
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organo-disulfide-based particles enable controlled stimulus-triggered cleaning of electrode surfaces†
Hongyi
Zhang
ab,
Garrett L.
Grocke
ab,
Samuel S.
Kopfinger
a,
Yilin
Wang
 bef,
Arnav
Brahmasandra
a,
Randy H.
Ewoldt
bef,
Stuart J.
Rowan
*abcd and
Shrayesh N.
Patel
*abc
bef,
Arnav
Brahmasandra
a,
Randy H.
Ewoldt
bef,
Stuart J.
Rowan
*abcd and
Shrayesh N.
Patel
*abc
aPritzker School of Molecular Engineering, University of Chicago, Chicago, Illinois 60637, USA. E-mail: shrayesh@uchicago.edu; stuartrowan@uchicago.edu
bJoint Center for Energy Storage Research, Argonne National Laboratory, Argonne, IL 60439, USA
cChemical Sciences and Engineering Division, Argonne National Laboratory, Argonne, IL 60439, USA
dDepartment of Chemistry, University of Chicago, Chicago, Illinois 60637, USA
eDepartment of Mechanical Science and Engineering, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
fBeckman Institute for Advanced Science and Technology, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 11th September 2024
Abstract
Electrode fouling resulting in reduced performance is an ongoing challenge in electrochemical flow cells based on redox active polymers (RAPs). An avenue that holds substantial promise yet remains relatively unexplored involves the strategic design of RAPs capable of undergoing electrochemical stimulation to facilitate in situ electrode cleaning within a flow cell. Herein, a new electrode cleaning strategy is demonstrated through the application of redox-active poly(glycidyl methacrylate) particles crosslinked with 2-amino-1,3,4-thiadiazole disulfide (PGMA–ATDDS). The resulting particles can de-crosslink through cleavage of the disulfide bond using stimuli, such as electrochemical reduction or UV photoexcitation. Using a custom flow cell, applying such a stimulus to an ITO electrode artificially fouled with PGMA–ATDDS in the presence of a fluid flow leads to a significant particle removal (80%) that is over six times more efficient relative to the case when no stimulus is applied. Confocal fluorescence imaging of the electrochemically stimulated electrode highlighted localized disulfide reduction of particles near the electrode surface. It is posited that this selective de-crosslinking and concomitant electrolyte swelling at the particle/electrode interface facilitate particle removal in the presence of a fluid flow. In addition, the regeneration of electrode performance upon cleaning was demonstrated through charging of a redox-active particle suspension of poly(vinylbenzyl chloride) functionalized with dimethylaminoferrocene (PVBC–Fc). Upon electrochemical cleaning of the fouled ITO electrode, the accessible charge of PVBC–Fc was statistically equivalent to the accessible charge measured using a pristine ITO electrode. Overall, this study introduces a new approach for leveraging stimulus-responsive chemistries for RAPs to impart inherent functionality to facilitate in-line electrode cleaning in electrochemical flow cells.
1. Introduction
Electrode fouling can significantly impact the function of electrochemical-based sensors and energy storage devices. It occurs when an irreversible inactive layer forms, preventing redox active species from effectively interacting with the electrode surface. Electrochemical systems that employ small molecules to polymeric redox active species (redoxmers) are particularly prone to experiencing electrode fouling issues. The irreversible inactive layer may form through the precipitation of insoluble species from normal operation or those that have undergone electrochemical charging and/or degradation.1,2 In turn, various anti-fouling or electrode cleaning strategies have been investigated, including protective layers, electrode surface modifications, and electrochemical activation.3 The conventional approach is preventing fouling during the electrochemical operation through electrode modification with carbon-based materials, metallic nanoparticle coatings, or anti-fouling polymers.3–5 However, the application of such anti-fouling layers can introduce additional impedance and reduce electrochemical accessibility of the desired redox active molecule (or redoxmer).6–8 An alternative and less explored approach is to impart functionality into the redoxmer that would allow for effective cleaning of an unmodified electrode surface after fouling has occurred. This approach may involve stimulus-responsive chemistries that allow for controlled cleaning. As such, designing redoxmers with the intrinsic ability to effectively and non-destructively clean an electrode surface in situ would allow for electrochemical systems with improved lifetimes and reduced waste products.One class of electrochemical devices that stands to benefit from in situ electrode cleaning strategies is the redox flow battery (RFB), which is highly scalable in its capacity and easily tunable in power delivery essential for long duration energy storage. RFBs based on organic redoxmers are especially interesting as a result of the high tunability of the redox active molecules, allowing for precise control over the redox potential, solubility, and supramolecular structure, among other properties.9–13 Of the possible redoxmer architectures, redox active polymers (RAPs) spanning soluble polymer chains to colloidal particles are particularly promising as they can enable high-performance batteries without the need for expensive, flux-inhibiting ion exchange membranes. In fact, all that is required for RAP-based RFBs are simpler size-exclusion membranes adequate for screening high molecular weight active species.14–19 This size-exclusion principle was demonstrated successfully in several soluble RAPs, where the hydrodynamic radius of the RAP exceeded the threshold for crossover across commercially-available porous battery separators.20,33 Despite these advantages, RAPs in flowing systems are susceptible to fouling of fluid-exposed solid surfaces. Over time, RAPs can form inactive layers on the electrode surfaces, limiting the RFB's ability to charge and discharge. Under electrochemical conditions of the RFB, the material comprising these blocking layers can degrade and the passivation layer can become permanently adhered, likely resulting in the need for expensive repairs and operational downtime.
Efforts are underway to mitigate the fouling process at different stages of the RFB operation. Coatings and pretreatments for surfaces susceptible to fouling can help mitigate or prevent the process before it occurs, but such approaches can be complex and costly, and modulating the substrate surface impacts the material performance.6,8,20 At the other end of the process flow, efforts have been made to regenerate surfaces once RAPs have already accumulated and decomposed to a nonfunctional state. Specifically, the focus has been on designing RAPs capable of being chemically or electrochemically deconstructed in a controlled manner. For example, Nguyen et al. demonstrated the design of RAPs by combining redox-active pendant groups with a polypeptide backbone, capable of being depolymerized under acidic conditions to amino acid small molecules potentially capable of recycling back to macromolecular materials later.21 Going one step further, Qian et al. demonstrated programmed degradation of RAPs using an electrochemical stimulus, wherein homobenzylic ethers could be controllably cleaved by applying an oxidative potential.22 This similarly allows for potential recapture of the degradation products, although both of these processes ultimately require separation of the degraded species from the greater system for further processing. These approaches, while innovative, generally require the separation and further processing of degradation products, adding an additional layer of complexity.
In contrast, the approach reported herein leverages a stimulus-responsive covalent disulfide bond within the RAP design to enable in situ, electrochemical-triggered cleaning of electrodes, without the need for additional processing steps. Disulfides are a class of stimulus-responsive dynamic bonds that can be cleaved with multiple external stimuli, including heat, UV light, electrochemical potential, and pH.23–29 Using such responsive properties of the disulfide bond has allowed access to a wide range of disulfide-containing polymeric materials that exhibit self-healing,26,30 reprogrammable shape-memory,31 rebondable adhesive capabilities,32,33 dynamic self-assembly,34etc. The objective of this study is to demonstrate electrochemical and UV stimulus triggered cleaning of electrodes in the presence of a fluid flow when artificially fouled with disulfide-crosslinked RAPs (Fig. 1a).
