
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxanorbornene-based phosphonium polymeric ionic liquids (PILs) and investigation of their electrical properties†
M. S.
M. Misenan
 a,
N. Ceren
Süer
a,
N.
Yılmaz Canli
b,
A. S.
A. Khiar
*c and
T.
Eren
*a
a,
N. Ceren
Süer
a,
N.
Yılmaz Canli
b,
A. S.
A. Khiar
*c and
T.
Eren
*a
aDepartment of Chemistry, College of Arts and Science, Yildiz Technical University, Istanbul, Turkey. E-mail: teren@yildiz.edu.tr
bDepartment of Physics, College of Arts and Science, Yildiz Technical University, Istanbul, Turkey
cFaculty of Science and Technology, Universiti Sains Islam Malaysia, Nilai, Malaysia. E-mail: azwanisofia@usim.edu.my
First published on 9th January 2024
Abstract
In this study, a series of phosphonium polymeric ionic liquids (PILs) was synthesized via ring-opening metathesis polymerization (ROMP), and the trifluorosulfonylimide anion successfully replaced the bromide anion via ion exchange. Subsequently, the effect of various substituent groups (including triphenyl, tripropyl, tertbutyl and trifluorophenyl) on the conductivity of the PILs was studied. The results showed that an alky substituent backbone resulted in higher conductivity compared to the aromatic functional group-bearing polymer architecture. The optimum conductivity of PILs at room temperature was found to be 6 × 10−4 S cm−1 for the propyl substituent group PILs (SM2) doped with 15 wt% LiTFSI salt. Furthermore, the effect of grafting polyethylene glycol (PEG) units to the polymer backbone on the conductivity value was also investigated.
1. Introduction
Due to the growing concerns regarding fossil fuel depletion, pollution and greenhouse gas emission, there is a strong call for electrified land and air transportation as well as the use of large-scale renewable energy sources, which has increased the need for the creation of more effective and dependable electrochemical energy storage devices,1,2 Consequently, rechargeable lithium-ion batteries (LIBs) have emerged as the most beneficial storage devices since Sony released its first commercial product in 1991.3 The market for LIBs, which was valued at 30 billion USD in 2017 is expected to reach 129.3 billion USD by 2027 and is projected to increase at a compound annual growth rate of 31% over the forecast period (2019–2027).4 Furthermore, due to the increasing interest in electric and hybrid vehicles, it is forecasted that the automotive end-use industry will dominate the global market.LIBs are composed of three primary parts, i.e., anode, cathode and electrolyte. Essentially, an electrolyte is a substance that exhibits electrical conductivity because it contains concentrated free ionic species.5 Nonetheless, there are numerous safety concerns resulting from the use of liquid electrolytes in conventional LIBs, such as the flammability of organic solvents and electrolyte leakage, hindering the wide application of LIBs.6–8
In this case, as an alternative, solid polymer electrolytes (SPEs) and ionic liquids utilizing both organic and inorganic materials have emerged as the most advantageous electrolyte systems for the construction of highly safe and dependable large-format batteries to overcome the above-mentioned issues. Wright et al.9 initially produced a polymer electrolyte (PE) by the complexation of polyethylene oxide (PEO) and alkali metal salts. PEs have been thoroughly investigated as a safer alternative to liquid electrolytes to address these issues.10 This is because PEs are more flexible and machinable than liquid electrolytes and enhance the safety performance of batteries,11,12
There is a growing interest in PEs, and the research on their application in various electrochemical devices especially in LIB technology is increasing. However, it is commonly known that PEs have a low lithium transference number and ionic conductivity at room temperature.13 Thus, several methods have been reported to increase the lithium-ion dissociation in the polymer matrix and increase the ionic conductivity. A new type of single lithium-ion polymer electrolyte with –SO2–N(−)–SO2–CF3 anionic groups and PEO was proposed by Armand et al.14 The ionic conductivity of these mixed polymer electrolytes can reach up to 10−6 S cm−1 at 70 °C. In particular, the unusual structure of –SO2–N(−)–SO(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NSO2CF3)–CF3 with both super-delocalized negative charge distribution and very flexible characteristics was reported by Zhou et al.15 This lithium-ion conducting polymer exhibited the optimum conductivity of 1.35 × 10−4 S cm−1 at 90 °C. The larger delocalized size of the anion further improves the ionic conductivity due to the increased negative charge distribution and lithium-ion dissociation. However, the process for the preparation of these lithium salts with super-delocalized structures is laborious and complicated. Additionally, due to the stiffness and absence of Li-ion conductors near the anionic centers in homopolymers, the homologous PEs produced by the polymerization of a salt monomer often cannot achieve high ionic conductivity.16 Thus, lithium salt-containing homopolymers are frequently blended with polymers having a soft segment, such as PEO, to increase their ionic conductivity. In PEO, the Lewis base groups act as a Li-ion conductor to enable the solvation and desolvation of Li+ ions.17 Another strategy involves copolymerizing a monomer of lithium salt with a co-monomer that has flexible segments containing a Lewis base group. The advantage of the soft chains is that through Lewis acid–base interactions, they can serve as a transporter of Li+ ions.18
NSO2CF3)–CF3 with both super-delocalized negative charge distribution and very flexible characteristics was reported by Zhou et al.15 This lithium-ion conducting polymer exhibited the optimum conductivity of 1.35 × 10−4 S cm−1 at 90 °C. The larger delocalized size of the anion further improves the ionic conductivity due to the increased negative charge distribution and lithium-ion dissociation. However, the process for the preparation of these lithium salts with super-delocalized structures is laborious and complicated. Additionally, due to the stiffness and absence of Li-ion conductors near the anionic centers in homopolymers, the homologous PEs produced by the polymerization of a salt monomer often cannot achieve high ionic conductivity.16 Thus, lithium salt-containing homopolymers are frequently blended with polymers having a soft segment, such as PEO, to increase their ionic conductivity. In PEO, the Lewis base groups act as a Li-ion conductor to enable the solvation and desolvation of Li+ ions.17 Another strategy involves copolymerizing a monomer of lithium salt with a co-monomer that has flexible segments containing a Lewis base group. The advantage of the soft chains is that through Lewis acid–base interactions, they can serve as a transporter of Li+ ions.18
Ionic liquids (ILs) have received significant attention due to their intriguing physicochemical characteristics, including non-flammability, minimal vapor pressure, particularly high conductivity, and broad electrochemical windows.19 However, the liquid nature of room-temperature ionic liquids typically results in issues in terms of leakage and fabrication.20 Hence, a new class of materials where the IL molecules are combined with polymer chains, known as polymeric ionic liquids (PILs), has emerged as an alternative. These materials are also known as polymerized ionic liquids or poly(ionic liquids).21 PILs are bound to the polymer backbone as either cationic or anionic species. They provide a new family of functional polymers that combines the distinct properties of room-temperature ionic liquid (RTILs) with the macromolecular architecture of polymers.
Accordingly, extensive research has been conducted on PILs for use as tailored electrolytes in various applications, such as energy storage (batteries and super capacitors), conversion (fuel cells), and electromechanical (actuators and sensors) devices.22–26 PILs are endowed with tunable features (such as glass transition temperature and solubility) due to their unique combination of polymers and counter-ions, enabling a wide range of sophisticated applications. The main benefits of adopting PILs over ILs include their greater mechanical stability, improved processability, robustness, and spatial controllability as compared to IL species.
Imidazolium, ammonium, and phosphonium-based PIL materials have received significant attention as ion-conductive components for electrochemical devices such as fuel cells, lithium batteries, actuators, and solar cells.27 However, compared to imidazolium- and ammonium-based compounds, phosphonium-based ILs and PILs are scarce in the literature despite their substantial advantages compared to their nitrogen-based analogues. One notable exception is the report in 2022 by Hofmann et al., who studied a phosphonium-based ionic liquid electrolyte for battery application.28
In ammonium cations, nitrogen carries a partial negative charge due to its higher electronegativity compared to the α-carbon. The electrostatic interaction primarily occurs between the counter anion and the positively charged α-carbons. Alternatively, in phosphonium cations, phosphorus carries a partially positive charge given that its electronegativity is lower than that of carbon, resulting in slightly negatively charged α-carbons. These negatively charged α-carbons effectively shield the positive charge on phosphorus, reducing the electrostatic interactions and allowing more single ions to freely diffuse, thereby resulting in higher ionic conductivity for phosphonium-based polyelectrolytes. Colby et al. calculated the charge distribution of Bu4P+, Bu4N+ and BuMeIm+ based on ab initio calculation.29
Phosphonium-based ILs and PILs exhibit greater thermal stability, base stability and ion conductivity compared to their nitrogen-based analogues.30–32 For instance, poly(4-vinylbenzyl ammonium) and poly(4-vinylbenzyl phosphonium) homopolymers with different alkyl replacements were examined by Long et al. and it was observed that the phosphonium-based PILs showed an increase in the onset of thermal deterioration of over 100 °C.30 Hoffman elimination and/or reversible Menshutkin degradation occur in ammonium salts (for benzylic protons). In contrast, phosphonium salts have higher thermal stability because they are less susceptible to both breakdown processes.30,33 Alternatively, alkaline anion exchange membranes made of polyethylene functionalized with phosphonium pendant groups were created in the study by Coates and colleagues.32 In comparison to their ammonium counterparts, phosphonium PILs also displayed higher ion conductivity of 22 × 10−3 S cm−1 (normalized with Tg).
According to the comparison of the major PILs, phosphonium cation-based PILs exhibit better properties, such as higher thermal stability, higher conductivity, and lower viscosity, although this family of PILs is the least studied.34 The majority of research focused on the flame retardancy,35 adjustable hydrophilicity,36 lower humidity sensitivity,26 and antibacterial properties37 of PILs.
