
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCathodic electrodeposition of polymer networks as ultrathin films on 3-D micro-architected electrodes†
Zhaoyi
Zheng
 a,
Anton B.
Resing
a,
Wenlu
Wang
a and
Jörg G.
Werner
*abc
a,
Anton B.
Resing
a,
Wenlu
Wang
a and
Jörg G.
Werner
*abc
aDivision of Materials Science and Engineering, Boston University, Boston, MA 02215, USA. E-mail: jgwerner@bu.edu
bDepartment of Mechanical Engineering, Boston University, Boston, MA 02215, USA
cDepartment of Chemistry, Boston University, Boston, MA 02215, USA
First published on 18th September 2024
Abstract
Advances in precision coatings are critical in enhancing the functionality of porous materials and the performance of three-dimensionally (3-D) micro-architected devices in applications ranging from molecular sorption and separation to energy storage and conversion. To address this need, we report the cathodic electrodeposition of polymer networks (EPoN) that utilizes the coupling between pre-synthesized polymers with electrochemically active end groups and a complementary crosslinker to form a step-growth polymer network. The electrochemically mediated crosslinking reaction confines the network formation to the electrode surface in a passivating and self-limiting film growth, preventing uncontrolled precipitation and deposition away from the surface. The cathodic electrodeposition is compatible with a variety of conductive substrates, which is demonstrated for 3-D carbons and metals with micron-scale pores of high aspect ratio. The entire pore surface of the 3-D electrodes is enveloped by a conformal polymer thin film that is free of detectable defects and highly electronically insulating for its potential use as an ultrathin artificial electrolyte interphase or solid polymer electrolyte. Since our EPoN concept decouples the polymer functionality from its electrodeposition chemistry, we envision it to be a widely applicable method to coat various conductive non-planar and micro-architected 3-D substrates with polymers of broad functionalities.
Introduction
Non-planar or porous three-dimensional (3-D) mesoscaled materials are essential in applications that require efficient mass transport coupled to interfacial processes, such as energy storage, electrocatalytic conversions, or sorption and separation technologies.1–4 Coating such porous architected materials with functional thin films can lead to improved performance due to the combination of large surface areas with bulk material properties of an interphase that exhibits a finite thickness compared to a volume-less interface, including electronic insulation, mechanical and chemical protection, or molecular selectivity, while ensuring fast mass transport due to ultrashort diffusion distances through the thin film coating.5,6 Polymer networks are of particular interest as application-tailored coatings on high surface area materials for their large parameter space of chemical and physical properties through monomer functionality and network topology, respectively, combined with molecular and ionic permeability.7–11 While fabrication methods for micro/nano-scaled thin films of polymer networks on planar substrates are well established, such as spin-coating of reactive macro/monomers and subsequent crosslinking, as well as (self-limiting) electrospray deposition on macroscale architectures, they face severe limitations on non-planar substrates and in 3-D materials with micron-sized and high aspect-ratio features and pores.12,13 For example, a major challenge in realizing 3-D micro-interdigitated batteries is the fabrication of an ultrathin but uniform and defect-free solid electrolyte layer that separates the two interdigitated electrodes.14 Thus, the development of new deposition methods applicable to emerging 3-D architected materials is critical in the effort to transition from traditional randomly distributed composites to next-generation interdigitated multi-layer designs with tailor-made and uniform functional interphases.Most established coating techniques for 3-D microstructured substrates utilize vapor deposition routes such as atomic layer deposition (ALD), molecular-layer deposition (MLD), chemical vapor deposition (CVD), vapor-deposition polymerization (VDP), and vapor phase infiltration (VPI).15–19 However, the need to maintain and operate vacuum deposition equipment limits their accessibility to a broad user base.16,17 Additionally, vapor depositions that exhibit non-self-limiting mechanisms such as CVD and VDP lead to under-deposition deep into porous materials and over-deposition at pore entrances, causing non-uniformity and ill-defined coatings. For example, initiated or oxidative chemical vapor phase deposition (i/o-CVD) has emerged for coating non-planar substrates with polymers, but restricted mass transport and competing side reactions result in limitations at smaller pore sizes and higher aspect ratios.20,21 Similarly, VPI is effective for introducing inorganics into polymer films and is particularly well-suited for hybrid materials and coatings, as it can uniformly infiltrate and modify existing polymer structures, but it requires the presence of polymer films in the first place.19 Solely ALD offers a high degree of conformality of the obtained coatings up to a certain limit of pore sizes and aspect ratios due to its cyclic self-limiting growth mechanism, but it is so far limited to inorganic materials.15,16 Analogous to ALD, MLD is a nanoscale deposition technique with self-limiting layer-by-layer growth of polymers, but the library of available precursors is limited and precursor diffusion and reaction kinetics can be challenging in high aspect ratio structure.17,18 Until now, the layer-by-layer (LbL) deposition of alternating positively and negatively charged polyelectrolytes, or strong hydrogen-bond donors/acceptors, is the only method to enable the precise fabrication of polymer thin films on complex substrate shapes and porous materials with nanoscale control over film thickness.22 However, due to the LbL deposition mechanism that is coupled to the main polymer functionality, single-component or low-polarity polymers are not applicable to this method, limiting the range of polymeric coatings and functional interphases by LbL.22,23
Electrochemical deposition approaches are promising for the fabrication of uniform thin films on conducting materials regardless of the substrate architecture due to the surface confinement of the charge transfer and subsequent chemical reaction.14 Meanwhile, electrodeposition approaches are solution based and, therefore, more accessible, scalable, and adaptive compared to vapor-based methods.24 Electropolymerization of (semi)conducting polymers and electrochemically initiated polymerization of vinylic monomers such as acrylates have been reported on various non-planar electrodes. However, both methods fail at fabricating conformal and uniform thin films due to mass transfer effects and their non-self-limiting nature: electropolymerization yields conductive coatings that grow fastest in areas with the least mass transfer restrictions (pore entrances), while electrochemically initiated polymerization only confines the initiation to the surface and chain growth continues in the bulk solution causing random material deposition and pore blockage.25 Truly self-limiting and, therefore, conformal electropolymerization has only been achieved in electrochemically mediated crosslinking reactions of molecules such as phenol and ortho-phenylene diamine to yield ultrathin (<50 nm) dense molecular networks as uniform coatings.26–29 The enabling design criteria for this conformal electrodeposition approach are (1) a charge-transfer event that is necessary for each crosslinking reaction and (2) the formation of electronically insulating coatings that eventually become impermeable to the monomers.
