
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContrasting interchain order and mixed ionic–electronic conduction in conjugated polymers: an isoindigo case study†
Rebecca F.
Meacham
a,
Heejung
Roh
a,
Camille E.
Cunin
a,
Eric R.
Lee
a,
Wenhao
Li
c,
Yan
Zhao
 c,
Sanket
Samal
*ab and
Aristide
Gumyusenge
*a
c,
Sanket
Samal
*ab and
Aristide
Gumyusenge
*a
aDepartment of Materials Science & Engineering, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA. E-mail: aristide@mit.edu; ssamal@mit.edu
bJames Tarpo Jr. and Margaret Tarpo Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, IN 47907, USA
cDepartment of Materials Science, Laboratory of Molecular Materials and Devices, State Key Laboratory of Molecular Engineering of Polymers, Fudan University, 220 Handan Rd, Shanghai, 200433, China
First published on 22nd October 2024
Abstract
In mixed ionic–electronic conductive polymers, electronic conduction is optimal in tightly packed flat chains, while ionic conduction benefits from free volume accommodating large ions. To this end, polymers with high crystallinity are often excluded from structure–property studies of high-performing mixed conductors due to their unbalanced transport, which favors electronic charges over ionic ones. Herein, we investigated how mixed conduction can be achieved in ordered conjugated polymers by systematically combining interchain order with side chain engineering. We synthesized a series of isoindigo (IID)-based copolymers with varying amounts of aliphatic and hydrophilic side chains and examined the impact of interchain order on mixed conduction. Through crystallographic, spectro-electrochemical, and molecular dynamics studies, we demonstrated that systematically introducing hydrophilic side chains reduces the bulk order and long-range aggregation by increasing chain flexibility while preserving the interchain stacking distances within crystalline domains. Testing these IID polymers in transistor devices revealed that ion insertion and device transconductance strongly depend on the amount of hydrophilic side chains. We demonstrated that glycol side chains can enhance mixed conduction while maintaining interchain order. Our findings suggest that the IID system is promising for designing polymers that can accommodate ionic species without compromising the chain ordering required for electronic conduction.
Introduction
Organic mixed ionic–electronic conductors (OMIECs) are of particular interest for applications that involve interfacing electronics with electrolytic media, such as biological fluids.1,2 Particularly, mixed conduction is leveraged in organic electrochemical transistors (OECTs), three-electrode devices in which, upon the application of a gate voltage, ion insertion into the semiconducting channel alters the concentration of charge carriers, thus the conductivity.3 This electrochemical charge modulation, which occurs at low power, is desirable for a variety of applications including the abovementioned bio-interfacing and continues to demand high performance materials.4,5 Conjugated polymers, owing to their structural tunability and mechano-physical properties, are attractive for engineering mixed conductive interfaces.1,5–7 While selecting conjugated cores allows for electronic conduction along the polymer backbone, side chains are used to endow ionic conduction in the presence of an electrolyte. However, performance metrics in these materials remain sub-optimal as co-optimization of ion uptake and electronic conduction in a single material is fundamentally challenging, thus necessitating systematic iterations of polymer architectures.It is difficult to concomitantly meet the requirements for ionic and electronic conduction when designing new mixed conductive polymers. Electronic conduction requires close packing and high molecular weight for charges to travel along the polymer backbone and avoid trapping in disordered regions.8,9 Ionic conduction, on the other hand, requires swelling of the polymer film upon contact with the electrolyte and having sufficient space throughout the film bulk for ions and their solvation shells.10 Swelling and deswelling of the polymer film require dynamic creation of spaces between polymer chains and can disrupt the packing needed for electronic conduction leading to decreased electronic performance. Additionally, hydrophilic side chains, such as ethylene glycol which are common in OMIECs,11–14 are flexible and can disrupt packing. It is thus important to understand how polymer morphology and interchain ordering impact mixed conduction to better inform polymer design and device performance.
One of the understudied cores in mixed conductive polymers is isoindigo (IID). This electron-deficient core is attractive particularly in donor–acceptor type of polymers due to its simple synthesis and stability in ambient conditions,15,16 but only recently it has become of interest in designing OMIEC materials.17,18 This is partly due to the highly planar nature of IID which imparts strong π–π stacking and backbone planarity into polymers, advantageous for electronic conduction but unfavorable for ion insertion.19 This strong stacking and aggregation have often excluded IID from OMIEC designs since an ideal mixed conductor must concomitantly accommodate both ionic species and electronic charge carriers. As a result, IID has been less studied compared to other less crystalline cores such as ethylenedioxythiophene (EDOT) and diketopyrrolopyrrole (DPP).19,20 With these acceptor cores, copolymerization offers a method to incorporate monomers with different side chains and create polymer structures capable of mixed conduction.21,22 A mix of hydrophobic long alkyl side chains and hydrophilic side chains such as ethylene glycol are used to impart solubility for solution processing as well as enhanced solvation of electrolyte and ionic conduction.8–11 In the case of IID, the copolymerization often leads to greater degrees of π–π stacking,23–28 which makes its use in OMIECs challenging. In fact, the successful examples of mixed conductors based on IID have been based on copolymerizing with monomers which intentionally disrupt the order and chain planarity to maximize electrolyte uptake and redox activity.17,18,29
In this work, we aimed to investigate side chain engineering as a versatile approach to modulate the electrochemical response in conjugated polymers without sacrificing their inherent interchain order. We designed a series of IID-based copolymers varying the ratio of aliphatic and hydrophilic side chains to systematically modulate bulk crystallinity and ion uptake. We used thin film morphology analyses and molecular dynamics simulations to understand the relationship between structure and crystallization behavior with increasing content of hydrophilic side chains. The IID series revealed that tethering branched glycol side chains onto backbones with inherent interchain order leads to tunable morphology and electrochemical performance resulting from (i) a change in the π–π stacking from an edge-on to a face-on orientation; (ii) reduced long-range aggregation in the film bulk; and (iii) an overall coiling of chain bundles, while conserving the π–π distance of the parent IID core within the crystalline regions. This conservation of inherent interchain distances while accommodating ions through side chains leads to enhanced transconductance in OECT devices, which proved tunable based on the amount of hydrophilic side chains present, as determined by morphology characterization and electrochemical operation in OECTs. With this study, we garnered insights that we believe will spark further investigations on highly crystalline organic mixed conductors, where the balance between interchain order to favor electronic transport and bulk ion transport is attainable through side chain engineering. Such investigations would thus tap into the unexploited potential of order polymers as mixed conductive materials.