A thorough understanding of the interactions between the length of the alkyl chain substituent and characteristics will be crucial in the development of phosphonium-containing polymers for practical applications such as fuel cell membranes, solid-state polyelectrolytes for lithium-ion batteries, and biosensors. Nowadays, the flammability of polymer electrolytes has the potential to endanger battery safety, although this issue has been neglected to date.38 Thus, the production of noncombustible polymer hosts, such as fluoropolymers and polymers containing phosphorus and/or nitrogen for self-extinguishing properties, is also important for polyelectrolytes that can be used in lithium ion batteries.39 Presently, due to the increasing public awareness regarding the environment, phosphonium-containing polymer flame retardants have become more significant.40
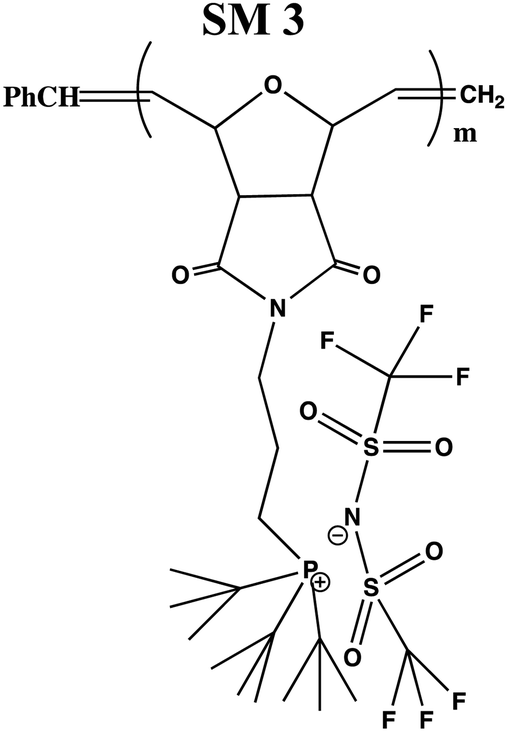
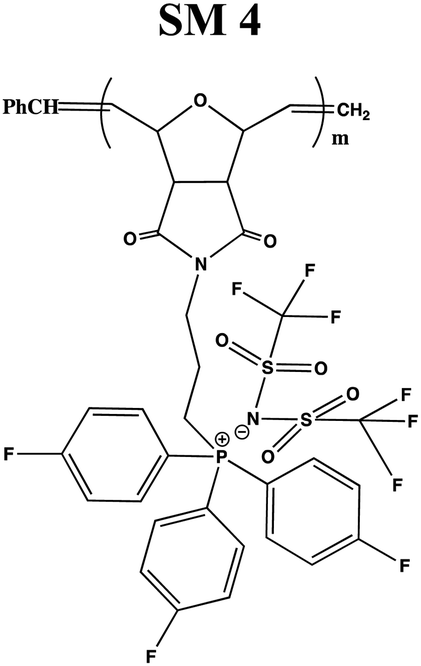
In this study, a series of phosphonium PILs with alkyl substitution with different lengths were synthesized including triphenyl (SM1), tripropyl (SM2), tertbutyl (SM3), and trifluorophenyl (SM4) functional groups. Subsequently, the effect of the substituent groups towards ionic conductivity was investigated. The quaternary phosphonium functionality attached to oxanorbornene monomers (Scheme 1) was polymerized using a Grubbs third-generation catalyst, as shown in Scheme 2. The bromine anion was replaced with the TFSI anion via ion exchange. The trifluorosulfonylimide (TFSI−) anion exhibits higher conductivity compared to the bromide ion (Br−) due to its larger size and relatively delocalized negative charge, which weakens the interactions between the TFSI anion and the phosphonium cation.28 Furthermore, the structure-conductance property relationship was examined using a controlled polymerization technique.
 | ||
| Scheme 1 Schematic illustration of the synthesis of the monomers. | ||
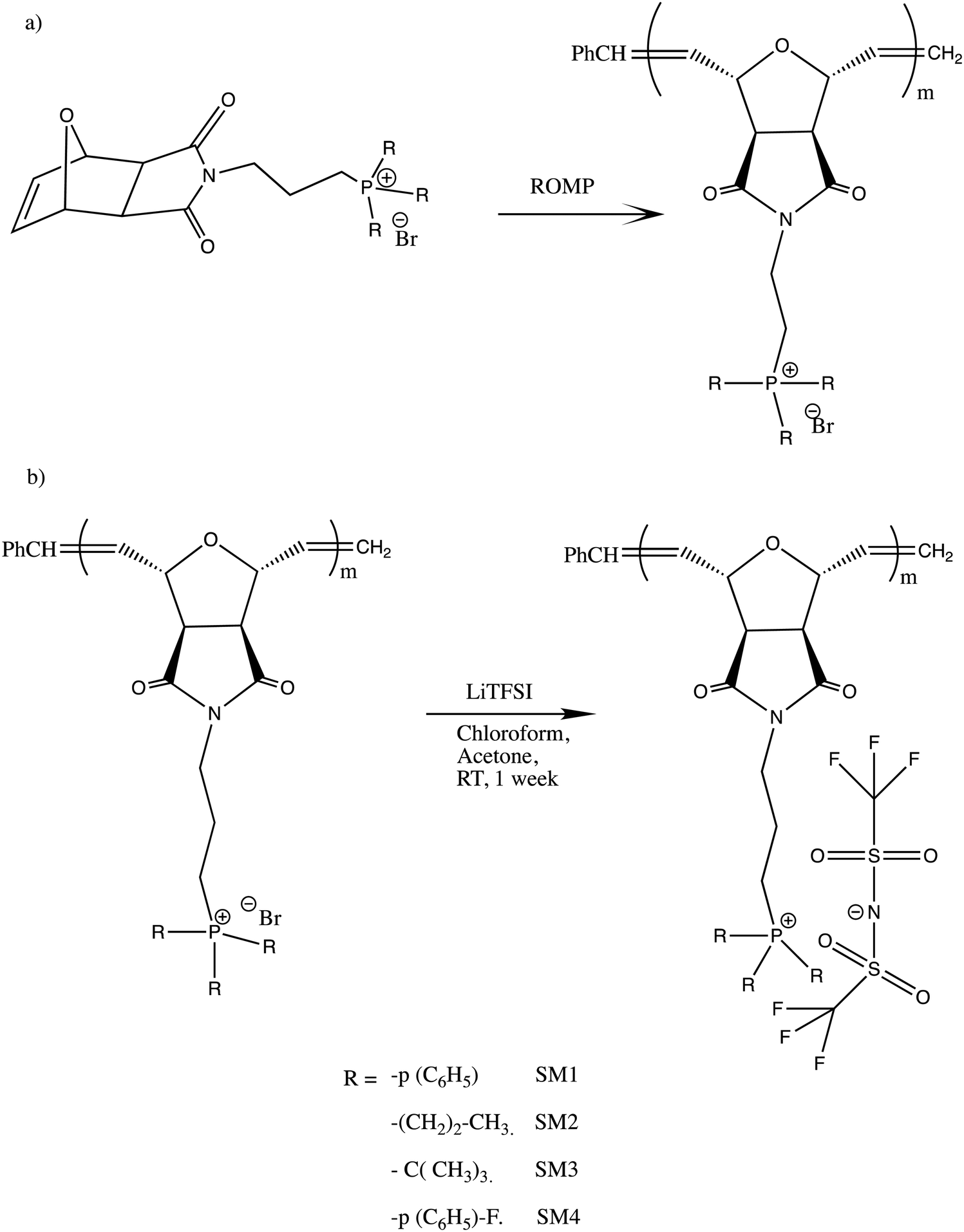 | ||
| Scheme 2 (a) Polymerization of phosphonium ionic liquid monomers via ROMP and (b) ion exchange procedure for the preparation of the phosphonium polymeric ionic liquid. | ||
2. Experimental
2.1 Materials
A series of phosphonium monomers (triphenyl, tripropyl, tertbutyl and trifluoro phenyl) was synthesized according to the literature.41 The Grubbs 2nd generation catalyst was purchased from Aldrich, which was used to freshly prepare the Grubbs 3rd generation catalyst [(H2-Imes)(3-Br-py)2-(Cl)2RuCHPh] according to the literature.42 Poly(ethylene glycol)dithiol (average Mn = 8000 g mol−1) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) (99%) was purchased from Aldrich and used without further purification. All solvents including dichloromethane (DCM), petroleum ether, diethyl ether, ethyl acetate, hexane, chloroform, N,N-dimethylformamide (DMF), pentane, ethyl vinyl ether, acetic anhydride, 2,2,2-trifluoroethanol, and acetone were also purchased from Aldrich and used without further purification.
2.2 Instrumentation
A PerkinElmer Fourier transform infrared (FTIR) spectrophotometer (Model FTIR – Spectrum 400) equipped with attenuated total reflection (ATR) was used to conduct the FTIR analysis. The infrared beam was passed through with a resolution of 4 cm−1 at a frequency in the range of 4000 to 400 cm−1.1H NMR (500 MHz) spectra were collected in CDCl3 or DMSO-d6 on a Bruker Avance III 500 MHz spectrometer. A Varian Mercury VX 400 MHz BB spectrometer was used to obtain 31P NMR spectra.
Field-emission scanning electron microscopy (FESEM) on a JEOUL 7600f was used to characterize the structural properties of the solids. The PILs were placed on black cellophane tape prior to image collection and transferred to the sample holder imaging and characterization. Images were acquired at 1 kV under the magnification of 5 μm–10 μm.
A DSC instrument (HP DSC3, Mettler-Toledo Co.) was used to examine the standard PIL samples and LiTFSI-doped PILs. The electrolyte content in the sample pan was 6.5 ± 0.5 mg. The samples were heated from 0 °C to 200 °C at a heating rate of 10 °C min−1 under nitrogen gas flow.
Impedance spectroscopy was carried out for conductivity measurement on a HIOKI 3531-01 LCR Hi Tester interfaced to a computer in the frequency range of 50 Hz–1 MHz. The cold press technique was used to press all the polymeric ionic liquid electrolyte films. The samples were 1 cm in diameter with a thickness of 0.05 ± 0.003 μm. Under spring pressure, the samples were placed between two stainless steel blocking electrodes. The impedance of the polymer electrolyte was determined using the complicated impedance approach. The conductivity of the samples was determined using the following equation:
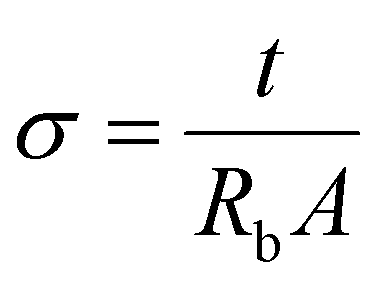 | (1) |
2.3 Monomer and polymer synthesis
The monomers and polymers used in this investigation were prepared using the process described in the literature.41,43 The synthesis of the monomers was initiated by the Diels–Alder reaction between maleic anhydride and furan to yield exo-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride (compound 1). Then, compound 1 was reacted with 3-bromopropyl amine hydrobromide to produce 4-(3-bromopropyl) 10-oxa-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione (compound 2). Compound 2 was purified by column chromatography using ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (1:1, v/v) as the eluent. A scheme of the monomer synthesis is presented in Fig. S1 (ESI†).
