
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(ionic liquid) ABC triblock and ABCBA pentablock terpolymer electrolytes for lithium metal batteries†
Dohyun
Kim
 a,
Rui
Sun
a,
Roger
Tocchetto
b,
Carl
Willis
b,
Bert
Krutzer
b,
Frederick L.
Beyer
c and
Yossef A.
Elabd
*a
a,
Rui
Sun
a,
Roger
Tocchetto
b,
Carl
Willis
b,
Bert
Krutzer
b,
Frederick L.
Beyer
c and
Yossef A.
Elabd
*a
aDepartment of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: elabd@tamu.edu
bKraton Polymers, LLC, Houston, TX 77032, USA
cU.S. Army Research Laboratory, Aberdeen Proving Ground, MD 21005, USA
First published on 26th August 2024
Abstract
In this study, poly(ionic liquid) (PIL) ABC triblock and ABCBA pentablock terpolymers (PILTTP and PILPTP, respectively) were synthesized to investigate the impact of chain architecture (ABC versus ABCBA) on properties. Specifically, the morphology, ionic conductivity, mechanical properties, electrochemical stability, and lithium metal battery performance of the PILTTP and PILPTP as ternary solid polymer electrolytes (SPEs) containing corresponding lithium salt and ionic liquid (IL) (at various IL concentrations) were measured. Interestingly, the PILPTP SPEs show one order of magnitude higher Young's modulus compared to the PILTTP at the same IL concentration due to bridged conformations and interlocked entanglements of the PILPTP. The improved mechanical properties of the PILPTP SPE lead to enhanced electrochemical stability and stable battery performance over 50 cycles at room temperature, exhibiting dendrite suppressing ability. This study highlights the importance of the symmetric ABCBA structure of PIL multiblock polymers on cycling stability for solid-state lithium metal batteries.
1. Introduction
Rechargeable lithium-ion batteries (LIBs) have rapidly developed in the past decades due to the growing global demand for efficient energy storage devices that allow flexible renewable energy production.1 Current commercial LIBs are widely utilized in large stationary energy storage systems, electric vehicles, and personal mobile devices.2,3 However, the energy density of LIBs is limited by low theoretical capacity of the conventional graphite anode (372 mA h g−1).4,5 Lithium metal, however, is an ideal anode possessing an extremely low electrode potential (−3.04 V vs. standard hydrogen electrode) and a high theoretical specific capacity (3860 mA h g−1), which allows broadening the operating cell voltage and reducing the active material loadings, effectively offering an enhanced energy density.6 Therefore, lithium metal batteries (LMBs) have been considered as a promising alternative to LIBs for next-generation rechargeable batteries. Nonetheless, the commercialization of LMBs paired with flammable liquid electrolytes has been hindered due to safety concerns resulting from electrolyte leakage and lithium dendrite growth. The leakage of organic liquid electrolytes with low flash points that are intrinsically volatile and flammable results in cell damage along with fire. Uncontrollable lithium dendritic growth during electrochemical stripping/plating can penetrate the separator and then reach the cathode, leading to short-circuit and potential explosion.7Replacing liquid electrolytes with solid polymer electrolytes (SPEs) has the benefit of addressing major safety issues of LMBs by avoiding electrolyte leakage and mitigating dendrite formation.8–10 SPEs that are composed of alkali metal salts dispersed in a polymer matrix in the absence of organic solvents offer low flammability, no leakage, high flexibility, and a stable interface between the electrode and electrolyte. In particular, SPEs afford the properties that affect dendrite nucleation and growth rate, which has led to extensive investigations towards fabricating SPEs that can suppress dendrite growth for battery safety.11–14 Chazalviel15 demonstrated that anion depletion near Li electrode leads to a large electric field, consequently resulting in dendrite formation. This study suggests that high ionic conductivity and high lithium-ion transference number (i.e., low anion mobility where fewer anions participate in overall conductivity) can impede dendrite nucleation. Monroe et al.16 reported that Li dendrite growth can be mechanically suppressed when a shear modulus of SPEs is twice as high as that of Li metal (ca. 7 GPa). Therefore, a number of studies have focused on enhancing both ionic conductivity and mechanical properties of SPEs using a variety of strategies, including blending,17–19 copolymerization,20–23 crosslinking,13,24,25 and composite polymer electrolytes with various kinds of fillers.26–28 However, most SPEs still suffer from inherently low room temperature ionic conductivity and a trade-off between ionic conductivity and mechanical strength.
Poly(ionic liquid) block copolymers (PILBCPs) have drawn considerable attention as SPEs for battery applications, given that they combine favorable properties of both ILs and block copolymers and offer orthogonal properties of high ionic conductivity (from the PIL block) and high modulus (from the neutral rigid block). The PIL block bearing covalently attached ionic moieties and mobile counter ions also has strong affinity with their corresponding ILs and Li salt, which allows for the creation of stable ternary SPEs without IL leakage. Furthermore, PILBCPs microphase separate into periodic morphologies, which is beneficial for ion transport by providing continuous conductive nanostructured channels. Mendes et al.29 synthesized a PILBCP, poly(styrene-b-1-((2-acryloyloxy)ethyl)-3-butylimidazolium bis(trifluoromethanesulfonyl)imide), and fabricated a ternary system using PILBCP with Li salt and ethylene carbonate. A moderate ionic conductivity of 0.01 mS cm−1 at 30 °C and enhanced mechanical integrity imposed by the presence of styrene blocks improved the interfacial stability between the SPE and the lithium metal, exhibiting dendrite suppression ability. Although a few studies report on PILBCPs as SPEs for LMBs,30,31 these are still limited on PILBCPs as SPEs, where PIL multiblock polymer-based SPEs have been rarely explored. Compared to PILBCPs, PIL multiblock polymers involve more than two block chemistries and enable to create diverse complex network structures with a combination of wanted mechanical and transport properties.32–34 In particular, PIL multiblock polymers allow SPEs with desired properties by tailoring and combining those of each block in a microphase-separated system. The properties of PIL multiblock polymers can be further enhanced by incorporating additives such as salts, ILs, and organic solvents, thereby imparting desired characteristics.35–37 However, numerous tuning parameters (e.g., block chemistry, block order, chain architecture, and cation/anion chemistry, etc.) of multiblock polymers increase the complexity on optimizing the properties of SPEs, which warrants a systematic study on property-tuning parameters of PIL multiblock polymer.
Chain architecture is an important parameter that affects the properties (e.g., morphology, conductivity, mechanical properties, etc.) of multiblock polymers.38–42 Symmetric multiblock polymers (i.e., ABA triblock copolymer and ABCBA pentablock terpolymer) and their analogous asymmetric multiblock polymers (i.e., AB diblock copolymer and ABC triblock terpolymer) have been studied to investigate the impact of chain architecture on overall properties of neutral multiblock polymers.43–47 Liu and coworkers48 compared self-assembled morphologies of ABCBA pentablock and corresponding ABC triblock terpolymers via three-dimensional self-consistent field theory (SCFT) method. They observed that pentablocks exhibit more diverse network structures than triblocks due to relieved packing frustrations within the structures. Meuler et al.49 experimentally compared the properties of ABCBA pentablocks and the homologous ABC triblocks with comparable compositions consisting of ethylene oxide (A block), styrene (B block), and isoprene (C block). Identical morphologies (i.e., orthorhombic Fddd network (O70), two-domain lamellae, and three-domain lamellae) were observed in both polymers at similar compositions; however, pentablocks exhibited higher mechanical properties (i.e., tensile strength, Young's modulus, and elongation at break) than triblocks, because the chain architecture of pentablocks prevents crack propagation. These property changes resulting from chain architecture are pivotal parameters for SPEs to mitigate dendrite growth and afford longer lifespan of LMBs, which provides an insight into how to design PIL multiblock polymer-based SPEs. However, the impact of chain architecture of PIL multiblock polymers for SPEs remains unexplored.
In this study, we synthesized ion conducting PIL ABC triblock and ABCBA pentablock terpolymers containing styrene (S) as the A block, ethylene-r-propylene (EP) as the B block, and imidazolium functionalized PIL as the C block with the same block composition. Ternary SPEs consisting of the polymers and the corresponding IL and Li salt were fabricated over a broad range of IL concentrations. The effect of chain architecture (ABC triblock vs. ABCBA pentablock) on the self-assembled morphologies, ion transport, and mechanical properties of the SPEs, as well as the electrochemical stability and battery cycling performance in LMBs was explored. To the best of our knowledge, this is the first report investigating the chain architecture impact of ion conducting PIL multiblock polymers as SPEs. This work reveals the importance of chain architecture, as well as the structure of symmetric PIL multiblock polymer-based SPEs and provides insight into SPE design with high mechanical strength and electrochemical stability for LMBs.
2. Experimental section
2.1. Materials
The non-ionic precursor ABC triblock terpolymers consisting of styrene (S) as the A block, ethylene-r-propylene random copolymer (EP) as the B block, and para-substituted methyl styrene (pmS) as the C block, poly(S-b-EP-b-pmS), and ABCBA pentablock terpolymers, poly(S-b-EP-b-pmS-b-EP-b-S), with the same composition were synthesized and provided by Kraton Polymers, LLC. The ABCBA pentablock terpolymer has an Mn ∼ 113.6 kg mol−1 with Mn of respective blocks equal to 33.6–2.6–41.2–2.6–33.6 kg mol−1. The ABC triblock terpolymer has an Mn ∼ 56.8 kg mol−1 with Mn of respective blocks equal to 33.6–2.6–20.6 kg mol−1.
N-Bromosuccinimide (NBS, 99%), 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%), 1-methylimidazole (99%), bis(trifluoromethylsulfonyl)imide lithium salt (Li-TFSI, 99.95% trace metals basis), chlorobenzene (anhydrous, 99.8%), methanol (ACS reagent, ≥99.8%), toluene (anhydrous, 99.8%), N,N-dimethylacetamide (DMAc; anhydrous, 99.8%), 1-methyl-2-pyrrolidinone (NMP; anhydrous, 99.5%), dichloromethane-d2 (CD2Cl2; 99.9% D, contains 0.1% (v/v) tetramethylsilane (TMS)), dimethyl sulfoxide-d6 (DMSO-d6; 99.9% D, contains 0.03% (v/v) TMS) were purchased from Sigma-Aldrich. Industrial grade liquid nitrogen was purchased from Airgas. 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMIm-TFSI; 99%) was purchased from IoLiTec. Mylar PET release liner substrates (GC 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 965, 0.0762 mm) were used as received from Loparex LLC. Li foil (99.9% purity, 60 mm width × 0.5 mm thickness) was purchased from MSE supplies and stored in an argon-filled glove box (UNIlab pro, mBraun). Lithium nickel manganese cobalt oxide (LiNi0.8Co0.1Mn0.1O2, NMC811), poly(vinylidene fluoride) (PVDF; ≥99.5%, Mw = 600000 g mol−1), conductive carbon black (super C45), conductive carbon coated aluminum foil (C–Al foil; double side coating with 1 μm thickness each side), stainless steel coin cell cases (CR2032; 20 mm diameter × 3.2 mm thickness) with polypropylene sealing gasket, stainless steel spacers for CR2032 cells (15.8 mm diameter × 1.0 mm thickness, 15.5 mm diameter × 0.5 mm thickness), and stainless steel wave springs for CR2032 cases (1.2 ± 0.05 mm height × 0.3 mm thickness) were purchased from MTI Corporation. All materials were used as received without further treatment.
