
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput prediction of oxygen vacancy defect migration near misfit dislocations in SrTiO3/BaZrO3 heterostructures†
William Ebmeyer and
Pratik P. Dholabhai *
*
School of Physics and Astronomy, Rochester Institute of Technology, Rochester, NY 14623, USA. E-mail: pratik.dholabhai@rit.edu
First published on 23rd November 2023
Abstract
Among their numerous technological applications, semi-coherent oxide heterostructures have emerged as promising candidates for applications in intermediate temperature solid oxide fuel cell electrolytes, wherein interfaces influence ionic transport. Since misfit dislocations impact ionic transport in these materials, oxygen vacancy formation and migration at misfit dislocations in oxide heterostructures are central to their performance as an ionic conductor. Herein, we report high-throughput atomistic simulations to predict thousands of activation energy barriers for oxygen vacancy migration at misfit dislocations in SrTiO3/BaZrO3 heterostructures. Dopants display a noticeable effect as higher activation energies are uncovered in their vicinity. Interface layer chemistry has a fundamental influence on the magnitude of activation energy barriers since they are dissimilar at misfit dislocations as compared to coherent terraces. Lower activation energies are uncovered when oxygen vacancies migrate toward misfit dislocations, but higher energies when they hop away, revealing that oxygen vacancies would get trapped at misfit dislocations and impact ionic transport. The results herein offer atomic scale insights into ionic transport at misfit dislocations and fundamental factors governing the ionic conductivity of thin film oxide electrolytes.
1. Introduction
Complex oxide heterostructures,1–3 an intriguing class of materials fabricated by combining two dissimilar oxides, exhibit superior and emergent properties4–6 than their individual constituents. They have found promising applications in a wide range of technologies that include solid oxide fuel cells (SOFCs),7,8 radiation tolerant nuclear materials,9 information storage,10 batteries,11 and catalysts.12 SOFCs are electrochemical conversion devices that produce electricity directly by oxidizing a fuel.13–15 They are highly efficient and have the potential to achieve our society's ambition of transitioning from fossil fuels to clean generation of electrical power. Due to the ever-growing miniaturization of advanced nanotechnologies such as SOFCs, the role of surfaces and interfaces has never been more critical.In SOFCs, solid ceramic oxide electrolyte is one of the most important components since it facilitates the passage of oxide ions from the cathode to the anode. Since high ionic conductivity is imperative for the development of next-generation SOFCs, significant research efforts have been dedicated toward the discovery and design of electrolytes that allow faster ionic diffusion.15 Since high operating temperatures lead to issues with stability and durability, widespread deployment of SOFC technology necessitates lowering the operating temperatures to the intermediate-temperature (IT-SOFC) range of 773–973 K.7,16 One promising option to lower the operating temperature is via implementation of thin film oxide electrolytes (oxide heterostructures), which exhibit superior performance at lower temperatures and are poised to take over as the next-generation IT-SOFC electrolytes.17 However, their widespread use requires the basic understanding of the role of interfaces in shaping enhanced performance.15,16
Depending on the lattice mismatch, in semi-coherent oxide heterostructures, misfit dislocations originate at the heterointerface to mitigate the misfit strain between the two mismatched oxides. Since misfit dislocations are inevitable and responsible for controlling vital material properties,8,18–20 several studies have focused on investigating their role in semi-coherent oxide heterostructures. Nonetheless, their impact on ionic transport is not well understood since studies focused on elucidating the atomic and electronic structure of misfit dislocations are limited due to challenges in studying and characterizing individual columns of atoms at the heterointerface. Owing to their fundamental character and influence on material properties, recent computational and experimental studies have focused on thorough investigation of the structure–property relationships at misfit dislocations in oxides.