Here, we apply these design criteria to pre-synthesized polymers in a novel polymer coating paradigm: the end-group assisted electrodeposition of polymer networks (EPoN). Our approach decouples the polymer network functionality from its electrodeposition chemistry, mitigates overgrowth at easily accessible surfaces due to a self-limiting mechanism, and prevents unwanted polymerization and material formation in the bulk solution. The iteration of EPoN reported here utilizes a pre-synthesized polymeric extender of arbitrary functionality with two electrochemically active groups at its chain ends. Upon surface-confined electrochemical reduction, this end group yields an activated species that undergoes a coupling reaction with a multifunctional crosslinker as a second complementary component, as illustrated in Scheme 1 (top). This electrochemically mediated coupling reaction of polymeric extender and multifunctional crosslinker generates a polymer network exclusively within the nanoscale vicinity of the electrode surface. The electrochemically generated polymer network deposits onto the electrode surface by adhesion due to its insolubility and precipitation. We demonstrate the two-component cathodic EPoN with the electrodeposition of a poly(ethylene glycol) (PEG) network on porous 3-D carbon electrodes and copper foams.
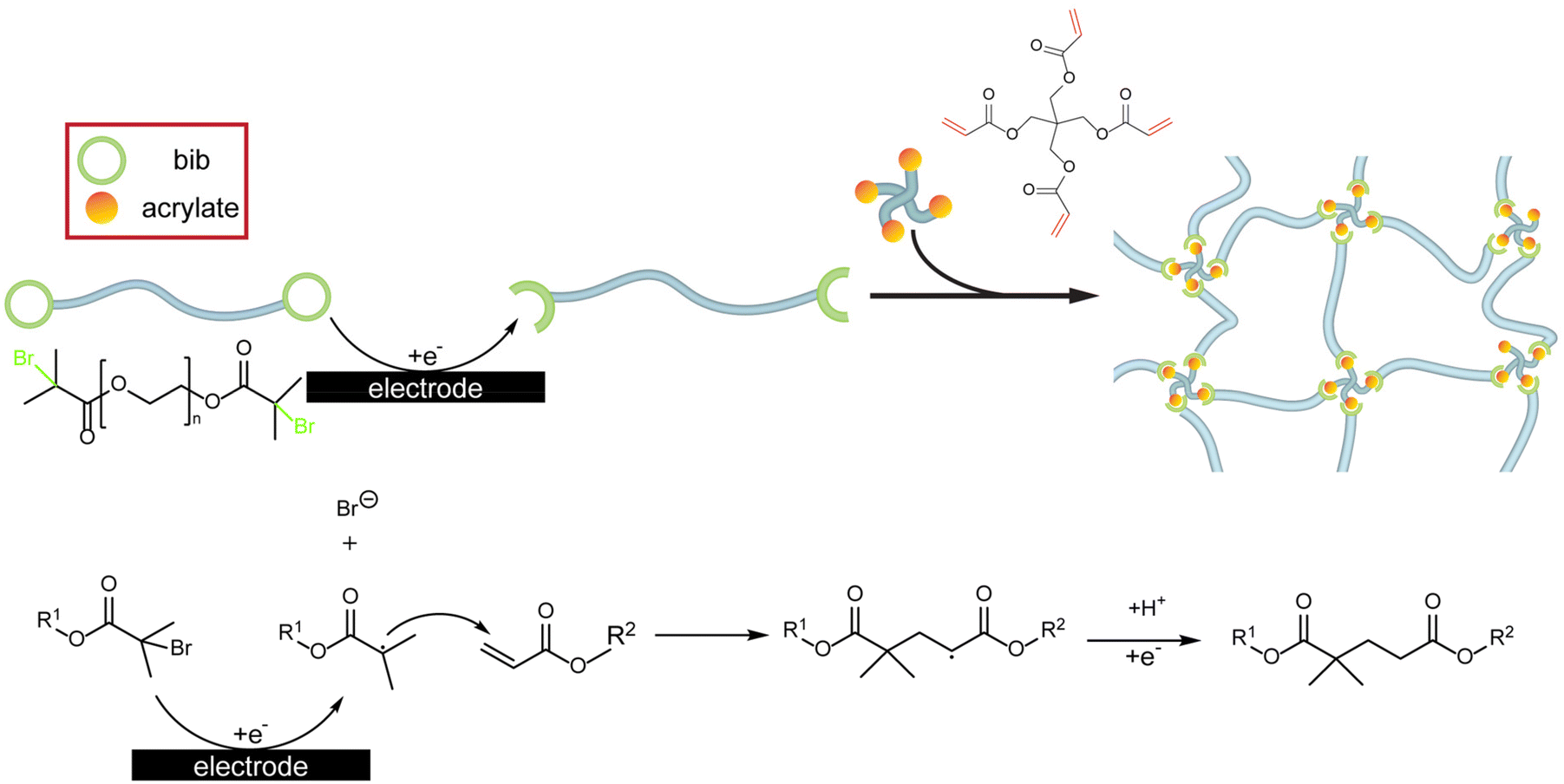 | ||
| Scheme 1 Schematic illustration (top) and chemical reaction (bottom) of the end-group-assisted two-component electrodeposition of polymer networks (EPoN). | ||
Experimental
Detailed experimental procedures are available in the ESI.†Synthesis
Hydroxy-terminated poly(ethylene glycol) is reacted with α-bromoisobutyryl bromide (bib) catalyzed by triethylamine in anhydrous toluene under nitrogen atmosphere at 0 °C for 30 minutes followed by room temperature for 10 hours. The resulting mixture is filtered to remove the triethyl ammonium bromide and the polymer product is precipitated in hexane. The precipitate is redissolved in chloroform, washed three times with deionized water and a saturated solution of NaCl, and precipitated twice in cold hexane to yield the end-group functionalized PEG-2bib. The successful modification is confirmed by proton-nuclear magnetic resonance (1H-NMR) spectroscopy in deuterated chloroform on an Agilent 400 MHz VNMRS unity plus spectrometer (Fig. S1†).3-D carbon electrode fabrication
3-D carbon electrodes are prepared with non-solvent induced phase inversion of a poly(acrylonitrile) (PAN) solution that is cast onto a dimethyl sulfoxide (DMSO)-swollen organogel and subsequently immersed in DI-water. The phase-inverted 3-D PAN film is stabilized at 250 °C in air and carbonized at 750 °C under argon for 1 hour. The 3-D carbon electrode is attached to a stainless steel disc with its dense side by applying a mixture of carbon black and poly(vinyl alcohol) in water between them and subsequent drying.Electrodeposition
All electrochemical experiments are performed on a Gamry Reference 600+ potentiostat. A coiled platinum wire serves as the counter electrode and a silver wire immersed in 0.1 M silver perchlorate solution with 0.1 M tetraethylammonium p-toluenesulfonate (TEA-Tos) in dimethylformamide (DMF) as the reference electrode separated by a Gamry glass frit from the deposition solution. All potentials are measured and displayed vs. this Ag/AgClO4 reference electrode. The electrodeposition solvent DMF is degassed with three freeze–pump–thaw cycles before use and the concentration of the supporting electrolyte TEA-Tos is 1 M for all electrodepositions. Potentiostatic electrodeposition is performed at −2 V vs. Ag/AgClO4. All coated electrodes are cleaned with acetonitrile and dried at room temperature.Film characterization
Scanning electron microscopy (SEM) is performed on a Zeiss Supra 55 field-emission SEM with an electron beam accelerating voltage of 3 kV using a secondary electron detector. Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy is performed on a Bruker FT-IR Microscope. Two-electrode electrochemical impedance spectroscopy (EIS) measurements with a signal amplitude of 100 mV are performed with a Gamry Reference 600+ potentiostat using a eutectic gallium indium drop sandwiched between the coated 3-D carbon (working electrode) and a bare ITO-coated glass slide (counter and reference electrode) separated by a 1 mm silicone rubber spacer resulting in a contact area of approximately 3.1 mm2.Results and discussion