965, 0.0762 mm) were used as received from Loparex LLC. Li foil (99.9% purity, 60 mm width × 0.5 mm thickness) was purchased from MSE supplies and stored in an argon-filled glove box (UNIlab pro, mBraun). Lithium nickel manganese cobalt oxide (LiNi0.8Co0.1Mn0.1O2, NMC811), poly(vinylidene fluoride) (PVDF; ≥99.5%, Mw = 600000 g mol−1), conductive carbon black (super C45), conductive carbon coated aluminum foil (C–Al foil; double side coating with 1 μm thickness each side), stainless steel coin cell cases (CR2032; 20 mm diameter × 3.2 mm thickness) with polypropylene sealing gasket, stainless steel spacers for CR2032 cells (15.8 mm diameter × 1.0 mm thickness, 15.5 mm diameter × 0.5 mm thickness), and stainless steel wave springs for CR2032 cases (1.2 ± 0.05 mm height × 0.3 mm thickness) were purchased from MTI Corporation. All materials were used as received without further treatment.
2.2. Synthesis and fabrication of PILTTP and PILPTP SPEs
PILTTP and PILPTP were synthesized via three steps. PILPTP synthesis is shown in Scheme 1; PILTTP was synthesized similarly to PILPTP. Initially, the pmS block of the non-ionic precursor terpolymers was brominated to synthesize PTP–Br consisting of vinylbenzyl bromide as the third block (step 1). 1-Methylimidazole was then converted into imidazolium cations by covalently bonding to the vinylbenzyl bromide block via quaternization (step 2). The bromide ion was then exchanged to TFSI ion via anion exchange reaction to achieve PILPTP containing poly(vinylbenzylmethylimidazolium bis(trifluoromethylsulfonyl)imide) (PVBMIm-TFSI) as the PIL block (step 3). The ion conducting PILTTP and PILPTP are referred to as TTP-TFSI and PTP-TFSI, respectively. Details of each synthetic step are described in our previous study.50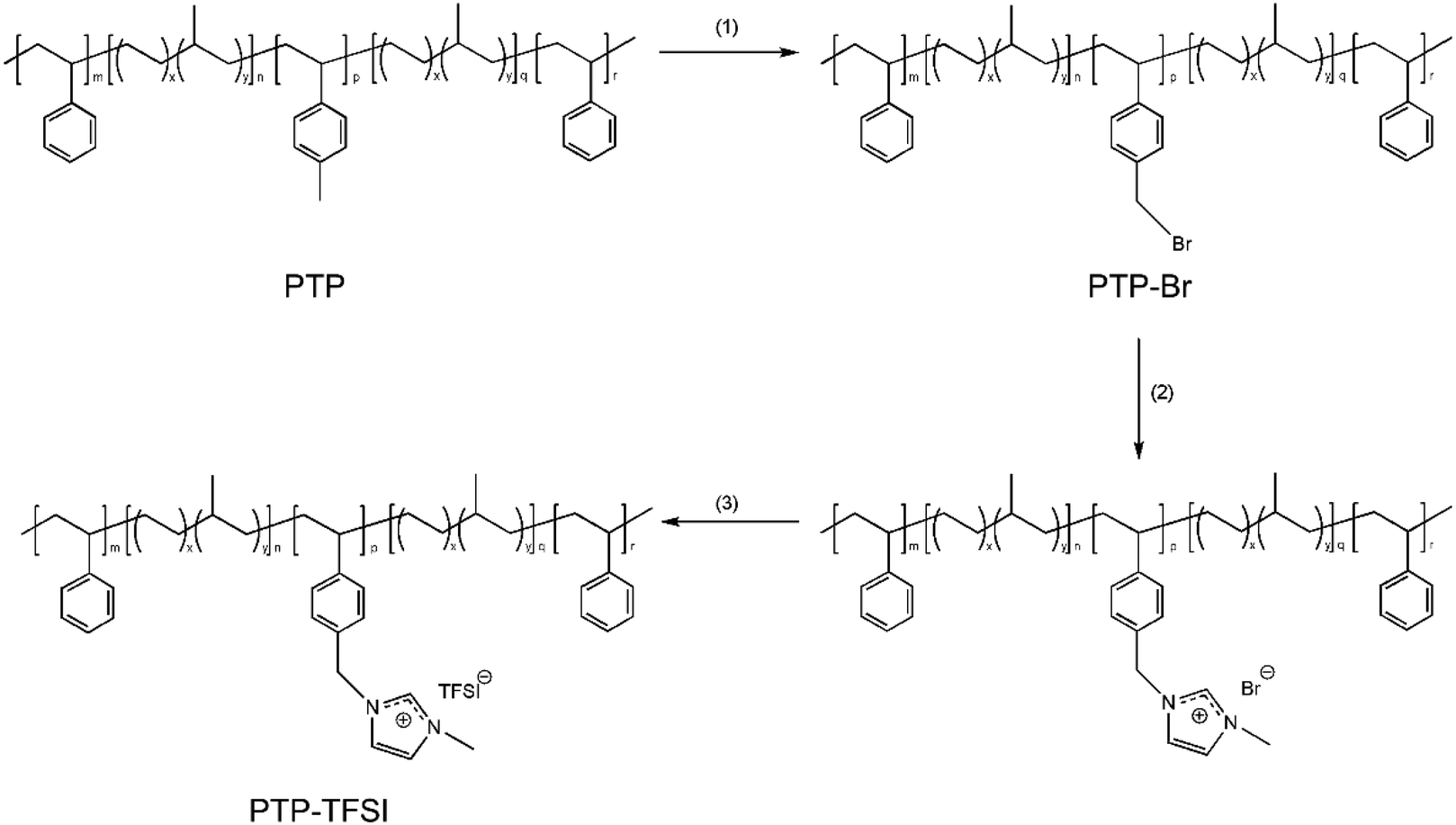 | ||
| Scheme 1 Synthesis of PILPTP. (1) NBS, AIBN, chlorobenzene, 70 °C, 5 h; (2) 1-methylimidazole, toluene, methanol, RT, 48 h; (3) Li–TFSI, toluene, methanol, RT, 48 h. | ||
The PILTTP and PILPTP SPEs were prepared by casting ternary mixture solutions (polymer, salt, IL dissolved in solvent) onto a silicon-coated Mylar PET film, as shown in Fig. 1. Dried TTP-TFSI and PTP-TFSI polymers were dissolved in DMAc to obtain 20 wt% polymer solutions, respectively. Li salt (Li–TFSI) and IL (EMIm–TFSI) were added to each polymer solution to achieve desired salt and IL concentrations as shown in Table 1. The molar ratio of Li–TFSI to PIL block was kept constant at 0.1 for all SPEs and the molar ratios of EMIm–TFSI to PIL block (r) were varied over a range of 0.2–0.9 to investigate the impact of IL on the properties of PILTTP and PILPTP SPEs. These ternary mixtures were mixed overnight at room temperature to achieve homogeneous solutions. To fabricate free-standing SPEs, the solutions were cast onto a silicon-coated Mylar PET film via doctor blade method using an automatic film applicator (Elcometer 4340) at gauge height of 350 μm and speed of 60 mm s−1. The films were dried under vacuum at ambient temperature for 24 h and then at 120 °C for 48 h to remove any residual solvent. The films were then stored in an argon-purged glove box. The final films were denoted as TTP-Li-r and PTP-Li-r, where r represents the molar ratio of IL to PIL block. The PTP-Li-r produced films regardless of r, whereas the TTP-Li-r produced free-standing films at limited IL contents (i.e., r = 0.3 and 0.5). The thickness of these films was measured with a digital indicator thickness gauge (Mitutoyo, 547–400S), where the thicknesses were ca. 35 μm.
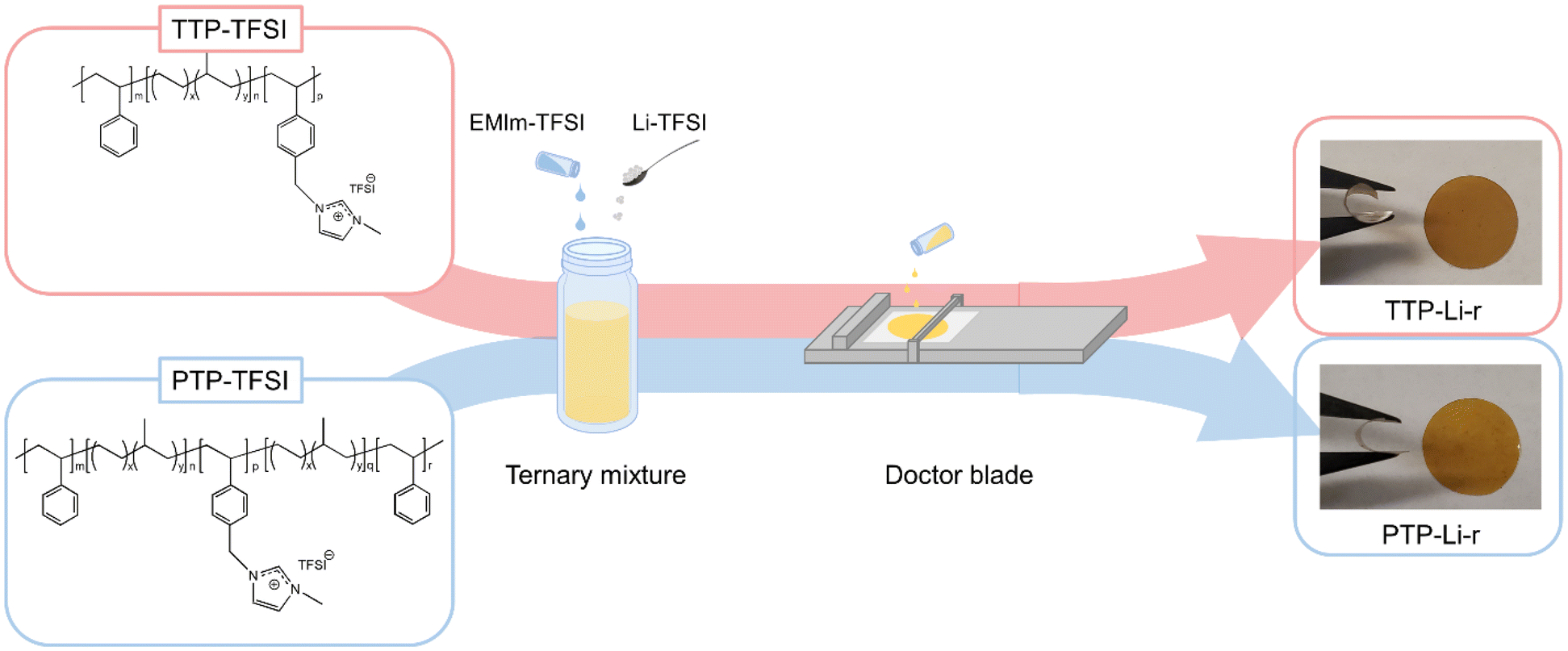 | ||
| Fig. 1 Fabrication of TTP-Li-r and PTP-Li-r SPEs. | ||
| TTP-Li-ra SPE | r | Chemical structure |
|---|---|---|
| TTP-Li-0.2 | 0.2 |
|
| TTP-Li-0.3 | 0.3 | |
| TTP-Li-0.5 | 0.5 | |
| TTP-Li-0.7 | 0.7 | |
| TTP-Li-0.9 | 0.9 |
2.3. Characterization
The chemical structures of non-ionic precursor polymers (TTP and PTP), brominated polymers (TTP–Br and PTP–Br), and anion exchanged polymers (TTP–TFSI and PTP–TFSI) were verified by proton nuclear magnetic resonance spectroscopy at 23 °C (1H NMR; AVANCE NEO 400, Bruker). Two types of deuterated solvents were used for NMR experiments: CD2Cl2 for the non-ionic precursors and brominated polymers, where chemical shifts were referenced to dichloromethane at 5.32 ppm, and DMSO-d6 for the anion exchanged polymers, where chemical shifts were referenced to DMSO at 2.50 ppm.Morphology of the anion exchanged polymers (TTP–TFSI and PTP–TFSI) and SPEs (TTP-Li-r and PTP-Li-r) was investigated by small-angle X-ray scattering (SAXS). The samples were dried under vacuum overnight at room temperature prior to characterization. SAXS data were collected using a Xenocs SAXS instrument (model Xeuss 3.0 HR). X-rays were generated with a Rigaku MicroMax-007HF rotating copper anode X-ray generator operated at 40 kV and 30 mA. Characteristic Cu-Kα photons were monochromated and collimated using a focusing optic and two scatterless slit apertures, producing a well-aligned incident beam with wavelength (λ) of 1.5418 Å. Data were collected using a Dectris Pilatus3R 300k solid-state X-ray detector. Isotropic two-dimensional data were azimuthally averaged to generate one-dimensional (1-D) data, I(q), for analysis. Silver behenate was used to calibrate the beam center and sample-to-detector distance.51 Transmitted flux was used to correct the data for absorption, which when combined with sample thickness, allows data to be placed on an absolute intensity scale.52 Data processing and analysis were performed using Wavemetrics Igor Pro v8 and procedures available from Argonne National Laboratory.53,54
Differential scanning calorimetry (DSC) measurements were conducted by a differential scanning calorimeter (Q200, TA Instruments) from −140 °C to 200 °C at a heating/cooling rate of 10 °C min−1 under nitrogen atmosphere. The samples were prepared in an argon atmosphere in a glove box to prevent SPEs from being exposed to moisture. The glass transition temperatures (Tgs) of SPEs were determined from the second heating cycle.