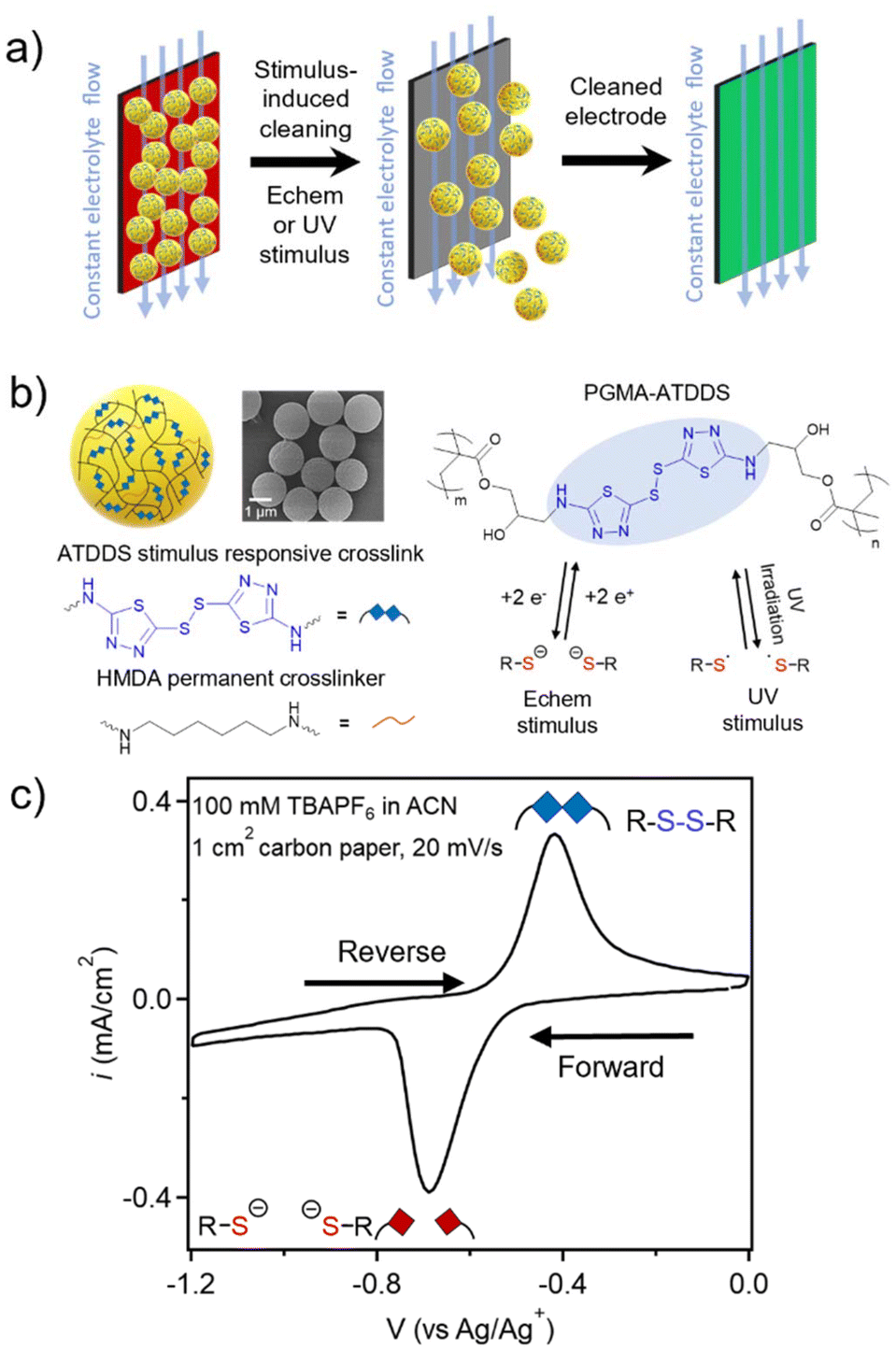 | ||
| Fig. 1 (a) Schematic of the proposed stimulus-triggered cleaning of a particle-fouled electrode in the presence of electrolyte fluid flow. (b) Left: Schematic drawing of PGMA–ATDDS particles bearing stimulus-responsive disulfide (ATDDS) and permanent (HMDA) crosslinkers. Representative SEM image of PGMA–ATDDS particles (ca. 1500 nm dry particle diameter). Right: Chemical structure for PGMA–ATDDS particles and their reversible reaction under either electrochemical or UV stimulus. (c) Cyclic voltammogram (CV) of PGMA–ATDDS particles drop-cast on a carbon paper electrode demonstrating electrochemical reduction and oxidation of disulfide bonds (20 mV s−1, 1 cm2 carbon paper working electrode, 100 mM tetrabutylammonium hexafluorophosphate (TBAPF6) in ACN supporting electrolyte). | ||
2. Results and discussion
2.1. Particle synthesis and characterization
Monodisperse PGMA particles (P1) were synthesized using dispersion polymerization followed by functionalization with 1 wt% of hexamethylenediamine (HMDA) as a permanent crosslinker to ensure particle integrity to yield lightly crosslinked PGMA particles (P2) (2200 ± 140 nm in acetonitrile (ACN) measured by dynamic light scattering (DLS), Fig. S1†). The P2 particles were then functionalized with an excess of 2-amino-1,3,4-thiadiazole disulfide (ATDDS) followed by a photoannealing step, as had been reported in prior work.27 The resulting crosslinked particles (PGMA–ATDDS) are therefore predominantly crosslinked with the stimulus-responsive ATDDS moiety (Fig. 1b) as confirmed by FT-IR and UV-Vis spectroscopy (Fig. S2 and S3†). The increase in crosslinking upon addition of the ATDDS moiety is further supported by the decrease in the size of the ACN swollen particles (to ca. 1731 ± 109 nm in diameter, Fig. S1†). Fig. 1c shows the cyclic voltammetry (CV) of PGMA–ATDDS. A reduction peak potential at −0.69 V vs. Ag/Ag+ and an oxidation peak potential at −0.48 V vs. Ag/Ag+ were observed. Control particles with no redox active disulfide bonds and only HMDA crosslinking moieties (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 amine:epoxy feed ratio) were synthesized from the same P2 particles to yield PGMA–HMDA particles (Fig. S4†), which reached the same ACN swollen hydrodynamic diameter as PGMA–ATDDS (ca. 1726 ± 106 nm).
:1 amine:epoxy feed ratio) were synthesized from the same P2 particles to yield PGMA–HMDA particles (Fig. S4†), which reached the same ACN swollen hydrodynamic diameter as PGMA–ATDDS (ca. 1726 ± 106 nm).