Results and discussion
Materials design and synthesis
To investigate the impact of inter-chain ordering on mixed conduction, IID was used owing to its highly planar nature and strong π–π stacking to induce order in the polymer thin films.30 In addition to the favorable packing of IID, we used thiophene-vinyl-thiophene (TVT) as the donor co-monomer to reduce chain rotational freedom and further increase backbone planarity and stacking.31 We then introduced branched side chains onto the conjugated backbone to increase both solubility and processability, and to promote ion conduction.7,24 To this end, we synthesized IID-TVT co-monomers with both hydrophobic (alkyl) and hydrophilic (glycol) side chains and formed copolymers with tuned alkyl/glycol ratios. Fig. 1A shows the IID-based copolymer structure that we investigated for the current work, where the content of branched alkyl and glycol side chains is tuned by controlling respective monomer feed ratios (n and m). By varying the amount of each type of side chain present in the polymer chain, we aimed to tune both ionic conduction and solid-state chain packing. That is, the long and branched alkyl chains were utilized to promote solubility and processability of IID polymers from common solvents, while the long and branched glycol side chains were used to enable electrochemical ion insertion and hence mixed conductive behaviors in the IID series.17,18 More synthetic details for the monomers and the polymers can be found in the ESI.† For simplicity, the copolymers are named mg-IID, where m symbolizes the content (percentage) of glycol side chains present in the polymer.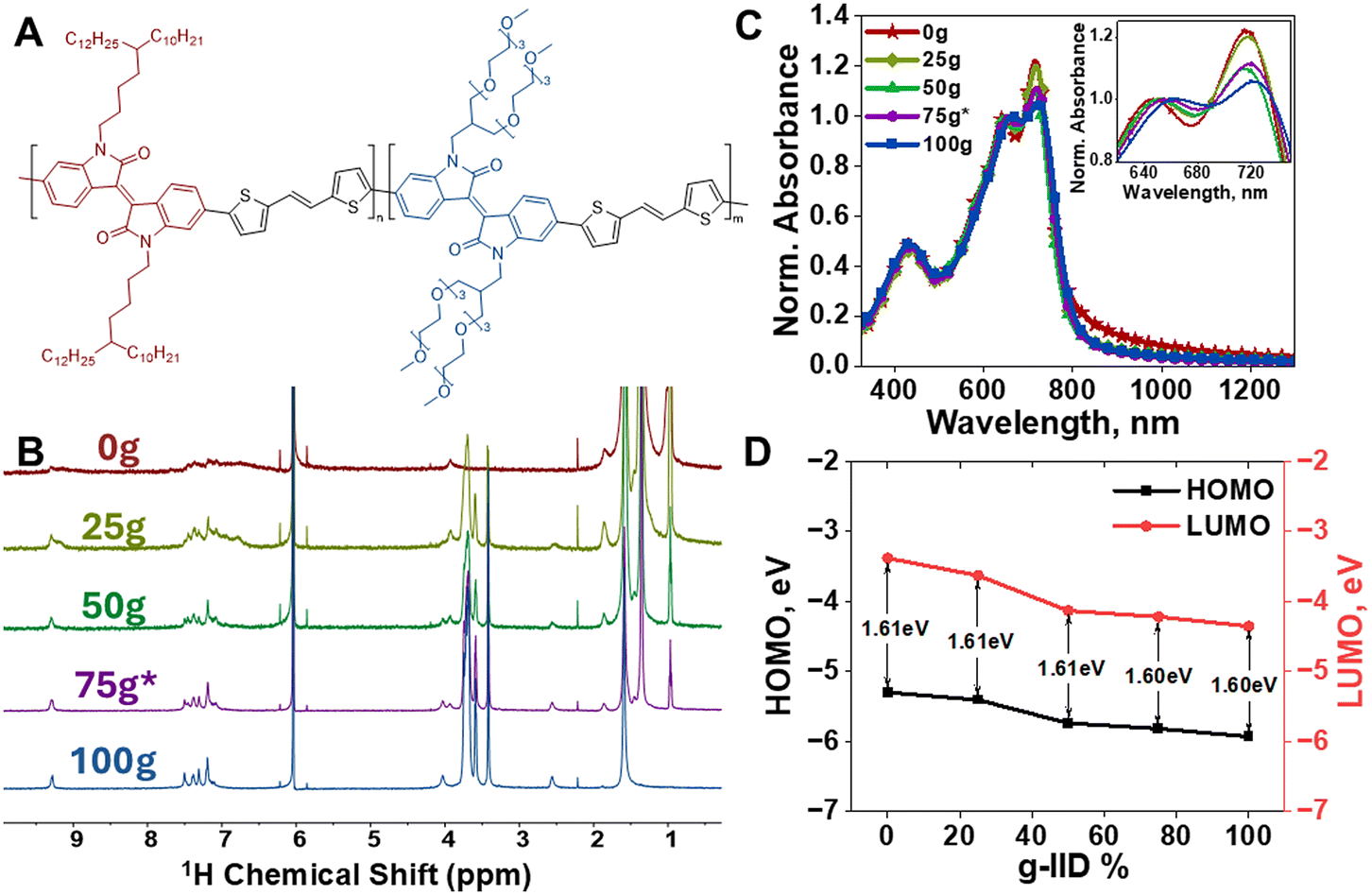 | ||
| Fig. 1 (A) Chemical structure of the IID-TVT co-polymer series – 0g-IID: n = 100, m = 0; 25g-IID: n = 75, m = 25; 50g-IIDn = 50, m = 50; 75g*-IID n = 25, m = 75; and 100g-IIDn = 0, m = 100. (B) 1H NMR spectra of the synthesized polymer series showing the increased incorporation of glycol side chains. (C) Solid-state thin film UV-Vis-NIR absorption spectra of mg-IID polymers. (D) Extracted HOMO, LUMO, and optical bandgap from UV-Vis-NIR and XPS spectroscopy. | ||
The target mg-IID polymer series was synthesized using Stille polycondensation and obtained as dark reddish, shiny solids (full synthetic details in the ESI†). 1H NMR was used to confirm the polymer structures as well as the ratios of a-IID and g-IID incorporated (Fig. 1B). The signature chemical shifts for the alkyl side chain appear at 0.97–1.85 ppm and at 3.93 ppm, while that of glycol side chain appear at 2.55 ppm, 3.41–3.74 ppm and 4.01 ppm. As the amount of g-IID in the feedstock increased, the corresponding NMR peaks for the glycol side chain also increased, while the peaks for the alkyl side chain decreased indicating proportional and gradual incorporation of g-IID into the polymers. The ratios of a-IID and g-IID in each polymer were consistent with the feedstock ratio within 2% for 25g-IID and 50g-IID. The 75g*-IID feedstock (25% a-IID and 75% g-IID) resulted in 34% a-IID and 66% g-IID incorporated into the 75g*-IID polymer, denoted throughout as 75g*-IID, given the target ratio was 75% g-IID. The NMR peaks became sharper with increased g-IID concentration possibly due to increased solubility in the TCE-D2 solvent. To confirm the respective molecular weights, high-temperature gel permeation chromatography (HT-GPC) was used, but due to poor solubility of the mg-IID polymers containing glycol side chains in the mobile phase, the molecular weight estimation was only successful for 0g-IID. Such poor solubility is common in IID-based structures which complicates accurate characterization of the polymers.18,28 We then employed Diffusion Ordered Spectroscopy (DOSY), as described in the ESI,† to estimate the molecular weight of g-IID containing polymers. The molecular weights of the mg-IID polymers are found to be about 14–65 kg mol−1 (Table 1), which are in good agreement with previously reported values.18 Note that the DOSY method yields values as a singular average, as opposed to a conventional distribution, but it does circumvent the solubility limitations and was thus used herein to confirm the polymer structure and compare the molecular weights of mg-IID polymers.