Dynamic mechanical analysis (DMA) experiments were performed with a dynamic mechanical analyzer (Q800, TA Instruments) with a custom-designed humidity and temperature chamber. The SPEs with dimensions of 20 mm × 5 mm × 0.03 mm were strained using a tensile clamp with a constant rate of 0.2% min−1 at 25 °C under a dry nitrogen atmosphere of ca. 5% RH.
Ionic conductivity was investigated via electrochemical impedance spectroscopy (EIS) with an impedance analyzer (Solartron SI 1260A) combined with potentiostat/galvanostat (Solartron SI 1287A). The circular SPE film (1.54 cm2 area) was placed between two stainless steel solid blocking electrodes (1.2161 cm2 area) encapsulated by a custom-made Teflon holder in argon-filled glove box with oxygen and water concentration below 1 ppm. For temperature-dependent ionic conductivity measurement, heat was applied to stainless steel blocking electrodes by heating tape (BIH051040, BriskHeat) with a digital temperature controller (SDC120JC-A, BriskHeat) coupled with J type thermocouple (Model 650, OMEGA). Impedance spectra were collected by sweeping frequency from 1 MHz to 0.1 Hz with an AC perturbation of 10 mV at open circuit potential at a temperature range from 30 to 100 °C. SPEs were held at each temperature for 1 h to reach equilibrium, followed by five measurements at equilibrium. The conductivity was calculated by using the following equation: σ = L/(AR), where L is the film thickness, A is the cross-sectional area of the blocking electrode (ca. 1.2161 cm2), and R is the film resistance of SPE, which was determined by the equivalent circuit regression of the Nyquist data.
2.4. Coin cell assembly with PILTTP and PILPTP SPEs
The cathode was prepared by dispersing NMC811 (80 wt%), carbon black (10 wt%), PVDF (10 wt%) in NMP and casting this cathode slurry onto a C–Al foil using the film applicator (Elcometer 4340) at gauge height of 70 μm and speed of 80 mm s−1. The cathode was dried at ambient conditions overnight, followed by drying at 120 °C under vacuum for 6 h to remove residual NMP. The cathode was punched out using a hollow punch (12 mm diameter, 66004, Mayhew Pro), with an active material loading was ca. 1.31 mg cm−2. The cathode was used without further treatment (e.g., calendaring process). Li/SPE/NMC811 cells were assembled by placing the SPE between lithium metal and NMC cathode in the cell cases and pressing them by an electric crimper (MSK-160E, MTI Corporation) under argon in the glove box. A drop of 1 M Li–TFSI/EMIm–TFSI (ca. 10 mg) was added to each electrode to improve contact between electrodes and SPE during the assembly. Symmetric Li/SPE/Li cells were assembled by the same process. Prior to measurement, each cell was allowed to rest for 24 h to form a stable interface between SPE and electrodes.
Galvanostatic stripping/plating cycling of the symmetric cells was evaluated using a battery tester (4200 M, MACCOR) at room temperature at a constant current density of 0.02 mA cm−1. The resistances between SPE and lithium electrodes were recorded by EIS every 10th polarization cycle at a frequency ranging from 100 kHz to 1 Hz with the amplitude of the voltage disturbance of 10 mV.
Battery cycling experiments on Li/SPE/NMC811 coin cells were performed between 3.0 and 4.2 V at room temperature at a C-rate of C/20 where C is the theoretical capacity of cathode material. Constant current (CC) charging that uses a constant current for charging the battery was used for the whole charging process. Constant current constant voltage (CC–CV) charging where a predetermined voltage is used to charge the battery followed by CC charging (e.g., 4.2 V for 1 h) was used as formation prior to cycling.
3. Results and discussion
Ion conducting ABC triblock and ABCBA pentablock terpolymers were synthesized via bromination, quaternization, and anion exchange reactions, as illustrated in Scheme 1. The resulting polymers were characterized by 1H NMR as shown in Fig. S1(a) and S1(b),† where the triblock and pentablock terpolymers exhibit similar resonances at each synthesis step. After bromination, the intensity of methyl proton resonance (a) at 2.26 ppm decreases, and a new resonance corresponding to the methylene protons (b) in the brominated block appears at 4.47 ppm. The degree of bromination (DB) for TTP–Br and PTP–Br was determined by the integration of protons on unreacted methyl groups (a′) and the resulting methylene group (b) contributions using the following equation: DB (%) = b/(b + a′). The DBs for TTP–Br and PTP–Br are 86 and 85%, respectively. After quaternization and anion exchange reactions, proton resonances of the imidazolium ring (e, f, g, and h) are clearly observed at 3.85, 7.69, and 9.24 ppm. The resonance of the methylene proton in the PIL block (b) shifts from 4.47 ppm to 5.29 ppm due to introduction of the imidazolium cation group. These successfully synthesized polymers, TTP–TFSI and PTP–TFSI, were used to fabricate ternary SPEs composed of Li salt and varying IL concentrations (r), which are referred to as TTP-Li-r and PTP-Li-r, respectively, as shown in Fig. 1 and Table 1.To investigate the impact of chain architecture and IL concentration on chain mobility, DSC thermograms of ternary SPEs at various IL concentrations (TTP-Li-r and PTP-Li-r) were collected and compared to the TTP–TFSI and PTP–TFSI polymers (Fig. S2†). Both TTP–TFSI and PTP–TFSI have two glass transition temperatures (Tgs) at around 63 °C and 120 °C, corresponding to the PIL block (C block) and S block (A block), respectively. This is a clear indication of the microphase-separated structure of the non-conductive S domain and the conductive PIL domain.31,55,56 The Tg for EP block (B block), which is generally located at approximately −53 °C,57 does not appear in the DSC data because the number average molecular weight for EP block is significantly smaller than other blocks (Mn,B = 2.6 kg mol−1 ≪ Mn,A = 33.6 kg mol−1 or Mn,C = 41.2 kg mol−1). Despite the incorporation of IL and salt, thermal transitions corresponding to IL and salt do not appear, suggesting good compatibility of IL and salt with the host polymers.
Fig. 2 shows the Tgs of PS and PIL phases for the TTP-Li-r and PTP-Li-r SPEs as a function of IL composition, r (DSC thermograms shown in Fig. S2†). With increasing r from 0 to 0.9, the Tgs of the PIL block decrease from 40 °C to −52 °C for TTP-Li-r and from 47 °C to −47 °C for PTP-Li-r, while the Tgs of the S block remain unchanged at 120 °C for both SPEs. The difference in PIL Tgs between triblock and pentablock is not significant, indicating that chain mobility of the PIL block as midblock in the pentablock polymer is similar to that of the PIL block as end block (i.e., dangling chains) in the triblock polymer, i.e., restrictions on chain mobility of the PIL block for the pentablock imposed by near blocks (i.e., A and B blocks) are negligible. The Tg depression with IL implies that IL is preferentially soluble in the PIL block due to its high affinity for the PIL block, indicating that the segmental mobility of the PIL block increases with increasing r and IL behaves as a plasticizer. The experimental data were compared to model predictions by the Fox equation, which describes the Tg behavior of a plasticized polymer using the following equation:
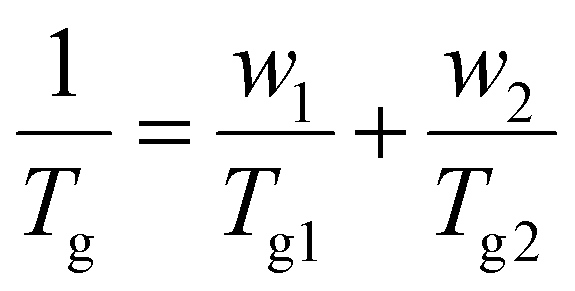 | (1) |
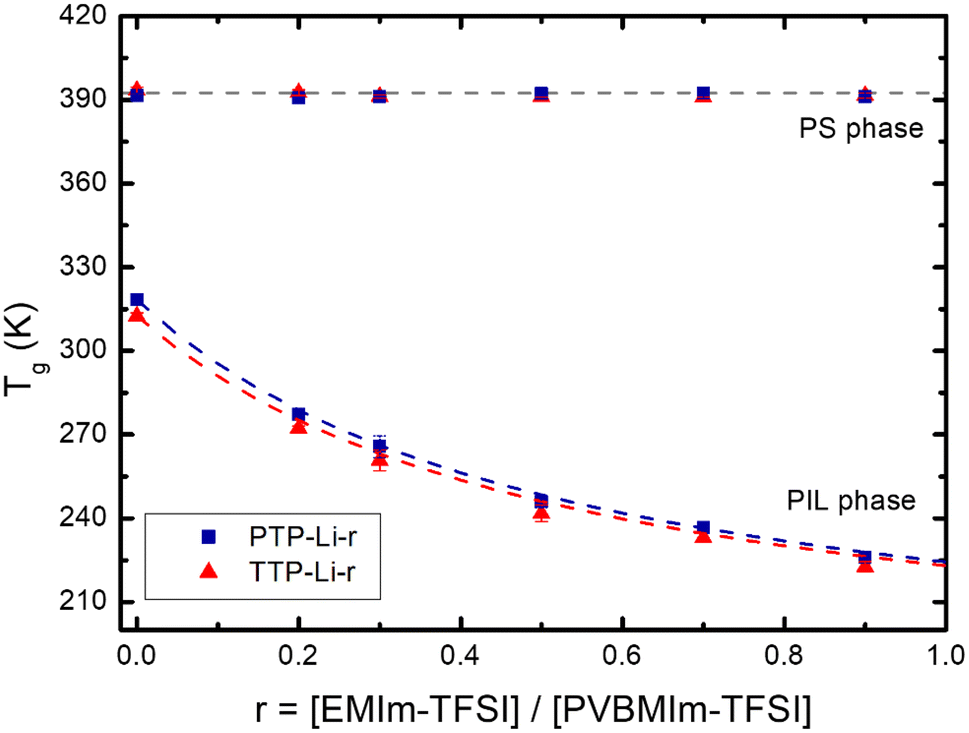 | ||
| Fig. 2 Glass transition temperatures (Tgs) of PS and PIL phases for the TTP-Li-r (red triangle) and PTP-Li-r (blue rectangle) SPEs as a function of IL composition (r) from 0 to 0.9. The theoretical Tgs estimated by Fox equation for TTP-Li-r and PTP-Li-r are indicated by red and blue dashed lines, respectively. | ||
Morphological behavior of the TTP–TFSI and PTP–TFSI with IL concentration (r = 0.3 and 0.5) was determined from the SAXS data shown in Fig. 3. Both TTP–TFSI and PTP–TFSI show strong correlation peaks centered at approximately 0.13 nm−1, and higher-order scattering features between 0.2 nm−1 and 1 nm−1. Modeling using the form factor and the structure factor for a sphere as shown in ESI (SI.1.†) suggests that these samples share a morphology of microphase-separated spheres, and that the primary peak is a correlation peak due to interdomain scattering. The less-organized structures may be attributed to high polydispersity for both polymers. The similarity in morphological structure between TTP–TFSI and PTP–TFSI supports Matsen and Thompson's hypothesis that the phase behavior of neutral AB and ABA systems becomes analogous due to negligible change in free energy of the melt for both systems,46,59 which also applies to higher-order multiblock polymers (e.g., ABCBA pentablocks and homologous ABC triblocks,49,60 and ABABAB and AB systems61). With the addition of IL and salt (r = 0.3), the SAXS data show slightly better defined features in both SPEs, suggesting improved ordering possibly due to plasticization by IL and Li salt as verified by the DSC results (Fig. S2†). The PIL domains are favorably swelled by IL and Li salt, improving microphase separation between the PIL and other domains, in agreement with published results.62,63 However, with additional IL (r = 0.5), the SAXS data contain less evidence of form factor scattering and a broader correlation peak, suggesting that further increase in IL content for both SPEs increases the distance between microphase domains and increase the size dispersity of those domains. Compared to TTP–TFSI (d = 47.5 nm) and PTP–TFSI (d = 48.9 nm), both domain spacings at r = 0.5 increased to 57.5 nm and 55.6 nm for TTP-Li-0.5 and PTP-Li-0.5, respectively, due to the swelling of the PIL block. The TTP-Li-r SPE has a larger domain spacing than the PTP-Li-r SPE, indicating that TTP is likely to swell more easily than PTP. The similar scattering from both SPEs as shown in Fig. S3 and S4† suggests that chain architecture (ABC vs. ABCBA) does not result in a significant change in morphology. The distorted shapes of the correlation peak in all the SAXS data suggest that the morphology is kinetically trapped.