2.2. Stimulus-triggered controlled particle release from the electrode surface
To create an artificially fouled surface, particles were coated onto an indium tin oxide (ITO)-coated glass substrate, by drop-casting 100 μL sonicated particle dispersion in ethanol (5 mg mL−1) (Fig. 2a). This process resulted in the particles coating a 17 mm by 15 mm area of the ITO substrate with an approximate particle loading of 0.2 mg cm−2. The substrates were imaged via an optical microscope in the region of interest and ImageJ analysis was used to count the number of particles (Fig. S5†). This served as the initial particle count before the electrode cleaning steps were applied.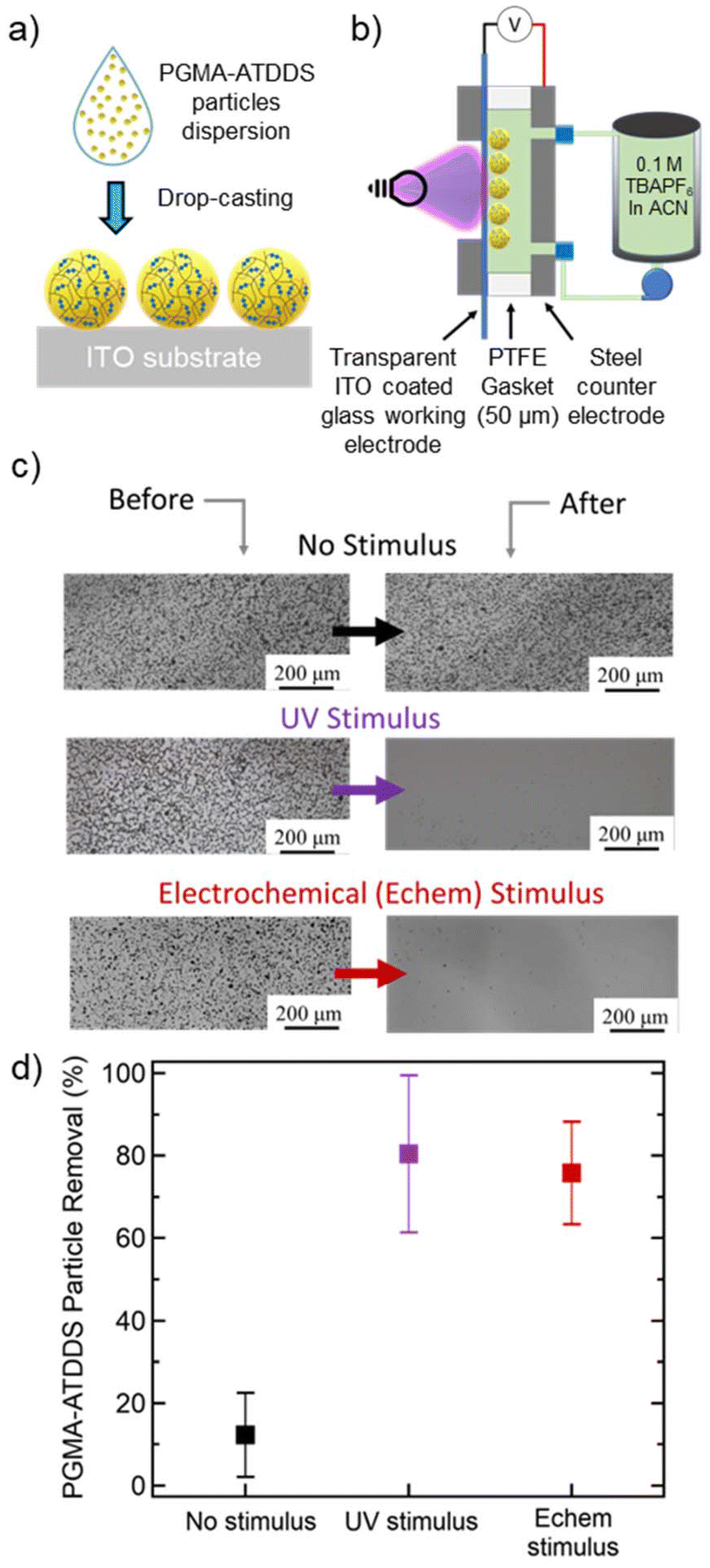 | ||
| Fig. 2 (a) An artificially fouled ITO electrode prepared by drop-casting PGMA–ATDDS particles from ethanol dispersion and drying. (b) A custom designed and fabricated flow cell for stimulus-triggered (UV or E-chem) electrode cleaning. (c) Before and after optical microscope images of the artificially-fouled ITO-electrode surface for the cases of no stimulus, UV stimulus, and electrochemical stimulus cleaning methods. (d) Corresponding quantified percent particle removal (at a flow rate of 30 mL min−1, stimulus application time of 30 min) for each case (no stimulus, UV stimulus, and electrochemical stimulus). | ||
Controlled stimulus-triggered cleaning of the artificially fouled substrate was demonstrated using a custom designed and fabricated flow cell, depicted in Fig. 2b and Fig. S6†, based on a previously reported flow cell.35,36 The flow cell allows for the liquid electrolyte (100 mM TBAPF6 in ACN) from an external reservoir to flow between two interchangeable parallel plates separated by a fixed Teflon spacer. A stainless-steel plate served as a counter electrode (CE) and as the inlet/outlet side for the flowing liquid electrolyte. A second stainless-steel plate supported the artificially fouled ITO-coated glass substrate, where the particle-bearing surface can be simultaneously exposed to a stimulus and flowing liquid electrolyte. Both electrochemical and UV stimuli were investigated for controlled electrode cleaning. After the electrode cleaning procedure, the flow cell was disassembled, and the substrate was collected for imaging. The extent of particles removed was then determined by the difference in the number of particles counted before and after exposure to the stimulus in the flow cell (see the ESI for more details, Fig. S7†).
For the electrochemical-based cleaning studies, a constant potential was applied to the ITO-coated substrate as the working electrode (WE) relative to the stainless-steel CE plate (Fig. S6b†). The potential was set beyond the reduction potential of PGMA–ATDDS to ensure sufficient cleavage of the disulfide bonds. Due to the configuration of the flow cell, a potential is applied across the entire cell without using a reference electrode. The applied 2 V is approximately −1.7 V vs. Ag/Ag+, which is in the reduction potential regime of PGMA–ATDDS (Fig. 1c) and within the electrochemical stability window of the liquid electrolyte.37 For the UV-based cleaning studies, a light source (≈1 W cm−2 with UV Bandpass (BP) filters from 320 nm to 500 nm) was secured into the optical viewport on the backside of the stainless steel plate supporting the artificially-fouled substrate (Fig. S6b†).
Initial cleaning studies focused on determining the influence of flow rate on the extent of electrode cleaning when no stimulus was applied. A series of flow rates were tested spanning 8 mL min−1, 30 mL min−1, and 40 mL min−1 (the maximum flow rate of the pump), with comparable shear rates/stresses to other systems, which are within the typical operating flow rates of RFBs (1–100 mL min−1).38–42 The samples were exposed to fluid flow for 30 min to match the stimulus exposure time (vide infra). Based on calculations and experimental validation of different flow rates in this cell geometry, the particles experience a laminar flow of electrolyte parallel to the substrate within the custom flow cell, with a Reynolds number, Re, calculated to be well below the critical transition number of 2300 (see the ESI for more details, Fig. S8†). The resulting viscous shear stress at the surface is estimated to range from 10 to 54 Pa across this range of flow rates. This range of stress can cause yielding in a wide range of soft viscoplastic materials,43 which explains why it may be sufficient to remove these soft polymeric particles from the surface.
As shown in Fig. 2c and d, a minimal level of cleaning (around 12%) was observed at a flow rate of 30 mL min−1. A similar extent of particle removal (around 10%) was achieved at the slowest flow rate of 8 mL min−1 (Fig. S7a†). At the highest flow rate of 40 mL min−1, the percent of particles removed increased to ≈50% likely arising from the increased shear force of the flowing fluid (Fig. S7a†). These results reveal that the fluid flow alone does not significantly clean the electrode surface, especially at flow rates 30 mL min−1 and below. For the remainder of the study, the flow rate was fixed to 30 mL min−1 (wall shear stress 41 Pa) to clearly test the influence of the applied stimulus on further removal of PGMA–ATDDS particles from the electrode surface.
The extent of particle removal via electrochemical stimulus was investigated by exposing the ITO-coated substrate to a constant reductive potential across different periods of time. As shown in Fig. S7b,† the percent of particle removal increased monotonically as a function of electrochemical stimulus time. At 1 min, the percent of particle removal was around 23%. Notably, this value increased and plateaued around 76% at an application time of 30 min (Fig. S7b†), which demonstrates significant electrode cleaning as visually demonstrated in the before and after images in Fig. 2c. Compared to fluid flow alone, this percent of particle removal was significantly enhanced by a factor of about 6.5 (Fig. 2d).
The application of a UV stimulus had a similar effect on electrode cleaning (Fig. 2c and d). As shown in Fig. S7c,† the percent of particle removal monotonically increased as a function of UV exposure time and plateaued to around 80% at 30 min, similar to the electrochemical stimulus. A notable difference was observed at a lower stimulus exposure time. A larger percentage of electrode cleaning occurred at the earliest time point (ca. 42% at 1 minute), attributed to the relatively faster kinetics of the photo-induced disulfide cleavage compared to the electrochemical stimulus. As a final control experiment, PGMA particles crosslinked with the non-dynamic HDMA (PGMA–HDMA) with comparatively similar solvent swollen size to PGMA–ATDDS were drop-cast onto a substrate and subjected to the same cleaning procedure using the UV stimulus. The percent of particle removal was minimal (≈9%) (Fig. S7d†), akin to the results of PGMA–ATDDS particle removal with fluid flow alone. Overall, the combined cleaning results of both stimuli and control experiments emphasize the importance of disulfide bond cleavage in the PGMA–ATDDS particle as a necessary component in facilitating particle removal for cleaning.