:hexane (1:1, v/v) as the eluent. A scheme of the monomer synthesis is presented in Fig. S1 (ESI†).
2.4 Anion-exchange reaction of phosphonium-containing polymeric ionic liquid (PILs)
The typical anion exchange procedure for the phosphonium-based polymeric ionic liquid was as follows: 200 mg of phosphonium PIL was dissolved in a mixture of 3 mL chloroform and 2.5 mL acetone and 0.5 g LiTFSI salt was dissolved in 2.5 mL acetone. Then the two solutions were mixed homogenously. After stirring at room temperature for 1 week, all the solvent was evaporated using a Rotavapor. Then, the samples were redissolved in 3 mL acetone. The solutions were precipitated with distilled water. The precipitate was washed until it was free from bromide ions. Specifically, the resulting PILs were tested with silver nitrate until no silver bromide precipitate was seen (clear solution), indicating that bromide anions were not present (see ESI,† Fig. S2). Finally, the product was dried in an oven at 60 °C.2.5 PEGylation of phosphonium-containing PILs
Briefly, 0.2 g of propyl PILs and 1.36 g PEG di-thiol (average Mn = 8000 g mol−1) in a molar ratio of 1 to 20 were dissolved in a mixture of 4 mL chloroform and 1 mL acetone. Then, 0.033 g Irgacure 2959 was added to the solution. The obtained solution was placed in a photoreactor (equipped with a 365 nm, 100 W lamp) and left overnight. After the reaction was completed, all the solvent was evaporated using a Rotavapor. The peg-propyl-substituted phosphonium PILs were redissolved in water and subjected to dialysis for further purification. The sample in the dialysis bag was further added to a Petri dish and the water was removed at 60 °C in an oven. After drying, the residue was redissolved in a 1:1 (v/v) mixture of acetone and chloroform, and then reprecipitated with a 1:1 (v/v) mixture of diethyl ether and petrol ether, producing a creamy rubbery residue.
2.6 Lithium ion-salt doping
The PIL–LiTFSI composition was prepared by dissolving PILs in acetone and 15 wt% of LiTFSI salt was doped in the solution and stirred homogenously for 5 min. Then, the solution was poured in a Petri dish and the solvent evaporated under a hood overnight.2.7 Film formation for conductivity analysis
Thin films of PILs were prepared via the cold press technique using a hydraulic press. All the samples were pressed at a pressure of 2 MPa for 15 s; The PILs film obtained had the diameter of 1 cm radius and 0.05 μm. A typical picture of the film is shown in the Graphical abstract.3. Results and discussion
3.1 Synthesis of trifluoromethanesulfonimide (TFSI) anion bearing phosphonium PILs
In the first part of this study, bromide anion-bearing phosphonium PILs were synthesized and characterized according to the literature,41 as shown in Scheme 1(a). Subsequently, the bromide anion was exchanged with the TFSI anion via the anion exchange method.34In preliminary studies, the bromide anion in phosphonium oxanorbornene monomer was readily exchanged with the trifluorosulfonylimide (TFSI) anion as the model compound. After workup, the model compound was subjected to thin layer chromatography (Fig. S3, ESI†) and NMR analysis (Fig. S4 and S5, ESI† respectively). The 1H NMR spectra (Fig. S4, ESI†) showed the chemical shift of methylene near the phosphonium cation (peak e), revealing an up-field shift for the anion from 3.6 ppm to 3.3 ppm, which indicated enhanced the shielding effects of the larger counterion. This finding was also supported by the 31P NMR (Fig. S5, ESI†) spectra, which showed the chemical shift in phosphonium from 24.8 to 24.1 ppm.
The series of phosphonium-bearing PILs was synthesized via the ring-opening metathesis polymerization (ROMP). The side chain attached to the phosphonium was varied with aliphatic and aromatic groups to study their influence on the conductivity. A targeted number average molecular weight (Mn) of 10000 g mol−1 was applied using the proper ratio of monomer to catalyst concentration. Then, the bromide anion-bearing phosphonium polymers were dissolved with acetone and chloroform solvent and LiTFSI salt for anion exchange from the bromide anion to TFSI anion.