Perovskite oxide heterostructures have gained popularity due to their versatility in diverse technologies, including SOFCs. Depending on the desired application, perovskites are often doped to enhance magnetic,21 ionic,22 electronic,23 optical, and catalytic24 properties. Among various perovskite oxides, SrTiO3 (STO)25 and BaZrO3 (BZO) have been frequently studied, wherein doping is used to tune their functionality. For instance, perovskite oxide heterostructures have been studied as model systems for understanding ionic transport across oxide interfaces. Due to the compositional flexibility of perovskite lattice (ABO3), dopants can replace either A-site, B-site, or O-site ions. Since our present work is mainly focused on the fundamental aspects of oxygen vacancy migration in STO/BZO heterostructures, we will limit the discussion to acceptor doping (p-type).26 Acceptor doping, wherein the B-site cations, either Ti4+ in STO or Zr4+ in BZO, could be replaced with trivalent dopants has been extensively studied in STO27 and BZO28 since it leads to the formation of oxygen vacancies to maintain the defect equilibria.
Widespread use of the acceptor doping strategy in perovskites is evident from the literature, wherein a multitude of aliovalent dopants have been added to improve the properties of STO29–36 and BZO.28,37,38 Unfortunately, the added impurities do not come without challenges since they induce physical and chemical changes in the materials by segregating at surfaces and interfaces. Segregation behavior depends on size mismatch and charge interactions,39 formation of space-charge layers at interfaces,40 and on growth conditions and post-deposition annealing procedures.41 Since segregated additives could lead to deleterious effects on material stability and performance, the role of dopants and associated vacancies in complex oxides have been studied extensively. In fast ion conductors, the influence of dopants is even more critical since they are primarily introduced to increase the oxygen vacancy concentration, but their presence on the cation sublattice alters the oxygen vacancy migration barriers on the anion sublattice. Oxygen vacancy formation42 and migration43–49 in various perovskite oxides have been reported, wherein density functional theory (DFT) calculations and simulations based on interatomic potentials have been the method of choice. These results demonstrate how dopants can influence oxygen vacancy migration in bulk perovskites.
From a fundamental perspective, acceptor doping has been extensively used to improve the properties of bulk STO and bulk BZO. Nonetheless, in perovskite heterostructures, especially near misfit dislocations, how dopants influence the migration pathways of these vacancies is not well understood since the majority of reported work is focused on either bulk STO or bulk BZO. At interfaces, it is imperative to understand the basic role of trivalent dopants and associated oxygen vacancies that are responsible for ionic conduction. To the best of our knowledge, there are no studies in the literature that report oxygen vacancy migration near misfit dislocations in STO/BZO heterostructures. Since STO50 and BZO28 have key technological applications that include SOFC electrolytes,8,51–53 oxide heterostructures synthesized using either of them have garnered significant research interest.54–56 To understand the ionic transport mechanisms in a model oxide heterostructure used for SOFC electrolytes, basic knowledge of oxygen vacancy migration pathways in the vicinity of misfit dislocations is crucial. One of the main reasons for the lack of understanding of ionic transport mechanisms is the challenges in experimentally visualizing dopant atoms, characterizing oxygen vacancies, and resolving the structure and chemistry of misfit dislocations. The problem is further compounded as even from a theoretical perspective, calculations that utilize DFT are limited to coherent interfaces to keep the supercell size tractable.57 As a result, DFT calculations are not optimal to study oxygen vacancy migration at misfit dislocations, which necessitate very large supercells. In this regard, atomistic simulations based on empirical interatomic potentials are valuable in offering atomic scale insights into dopants and defects at misfit dislocations in oxide heterostructures.29
In perovskite oxides, defects could typically introduce ligand holes, which could further lead to altered magnetic properties as observed in STO.58 In addition, oxygen vacancy formation and migration in perovskites could be influenced by changes in oxidation states of added impurities having mixed valence, which could offer routes for tailoring mixed ionic electronic conductors in SOFCs.59 However, we strictly focus on studying the role of defects (oxygen vacancies) that are formed to maintain the defect equilibria after incorporation of trivalent dopants that do not exhibit mixed valence. As the current work is focused on studying the qualitative behavior of the interface governed ionic transport, emphasis on fixed ionic charges of metals (dopant and host) and resulting oxygen vacancies is rational since the oxygen vacancy diffusion mechanism is the primary mechanism for ionic conductivity in SOFC electrolytes.7,8
In cases where perovskite oxide heterostructures are implemented as model systems for SOFC electrolytes, knowledge of oxygen vacancy migration pathways in influencing the ionic transport would be beneficial. To achieve this goal, we used atomistic simulations in conjunction with high-throughput computing to predict thousands of activation energy barriers for oxygen vacancy migration in the model system of the STO/BZO heterostructure,60 with emphasis on misfit dislocations and the interface region. We studied oxygen vacancy migration pathways at SrO–ZrO2, BaO–TiO2, and ZrO2–TiO2 interfaces having dissimilar misfit dislocation structures.60 The main goal of this work is to offer atomic scale insights pertaining to the migration behavior of oxygen vacancies near misfit dislocations and shed light on the potential influence of differing misfit dislocation structures on ionic conductivity.