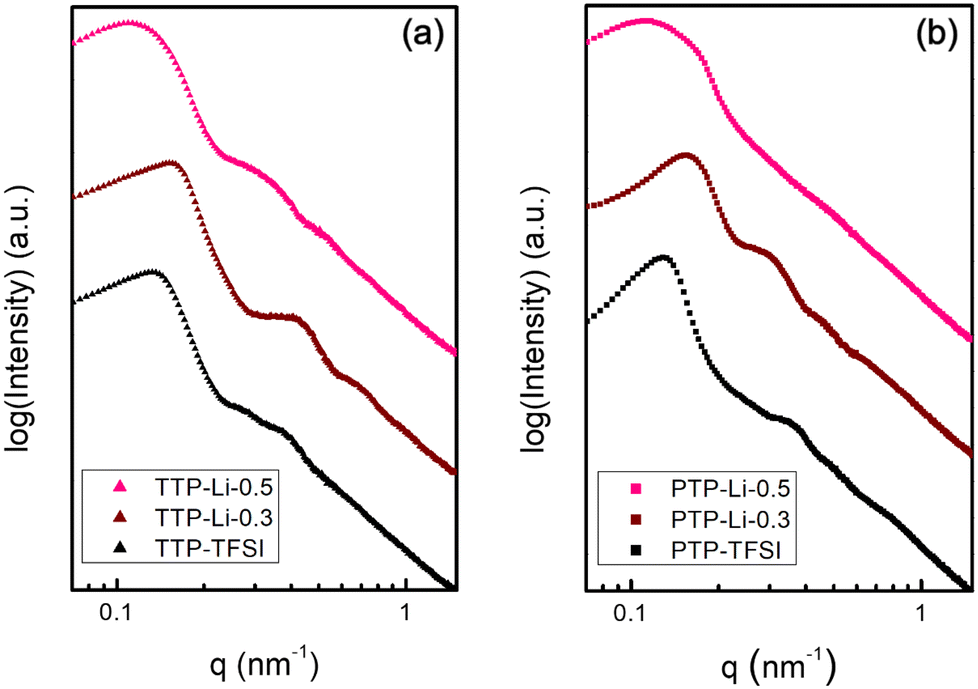 | ||
| Fig. 3 SAXS profiles of (a) TTP–TFSI and TTP-Li-r and (b) PTP–TFSI and PTP-Li-r SPEs (r = 0.3 and 0.5) at room temperature. Data are scaled vertically for comparison. | ||
The ionic conductivity was measured from 30 to 100 °C under argon atmosphere to investigate the impact of chain mobility on the conductivity for both SPEs as shown in Fig. 4. Both TTP-Li-r and PTP-Li-r SPEs exhibit comparable ionic conductivities at the same r ratio (e.g., 3.0 × 10−6 S cm−1 for TTP-Li-0.5 and 2.9 × 10−6 S cm−1 for PTP-Li-0.5 at 30 °C) due to similar morphology and chain mobility as observed in Fig. 2 and 3, suggesting that the difference in chain architecture has a negligible impact on conductivity. As r increases, the conductivity for both TTP-Li-r and PTP-Li-r increases, and the PTP-Li-0.9 SPE shows the highest ionic conductivity of 8.2 × 10−5 S cm−1 at 30 °C and 1.7 × 10−3 S cm−1 at 100 °C. This improvement is attributed to the coupling of enhanced chain mobility (Tg depression for PIL block) and increased number of mobile ions in the SPE with increasing IL concentration.
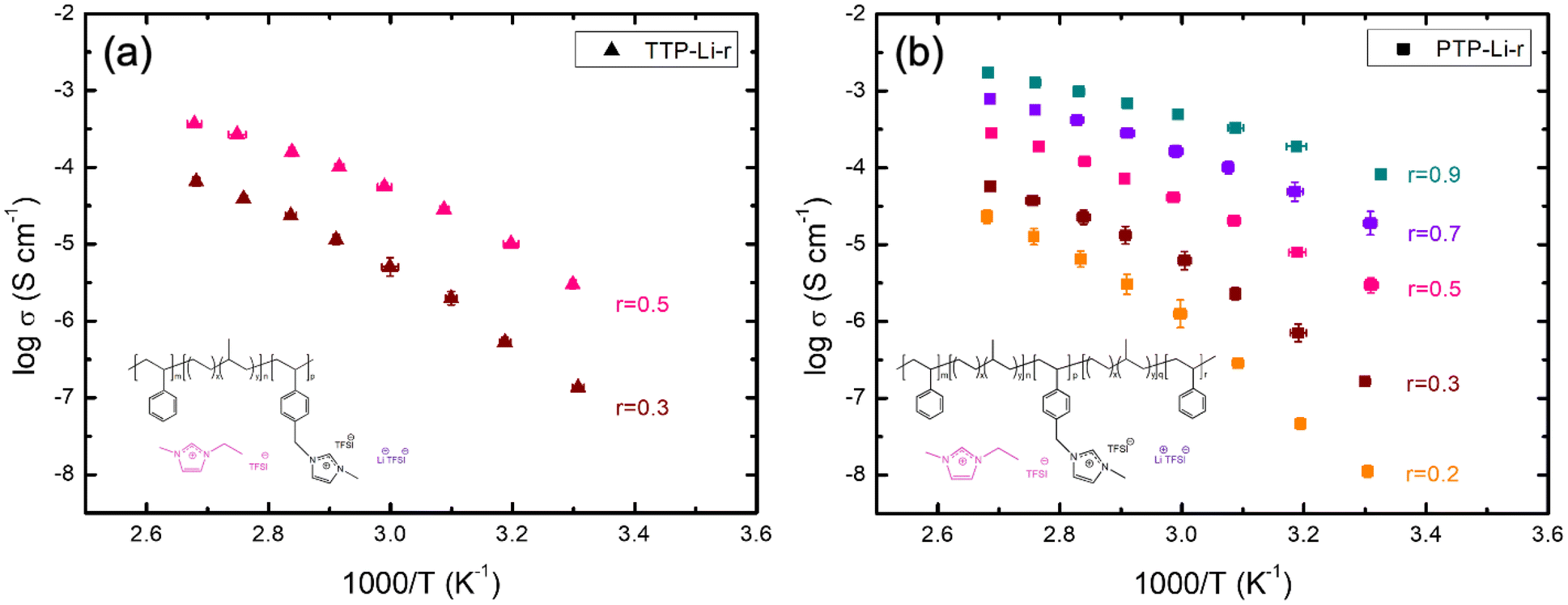 | ||
| Fig. 4 Temperature-dependent ionic conductivity of the SPEs at various r = [IL]/[PIL block] ratios: (a) TTP-Li-r and (b) PTP-Li-r. | ||
The ionic conductivity for both SPEs increases with increasing temperature and this conductivity trend as a function of temperature provides underlying mechanisms for ion transport. The kinetics of ion movement were further explored with regressions to Vogel–Fulcher–Tammann (VFT) (Fig. 5(a) and (b)) and Arrhenius (Fig. 5(c) and (d)) equations, VFT equation follows:
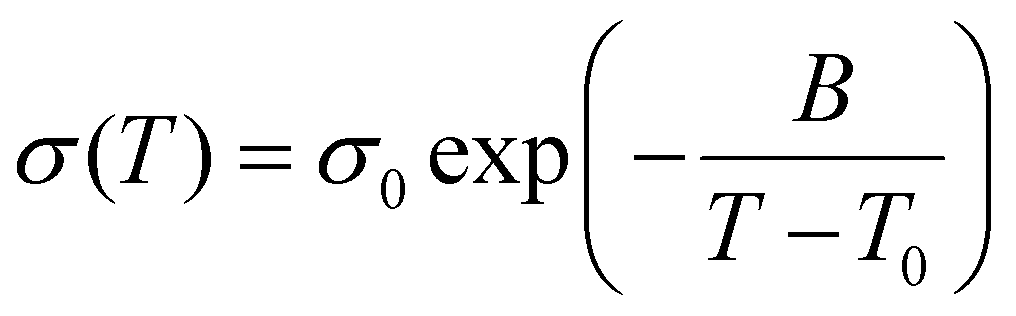 | (2) |
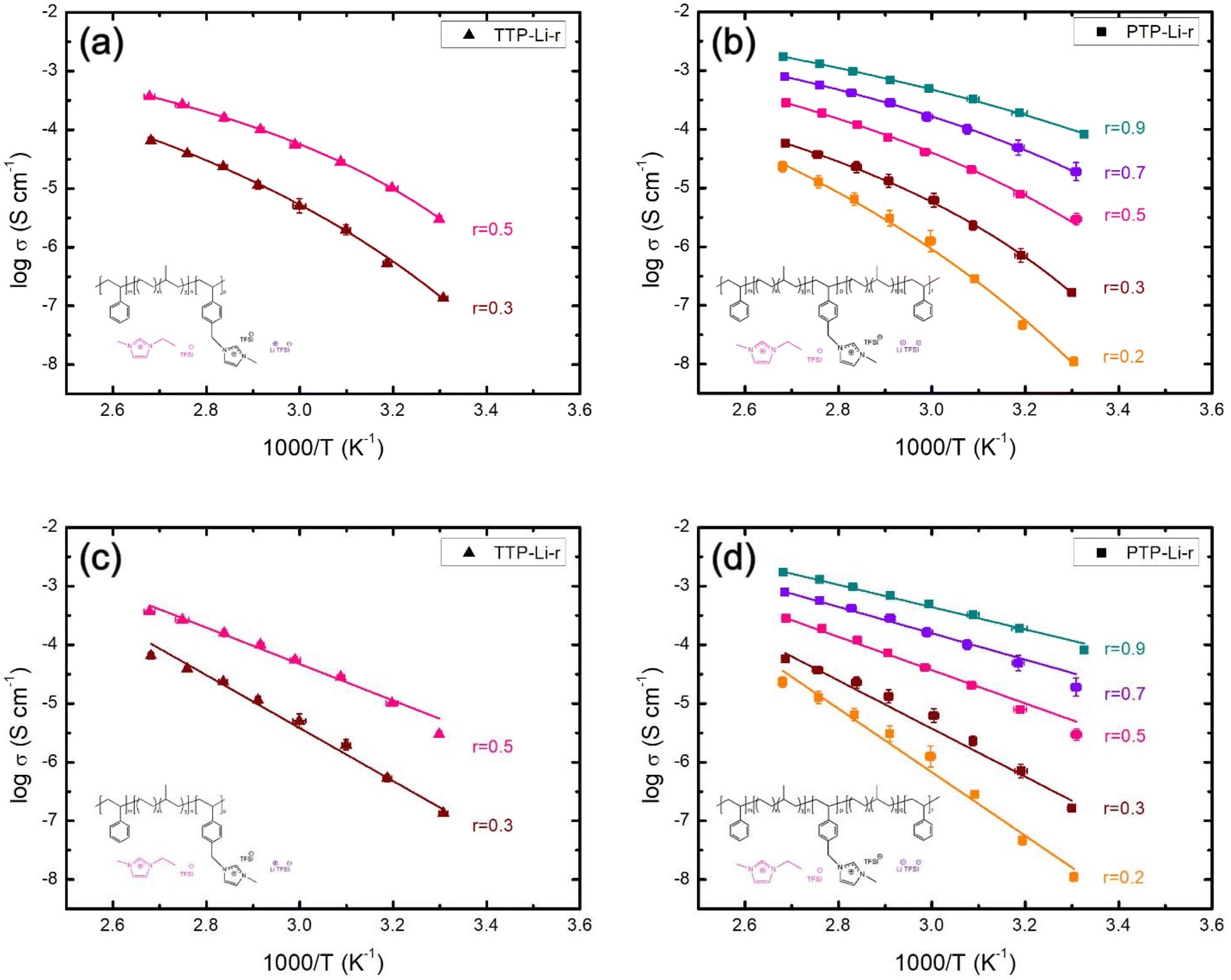 | ||
| Fig. 5 Temperature-dependent ionic conductivity of the (a) TTP-Li-r, (b) PTP-Li-r with VFT regressions, (c) TTP-Li-r, and (d) PTP-Li-r with Arrhenius regressions at various r = [IL]/[PIL block]. | ||
Arrhenius equation follows:
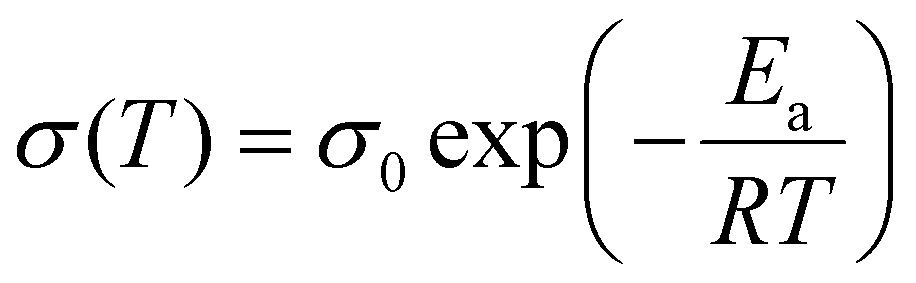 | (3) |