After workup, the full elimination of the monomer olefin proton peaks at 6.4 ppm in the 1H NMR spectra indicated that the polymerization process was complete and the occurrence of cis or trans signals for the polymer backbone double bond protons at 5.7–5.9 ppm for the triphenyl side chain phosphonium PILs (SM1), as depicted in Fig. 1. The production of both cis- and trans-double bonds was caused by the appearance of vinylic hydrogens produced by the ROMP pathway and the integration of these two signals in the 1H NMR spectra revealed the relative cis/trans ratio in the polymer backbone. The PILs contained 65% trans residue. The NMR spectra of the other phosphonium PILs are provided in the ESI† (Fig. S6–S8). Then, the 31P NMR spectra exhibited a peak at 24.1 ppm, which is the same as monomer model compound, indicating the anion exchange successfully occurred.
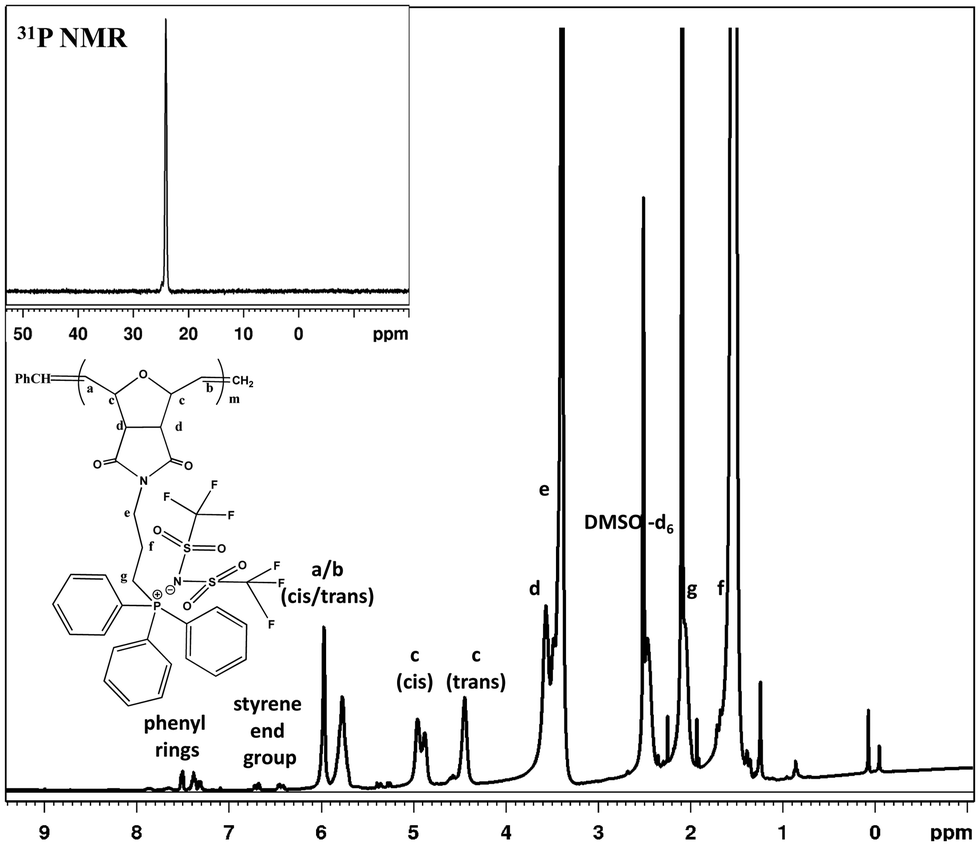 | ||
| Fig. 1 1H NMR spectrum of triphenyl–trifluoromethylsulfonylimide salt (SM1). Inset shows 31P NMR spectrum. | ||
The FTIR analysis also confirmed the anion exchange in the PIL series. Various bands were monitored to examine the chemical structural changes that occurred in the backbone of the phosphonium PILs after the Br anion was changed to the –TFSI anion (Fig. 2). The phosphonium PILs containing the bromide anion showed the characteristic CO, –CH3 asymmetric, –CH3 symmetric, and C–O–C stretching frequencies at 1773 cm−1, 2924 cm−1, 2857 cm−1 and 1110 cm−1, respectively. The peaks for the –CH3 asymmetric and –CH3 symmetric bending frequencies were identified at 1450 and 1399 cm−1, respectively.44 The medium peaks at 722–755 cm−1 correspond to the P–C stretching.45
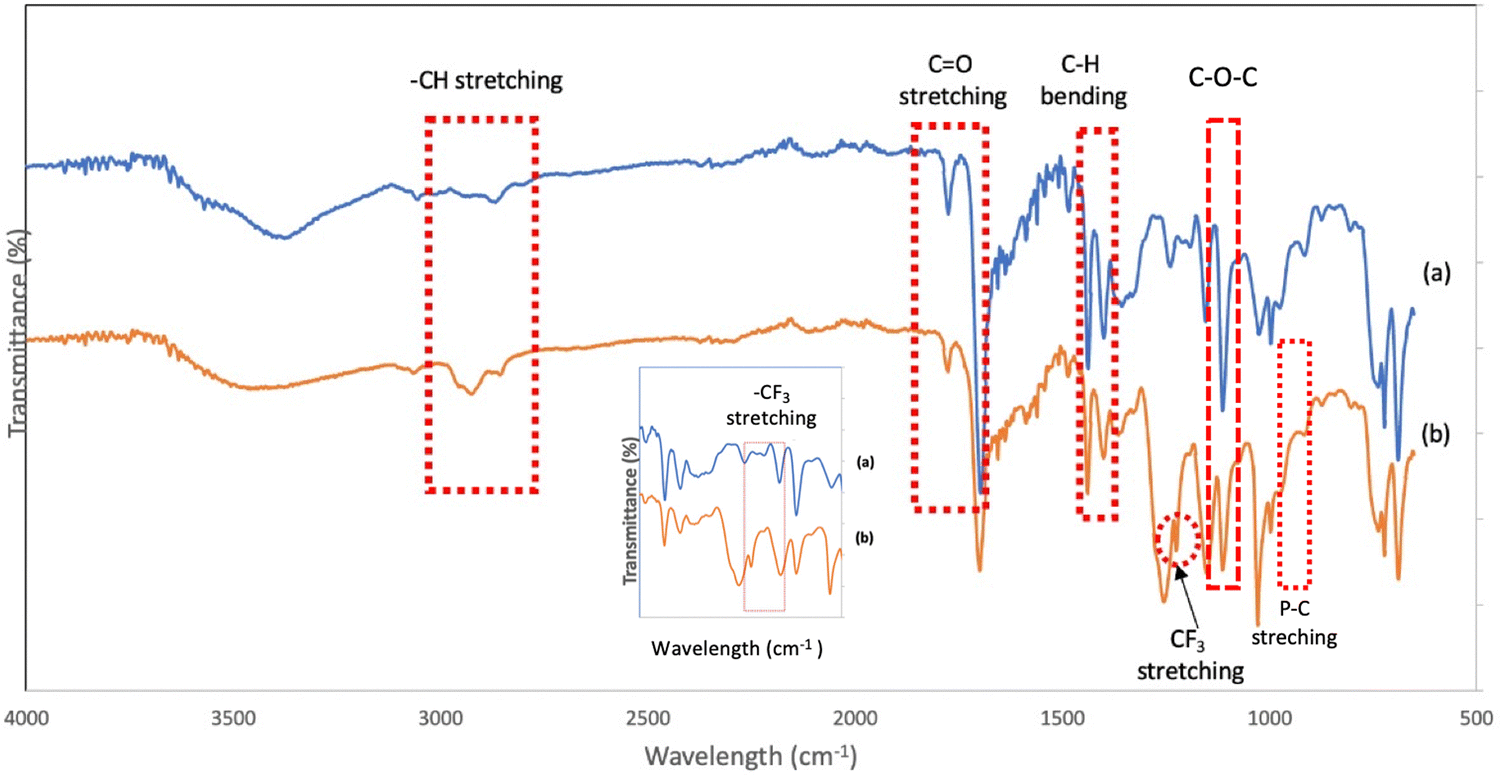 | ||
| Fig. 2 FTIR spectra for polymers possessing (a) triphenyl-Br salt and (b) triphenyl-TFSI salt (SM1) in the range of 4000 cm−1 to 550 cm−1. Inset shows FTIR spectra for (a) triphenyl-Br salt and (b) triphenyl-TFSI salt (SM1) in the range of 1500 cm−1 to 1000 cm−1. | ||
The phosphonium PILs consisting of the –TFSI anion showed almost the same absorption peaks with the bromide anion-containing PILs, thus indicating that there no new bonds were formed or strong chemical interaction occurred in the polymer backbone during the ion exchange procedure. However, a new peak was observed at 1222 cm−1 (depicted in Fig. 2) due to the stretching characteristic of –CF3 from the –TFSI anion.46 Thus, according to the NMR and FTIR analysis, the anion exchange procedure successfully achieved the substitution of the bromide anion for the –TFSI anion.
3.3 EDX analysis
The surface of PIL–TFSI was subjected to EDX examination (Fig. 3) and elemental mapping analysis (Fig. 4). The expected elements were found in atomic percentage%, including carbon (86%), oxygen (21%), fluorine (7.7%), phosphorus (0.9%) and sulphur (0.8%). The X-ray elemental mapping showed the even distribution of phosphorus and fluorine near the film surface.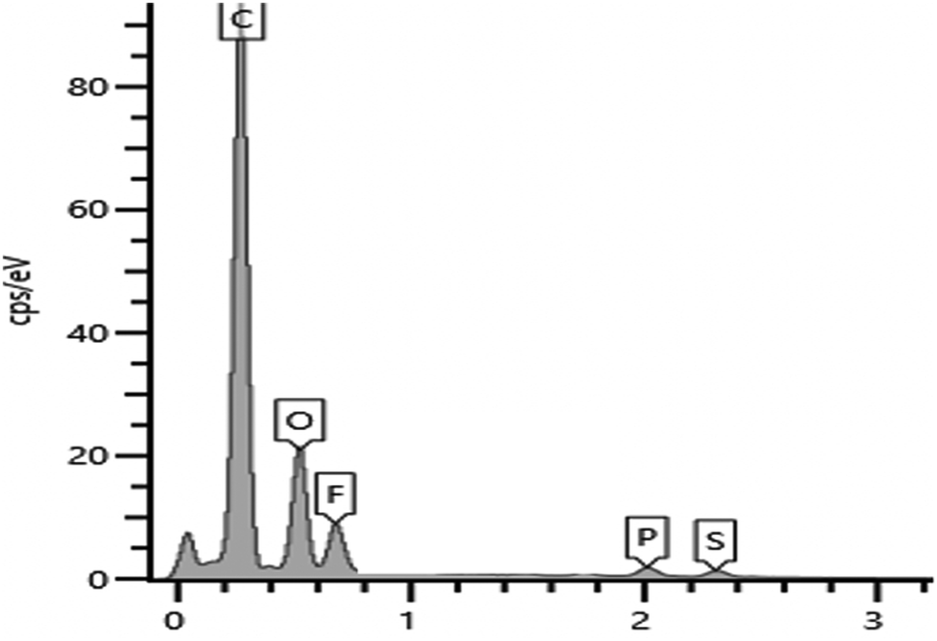 | ||
| Fig. 3 Surface elemental composition of SM1. | ||
 | ||
| Fig. 4 SEM image and elemental mapping images of the constituent elements in SM1 (C, O, F, S, and P), respectively. | ||
3.4 Conductivity studies
In the conductivity studies, the PIL powder was pressed using a cold press and analyzed by impedance spectroscopy operating at 303 K to 353 K. Fig. 5 displays the Cole–Cole plot for SM1 at ambient temperature. The point where the semicircle line and the spike intersect can be used to calculate the bulk resistance (Rb). The slanted spikes represent the capacitance nature of the border and absence of electronic conductivity, while the semicircle in the plot represents the characteristic of comparable combinations of bulk resistance, Rb, and bulk capacitance.47,48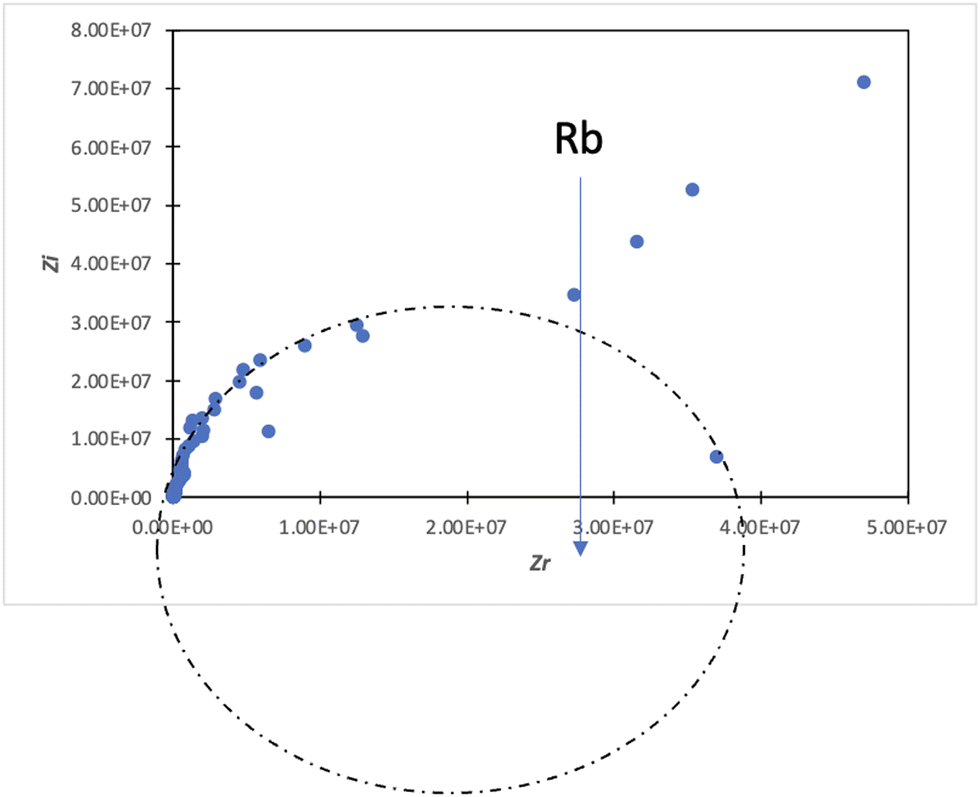 | ||
| Fig. 5 Cole–Cole plot of SM1. | ||
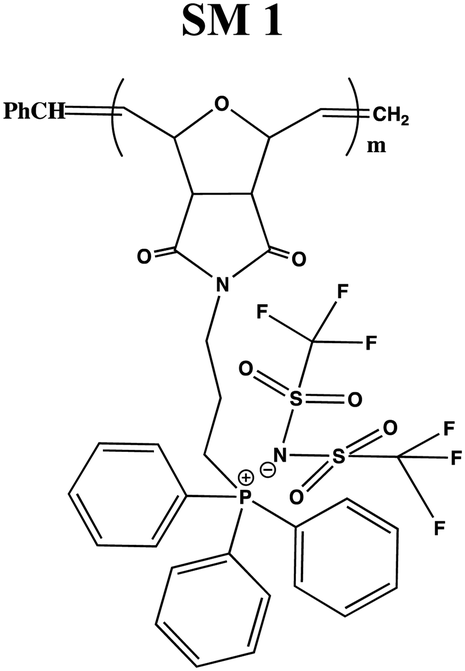
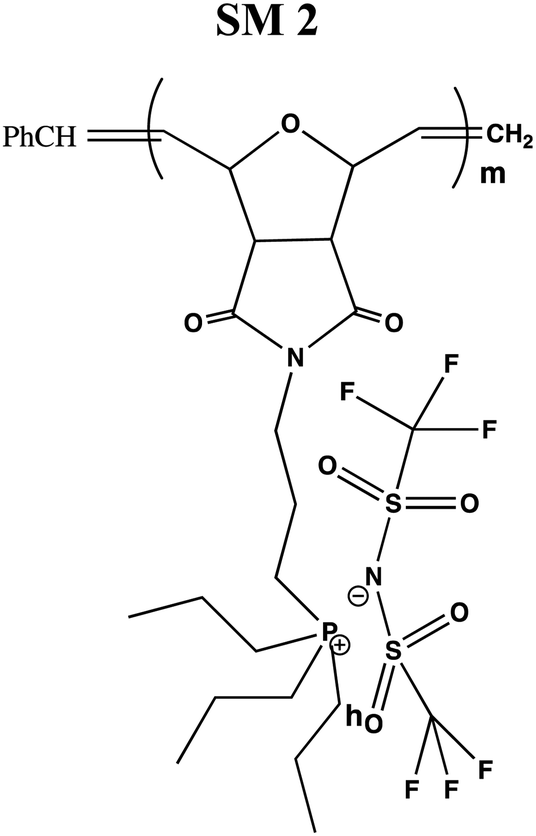
The conductivity and glass transition (Tg) values of the phosphonium PIL samples containing the LiTFSI anion are reported in Table 1. LiTFSI was also doped in the PILs in 15 wt% to enhance their ionic conductivity. It was reported that doping LiTFSI in the polymer matrix results in improved thermal and electrochemical stability, good ionic conductivity, and significantly reduced moisture sensitivity.49Table 1 shows that all samples exhibited significantly higher conductivity after being doped with 15 wt% LiTFSI salt, with an increase ranging from 2 to 3 magnitude. The highest conductivity was obtained at 6.00 × 10−7 S cm−1 for SM2, which possessed propyl subunits. Doping 15 wt% LiTFSI in this polymer resulted in the highest increase in the conductivity value (6.00 × 10−4 S cm−1) in the polymer series.