2. Methodology
Atomistic simulations with 3D periodic boundary conditions (PBC) were conducted within the framework of large-scale atomic/molecular massively parallel simulator (LAMMPS).61 The simulations are based on energy minimization using a classical Born-like description of an ionic solid. Parameterized Buckingham62 pair potentials given by eqn (1) were utilized to describe the two-body short-range interactions, whereas interactions due to the long-range coulombic (electrostatic) forces were calculated by means of Ewald's method.63
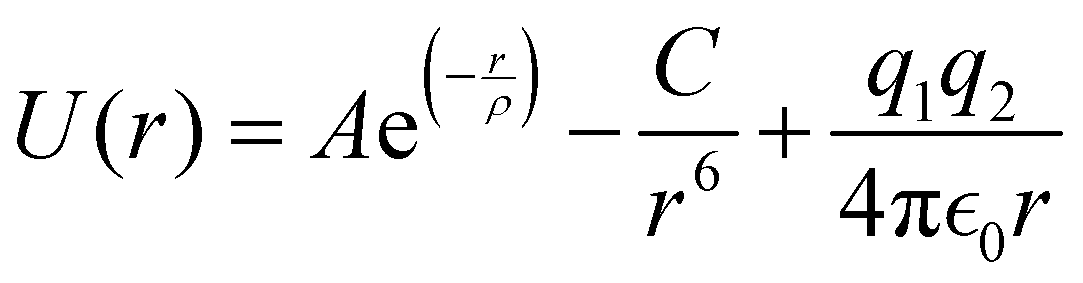 | (1) |
Here, A, ρ, and C are the potential parameters fitted to match the experimental data for a given pair of atoms. The first two terms model Pauli repulsion and van der Waals attraction, respectively, whereas the last term represents the Coulomb potential between ions. These pair potentials are not polarizable, so charge fluctuations are not allowed within this formalism.
Parameters for the Buckingham pair potential as derived by Busker et al.64 and Zacate et al.65 were used for Sr2+, Ti4+, Ba2+, Zr4+, O2−, and Gd3+. These interatomic potential parameters were chosen since all the divalent, trivalent, and tetravalent cation interactions were fitted against the same O2−–O2− potential. Though the potential parameters used in this work were derived for bulk perovskites, they are expected to qualitatively capture the interface effects. The methodology used in this work has been successfully used to study a wide variety of oxide heterostructures.29,54–57,60
Atomic models of the STO/BZO heterostructure were constructed for the experimentally observed cube-on-cube orientation relationship, wherein (001)STO‖(001)BZO‖interface and [010]STO‖[010]BZO.66 Since lattice parameters are aSTO = 3.905 Å and aBZO = 4.197 Å, the lattice mismatch (∼7.0%, computed with respect to a BZO substrate) resulting from this orientation relationship is accommodated via the formation of misfit dislocations at the heterointerface. In these atomic models, 14 unit cells of STO were matched with 13 unit cells of BZO to ensure a heterostructure with a negligible amount of extrinsic strain. The supercell model for heterostructures comprises of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 atoms. Via cleaving STO at either the neutral SrO or the TiO2 layer and BZO at either the neutral BaO or the ZrO2 layer, there are four combinations of heterointerfaces possible, namely SrO–BaO, SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2. For these four fully stoichiometric heterostructures, constant temperature and pressure (NPT) simulations were performed using the conjugate gradient method for internal coordinate and stress minimization using the Nosé–Hoover thermostat at 0 K and zero pressure to obtain a relaxed structure free from internal stresses. The 0 K relaxed structures were further annealed at 1000 K.