The surface-confined formation and deposition of a PEG network is induced with the cathodic electrocoupling reaction between bromoisobutyrate (“bib”) and acrylate (“A”): the electrochemically reduced bib undergoes a nucleophilic addition to the electron-poor double bond of the acrylate (Scheme 1, bottom). To utilize this electrochemically activated coupling reaction, the chain ends of a linear PEG, a common polymer electrolyte exhibiting good lithium-ion conductivity, are functionalized with the electrochemically active bib group to yield the difunctional PEG-2bib (Fig. 1 and Fig. S1†).30,31 As the complementary crosslinker, pentaerythritol tetraacrylate (“4A”) with four acrylate groups is employed. The 4A crosslinker couples to the reduced bib to form a crosslinked PEG step-growth network either as a radical or an anion in a nucleophilic addition reaction.32 The coupled product itself is a radical or anion that can be quenched by protons, for example. Simultaneously, the coupled product could initiate the radical/anionic polymerization of 4A, leading to an uncontrolled chain reaction that would produce a coating with non-uniform composition and crosslink density, as PEG chains would inhomogenously incorporate into the network. To prevent this, we optimize the deposition conditions to ensure that the one-to-one coupling between bib and acrylate is the predominant reaction. Importantly, the growing and depositing PEG network is electronically insulating, preventing reduction at the coating-electrolyte interface, and it eventually becomes impermeable towards PEG-2bib, preventing the macromer from reaching the electrode surface and stopping its continued reduction and activation. Thus, the deposition is self-limiting and leads to full passivation of the electrode surface towards further growth at a critical point. | ||
| Fig. 1 (a) Cyclic voltammograms of solutions of PEG-2bib (blue curve), 4A (red curve), and pure electrolyte (green curve), respectively. The shaded region indicates the bib-based electrodeposition window without acrylate reduction. The dashed vertical line indicates the potential used for deposition. (b) Scanning electron microscopy (SEM) image of a 3-D carbon electrode at approximately 30° to its cross-section showing cylindrical micron-scale pores open on one side of the electrode. (c) Chronoamperometry during the application of a constant reducing potential (−2 V vs. Ag/AgClO4) to a 3-D carbon electrode immersed in solutions containing only PEG-2bib (red curve) and PEG-2bib with 4A (black curve), indicating passivating EPoN only with both components present. | ||
Individual cyclic voltammograms (CV) of PEG-2bib, 4A, and the pure DMF/TEA-Tosylate electrolyte solution are obtained to determine their reduction onset potential and probe the stability of the electrolyte solution (Fig. 1a). All solutions exhibit a small reduction peak around −1.3 V vs. Ag/AgClO4 from the reduction of residual oxygen.33 The onset potential of the PEG-2bib reduction is identified at −1.1 V and reaches its peak current at −2.2 V vs. Ag/AgClO4. The broad reduction wave indicates an irreversible multi-electron transfer to the bib group, forming butyric radicals and anions under bromide generation (Scheme 1, bottom). The crosslinker 4A only exhibits cathodic currents at potentials more negative than −2.3 V vs. Ag/AgClO4, associated with acrylate chemisorption, and the onset of its reduction wave is found at −2.6 V vs. Ag/AgClO4, which electrochemically initiates its polymerization. Therefore, the functional bib end-group is selectively reducible at less negative potentials than 4A. This establishes an electrodeposition window between −1.5 V and −2.3 V vs. Ag/AgClO4 for the exclusive reduction of the bib end group without electrochemically initiated acrylic chain-growth polymerization. Here, −2.0 V vs. Ag/AgClO4 is selected to achieve mass-transfer-limited electrodeposition without the electrochemical adsorption and reduction of the acrylate crosslinker.