| Sample | T g (°C) | Conductivity value (S cm−1) | |
|---|---|---|---|
| Bare | Doping 15 wt% LiTFSI | ||
|
122 | 1.01 × 10−9 | 1.21 × 10−6 |
|
119 | 2.91 × 10−7 | 6.00 × 10−4 |
|
120 | 1.18 × 10−8 | 3.20 × 10−6 |
|
121 | 1.20 × 10−8 | 4.30 × 10−5 |
A polymer electrolyte with high ionic conductivity should have a low glass transition temperature (Tg) to promote the movement of the conductive polymer chains. Thus, DSC analysis was performed to determine the Tg of the polymers. The Tg of the polymers appeared to be close; however, the Tg of the propyl-based polymer was observed at a slightly lower temperature (119 °C). Also, the polymers did not show any melting transition. The polymer with the highest conductivity, SM2, was selected to examine the effect of LiTSF doping on the thermal relaxation. The introduction of 15 wt% LiTFSI salt resulted in a melting temperature of 52 °C. Thus, the addition of extra LiTFSI salt to the polymer matrix can reduced the thermal relaxation of the matrix and increase the ionic conductivity.
The Cole–Cole plots of the 15 wt% of LiTFSI-doped PILs, i.e., SM1, SM2, SM3 and SM4, are depicted in Fig. 6.
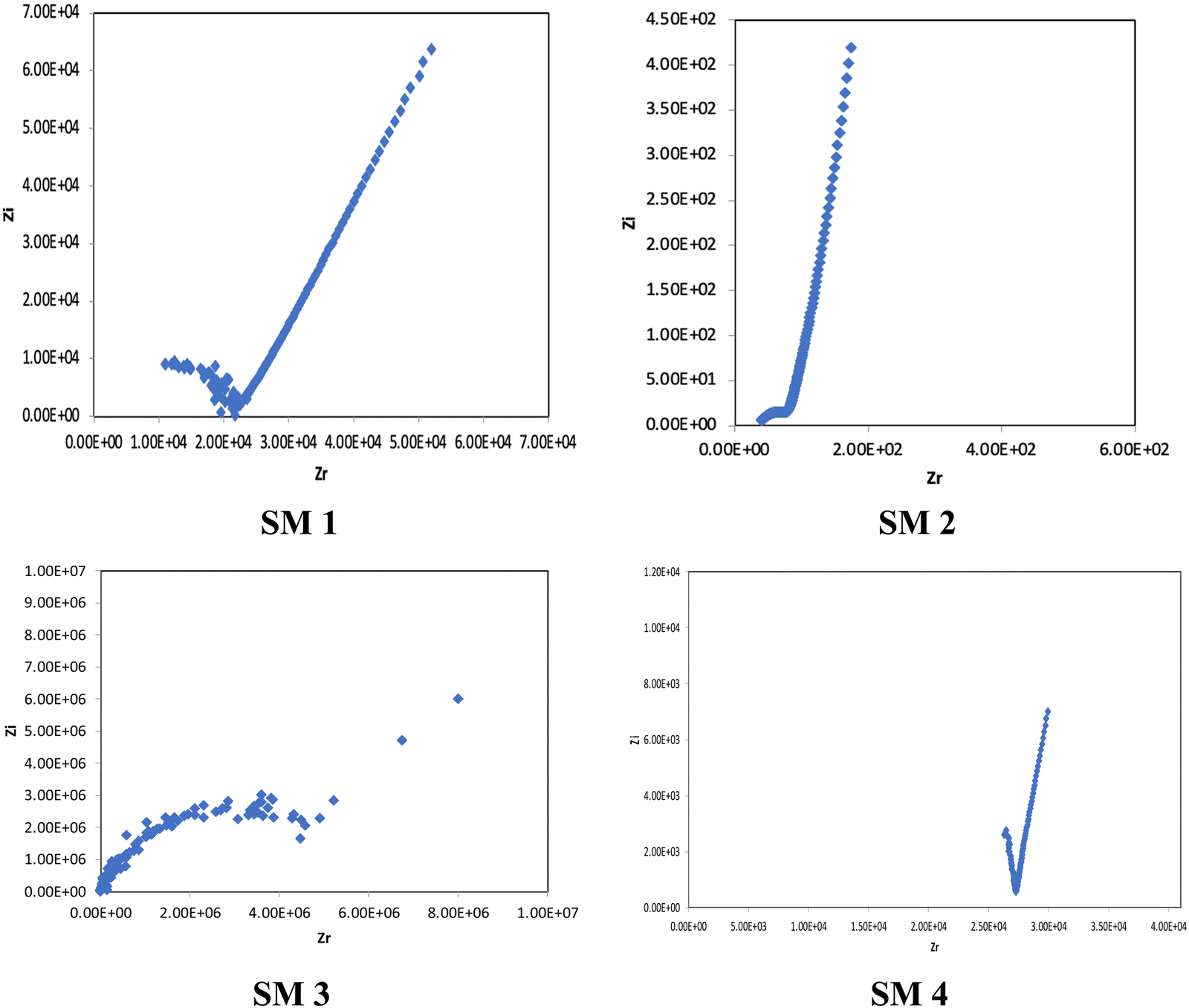 | ||
| Fig. 6 Cole–Cole plots of SM1, SM2, SM3 and SM4 doped with 15 wt% LiTFSI salt, respectively. | ||
In general, four types of phosphonium PILs containing LiTFSI salt (SM1, SM2, SM3 and SM4) were synthesized, demonstrating potential to be good candidates as polyelectrolytes for application in LIBs. The substituent group was varied with two classes, i.e., aromatic (SM1 and SM4) and aliphatic (SM2 and SM3) units. The aliphatic alkyl chains showed greater conductivity compared to the aromatic groups due to their better flexibility, and thus lower hindrance to the bond rotation.50
Aziz et al. reported that51 two main factors need to be considered when choosing a polymer host. One is that the backbone of the polymer must have polar functional groups with large electron-donating power and the other is that the polymer must possess low hindrance for bond rotation. It was observed that SM2, possessing an aliphatic tripropyl side chain group, exhibited the highest conductivity. Surprisingly, the polymer bearing an aromatic fluorophenyl side chain group, SM4, exhibited a higher conductivity value compared to the polymer possessing an aliphatic tertbutyl side chain, i.e., SM3. This is probably because the presence of fluoride groups in SM4 resulted in H bonding between the polymer matrix, which enhanced the molecular packing efficiency in the PIL backbone. Besides this, the steric effect cannot be ruled out as well. The tertbutyl side chains in SM3 may also cover and pack the anions, and thus decreased the conductivity. Previous research revealed that depending on the ion concentration, polyelectrolytes contain three separate ionic constituents, i.e., free ions, contact ion pairs, and ion aggregates.52 In this case, SM3 (possessing aliphatic tertbutyl side chain group) exhibited lower conductivity compared to SM4 (possessing aromatic fluorophenyl side chain group), which can be clearly attributed to the decrease in free ions given that the dissociation constant of the tertbutyl phosphine salt is lower than that of the tris(4-fluorphenyl) phosphine-based salt.53
To fully comprehend the potential ionic conduction process, the ionic conductivity was further studied at various temperatures in the range of 303 K to 353 K. Fig. 7(a) depicts the plot of conductivity as a function of temperature, demonstrating how the increase in temperature can increase the conductivity of the samples. This can be attributed to the ion mobility in the polyelectrolytes, which can be accelerated by an increase in temperature. Thus, an increase in temperature increased the conductivity by increasing the cation hoping phenomenon.54,55 The Arrhenius behaviour was confirmed by the conductivity trend, with the regression value (R2) getting closer to unity.