| MWa (kg mol−1) |
T
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (°C)
(°C) |
θ (°) | γ (mJ m−2) |
E
g
(eV) |
|
|---|---|---|---|---|---|
| a 0g-IID Mn obtained from GPC, others M obtained from DOSY. b Decomposition temperature defined as 1% mass loss. c Optical band gap calculated using Tauc plot with UV-Vis-NIR absorption spectroscopy. | |||||
| 0g-IID | 65.5 | 390 | 104.3 | 20.45 | 1.61 |
| 25g-IID | 33.7 | 358 | 99.9 | 23.06 | 1.61 |
| 50g-IID | 14.8 | 338 | 82.7 | 33.78 | 1.61 |
| 75g*-IID | 23.5 | 334 | 80.6 | 35.14 | 1.60 |
| 100g-IID | 34.1 | 325 | 61.6 | 46.85 | 1.60 |
All polymers were then fully characterized and key parameters are shown in Table 1. Notably, all mg-IID polymers showed little or no melting and recrystallization peaks in DSC and were thermally stable up to 300 °C as determined by TGA with decreasing decomposition temperatures as the g-IID content was increased (Fig. S1†). UV-Vis-NIR absorption spectra were recorded for thin films of the mg-IID series to probe the inter-chain order and π–π stacking of the copolymers. UV-Vis-NIR absorption spectra of all polymers showed prominent 0–0 and 0–1 vibronic peaks, characteristic of IID units (Fig. 1C).16 The high relative intensity of the 0–0 peak (∼720 nm) in the 0g-IID and 25g-IID thin films indicated strong inter-chain stacking. A decrease in intensity was observed for the 50g-IID and 75g*-IID thin films, indicating a disruption in order. As the concentration of g-IID increased, the relative intensity at ∼720 nm continued to decrease, however remained well defined even in the 100g-IID thin film. This indicates that the introduction of large, flexible glycolated side chains does disrupt the packing in polymer thin films containing g-IID as compared to the parent 0g-IID polymer thin film. However, some order in the thin film is still retained even at the highest g-IID concentration (100g-IID). UV-Vis-NIR absorption of the polymers in chloroform showed some aggregation, indicating stacking behavior even in solution. The 0–0 peak remained prominent for 0g-IID in solution, was less prominent in the 25g-IID solution, and did not appear in the 50g-IID, 75g*-IID, and 100g-IID solutions indicating that formation into thin films enhanced ordering and π–π stacking particularly for the less aggregated solutions with higher g-IID concentration (Fig. S2†). Optical band gaps were extracted from thin film UV-Vis-NIR absorption profiles and used with X-ray Photoelectron Spectroscopy (XPS) to calculate HOMO and LUMO energy levels to determine the impact of copolymerization (Fig. 1D).
Film morphology
To evaluate the impact of glycol side chains on the solid-state properties, we carried out topology imaging as well as grazing incidence X-ray diffraction (GIXRD) on dried and annealed thin films. Previously reported IID-based polymers have often revealed high degree of ordering and highly crystalline thin film morphologies. In fact, IID-polymers tend to exhibit scale-like texture and the formation of large aggregates.16,23,28 As discussed above, such crystalline nature, though desirable in dried-state devices, is less favorable in electrochemical counterparts. AFM height images revealed homogeneous film topology for all copolymers in the series indicating no obvious phase separation between alkyl- and glycol-rich segments (Fig. S4†). This agreed with previous works on the co-polymerization strategy between glycol-rich segments and alky-rich co-monomers to prevent phase separation.22 This homogeneous distribution was also supported by contact angle measurements which revealed a gradual increase in film hydrophilicity and increased surface energy with increasing g-IID percentages (Table 1 and Fig. S5†). Furthermore, the extracted roughness values indicated that all copolymers yielded smooth film surfaces, with roughness (Rq) values around 0.20 nm (apart from 50g-IID). Notably, the scale-like morphology, more obvious in 0g-IID thin films, gradually became smoother as the amount of glycol side chains increased (Fig. S4†). This morphology trend was in good agreement with the UV-Vis-NIR results discussed above, where increase in glycol side chains beyond 25% led to lower aggregation behavior and reduced inter-chain interaction, as evidenced by the decrease in the 0–0 vibronic peak intensity.X-ray diffraction patterns further revealed that glycol side chains have a significant impact on the preferential orientation of the IID backbone relative to the substrate surface. Fig. 2A illustrates different modes of packing observed in the IID polymer thin films, and Fig. 2B–F shows the respective 2D diffraction patterns of mg-IID polymer thin films. As expected, 0g-IID yielded well-ordered and highly crystalline films, indicated by high intensity diffraction peaks (between 0.1 and 1.2 Å−1) detected in the out-of-plane direction corresponding to the lamellae stacking (Fig. 2B and G). Additionally, 0g-IID chains exhibit a preferentially edge-on stacking behavior, as the in-plane direction shows no lamellae peaks, and instead, a sharp (010) peak centered at 1.75 Å−1, assigned to the π–π stacking. Additionally, the typical halo between Q values of 1.4–1.5 Å−1, assigned to the amorphous region, is very dim indicating high film crystallinity.