2.3. Examining surface-bound particle stimulus response
To examine the mechanism behind the electrochemical cleaning process in more detail, studies were carried out to probe and visualize the electrochemical reduction of PGMA–ATDDS at the electrode/particle interface. To this end, 1-(bromomethyl)pyrene (BrMePyr) was used as an electrophilic fluorescence tag to trap any thiolates that were formed upon reduction of the disulfide bond (Fig. 3a). Initial studies verified this electrochemically-induced reaction by carrying out CV experiments on a dimethylsulfoxide (DMSO) solution of the model compound ethylaminothiadiazole disulfide (EATDDS) with excess BrMePyr. DMSO was chosen due to its ability to offer adequate solubility for BrMePyr. As shown in Fig. 3b, CV sweeps were performed from 0 to −1.50 V vs. Ag/Ag+, where the forward sweep leads to the reduction of the EATDDS dimer to thiolate monomers, with a reduction peak potential at −1.05 V vs. Ag/Ag+. In a solution devoid of BrMePyr, an oxidation peak is observed at −0.34 V on the reverse sweep, indicating the re-oxidation of thiolates back to disulfides. However, in the presence of excess BrMePyr in the solution, there is a distinct lack of the thiolate to disulfide re-oxidation peak, verifying the reaction of thiolates with BrMePyr (Fig. 3b).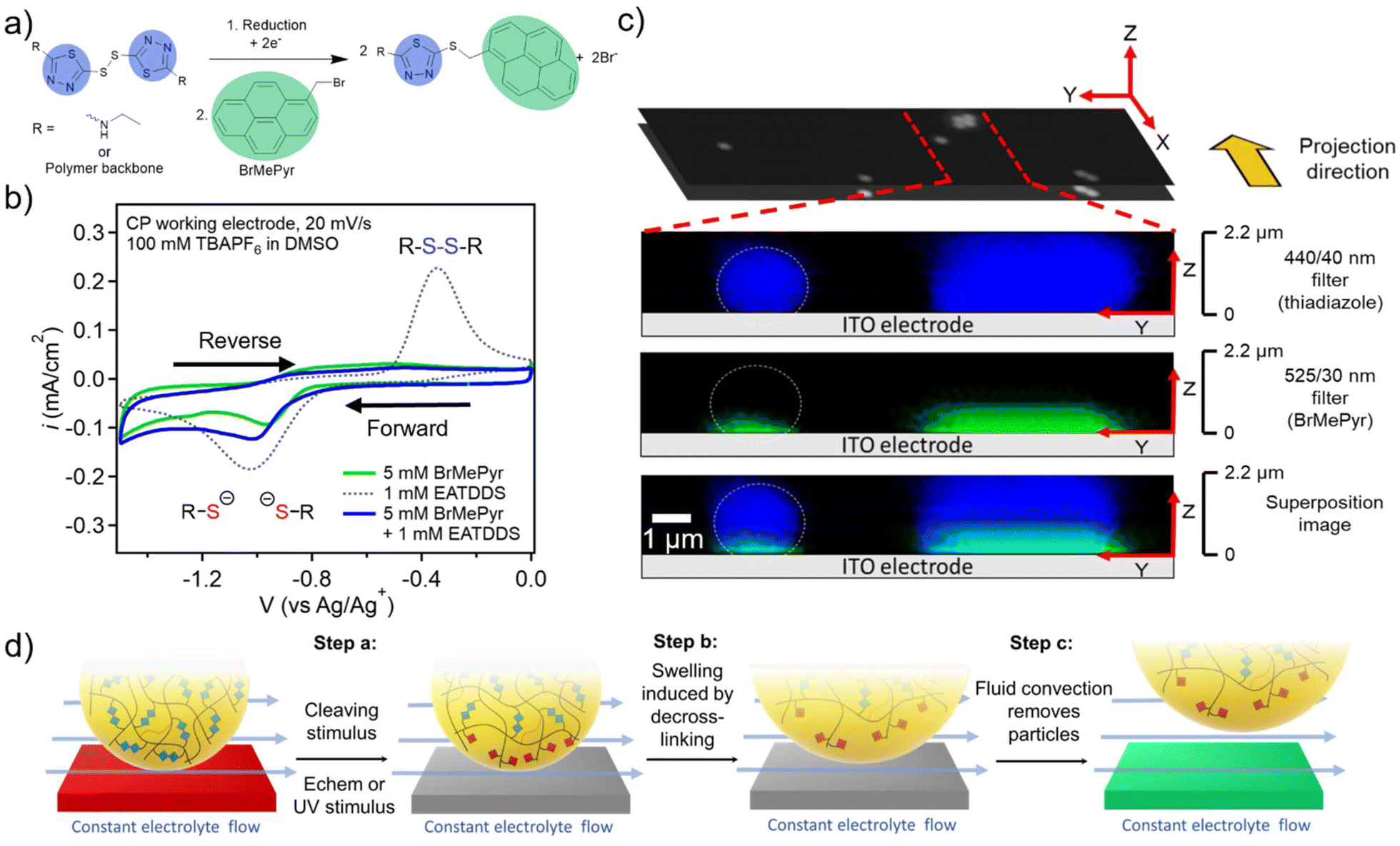 | ||
| Fig. 3 (a) Fluorescence tagging reaction scheme indicating the reduction of thiadiazole disulfide and subsequent reaction with BrMePyr. (b) Solution CV in DMSO with 100 mM TBAPF6 for 5 mM BrMePyr, 1 mM EATDDS, and a 5 mM BrMePyr + 1 mM EATDDS mixture. The lack of oxidation peak in BrMePyr:EATDDS highlights the reaction of BrMePyr with thiolate anions described in (a). (c) Confocal fluorescence microscopy of PGMA–ATDDS particles tagged with BrMePyr after electrochemical reduction (blue = thiadiazole fluorescence, green = pyrene fluorescence). Bottom-up view of isolated particles on an ITO-coated cover slip and side view of the vertical projection of particles on the substrate using different fluorescent filters. (d) Proposed cleaning mechanism for the stimulus-triggered particle removal in the presence of fluid flow. | ||
To examine how this reaction propagates in the particles, a PGMA–ATDDS particle dispersion was drop-cast onto an ITO-coated substrate as detailed in the Materials and methods section. The substrates bearing PGMA–ATDDS particles were used as the WE in a 3-probe electrochemical cell. Electrodes were submerged in a DMSO electrolyte solution of 1 mM BrMePyr and 100 mM TBAPF6. A constant reduction potential of −1.0 V vs. Ag/Ag+ was applied to the particle-bearing ITO-coated substrate for 5 minutes. Afterward, the potential was removed, and the system was allowed to relax at open circuit. The substrate bearing PGMA–ATDDS particles was removed from the solution and washed by soaking in DMSO and then acetone to remove any unreacted BrMePyr. Subsequently, confocal fluorescence images were acquired from a bottom-up view, taking progressive focal points orthogonal to the substrate surface at 0.1 μm intervals (Z direction), one layer of which is shown in the black and white image in Fig. 3c. A side-view projection (YZ plane) of the reconstructed confocal image stacks allows for visualization of fluorescence in the direction perpendicular to the ITO substrate. The image from the blue 440/40 nm filter corresponds to the fluorescence of the thiadiazole moiety of PGMA–ATDDS, which can be seen evenly distributed throughout the particle height, providing evidence for relatively homogeneous particle functionalization with ATDDS. The image from the green 525/30 nm filter corresponds to the fluorescence of the pyrene moiety arising from the reaction of BrMePyr with thiolate anions. Notably, the green fluorescence of the pyrene moiety is localized near the electrode surface, consistent with only the disulfides closest to the electrodes being reduced.
