
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilities of bis(thienyl)ethenes in polymethyl methacrylate (PMMA) coatings as absorbance modulation layers for nanoscale imaging†
Sven
Nagorny
a,
Marvin
Schewe
 b,
Thea
Weingartz
a,
André
Eitzeroth
c,
Jörg
Adams
c,
Christian
Rembe
b and
Andreas
Schmidt
*a
b,
Thea
Weingartz
a,
André
Eitzeroth
c,
Jörg
Adams
c,
Christian
Rembe
b and
Andreas
Schmidt
*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de
bClausthal University of Technology, Institute for Electrical Information Technology, Leibnizstraße 28, D-38678 Clausthal-Zellerfeld, Germany
cClausthal University of Technology, Institute of Physical Chemistry, Arnold-Sommerfeld-Straße 4, D-38678 Clausthal-Zellerfeld, Germany. E-mail: adams@pc.tu-clausthal.de
First published on 15th November 2023
Abstract
For absorption modulation layers for nanoscale imaging, a series of photochromic 1,2-bis(thienyl)ethenes (BTEs) was prepared, spectroscopically characterized in solution, and fatigue resistancies in PMMA films were investigated. The BTEs examined possess hydrogen or methyl groups in the β-position (C4/C4′) of the thiophene wings. The non-symmetrically substituted BTEs studied are substituted at the C5/C5′-positions of the thiophenes with combinations of pyridin-3-yl, pyridin-4-yl, 3-PhOMe, 4-PhOMe, 3-PhOH, and 4-PhOH groups. The substitution pattern of the symmetric BTEs studied was varied over a broad range (Ph, 4-tol, 4-PhOMe, 4-PhOH, pyridin-4-yl, 5-Me-thiophen-2-yl, 6-MeO-naphthalen-2-yl). In addition, a benzothiophene derivative was examined. It was shown that the stabilities in PMMA films cannot be derived from results in solution. Thus, the β-methylated BTEs proved to be less stable in PMMA films than their non-methylated counterparts. Best fatigue resistancies were obtained when a BTE with cross-conjugated 3,3-connectivity of a pyridin-3-yl at C5 and a 3-methoxyphenyl group at C5′ without methyl groups at C4/C4′ was applied. Substitution by 4-anisyl groups at C5/C5′ gave the best fatigue resistancies of the series of symmetrically substituted BTEs. The benzothiophene derivative proved to be less stable in PMMA films than its thiophene analog.
Introduction
By the absorption of light, photochromic molecules can reversibly be transformed between two forms which differ in their optical and spectroscopic properties.1 Photochromic molecules encompass a variety of different structures, and consequently, their photochromism is accomplished by several distinct mechanisms. Thus, switchable molecules possessing the 5-(iminio)penta-1,3-dien-1-olate chromophor such 2-(indolium-2-yl)vinyl)phenolates 1 (open) undergo reversible ring-closure reactions to form 6-oxa-1-azaspiro[4.5]decadienes 1 (closed) (Scheme 1).2Cis–trans isomerism cause the photochromism of azobenzenes 2,3 and light-induced redox reactions form radical cations and radical anions of photochromic punicines 3.4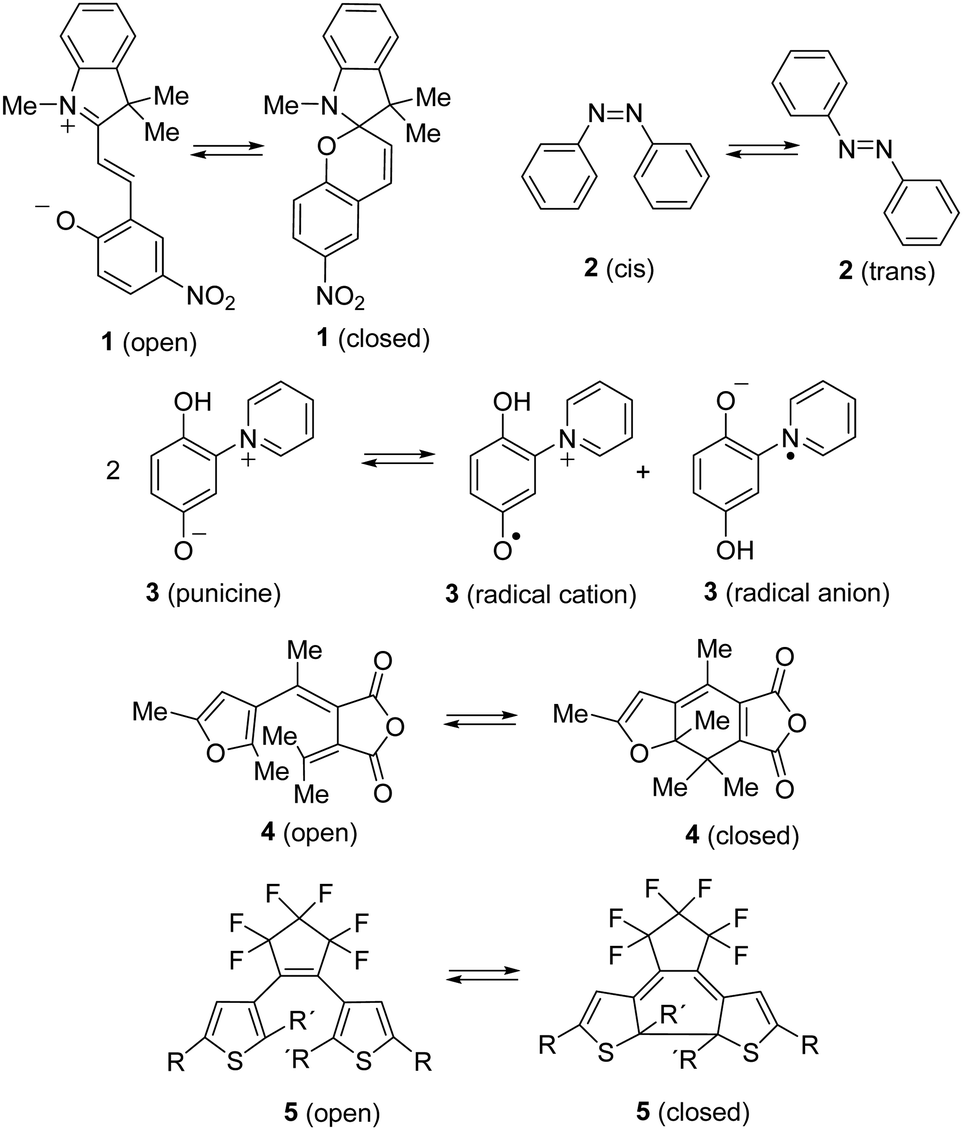 | ||
| Scheme 1 Different types of photochromic molecules. | ||
By contrast, 6π-electrocyclizations of 1,3,5-triene moities induce the photochromism of the 3-(1-(furan-3-yl)ethylidene)-4-(propan-2-ylidene)dihydrofuran-2,5-dione 45 or diarylethenes (DAEs) and their subclass of bis(thienyl)ethenes (BTEs) such as 5.1 The latter mentioned class of switchable molecules is well known for more than three decades and is still further investigated due to its high thermal stability, good fatigue resistance and fast response upon irradiation. They are promising candidates for the application as optical storage media,6 as building block of organic photodetectors,7 or to conduct electrical charges when the switchable molecule is part of a polymer structure like PEDOT,8 among others. Recently, we reported on linear free enthalpy relationships on applying the spectroscopic Hammett–Brown equation to correlate substituent effects and absorption maxima of BTEs.9 In addition, we described BTEs which are part of prolonged push–pull-chromophors to induce bathochromic absorption maxima,10 and BTEs with α/α′-MOM groups.11 According to the literature several aspects should be considered on designing fatigue resistant BTEs for technical applications. Thus, it was reported that aromatic substituents which enlarge the π-electronic system increase the stabilities.12 Functional groups such as SF5 or CF3 were identified as stabilizing substituents,13 and the same is true for additional methyl groups in the β-positions of the thiophene units.14 In addition, BTEs possessing benzothiophene moieties are considered more stable than BTEs with thiophene rings.15Fig. 1 shows the stability of the aforementioned BTE-cores as determined by experiments in solution.
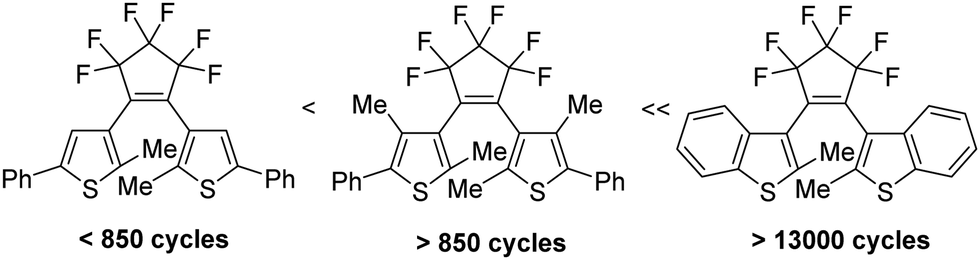 | ||
| Fig. 1 Fatigue resistance of different BTE backbones in solution.13 | ||
As part of an ongoing project dealing with nanoscale imaging techniques based on the RESOLFT principle16 we became interested in these molecules. This technique is derived from the STED microscopy17 and aims at breaking the diffraction limit of a confocal reflection microscope. For this purpose homogenous films containing bis(thienyl)ethenes as photochromic molecules are coated onto the surface to be examined. In absorbance modulation imaging (AMI) the homogenous film, the so-called absorbance modulation layer (AML), is then irradiated with a doughnut shaped UV-beam to convert the BTE molecules into their closed, intensely colored forms (Fig. 2). HeNe-lasers or others are used for the data acquisition as the laser hits the sample through the small transparent area of the doughnut. As the measurement laser is subject to the diffraction limit themselves, data can only be obtained within a short period of time before the BTEs undergo conversions into their open forms and the film becomes transparent again. A sufficient number of switching cycles of the BTE film, at least in the diameter range of the used lasers, is therefore mandatory to achieve efficient data acquisitions. In first attempts, we were able to increase the diffraction limit by the factor of 2.418 and thus to verify theoretical predictions.19 For these studies we used BTE 12 from the literature.20
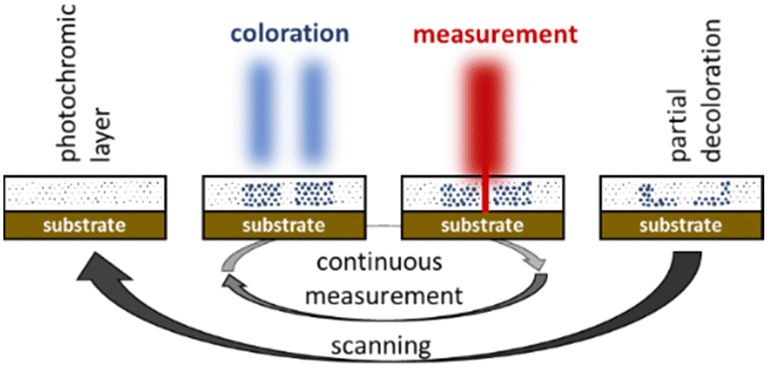 | ||
| Fig. 2 Nanoscale imaging techniques based on the RESOLFT principle. | ||
In order to improve the efficiency of this nanoscale imaging technique by identification of the most suitable photochromic dye, we studied the behaviour of a series of new bis(thienyl)ethenes in solution as well as in polymethyl methacrylate (PMMA) films. Syntheses and results of spectroscopic investigations are presented here. We will show that stabilities in PMMA films cannot be derived from results in solution.