900 atoms. Via cleaving STO at either the neutral SrO or the TiO2 layer and BZO at either the neutral BaO or the ZrO2 layer, there are four combinations of heterointerfaces possible, namely SrO–BaO, SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2. For these four fully stoichiometric heterostructures, constant temperature and pressure (NPT) simulations were performed using the conjugate gradient method for internal coordinate and stress minimization using the Nosé–Hoover thermostat at 0 K and zero pressure to obtain a relaxed structure free from internal stresses. The 0 K relaxed structures were further annealed at 1000 K.
A wide variety of trivalent dopants can be added to improve the properties of perovskites, as they have a net effective charge that affects the electroneutrality condition, and thereby the defect equilibria.29–31,37 In perovskites, trivalent dopants can replace either of the cations on the A and B sublattices. For instance, in STO and BZO, donor doping (n-type) can be carried out by replacing Sr2+ or Ba2+ with trivalent dopants, whereas replacing Ti4+ or Zr4+ with trivalent dopants leads to acceptor doping (p-type).26 Since the current work is focused on ionic transport in perovskite heterostructures, we have exclusively studied acceptor doping. The defect reaction governing the addition of trivalent dopants to the TiO2 (STO) or ZrO2 layer (BZO) can be expressed in Kröger–Vink notation as:
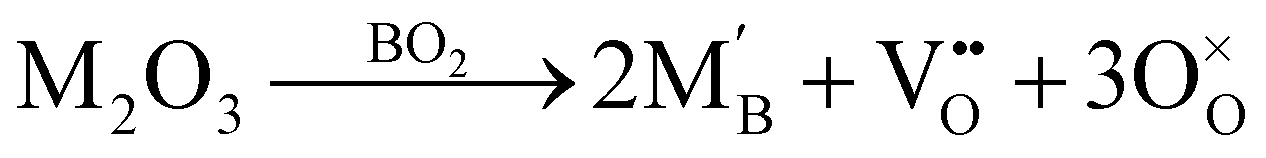 | (2) |
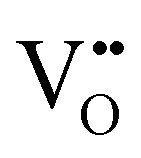 , B, and M correspond to the oxygen vacancy, B-site cations (either Ti4+ or Zr4+) and trivalent dopants (Gd3+), respectively. Owing to ionic compensation, this reaction indicates that the substitution of two B-site cations with trivalent dopants will result in the formation of one oxygen vacancy. The formation and migration of oxygen vacancy are at the heart of our work, so we will strictly focus on its migration in the vicinity of Gd3+ dopants at various interfaces for diverse nearest neighbor scenarios. In p-type oxides used for catalysis, surface oxygen vacancies become doubly positively charged, which lowers their formation energy and results in a higher defect concentration.67 In addition, this study reports that space-charge regions impact the concentration of oxygen vacancies at oxide surfaces. The type of vacancy in this study67 is consistent with the current work. However, studying the fundamental impact of space-charge regions at such complex interfaces is beyond the scope of this work since we study doping scenarios that result in dilute oxygen vacancy concentrations. Furthermore, we explicitly study the role of oxygen vacancies in the neighborhood of trivalent dopants, which is not the case in the study by Richter et al.,67 since it neglects the role of dopant-defect clusters.