Potentiostatic electrodeposition with chronoamperometry (“CA”) is employed to coat a (PEG-2bib)x-(4A)y thin film on the surfaces of a 3-D micro-architected carbon electrode obtained from our customized phase-inversion of poly(acrylonitrile) with subsequent carbonization.4 The 3-D carbon electrode of 80 μm thickness exhibits vertical cylindrical pores that are open and accessible on one side and closed on the other, with a pore diameter gradient from 5 to 20 μm towards the open pore entrance (Fig. 1b). During potentiostatic electrodeposition from a solution containing PEG-2bib and 4A at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of the functional groups (bib:A), the current density continuously decreases to near zero from initially 1 mA cm−2 over the course of 20 minutes, demonstrating passivation of the 3-D carbon electrode towards further reduction of the PEG-2bib (Fig. 1c). In contrast, a solution containing only PEG-2bib without 4A exhibits an initial decrease in current from 1 mA cm−2 but reaches a steady state after 200 s at over 0.4 mA cm−2, indicating no passivation upon bib reduction in the absence of a complementary crosslinker. The self-passivation nature of this electrodeposition is attributed to two factors: (1) the depositing polymer network is electrically insulating, preventing electron transfer from the carbon surface to the solution through the coating, and (2) the polymer network increases in density and eventually turns impermeable to the macromer PEG-2bib, preventing its diffusion from the bulk solution to the carbon surface and stopping its electrochemical reduction.
:1 molar ratio of the functional groups (bib:A), the current density continuously decreases to near zero from initially 1 mA cm−2 over the course of 20 minutes, demonstrating passivation of the 3-D carbon electrode towards further reduction of the PEG-2bib (Fig. 1c). In contrast, a solution containing only PEG-2bib without 4A exhibits an initial decrease in current from 1 mA cm−2 but reaches a steady state after 200 s at over 0.4 mA cm−2, indicating no passivation upon bib reduction in the absence of a complementary crosslinker. The self-passivation nature of this electrodeposition is attributed to two factors: (1) the depositing polymer network is electrically insulating, preventing electron transfer from the carbon surface to the solution through the coating, and (2) the polymer network increases in density and eventually turns impermeable to the macromer PEG-2bib, preventing its diffusion from the bulk solution to the carbon surface and stopping its electrochemical reduction.
To evaluate the conformality and topography of the electrodeposited PEG network thin film, scanning electron microscopy (SEM) images of the cross section of uncoated and coated 3-D carbon electrodes are compared in Fig. 2a. Evidently, a smooth EPoN-derived submicron coating on the surface is obtained at all locations along the 80-micron long pore. The thickness of the thin film is evaluated from the SEM images of 15 pores at the pore entrance, middle, and end, and summarized in Fig. 2b. Interestingly, the average thickness of the PEG network film at the entrance and middle of the pores is relatively uniform at 175–200 nm, and increases to 350 nm at the pore end, while exhibiting a fairly narrow pore-to-pore variance at the pore entrances and ends. This coating thickness distribution is opposite to traditional deposition techniques which exhibit overgrowth at the pore entrance and undergrowth deeper into the pore.20 We speculate that this result stems from a balance between the bib-A coupling kinetics and the diffusion of the reduced PEG-2bib within the confinement of the pore, leading to an initially denser and thinner coating at the pore entrance compared to deeper into the pore. We note that the pores in the 3-D carbon electrode are not perfectly uniform in diameter or aspect ratio, and that the “middle” of the electrode is not an equivalent spot for each pore, likely leading to larger variations in thickness measurements at that nominal spot over the many measured pores summarized in Fig. 2b. Varying the polymer concentration and end-group ratio (bib:A) has a substantial effect on the topography of the EPoN-derived coatings: films formed at lower concentrations are thinner and non-uniform, and become porous at bib concentrations below 0.1 M, potentially due to insufficient crosslinking at low concentrations leading to cracking of the coating during post-deposition drying and deswelling (Fig. S2†). Higher relative content of 4A also leads to less homogeneous coatings, likely due to an increased contribution of bib-initiated radical chain-growth polymerization of 4A.