Results and discussion
For our investigations, we selected a series of BTEs as illustrated in Fig. 3. We prepared both non-symmetric (6a–11a; 6b–11b) and symmetric BTEs (12a–g; 13a–e), varying the β-positions of the thiophene wings between unsubstituted and methylated forms to investigate the influence of the latter group on spectroscopic properties and stabilities in PMMA films.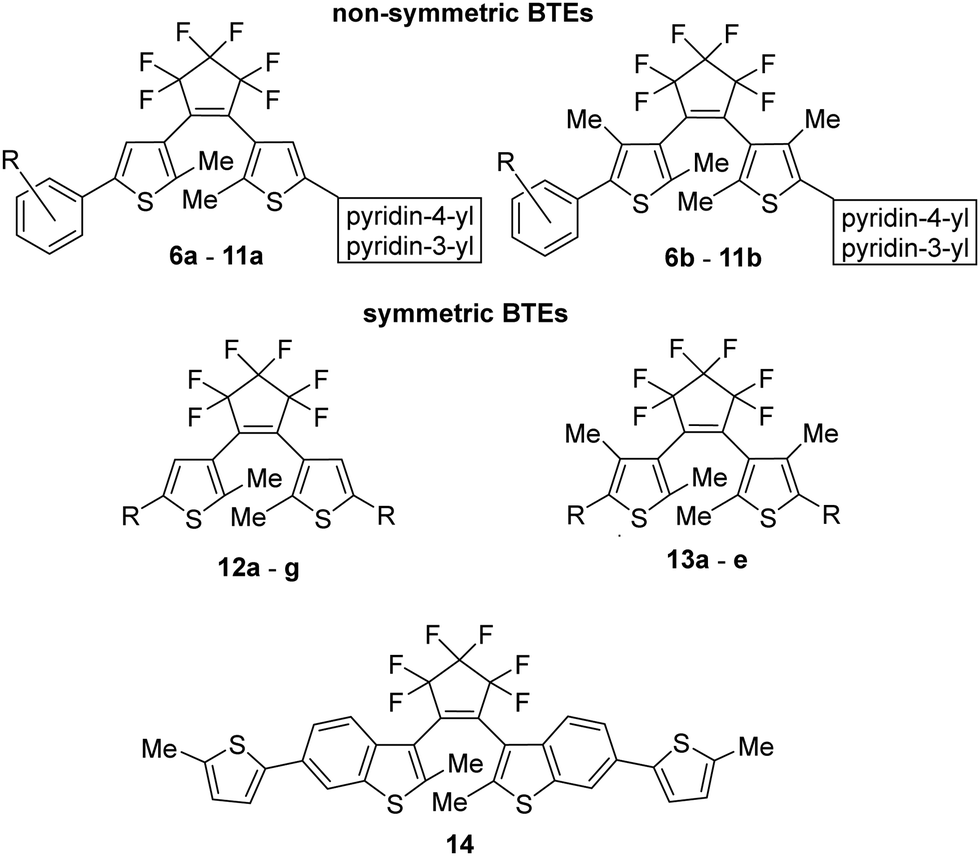 | ||
| Fig. 3 Target molecules for the studies presented here. | ||
With the exception of 13b and 13c,27 all β-methylated species are new compounds, whereas BTEs 12a,2912b,1312d,30 and 12g27 have been previously documented. The non-symmetric BTEs feature either pyridin-4-yl or pyridin-3-yl substituents, exhibiting intriguing spectroscopic characteristics such as bathochromic shifts of the absorption maxima.10 To provide a basis for comparison, we also included the 2,2′-bithiophene derivative 12e, which served previously as a reference BTE,18,19 a newly synthesized naphthyl-substituted BTE 12f to explore the impact of an extended π-electron system on properties, and an hitherto unknown benzothiophene derivative 14 in our investigations. The latter compound serves as the benzothiophene counterpart to the reference BTE 12e.
To assess the influence of the β-methyl group of the thiophene moieties on the properties of the non-symmetric BTEs, we synthesized the molecules 6b–11b maintaining the same substitution pattern as described earlier for 6a–11a.10 We started the syntheses from the boronic acid 15, following procedures reported previously21 and our own work10 (Scheme 2).
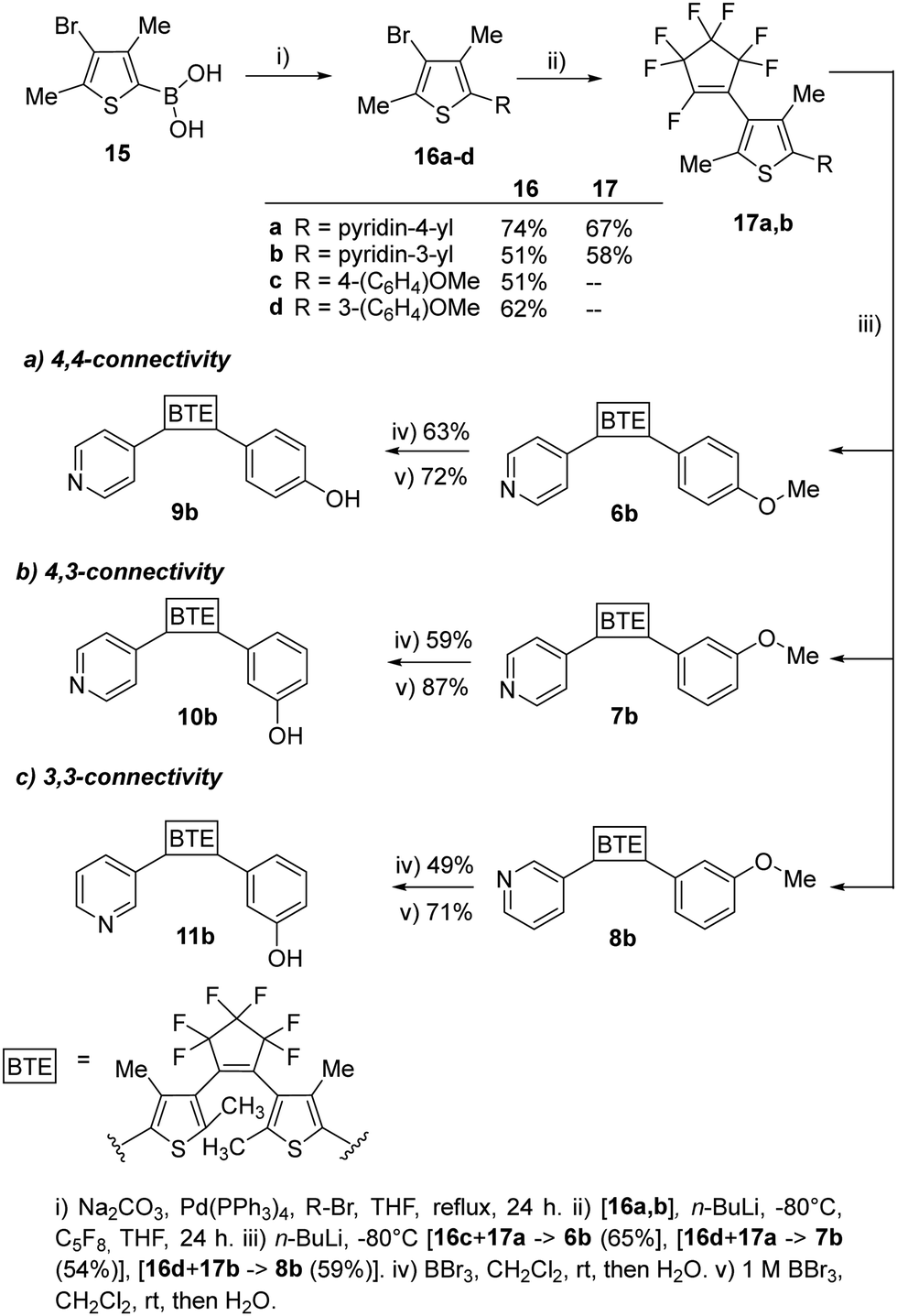 | ||
| Scheme 2 Synthesis of the BTEs 6b–11b, possessing the same substitution pattern as the previously reported BTEs 6a–11b, except for their β-methyl groups. | ||
To increase the yields of the Suzuki–Miyaura reactions (method i), we degassed both THF and bidistilled water before addition of the Pd-catalyst and obtained 16a–d in acceptable yields. The reactions with octafluorocyclopentene to give 17a,b (method ii) as well as 6b–8b (method iii) were optimized by using an ethylacetate-liquid nitrogen bath. Nitrogen was added until the ethylacetate solidified. The yields of those reactions also increased by using a septum to seal the reaction flask during the reaction so that 54% of 6b, 54% of 7b, and 61% of 8b were obtained as colorless foams, respectively. For demethylating the methoxy groups of the BTEs 6b–8b to yield the phenols 9b–11b, borontribromide was used. It was observed that the reaction outcomes were optimized when a 1 M BBr3 solution in dichloromethane was used instead of pure BBr3. This was due to more accurate dosing, as less BBr3 could react with the moisture in the surrounding air, resulting in yields ranging from 71% (11b) and 87% (10b) of the target BTEs.
