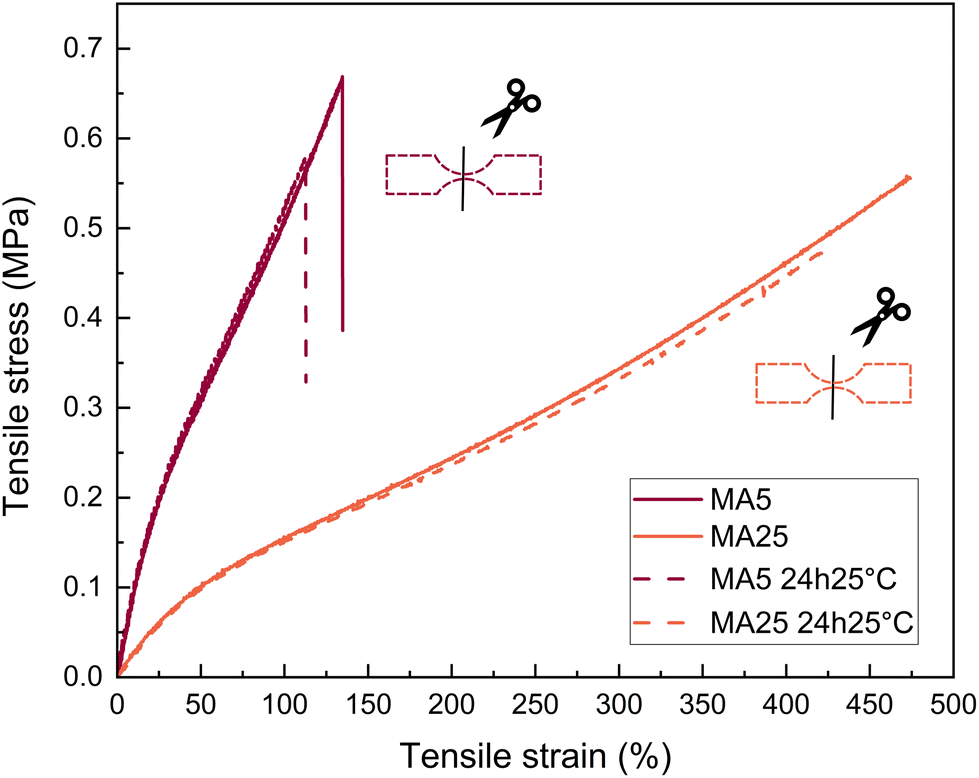 | ||
| Fig. 7 Mechanical test comparison of pristine and self-healed over 24 h sample of MA5 and MA25. | ||
The ability of MA networks to self-heal to high efficiency is explained by the changed intermolecular organisation of the material. The network has both the traditional cross-linked network's advantages while allowing enhanced mobility to enable self-healing. The strong hydrogen bonding domains preserve the structural integrity and high strength of the network. Conversely, MI networks do not have this ability.
While some hydrogen bonding systems show fast healing without external stimulus, occurring in seconds,53 MA networks seem to require prolonged healing times. This is likely due to the constraints in the network's mobility. However, after healing for 1 h at 75 °C, a level of recovery can be achieved (Fig. S13†). This correlates well with the rheological and electrical data presented, suggesting that the MA networks are temperature dependant systems due to the equilibrium of the hydrogen bonding domains that govern its properties.
Notably, upon mechanical testing of the self-healed samples, the inflicted cut was not the point of failure (Fig. S14†). This highlights the ability of the strong hydrogen bonding domains to retain the material's original strength. Optical microscopy shows that the cut remains visible in the material, despite some diffusive processes beginning to bridge it (Fig. 8 and S13†). This interdiffusion characteristic of MA networks further points out the difference between the organisation of MA and MI networks, which do not heal at all nor show any extent of crack closure.
 | ||
| Fig. 8 Optical microscopy at 400× magnification of MA25 cross-linked network with an imposed cut visible but healed after self-healing for 24 h under room temperature. | ||
Conclusion
Two different cross-linked networks based on Michael addition were prepared. MI networks show fast and facile cross-linking, and improved dielectric permittivity, but limitations in terms of mechanical properties which require future optimisation. MA networks exhibit behaviour indicative of strong intermolecular interactions that lead to significantly different properties. The increased presence of both H-bond donating and accepting groups leads to intermolecular arrangements like loops and threads that make the material more resilient, enable self-healing, and significantly improve dielectric permittivity. Self-healing of MA networks is efficient and does not require any specific conditions. Rheological measurements and dielectric spectroscopy were used as primary methods for differentiating these two chemically similar, yet ultimately different systems.Potential future research will further explore the maleic amide/maleimide networks with a stoichiometric variation of components and consider integrating common and/or specialised additives to enhance the properties of the ultimate composites. Due to their self-healing properties, high permittivity and electric breakdown strength, these materials can be of great use for wearable electronics in the form of dielectric elastomers.