Based on combined cleaning and confocal fluorescence imaging results, Fig. 3d lays out the proposed stages for the stimulus-mediated electrode cleaning. Prior to the stimulus, the majority of the particles remain adhered to the electrode surface under the continuous flow of the electrolyte. Upon the application of the stimulus, partial de-crosslinking of the PGMA–ATDDS particles occurs near the electrode surface (Fig. 3d Step a). As a result of the reduced crosslinking density, the particles will swell at the interface with an additional electrolyte, which weakens the interaction/adhesive strength between the particle and electrode surface (Fig. 3d Step b). As a frame of reference, full reduction of disulfide bonds via a chemical reducing agent leads to a 20% increase in particle diameter (73% increase by volume), consistent with the expected higher swelling of lower crosslinked particles (Fig. S1†). Lastly (Fig. 3d Step c), the continuous electrolyte flow generating shear stress parallel to the electrode surface provides sufficient force to facilitate final removal of partially-reduced particles from the electrode surface. In Table S1,† the shear force experienced by the particles was calculated by multiplying the shear stress by the cross-sectional area of the particles. The calculation results indicate that at a flow rate of 30 mL min−1, a very minimal shear force of 9.5 × 10−11 N is sufficient to facilitate the removal of particles from the electrode surface.
2.4. Demonstrating regeneration of electrode performance via stimulus-induced electrode cleaning
To investigate if the electrode cleaning process is effective in restoring the performance of the regenerated electrode, a model test was performed to probe the electrochemical accessibility of the ITO-coated electrode using a ferrocene-bearing RAP (PVBC–Fc). PVBC–Fc particles were synthesized by functionalizing poly(vinylbenzyl chloride) (PVBC) microparticles with dimethylaminoferrocene, as reported by Montoto and coworkers38 (Fig. 4a and Fig. S9†). These tests were performed in the presence of the pristine ITO-coated electrode, the ITO-coated electrode artificially fouled with PMGA–ATDDS, and the regenerated ITO-coated electrode after stimulus-controlled cleaning. As seen in Fig. 4a, PVBC–Fc was selected as its electrochemical window was sufficiently distinct from that of PGMA–ATDDS. This allowed for a linear sweep voltammetry (LSV) test from 0 to 0.6 V vs. Ag/Ag+ for oxidative charging of Fc moieties to Fc+ while not electrochemically activating the oxidation or reduction of PGMA–ATDDS.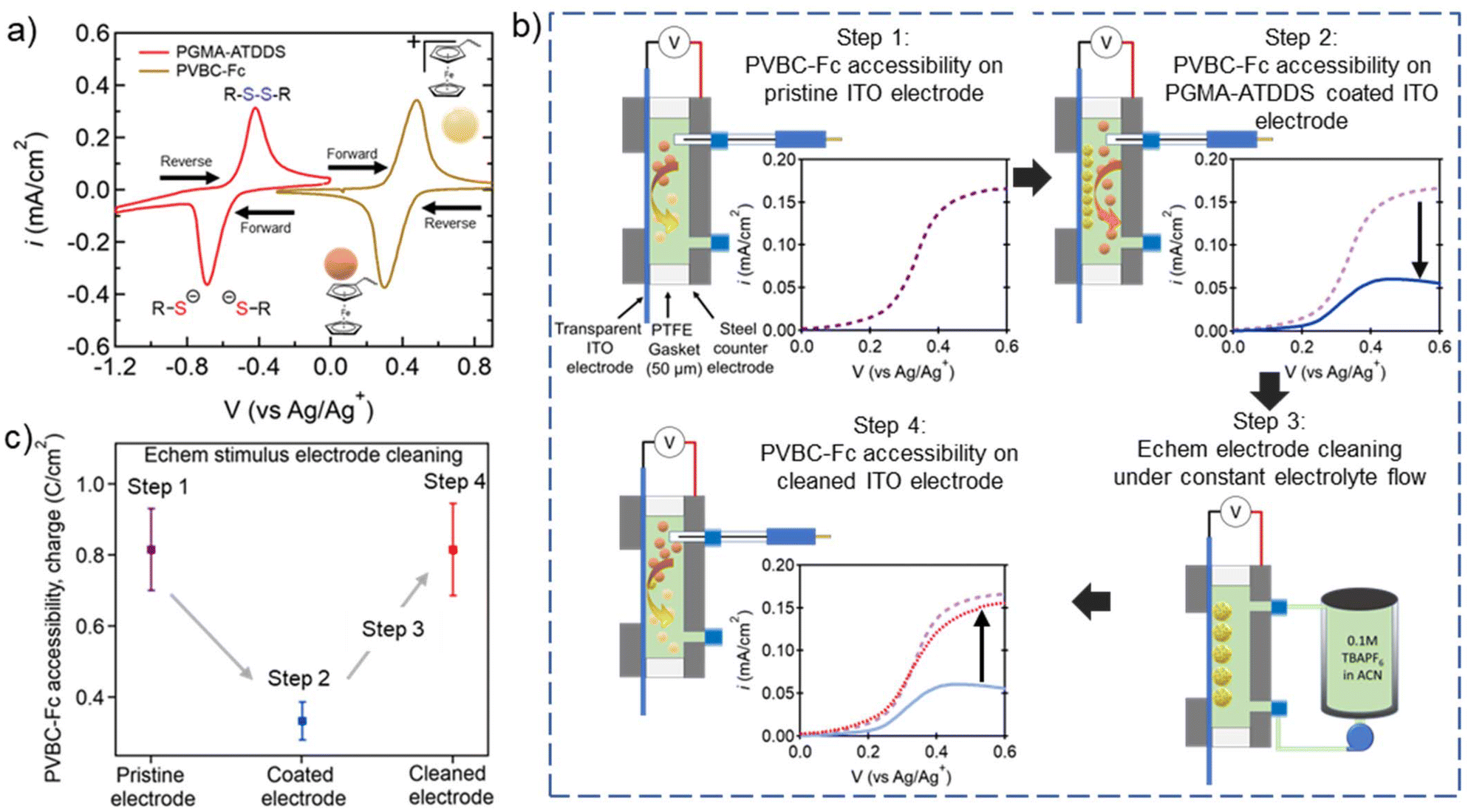 | ||
| Fig. 4 Examining the impact of fouling and electrode cleaning on regenerated ITO electrode accessibility. (a) CVs of PGMA–ATDDS and PVBC–Fc demonstrating no overlap in redox active regions (20 mV s−1, 1 cm2 carbon paper WE, carbon felt CE, Ag/AgNO3 RE, 0.1 M TBAPF6 in ACN as the supporting electrolyte). (b) Process of flow electrode regeneration experiments (Fig. S10† for more details). Step 1: Schematic of the custom static 3 probe cell and LSV testing of PVBC–Fc accessibility on pristine ITO; Step 2: schematic of the custom static 3 probe cell and LSV testing of PVBC–Fc accessibility on a PGMA–ATDDS coated ITO electrode; Step 3: schematic of the custom flow cell connected to the electrolyte reservoir and pump for the electrochemical stimulus cleaning procedure; Step 4: schematic of the custom static 3 probe cell and LSV testing of PVBC–Fc accessibility on a cleaned ITO electrode. (c) Integrated charge density (expressed in C cm−2) of PVBC–Fc from the LSV profiles averaged across electrode regeneration experiments using electrochemical stimulus. | ||
Fig. 4b highlights the key steps in the experiments to demonstrate the regeneration of electrode performance via stimulus-induced electrode cleaning (a more detailed scheme is shown in Fig. S10†). In step 1, a 5 mg mL−1 dispersion of PVBC–Fc particles in ACN with 100 mM TBAPF6 supporting electrolyte was injected into a custom static electrochemical cell consisting of a pristine ITO-coated WE and a stainless-steel CE (Fig. 4b Step 1) and the PVBC–Fc suspension was oxidatively charged as shown in the LSV profile on the lower right side. Note that the inclusion of the Ag/Ag+ reference electrode allows for precise control over the potential of the ITO electrode during the LSV measurement. In step 2, the same ITO electrode was artificially fouled with PGMA–ATDDS particles and the LSV measurement was performed on the PVBC–Fc suspension. As seen in the LSV profile, the measured current density (i) across the sweep is lower compared to the pristine ITO WE on account of the adsorbed PGMA–ATDDS particles limiting PVBC–Fc particles’ accessibility to the WE (Fig. 4b Step 2). In step 3, the electrochemical cell was reconfigured to a cleaning flow cell and the same fouled electrode was put through the cleaning procedure using an electrochemical stimulus as described earlier (Fig. 4b Step 3). Finally, in step 4, the cell containing the cleaned ITO electrode was returned to the static electrochemical cell with fresh PVBC–Fc suspension injected to determine the recovery of electrode performance (Fig. 4b Step 4). Notably, a substantial increase in i was observed in the LSV profile, which closely resembled the LSV result of the pristine ITO electrode, thus providing clear evidence of the effective regeneration of the electrode after the cleaning process. To quantitatively illustrate the PVBC–Fc accessibility, Fig. 4c provides the integrated charge density (expressed in C cm−2) from the LSV profiles averaged across multiple electrode regeneration experiments. Note that the electrode generation demonstration was also performed with the UV cleaning procedure and yielded results similar to those of the electrochemical cleaning (Fig. S11†). Overall, this collective analysis highlights the consistent restoration of electrode performance subsequent to the stimulus-triggered cleaning of the PGMA–ATDDS-fouled ITO electrode.