The ion transport by ion hopping and ion diffusion at high IL contents is also clearly observed in Fig. 6 where temperature has been normalized by the Tg of PIL block. Both SPEs at IL concentration below 0.5 exhibit a similar conductivity trend at the same normalized temperature (Tg/T), i.e., collapse onto one master curve, indicating that ion movement is coupled to the polymer segmental relaxation up to certain concentration of IL (i.e., r = 0.5). However, conductivities for SPEs at high IL contents deviate from the single master curve, indicating that ion transport is decoupled from the segmental mobility of polymer chains and is more dependent on ion hopping and ion diffusion, which corroborates with results from Fig. 5.
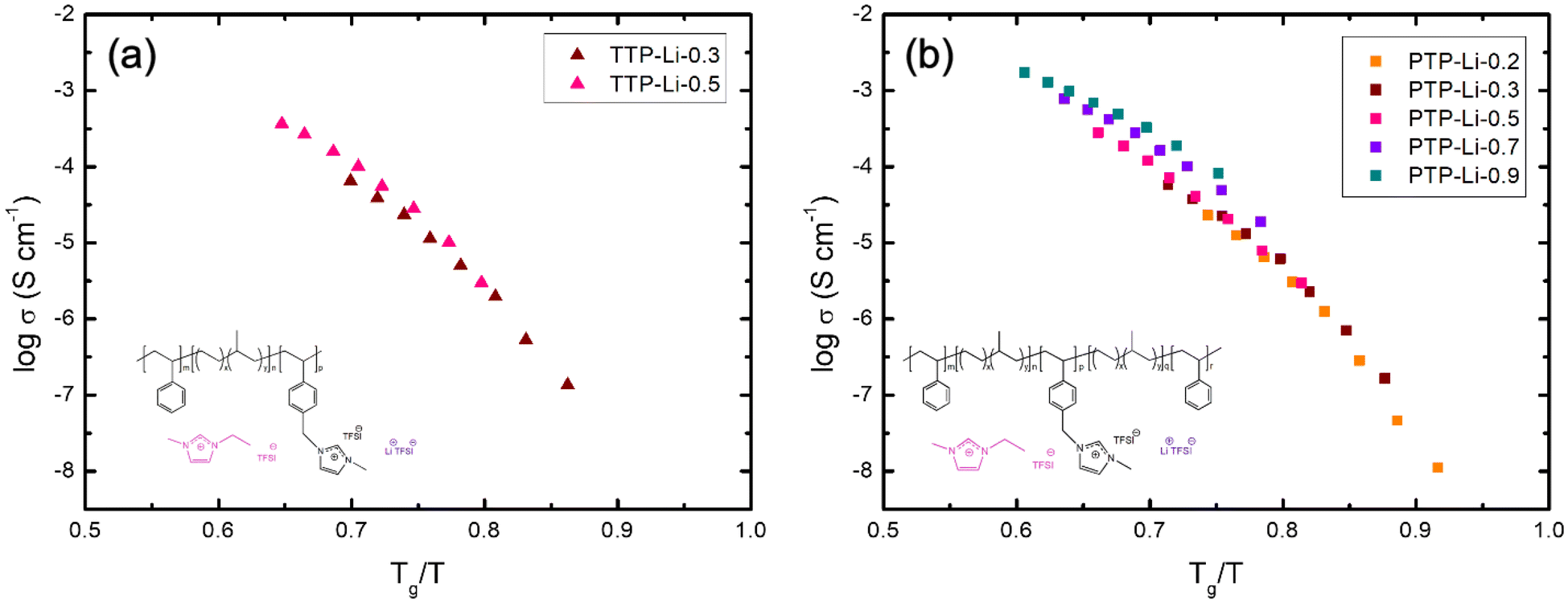 | ||
| Fig. 6 Ionic conductivity of the SPEs as a function of Tg/T: (a) TTP-Li-r and (b) PTP-Li-r. | ||
The high mechanical properties of SPEs are a key factor for LMBs due to its dendrite-suppressing ability that can improve cyclability of batteries.16,71 In order to understand the difference in mechanical properties between ABC triblock and ABCBA pentablock SPEs, (i.e., film fabrication at limited r ratio for TTP-Li-r), the mechanical strength of TTP-Li-r and PTP-Li-r was investigated with tensile measurements at a constant strain rate of 0.2 mm min−1. Fig. 7 shows stress–strain profiles of the TTP-Li-r and PTP-Li-r and Table 2 summarizes the mechanical properties of the SPEs. The PTP-Li-0.2 SPE exhibits the highest Young's modulus of 78.95 MPa and tensile strength of 8.15 MPa. As r increases, mechanical properties reduce for both SPEs due to an increasing volume fraction of IL. Interestingly, the PTP-Li-0.9 SPE exhibits slightly higher Young's modulus (0.88 MPa) and tensile strength (1.21 MPa) than the TTP-Li-0.5 SPE despite higher IL content in the former. Moreover, at the same r (i.e., 0.3 and 0.5), an order of magnitude higher Young's modulus is achieved for the PTP-Li-r compared to the TTP-Li-r, suggesting that chain architecture (ABC vs. ABCBA) has a substantial impact on mechanical properties.