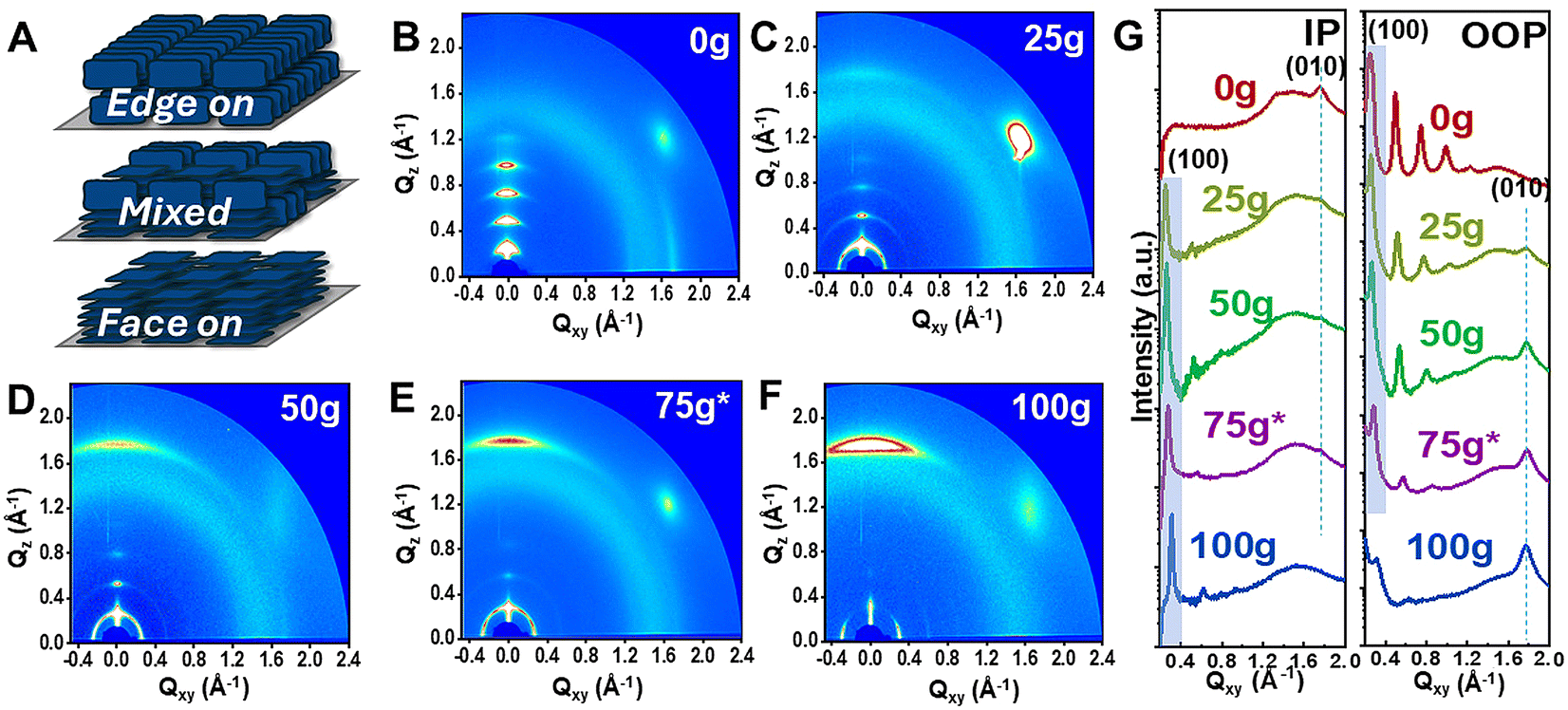 | ||
| Fig. 2 (A) Illustration of edge-on and face-on orientations in polymers. (B–F) 2D diffractions and (G) corresponding 1D line cuts in both the in-plane (IP) and out-of-plane (OOP) directions from annealed thin films of mg-IID polymers. | ||
Compared to the parent 0g-IID polymer, the glycolated copolymers showed an overall loss in film crystallinity, as hinted by UV-Vis-NIR absorption spectra (Fig. 1C & Table S3†). With the increase in g-IID content within the polymer two key features emerged from the GIXRD data: film crystallinity decreases and the chain orientation flips. Starting with 25g-IID, the polymers exhibited a gradual decrease in the intensity of the lamellar stacking peaks in the out-of-plane direction. Additionally, the π–π stacking peak in the out-of-plane direction gradually decreases in intensity, and eventually vanishes in the case of 100g-IID (Fig. 2C–G). Concomitantly, a new (010) peak centered at 1.75 Å−1 starts to emerge in the out-of-plane direction indicating a predominantly face-on π–π stacking as the amount of glycol side chains increases. This orientation was also supported by the presence of new lamellar stacking peaks in the in-plane direction (Fig. 2C–G). It was thus obvious that the branched glycol side chains not only impacted the overall film crystallinity, but also the packing behavior between neighboring chains. Also noteworthy was the mixed orientation observed when the polar and non-polar side chains are systematically well balanced, i.e., 50g-IID and 75*g-IID thin films exhibit a mixed phase, which might be of interest for directional ion insertion.32 As ion injection is fastest in parallel to lamellar stacking, changing from edge-on to face-on can have large impacts on ionic response time depending on vertical or lateral ion injection operation, with films of edge-on morphology responding faster when ion injection is vertical.32 This ability to tune the film crystallinity, the lamellar spacing, as well as the chain orientation is of interest, especially when designing materials with co-optimized electronic and ionic transport. Note that despite the lowered degree of crystallinity and change in orientation, the π–π stacking distance (around 3.56 Å, similar to previously reported IID-based structures16,23,28,33,34) remains unchanged, and the variations are accommodated through change in lamellar spacing (Table S3†). This modular behavior thus shows the versatility of the IID core and its ability to offer tunable mixed conduction properties.
Molecular dynamics simulations
To gain a more comprehensive understanding of how different side chains impact polymer packing, we performed molecular dynamics (MD) simulations. The supercell was packed with 16 polymer chains, with 8 polymers on each plane. The polymers were annealed to 375 K before cooling down to 300 K at 1 atm to achieve an equilibrated packing structure. Although both the polymers (alkyl – 0g-IID and glycolated – 100g-IID) initially exhibited flat backbones in DFT calculations (Fig. S6†), MD simulations revealed distinct differences. The 0g-IID polymer chains exhibited a more ordered backbone arrangement with a better π–π stacking arrangement (Fig. 3A). Conversely, the 100g-IID polymer chains displayed a wavy arrangement attributed to the increased twist originating at the IID core. Notably, despite this increased twist at the IID core in 100g-IID polymers, the polymers still demonstrated good π–π packing in accordance with the GIXRD results.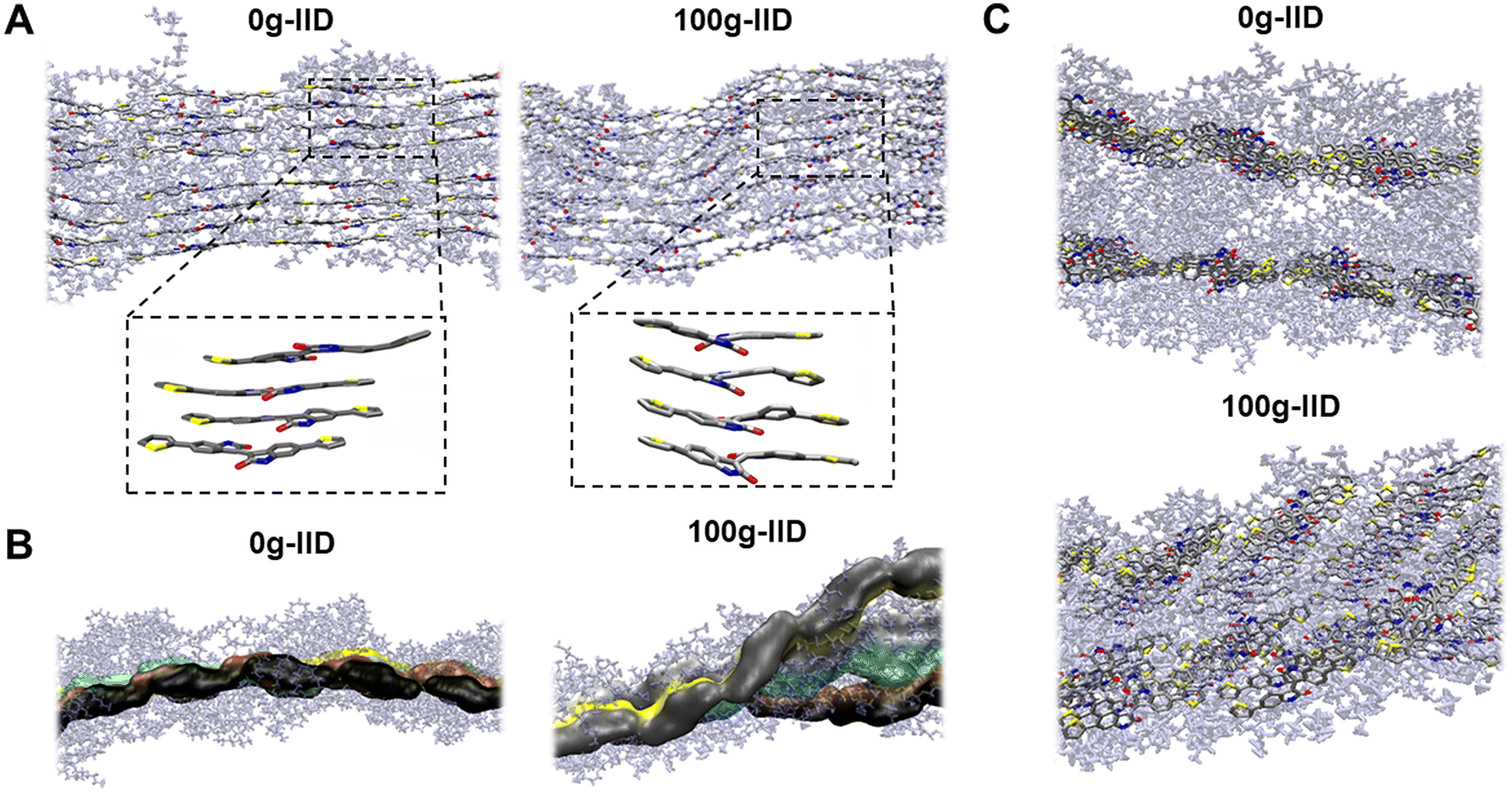 | ||
| Fig. 3 MD simulation snapshots of the mg-IID polymers with highlighted backbones. (A) Difference in the π-stacking of the two polymers, with zoomed packing of the IID cores. (B) Arrangement of the packed polymer chains in a plane. (Each color represents the backbone for the same chain) (C) Difference in the lamellae stacking of the two polymers. | ||
The 0g-IID polymer chains exhibited a more orderly packing arrangement, with all the chains situated in the same plane. In contrast, the 100g-IID polymer chains deviated away from the plane, leading to a coiled architecture (Fig. 3B). However, despite this deviation, they still maintain the π–π interaction, which is also consistent with GIXRD results, where a strong π–π interaction is still observed as the glycol percentage is increased even though the overall film crystallinity is decreased. Due to this reason, the 0g-IID chains arrange parallelly with tight side chain interdigitation, whereas the 100g-IID arrange themselves in a disordered and twisted manner (Fig. 3C). We believe that this disorder in the 100g-IID polymers arises due to the closely positioned branched glycol side chains near the polymer backbone with only one linker carbon, forcing the IID core to twist while maintaining the strong π–π interaction.