3. Conclusions
In summary, this study presents a new and effective electrode cleaning strategy employing redox-active poly(glycidyl methacrylate) particles crosslinked with 2-amino-1,3,4-thiadiazole disulfide (PGMA–ATDDS). These RAP particles exhibit the ability to undergo de-crosslinking by breaking the disulfide bond upon exposure to either electrochemical reduction or UV photoexcitation. The application of these stimuli to an ITO electrode fouled with PGMA–ATDDS within a customized flow cell resulted in a significant 80% removal of particles, an over six-fold enhancement compared to non-stimulated conditions. Importantly, the investigation employing confocal fluorescence imaging post electrochemical stimulation revealed targeted disulfide reduction in particles near the electrode surface. This phenomenon is believed to result from electrolyte swelling selectively at the particle/electrode interface, thereby promoting effective particle removal in the presence of a fluid flow. Moreover, the successful regeneration of electrode performance post-cleaning was exemplified by testing the charge accessibility of poly(vinylbenzyl chloride) functionalized with dimethylaminoferrocene (PVBC–Fc). Following the electrochemical cleaning of the fouled ITO electrode, the attainable charge of PVBC–Fc closely matched the charge observed on a pristine ITO electrode. A similar result was achieved through UV cleaning as well. In essence, this investigation introduces a new approach that harnesses stimulus-responsive chemistries within RAPs to endow them with the inherent functionality to facilitate real-time electrode cleaning in electrochemical flow cells such as redox flow batteries.4. Materials and methods
4.1. Materials
Glycidyl methacrylate (GMA) containing inhibitor, hydrogen peroxide solution (30% w/w) in an aqueous solution containing an inhibitor, polyvinylpyrrolidone (PVP) (with a number average molecular weight Mn of 40000 g mol−1), HMDA, (dimethylaminomethyl)ferrocene, 4-vinyl benzyl chloride, divinyl benzene, ammonium hexafluorophosphate, and TBAPF6 were purchased from Sigma-Aldrich. All other reagents, including DMSO, ACN, methanol, tetrahydrofuran (THF) and 200 proof ethanol, were purchased from Fisher Scientific. BrMePyr was purchased from Ambeed, Inc. All reagents were used without further purification unless mentioned otherwise. ITO coverslips were sourced from SPI Supplies (06480-AB, #1.5 thickness, 15–30 ohms).
4.2. Synthesis of P1 and P2 particles
P1 and P2 particles were synthesized following the same procedure previously reported by the same authors.37 The inhibitor in the GMA monomer was removed by running through an alumina basic column. In a 250 mL round bottom flask, 2.1 g of PVP was dissolved in 83 g of ethanol and 7.7 g of DI water. The reaction vessel was heated to 70 °C with a stir bar rotating at a speed of 165 rpm while purging with argon gas for 20 min. 0.24 g of AIBN was dissolved in 12 g of purified GMA monomer. The monomer and initiator were added dropwise into the reaction vessel. The system was continued to be purged with argon for 15 min to ensure thorough removal of oxygen and then allowed to react for 12 hours. The product was centrifuged at 4000 rpm for 10 minutes, decanted, and then washed three times with methanol prior to drying in a vacuum oven at 55 °C. 10.2 g of P1 particles was obtained with a yield of 85%.A total of 4 g of P1 particles were re-distributed in 50 mL of ethanol using a bath sonicator. Following this, 1% by weight (40 mg) of HMDA was added to the dispersion. This addition was made by injecting 10 mg mL−1 of HMDA dissolved in ethanol. The reaction was proceeded at a temperature of 50 °C for 24 hours to yield lightly crosslinked P2 particles. P2 particles were centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with methanol. Subsequently, P2 particles were dried under vacuum at 55 °C overnight.
4.3. Functionalization of P2 with ATDDS to produce PGMA–ATDDS particles
The PGMA–ATDDS particles were synthesized following the same procedure previously reported by the same authors.37 0.5 g (∼3.5 mmol of GMA repeating units) of P2 particles were dispersed in 25 mL of THF via bath sonication, followed by ATDDS (4.65 g, 17.5 mmol) dissolved in DMSO (100 mL). The reaction vessel was purged with argon gas and heated to 66 °C at 300 rpm for 6 days. Post functionalization, the particles were centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with DMSO. The particles were then centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with methanol. The particles were then dispersed in DMSO and irradiated with a UV source (320 nm to 500 nm filter) at 350 mw cm−2 for three hours as described in the previous work to remove mono-functionalized ATDDS to produce PGMA–ATDDS.37 The particle dispersion was centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with a methanol and DMSO co-solvent (mix ratio 1:1 by volume). The particles were dried under vacuum at 55 °C overnight.
4.4. Synthesis of PGMA–HMDA particles
0.5 g of P2 particles (∼3.5 mmol of GMA repeating units) were dispersed in 40 mL of ethanol via bath sonication. 200 mg (1.75 mmol) of HMDA dissolved in 10 mL of ethanol was added to the reaction vessel. The reaction was carried out at 50 °C with stirring for 2 days. The particles were centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with methanol. The PGMA–HMDA particles were then dried under vacuum at 55 °C overnight.4.5. Synthesis of PVBC–Fc particles
The PVBC particles were synthesized following the procedure reported by Montoto and coworkers.38 PVP was dissolved in 95 mL of ethanol with a magnetic stir bar at 165 rpm. Inhibitors were removed from vinyl benzyl chloride and divinyl benzene monomers by passing through a basic alumina column, separately. 0.1 g of AIBN was dissolved in 4.9 mL of 4-vinyl benzyl chloride and 0.1 mL of divinylbenzene. The monomer mixture with AIBN was added into the reaction vessel. The reaction vessel was sealed and purged with (filtered dry) Ar for 30 min before being heated up to 70 °C and left to react for 12 h. The resulting particles were centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with methanol prior to drying in a vacuum oven at 55 °C for 1 day.1 g of dried PVBC particles (∼6.5 mmol of 4-vinyl benzyl chloride repeating unit) was redispersed in 30 mL DMF and 30 mL THF solvent mixture using a bath sonicator for one hour. 10 mL (33.5 mmol) of (dimethylaminomethyl)ferrocene was added to the reaction vessel followed by 30 min purging with filtered Ar. The reaction vessel was heated to 50 °C and the reaction was carried out for 5 days. 12 g of ammonium hexafluorophosphate was dissolved in 30 mL of water and added into the reaction vessel to conduct the ion-exchange reaction. The ion-exchange reaction was carried out for 1 day at RT with stirring. The resulting particles were centrifuged at 4000 rpm for 10 minutes, decanted, and washed three times with THF and three times with methanol prior to drying in a vacuum oven at 50 °C for 1 day.