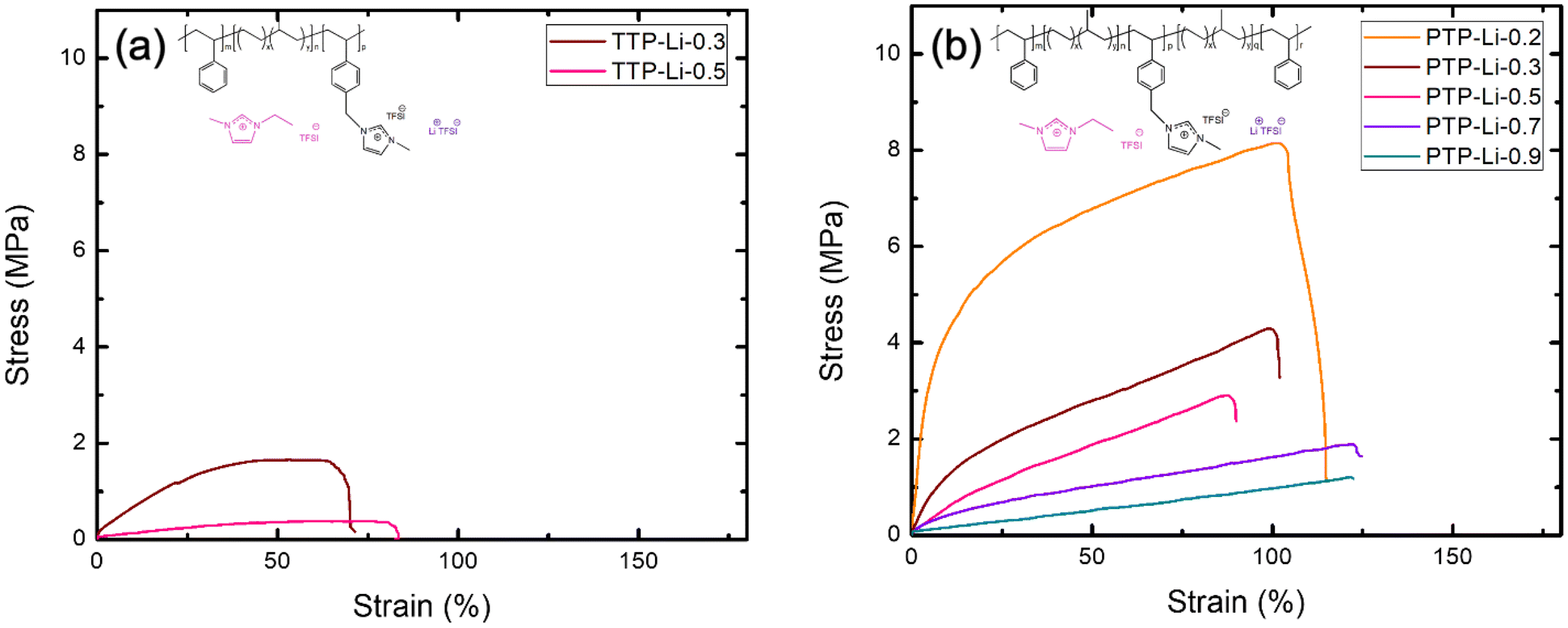 | ||
| Fig. 7 Stress–strain profiles for the SPEs at various r = [IL]/[PIL block] ratios: (a) TTP-Li-r and (b) PTP-Li-r. | ||
| SPE | Young's modulus (MPa) | Ultimate tensile strength (MPa) | Elongation at break (%) |
|---|---|---|---|
| TTP-Li-0.3 | 4.49 ± 0.27 | 1.62 ± 0.04 | 70.18 ± 2.61 |
| TTP-Li-0.5 | 0.65 ± 0.04 | 0.38 ± 0.02 | 83.48 ± 5.19 |
| PTP-Li-0.2 | 78.95 ± 2.92 | 8.15 ± 0.07 | 114.88 ± 2.67 |
| PTP-Li-0.3 | 14.45 ± 0.61 | 4.12 ± 0.18 | 99.60 ± 2.40 |
| PTP-Li-0.5 | 5.18 ± 0.19 | 2.49 ± 0.42 | 88.19 ± 1.73 |
| PTP-Li-0.7 | 3.70 ± 0.01 | 2.01 ± 0.12 | 120.22 ± 5.99 |
| PTP-Li-0.9 | 0.84 ± 0.05 | 1.21 ± 0.00 | 130.33 ± 6.87 |
The mechanical property difference between triblock and pentablock despite similar morphology can be attributed to different chain conformations of these two polymers. Compared to the TTP-Li-r, where only one chain conformation exists, the PTP-Li-r SPEs have two types of chain conformations, looped and bridged conformations, as shown in Fig. 8(a).72–74 In a bridged conformation, A end blocks reside in two different A domains, linking separate domains together. The presence of bridged conformations significantly enhances the mechanical strength of polymers.46,49,75 Moreover, the bridging chains efficiently restrict the swelling,75–77 possibly improving the mechanical strength of PTP-Li-r SPEs compared to TTP-Li-r SPEs at the same IL uptake. In a looped conformation, A end blocks reside in the same A domain, which does not substantially improve mechanical strength by itself. However, the looped chains are able to form interlocked entanglement with other looped chains or bridged chains as shown in Fig. 8(b). These looped chains with interlocked entanglement may act as bridged chains under deformation, further enhancing mechanical strength of the PTP-Li-r SPEs.78,79 This demonstrates that ion conducting PIL multiblock polymers abide by the behavior of neutral block copolymers, where symmetric polymers (e.g., ABA triblock and ABCBA pentablock polymers) have higher mechanical properties than asymmetric polymers (e.g., AB diblock and ABC triblock polymers).
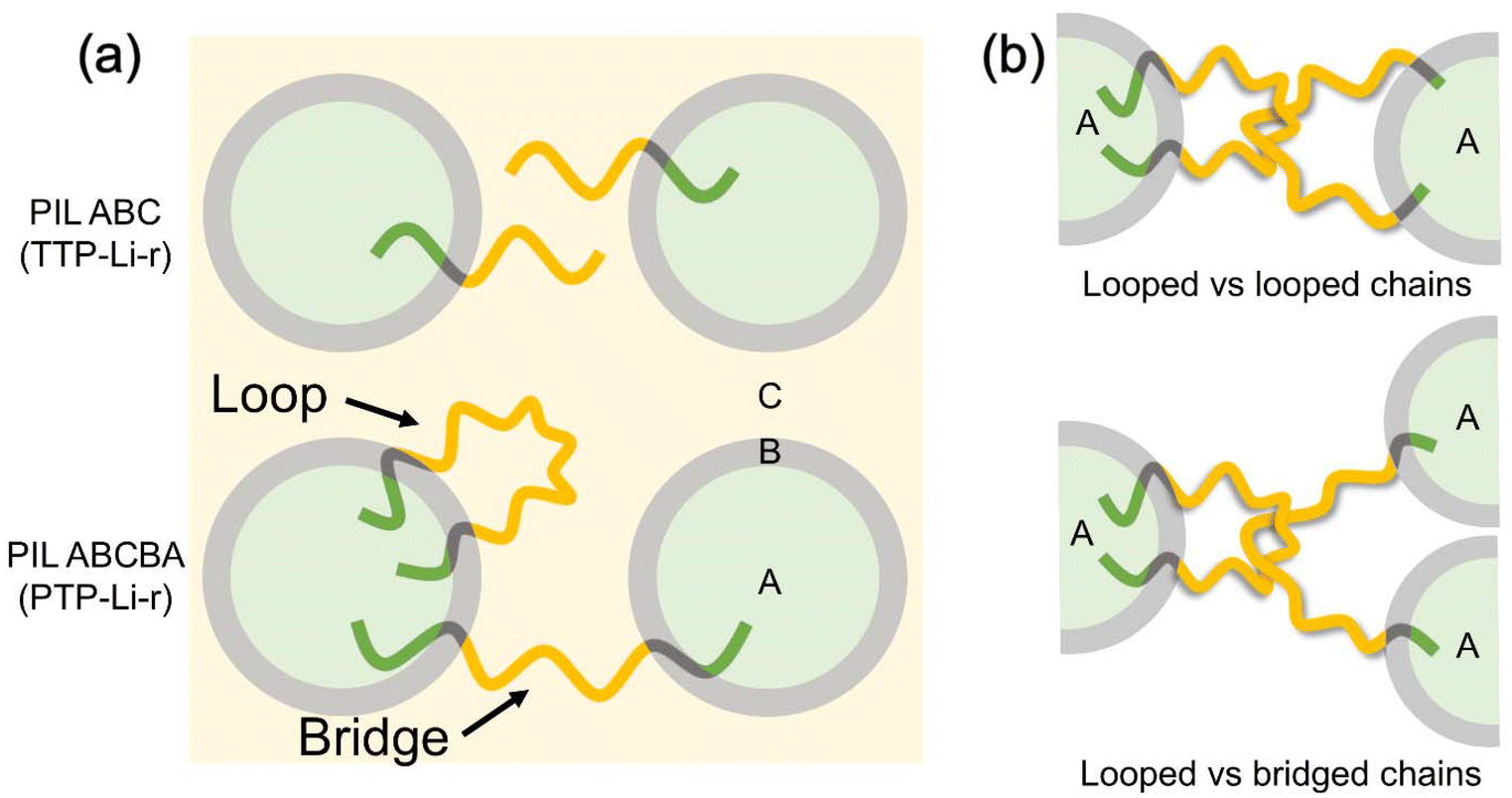 | ||
| Fig. 8 (a) Chain conformation of the TTP–TFSI and PTP–TFSI in the ternary SPEs and (b) interlocked entanglement of the PTP–TFSI by looped vs. looped chains and looped vs. bridged chains. | ||
Fig. 9 summarizes the ionic conductivity and Young's modulus of the TTP-Li-r and PTP-Li-r SPEs as a function of IL concentration. As r increases, the ionic conductivity for both SPEs increases, whereas Young's modulus decreases. At r = 0.5, the PTP-Li-0.5 SPE displays a good balance between both properties. As discussed previously, the TTP-Li-r and PTP-Li-r SPEs achieve similar ionic conductivity at all IL contents, whereas a significant difference between TTP-Li-r and PTP-Li-r is observed in Young's modulus, suggesting that chain architecture has a significant impact on mechanical properties of SPEs.
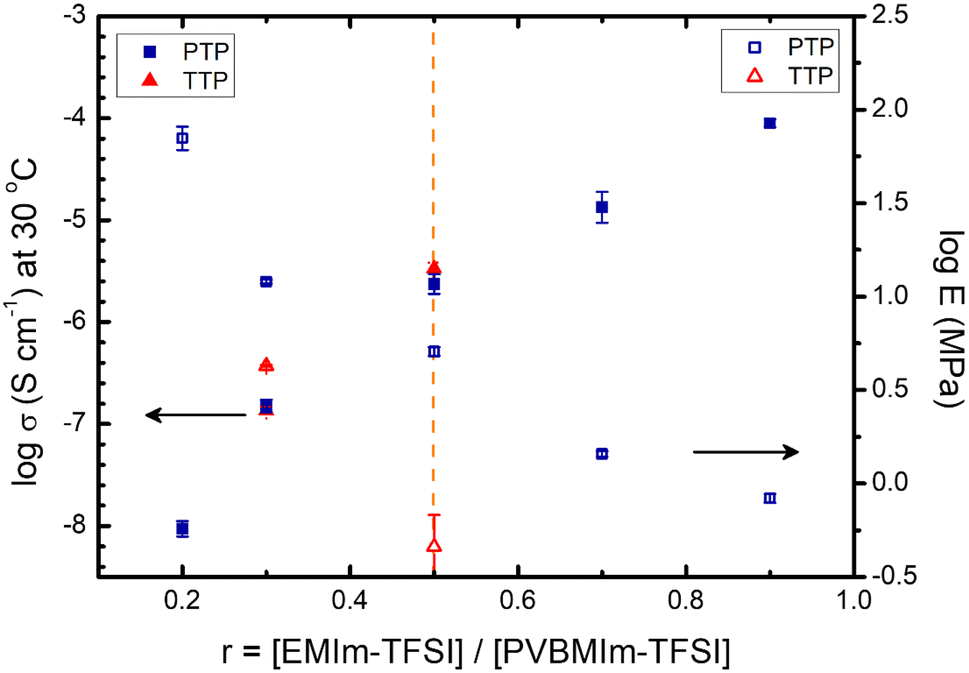 | ||
| Fig. 9 Comparison of ionic conductivity at 30 °C (solid) and Young's modulus (open) for TTP-Li-r (red) and PTP-Li-r (blue) SPEs with various r ratios. | ||
To investigate the impact of Young's modulus difference imposed by chain architecture on electrochemical stability of the SPE with lithium metal, the TTP-Li-0.5 and PTP-Li-0.5 SPEs were selected for cycling stability test. Lithium stripping and plating was conducted under a current density of 0.02 mA cm−2 at room temperature. As shown in the inset of Fig. 10(a), the TTP-Li-0.5 and PTP-Li-0.5 SPEs exhibit similar initial overpotential of 0.1 V at the beginning, where the voltage plateau is rapidly reached. The TTP-Li-0.5 displays pronounced overpotentials after 250 h and its voltage profile becomes sharper over cycle, indicating fast dendrite growth due to increased cell polarization.80,81 In contrast, the PTP-Li-0.5 exhibits less pronounced and more stable voltage profiles compared to the TTP-Li-0.5, leading to no short circuit during the measurement. This enhancement in cyclability may result from the higher mechanical properties of the PTP-Li-0.5 SPE.