, B, and M correspond to the oxygen vacancy, B-site cations (either Ti4+ or Zr4+) and trivalent dopants (Gd3+), respectively. Owing to ionic compensation, this reaction indicates that the substitution of two B-site cations with trivalent dopants will result in the formation of one oxygen vacancy. The formation and migration of oxygen vacancy are at the heart of our work, so we will strictly focus on its migration in the vicinity of Gd3+ dopants at various interfaces for diverse nearest neighbor scenarios. In p-type oxides used for catalysis, surface oxygen vacancies become doubly positively charged, which lowers their formation energy and results in a higher defect concentration.67 In addition, this study reports that space-charge regions impact the concentration of oxygen vacancies at oxide surfaces. The type of vacancy in this study67 is consistent with the current work. However, studying the fundamental impact of space-charge regions at such complex interfaces is beyond the scope of this work since we study doping scenarios that result in dilute oxygen vacancy concentrations. Furthermore, we explicitly study the role of oxygen vacancies in the neighborhood of trivalent dopants, which is not the case in the study by Richter et al.,67 since it neglects the role of dopant-defect clusters.
It is anticipated that DFT calculations would offer a better description of migration pathways by including electronic effects in perovskites. However, these calculations are beyond the scope of this work primarily due to computational time and system size, both of which enter the realm of the impossible when it comes to employing DFT since the supercell size in this work is ∼9.88 × 5.47 × 5.47 nm3 (21900 atoms). Furthermore, we recently computed the electronic structure and predicted the atomic scale structure of misfit dislocations at CeO2/MgO heterostructures using DFT68 and compared it with atomistic simulations.69 We found that the atomic structure and qualitative trends for dopant-defect cluster stability predicted using DFT and atomistic simulations agree very well. This agreement not only validates the current approach, but also reveals that the underlying assumptions of the method remain truthful.
A usual critique of the methodology based on interatomic potentials used in this work is that they are unsuitable for chemical environments for which they are not fitted, such as interfaces and transition states for migration barrier calculations. However, it is important to note that we have rigorously tested the methodology implemented in this work and successfully utilized it to understand the thermodynamics and kinetics of defects at interfaces,29,54,55,60,69,70 surfaces,71,72 steps,56,73 and grain boundaries74–76 in complex oxides. In addition, we have effectively used this methodology to study structure–property relationships at misfit dislocations, as well as computed migration barriers in an intricate chemical environment.55 Crucially, we have used classical forcefields to interpret the experimental observations by predicting qualitative structure–property trends and basic mechanisms at oxide interfaces, which includes perovskites.
To tackle the computation of a vast amount of activation energy barriers at asymmetric interfaces, hundreds for a single interface, and hundreds of thousands for an entire structure, we developed a high-throughput framework using both custom software tools and LAMMPS. High-throughput nudged elastic band (NEB) simulations were performed to map out energy barriers for oxygen vacancy jumps in many possible scenarios. Initial effort was focused on mapping out all nearest-neighbor jumps within one doped interface layer at a time. However, later scenarios eventually moved to mapping such barriers across the entire structure. This framework enabled rapid preparation and mapping of entire scenarios with varying interface chemistry and placement of dopants and vacancies. The approach starts by assigning ten dopants and five resulting vacancies. Later, energy minimization was performed before passing the result onto high-throughput NEB simulations, wherein one moving and four static oxygen vacancies were assigned to the structure, and the NEB simulation was allowed to run its course. Each NEB trajectory consisted of seven replicas with energy and force tolerances of 10−6 eV and 10−6 eV Å−1, respectively.