Author contributions
P. R. conducted the synthesis, analyzed data, and wrote the draft. L. Y. assisted with all measurements and the data analysis, A. L. S. and A. E. D. supervised the work and assisted with corrections to the draft. The experimental plan was developed by all authors.Data availability
Data for this article, including unprocessed files of all the measurements performed, are available at Open Science Framework (https://doi.org/10.17605/OSF.IO/UNEQ8/).Conflicts of interest
There are no conflicts of interest to declare by any author.Acknowledgements
The authors gratefully acknowledge the funding from the Novo Nordisk Foundation through the Challenge Program, grant no NNF22OC0071130.References
- P. A. Klonos, Crystallization, Glass Transition, and, Molecular Dynamics in PDMS of Low Molecular Weights: A Calorimetric and Dielectric Study, Polymer, 2018, 159, 169–180, DOI:10.1016/j.polymer.2018.11.028.
- E. Hajiesmaili and D. R. Clarke, Dielectric Elastomer Actuators, J. Appl. Phys., 2021, 129(15), 151102, DOI:10.1063/5.0043959.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, The Current State of Silicone-Based Dielectric Elastomer Transducers, Macromol. Rapid Commun., 2016, 37(5), 378–413, DOI:10.1002/marc.201500576.
- I. Miranda, A. Souza, P. Sousa, J. Ribeiro, E. M. S. Castanheira, R. Lima and G. Minas, Properties and Applications of PDMS for Biomedical Engineering: A Review, J. Funct. Biomater., 2022, 13(1), 2, DOI:10.3390/jfb13010002.
- R. Sønderbæk-Jørgensen, S. Meier, K. Dam-Johansen, A. L. Skov and A. E. Daugaard, Reactivity of Polysilazanes Allows Catalyst-Free Curing of Silicones, Macromol. Mater. Eng., 2022, 307(9), 2200157, DOI:10.1002/mame.202200157.
- A. H. A. Razak, L. Yu and A. L. Skov, Voltage-Stabilised Elastomers with Increased Relative Permittivity and High Electrical Breakdown Strength by Means of Phase Separating Binary Copolymer Blends of Silicone Elastomers, RSC Adv., 2017, 7(29), 17848–17856, 10.1039/C7RA02620J.
- H. Silau, N. B. Stabell, F. R. Petersen, M. Pham, L. Yu and A. L. Skov, Weibull Analysis of Electrical Breakdown Strength as an Effective Means of Evaluating Elastomer Thin Film Quality, Adv. Eng. Mater., 2018, 20(9), 1800241, DOI:10.1002/adem.201800241.
- T. Rambarran, F. Gonzaga, M. A. Brook, F. Lasowski and H. Sheardown, Amphiphilic Thermoset Elastomers from Metal-Free, Click Crosslinking of PEG-Grafted Silicone Surfactants, J. Polym. Sci., Part A: Polym. Chem., 2015, 53(9), 1082–1093, DOI:10.1002/pola.27539.
- D. Damiron, N. Okhay, S. A. Akhrass, P. Cassagnau and E. Drockenmuller, Crosslinked PDMS Elastomers and Coatings from the Thermal Curing of Vinyl-Functionalized PDMS and a Diazide Aliphatic Crosslinker, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(1), 98–107, DOI:10.1002/pola.24991.
- A. S. Fawcett, J. B. Grande and M. A. Brook, Rapid, Metal-Free Room Temperature Vulcanization Produces Silicone Elastomers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(3), 644–652, DOI:10.1002/pola.26414.
- H. P. Xiang, M. Z. Rong and M. Q. Zhang, A Facile Method for Imparting Sunlight Driven Catalyst-Free Self-Healability and Recyclability to Commercial Silicone Elastomer, Polymer, 2017, 108, 339–347, DOI:10.1016/j.polymer.2016.12.006.