Electrochemical performance
To evaluate the mixed ionic-electronic conduction performance of the mg-IID polymers, given their inherent difference in thin film morphology and crystallinity, we used cyclic voltammetry, spectro-electrochemical analysis, as well as organic electrochemical transistor (OECT) devices using aqueous electrolytes to further characterize them. From our previous works,22,35 we selected LiPF6 as the model electrolyte owing to its effective insertion into polymer thin films and redox activity thereof. As shown in Fig. 4A, the cyclic voltammograms of the mg-IID series in 0.1 M LiPF6 showed increased current density and capacitance as the concentration of g-IID increases. With the increase in the amount of hydrophilic side chain, we indeed expected an increased ability to uptake ions than the fully hydrophobic counterpart. Note that as we continued to increase the amount of glycol side chains, the cycling stability of the polymer thin films gradually declined, with significant decrease in performance after 100 cycles in the more hydrophilic thin films (75g*-IID and 100g-IID) (Fig. S7†). The performance degradation seen in the hydrophilic polymers upon cycling is likely a result of non-reversible morphology alteration due to swelling when cycled in aqueous electrolyte, as previously reported in mixed conductive polymers.17 As a result, for optimal cycling stability, a balance of hydrophobic and hydrophilic regions are needed to prevent irreversible morphology disruption.17 Additionally, a systematic selection of electrolytic species that are suitable for the interchain spacing becomes crucial. For the IID series studied herein, we found that smaller hydration radii were more favorable for preventing the disruption of the inherent interchain order. Nonetheless, all polymers showed reversible redox behavior, with relatively low on-set potentials (lower with greater amount of g-IID) (Fig. 4A). This tuned ease of ion insertion is desirable in mixed conductors and is herein attained, even in relatively crystalline polymer films.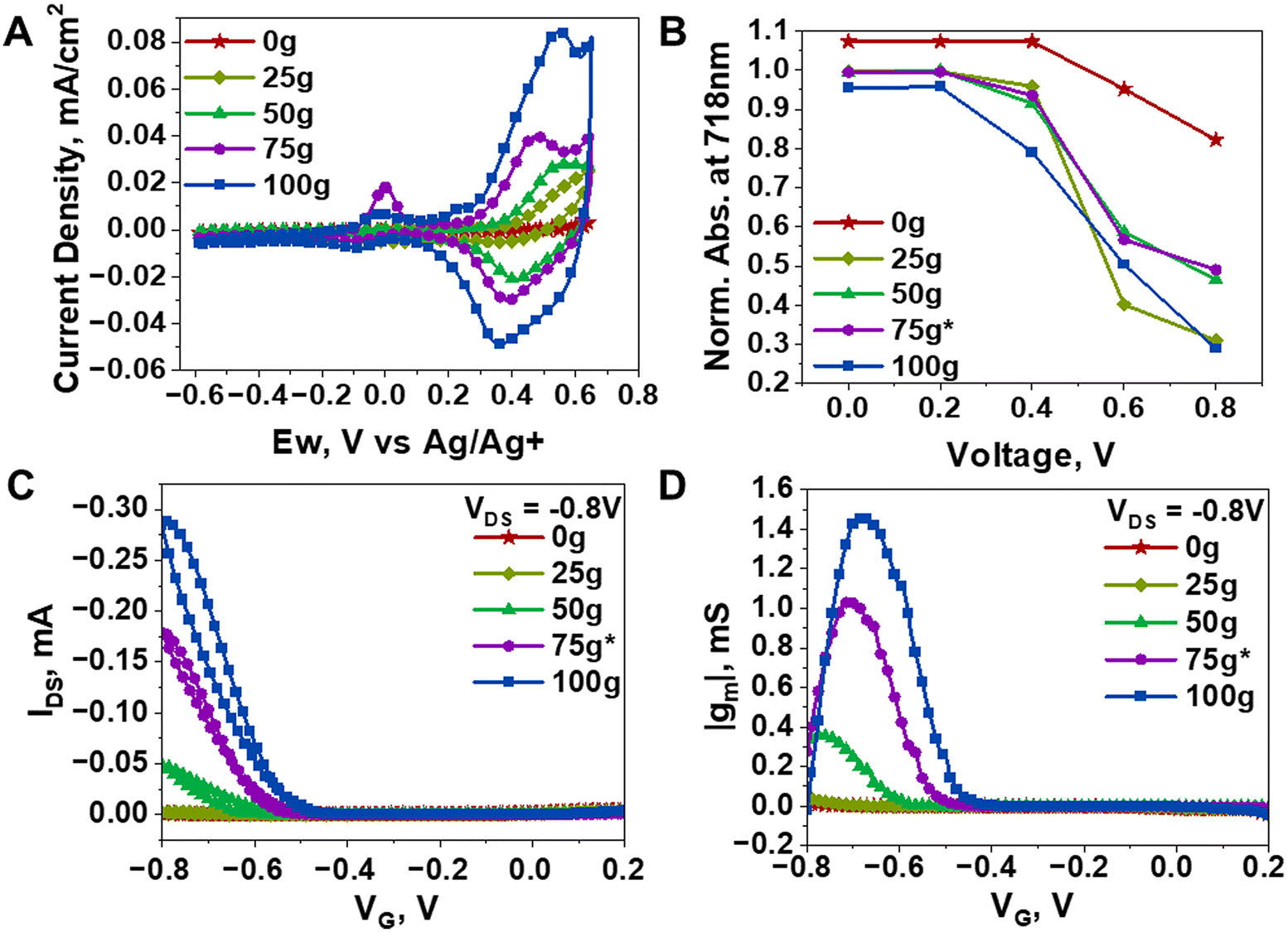 | ||
| Fig. 4 Electrochemical performance of polymers (A) cyclic voltammograms in 0.1 M LiPF6, and (B) corresponding spectro-electrochemical absorbance at 718 nm. (C) Characteristic transfer curves and (D) corresponding transconductance values from OECT devices based on mg-IID polymers with 0.1 M LiPF6 as the electrolyte. | ||
Further insights into ion insertion were garnered from the bleaching behavior of the polymer thin films during spectro-electrochemical analysis. We thus tracked the absorption spectra of the annealed mg-IID thin films during the cyclic voltammetry. Full absorption spectra from all polymers are shown in Fig. S8† and more details of the measurement setup can be found in the ESI.† As shown in Fig. 4B, 0g-IID thin films did not begin to bleach until >0.4 V was applied onto the polymer-coated working electrode. Even with 0.8 V applied, the thin film still showed strong absorption in the visible region. On the contrary, 25g-IID, 50g-IID, 75g*-IID, and 100g-IID thin films showed observable drop in the absorption peak intensities with as low as 0.2 V, and significant bleaching above 0.4 V, indicative of low on-set potentials when the glycol content is increased in the polymer thin film, as discussed above. Note that the bleaching in the visible region was accompanied by the rise in the NIR absorption peaks associated with the formation of polarons in all polymers upon electrochemical doping (Fig. S8†). We could thus conclude that increasing the amount of hydrophilic side chain in the polymer enables greater ion insertion, as indicated by the bleaching behavior and the formation of polarons, even in highly stacked cores such as IID. This is further supported by ionic conductivity of each film determined through electrochemical impedance spectroscopy (EIS) (Fig. S9†). The ionic conductivity at 1 kHz of the 0g-IID was 1.73 × 10−8 S cm−1 and was highest in the 100-gIID film at 4.22 × 10−8 S cm−1 showing a 4× increase with the presence of branched glycol side chains as compared to the branched alkyl side chains present in the 0g-IID film. The hydrophobic alkyl side chains increase the resistance to ion insertion in the films, and increasing the amount of glycol side chain decreases the resistance, leading to higher ionic conductivity.