4.6. Dynamic light scattering (DLS)
DLS measurements were performed using a Brookhaven Instruments BI-200SM. The dry particle samples were dispersed in ACN to 1 mg mL−1 concentration using bath sonication for a minimum of 1 hour and then diluted to 0.1 mg mL−1 dispersion prior to the measurement.4.7. Scanning electron microscopy (SEM)
Silicon substrates measuring 1 cm × 1 cm were sectioned and subjected to a cleaning process involving sonication in acetone followed by methanol. Subsequently, 50 microliters of particle dispersions at a concentration of 1 mg mL−1 in ethanol were applied onto the silicon substrates and left to air dry. Before performing SEM imaging, a thin layer of platinum/palladium measuring 8 nm in thickness was applied to the samples using a sputter coater (Cressington 208HR).4.8. Cyclic voltammetry (CV)
A Biologic SP-200 potentiostat was used for all CV measurements. The experiments were carried out using a conventional 3-electrode electrochemical cell in an Ar glovebox. To serve as the WE, a porous carbon substrate called Sigracet GDL 39 AA carbon paper (CP) with an area of 1 cm2 was chosen. To attach the CP to a Pt wire electrode (CH Instruments CHI115), the wire was threaded through two small holes near opposing edges of the CP sample. For particle measurement, the active material was deposited onto CP electrodes through drop-casting from an ethanol suspension, resulting in an active material loading of 0.2 mg cm−2. The counter electrode was a plain Pt wire, and a non-aqueous Ag/Ag+ reference electrode was used. The reference electrode consisted of a silver wire immersed in 0.01 M AgNO3 and 0.1 M TBAPF6 in ACN. The data were obtained using a scan rate of 20 mV s−1. For the electrochemical experiments, the solvents were degassed using Ar gas.4.9. Drop-casting particles onto transparent ITO substrates
ITO substrates were sonicated in acetone and methanol respectively for 5 minutes. The substrates were then blow-dried with nitrogen. 5 mg mL−1 of particles were dispersed in ethanol via bath sonication for 30 min prior to drop-casting. 20 microliters of particle dispersion were drop-cast onto the center of the substrate and allowed to dry overnight.4.10. Process flow for the electrode cleaning procedure
The backing plate was positioned on a flat surface, followed by placing the ITO-coated substrate with particles facing upwards (Fig. S6†). The 50 μm thick PTFE gasket, featuring a central hollow opening measuring 17 mm × 15 mm, was then positioned atop the ITO substrate. The positioning of the ITO substrate was adjusted to ensure that all the drop-cast particles were contained within the hollow opening of the PTFE gasket. Subsequently, a positive electrode was affixed along with flow channels, followed by attaching a flow cell body complete with connectors. As depicted in Fig. S6b,† the flow cell was subsequently linked to an external reservoir and a volumetric flow rate controlled Masterflex L/S Series peristaltic pump containing a liquid electrolyte (100 mM TBAPF6 in ACN) through the inlet/outlet connections. For the electrochemical stimulus, a 2 V (approximately −1.7 V vs. Ag/Ag+) constant potential was applied to the ITO-coated substrate functioning as the WE, with the stainless-steel CE plate connected via electrical connections. For the UV stimulus, a UV fiber optic was secured into the optical viewport on the reverse side of the stainless-steel backing plate (Fig. S6b†). Particle imaging and counting were conducted both prior to and after the electrode cleaning procedure, in accordance with the subsequent procedure.4.11. Process flow for quantitative determination of particle removal
The data presented in the manuscript were obtained from experiments with 5 samples of substrates coated with particles under various flow cell conditions. On each sample, 5 spots were chosen within the yellow dashed circle shown in Fig. S5.† Using an optical microscope, predefined spots were imaged at 10× and 50× magnification. Markers etched into the back (uncoated) side of the substrates, as well as any visible landmarks (specific particle arrangements, defects in the glass surface, etc.) were used to ensure exact alignment of the image field before and after cleaning. Landmarks were obtained and aligned, and then the image was focused to bring particles into view. ImageJ's particle analysis tool was used to count the number of particles visible in the field of view. On average, approximately 400 particles were counted on each spot before the cleaning procedure.4.12. Sample preparation for confocal fluorescence microscopy imaging
PGMA–ATDDS particles were dispersed in ethanol (1 mg mL−1) using a bath sonicator for 1 hour. 40 μL of the dispersion was drop-cast onto ITO coverslips (SPI Supplies 06480-AB, #1.5 thickness, 15–30 ohms). The particles were dried in air and then mounted onto the electrolytic PTFE electrode holder with clip design. The electrode holder was used as a WE and a Pt wire electrode (CH Instruments CHI115) was used as the CE. The electrodes were then pre-soak in 100 mM TBAPF6 in DMSO electrolyte to swell the particles. This pre-soaking step helps avoid the uptake of the fluorescence dye by absorption during particle swelling. In the electrochemical cell, 3 mL of DMSO electrolyte with 1 mM BrMePyr was added followed by the attachment of the the custom-made electrode holder. A non-aqueous Ag/Ag+ reference electrode was used, consisting of an Ag wire immersed in 0.01 M AgNO3 and 0.1 M TBAPF6 in ACN. A constant voltage of −1 V was applied to the system for 5 min. After the reaction, ITO was immersed in fresh DMSO for 1 day to remove the unreacted BrMePyr from the particles. The particle-bearing ITO substrate was then washed with acetone to remove residual DMSO prior to mounting onto a glass slide with 5 microliters of Vectashield antifade mounting medium.4.13. Confocal fluorescence imaging
An Olympus “fixed cell” DSU spinning disk confocal microscope was used for confocal fluorescence imaging. An inverted platform (model IX81) with a 100×/1.35 oil (UPlanApo) objective lens was used for imaging through slides with a Z-galvo stage for Z-sectioning. The fluorescence of the thiadiazole moiety was observed as blue emission using a 440/40 nm fluorescence filter, and BrMePyr was observed as green emission using a 525/30 nm fluorescence filter. 22 images were acquired for each sample, ranging from the particle–electrode surface to slightly above the uppermost point of the particles, with an increment of 0.1 μm along the z-axis, covering a total span of 2.2 μm. Subsequently, the images underwent conversion into 3D projections using ImageJ, incorporating a total rotation angle of 90 degrees to yield a side view from the XY plane. The same procedure was replicated for the images obtained through both the 440/40 nm fluorescence filter for the thiadiazole moiety and the 525/30 nm fluorescence filter for functionalized MePyr. The superposition images, as shown in the bottom figure of Fig. 3c, were generated by utilizing the ‘Merge channels’ functionality within ImageJ.4.14. Electrode performance regeneration via stimulus-induced surface electrode cleaning
After performing LSV on the coated electrode, it was transferred to the cleaning flow cell and subjected to the previously described electrode cleaning procedure: 30 mL min−1 flow under the applied 2 V (approximately −1.7 V vs. Ag/Ag+) for 30 min (Fig. S9† Step 3). After the cleaning procedure, the cell was disconnected from the pump and charged with a new aliquot of the PBVC–Fc electrolyte dispersion. The cell was then connected to the potentiostat and tested with the same LSV test using the cleaned electrode as the working electrode (Fig. S9† Step 4).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Joint Center for Energy Storage Research (JCESR), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (BES). This work made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center, supported by the National Science Foundation under award number DMR-2011854. Parts of this work were carried out at the Soft Matter Characterization Facility of the University of Chicago. Fluorescence microscopy was performed at the Integrated Light Microscopy Core at the University of Chicago, which receives financial support from the Cancer Center Support Grant (P30CA014599), RRID: SCR_019197. S. S. K. acknowledges support from the National Science Foundation (NSF) Graduate Research Fellowship under Grant No. DGE-1746045.References
- M. Gattrell and D. W. Kirk, J. Electrochem. Soc., 1992, 139, 2736–2744 CrossRef CAS.