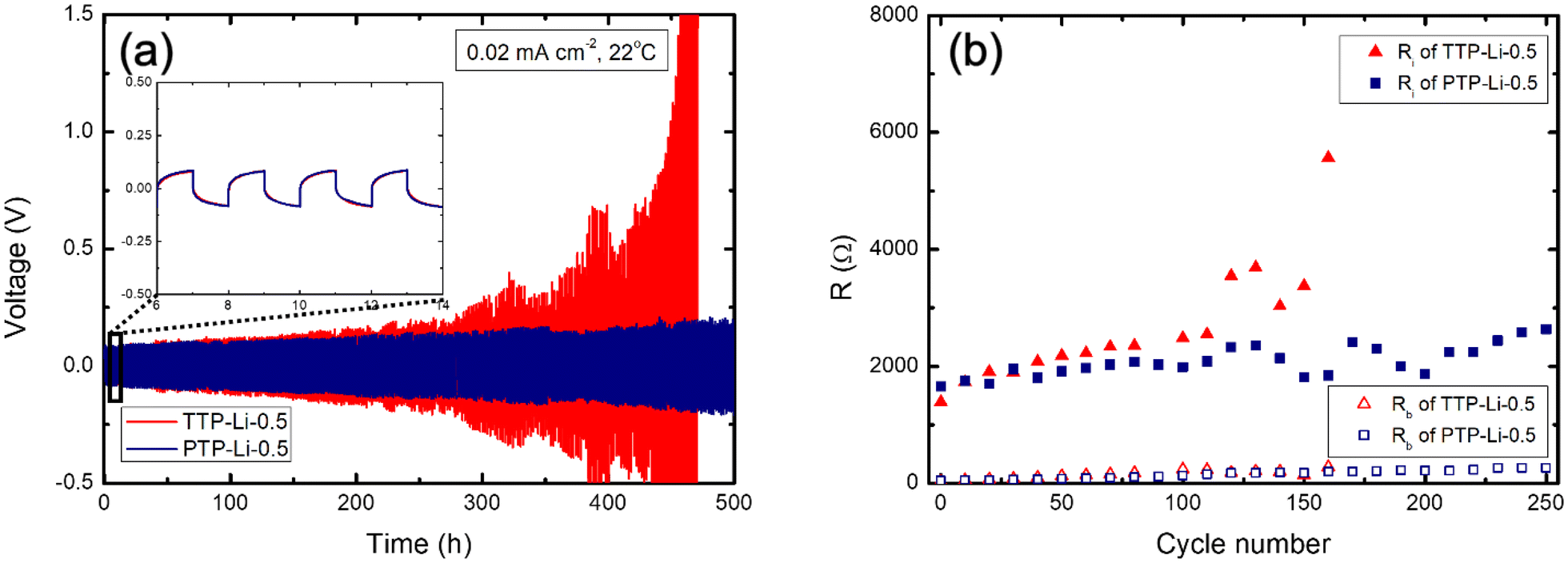 | ||
| Fig. 10 (a) Stripping/plating voltage profiles of symmetric Li/SPE/Li cells cycled with the TTP-Li-0.5 and PTP-Li-0.5 SPEs under 0.02 mA cm−2 at room temperature and (b) resistances at selected cycles for the TTP-Li-0.5 and PTP-Li-0.5 SPEs. | ||
The impedance response was recorded every 10 cycles and analyzed using an equivalent circuit model illustrated in Fig. S5† to determine the bulk electrolyte resistance (Rb) and the interfacial resistance (Ri) of the cells. Specifically, Ri consists of the passivation layer resistance (Rp) from solid electrolyte interphase (SEI) and the charge transfer resistance (Rct) of the electrode/electrolyte interface, where Ri = Rp + Rct. Fig. 10(b) shows Rb and Ri as a function of the number of cycles. The Ri of the TTP-Li-0.5 remains constant up to 100 cycles and then increases, which is consistent with the voltage profiles of the TTP-Li-0.5. This indicates that increased overpotential is associated with the increased interfacial resistance. Contrary to the TTP-Li-0.5, the Ri of PTP-Li-0.5 remains steady at around 2000 Ω during the whole cycle. The stable voltage profiles and constant Ri for the PTP-Li-0.5 suggest that high mechanical strength of the PTP-Li-r SPE efficiently mitigates dendrite formation, enhancing electrochemical interfacial stability with lithium metal.
The PTP-Li-0.5 possessing high mechanical strength and good compatibility with lithium metal was selected as SPE for a Li/NMC811 2032 coin cell battery. The cell was cycled between 3.0 and 4.2 V at room temperature at C/20 as shown in Fig. 11(a). The initial discharge capacities of the battery with TTP-Li-0.5 and PTP-Li-0.5 SPEs are similar at around 100 mA h g−1, which is ca. 48% of the theoretical capacity of NMC811 (210 mA h g−1) at C/20.82 The cell coupled with TTP-Li-0.5 SPE exhibits significant capacity fading showing 40.8% of capacity retention at 50 cycle, which is consistent with electrochemical stability result. On the contrary, the cell with PTP-Li-0.5 SPE retains 83.7% of the initial specific capacity over 50 cycle, which is comparable to other LMBs with PIL-based SPEs,83–85 where batteries were cycled at elevated temperature or less than 50 cycles. The coulombic efficiency for both SPEs is similar to each other, which is relatively constant above 91.0% for the entire 50 cycles. Fig. 11(b) shows selected voltage profiles during charge and discharge for Li/NMC811 cell with PTP-Li-0.5 SPE. The charge and discharge profiles at the first cycle remains steady over 30 cycles. After that, the slope of voltage profiles becomes slightly steeper, indicating a cell polarization that explains the slight capacity fading of PTP-Li-0.5 SPE.
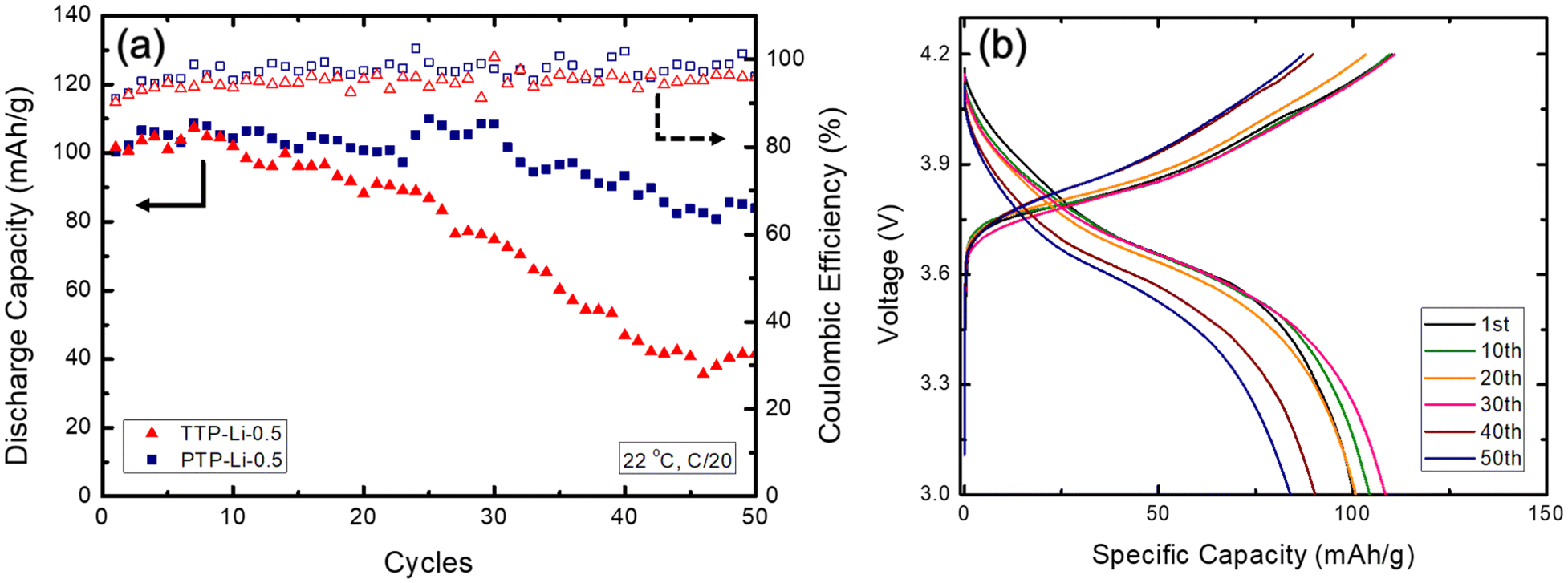 | ||
| Fig. 11 (a) Discharge capacity and coulombic efficiency of Li/NMC811 cell with TTP-Li-0.5 and PTP-Li-0.5 SPEs at 22 °C cycled at C/20 and (b) selected charge and discharge profiles of Li/NMC811 cell with PTP-Li-0.5 SPE at different cycles. | ||
The PTP-Li-0.5 SPE outperforms the TTP-Li-0.5 in interfacial compatibility with lithium metal and displays promising room temperature cycling stability in a LMB with final capacity retention of 83.7% over 50 cycles. It demonstrates that chain architecture in PIL multiblock copolymer SPEs has a significant impact on cycling performance of LMBs. The relatively low specific capacity (83.9 mA h g−1 at the 50 cycle) may be due to moderate room temperature ionic conductivity of ca. 2.9 × 10−6 S cm−1, which may be improved by exploring other chemistries (e.g., cations and anions of PIL block, Li salt, and IL). This study reveals that the chain architecture significantly affects mechanical properties of PIL multiblock polymers, suggesting that the structure of symmetric PIL multiblock polymers benefits electrochemical stability and battery cycling performance for LMBs.
4. Conclusions
PIL ABC triblock and ABCBA pentablock terpolymers were synthesized to investigate the impact of chain architecture on the properties of SPEs for LMBs. Ternary SPEs containing PIL multiblock polymers with corresponding IL and Li salt were fabricated at different IL ratios, and systematically investigated with respect to morphology, ionic conductivity, mechanical properties, electrochemical stability, and battery cycling performance. TTP-Li-r and PTP-Li-r SPEs exhibit similar morphology and the morphology remains unchanged regardless of IL concentration. The ionic conductivities for both TTP-Li-r and PTP-Li-r SPEs are comparable at each IL concentration due to the identical morphology with similar chain mobility. The PTP-Li-0.9 SPE exhibits the highest ionic conductivity among the SPEs (i.e., 8.2 × 10−5 S cm−1 at 30 °C). In addition, the PTP-Li-r displays an order of magnitude higher Young's modulus than the TTP-Li-r at the same r due to its unique chain conformations: bridged chains and interlocked entanglement of bridged chains and looped chains. This increase in mechanical properties reflects the excellent electrochemical stability of the PTP-Li-0.5 SPE and effectively mitigates dendrite formation during cycles. The Li/NMC811 battery paired with PTP-Li-0.5 SPE exhibits a promising specific capacity of 83.9 mA h g−1 with capacity retention of 83.7% over 50 cycles. This work promotes the importance of chain architecture of PIL multiblock polymers on the properties of SPEs and provides insight into the design of PILBCP-based SPEs for lithium metal batteries.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Kraton Polymers, LLC.References
- A. Yoshino, in Lithium-ion batteries, ed. G. Pistoia, Elsevier, 2014, pp. 1–20 Search PubMed.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621–630 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li and N. Tavajohi, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- Q. Wang, P. Ping, X. Zhao, G. Chu, J. Sun and C. Chen, J. Power Sources, 2012, 208, 210–224 CrossRef CAS.
- K. Qin, K. Holguin, M. Mohammadiroudbari, J. Huang, E. Y. S. Kim, R. Hall and C. Luo, Adv. Funct. Mater., 2021, 31, 2009694 CrossRef CAS.
- A. Jana and R. E. García, Nano Energy, 2017, 41, 552–565 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, Phys. Chem. Chem. Phys., 2017, 19, 20493–20505 RSC.
- X. He, S. Schmohl and H. D. Wiemhöfer, ChemElectroChem, 2019, 6, 1166–1176 CrossRef CAS.