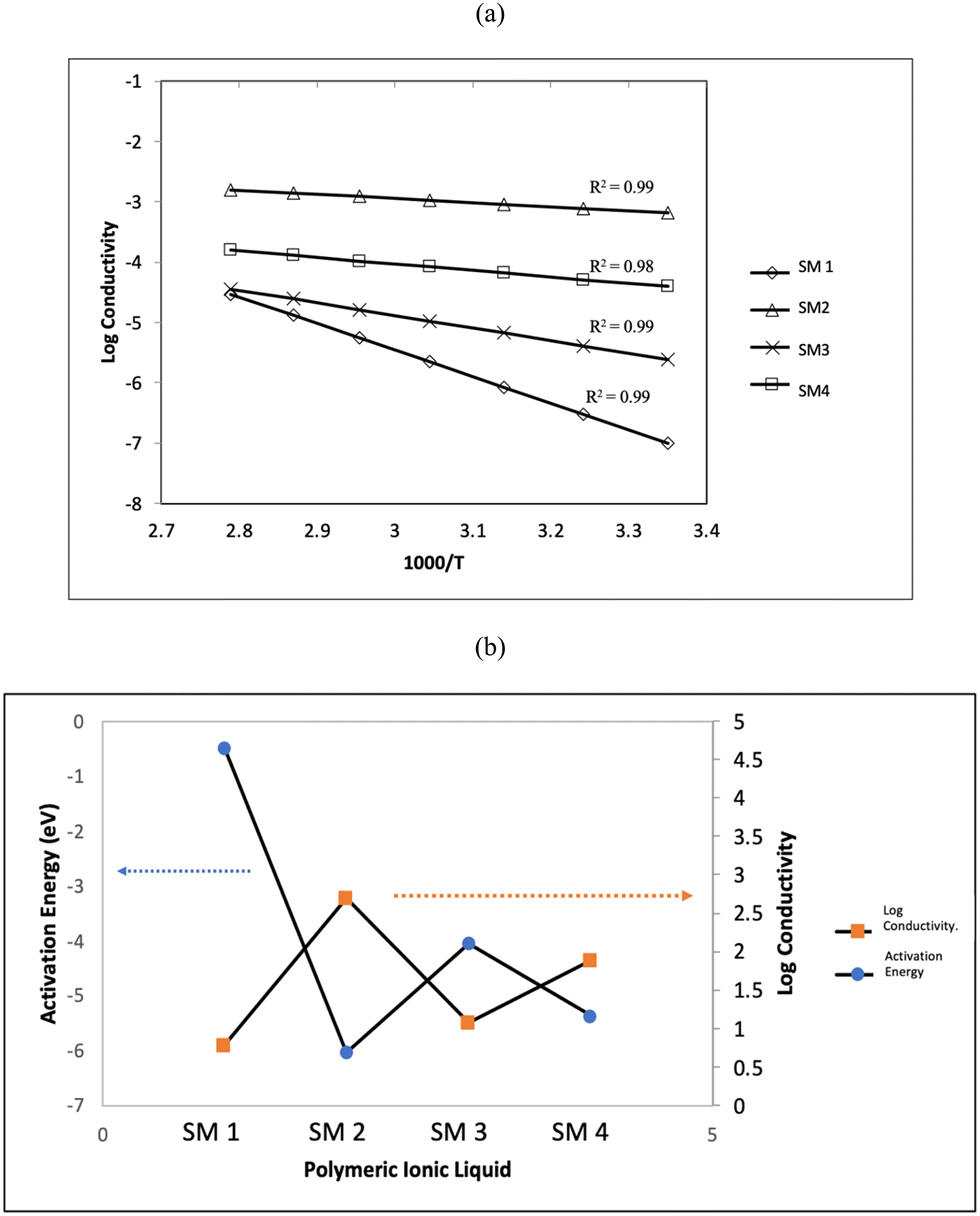 | ||
| Fig. 7 (a) Log conductivity versus 1000/T and (b) relationship between activation energy and conductivity at ambient. | ||
The activation energy, Ea, was calculated using the gradient of log conductivity versus 1000/T, as follows:
| σ = σ0exp(−Ea)/KT |
Fig. 7(b) illustrates the relationship between the activation energy and the conductivity at room temperature. It can be seen clearly from this graph that the relationship between activation energy and the ionic conductivity is inverse. According to the reported studies, a lower activation energy enables the faster segmental movement of the polymer backbone, resulting in a larger free volume.56 Consequently, this promotes the passage of ions through the host matrix, increasing the conductivity. It is evident that the sample with the lowest activation energy (SM2) possessed the highest conductivity. This indicates that the flexibility of the tripropyl substituent group enhanced the ionic conductivity, which requires less energy to move more ions through the backbone of the polymer blend. As discussed above, the lower Tg value of SM2 compared to the other polymers also contributed to its higher conductivity.
4. PEGylation of phosphonium PILs
Given that the SM2 sample showed the optimum conductivity, polyethylene glycol (PEG) units were attached to the SM2 backbone to investigate the conductivity values with an increase in the flexibility of the polymer backbone. PEGylation is one of the strategies to reduce the rigidity of the oxanorbornene polymer backbone due to the high flexibility of the PEG blocks.57 The SM2 samples were subjected to the PEGylation process via thiol–ene coupling using polyethylene glycol dithiol under 365 nm irradiation for 24 h, as depicted in Scheme 3.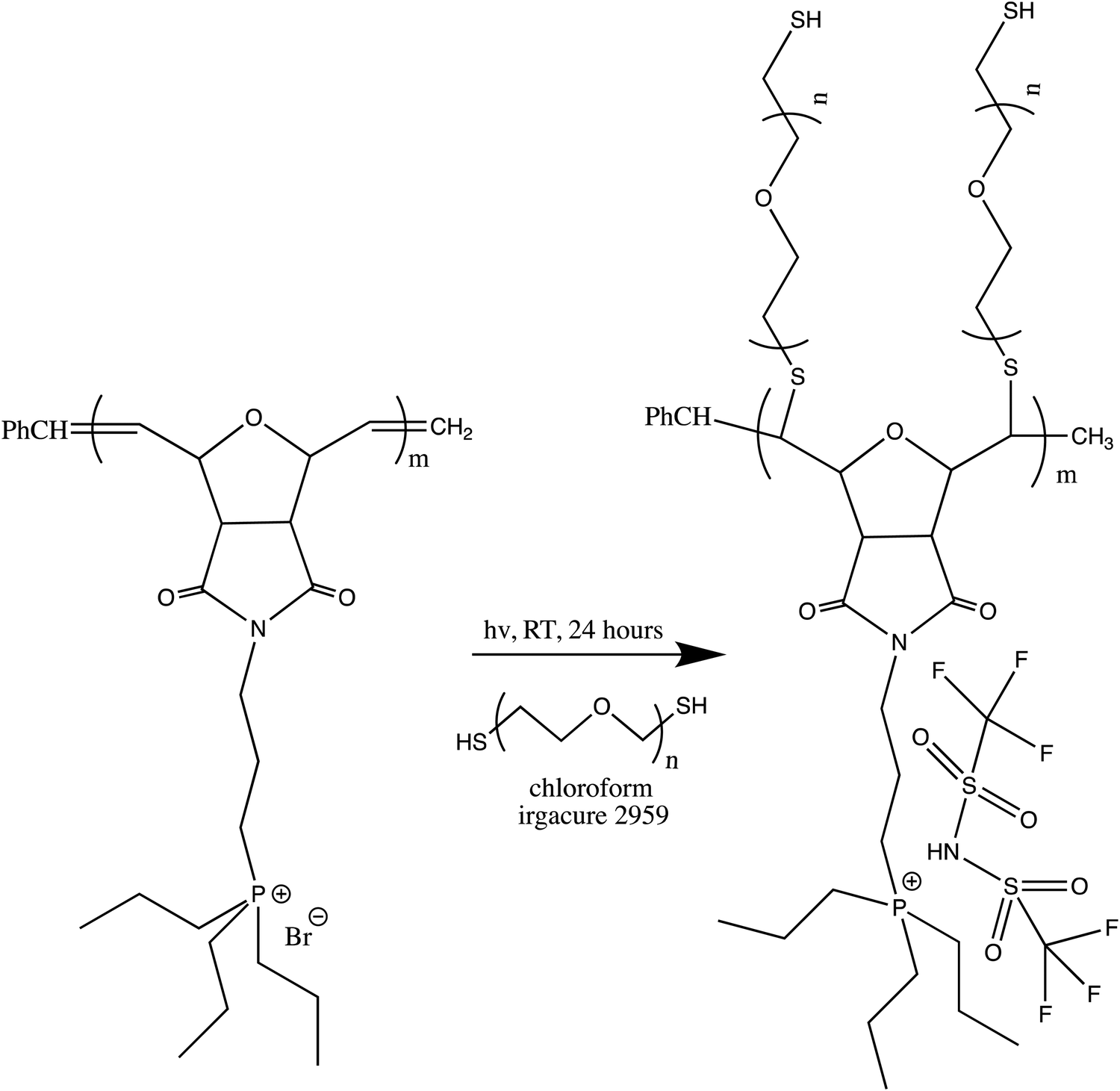 | ||
| Scheme 3 PEGylation process of SM2 sample. | ||
After dialysis of the product to remove the unsubstituted PEG units, a rubbery residue was obtained. Initially, SM2 was insoluble in water; however, after the PEGylation process, the polymer was found to be soluble in water. Additionally, the 1H NMR spectrum (Fig. 8) showed new peaks at 3.7 (l) and 2.7 (k) ppm, which are attributed to the protons in the PEG units.
 | ||
| Fig. 8 1H NMR spectrum of PEGylated SM2. | ||
The FTIR spectra of SM2, SM2-PEG and pure polyethylene glycol dithiol are depicted in Fig. S10 (ESI†). The peaks observed at 1690 cm−1 and 1342 cm−1 for SM2 and SM2–PEG are attributed to the CO stretching and aromatic imine C–N stretching, respectively. Additionally, in the PEGylated SM2 sample, a new peak was observed at 1095 cm−1, which is attributed to the stretching of the C–O group from polyethylene glycol dithiol.58 After PEGylation, the peaks attributed to C–H alkene stretching at 3000 cm−1 and CC stretching at 1638 cm−1 were found to disappear in the spectrum of SM2. Thus, it can be concluded that the polyethylene glycol dithiol reacted at the double bond of the SM2 backbone.
DSC analysis was also conducted to examine the impact of PEGylation on the thermal relaxation of SM2, showing a melting transition at 39 °C and 137 °C. Due to fact that the polymer molecular weight and PEG molecular weight were close, the PEG units were dominant as a result of the thiol–ene chemistry.