Quantum yields were determined following the method outlined by Maafi et al.22a,b Solutions of the respective BTEs were irradiated at the wavelength of their highest wavelength isosbestic points. For instance, BTE 6a was irradiated at λisos = 325.6 nm (Fig. 4). The calculation of quantum yields then required only information such as molecular weight, the concentration of the solution, isosbestic point wavelength, absorbance at this point as well as the number of photons per cm2. Details are summarized in the ESI.†
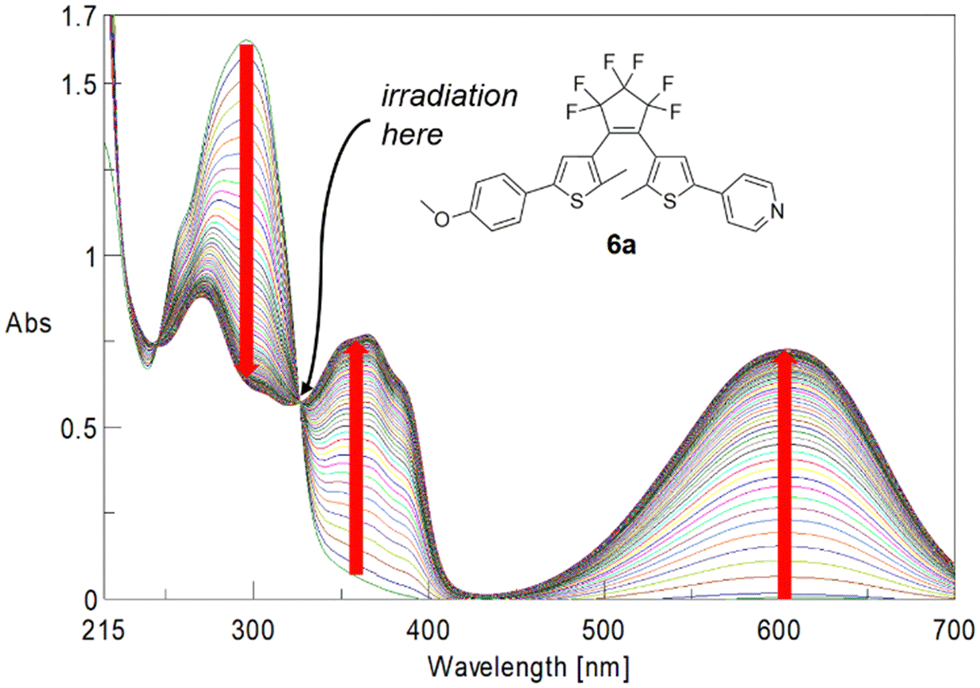 | ||
| Fig. 4 Time-dependent UV-spectra of BTE 6a. | ||
For the ring opening reaction, the BTEs were irradiated at 635 nm with an LED lamp until the BTEs were completely converted into their open forms. A program provided by Stranius and Börjesson was used to determine the quantum yields of the cycloreversion reaction.22c
The measured quantum yields of the BTEs 6a–11b are summarized in Table 1. Due to the steric hindrance caused by the β-methyl group, the BTEs 6b–11b have lower quantum yields than their unsubstituted derivatives 6a–11a.
| BTE | λ max [nm] (ε/103[M−1 cm−1]) | λ isos [nm] | φ isosAB [%] | φ 635nmBA [%] | x 50 | |
|---|---|---|---|---|---|---|
| Open form | Closed form | |||||
| All spectra were measured in THF, for ring-opening a 3 Watt LED (630–640 nm, Bridgelux/Epistar) was used. For the solution measurements light intensity was measured with a PM100D powermeter from THORLabs. | ||||||
| 6a | 296.6 (35.9) | 604.0 (17.7) | 325.6 | 62.2 | 0.42 | 133 |
| 6b | 279.6 (23.8) | 581.8 (9.6) | 313.4 | 44.3 | 0.71 | 20 |
| 7a | 296.6 (30.2) | 595.4 (13.8) | 322.0 | 55.4 | 0.72 | 100 |
| 7b | 273.4 (23.7) | 575.4 (8.7) | 313.4 | 34.9 | 1.21 | 26 |
| 8a | 277.6 (36.5) | 588.6 (13.8) | 316.6 | 47.0 | 0.56 | 158 |
| 8b | 269.2 (28.1) | 574.8 (9.8) | 303.2 | 36.7 | 1.37 | 41 |
| 9a | 292.0 (26.6) | 604.4 (13.5) | 327.0 | 76.4 | 0.52 | 111 |
| 9b | 280.2 (29.4) | 583.2 (12.5) | 314.4 | 34.3 | 0.69 | 25 |
| 10a | 296.4 (38.4) | 593.6 (14.3) | 323.3 | 45.7 | 0.61 | 97 |
| 10b | 273.3 (21.0) | 576.4 (8.9) | 313.0 | 40.6 | 1.39 | 21 |
| 11a | 295.6 (23.4) | 590.2 (9.2) | 318.4 | 50.3 | 0.90 | 53 |
| 11b | 286.6 (23.3) | 574.0 (9.5) | 303.8 | 35.2 | 2.00 | 49 |
This outcome aligns with expectations, as fewer molecules are capable of adopting the antiparallel form, which is the only form conducive to undergoing the 6π-electrocyclization. For the same reasons, the quantum yields for cycloreversion are also slightly higher for the β-methylated species. The presence of a methyl group in the β-position also leads to a hypsochromic shift, attributed to the positive inductive effect of the alkyl group [σp(Me) = −0.17].23 Additionally, it reduces the absorbance of the BTEs in solution.
Thin films of the BTEs were prepared starting from a PMMA-anisole solution with a concentration of 180 mg mL−1. A sample of 50 μL of this solution was mixed with 3 mg of the corresponding BTEs until the dye was completely dissolved. This corresponds to a 24 wt% solution of BTE/PMMA with a ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75. Using a spin coater, 40 μL of this solution were coated on a sapphire glass plate (∅ = 10 mm) with a rotation speed of 2500 rpm for 60 s, respectively. This process yielded homogenously coated films with a thickness of 1.6 μm, measured using a confocal laser scanning microscope (CLSM). In the ESI,† several CLSM images of BTE 6a are provided as examples, illustrating the absence of aggregation or crystallization using this technique. Another image shows a magnified degraded spot from the cyclization experiment, which was bleached by laser treatment, confirming the absence of phase separation. To quantify the number of cycles of the BTE-PMMA films, a setup employing two aligned lasers was used. The setup is shown in the ESI.† For the 6π-electrocyclization forming the ring-closed isomers, a HeCd laser (325 nm) was employed. For cycloreversion, two different lasers were used. To examine the β-unsubstituted BTEs, a HeNe laser (633 nm) was used, whereas a yellow laser was used (560 nm, CW Fiber-Laser) to study the β-methyl-substituted BTEs, as they absorb at shorter wavelengths. To avoid overexposing of the sensor during the ring opening reaction, data were only measured during the switching reaction into the closed form, i.e. the opaque state. To measure the switching cycles, the laser for the cycloreversion (at 560 nm or 633 nm respectively) was also activated during the switching reaction and acted as the readout laser. However, the intensity was set sufficiently low to make backswitching reactions negligible. An edge filter blocked the UV-light, ensuring that only the light from the readout laser was detected by the sensor. To avoid irradiating the samples beyond their photostationary states (PSS), the irradiation time was adjusted individually for each individual BTE. To determine the exposure time to reach the PSS, one spot was irradiated well beyond the PSS for each sample. For accurate measurement of the number of cycles, a new spot on the film was selected, and the irradiation time was set to 90% before the PSS was reached. Additionally, a value denoted as x50 was defined for comparing different BTEs (cf.Table 1). This value represents a 50% decomposition of the original absorption after a specific number of cycles. The absorbance at the start is based on the difference between the fully opened and the closed form in the first cycle of the cyclization shown in Fig. 5. Initially, the absorption is slightly above 75%, or about 0.65. In the plot below, this has been normalized to 1.
:75. Using a spin coater, 40 μL of this solution were coated on a sapphire glass plate (∅ = 10 mm) with a rotation speed of 2500 rpm for 60 s, respectively. This process yielded homogenously coated films with a thickness of 1.6 μm, measured using a confocal laser scanning microscope (CLSM). In the ESI,† several CLSM images of BTE 6a are provided as examples, illustrating the absence of aggregation or crystallization using this technique. Another image shows a magnified degraded spot from the cyclization experiment, which was bleached by laser treatment, confirming the absence of phase separation. To quantify the number of cycles of the BTE-PMMA films, a setup employing two aligned lasers was used. The setup is shown in the ESI.† For the 6π-electrocyclization forming the ring-closed isomers, a HeCd laser (325 nm) was employed. For cycloreversion, two different lasers were used. To examine the β-unsubstituted BTEs, a HeNe laser (633 nm) was used, whereas a yellow laser was used (560 nm, CW Fiber-Laser) to study the β-methyl-substituted BTEs, as they absorb at shorter wavelengths. To avoid overexposing of the sensor during the ring opening reaction, data were only measured during the switching reaction into the closed form, i.e. the opaque state. To measure the switching cycles, the laser for the cycloreversion (at 560 nm or 633 nm respectively) was also activated during the switching reaction and acted as the readout laser. However, the intensity was set sufficiently low to make backswitching reactions negligible. An edge filter blocked the UV-light, ensuring that only the light from the readout laser was detected by the sensor. To avoid irradiating the samples beyond their photostationary states (PSS), the irradiation time was adjusted individually for each individual BTE. To determine the exposure time to reach the PSS, one spot was irradiated well beyond the PSS for each sample. For accurate measurement of the number of cycles, a new spot on the film was selected, and the irradiation time was set to 90% before the PSS was reached. Additionally, a value denoted as x50 was defined for comparing different BTEs (cf.Table 1). This value represents a 50% decomposition of the original absorption after a specific number of cycles. The absorbance at the start is based on the difference between the fully opened and the closed form in the first cycle of the cyclization shown in Fig. 5. Initially, the absorption is slightly above 75%, or about 0.65. In the plot below, this has been normalized to 1.
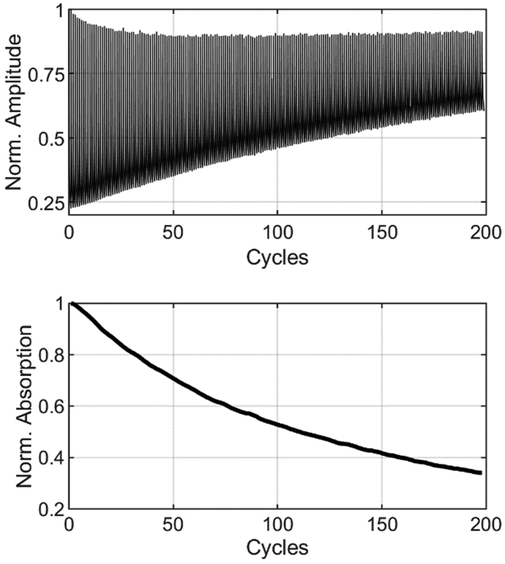 | ||
| Fig. 5 Determination of x50 of BTE 6a in a PMMA film. Amplitudes as function of number of cycles (above), normalized absorption derived from the amplitudes by logarithmizing (below). | ||
The x50 value indicates the point at which 50% of the original absorption is attained. Control experiments, involving both lasers mentioned above for the ring-opening reaction, yielded similar x50 results and only slightly different absorbances.
Interestingly, the β-methylated BTEs proved to be less stable in PMMA films than their non-methylated counterparts. As indicated by the x50 values, they endure a significant lower number of switching cycles without decomposition in comparison to their non-methylated analogs. Among the BTEs examined in this study, only derivatives 6a–8a were able to undergo more than 100 cycles, with the highest number achieved by derivative 8a. The latter possesses a 3,3-connectivity, i.e. a cross-conjugated substitution pattern with restricted conjugation on the central BTE-core. Obviously, results regarding the fatigue resistancies of BTEs in solution (cf.Fig. 1) differ from those obtained in PMMA films.