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Nat. Rev. Mater., 2019, 4, 312–330 CrossRef CAS.
- Y. Zhang, Y. He, K. Ma, P. Li, Z. Wang, X. Li and Y. Zhang, Energy Fuels, 2021, 35, 5333–5341 CrossRef CAS.
- Y. Zhong, J. Zhang, S. Wang, D. Han, M. Xiao and Y. Meng, Mater. Adv., 2020, 1, 873–879 RSC.
- R. Khurana, J. L. Schaefer, L. A. Archer and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7395–7402 CrossRef CAS PubMed.
- S. Stalin, H. E. Johnson, P. Biswal, D. Vu, Q. Zhao, J. Yin, B. A. Abel, Y. Deng, G. W. Coates and L. A. Archer, Macromolecules, 2020, 53, 5445–5454 CrossRef CAS.
- J.-N. Chazalviel, Phys. Rev. A, 1990, 42, 7355 CrossRef CAS PubMed.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396 CrossRef CAS.
- L. Caradant, N. Verdier, G. Foran, D. Lepage, A. Prébé, D. Aymé-Perrot and M. Dollé, ACS Appl. Polym. Mater., 2021, 3, 6694–6704 CrossRef CAS.
- R. P. Doyle, X. Chen, M. Macrae, A. Srungavarapu, L. J. Smith, M. Gopinadhan, C. O. Osuji and S. Granados-Focil, Macromolecules, 2014, 47, 3401–3408 CrossRef CAS.
- X. Ye, W. Xiong, T. Huang, X. Li, Y. Lei, Y. Li, X. Ren, J. Liang, X. Ouyang and Q. Zhang, Appl. Surf. Sci., 2021, 569, 150899 CrossRef CAS.
- D. Devaux, D. Glé, T. N. Phan, D. Gigmes, E. Giroud, M. Deschamps, R. Denoyel and R. Bouchet, Chem. Mater., 2015, 27, 4682–4692 CrossRef CAS.
- Z. Lin, X. Guo, Y. Yang, M. Tang, Q. Wei and H. Yu, J. Energy Chem., 2021, 52, 67–74 CrossRef CAS.
- W. S. Young, W. F. Kuan and T. H. Epps III, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1–16 CrossRef CAS.
- H.-D. Nguyen, G.-T. Kim, J. Shi, E. Paillard, P. Judeinstein, S. Lyonnard, D. Bresser and C. Iojoiu, Energy Environ. Sci., 2018, 11, 3298–3309 RSC.
- S. Chen, Y. Li, Y. Wang, Z. Li, C. Peng, Y. Feng and W. Feng, Macromolecules, 2021, 54, 9135–9144 CrossRef CAS.
- Q. Lu, Y. B. He, Q. Yu, B. Li, Y. V. Kaneti, Y. Yao, F. Kang and Q. H. Yang, Adv. Mater., 2017, 29, 1604460 CrossRef PubMed.
- Y. Zhai, G. Yang, Z. Zeng, S. Song, S. Li, N. Hu, W. Tang, Z. Wen, L. Lu and J. Molenda, ACS Appl. Energy Mater., 2021, 4, 7973–7982 CrossRef CAS.
- Z. Tu, Y. Kambe, Y. Lu and L. A. Archer, Adv. Energy Mater., 2014, 4, 1300654 CrossRef.
- X. Li, Z. Zhang, S. Li, L. Yang and S.-I. Hirano, J. Power Sources, 2016, 307, 678–683 CrossRef CAS.
- T. C. Mendes, N. Goujon, N. Malic, A. Postma, J. Chiefari, H. Zhu, P. C. Howlett and M. Forsyth, J. Electrochem. Soc., 2020, 167, 070525 CrossRef CAS.
- N. Goujon, T. V. Huynh, K. J. Barlow, R. Kerr, K. Vezzu, V. Di Noto, L. A. O'Dell, J. Chiefari, P. C. Howlett and M. Forsyth, Batteries Supercaps, 2019, 2, 132–138 CrossRef CAS.
- T.-L. Chen, P. M. Lathrop, R. Sun and Y. A. Elabd, Macromolecules, 2021, 54, 8780–8797 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 CrossRef CAS.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- I. Lee and F. S. Bates, Macromolecules, 2013, 46, 4529–4539 CrossRef CAS.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473–13476 RSC.
- C. Jangu, J.-H. H. Wang, D. Wang, G. Fahs, J. R. Heflin, R. B. Moore, R. H. Colby and T. E. Long, J. Mater. Chem. C, 2015, 3, 3891–3901 RSC.
- M. L. Lehmann, G. Yang, J. Nanda and T. Saito, Macromolecules, 2022, 55, 7740–7751 CrossRef CAS.
- L. Wu, E. W. Cochran, T. P. Lodge and F. S. Bates, Macromolecules, 2004, 37, 3360–3368 CrossRef CAS.
- T. R. Panthani and F. S. Bates, Macromolecules, 2015, 48, 4529–4540 CrossRef CAS.
- B. Arman, A. S. Reddy and G. Arya, Macromolecules, 2012, 45, 3247–3255 CrossRef CAS.
- J. Park, A. Staiger, S. Mecking and K. I. Winey, Macromolecules, 2021, 54, 4269–4279 CrossRef CAS.
- G. P. Baeza, J. Polym. Sci., 2021, 59, 2405–2433 CrossRef CAS.
- C. Y. Ryu, J. Ruokolainen, G. H. Fredrickson, E. J. Kramer and S. F. Hahn, Macromolecules, 2002, 35, 2157–2166 CrossRef CAS.
- H. Fujita, T. Michinobu, S. Fukuta, T. Koganezawa and T. Higashihara, ACS Appl. Mater. Interfaces, 2016, 8, 5484–5492 CrossRef CAS PubMed.
- S. Ji, U. Nagpal, G. Liu, S. P. Delcambre, M. Müller, J. J. de Pablo and P. F. Nealey, ACS Nano, 2012, 6, 5440–5448 CrossRef CAS PubMed.
- M. W. Matsen and R. Thompson, J. Chem. Phys., 1999, 111, 7139–7146 CrossRef CAS.
- L. Sun, L. J. Shen, M. Q. Zhu, C. M. Dong and Y. Wei, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4583–4593 CrossRef CAS.
- H.-H. Liu, C.-I. Huang and A.-C. Shi, Macromolecules, 2015, 48, 6214–6223 CrossRef CAS.
- A. J. Meuler, G. Fleury, M. A. Hillmyer and F. S. Bates, Macromolecules, 2008, 41, 5809–5817 CrossRef CAS.
- T.-L. Chen, R. Sun, C. Willis, B. Krutzer, B. F. Morgan, F. L. Beyer, K. S. Han, V. Murugesan and Y. A. Elabd, Polymer, 2020, 209, 122975 CrossRef CAS.
- T. Huang, H. Toraya, T. Blanton and Y. Wu, J. Appl. Crystallogr., 1993, 26, 180–184 CrossRef CAS.
- B. R. Pauw, J. Condens. Matter Phys., 2013, 25, 383201 CrossRef PubMed.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- S. Sharick, J. Koski, R. A. Riggleman and K. I. Winey, Macromolecules, 2016, 49, 2245–2256 CrossRef CAS.
- A. J. Butzelaar, P. Röring, M. Hoffmann, J. Atik, E. Paillard, M. Wilhelm, M. Winter, G. Brunklaus and P. Theato, Macromolecules, 2021, 54, 11101–11112 CrossRef CAS.
- T.-L. Chen, R. Sun, C. Willis, B. F. Morgan, F. L. Beyer and Y. A. Elabd, Polymer, 2019, 161, 128–138 CrossRef CAS.
- S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS.
- M. Matsen, J. Chem. Phys., 1995, 102, 3884–3887 CrossRef CAS.
- Y. T. Lo, C. H. Chang, H. H. Liu, C. I. Huang and A. C. Shi, Macromol. Theory Simul., 2021, 30, 2100014 CrossRef CAS.
- M. Xenidou, F. L. Beyer, N. Hadjichristidis, S. P. Gido and N. B. Tan, Macromolecules, 1998, 31, 7659–7667 CrossRef CAS.
- D. Y. Liu and D. V. Krogstad, Macromolecules, 2021, 54, 988–994 CrossRef CAS.
- K. Hashimoto, M. Hirasawa, H. Kokubo, R. Tamate, X. Li, M. Shibayama and M. Watanabe, Macromolecules, 2019, 52, 8430–8439 CrossRef CAS.
- M. Ghelichi, N. T. Qazvini, S. H. Jafari, H. A. Khonakdar, Y. Farajollahi and C. Scheffler, J. Appl. Polym. Sci., 2013, 129, 1868–1874 CrossRef CAS.
- R. J. Klein and J. Runt, J. Phys. Chem. B, 2007, 111, 13188–13193 CrossRef CAS PubMed.
- H. Yang and N. Wu, Energy Sci. Eng., 2022, 10, 1643–1671 CrossRef CAS.
- Z. Li, J. Fu, X. Zhou, S. Gui, L. Wei, H. Yang, H. Li and X. Guo, Adv. Sci., 2023, 10, 2201718 CrossRef CAS PubMed.
- S. Mogurampelly and V. Ganesan, Macromolecules, 2018, 51, 9471–9483 CrossRef CAS.
- S. B. Aziz, T. J. Woo, M. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Devices, 2018, 3, 1–17 Search PubMed.
- S. Seki, M. A. B. H. Susan, T. Kaneko, H. Tokuda, A. Noda and M. Watanabe, J. Phys. Chem. B, 2005, 109, 3886–3892 CrossRef CAS PubMed.
- G. Stone, S. Mullin, A. Teran, D. Hallinan, A. Minor, A. Hexemer and N. Balsara, J. Electrochem. Soc., 2011, 159, A222 CrossRef.
- S. T. Milner and T. A. Witten, Macromolecules, 1992, 25, 5495–5503 CrossRef CAS.
- E. Zhulina and A. Halperin, Macromolecules, 1992, 25, 5730–5741 CrossRef CAS.
- M. Matsen and M. Schick, Macromolecules, 1994, 27, 187–192 CrossRef CAS.
- C. Guo and T. S. Bailey, Soft Matter, 2010, 6, 4807–4818 RSC.
- E. M. Tsang, Z. Zhang, Z. Shi, T. Soboleva and S. Holdcroft, J. Am. Chem. Soc., 2007, 129, 15106–15107 CrossRef CAS PubMed.
- B. Li and E. Ruckenstein, Macromol. Theory Simul., 1998, 7, 333–348 CrossRef CAS.
- A. Takano, I. Kamaya, Y. Takahashi and Y. Matsushita, Macromolecules, 2005, 38, 9718–9723 CrossRef CAS.
- L. A. Rankin, B. Lee and K. P. Mineart, J. Polym. Sci., 2021, 59, 34–42 CrossRef CAS.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- J. Atik, D. Diddens, J. H. Thienenkamp, G. Brunklaus, M. Winter and E. Paillard, Angew. Chem., Int. Ed., 2021, 60, 11919–11927 CrossRef CAS PubMed.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132 CrossRef CAS.
- Y. Yu, F. Lu, N. Sun, A. Wu, W. Pan and L. Zheng, Soft Matter, 2018, 14, 6313–6319 RSC.
- G. M. Girard, X. Wang, R. Yunis, D. R. MacFarlane, A. J. Bhattacharyya, M. Forsyth and P. C. Howlett, Batteries Supercaps, 2019, 2, 229–239 CrossRef CAS.
- F. Lu, X. Gao, A. Wu, N. Sun, L. Shi and L. Zheng, J. Phys. Chem. C, 2017, 121, 17756–17763 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lp00204k |
| This journal is © The Royal Society of Chemistry 2024 |