As channel materials in OECT devices, all mg-IID polymers exhibited relatively modest performances as shown in Fig. 4C & D. Details on the devices fabrication and characterization are provided in the ESI and corresponding device metrics are summarized in Table S3.† Also found in the ESI are the characteristic transfer curves, exhibiting conventional p-type turn-on behaviors in all polymers (Fig. S10†). OECT device operation varied depending on hydrophilic sidechain concentration within the polymer. Higher voltage was required to turn on devices with low g-IID concentration, consistent with the spectro-electrochemical analysis indicating higher voltage needed for ion insertion (Fig. 4A–D). Increasing the concentration of glycol side chains increased the device current and transconductance. Increasing the amount of g-IID increased device performance with the 100g-IID film reaching 100 times more device current than 25g-IID and the maximum transconductance of the series studied (Fig. 4C, D & Table S3†). Higher concentration of g-IID increased ion insertion due to the hydrophilicity and disorder created by the flexible chains. Note that as expected, the 0g-IID and 25g-IID films did not show high performance (compared to state-of-the-art and less crystalline counterparts achieving μC* up to 522 F cm−1 V−1 s−1 (ref. 4 and 20)) in OECTs due to the lack of space for ions in highly ordered regions but still showed to outperform previously reported IID-based analogues, achieving comparable normalized transconductance17 and improved μC*.18
To further quantify the impact of interchain order on mixed ionic-electronic conduction, the characteristic metrics, namely μC*, were extracted from OECT measurements, where μ is the charge carrier mobility and C* is the volumetric capacitance. More details on the extraction and calculations of the device metrics are available in the ESI.† As mentioned above, the mg-IID series performed rather modestly; the extracted μC* values could only reach as high as 12.26 F cm−1 V−1 s−1 as the g-IID ratio increased to 100%. However, as we hypothesized and in agreement with our morphological and electrochemical results, the ionic-electronic conduction increases with the glycol content, without sacrificing electronic conduction (Table S3†). We calculated C* values from electrochemical measurements discussed above and extracted OECT mobilities to decouple the contributions from the two modes of transport across the IID series. The extracted OECT mobility was used as a parameter to compare electronic conduction due to low performance in dry-state OFET devices. Among the polymer series, 75g*-IID film shows the most balanced performance, with the highest OECT mobility and second highest C*. We found that by increasing the concentration of glycol side chains, the volumetric capacitance increases by over 10× upon the incorporation of polar and branched sidechains, while minimal changes occur in the extracted μOECT, particularly when comparing the 0g-IID and 100g-IID films (Table S3†). This increase was in contrast with the sidechain-insensitive π–π stacking distances as revealed by the GIXRD results. Instead of driving the chains further apart,18 the chain coiling and lowered long-range aggregation (upon the incorporation of polar glycol sidechains) improve the ability of IID films to uptake ions more efficiently. This is in contrast with the common understanding for optimizing redox activity where the addition of polar sidechains tends to greatly disrupt chain-stacking, or the parent systems are designed to be inherently amorphous. Here, to accommodate for the ionic species, the IID chains can maintain their favored spacing, and instead adapt to a more coiled configuration. As result, the thin films reveal a face-on orientation and an overall lowered crystallite aggregation. Such chain flexibility and in-bulk reconfiguration, associated to the incorporation of the branched glycol sidechains, could thus rationalize the observed mixed conduction behaviors across the IID series, albeit the conserved and close interchain distances.