To evaluate these results even further, symmetric BTEs without (12a–g) and with β-methyl group (13a–e) have been synthesized (Scheme 3). In addition, we prepared two BTEs with 2,2′-bithiophene moieties (12e) and 2-(naphthalen-2-yl)thiophene substituent (12f). In view of their potential application in nanoscopy, substituents were chosen which are known to result in high absorbances of the closed form.24 The syntheses of the non-methylated BTEs are shown in Scheme 3. The reactions were conducted under similar conditions as for the non-symmetric BTEs shown above, however, the species of the boronic acid and the halide were changed. Of course, the amounts of octafluorocyclopentene were adjusted to form the symmetric species, however, only quite low yields were obtained.
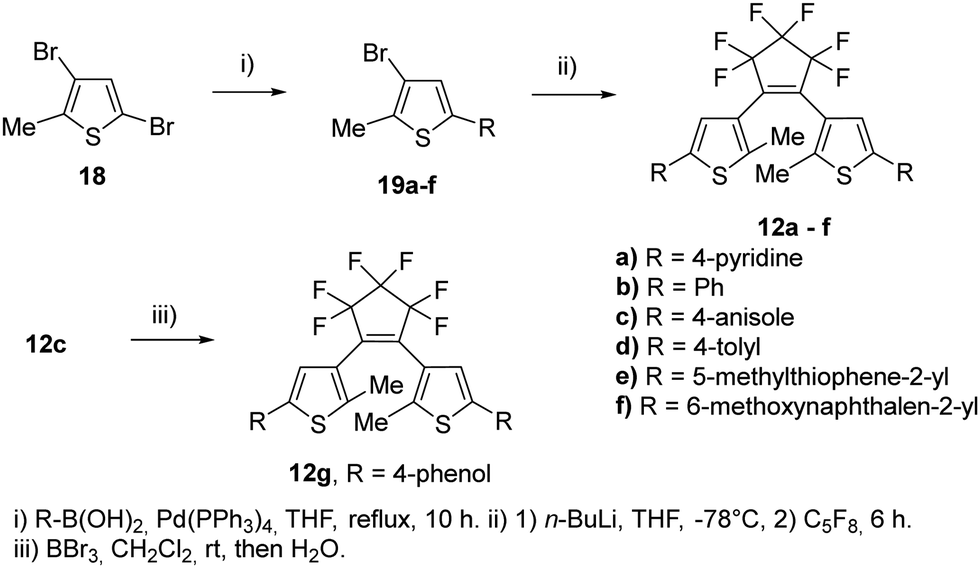 | ||
| Scheme 3 Syntheses of the symmetric BTEs without β-methyl groups. | ||
The syntheses of the corresponding β-methylated BTEs are shown in Scheme 4. The precursors 22b and 22d were prepared from 2,4-dimethylthiophene 20 and phenyliodide as well as tolyl iodide in the presence of copper(I) iodide to give 21b and 21d, respectively. Iodination of the 3-position was accomplished by elemental iodine in the presence of iodic acid, so that 22b and 22d were obtained in 54% and 73% yields, respectively. BTE formation started from 16a,c, which were prepared before,10 as well as 22b and 22d, and yielded the target dyes 13a–d. BTE 13c was demethylated by boron tribromide as described above, and 13e was obtained in 84% yield. Iodide and bromide were changed at the β-position to investigate a potential influence on the reaction outcome due to the different leaving group tendencies. Introducing the iodine required an additional step, however, the final reaction to obtain the BTE seemingly is not dependent on the halide as similar yields were obtained (39–43%).
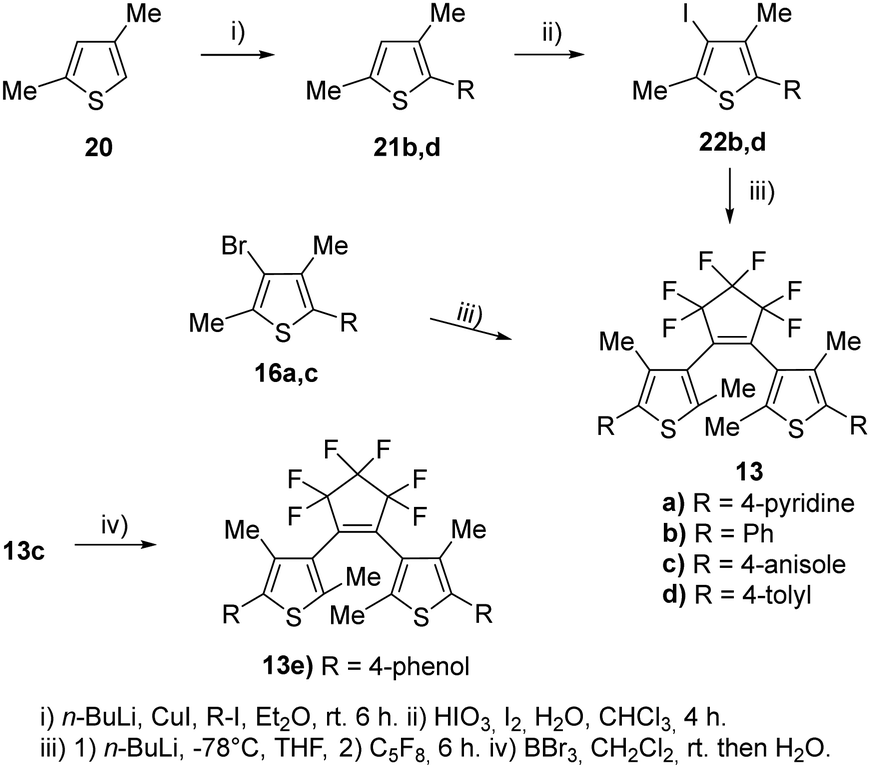 | ||
| Scheme 4 Syntheses of the β-methylated symmetric BTEs. | ||
The bis(thienyl)ethene 14 was also prepared as we anticipated a high absorbance similar to BTE 12e which has already been successfully applied in AMI technology18 (Scheme 5). BTE 14 differs from BTE 12e solely in the benzothiophene moiety. According to the properties mentioned in the introduction, this should lead to a more stable BTE with a high absorbance. The benzo[b]thiophene unit is known to induce a redshift in the UV-range but a blueshift of the visible part of the closed form. However, this particular BTE was found to be incapable of undergoing 6π-electrocyclizations with visible light >350 nm. These wavelengths induce 6π-cycloreversions, rendering the absorbance inadequate for the intended purpose.
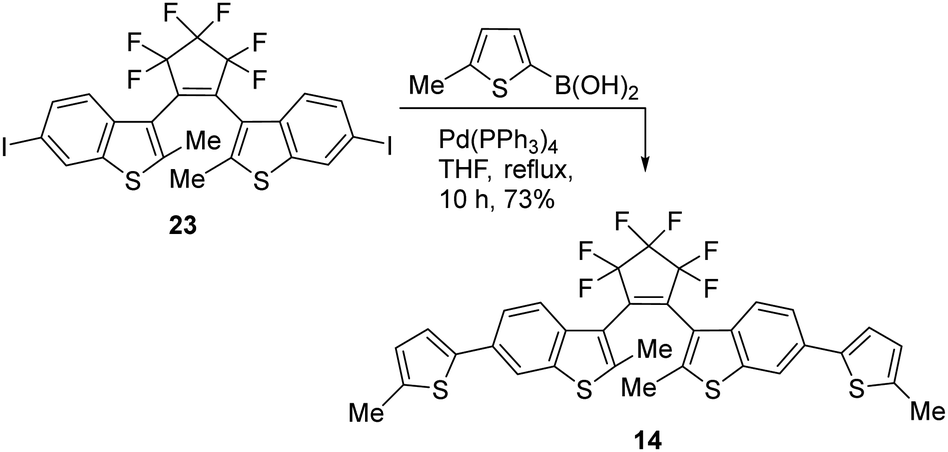 | ||
| Scheme 5 Synthesis of BTE 14. | ||
The series of symmetric BTEs were analyzed under the same conditions as the non-symmetric described above. The quantum yields and number of cycles are shown in Table 2. Comparing these results with those in Table 1, it is evident that the β-methylated species once again exhibit lower quantum yields for the switching reaction and a lower absorbance. However, in general, the overall reached absorbance is higher than for the non-symmetric BTEs. It appears that the quantum yields increase within the same type of BTE with the wavelength of the isosbestic point. The highest number of cycles x50 is achieved by the symmetric anisole-BTE. The phenol derivative 12g also demonstrated good fatigue resistance with an acceptable absorbance, despite the measurements being conducted under atmospheric conditions without the film's protection. As phenol is more reactive than anisole we would have expected a bigger difference in their number of cycles. The most interesting results stem from BTEs 12f and 14. When an annulated substituent is used (methoxynapthalene for 12f) the absorbance is increased in comparison to the anisole derivative as the π-system is more extended. This compound has the highest cyclization quantum yields. However, as a member of the non-β-methylated thiophene moieties investigated in this work, it exhibits a low number of cycles. In view of the measured stabilities in solutions, a higher stability in comparison to the double methylated BTEs would have been expected for BTE 14. However, the number of cycles up to which 50% of the BTE has decomposed does not exceed 125, whereas BTEs with a benzothiophene-core can reach over 10000 cycles when measured in solution.15a On the other hand, BTE 14 displayed the highest extinction coefficient ε and the highest quantum yield for the cycloreversion reaction. Being soluble in anisole and forming defect-free films, it exhibits a crucial property – the highest wavelength isosbestic point, located near 350 nm. This implies that BTE 14 (and 12f) can be switched using visible light, resulting in a closed form with a high absorbance. These properties make them highly desirable for potential applications.