 | ||
| Fig. 2 (a) Cross-sectional SEM images at different locations along an uncoated (left) and a coated (right) 80 μm long pore of a 3-D carbon electrode. (b) Distribution of EPoN-derived PEG coating thickness measured at the three different locations of 15 pores within the 3-D carbon. | ||
Having established the proof-of-concept for EPoN to achieve conformal submicron coatings on 3-D electrodes, the films are evaluated for defects, permeability, composition, and electronic properties. The molecular permeability and coverage of the films is deduced from the electrochemical signals of probe molecules dissolved in solution and in contact with the electrodes before and after polymer network electrodeposition. Decamethyl ferrocene (DmFc) and polyaniline (PANI, Mw = 20 kg mol−1) are used as electrochemical probe molecules. Since PANI is an order of magnitude larger than the polymeric PEG-2bib extender, one would expect a defect-free film to block its electrochemical activity, while the small-molecule DmFc may permeate through the PEG network. This behavior is indeed observed, as shown by the CVs in Fig. 3a and b for PANI and DmFc, respectively: the reversible oxidation and reduction peaks of PANI diminish after polymer network electrodeposition on the 3-D carbon electrode and double-layer capacitance dominates the CV. The absence of the PANI redox peaks above 0 V after coating of the carbon, as shown in Fig. 3a, suggests the absence of physical defects above a few nanometers in size at a quantity within the electrochemical detection limit, and that the mesh size of the polymer film is smaller than the size of the PANI. The redox peaks for DmFc remain for the 3-D carbon coated with the PEG networks with a larger peak splitting and slightly lower peak current, indicating the permeability of the electrodeposited polymer network to DmFc with an additional resistance of DmFc diffusion through the polymeric coating. The retention of DmFc electrochemistry further demonstrates that the polymer network film consists of a crosslinked PEG network rather than a purely electropolymerized poly(4A) coating, which exhibits no DmFc redox peaks when intentionally and exclusively electrochemically initiated polymerization of 4A is performed at −2.75 V vs. Ag/AgClO4 (Fig. S3†).
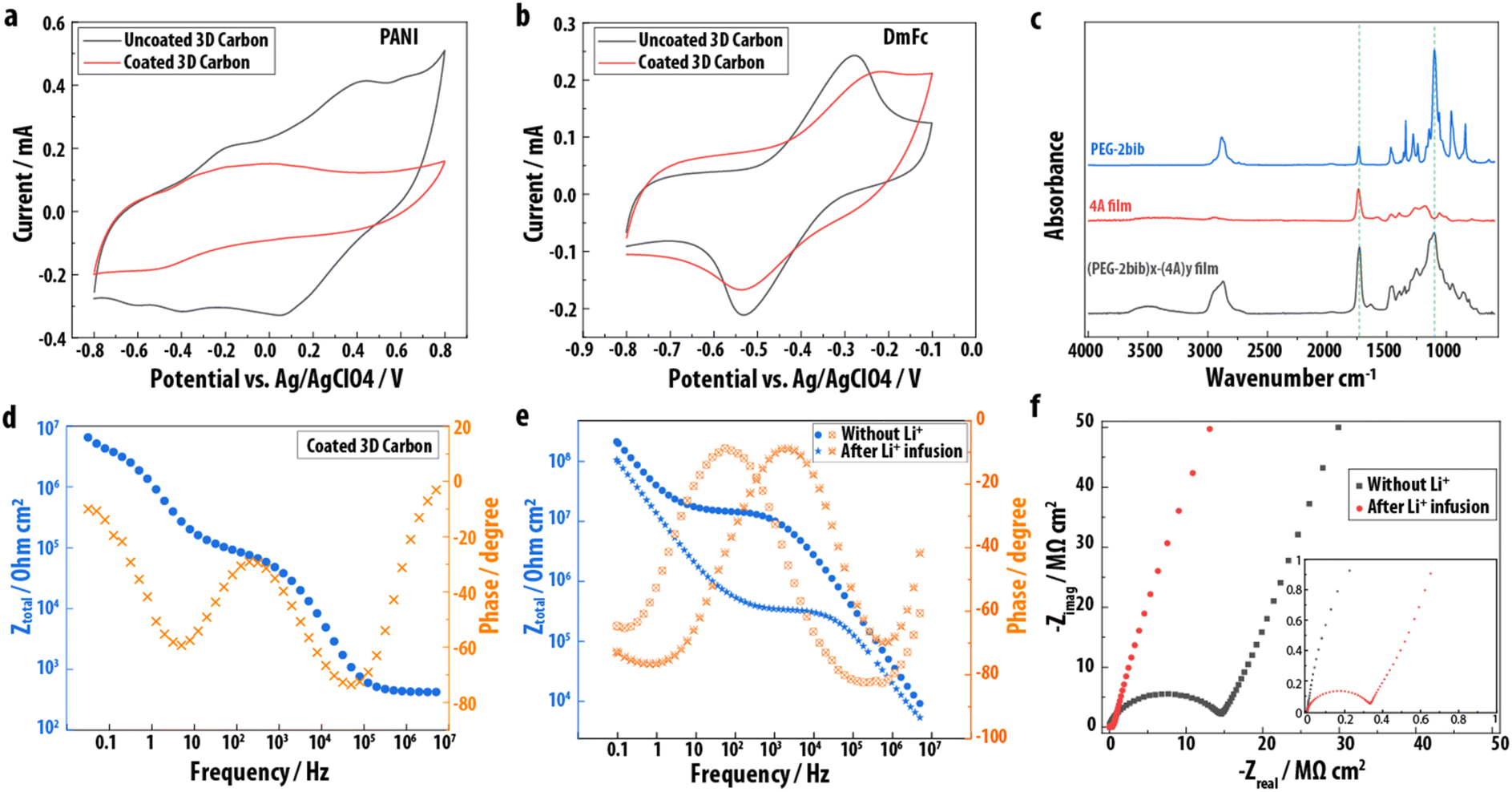 | ||
| Fig. 3 (a and b) CVs of PANI (a) and DmFc (b) before and after electrodeposition on 3-D carbon. (c) ATR-FTIR spectra of PEG-2bib (blue), an electropolymerized 4A film (red), and an EPoN-derived (PEG-2bib)x-(4A)y films (black). Dashed lines indicate position of carbonyl (1730 cm−1) and ether (1100 cm−1) absorption bands. Spectra are shifted vertically for clarity. (d) Bode plot of the solid-state EIS on the top surface of a 3-D carbon coated with a (PEG-2bib)x-(4A)y thin film. (e and f) EIS spectra of (PEG-2bib)x-(4A)y films on a planar gold substrate before and after lithium infusion: (e) Bode plot, (f) Nyquist plot. | ||
The chemical composition of the polymer network coatings is assessed by ATR-FTIR spectroscopy on planar gold electrodes. Fig. 3c shows the ATR-FTIR spectrum of the starting material PEG-2bib, an electrodeposited (PEG-2bib)x-(4A)y thin film, and an electrochemically polymerized poly(4A). The presence of a strong absorption band at 1100 cm−1 related to the C–O stretching of the ether group, which is abundant in the PEG chain but not in 4A, confirms the electrodeposition of the PEG macromer and its incorporation into the polymer network thin film. The material obtained from the electrochemically initiated polymerization of 4A exhibits a strong absorbance band at 1730 cm−1, related to its four carbonyl groups. The absorbance at 1730 cm−1 is stronger for the (PEG-2bib)x-(4A)y than for the PEG-2bib starting material, confirming the co-deposition of 4A and PEG-2bib. Additionally, we observe a higher PEG incorporation with increased bib:acrylate ratio during EPoN, as determined by comparing the IR absorbance of carbonyl and ether groups in films deposited with different end-group ratios (Fig. S4 and Table S1†). Based on these results, we conclude that the bib-acrylate coupling reaction under our optimized conditions and end-group ratio is the dominant mechanism to form the polymer network coating, as opposed to the electrochemically initiated acrylate chain polymerization.