Conclusions
A series of IID copolymers was synthesized, varying the amount of hydrophobic (alkyl) and hydrophilic (glycol) side chains. The structure and ratio of side chains were confirmed with 1H NMR spectroscopy. The introduction of hydrophilic side chain was linked to lowered long-range aggregation and flipped chain orientation within the polymer thin films, as demonstrated with UV-Vis-NIR and GIXRD results without driving the π–π stacking distances further apart. MD calculations verified the increased chain coiling and decreased crystallinity when the alkyl side chains are changed to branched glycol side chains, however strong π–π stacking was observed in both cases. Additionally, ion insertion, device current and transconductance were highly dependent on the amount of hydrophilic side chain as shown by spectro-electrochemical and OECT device behaviors, resulting from the openness of the film bulk and enhanced wettability. The introduction of flexible and hydrophilic side chains was used as a method to create space in an ordered polymer thin film to allow for easier ion insertion, needed for mixed conduction while maintaining the inherent chain to chain stacking distances within the crystalline domains. Without the flexible and hydrophilic side chains, the polymer films showed inefficient ion insertion due to lack of spatial and chemical compatibility with ions. With this side chain engineering method, a detailed structure–function relationship was investigated for achieving a balance between the ordered and disordered domains within the polymer film to allow for ionic insertion in disordered regions while maintaining efficient electronic conduction in the ordered regions.Data availability
All data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. M. acknowledges support by the National Science Foundation Graduate Research Fellowship Program under grant no. 2141064. A. G. and C. C. acknowledge support from the K. Lisa Yang Brain-Body Center at the Massachusetts Institute of Technology (MIT). Y. Z. and W. L. acknowledge support from the Natural Science Foundation of Shanghai (grant no. 22ZR1407800). Part of this work was carried out in the shared experimental facilities at MIT supported by the National Science Foundation (grant no. DMR-14-19807) and the MIT Department of Chemistry Instrumentation Facility (DCIF).References
- Y. Wang, S. Wustoni, J. Surgailis, Y. Zhong, A. Koklu and S. Inal, Designing Organic Mixed Conductors for Electrochemical Transistor Applications, Nat. Rev. Mater., 2024, 1–17, DOI:10.1038/s41578-024-00652-7.
- X. Strakosas, M. Bongo and R. M. Owens, The Organic Electrochemical Transistor for Biological Applications, J. Appl. Polym. Sci., 2015, 132(15), 41735, DOI:10.1002/app.41735.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Organic Electrochemical Transistors, Nat. Rev. Mater., 2018, 3(2), 1–14, DOI:10.1038/natrevmats.2017.86.
- H. Roh, C. Cunin, S. Samal and A. Gumyusenge, Towards Organic Electronics That Learn at the Body-Machine Interface: A Materials Journey, MRS Commun., 2022, 12(5), 565–577, DOI:10.1557/s43579-022-00269-3.
- S. T. M. Tan, A. Gumyusenge, T. J. Quill, G. S. LeCroy, G. E. Bonacchini, I. Denti and A. Salleo, Mixed Ionic–Electronic Conduction, a Multifunctional Property in Organic Conductors, Adv. Mater., 2022, 34(21), 2110406, DOI:10.1002/adma.202110406.
- S. Inal, G. G. Malliaras and J. Rivnay, Benchmarking Organic Mixed Conductors for Transistors, Nat. Commun., 2017, 8(1), 1767, DOI:10.1038/s41467-017-01812-w.
- B. D. Paulsen, K. Tybrandt, E. Stavrinidou and J. Rivnay, Organic Mixed Ionic–Electronic Conductors, Nat. Mater., 2020, 19(1), 13–26, DOI:10.1038/s41563-019-0435-z.
- R. Noriega, J. Rivnay, K. Vandewal, F. Koch, N. Stingelin, P. Smith, M. Toney and A. Salleo, A General Relationship between Disorder, Aggregation and Charge Transport in Conjugated Polymers, Nat. Mater., 2013, 12, 1038–1044, DOI:10.1038/nmat3722.
- L. Balhorn, Q. MacPherson, K. C. Bustillo, C. J. Takacs, A. J. Spakowitz and A. Salleo, Closing the Loop between Microstructure and Charge Transport in Conjugated Polymers by Combining Microscopy and Simulation, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(46), e2204346119, DOI:10.1073/pnas.2204346119.
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, Polymer Crystallinity Controls Water Uptake in Glycol Side-Chain Polymer Organic Electrochemical Transistors, J. Am. Chem. Soc., 2019, 141(10), 4345–4354, DOI:10.1021/jacs.8b12640.
- S. Moro, N. Siemons, O. Drury, D. A. Warr, T. A. Moriarty, L. M. A. Perdigão, D. Pearce, M. Moser, R. K. Hallani, J. Parker, I. McCulloch, J. M. Frost, J. Nelson and G. Costantini, The Effect of Glycol Side Chains on the Assembly and Microstructure of Conjugated Polymers, ACS Nano, 2022, 16(12), 21303–21314, DOI:10.1021/acsnano.2c09464.
- N. Siemons, D. Pearce, H. Yu, S. M. Tuladhar, G. S. LeCroy, R. Sheelamanthula, R. K. Hallani, A. Salleo, I. McCulloch, A. Giovannitti, J. M. Frost and J. Nelson, Controlling Swelling in Mixed Transport Polymers through Alkyl Side-Chain Physical Cross-Linking, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(35), e2306272120, DOI:10.1073/pnas.2306272120.
- A. Savva, R. Hallani, C. Cendra, J. Surgailis, T. C. Hidalgo, S. Wustoni, R. Sheelamanthula, X. Chen, M. Kirkus, A. Giovannitti, A. Salleo, I. McCulloch and S. Inal, Balancing Ionic and Electronic Conduction for High-Performance Organic Electrochemical Transistors, Adv. Funct. Mater., 2020, 30(11), 1907657, DOI:10.1002/adfm.201907657.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, Molecular Design of Semiconducting Polymers for High-Performance Organic Electrochemical Transistors, J. Am. Chem. Soc., 2016, 138(32), 10252–10259, DOI:10.1021/jacs.6b05280.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Synthesis of Isoindigo-Based Oligothiophenes for Molecular Bulk Heterojunction Solar Cells, Org. Lett., 2010, 12(4), 660–663, DOI:10.1021/ol902512x.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, High-Performance Air-Stable Organic Field-Effect Transistors: Isoindigo-Based Conjugated Polymers, J. Am. Chem. Soc., 2011, 133(16), 6099–6101, DOI:10.1021/ja111066r.