| DAE | Moiety | β-Pos. | λ max [nm] (ε/103[M−1 cm−1]) | λ isos [nm] | φ isosAB [%] | φ 635nmBA [%] | x 50 | |
|---|---|---|---|---|---|---|---|---|
| Open form | Closed form | |||||||
| All spectra were measured in THF, for ring-opening a 3 Watt LED (630–640 nm, Bridgelux/Epistar) was used. For the solution measurements light intensity was measured with a PM100D powermeter from THORLabs. | ||||||||
| 12a | 4-Pyridine | H | 283.6 (38.1) | 587.2 (15.8) | 307.6 | 52.9 | 0.45 | 199 |
| 13a | 4-Pyridine | Me | 271.4 (26.9) | 574.2 (9.8) | 290.4 | 43.9 | 1.63 | 103 |
| 12b | Phenyl | H | 290.6 (36.3) | 590.0 (15.9) | 312.8 | 60.2 | 0.35 | 253 |
| 13b | Phenyl | Me | 272.6 (31.7) | 579.2 (11.8) | 295.0 | 38.6 | 0.51 | 52 |
| 12c | 4-Anisole | H | 296.6 (36.3) | 595.0 (17.7) | 322.4 | 62.0 | 0.31 | 281 |
| 13c | 4-Anisole | Me | 279.0 (27.9) | 582.4 (11.4) | 304.4 | 41.8 | 0.61 | 57 |
| 12d | 4-Tolyl | H | 297.0 (32.1) | 596.0 (15.0) | 323.8 | 69.3 | 0.29 | 207 |
| 13d | 4-tolyl | Me | 278.1 (27.4) | 583.4 (12.1) | 305.8 | 43.1 | 0.44 | 40 |
| 12g | 4-Phenol | H | 299.6 (26.0) | 593.0 (10.1) | 324.4 | 72.3 | 1.40 | 76 |
| 13e | 4-Phenol | Me | 283.3 (20.3) | 575.0 (7.5) | 320.8 | 48.2 | 1.85 | 33 |
| 12e | 5-Methylthiophene | H | 282.8 (33.7) | 603.6 (20.9) | 335.0 | 62.5 | 0.004 | 210 |
| 12f | 6-Methoxynaphthalene | H | 322.0 (50.4) | 608.6 (19.9) | 345.8 | 72.8 | 0.25 | 64 |
| 14 | 5-Methylthiophene | — | 320.0 (42.4) | 569.0 (24.4) | 349.9 | 53.8 | 3.93 | 125 |
In summary, we investigated different non-symmetric and symmetric BTEs which differ by β-methyl groups, focusing on their optical properties and their fatigue resistance in PMMA-films. The predicted properties in terms of the number of cycles based on the results in solution were not confirmed for PMMA films. In fact, the BTEs examined in our studies have an opposite behavior in films compared to solution, so that the design of photoresistent BTEs for applications in polymer films have to be reevaluated. Important factors like the solubility in the common solvents for the coating process such as anisole should be considered, as this affects the homogeneity of the final films. As examples, BTE 12e has a high absorbance but to cast defect-free films is more difficult than for BTE 14 which forms a foam and has a better solubility in anisole. In addition, it has a higher absorbance and can be switched with light sources larger than 350 nm. Our next steps involve the application of the most promising candidates in different polymer matrices and improve the number of cycles to have an even better dye to be used in the absorbance modulation imaging technique.
Experimental section
General
All reactions were carried out under an atmosphere of nitrogen in oven-dried glassware. Unless otherwise mentioned, all chemicals were purchased and used without further purification. Anhydrous solvents were dried according to standard procedures before usage. Melting points are uncorrected and were determined in an apparatus according to Dr Tottoli (Büchi). The ATR-IR spectra were obtained on a Bruker Alpha in the range of 400 to 4000 cm−1. 1H NMR spectra were recorded at 400 MHz or 600 MHz. 13C NMR spectra were recorded at 100 MHz or 150 MHz, with the solvent peak used as the internal reference. Multiplicities are described by using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. Signal orientations in DEPT experiments were described as follows: o = no signal; + = up (CH, CH3); − = down (CH2). The electrospray ionization mass spectra (ESIMS) were measured with a Bruker Impact-II mass spectrometer. Samples were sprayed from MeCN. Chromatography: The reactions were traced by thin layer chromatography with silica gel 60 (F254, MERCK KGAA). For the detection of substances, quenching was used at either 254 nm or 366 nm with a mercury lamp. The preparative column chromatography was conducted through silica gel 60 (230–400 mesh).For the photostability measurements we used sapphire substrates (10.0 mm ∅, 1 mm thickness), SaphTec. For building up a matrix we used poly(methyl methacyrylate) (PMMA, average Mw 120000 by GPC, SIGMA-ALDRICH). For producing the coatings a spin-coater (LAURELL WS-650 MZ-23MPP) was used. The produced coatings were investigated by using CLSM (Keyence VK-X210).
The UV-Vis spectra were recorded with the UV-Vis-NIR spectrophotometer (type UV-670 JASCO GMBH, Germany), and the reviewed wavelengths were in the range of 200 and 800 nm. The used quartz cuvettes had a path length of 1 cm. For the switching procedure the light of a fluorescence spectrometer (FP-8500, JASCO) was used and guided via a liquid light guide (250-series, Lumatec) into the UV-Vis-spectrometer. A self-made cell holder allows irradiate the solutions while simultaneously acquire spectra.
To determine the quantum yields, the UV-Vis-NIR spectrophotometer mentioned above was used. As solvent, THF (99.9%, anhydrous, inhibitor-free) was used.
To determine the photochrome's stabilities in films, we used a setup as shown in the ESI.† We applied a red laser with a wavelength of 638 nm and a power of approx. 180 mW (Hübner Photonics, Model Cobolt 06-mld 638 nm) and a yellow laser with a wavelength of 565 nm and a power of approx. 150 mW (MPB Communications 565 nm laser, Model F-04306-2) and a UV laser with a wavelength of 325 nm (Kimmon Koha HeCd Laser IK 3083 R-D). To control the intensity of the yellow or red laser, we employed a rotation stage with a half wave plate and a polarizing beamsplitter. An acousto-optic modulator (MQ110-A3-UV) was used to regulate the light power of the UV laser. A photosensor (Hamamatsu H10722-20 with 740 mV gain) was employed as detector, with an edge filter positioned in front to block UV light.
The compounds 12a, 12e, 13a, 16a, 18, 19a, 19c and 20 are described in our previous paper.9,10
General procedure of the Suzuki–Miyaura coupling (procedure 1)
The reaction is carried out under nitrogen atmosphere. If not otherwise mentioned, the boronic acid (15) is dissolved in anhyd. tetrahydrofuran. To this solution was added a solution of Na2CO3 in water followed by 1 eq. of the halide and 5 mol% of Pd(PPh3)4. The reaction outcome is monitored by TLC. After the conversion water is added to the solution, the solution is then filtered through Celite and extracted with diethylether. The organic phase is then washed with water and dried over MgSO4. The solvent is removed in vacuo followed by column chromatography to afford the desired product.:1). Yield 1.45 g (51%), a pale green solid. 1H NMR (600 MHz, CDCl3): δ = 8.71 (br, 1H, 2′-H), 8.60 (d, J = 4.0 Hz, 1H, 4′-H), 7.73 (dt, J1 = 1.9 Hz, J2 = 7.9 Hz, 1H, 6′-H), 7.37 (dd, J1 = 4.9 Hz, J2 = 7.9 Hz, 1H, 5′-H), 2.47 (s, 3H, 6-H), 2.29 (s, 3H, 7-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 149.5 (+, 2′-C), 148.4 (+, 4′-C), 136.2 (+, 6′-C), 133.8 (o, 3-C), 133.8 (o, 5-C), 130.7 (o, 1′-C), 130.6 (o, 2-C), 123.4 (+, 5′-C), 114.1 (o, 4-C), 15.5 (+, 7-C), 15.2 (+, 6-C) ppm. IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3035, 2981, 2943, 2910, 2852, 1757, 1559, 1441, 1404, 1324, 1258, 1000, 931, 798, 763, 703, 623, 550 cm−1. HRMS (ESI): calc.: 289.9610 [M + Na+], found: 289.9608 [M + Na+]. mp.: 47.8 °C.
= 3004, 2955, 2923, 2833, 1602, 1506, 1445, 1287, 1241, 1170, 1026, 824, 754, 529 cm−1. HRMS (ESI): calc.: 295.9870 [M+], found: 295.9870 [M+]. mp.: 72.1 °C.
= 3064, 2996, 2945, 2915, 2833, 1596, 1469, 1281, 1208, 1167, 1048, 1011, 826, 772, 694 cm−1. HRMS (ESI): calc.: 296.9943 [M + H+], found: 296.9939 [M + H+].
= 3064, 2907, 2847, 1518, 1443, 1209, 1157, 996, 870, 817, 781, 471 cm−1. HRMS (APCI): calc.: 272.9402 [M + H+], found: 272.9392. mp.: 81.1 °C.
:1). Yield 1.77 g (74%), a white solid. 1H NMR (600 MHz, CDCl3): δ = 7.87 (d, J = 1.7 Hz, 1H, 8-H), 7.73 (d, J = 4.0 Hz, 1H, 15-H), 7.72 (d, J = 3.6 Hz, 1H, 10-H), 7.60 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, 16-H), 7.18 (s, 1H, 3-H), 7.16 (dd, J1 = 8.9 Hz, J2 = 2.5 Hz, 1H, 11-H), 7.11 (d, J = 2.5 Hz, 1H, 13-H), 3.93 (s, 3H, 17-H), 2.44 (s, 3H, 6-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 158.0 (o, 12-C), 141.6 (o, 2-C), 134.1 (o, 7-C), 133.3 (o, 5-C), 129.6 (+, 15-C), 129.0 (o, 9-C), 128.8 (o, 14-C), 127.5 (+, 10-C), 125.4 (+, 3-C), 124.2 (+, 16-C), 123.7 (+, 8-C), 119.5 (+, 11-C), 109.9 (o, 4-C), 105.8 (+, 13-C), 55.4 (+, 17-C), 14.9 (+, 6-C) ppm. IR (ATR): = 3056, 3007, 2950, 2912, 2841, 1595, 1528, 1493, 1459, 1380, 1340, 1235, 1193, 1158, 1117, 1023, 951, 852, 814, 511, 466 cm−1. HRMS (ESI): calc.: 332.9943 [M + H+], found: 332.9937 [M + H+]. mp.: 161.3 °C.
= 3035, 2981, 2943, 2910, 2852, 1757, 1559, 1441, 1404, 1324, 1258, 1000, 931, 798, 763, 703, 623, 550 cm−1. HRMS (ESI): calc.: 289.9610 [M + Na+], found: 289.9608 [M + Na+]. mp.: 47.8 °C.
= 3004, 2955, 2923, 2833, 1602, 1506, 1445, 1287, 1241, 1170, 1026, 824, 754, 529 cm−1. HRMS (ESI): calc.: 295.9870 [M+], found: 295.9870 [M+]. mp.: 72.1 °C.
= 3064, 2996, 2945, 2915, 2833, 1596, 1469, 1281, 1208, 1167, 1048, 1011, 826, 772, 694 cm−1. HRMS (ESI): calc.: 296.9943 [M + H+], found: 296.9939 [M + H+].
= 3064, 2907, 2847, 1518, 1443, 1209, 1157, 996, 870, 817, 781, 471 cm−1. HRMS (APCI): calc.: 272.9402 [M + H+], found: 272.9392. mp.: 81.1 °C.