3. Results & discussion
3.1 Atomic scale structure of the interface
In STO/BZO heterostructures having experimentally observed cube-on-cube orientation relationship, four different types of interfaces are possible depending on the interface layer chemistries involved in the formation of the interface. Fig. 1 depicts the minimized and subsequently annealed structures of four possible STO/BZO interfaces, which are SrO–BaO, SrO–ZrO2, BaO–TiO2, and ZrO2–TiO2 interfaces. In Fig. 1, the heterostructures are shown along the interface normal direction with the relaxed supercells extended in the x and y direction (2 × 2 × 1) to clearly illustrate the misfit dislocation network indicated via black lines. We predict that SrO–BaO, SrO–ZrO2, and TiO2–BaO interfaces have a dislocation structure with Burgers vector of![[b with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0062_20d1.gif) = 〈100〉, whereas the TiO2–ZrO2 interface exhibits a misfit dislocation structure with Burgers vector of = 〈110〉, which is in agreement with the qualitative prediction reported using a heuristic approach.60 While experiments have reported perovskite heterostructures with cube-on-cube epitaxy,18,66,77 dependence of misfit dislocation structure on interface layer chemistry is not well understood. Since these differing misfit dislocation structures as a function of interface chemistry are expected to influence interface-governed properties,29,54 these results reveal the importance of understanding such fundamental aspects of interface formation.
= 〈100〉, whereas the TiO2–ZrO2 interface exhibits a misfit dislocation structure with Burgers vector of = 〈110〉, which is in agreement with the qualitative prediction reported using a heuristic approach.60 While experiments have reported perovskite heterostructures with cube-on-cube epitaxy,18,66,77 dependence of misfit dislocation structure on interface layer chemistry is not well understood. Since these differing misfit dislocation structures as a function of interface chemistry are expected to influence interface-governed properties,29,54 these results reveal the importance of understanding such fundamental aspects of interface formation.
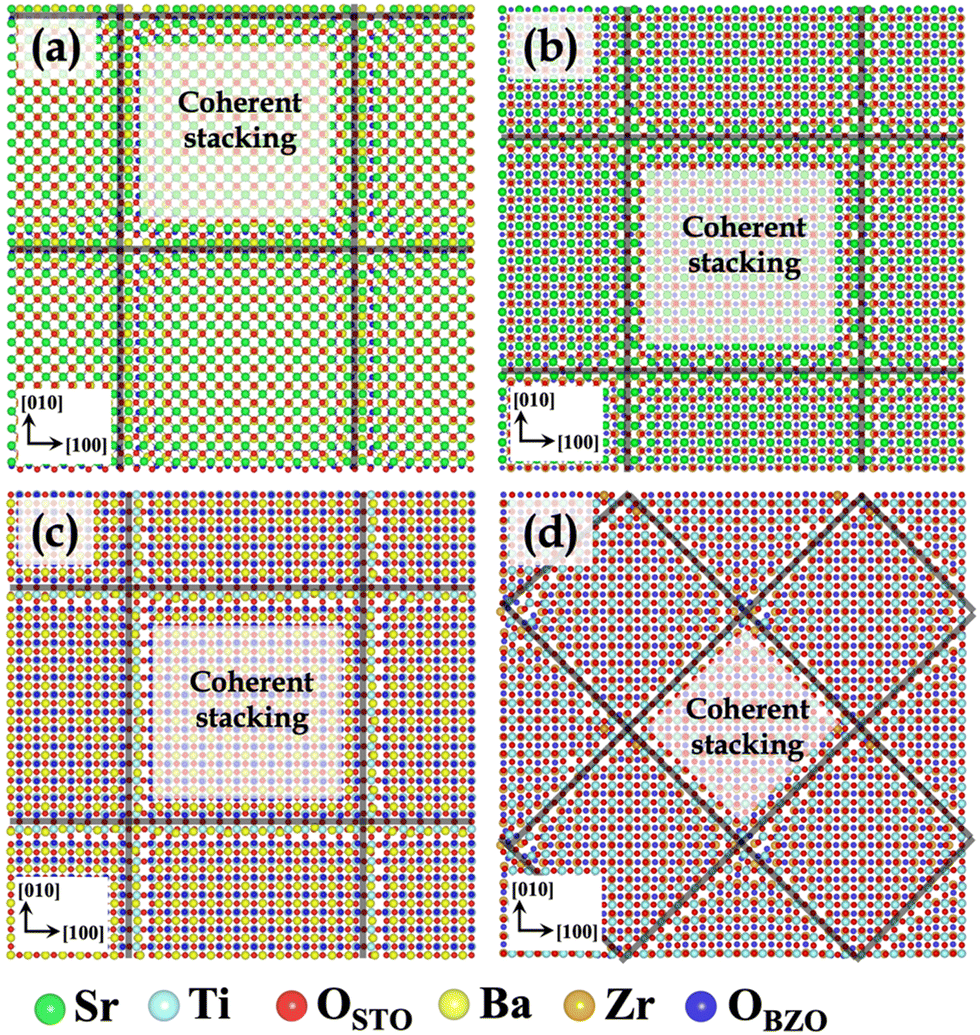 | ||
| Fig. 1 Minimized and subsequently annealed STO/BZO interfaces. (a) SrO–BaO interface (b) SrO–ZrO2 interface (c) BaO–TiO2 interface and (d) TiO2–ZrO2 interface. The view is normal to the interface plane. In (a), (b) and (d), the STO layer is shown on top, whereas in (c), BZO layer is shown on top for a better contrast. Coherent stackings are shown for clarity. Black lines indicate the misfit dislocation network. Atomic colors are given below the figure panel. Oxygen ions in STO and BZO are colored differently for clarity. | ||