- L. J. Karas, C.-H. Wu, R. Das and J. I.-C. Wu, Hydrogen Bond Design Principles, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10(6), e1477, DOI:10.1002/wcms.1477.
- S. Jeong, A. L. Skov and A. E. Daugaard, Recycling of Dielectric Electroactive Materials Enabled through Thermoplastic PDMS, RSC Adv., 2022, 12(14), 8449–8457, 10.1039/D2RA00421F.
- Z. Xie, B.-L. Hu, R.-W. Li and Q. Zhang, Hydrogen Bonding in Self-Healing Elastomers, ACS Omega, 2021, 6(14), 9319–9333, DOI:10.1021/acsomega.1c00462.
- F. B. Madsen, L. Yu and A. L. Skov, Self-Healing, High-Permittivity Silicone Dielectric Elastomer, ACS Macro Lett., 2016, 5(11), 1196–1200, DOI:10.1021/acsmacrolett.6b00662.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, A Highly Stretchable Autonomous Self-Healing Elastomer, Nat. Chem., 2016, 8(6), 618–624, DOI:10.1038/nchem.2492.
- P.-F. Cao, B. Li, T. Hong, J. Townsend, Z. Qiang, K. Xing, K. D. Vogiatzis, Y. Wang, J. W. Mays, A. P. Sokolov and T. Saito, Superstretchable, Self-Healing Polymeric Elastomers with Tunable Properties, Adv. Funct. Mater., 2018, 28(22), 1800741, DOI:10.1002/adfm.201800741.
- J. Kang, D. Son, G.-J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B.-H. Tok and Z. Bao, Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin, Adv. Mater., 2018, 30(13), 1706846, DOI:10.1002/adma.201706846.
- S. Ge, Y.-H. Tsao and C. M. Evans, Polymer Architecture Dictates Multiple Relaxation Processes in Soft Networks with Two Orthogonal Dynamic Bonds, Nat. Commun., 2023, 14(1), 7244, DOI:10.1038/s41467-023-43073-w.
- J. Xu, L. Zhu, Y. Nie, Y. Li, S. Wei, X. Chen, W. Zhao and S. Yan, Advances and Challenges of Self-Healing Elastomers: A Mini Review, Materials, 2022, 15, 5993, DOI:10.3390/ma15175993.
- B. H. Northrop, S. H. Frayne and U. Choudhary, Thiol–Maleimide “Click” Chemistry: Evaluating the Influence of Solvent, Initiator, and Thiol on the Reaction Mechanism, Kinetics, and Selectivity, Polym. Chem., 2015, 6(18), 3415–3430, 10.1039/C5PY00168D.
- L.-T. T. Nguyen, M. T. Gokmen and F. E. Du Prez, Kinetic Comparison of 13 Homogeneous Thiol–X Reactions, Polym. Chem., 2013, 4(22), 5527–5536, 10.1039/c3py00743j.
- A. Wall, A. G. Wills, N. Forte, C. Bahou, L. Bonin, M. T. Ma, V. Chudasama and J. R. Baker, One-Pot Thiol-Amine Bioconjugation to Maleimides; Simultaneous Stabilisation and Dual Functionalisation, Chem. Sci., 2020, 11, 11455–11460, 10.1039/D0SC05128D.
- W. Huang, X. Wu, X. Gao, Y. Yu, H. Lei, Z. Zhu, Y. Shi, Y. Chen, M. Qin, W. Wang and Y. Cao, Maleimide–Thiol Adducts Stabilized through Stretching, Nat. Chem., 2019, 11(4), 310–319, DOI:10.1038/s41557-018-0209-2.
- M. Lahnsteiner, A. Kastner, J. Mayr, A. Roller, B. K. Keppler and C. R. Kowol, Improving the Stability of Maleimide–Thiol Conjugation for Drug Targeting, Chem. – Eur. J., 2020, 26(68), 15867–15870, DOI:10.1002/chem.202003951.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Self-Hydrolyzing Maleimides Improve the Stability and Pharmacological Properties of Antibody-Drug Conjugates, Nat. Biotechnol., 2014, 32(10), 1059–1062, DOI:10.1038/nbt.2968.