:1). Yield 1.77 g (74%), a white solid. 1H NMR (600 MHz, CDCl3): δ = 7.87 (d, J = 1.7 Hz, 1H, 8-H), 7.73 (d, J = 4.0 Hz, 1H, 15-H), 7.72 (d, J = 3.6 Hz, 1H, 10-H), 7.60 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, 16-H), 7.18 (s, 1H, 3-H), 7.16 (dd, J1 = 8.9 Hz, J2 = 2.5 Hz, 1H, 11-H), 7.11 (d, J = 2.5 Hz, 1H, 13-H), 3.93 (s, 3H, 17-H), 2.44 (s, 3H, 6-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 158.0 (o, 12-C), 141.6 (o, 2-C), 134.1 (o, 7-C), 133.3 (o, 5-C), 129.6 (+, 15-C), 129.0 (o, 9-C), 128.8 (o, 14-C), 127.5 (+, 10-C), 125.4 (+, 3-C), 124.2 (+, 16-C), 123.7 (+, 8-C), 119.5 (+, 11-C), 109.9 (o, 4-C), 105.8 (+, 13-C), 55.4 (+, 17-C), 14.9 (+, 6-C) ppm. IR (ATR): = 3056, 3007, 2950, 2912, 2841, 1595, 1528, 1493, 1459, 1380, 1340, 1235, 1193, 1158, 1117, 1023, 951, 852, 814, 511, 466 cm−1. HRMS (ESI): calc.: 332.9943 [M + H+], found: 332.9937 [M + H+]. mp.: 161.3 °C.
General procedure for the Sonogashira coupling (procedure 2)
The reaction was carried out under nitrogen atmosphere. If not otherwise mentioned, 25 is dissolved in anhyd. Et2O and 1.1 eq. TEMED was added at room temperature. To this solution 1.1 eq. of 1.6 M n-BuLi (in hexane) solution was added. After one hour 1.1 eq. of CuI was added in portions and after 30 additional minutes 1 eq. of the halide was added as well as 5 mol% of Pd(PPh3)4. The reaction outcome was monitored by TLC. After the conversion, water was added to the solution, the solution was filtered through Celite and extracted with diethylether. The organic phase was then washed with water and dried over MgSO4. The solvent was removed in vacuo followed by column chromatography to afford the desired product. = 2966, 2919, 2860, 1516, 1446, 1379, 1137, 951, 813, 619, 533, 499 cm−1. HRMS (ESI): calc.: 203.0889 [M + H+], found: 203.0893 [M + H+].
= 2914, 2851, 1507, 1437, 1377, 1112, 988, 936, 814, 748, 645, 549, 504, 443 cm−1. HRMS (ESI): calc.: 327.9783 [M+], found: 327.9784 [M+]. mp.: 72.0 °C.
General procedure for the synthesis of a BTE-precursors 17a,b (procedure 3)
The reaction was carried out under nitrogen atmosphere. 1 eq. halide was dissolved in dry THF and the mixture was cooled to −80 °C. To this solution 1.1 eq. of 2.8 M n-BuLi in cyclohexane solution were added. After 30–60 min, octafluorocyclopentene (1.5 eq.) was added in one portion. After 2 h the reaction was allowed to warm up to room temperature and was stirred overnight. Water was added to the solution. Diethyl ether was used for extraction, the organic phase was washed with water. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuo followed by column chromatography to afford the target molecule.:1). Yield 1.80 g (67%), a yellow-green solid. 1H NMR (400 MHz, CDCl3): 8.66 (d, J = 6.2 Hz, 2H, 3′-H), 7.36 (d, J = 6.2 Hz, 2H, 2′-H), 2.41 (s, 3H, 6-H), 2.19 (s, 3H, 7-H) ppm. Spectroscopic data are in agreement with those reported in the literature.28
:1). Yield 1.57 g (58%), a orange-red solid. 1H NMR (600 MHz, CDCl3): δ = 8.68 (d, J = 1.8 Hz, 1H, 2′-H), 8.58 (dd, J1 = 1.8 Hz, J2 = 4.9 Hz, 1H, 4′-H), 7.71 (dt, J1 = 2.1 Hz, J2 = 7.9 Hz, 1H, 6′-H), 7.35 (dd, J1 = 4.9 Hz, J2 = 7.9 Hz, 1H, 5′-H), 2.40 (s, 3H, 6-H), 2.12 (s, 3H, 7-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 153.9 (o, 12-C), 149.8 (+, 2′-C), 148.8 (+, 4′-C), 141.4 (o, 5-C), 136.3 (+, 6′-C), 133.4 (o, 3-C), 132.7 (o, 2-C), 129.9 (o, 1′-C), 123.4 (+, 5′-C), 121.2 (o, 4-C), 120.1 (o, 8-C), 115.3 (o, 9-C), 112.0 (o, 11-C), 110.7 (o, 10-C), 14.0 (+, 6-C), 13.7 (+, 7-C) ppm. IR (ATR): = 3051, 2927, 2866, 1705, 1388, 1272, 1197, 1150, 1024, 965, 868, 802, 711, 553 cm−1. HRMS (ESI): calc.: 382.0495 [M + H+], found: 382.0490 [M + H+]. mp.: 42.3 °C.
General procedure for the synthesis of the non-symmetric BTEs
The reaction was carried out under nitrogen atmosphere. 1 eq. of the halide was dissolved in dry THF and the mixture was cooled to −80 °C. To this solution, 1.1 eq. of 2.8 M n-BuLi in cyclohexane solution was added. After 30–60 min a solution of the cyclopentene unit dissolved in dry THF at −80 °C was added. After 2 h the reaction was allowed to warm up to room temperature and was stirred overnight. Water was added to the solution. Diethyl ether was used for extraction, the organic phase was washed with water. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuo followed by column chromatography to afford the BTE.:1). Yield 1.05 g (54%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.60 (d, J = 5.2 Hz, 2H, 3′′′-H), 7.28–7.26 (m, 4H, 2′′′-H & 2′′-H), 6.92 (d, J = 8.9 Hz, 2H, 3′′-H), 3.81 (s, 3H, 6′′-H), 2.39 (s, 3H, 6′-H), 2.35 (s, 3H, 6-H), 2.17 (s, 3H, 7′-H), 2.07 (s, 3H, 7-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 159.2 (o, 4′′-C), 150.0 (+, 3′′′-C), 142.7 (o, 8-C), 141.7 (o, 1′′′-C), 141.1 (o, 8′-C), 141.0 (o, 2′-C), 138.2 (o, 5-C), 136.4 (o, 2-C9, 134.5 (o, 4′-C), 133.3 (o, 5′-C), 131.1 (o, 3-C), 130.3 (2′′-C), 126.8 (o, 3′-C), 126.3 (o, 1′′-C), 125.5 (o, 4-C), 123.1 (+, 2′′′-C), 116.2 (o, 9-C & 9′-C), 114.0 (+, 3′′-C), 111.4 (o, 10-C), 55.3 (+, 6′′-C), 14.9 (+, 6-C & 6′-C), 14.7 (+, 7′-C), 14.5 (+, 7-C) ppm. IR (ATR): = 3026, 2959, 2929, 2839, 1596, 1510, 1339, 1249, 1184, 1107, 1032, 986, 867, 822, 526 cm−1. HRMS (ESI): calc.: 602.1017 [M + Na+], found: 602.1019 [M + Na+]. mp.: 79.6 °C.
:1). Yield 951 mg (54%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.61 (d, J = 4.9 Hz, 2H, 3′′′-H), 7.30 (t, J = 8.0 Hz, 1H, 5′′-H), 7.29–7.27 (m, 2H, 2′′′-H), 6.94 (ddd, J1 = 0.7 Hz, J2 = 2.5 Hz, J3 = 7.9 Hz, 1H, 4′′-H), 6.90–6.89 (m, 1H, 2′′-H), 6.86 (ddd, J1 = 0.7 Hz, J2 = 2.5 Hz, J3 = 8.1 Hz, 1H, 6′′-H), 3.82 (s, 3H, 7′′-H), 2.39 (s, 3H, 6-H), 2.36 (s, 6′-H), 2.17 (s, 3H, 7-H), 2.11 (s, 3H, 7′-C) ppm. 13C NMR (150 MHz, CDCl3): δ = 159.6 (o, 3′′-C), 150.1 (+, 3′′′-C), 142.6 (o, 8-C), 141.7 (1′′′-C), 141.4 (o, 8′-C), 141.1 (o, 2′-C), 139.0 (o, 5-C), 136.4 (o, 2-C), 135.2 (o, 1′′-C), 134.4 (o, 4′-C), 133.3 (o, 5′-C), 132.0 (o, 3-C), 129.6 (+, 5′′-C), 126.8 (o, 3′-C), 125.8 (o, 4-C), 123.1 (+, 2′′′-C), 121.6 (+, 4′′-C), 116.1 (o, 9-C & 9′-C), 115.0 (+, 2′′-C), 112.9 (+, 6′′-C), 111.3 (o, 10-C), 55.3 (+, 7′′-C), 15.0 (+, 6-C & 7′-C), 14.9 (+, 6′-C & 7-C) ppm. IR (ATR): = 2960, 2930, 2837, 1593, 1442, 1339, 1264, 1189, 1141, 1107, 1049, 986, 868, 818, 779, 695, 501 cm−1. HRMS (ESI): calc.: 580.1198 [M + H+], found: 580.1201 [M + H+]. mp.: 68.3 °C.
:1). Yield 1.612 g (62.2%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.69–8.67 (m, 1H, 2′′′-H), 8.52 (d, J = 4.0 Hz, 1H, 4′′′-H), 7.72–7.69 (m, 1H, 6′′′-H), 7.36 (dd, J1 = 4.6 Hz, J2 = 7.2 Hz, 1H, 5′′′-H), 7.30 (t, J = 8.0 Hz, 1H, 5′′-H), 7.00–6.98 (m, 1H, 4′′-H), 6.95–6.94 (m, 1H, 2′′-H), 6.90 (ddd, J1 = 0.8 Hz, J2 = 2.5 Hz, J3 = 8.3 Hz, 1H, 6′′-H), 3.86 (s, 3H, 7′′-H), 2.43 (s, 3H, 6-H), 2.41 (s, 3H, 6′-H), 2.16 (s, 3H, 7′-H), 2.14 (s, 3H, 7-C) ppm. 13C NMR (150 MHz, CDCl3): δ = 159.6 (3′′-C), 149.7 (+, 2′′′-C), 148.5 (+, 4′′′-H), 142.3 (o, 8-C), 141.6 (o, 8′-C), 140.3 (o, 2′-C), 139.0 (o, 5-C), 136.6 (o, 2-C), 136.2 (+, 6′′′-C), 135.2 (o, 1′′-C), 133.5 (o, 3-C), 132.4 (o, 5′-C), 132.1 (o, 3-C), 130.2 (o, 1′′′-C), 129.6 (+, 5′′-C), 126.3 (o, 4-C), 125.8 (o, 3′-C), 123.3 (+, 5′′′-C), 121.6 (+, 4′′-C), 116.2 (o, 9-C & 9′-C), 114.9 (+, 6′′-C), 112.9 (+, 2′′-C), 111.5 (o, 10-C), 55.3 (+, 7′′-C), 15.0 (+, 6-C), 14.9 (+, 6′-C), 14.7 (+, 7-C), 14.6 (+, 7′-C) ppm. IR (ATR): = 2960, 2930, 2866, 1599, 1443, 1340, 1265, 1189, 1108, 1050, 986, 869, 780, 704, 534 cm−1. HRMS (ESI): calc.: 602.1017 [M + Na+], found: 602.1031 [M + Na+]. mp.: 63.3 °C.