Solid-state electrochemical impedance spectroscopy (EIS) was carried out on the surface of the coated 3-D carbon using a liquid metal contact to evaluate the electronic properties of the electrodeposited polymer network. The Bode plot shows three plateaus in the total impedance at low, medium, and high frequency, respectively, with corresponding phase shifts close to zero degree, indicating contributions of three ohmic resistances and two capacitive elements (Fig. 3d): system resistance (R1, high frequency), ionic resistance (R2, medium frequency), and electronic resistance (R3, low frequency), as well as double layer capacitance (C2) due to residual ions within the PEG network and plate capacitance (C1) of the conductor/insulator/conductor architecture (Fig. S5†). The fit of the EIS data to the respective circuit model reveals a high electronic resistance of approximately 109 Ω cm for the electrodeposited submicron PEG network coating, and an ionic resistance two orders of magnitude lower, likely from residual electrolyte salt.
To confirm that the EPoN-derived PEG thin films exhibit Li ion-conducting properties, (PEG-2bib)x-(4A)y thin films are deposited on planar gold electrodes, infused with a controlled amount of lithium bis(trifluoromethane)sulfonimide (LiTFSI) solution, and then dried. The solution is fully imbibed by the film and no excess LiTFSI salt was observed after drying, confirming its incorporation in the thin film. Bode plots of EIS spectra obtained before and after LiTFSI infusion reveal an impedance plateau at medium frequencies that is almost two orders of magnitude lower with lithium salt present (Fig. 3e), demonstrating the successful incorporation of lithium ions and the coating's ionic conductivity. Additionally, the Nyquist plots (Fig. 3f) show that both pre- and post-infusion films exhibit a half semi-circle at high frequencies, with a sharp rise in the negative imaginary component of the impedance at low frequencies. Notably, the reduced radius of the semi-circle after lithium infusion shows the increase in ionic conductivity by lithium infusion.
Since the two-component EPoN reported here utilizes electrochemical reduction, it is applicable to non-noble metal substrates. As a proof-of-concept, we deposited PEG networks on a porous copper foam following the same method, resulting in passivation and complete coverage of the copper surface by a conformal polymer network thin film of approximately 1.7 μm in thickness (Fig. S6†), demonstrating the substrate versatility of our reductive two-component EPoN process.
Conclusion
In summary, the electrodeposition of polymer networks (EPoN) as conformal thin films on conductive 3-D architected substrates has been demonstrated utilizing the electrocoupling reaction between a polymeric extender with electrochemically active end groups and a complementary crosslinker. The electron-transfer mediated crosslinking of pre-synthesized PEG-2bib and 4A is surfaced confined, passivating, and self-limiting, as demonstrated by their electrodeposition as a polymer film covering the entire surface of a porous 3-D carbon electrode and a copper foam. The deposited crosslinked PEG film exhibits high electronic resistance and decent ionic conductance, rendering it potentially useful as an ultrathin solid polymer electrolyte or artificial electrolyte interphase. The observed non-uniformity in coating thickness is likely due to the local differences in supply of reagents within the porous electrodes combined with the concentration-dependent film thickness. However, the self-limiting nature of EPoN guarantees deposition everywhere with a conformal yet not perfectly uniform coating. We postulate that a higher degree of uniformity could be achieved with an optimized deposition protocol such as pulsed EPoN, which may overcome variations in local concentrations.Since this cathodic EPoN is solution-based, vacuum-free, and accomplished at room temperature, it is accessible and widely applicable to coat a variety of conductive non-planar surfaces. Importantly, the end group and crosslinker used in EPoN make up only a small fraction of the resulting polymer thin film and could be combined with other arbitrary functional polymer chains, decoupling the deposition chemistry from the chemical coating properties and rendering this approach tunable and versatile. Additionally, the large potential window for electrodeposition and the two-component design offers additional degrees of freedom to fine-tune and tailor properties of the EPoN-derived coatings to applications of interest.