- L. N. Tumey, M. Charati, T. He, E. Sousa, D. Ma, X. Han, T. Clark, J. Casavant, F. Loganzo, F. Barletta, J. Lucas and E. I. Graziani, Mild Method for Succinimide Hydrolysis on ADCs: Impact on ADC Potency, Stability, Exposure, and Efficacy, Bioconjugate Chem., 2014, 25(10), 1871–1880, DOI:10.1021/bc500357n.
- L. Li, X. Qin, H. Mei, L. Liu and S. Zheng, Reprocessed and Shape Memory Networks Involving Poly(Hydroxyl Ether Ester) and Polydimethylsiloxane through Diels-Alder Reaction, Eur. Polym. J., 2021, 160, 110811, DOI:10.1016/j.eurpolymj.2021.110811.
- C. Pozos Vázquez, R. Tayouo, C. Joly-Duhamel and B. Boutevin, UV-Curable Bismaleimides Containing Poly(Dimethylsiloxane): Use as Hydrophobic Agent, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(10), 2123–2134, DOI:10.1002/pola.23980.
- J. Bai, H. Li, Z. Shi and J. Yin, An Eco-Friendly Scheme for the Cross-Linked Polybutadiene Elastomer via Thiol–Ene and Diels–Alder Click Chemistry, Macromolecules, 2015, 48(11), 3539–3546, DOI:10.1021/acs.macromol.5b00389.
- M. Yang, J. Mao, W. Nie, Z. Dong, D. Wang, Z. Zhao and X. Ji, Facile Synthesis and Responsive Behavior of PDMS-b-PEG Diblock Copolymer Brushes via Photoinitiated “Thiol-Ene” Click Reaction, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(10), 2075–2083, DOI:10.1002/pola.25985.
- J. Zhang, Y. Chen and M. A. Brook, Facile Functionalization of PDMS Elastomer Surfaces Using Thiol–Ene Click Chemistry, Langmuir, 2013, 29(40), 12432–12442, DOI:10.1021/la403425d.
- P. Lucas, E. Fleury, J.-F. Estur, V. Lapinte and J.-J. Robin, Peroxide-Grafted PDMS: Hydrosilylation Reaction and Thiol-Ene Chemistry as an Alternative Pathway, Macromol. Chem. Phys., 2009, 210(22), 1933–1941, DOI:10.1002/macp.200900310.
- K. D. Q. Nguyen, W. V. Megone, D. Kong and J. E. Gautrot, Ultrafast Diffusion-Controlled Thiol–Ene Based Crosslinking of Silicone Elastomers with Tailored Mechanical Properties for Biomedical Applications, Polym. Chem., 2016, 7(33), 5281–5293, 10.1039/C6PY01134A.
- I. Krizhanovskiy, M. Temnikov, Y. Kononevich, A. Anisimov, F. Drozdov and A. Muzafarov, The Use of the Thiol-Ene Addition Click Reaction in the Chemistry of Organosilicon Compounds: An Alternative or a Supplement to the Classical Hydrosilylation?, Polymers, 2022, 14(15), 3079, DOI:10.3390/polym14153079.
- Y. Pang, J. J. Koh, Z. Li and C. He, Reactive Functionally Terminated Polyorganosiloxanes, in Silicon Containing Hybrid Copolymers, ed. C. He and Z. Li, 2020. DOI:10.1002/9783527823499.ch2.
- M. Kajiyama, Y. Nishikata, M. Kakimoto and Y. Imai, Synthesis and Properties of Block Copolymers Based on Amine-Terminated Polydimethylsiloxane and Aromatic Polyamides, Polym. J., 1986, 18(10), 735–740, DOI:10.1295/polymj.18.735.
- A. Genest, S. Binauld, E. Pouget, F. Ganachaud, E. Fleury and D. Portinha, Going beyond the Barriers of Aza-Michael Reactions: Controlling the Selectivity of Acrylates towards Primary Amino-PDMS, Polym. Chem., 2017, 8(3), 624–630, 10.1039/C6PY01802E.
- M. A. Tallon, Reactions Involving Maleic Anhydride, in Handbook of Maleic Anhydride Based Materials: Syntheses, Properties and Applications, ed. O. M. Musa, Springer International Publishing, Cham, 2016, pp. 59–149. DOI:10.1007/978-3-319-29454-4_2.