- Y. Wang, E. Zeglio, H. Liao, J. Xu, F. Liu, Z. Li, I. P. Maria, D. Mawad, A. Herland, I. McCulloch and W. Yue, Hybrid Alkyl–Ethylene Glycol Side Chains Enhance Substrate Adhesion and Operational Stability in Accumulation Mode Organic Electrochemical Transistors, Chem. Mater., 2019, 31(23), 9797–9806, DOI:10.1021/acs.chemmater.9b03798.
- A. Hu, A. Nyayachavadi, M. Weires, G. Garg, S. Wang and S. Rondeau-Gagné, Unravelling the Influence of Side-Chain Symmetry on Device Performance: Insights from Isoindigo-Based Polymers in Thin-Film Transistors, RSC Appl. Polym., 2023, 1(2), 292–303, 10.1039/D3LP00104K.
- G. Krauss, F. Meichsner, A. Hochgesang, J. Mohanraj, S. Salehi, P. Schmode and M. Thelakkat, Polydiketopyrrolopyrroles Carrying Ethylene Glycol Substituents as Efficient Mixed Ion-Electron Conductors for Biocompatible Organic Electrochemical Transistors, Adv. Funct. Mater., 2021, 31(20), 2010048, DOI:10.1002/adfm.202010048.
- P. Li and T. Lei, Molecular Design Strategies for High-Performance Organic Electrochemical Transistors, J. Polym. Sci., 2022, 60(3), 377–392, DOI:10.1002/pol.20210503.
- A. Giovannitti, I. P. Maria, D. Hanifi, M. J. Donahue, D. Bryant, K. J. Barth, B. E. Makdah, A. Savva, D. Moia, M. Zetek, P. R. F. Barnes, O. G. Reid, S. Inal, G. Rumbles, G. G. Malliaras, J. Nelson, J. Rivnay and I. McCulloch, The Role of the Side Chain on the Performance of N-Type Conjugated Polymers in Aqueous Electrolytes, Chem. Mater., 2018, 30(9), 2945–2953, DOI:10.1021/acs.chemmater.8b00321.
- S. Samal, H. Roh, C. E. Cunin, G. G. Yang and A. Gumyusenge, Molecularly Hybridized Conduction in DPP-Based Donor–Acceptor Copolymers toward High-Performance Iono-Electronics, Small, 2023, 19(18), 2207554, DOI:10.1002/smll.202207554.
- G. Xue, X. Zhao, G. Qu, T. Xu, A. Gumyusenge, Z. Zhang, Y. Zhao, Y. Diao, H. Li and J. Mei, Symmetry Breaking in Side Chains Leading to Mixed Orientations and Improved Charge Transport in Isoindigo-Alt-Bithiophene Based Polymer Thin Films, ACS Appl. Mater. Interfaces, 2017, 9(30), 25426–25433, DOI:10.1021/acsami.7b07624.
- J. Mei and Z. Bao, Side Chain Engineering in Solution-Processable Conjugated Polymers, Chem. Mater., 2014, 26(1), 604–615, DOI:10.1021/cm4020805.
- X. Zhao, S. T. Chaudhry and J. Mei, Chapter Five - Heterocyclic Building Blocks for Organic Semiconductors. In Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Heterocyclic Chemistry in the 21st Century, Academic Press, 2017, Vol. 121, pp. 133–171. DOI:10.1016/bs.aihch.2016.04.009.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors, J. Am. Chem. Soc., 2013, 135(18), 6724–6746, DOI:10.1021/ja400881n.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Multifluorination toward High-Mobility Ambipolar and Unipolar n-Type Donor–Acceptor Conjugated Polymers Based on Isoindigo, Adv. Mater., 2017, 29(13), 1606217, DOI:10.1002/adma.201606217.
- Y. Jiang, Y. Gao, H. Tian, J. Ding, D. Yan, Y. Geng and F. Wang, Synthesis and Characterization of Isoindigo[7,6 g]Isoindigo-Based Donor–Acceptor Conjugated Polymers, Macromolecules, 2016, 49(6), 2135–2144, DOI:10.1021/acs.macromol.6b00004.
- Y. Zhao, C. Su, G. Shen, Z. Xie, W. Xiao, Y. Fu, S. Inal, Q. Wang, Y. Wang, W. Yue, I. McCulloch and D. He, Donor Engineering Tuning the Analog Switching Range and Operational Stability of Organic Synaptic Transistors for Neuromorphic Systems, Adv. Funct. Mater., 2022, 32(36), 2205744, DOI:10.1002/adfm.202205744.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Isoindigo, a Versatile Electron-Deficient Unit For High-Performance Organic Electronics, Chem. Mater., 2014, 26(1), 664–678, DOI:10.1021/cm402219v.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Highly π-Extended Copolymers with Diketopyrrolopyrrole Moieties for High-Performance Field-Effect Transistors, Adv. Mater., 2012, 24(34), 4618–4622, DOI:10.1002/adma.201201318.
- J. H. Kim, R. Halaksa, I.-Y. Jo, H. Ahn, P. A. Gilhooly-Finn, I. Lee, S. Park, C. B. Nielsen and M.-H. Yoon, Peculiar Transient Behaviors of Organic Electrochemical Transistors Governed by Ion Injection Directionality, Nat. Commun., 2023, 14(1), 7577, DOI:10.1038/s41467-023-42840-z.
- D. T. Tran, A. Gumyusenge, X. Luo, M. Roders, Z. Yi, A. L. Ayzner and J. Mei, Effects of Side Chain on High Temperature Operation Stability of Conjugated Polymers, ACS Appl. Polym. Mater., 2020, 2(1), 91–97, DOI:10.1021/acsapm.9b00999.
- A. Gumyusenge, X. Luo, H. Zhang, G. M. Pitch, A. L. Ayzner and J. Mei, Isoindigo-Based Binary Polymer Blends for Solution-Processing of Semiconducting Nanofiber Networks, ACS Appl. Polym. Mater., 2019, 1(7), 1778–1786, DOI:10.1021/acsapm.9b00321.
- H. Roh, S. Yue, H. Hu, K. Chen, H. J. Kulik and A. Gumyusenge, Unraveling Polymer–Ion Interactions in Electrochromic Polymers for Their Implementation in Organic Electrochemical Synaptic Devices, Adv. Funct. Mater., 2023, 33(45), 2304893, DOI:10.1002/adfm.202304893.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, synthetic procedures for monomers and polymers, 1H NMR spectra, thermogravimetric analysis, differential scanning calorimetry, UV-Vis-NIR solution absorption, DFT simulations, atomic force microscopy, grazing incidence wide angle X-ray diffraction spacing, cyclic voltammetry, spectroelectrochemical analysis, electrochemical impedance spectroscopy, OECT characteristic transfer curves. See DOI: https://doi.org/10.1039/d4lp00272e |
| This journal is © The Royal Society of Chemistry 2024 |