Author contributions
All authors have given approval to the final version of the manuscript. JGW devised the original idea and supervised this project. ZZ conducted the polymer synthesis, electrodeposition, and characterization. AR fabricated the 3-D carbon electrodes. WW instructed and aided the electrochemical characterization. JGW and ZZ wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
A patent application related to this research has been filed by Boston University.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. CBET-2146597. This work was performed in part at the Harvard University Center for Nanoscale Systems (CNS); a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. ECCS-2025158.References
- D. R. Rolison, J. W. Long, J. C. Lytle, A. E. Fischer, C. P. Rhodes, T. M. McEvoy, M. E. Bourg and A. M. Lubers, Multifunctional 3D Nanoarchitectures for Energy Storage and Conversion, Chem. Soc. Rev., 2009, 38(1), 226–252, 10.1039/B801151F.
- H. Li, Y. Huang, Y. Zhang, X. Zhang, L. Zhao, W. Bao, X. Cai, K. Zhang, H. Zhao, B. Yi, L. Su, A. K. Cheetham, S. Jiang and J. Xie, An Ultrathin Functional Layer Based on Porous Organic Cages for Selective Ion Sieving and Lithium–Sulfur Batteries, Nano Lett., 2022, 22(5), 2030–2037, DOI:10.1021/acs.nanolett.1c04838.
- Y. Chen, X. Zhang, W. Xue and Z. Xie, Three-Dimensional SiC/Holey-Graphene/Holey-MnO2 Architectures for Flexible Energy Storage with Superior Power and Energy Densities, ACS Appl. Mater. Interfaces, 2020, 12(29), 32514–32525, DOI:10.1021/acsami.0c04825.
- A. B. Resing, C. Fukuda and J. G. Werner, Architected Low–Tortuosity Electrodes with Tunable Porosity from Nonequilibrium Soft–Matter Processing, Adv. Mater., 2022, 2209694, DOI:10.1002/adma.202209694.
- Z. Li, J. Su and X. Wang, Atomic Layer Deposition in the Development of Supercapacitor and Lithium-Ion Battery Devices, Carbon, 2021, 179, 299–326, DOI:10.1016/j.carbon.2021.03.041.
- Y. Zhao, L. Zhang, J. Liu, K. Adair, F. Zhao, Y. Sun, T. Wu, X. Bi, K. Amine, J. Lu and X. Sun, Atomic/Molecular Layer Deposition for Energy Storage and Conversion, Chem. Soc. Rev., 2021, 50(6), 3889–3956, 10.1039/D0CS00156B.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing, Chem. Rev., 2021, 121(3), 1716–1745, DOI:10.1021/acs.chemrev.0c00938.
- S. Livi, L. C. Lins, L. B. Capeletti, C. Chardin, N. Halawani, J. Baudoux and M. B. Cardoso, Antibacterial Surface Based on New Epoxy-Amine Networks from Ionic Liquid Monomers, Eur. Polym. J., 2019, 116, 56–64, DOI:10.1016/j.eurpolymj.2019.04.008.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C–N Bonds, J. Am. Chem. Soc., 2015, 137(18), 6078–6083, DOI:10.1021/jacs.5b02653.
- J. G. Werner, B. T. Deveney, S. Nawar and D. A. Weitz, Dynamic Microcapsules with Rapid and Reversible Permeability Switching, Adv. Funct. Mater., 2018, 28(39), 1803385, DOI:10.1002/adfm.201803385.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Porous Polymer Networks: Synthesis, Porosity, and Applications in Gas Storage/Separation, Chem. Mater., 2010, 22(21), 5964–5972, DOI:10.1021/cm1021068.
- S. Mondal, Impact of the Process Conditions on Polymer Pattern Morphology during Spin Coating over Topological Surfaces, Soft Matter, 2021, 17(5), 1346–1358, 10.1039/D0SM01622E.
- A. Rouf, S. H. Park and J. P. Singer, Sub-Micron Thickness Self-Limiting Electrospray Deposition via Voltage Bias of Spray Target, Adv. Mater. Interfaces, 2024, 11(12), 2300982, DOI:10.1002/admi.202300982.
- J. W. Long, B. Dunn, D. R. Rolison and H. S. White, 3D Architectures for Batteries and Electrodes, Adv. Energy Mater., 2020, 10(46), 2002457, DOI:10.1002/aenm.202002457.
- S. M. George, Atomic Layer Deposition: An Overview, Chem. Rev., 2010, 110(1), 111–131, DOI:10.1021/cr900056b.
- J. G. Werner, M. R. J. Scherer, U. Steiner and U. Wiesner, Gyroidal Mesoporous Multifunctional Nanocomposites via Atomic Layer Deposition, Nanoscale, 2014, 6(15), 8736, 10.1039/C4NR01948B.
- X. Meng, An Overview of Molecular Layer Deposition for Organic and Organic–Inorganic Hybrid Materials: Mechanisms, Growth Characteristics, and Promising Applications, J. Mater. Chem. A, 2017, 5(35), 18326–18378, 10.1039/C7TA04449F.