- P. Schmidt and S. Eschig, An Industrial Applicable Method for the Synthesis of N-Alkylated Maleimides Based on Fatty Amines, Eur. J. Lipid Sci. Technol., 2019, 121(1), 1800320, DOI:10.1002/ejlt.201800320.
- P. A. Szijj, C. Bahou and V. Chudasama, Minireview: Addressing the Retro-Michael Instability of Maleimide Bioconjugates, Drug Discovery Today: Technol., 2018, 30, 27–34, DOI:10.1016/j.ddtec.2018.07.002.
- S. Kang, Y. Kim, Y. Song, J. U. Choi, E. Park, W. Choi, J. Park and Y. Lee, Comparison of PH-Sensitive Degradability of Maleic Acid Amide Derivatives, Bioorg. Med. Chem. Lett., 2014, 24(10), 2364–2367, DOI:10.1016/j.bmcl.2014.03.057.
- Y. Wang, F. Xie, L. Liu, X. Xu, S. Fan, W. Zhong and X. Zhou, Development of Applicable Thiol-Linked Antibody–Drug Conjugates with Improved Stability and Therapeutic Index, Drug Delivery, 2022, 29(1), 754–766, DOI:10.1080/10717544.2022.2039807.
- D. E. Ames and T. F. Grey, The Synthesis of Some N-Hydroxyimides, J. Chem. Soc., 1955, 631–636, 10.1039/JR9550000631.
- P. T. McKittrick and J. E. Katon, Infrared and Raman Group Frequencies of Cyclic Imides, Appl. Spectrosc., 1990, 44(5), 812–817 CrossRef CAS.
- C.-L. He, F.-C. Liang, L. Veeramuthu, C.-J. Cho, J.-S. Benas, Y.-R. Tzeng, Y.-L. Tseng, W.-C. Chen, A. Rwei and C.-C. Kuo, Super Tough and Spontaneous Water-Assisted Autonomous Self-Healing Elastomer for Underwater Wearable Electronics, Adv. Sci., 2021, 8(21), 2102275, DOI:10.1002/advs.202102275.
- K. Xing, M. Tress, P. Cao, S. Cheng, T. Saito, V. N. Novikov and A. P. Sokolov, Hydrogen-Bond Strength Changes Network Dynamics in Associating Telechelic PDMS, Soft Matter, 2018, 14(7), 1235–1246, 10.1039/C7SM01805C.
- D.-D. Zhang, Y.-B. Ruan, B.-Q. Zhang, X. Qiao, G. Deng, Y. Chen and C.-Y. Liu, A Self-Healing PDMS Elastomer Based on Acylhydrazone Groups and the Role of Hydrogen Bonds, Polymer, 2017, 120, 189–196, DOI:10.1016/j.polymer.2017.05.060.
- J. Li, C. L. Lewis, D. L. Chen and M. Anthamatten, Dynamic Mechanical Behavior of Photo-Cross-Linked Shape-Memory Elastomers, Macromolecules, 2011, 44(13), 5336–5343, DOI:10.1021/ma2004019.
- P. Hu, J. Madsen, Q. Huang and A. L. Skov, Elastomers without Covalent Cross-Linking: Concatenated Rings Giving Rise to Elasticity, ACS Macro Lett., 2020, 9(10), 1458–1463, DOI:10.1021/acsmacrolett.0c00635.
- N. B. Tito, C. Creton, C. Storm and W. G. Ellenbroek, Harnessing Entropy to Enhance Toughness in Reversibly Crosslinked Polymer Networks, Soft Matter, 2019, 15(10), 2190–2203, 10.1039/C8SM02577K.
- A. Mata, A. J. Fleischman and S. Roy, Characterization of Polydimethylsiloxane (PDMS) Properties for Biomedical Micro/Nanosystems, Biomed. Microdevices, 2005, 7(4), 281–293, DOI:10.1007/s10544-005-6070-2.
- Y. Yao, H. Tai, D. Wang, Y. Jiang, Z. Yuan and Y. Zheng, One-Pot Preparation and Applications of Self-Healing, Self-Adhesive PAA-PDMS Elastomers, J. Semicond., 2019, 40(11), 112602, DOI:10.1088/1674-4926/40/11/112602.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00169a |
| This journal is © The Royal Society of Chemistry 2024 |