General procedure for the demethylation of the anisole-substituted BTEs (procedure 5)
To a solution of the anisole-substituted BTE in dry DCM 2 eq. of 1M BBr3 in DCM solution was added at room temperature. The reaction was controlled via TLC. If necessary, more BBr3 was added till full completion of the reaction. Water was the added to the solution. Diethyl ether was used for extraction, the organic phase was washed with water. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuo followed by column chromatography to afford the phenolic BTE.:1). Yield 248 mg (72%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.64–8.57 (m, 2H, 3′′′-H), 7.39–7.35 (m, 2H, 2′′′-H), 7.20–7.17 (m, 2H, 2′′-H), 6.90 (d, J = 8.5 Hz, 2H, 3′′-H), 2.41 (s, 3H, 6′-H), 2.36 (s, 3H, 6-H), 2.18 (s, 3H, 7′-H), 2.04 (s, 3H, 7-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 157.0 (o, 4′′-C), 148.9 (+, 3′′′-C), 143.0 (o, 8-C), 143.0 (o, 1′′′-C), 141.7 (o, 2′-C), 140.9 (o, 8′-C), 138.0 (o, 5-C), 136.9 (o, 2-C), 135.2 (o, 4′-C), 132.7 (o, 5′-C), 131.0 (o, 3-C), 130.5 (+, 2′′-C), 127.1 (o, 3′-C), 125.4 (o, 4-C), 125.2 (o, 1′′-C), 123.6 (+, 2′′′-C), 116.1 (o, 9-C & 9′-C), 115.8 (+, 3′′-C), 111.4 (o, 10-C), 15.3 (+, 7′-C), 15.1 (+, 6′-C), 14.9 (+, 6-C), 14.7 (+, 7-C) ppm. IR (ATR): = 2962, 2925, 1599, 1510, 1441, 1339, 1265, 1141, 1105, 1052, 986, 867, 822, 528 cm−1. HRMS (ESI): calc.: 566.1042 [M + H+], found: 566.1039 [M + H+]. mp.: 124.8 °C.
:1). Yield 185 mg (87%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.60–8.56 (m, 2H, 3′′′-H), 7.33 (d, J = 5.3 Hz, 2H, 2′′′-H), 7.30 (t, J = 8.0 Hz, 1H, 5′′-H), 6.88–6.87 (m, 1H, 4′′-H), 6.87–6.87 (m, 1H, 2′′-H), 6.83–6.82 (m, 1H, 6′′-H), 2.39 (s, 3H, 6′-H), 2.35 (s, 3H, 6-H), 2.16 (s, 3H, 7′-H), 2.08 (s, 3H, 7-H), ppm. 13C NMR (150 MHz, CDCl3): δ = 156.9 (o, 3′′-C), 149.1 (+, 3′′′-C), 142.7 (o, 8-C), 142.7 (o, 1′′′-C), 141.5 (o, 2′-C), 141.1 (o, 8′-C), 138.8 (o, 5-C), 136.4 (o, 2-C), 135.2 (o, 1′′-C), 134.9 (o, 4′-C), 132.8 (o, 5′-C), 131.8 (o, 3-C), 129.7 (+, 5′′-C), 126.9 (o, 3′-C), 125.6 (o, 4-C), 123.5 (+, 2′′′-C), 120.7 (+, 4′′-C), 116.2 (+, 2′′-C), 116.1 (o, 9-C & 9′-C), 114.9 (+, 6′′-C), 111.2 (o, 10-C), 15.1 (+, 6-C & 7′-C), 14.9 (+, 6′-C), 14.8 (+, 7-C) ppm. IR (ATR): = 3046, 2975, 2926, 1596, 1443, 1339, 1267, 1192, 1142, 1109, 1055, 988, 869, 820, 780, 697, 534 cm−1. HRMS (ESI): calc.: 566.1042 [M + H+], found: 566.1044 [M + H+]. mp.: 123.5 °C.
:1). Yield 276 mg (71%), a colorless foam. 1H NMR (600 MHz, CDCl3): δ = 8.61–8.59 (m, 1H, 2′′′-H), 8.55–8.53 (m, 1H, 4′′′-H), 7.74–7.72 (m, 1H, 6′′′-H), 7.38 (dd, J1 = 4.8 Hz, J2 = 8.2 Hz, 1H, 5′′′-H), 7.23–7.19 (m, 1H, 5′′-H), 6.89–6.86 (m, 1H, 2′′-H), 6.88–6.87 (m, 1H, 6′′-H), 6.83–6.81 (m, 1H, 6′′-H), 2.38 (s, 3H, 6-H), 2.33 (s, 3H, 6′-H), 2.07 (s, 3H, 7-C), 2.06 (s, 3H, 7′-H), ppm. 13C NMR (150 MHz, CDCl3): δ = 156.8 (o, 3′′-C), 148.9 (+, 2′′′-C), 147.5 (+, 4′′′-C), 142.5 (o, 8-C), 141.4 (o, 8′-C), 140.6 (o, 5-C), 138.9 (o, 2′-C), 137.1 (+, 6′′′-C), 136.4 (o, 2-C), 135.3 (o, 1′′-C), 133.8 (o, 4′-C), 131.9 (o, 5′-C), 131.8 (o, 3-C), 130.8 (o, 1′′′-C), 129.8 (+, 5′′-C), 126.4 (o, 4-C), 125.7 (o, 4′-C), 123.8 (+, 5′′′-C), 120.9 (+, 6′′-C), 116.2 (+, 2′′-C), 116.2 (o, 9-C & 9′-C), 114.9 (+, 4′′-C), 112.9 (+, 2′′-C), 111.3 (o, 10-C), 14.9 (+, 6-C), 14.8 (+, 6′-C), 14.7 (+, 7′-C), 14.5 (+, 7′-C) ppm. IR (ATR): = 2961, 2924, 1582, 1443, 1339, 1266, 1190, 1141, 1108, 1053, 986, 867, 780, 701, 533 cm−1. HRMS (ESI): calc.: 588.0861[M + Na+], found: 588.0851 [M + Na+]. mp.: 97.6 °C.
General procedure for the synthesis of a symmetric BTE-(procedure 6)
The reaction was carried out under nitrogen atmosphere and with a septum-sealed glassware. One eq. of halide was dissolved in anhyd. THF and the mixture was cooled to −80 °C. To this solution, 1.1 eq of 2.8 M n-BuLi in cyclohexane solution were added. After 30–60 min octafluorocyclopentene (0.5 eq.) was added in small portions. After 2 h the reaction was allowed to warm up to room temperature and was stirred overnight. Water was added to the solution. Diethyl ether was used for extraction, the organic phase was washed with water. The combined organic layers were dried over MgSO4 and the solvent was removed in vacuo followed by column chromatography to afford the target molecule.:1). Yield 203 mg (25%), a white solid. 1H NMR (600 MHz, CDCl3): δ = 7.91 (d, J = 1.6 Hz, 2H, 2′-H), 7.75 (d, J = 4.3 Hz, 2H, 9′-H), 7.74 (d, J = 4.4 Hz, 2H, 4′-H), 7.65 (dd, J1 = 8.6 Hz, J2 = 1.8 Hz, 2H, 10′-H), 7.39 (s, 2H, 3-H), 7.18 (dd, J1 = 8.8 Hz, J2 = 2.6 Hz, 2H, 5′-H), 7.13 (d, J = 2.4 Hz, 2H, 7′-H), 3.93 (s, 6H, 11′-H), 2.03 (s, 6H, 6-H) ppm. 13C NMR (150 MHz, CDCl3): δ = 158.1 (o, 6′-C), 142.6 (o, 2-C), 141.1 (o, 5-C), 136.4–136.2 (o, m, 7-C), 134.1 (o, 1′-C), 129.6 (+, 4′-C), 129.0 (o, 8′-C), 128.7 (o, 3′-C), 127.6 (+, 9′-C), 126.0 (o, 4-C), 124.4 (+, 10′-C), 124.1 (+, 2′-C), 122.2 (+, 3-C), 119.6 (+, 5′-C), 116.4 (o, m, 8-C), 111.3 (o, m, 9-C), 105.8 (+, 7′-C), 55.4 (+, 11′-C), 14.7 (+, 6-C) ppm. IR (ATR): = 3057, 2957, 2926, 2848, 1626, 1600, 1461, 1388, 1332, 1269, 1193, 1110, 1027, 980, 882, 848, 808, 750, 519, 471 cm−1. HRMS (ESI): calc.: 681.1351 [M + H+], found: 681.1346 [M + H+]. mp.: 176.9 °C.
:1). Yield 93 mg (87%), a pale yellow solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.76 (s, 2H, 5′-H), 7.47 (d, J = 8.5 Hz, 4H, 2′-H), 7.30 (s, 2H, 3-H), 6.85 (d, J = 8.5 Hz, 4H, 3′-H), 1.98 (s, 6H, 6-H) ppm. Spectroscopic data are in agreement with those reported in the literature.27
:1). Yield 416 mg (42%), a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ = 7.29 (d, J = 8.7 Hz, 4H, 2′-H), 6.93 (d, J = 8.7 Hz, 4H, 3′-H), 3.83 (s, 6H, 5′-H), 2.35 (s, 6H, 6-H), 2.07 (s, 6H, 7-H) ppm. Spectroscopic data are in agreement with those reported in the literature.30
= 3056, 2913, 2849, 1471, 1382, 1234, 1056, 997, 740, 695, 502 cm−1. HRMS (ESI): calc.: 577.1453 [M+], found: 577.1455 [M+]. mp.: 139.5 °C.
:1). Yield 93 mg (87%), a pale yellow solid. 1H NMR (600 MHz, DMSO-d6): δ = 9.70 (s, 2H, 5′-H), 7.20 (d, J = 8.6 Hz, 4H, 2′-H), 6.81 (d, J = 8.6 Hz, 4H, 3′-H), 2.31 (s, 6H, 6-H), 2.03 (s, 6H, 7-H) ppm. 13C NMR (150 MHz, DMSO-d6): δ = 157.7 (o, 4′-C), 142.3–141.9 (o, m, 8-C), 138.3 (o, 5-C), 136.9 (o, 2-C), 130.7 (o, 3-C), 130.5 (+, 2′-C), 125.3 (o, 4-C), 124.1 (o, 1′-C), 116.2 (o, 9-C), 116.1 (+, 3′-C), 111.5 (o, 10-C), 14.9 (+, 6-C), 14.8 (+, 7-C) ppm. IR (ATR): = 3281, 2977, 2926, 1703, 1607, 1510, 1436, 1339, 1251, 1140, 1104, 1051, 985, 826, 523 cm−1. HRMS (ESI): calc.: 581.1038 [M + H+], found: 581.1021 [M + H+]. mp.: 96.7 °C.