- H. Zhu, M. H. Aboonasr Shiraz, L. Yao, K. Adair, Z. Wang, H. Tong, X. Song, T.-K. Sham, M. Arjmand, X. Song and J. Liu, Molecular-Layer-Deposited Tincone: A New Hybrid Organic–Inorganic Anode Material for Three-Dimensional Microbatteries, Chem. Commun., 2020, 56(86), 13221–13224, 10.1039/D0CC03869E.
- C. Z. Leng and M. D. Losego, Vapor Phase Infiltration (VPI) for Transforming Polymers into Organic–Inorganic Hybrid Materials: A Critical Review of Current Progress and Future Challenges, Mater. Horiz., 2017, 4(5), 747–771, 10.1039/C7MH00196G.
- W. Li, L. C. Bradley and J. J. Watkins, Copolymer Solid-State Electrolytes for 3D Microbatteries via Initiated Chemical Vapor Deposition, ACS Appl. Mater. Interfaces, 2019, 11(6), 5668–5674, DOI:10.1021/acsami.8b19689.
- H. O. Ford, B. L. Chaloux, B. Jugdersuren, X. Liu, C. A. Klug, J. B. Miller, X. Zuo, M. W. Swift, M. D. Johannes, J. W. Long, D. R. Rolison and M. B. Sassin, Non-Line-of-Sight Synthesis and Characterization of a Conformal Submicron-Thick Cationic Polymer Deposited on 2D and 3D Substrates, RSC Appl. Interfaces, 2024, 1, 531–543, 10.1039/D3LF00256J.
- J. J. Richardson, M. Björnmalm and F. Caruso, Technology-driven layer-by-layer assembly of nanofilms, Science, 2015, 348, aaa2491, DOI:10.1126/science.aaa2491.
- J. Chen, J. Duchet, D. Portinha and A. Charlot, Layer by layer H-bonded assembly of P4VP with various hydroxylated PPFS: impact of the donor strength on growth mechanism and surface features, Langmuir, 2014, 30(35), 10740–10750, DOI:10.1021/la502370h.
- X. Wang, J. Wu, H. Liu, F. Kang, F. Yan and Q. Zhang, Cathodic Polymerization through Electrochemical Dehalogenation, Macromolecules, 2023, 56(24), 10198–10205, DOI:10.1021/acs.macromol.3c01154.
- M. E. Abdelhamid, T. Rüther, J.-P. Veder, T. Rodopoulos, A. I. Bhatt, J. Lee, A. F. Hollenkamp, M. D. Horne, T. Huynh, A. Ong, K. J. Putman, G. Rowe and P. de Souza, Electrochemically Controlled Deposition of Ultrathin Polymer Electrolyte on Complex Microbattery Electrode Architectures, J. Electrochem. Soc., 2019, 166(3), A5462–A5469, DOI:10.1149/2.0601903jes.
- C. P. Rhodes, J. W. Long and D. R. Rolison, Direct Electrodeposition of Nanoscale Solid Polymer Electrolytes via Electropolymerization of Sulfonated Phenols, Electrochem. Solid-State Lett., 2005, 8(11), A579, DOI:10.1149/1.2050508.
- C. P. Rhodes, J. W. Long, M. S. Doescher, J. J. Fontanella and D. R. Rolison, Nanoscale Polymer Electrolytes: Ultrathin Electrodeposited Poly(Phenylene Oxide) with Solid-State Ionic Conductivity, J. Phys. Chem. B, 2004, 108(35), 13079–13087, DOI:10.1021/jp047671u.
- W. Wang, Z. Zheng, A. B. Resing, K. A. Brown and J. G. Werner, Conformal Electrodeposition of Ultrathin Polymeric Films with Tunable Properties from Dual-Functional Monomers, Mol. Syst. Des. Eng., 2023, 8(5), 624–631, 10.1039/D2ME00246A.
- L. F. D'Elia, R. L. Ortíz, O. P. Márquez, J. Márquez and Y. Martínez, Electrochemical Deposition of Poly(o-Phenylenediamine) Films on Type 304 Stainless Steel, J. Electrochem. Soc., 2001, 148(4), C297, DOI:10.1149/1.1354619.
- Z. Xue, Y. Zhang, Z. Zhao, X. Shi, H. Zhao, K. Cheng, J. Liu and L. Li, Completely Amorphous Poly(Ethylene Oxide)-Based Electrolyte Enables High Ionic Conductivity for Room-Temperature All-Solid-State Lithium Metal Batteries, ACS Appl. Energy Mater., 2023, 6(24), 12343–12352, DOI:10.1021/acsaem.3c02162.
- A. J. de Graaf, I. I. Azevedo Próspero dos Santos, E. H. E. Pieters, D. T. S. Rijkers, C. F. van Nostrum, T. Vermonden, R. J. Kok, W. E. Hennink and E. Mastrobattista, A Micelle-Shedding Thermosensitive Hydrogel as Sustained Release Formulation, J. Controlled Release, 2012, 162(3), 582–590, DOI:10.1016/j.jconrel.2012.08.010.
- K. Sugino and T. Nonaka, Cathodic Crossed Hydrocoupling XIII. Synthetic Aspect of the Cathodic Crossed Hydrocoupling Reaction of Aliphatic Carbonyl Compounds with Electrophiles in Aqueous Sulfuric Acid, J. Electrochem. Soc., 1977, 22(12), 271–277, DOI:10.1149/1.2423413.
- D. Li, T.-K. Ma, R. J. Scott and J. D. Wilden, Electrochemical Radical Reactions of Alkyl Iodides: A Highly Efficient, Clean, Green Alternative to Tin Reagents, Chem. Sci., 2020, 11(20), 5333–5338, 10.1039/D0SC01694B.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00180j |
| This journal is © The Royal Society of Chemistry 2024 |