- M. Narmadha, M. Noel and V. Suryanarayanan, J. Electroanal. Chem., 2011, 655, 103–110 CrossRef CAS.
- D. Tyagi, J. B. Perez, A. Nand, C. Zhiqiang, P. Wang, J. Na and J. Zhu, Anal. Biochem., 2015, 471, 29–37 CrossRef CAS PubMed.
- H. Vaisocherová, K. Mrkvová, M. Piliarik, P. Jinoch, M. Šteinbachová and J. Homola, Biosens. Bioelectron., 2007, 22, 1020–1026 CrossRef PubMed.
- H. Ma, J. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- A. Barfidokht and J. J. Gooding, Electroanalysis, 2014, 26, 1182–1196 CrossRef CAS.
- A. L. Gui, E. Luais, J. R. Peterson and J. J. Gooding, ACS Appl. Mater. Interfaces, 2013, 5, 4827–4835 CrossRef CAS PubMed.
- Y. Hu, G. Yang, B. Liang, L. Fang, G. Ma, Q. Zhu, S. Chen and X. Ye, Acta Biomater., 2015, 13, 142–149 CrossRef CAS PubMed.
- K. Wedege, E. Dražević, D. Konya and A. Bentien, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- J. D. Milshtein, J. L. Barton, T. J. Carney, J. A. Kowalski, R. M. Darling and F. R. Brushett, J. Electrochem. Soc., 2017, 164, A2487–A2499 CrossRef CAS.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS.
- V. Singh, S. Kim, J. Kang and H. R. Byon, Nano Res., 2019, 12, 1988–2001 CrossRef CAS.
- J. Chai, A. Lashgari and J. J. Jiang, Clean Energy Mater. Part 1 - Electroact. Mater. Next-Generation Redox Flow Batter. From Inorg. to Org., 2020, pp. 1–47 Search PubMed.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdörfer, B. Häupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
- J. Winsberg, T. Hagemann, S. Muench, C. Friebe, B. Häupler, T. Janoschka, S. Morgenstern, M. D. Hager and U. S. Schubert, Chem. Mater., 2016, 28, 3401–3405 CrossRef CAS.
- J. Winsberg, S. Muench, T. Hagemann, S. Morgenstern, T. Janoschka, M. Billing, F. H. Schacher, G. Hauffman, J. F. Gohy, S. Hoeppener, M. D. Hager and U. S. Schubert, Polym. Chem., 2016, 7, 1711–1718 RSC.
- E. C. Montoto, G. Nagarjuna, J. S. Moore and J. Rodríguez-López, J. Electrochem. Soc., 2017, 164, A1688–A1694 CrossRef CAS.
- M. J. Baran, M. N. Braten, E. C. Montoto, Z. T. Gossage, L. Ma, E. Chénard, J. S. Moore, J. Rodríguez-López and B. A. Helms, Chem. Mater., 2018, 30, 3861–3866 CrossRef CAS.
- K. H. Hendriks, S. G. Robinson, M. N. Braten, C. S. Sevov, B. A. Helms, M. S. Sigman, S. D. Minteer and M. S. Sanford, ACS Cent. Sci., 2018, 4, 189–196 CrossRef CAS PubMed.
- B. L. Hanssen, S. Siraj and D. K. Y. Wong, Rev. Anal. Chem., 2016, 35, 1–28 CAS.
- T. P. Nguyen, A. D. Easley, N. Kang, S. Khan, S. M. Lim, Y. H. Rezenom, S. Wang, D. K. Tran, J. Fan, R. A. Letteri, X. He, L. Su, C. H. Yu, J. L. Lutkenhaus and K. L. Wooley, Nature, 2021, 593, 61–66 CrossRef CAS PubMed.
- H. Qian, M. J. Counihan, H. A. Doan, N. A. Ibrahim, A. S. Danis, W. Setwipatanachai, N. S. Purwanto, J. Rodríguez-López, R. S. Assary and J. S. Moore, J. Mater. Chem. A, 2022, 10, 7739–7753 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- A. Ruiz De Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Mater. Horiz., 2016, 3, 241–247 RSC.
- G. K. Helmkamp, Organic chemistry of sulfur, Plenum Press, 1978, vol. 55 Search PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- S. Patai, Chem. Thiol Group, 2010, 1–479 Search PubMed.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31–45 RSC.
- A. W. Jackson and D. A. Fulton, Macromolecules, 2012, 45, 2699–2708 CrossRef CAS.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- B. T. Michal, C. A. Jaye, E. J. Spencer and S. J. Rowan, ACS Macro Lett., 2013, 2, 694–699 CrossRef CAS PubMed.
- B. T. Michal, E. J. Spencer and S. J. Rowan, ACS Appl. Mater. Interfaces, 2016, 8, 11041–11049 CrossRef CAS PubMed.
- R. Kato, P. Mirmira, A. Sookezian, G. L. Grocke, S. N. Patel and S. J. Rowan, ACS Macro Lett., 2020, 9, 500–506 CrossRef CAS PubMed.
- N. K. P. Samuel, M. Singh, K. Yamaguchi and S. L. Regen, J. Am. Chem. Soc., 1985, 42–47 CrossRef CAS.
- J. D. Milshtein, A. P. Kaur, M. D. Casselman, J. A. Kowalski, S. Modekrutti, P. L. Zhang, N. H. Attanayake, C. F. Elliott, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, Energy Environ. Sci., 2016, 9, 3531–3543 RSC.
- J. D. Milshtein, J. L. Barton, R. M. Darling and F. R. Brushett, J. Power Sources, 2016, 327, 151–159 CrossRef CAS.
- G. L. Grocke, H. Zhang, S. S. Kopfinger, S. N. Patel and S. J. Rowan, ACS Macro Lett., 2021, 10, 1637–1642 CrossRef CAS PubMed.
- E. C. Montoto, G. Nagarjuna, J. Hui, M. Burgess, N. M. Sekerak, K. Hernández-Burgos, T.-S. Wei, M. Kneer, J. Grolman, K. J. Cheng, J. A. Lewis, J. S. Moore and J. Rodríguez-López, J. Am. Chem. Soc., 2016, 138, 13230–13237 CrossRef CAS PubMed.
- D. K. Kim, S. J. Yoon, J. Lee and S. Kim, Appl. Energy, 2018, 228, 891–901 CrossRef CAS.
- T. Jyothi Latha and S. Jayanti, J. Appl. Electrochem., 2014, 44, 995–1006 CrossRef CAS.
- Y. K. Zeng, X. L. Zhou, L. Zeng, X. H. Yan and T. S. Zhao, J. Power Sources, 2016, 327, 258–264 CrossRef CAS.
- Y. (王怡琳) Wang, A. P. Kaur, N. H. Attanayake, Z. (于洲) Yu, T. M. Suduwella, L. (程蕾) Cheng, S. A. Odom and R. H. Ewoldt, Phys. Fluids, 2020, 32, 83108 CrossRef CAS.
- A. Z. Nelson, K. S. Schweizer, B. M. Rauzan, R. G. Nuzzo, J. Vermant and R. H. Ewoldt, Curr. Opin. Solid State Mater. Sci., 2019, 23, 100758 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of P1, P2, PGMA-ATDDS and PGMA-HMDA, size change of PGMA-ATDDS, FT-IR spectra of P2, PGMA-ATDDS and PGMA-HMDA, UV-Vis absorbance spectra of PGMA-ATDDS, the optical image and DLS of PGMA-HMDA, design of modular half flow cell electrode cleaning apparatus, image analysis and particle counting on substrates, particle removal via a controlled stimulus under shear flow conditions, geometry for a flow channel in an electrode cleaning flow cell, flow calculations for a narrow channel flow field, the synthetic scheme and characterization for PVBC-Fc particles, the approach used to determine the effect of particle removal, and the electrode performance regeneration procedure via electrode cleaning. See DOI: https://doi.org/10.1039/d4lp00250d |
| This journal is © The Royal Society of Chemistry 2024 |