:1). Yield 120 mg (73%), a yellow foam. 1H NMR (600 MHz, CDCl3): δ = 7.84 (s, 1.21H, 8-H, ap), 7.74 (s, 0.80H, 8-H, p), 7.63 (dd, J1 = 8.4 Hz, J2 = 1.30 Hz, 1.3H, 6-H, ap), 7.58 (dd, J1 = 8.5 Hz, J2 = 1.4 Hz, 1.39H, 5-H, ap), 7.52–7.51 (m, 0.69H, 6-H, p), 7.40–7.39 (m, 0.59H, 5-H, p), 7.14 (d, J = 3.5 Hz, 1.37H, 3′-H, ap), 7.05 (d, J = 3.5 Hz, 0.59H, 3′-H, p), 6.74 (d, J = 2.4 Hz, 1.32H, 4′-H, ap), 6.69 (d, J = 2.4 Hz, 0.66H, 4′-H, p), 2.53 (s, 1.70H, 6′-H, p), 2.52 (s, 1.9H, 10-H, p), 2.48 (s, 4.29H, 6′-H, ap), 2.22 (s, 4.10H, 10-H, ap) ppm. 13C NMR (150 MHz, CDCl3): δ = 142.9 (o, 2-C, ap), 142.4 (o, 2-C, p), 142.0 (o, 11-C, ap,p), 141.3 (o, 2′-C, ap, p), 139.9 (o, 5′-C, ap), 139.8 (o, 5′-C, p), 139.0 (o, 9-C, ap), 138.9 (o, 9-C, p), 137.2 (o, 4-C, ap, p), 131.4 (o, 7-C, ap, p), 126.4 (+, 4′-C, ap), 126.3 (+, 4′-C, p), 123.3 (+, 3′-C, ap, p), 122.9 (+, 6-C, ap), 122.8 (+, 6-C, p), 122.4 (+, 8-C, ap), 122.1 (+, 8-C, p), 119.3 (o, 3-C, p), 119.1 (o, 3-C, ap), 118.5 (+, 5-C, ap) 118.4 (+, 5-C, p), 116.2 (o, 12-C, ap, p), 111.5 (o, 13-C, ap, p), 15.5 (+, 10-C, p & 6′-C, p), 15.4 (+, 6′-C, ap), 15.3 (+, 10-C, ap) ppm. IR (ATR): = 3064, 2918, 2855, 1486, 1442, 1336, 1266, 1188, 1140, 1111, 1045, 992, 960, 863, 791, 564, 527, 492 cm−1. HRMS (ESI): calc.: 683.0401 [M + Na+], found: 683.0399 [M + Na+]. mp.: 97.3 °C. The ratio of the antiparallel (ap) and parallel (p) forms of the measured open form is 1.44:1.
Author contributions
Sven Nagorny performed all syntheses. Together with Thea Weingartz he did the spectroscopic characterizations and both produced the PMMA films. André Eitzeroth determined the absorption coefficients and quantum yields. Marvin Schewe constructed the experimental setup to measure the fatigue resistancies and investigated the stabilities of the films. Jörg Adams, Christian Rembe and Andreas Schmidt are group leaders. Together with other cooperation partners (Alexander Egner, Vladimir N. Belov, Stefan W. Hell), they have a common project funded by the German Research Foundation (Deutsche Forschungsgemeinschaft DFG), called “NanoVidere – Photochromic Layers for Nanoscopy”Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Prof. Dr h. c. mult. Stefan W. Hell and Dr Vladimir N. Belov, Max-Planck-Institute for Multidisciplinary Sciences, Department of NanoBiophotonics, Göttingen (Germany), is gratefully acknowledged for helpful discussions, measurements with the yellow laser, and the synthesis of compound 23. The Deutsche Forschungsgemeinschaft is gratefully acknowledged for financial support (project NanoVidere; grant numbers 504588119 and 412988268).Notes and references
- (a) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS; (b) M. Irie, Photochem. Photobiol. Sci., 2010, 9, 1535–1542 CrossRef CAS PubMed; (c) M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS.
- (a) S. Aiken, R. J. Edgar, C. D. Gabbutt, B. M. Heron and P. A. Hobson, Dyes Pigm., 2018, 149, 92–121 CrossRef CAS; (b) A. Towns, Phys. Sci. Rev., 2021, 6, 341–368 Search PubMed.
- (a) G. S. Hartley, Nat. Chem., 1937, 1937, 33 Search PubMed; (b) H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- (a) M. Albrecht, M. Gjikaj and A. Schmidt, Tetrahedron, 2010, 66, 7149–7154 CrossRef CAS; (b) M. Albrecht, M. Yulikov, T. Kohn, G. Jeschke, J. Adams and A. Schmidt, J. Mater. Chem., 2010, 20, 3025–3034 RSC; (c) M. Albrecht, O. Schneider and A. Schmidt, Org. Biomol. Chem., 2009, 7, 1445–1453 RSC; (d) A. Schmidt, M. Albrecht, T. Mordhorst, M. Topp and G. Jeschke, J. Mater. Chem., 2007, 17, 2793–2800 RSC; (e) C. F. Otto, M. Liu, C. Herzberger, J. C. Namyslo, M. Nieger, E. G. Hübner, F. Lederle, T. Freese and A. Schmidt, Tetrahedron, 2020, 76, 131627 CrossRef CAS.
- (a) H. G. Heller and S. Oliver, J. Chem. Soc., Perkin Trans. 1, 1981, 197–201 RSC; (b) G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741–1754 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- Y. Chen, Y. Zheng, Y. Jiang, H. Fan and X. Zhu, J. Am. Chem. Soc., 2021, 143, 4281–4289 CrossRef CAS PubMed.
- (a) H.-B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS; (b) I. Hnid, D. Frath, F. Lafolet, X. Sun and J.-C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed; (c) C. Yun, S. Seo and E. Kim, J. Nanosci. Nanotechnol., 2010, 10, 6850–6854 CrossRef CAS; (d) A. J. Kronemeijer, H. B. Akkerman, T. Kudernac, B. J. van Wees, B. L. Feringa, P. W. M. Blom and B. de Boer, Adv. Mater., 2008, 20, 1467–1473 CrossRef CAS.
- S. Nagorny, T. Weingartz, J. C. Namyslo, J. Adams and A. Schmidt, Eur. J. Org. Chem., 2023, e202200996 CrossRef CAS.
- S. Nagorny, F. Lederle, V. Udachin, T. Weingartz, E. G. Hübner, S. Dahle, W.-M. Friedrichs, J. Adams and A. Schmidt, Eur. J. Org. Chem., 2021, 3178–3189 CrossRef CAS.
- T. Weingartz, S. Nagorny, J. Adams, A. Eitzeroth, M. Schewe, C. Rembe and A. Schmidt, RSC Adv., 2023, 13, 25704–25716 RSC.
- (a) R. Wang, S. Pu, G. Liu, S. Cui and H. Li, Tetrahedron Lett., 2013, 54, 5307–5310 CrossRef CAS; (b) G. Liu, M. Liu, S. Pu, C. Fan and S. Cui, Dyes Pigm., 2012, 95, 553–562 CrossRef CAS.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS.
- H. Tian and J. Zhang, Photochromic Materials: Preparation, Properties and Applications, Wiley-VCH, Weinheim, Germany, 1st edn, 2016 Search PubMed.
- (a) M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS; (b) K. Uno, M. L. Bossi, M. Irie, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 16471–16478 CrossRef CAS; (c) D. Becker, N. Konnertz, M. Böhning, J. Schmidt and A. Thomas, Chem. Mater., 2016, 28, 8523–8529 CrossRef CAS; (d) Y.-C. Jeong, D. G. Park, I. S. Lee, S. I. Yang and K.-H. Ahn, J. Mater. Chem., 2009, 19, 97–103 RSC; (e) M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS.
- (a) S. W. Hell, Phys. Lett. A, 2004, 326(1–2), 140–145 CrossRef CAS; (b) D. Kim, A. Aktalay, N. Jensen, K. Uno, M. L. Bossi, V. N. Belov and S. W. Hell, J. Am. Chem. Soc., 2022, 144, 14235–14247 CrossRef CAS; (c) N. A. Jensen, J. G. Danzl, K. I. Willig, F. Lavoie-Cardinal, T. Brakemann, S. W. Hell and S. Jakobs, ChemPhysChem, 2014, 15, 756–762 CrossRef CAS PubMed.
- (a) S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780 CrossRef CAS; (b) T. A. Klar, S. Jakobs, M. Dyba, A. Egner and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8206–8210 CrossRef CAS PubMed.
- P. Jain, C. Geisler, D. Leitz, V. Udachin, S. Nagorny, T. Weingartz, J. Adams, A. Schmidt, C. Rembe and A. Egner, ACS Nanosci. Au, 2023, 3, 375–380, DOI:10.1021/acsnanoscienceau.3c00013.
- R. Kowarsch, C. Geisler, A. Egner and C. Rembe, Opt. Express, 2018, 26, 5327 CrossRef CAS PubMed.
- (a) T. L. Andrew, H. Y. Tsai and R. Menon, Science, 2009, 324, 917–921 CrossRef CAS; (b) N. Brimhall, T. L. Andrew, R. V. Manthena and R. Menon, Phys. Rev. Lett., 2011, 107, 1–5 CrossRef.
- M. Bossi, V. Belov, S. Polyakova and S. W. Hell, Angew. Chem., Int. Ed., 2006, 45, 7462–7465 ( Angew. Chem., Int. Ed. , 2006 , 118 , 7623–7627 ) CrossRef CAS.
- (a) M. Maafi, Molecules, 2008, 13, 2260–2302 CrossRef CAS; (b) M. Maafi and R. G. Brown, Photochem. Photobiol. Sci., 2008, 7, 1360–1372 CrossRef CAS; (c) K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- (a) S. Pu, C. Fan, W. Miao and G. Liu, Dyes Pigm., 2010, 84, 25–35 CrossRef CAS; (b) M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS; (c) S. Pu, H. Li, G. Liu, W. Liu, S. Cui and C. Fan, Tetrahedron, 2011, 67, 1438–1447 CrossRef CAS; (d) S. Pu, G. Liu, G. Li, R. Wang and T. Yang, J. Mol. Struct., 2007, 833, 23–29 CrossRef CAS.
- G. Liu, M. Liu, S. Pu, C. Fan and S. Cui, Dyes Pigm., 2012, 95, 553–562 CrossRef CAS.
- S. Pu, H. Li, G. Liu, W. Liu, S. Cui and C. Fan, Tetrahedron, 2011, 67, 1438–1447 CrossRef CAS.
- M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305–8309 CrossRef CAS.
- M. Bossi, V. Belov, S. Polyakova and S. W. Hell, Angew. Chem., Int. Ed., 2006, 45, 7462–7465 ( Angew. Chem., Int. Ed. , 2006 , 118 , 7623–7627 ) CrossRef PubMed.
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS.
- J. J. D. de Jong, L. N. Lucas, R. Hania, A. Pugzlys, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Eur. J. Org. Chem., 2003, 1887–1893 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and UV spectra. See DOI: https://doi.org/10.1039/d3ma00791j |
| This journal is © The Royal Society of Chemistry 2024 |