The Cole–Cole plot and conductivity value of the PEGylated SM2 and PEGylated SM2 doped with 15 wt% LiTFSI salt are presented in Table 2. The optimum conductivity for the PEGylated SM2 when doped with 15 wt% LiTFSI salt was 5.8 × 10−5 S cm−1 under ambient conditions. It was observed that this conductivity value was slightly lower than that of SM2. The conductivity seems to be dominated by the tendency of the high-molecular weight of the PEG unit in the overall polymer matrix to have a greater barrier to segmental motion of the chains, resulting in a decrease in conductivity.59 Additionally, the high molecular weight of PEG is typically correlated with less PEG-PIL phase boundary delineation, which is another element that influences the decrease in the conductivity value. It has been demonstrated that the transport characteristics of polymer electrolyte materials are adversely affected by poorly defined boundaries.60–64
| Sample | Cole–Cole plot | Conductivity value (S cm−1) |
|---|---|---|
| PEGylated SM2 |
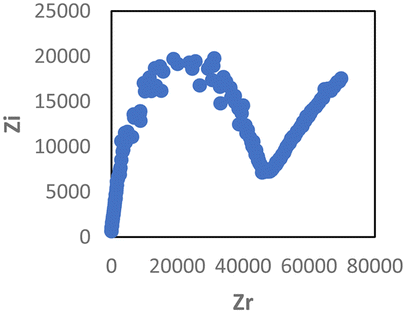
|
1.34 × 10−7 |
| PEGylated SM2 doped with 15 wt% LiTFSI |
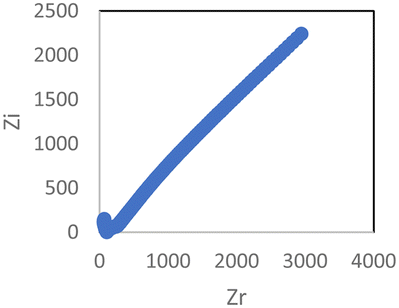
|
5.8 × 10−5 |
5. Conclusion
In this study, a series of polyoxanorbornene phosphonium PILs possessing various alkyl and aromatic substituents was synthesized and their counterions changed from the bromide anion to the trifluorosulfonylimide anion. Subsequently, the influence of the chemical structure of the polymers on the conductivity of the PILs was investigated using a controlled polymerization technique, i.e., ring-opening metathesis polymerization (ROMP). It was observed that the more flexible substituent group (SM 2–15 wt% LiTFSI) exhibited the highest conductivity value of 6.00 × 10−4 S cm−1 under ambient conditions. The rigid backbone of the nonaromatic groups was found to limit the conductivity value of the PILs. The introduction of PEG units in the polymer backbone of SM 2 resulted in a decrease in its conductivity. Nonetheless, continuing efforts to design flexible backbone polyethylene phosphorus ionic liquids are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge that this paper is submitted in partial fulfilment of the requirements for PhD degree at Yildiz Technical University. Thank you to Yildiz Technical University, Universiti Sains Islam Malaysia and National Defend University of Malaysia for the research facilities. One of the authors M. S. M. Misenan wishes to acknowledge Türkiye Scholarships (scholarship number: 19MY000187) for the scholarship given. This study was supported by the Yildiz Technical University (Istanbul, Turkey) Scientific Research Projects Coordinator project no: FBA 6111 Synthesis of Phosphonium based Polymer Electrolytes and Application in Lithium Ion Batteries. This study also partially supported by Ministry of Higher Education Malaysia under the Fundamental Research Grant Scheme under vote FRGS/1/2023/STG05/USIM/02/3.References
- G. G. Eshetu, D. Mecerreyes, M. Forsyth, H. Zhang and M. Armand, Polymeric ionic liquids for lithium-based rechargeable batteries, Mol. Syst. Des. Eng., 2019, 4(2), 294–309, 10.1039/c8me00103k.
- M. S. M. Misenan and A. S. A. Khiar, Conduction Mechanism of Chitosan/Methylcellulose/1-Butyl-3 Methyl Imidazolium Bis (Trifluoromethylsulfonyl) Imide (BMIMTFSI) Biopolymer Electrolyte Doped with Ammonium Triflate, Malays. J. Chem., 2020, 22(4), 1–13 CrossRef.
- M. S. M. Misenan, A. S. A. Khiar and T. Eren, Polyurethane-based polymer electrolyte for lithium ion batteries: a review, Polym. Int., 2022, 71, 751–769, DOI:10.1002/pi.6395.
- M. S. M. Misenan, R. Hempelmann, M. Gallei and T. Eren, Phosphonium-Based Polyelectrolytes: Preparation, Properties, and Usage in Lithium-Ion Batteries, Polymer, 2023, 15(13), 2920, DOI:10.3390/POLYM15132920.
- M. S. M. Misenan and A. S. A. Khiar, Structural Studies and Ionic Transport Properties of Solid Biopolymer Electrolytes Based on Chitosan/Methyl Cellulose Blend Doped with BMIMTFSI, Solid State Phenom., 2020, 307, 119–124, DOI:10.4028/www.scientific.net/SSP.307.119.
- A. H. Shaffie, M. S. M. Misenan, M. I. N. Isa and A. S. A. Khiar, Effect of ionic liquid bmimno3 to chitosan-starch blend biopolymer electrolyte system, 290 SSP. 2019.
- M. S. M. Misenan, E. S. Ali and A. S. A. Khiar, Conductivity, dielectric and modulus study of chitosan-methyl cellulose – BMIMTFSI polymer electrolyte doped with cellulose nano crystal, AIP Conf. Proc., 2018, 1972, 030010-1–030010-5, DOI:10.1063/1.5041231.
- D. Z. Zhang, Y. Yuan Ren, Y. Hu, L. Li and F. Yan, Ionic Liquid/Poly(ionic liquid)-based Semi-solid State Electrolytes for Lithium-ion Batteries, Chin. J. Polym. Sci., 2020, 38(5), 506–513, DOI:10.1007/s10118-020-2390-1.
- P. V. Wright, Electrical conductivity in ionic complexes of poly(ethylene oxide), Br. Polym. J., 1975, 7(5), 319–327, DOI:10.1002/pi.4980070505.
- A. Szczęsna-Chrzan, M. Marczewski, J. Syzdek, M. K. Kochaniec, M. Smoliński and M. Marcinek, Lithium polymer electrolytes for novel batteries application: the review perspective, Appl. Phys. A: Mater. Sci. Process., 2023, 129(1), 1–20, DOI:10.1007/s00339-022-06269-3.
- T. K. Lee, et al., PEO based polymer electrolyte comprised of epoxidized natural rubber material (ENR50) for Li-Ion polymer battery application, Electrochim. Acta, 2019, 316, 283–291, DOI:10.1016/j.electacta.2019.05.143.
- M. S. M. Misenan, A. H. Shaffie and A. S. A. Khiar, Effect of BMITFSI to the electrical properties of chitosan/methylcellulose based polymer electrolyte, AIP Conf. Proc., 2018, 1972, 030001-1–030001-6, DOI:10.1063/1.5041222.
- S. Noor, A. Ahmad, M. Rahman and I. Talib, Solid polymeric electrolyte of poly(ethylene) oxide-50% epoxidized natural rubber-lithium triflate, Nat. Sci., 2010, 2(3), 190–196 Search PubMed.
- R. Meziane, J. P. Bonnet, M. Courty, K. Djellab and M. Armand, Single-ion polymer electrolytes based on a delocalized polyanion for lithium batteries, Electrochim. Acta, 2011, 57(1), 14–19, DOI:10.1016/j.electacta.2011.03.074.
- Q. Ma, et al., Single lithium-ion conducting polymer electrolytes based on a super-delocalized polyanion, Angew. Chem., Int. Ed., 2016, 55(7), 2521–2525, DOI:10.1002/anie.201509299.
- H. I. E. Tsuchida, H. Ohno, N. Kobayashi and H. Ishizaka, Poly[(L-carboxy)oligo(oxyethylene) methacrylate] as a New Type of Polymeric Solid Electrolyte for Alkali-Metal Ion Transport, Macromolecules, 1989, 1775(4), 1771–1775 CrossRef.
- K. Ueno, et al., Li+ Solvation and Ionic Transport in Lithium Solvate Ionic Liquids Diluted by Molecular Solvents, J. Phys. Chem. C, 2016, 120(29), 15792–15802, DOI:10.1021/acs.jpcc.5b11642.
- H. Zhang, et al., Single lithium-ion conducting solid polymer electrolytes: advances and perspectives, Chem. Soc. Rev., 2017, 46(3), 797–815, 10.1039/C6CS00491A.
- D. R. MacFarlane, et al., Ionic liquids and their solid-state analogues as materials for energy generation and storage, Nat. Rev. Mater., 2016, 1(2), 1–15, DOI:10.1038/natrevmats.2015.5.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, Safer electrolytes for lithium-ion batteries: State of the art and perspectives, ChemSusChem, 2015, 8(13), 2154–2175, DOI:10.1002/cssc.201500284.
- M. R. Gao, J. Yuan and M. Antonietti, Ionic Liquids and Poly(ionic liquid)s for Morphosynthesis of Inorganic Materials, Chem. – Eur. J., 2017, 23(23), 5391–5403, DOI:10.1002/chem.201604191.
- D. Mecerreyes, Polymeric ionic liquids: Broadening the properties and applications of polyelectrolytes, Prog. Polym. Sci., 2011, 36(12), 1629–1648, DOI:10.1016/j.progpolymsci.2011.05.007.
- J. Yuan and M. Antonietti, Poly(ionic liquid)s: Polymers expanding classical property profiles, Polymer, 2011, 52(7), 1469–1482, DOI:10.1016/j.polymer.2011.01.043.
- N. Nishimura and H. Ohno, 15Th Anniversary of Polymerised Ionic Liquids, Polymer, 2014, 55(16), 3289–3297, DOI:10.1016/j.polymer.2014.02.042.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Recent Advances in Innovative Polymer Electrolytes based on Poly(ionic liquid)s, Electrochim. Acta, 2015, 175, 18–34, DOI:10.1016/j.electacta.2015.03.038.
- F. N. Ajjan et al. , Innovative polyelectrolytes/poly(ionic liquid) s for energy and the environment, 2017 DOI:10.1002/pi.5340.
- M. G. Cowan, M. Masuda, W. M. McDanel, Y. Kohno, D. L. Gin and R. D. Noble, Phosphonium-based poly(Ionic liquid) membranes: The effect of cation alkyl chain length on light gas separation properties and Ionic conductivity, J. Membr. Sci., 2016, 498, 408–413, DOI:10.1016/j.memsci.2015.10.019.
- A. Hofmann, D. Rauber, T. M. Wang, R. Hempelmann, C. W. M. Kay and T. Hanemann, Novel Phosphonium-Based Ionic Liquid Electrolytes for Battery Applications, Molecules, 2022, 27(15), 1–25, DOI:10.3390/molecules27154729.
- S.-W. Wang, W. Liu and R. H. Colby, Counterion Dynamics in Polyurethane-Carboxylate Ionomers with Ionic Liquid Counterions, Chem. Mater., 2011, 23(7), 1862–1873, DOI:10.1021/cm103548t.
- S. T. Hemp, M. Zhang, M. H. Allen, S. Cheng, R. B. Moore and T. E. Long, Comparing Ammonium and Phosphonium Polymerized Ionic Liquids: Thermal Analysis, Conductivity, and Morphology, Macromol. Chem. Phys., 2013, 214(18), 2099–2107, DOI:10.1002/macp.201300322.
- K. Tsunashima, E. Niwa, S. Kodama, M. Sugiya and Y. Ono, Thermal and Transport Properties of Ionic Liquids Based on Benzyl-Substituted Phosphonium Cations, J. Phys. Chem. B, 2009, 113(48), 15870–15874, DOI:10.1021/jp908356c.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruña and G. W. Coates, Phosphonium-Functionalized Polyethylene: A New Class of Base-Stable Alkaline Anion Exchange Membranes, J. Am. Chem. Soc., 2012, 134(44), 18161–18164, DOI:10.1021/ja307466s.
- A. Wang, et al., The synthesis of a hyperbranched star polymeric ionic liquid and its application in a polymer electrolyte, Polym. Chem., 2017, 8(20), 3177–3185, 10.1039/C7PY00499K.
- S. Cheng, et al., Ionic aggregation in random copolymers containing phosphonium ionic liquid monomers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(1), 166–173, DOI:10.1002/pola.25022.
- I. C. Çetinkaya, G. Yüksel, C. Macit, M. E. Üreyen and T. Eren, Synthesis and Characterization of Polyphosphonates and Polyurethanes Using Chalcone and DOPO-Chalcone as a Flame Retardant, ACS Appl. Polym. Mater., 2021, 3(10), 5277–5290, DOI:10.1021/acsapm.1c01054.
- Y. Men, H. Schlaad and J. Yuan, Cationic Poly(ionic liquid) with Tunable Lower Critical Solution Temperature-Type Phase Transition, ACS Macro Lett., 2013, 2(5), 456–459, DOI:10.1021/mz400155r.
- C. Demir, et al., Biocidal activity of ROMP- polymer coatings containing quaternary phosphonium groups, Prog. Org. Coat., 2019, 135, 299–305, DOI:10.1016/j.porgcoat.2019.06.008.
- P. Lv, S. Xie, Q. Sun, X. Chen and Y. He, Flame-Retardant Solid Polymer Electrolyte Based on Phosphorus-Containing Polyurethane Acrylate/Succinonitrile for Lithium-Ion Batteries, ACS Appl. Energy Mater., 2022, 5(6), 7199–7209, DOI:10.1021/acsaem.2c00751.
- J. Xie, et al., Three-in-one fire-retardant poly(phosphate)-based fast ion-conductor for all-solid-state lithium batteries, J. Energy Chem., 2023, 324–334, DOI:10.1016/j.jechem.2022.12.053.
- J. Gao, et al., A review of the recent developments in flame-retardant nylon composites, Compos., Part C: Open Access, 2022, 9, 100297, DOI:10.1016/j.jcomc.2022.100297.
- N. C. Süer, C. Demir, N. A. Ünübol, Ö. Yalçin, T. Kocagöz and T. Eren, Antimicrobial activities of phosphonium containing polynorbornenes, RSC Adv., 2016, 6(89), 86151–86157, 10.1039/c6ra15545f.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, A Practical and Highly Active Ruthenium-Based Catalyst that Effects the Cross Metathesis of Acrylonitrile, Angew. Chem., Int. Ed., 2002, 41(21), 4035–4037, DOI:10.1002/1521-3773(20021104)41:21<4035::AID-ANIE4035>3.0.CO;2-I.
- N. Ceren Süer, T. Arasoglu, H. Cankurtaran, M. Okutan, M. Gallei and T. Eren, Detection of bacteria using antimicrobial polymer derived via ring-opening metathesis (romp) pathway, Turk. J. Chem., 2021, 45(4), 986–1003, DOI:10.3906/kim-2012-14.
- B. W. Chieng, N. A. Ibrahim, W. M. Z. W. Yunus and M. Z. Hussein, Poly(lactic acid)/poly(ethylene glycol) polymer nanocomposites: Effects of graphene nanoplatelets, Polymers, 2014, 6(1), 93–104, DOI:10.3390/polym6010093.
- Z. Ullah, M. Azmi Bustam, Z. Man and A. S. Khan, Phosphonium-based ionic liquids and their application in separation of dye from aqueous solution, ARPN J. Eng. Appl. Sci., 2016, 11(3), 1653–1659 CAS.
- I. Rey, P. Johansson, J. Lindgren and J. C. Lasse, Spectroscopic and Theoretical Study of (CF3SO2)2N-(TFSI-) and (CF3SO2)2 NH (HTFSI), J. Phys. Chem. A, 1998, 102(98), 3249–3258 CrossRef CAS.
- M. S. M. Misenan, M. I. N. Isa and A. S. A. Khiar, Electrical and structural studies of polymer electrolyte based on chitosan/methyl cellulose blend doped with BMIMTFSI, Mater. Res. Express, 2018, 5(5), 55304 CrossRef , [Online]. Available: https://stacks.iop.org/2053-1591/5/i=5/a=055304.
- S. Misenan, A. Sofia Ahmad Khiar, M. S. M. Misenan and A. S. A. Khiar, Conductivity, Dielectric And Modulus Studies of Methylcellulose-NH4TF Polymer Electrolyte, Chem. Sci., 2018, 1(2), 59–62 Search PubMed , [Online]. Available: https://www.dergipark.gov.tr/ejbcs.
- J. Kalhoff, D. Bresser, M. Bolloli, F. Alloin, J. Y. Sanchez and S. Passerini, Enabling LiTFSI-based electrolytes for safer lithium-ion batteries by Using linear fluorinated carbonates as (Co)solvent, ChemSusChem, 2014, 7(10), 2939–2946, DOI:10.1002/cssc.201402502.
- N. Soszka, et al., Aromaticity effect on supramolecular aggregation. Aromatic vs. cyclic monohydroxy alcohols, Spectrochim. Acta, Part A, 2022, 276, 1–13, DOI:10.1016/j.saa.2022.121235.
- S. B. Aziz and Z. H. Z. Abidin, Electrical Conduction Mechanism in Solid Polymer Electrolytes: New Concepts to Arrhenius Equation, J. Soft Matter, 2013, 2013, 1–8, DOI:10.1155/2013/323868.
- S. B. Aziz, R. M. Abdullah, M. A. Rasheed and H. M. Ahmed, Role of ion dissociation on DC conductivity and silver nanoparticle formation in PVA:AgNt based polymer electrolytes: deep insights to ion transport mechanism, Polymers, 2017, 9(8), 1–19, DOI:10.3390/polym9080338.
- W. A. Henderson and C. A. Streuli, The Basicity of Phosphines, J. Am. Chem. Soc., 1960, 82(22), 5791–5794, DOI:10.1021/ja01507a008.
- M. I. H. A. Sohaimy and M. I. N. M. Isa, Natural inspired carboxymethyl cellulose (Cmc) doped with ammonium carbonate (ac) as biopolymer electrolyte, Polymers, 2020, 12(11), 1–14, DOI:10.3390/polym12112487.
- M. I. H. A. Sohaimy, N. I. Zainuddin and M. I. N. M. Isa, Plasticized Cmc-Ammonium Acetate Based Solid Biopolymer Electrolyte: Ionic Conductivity and Transport Study, J. Sustainability Sci. Manage., 2022, 17(5), 139–148, DOI:10.46754/jssm.2022.05.011.
- H. T. Ahmed, V. J. Jalal, D. A. Tahir, A. H. Mohamad and O. G. Abdullah, Effect of PEG as a plasticizer on the electrical and optical properties of polymer blend electrolyte MC-CH-LiBF4 based films, Results Phys., 2019, 15, 1–7, DOI:10.1016/j.rinp.2019.102735.
- Y. Baimark, W. Rungseesantivanon and N. Prakymoramas, Synthesis of flexible poly(L-lactide)-b-polyethylene glycol-b-poly(L-lactide) bioplastics by ring – opening polymerization in the presence of chain extender, e-Polymers, 2020, 423–429 CrossRef CAS.
- W. Guan, F. Ji, Q. Chen, P. Yan and L. Pei, Synthesis and enhanced phosphate recovery property of porous calcium silicate hydrate using polyethyleneglycol as pore-generation agent, Materials, 2013, 6(7), 2846–2861, DOI:10.3390/ma6072846.
- E. Coletta, M. F. Toney and C. W. Frank, Impacts of polymer–polymer interactions and interfaces on the structure and conductivity of PEG-containing polyimides doped with ionic liquid, Polymer, 2014, 55(26), 6883–6895, DOI:10.1016/j.polymer.2014.10.075.
- K. Okamoto, M. Fuji, S. Okamyo, H. Suzuki, K. Tanaka and H. Kita, Gas permeation properties of poly(ether imide) segmented copolymers, Macromolecules, 1995, 28(20), 6950–6956 CrossRef CAS.
- K. Okamoto, N. Umeo, S. Okamyo, K. Tanaka and H. Kita, Selective permeation of carbon dioxide over nitrogen through polyethyleneoxide-containing polyimide membranes, Chem. Lett., 1993,(2), 225–228 CrossRef CAS.
- Y. N. Lazareva, M. N. Vidyakin, A. Y. Alentiev, M. Y. Yablokova, A. A. Kuznetsov and I. A. Ronova, Transport properties of polyimides derived from benzophenonetetracarboxylic dianhydride and other diamines, Polym. Sci., Ser. A, 2009, 51, 1068–1074 CrossRef.
- B. T. Low, Y. Xiao and T. S. Chung, Amplifying the molecular sieving capability of polyimide membranes via coupling of diamine networking and molecular architecture, Polymer, 2009, 50(14), 3250–3258 CrossRef CAS.
- T. Nakano, S. Nagaoka and H. Kawakami, Preparation of novel sulfonated block copolyimides for proton conductivity membranes, Polym. Adv. Technol., 2005, 16(10), 753–757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00630a |
| This journal is © The Royal Society of Chemistry 2024 |