A closer inspection of the minimized interface atomic arrangement reveals that there are commonalities as well as dissimilarities between the four interfaces. The four STO/BZO heterostructures (Fig. 1) constitute different types of bonding and atomic stacking at the interface. The coherent stackings were distinguished from the misfit dislocations based on disregistry of atoms in a single layer across the interface. In all the cases, coherent stackings were identified roughly within 1–2 lattice constants of the visibly dislocated area. From here on, we will refer to OSTO (red) and OBZO (blue) as oxygen atoms in STO and BZO layers, respectively. To describe the various stackings, atomic pairs across the interface will be denoted by the {/} notation. That is, {Sr/OBZO} indicates that the Sr atom from STO is coordinated (paired) with an oxygen atom in BZO. Notation [ ] indicates that the particular atomic species does not have any coordinating atom (nearest neighbor bonding species) across the interface.
For the four interfaces (Fig. 1), the coherent terrace comprises of the following nearest neighbor environments: (i) SrO–BaO interface: {Sr/OBZO} and {OSTO/Ba} (ii) SrO–ZrO2 interface: {Sr/[ ]}, {OSTO/Zr}, and {[ ]/OBZO} (iii) TiO2–BaO interface: {Ti/OBZO}, {OSTO/[ ]}, and {[ ]/Ba} (iv) TiO2–ZrO2 interface: {Ti/OBZO}, {OSTO/Zr}, {[ ]/OBZO}, and {OSTO/[ ]}. Evidently, the coherent terraces constitute favorable electrostatic interactions at the four interfaces. The intermediate regions separating the coherent terraces consist of the misfit dislocation lines and intersections. In contrast to the coherent terraces, they exhibit favorable (anion–cation) as well as unfavorable (cation–cation and anion–anion) electrostatic interactions. Repulsive interactions lead to chemical frustration at the interfaces, which are often conducive to defect formation.54,56 Such intricate interface chemical bonding due to the existence of misfit dislocations is observed in other semi-coherent oxide interfaces but has not received much attention in semi-coherent perovskite–perovskite interfaces.60
3.2 Defect migration at interfaces
Oxygen vacancy formation and migration in SOFC electrolytes are fundamental to their performance as an ionic conductor. We have used STO/BZO as a model system to study oxygen vacancy migration at interfaces and elucidate the influence of misfit dislocations on ionic transport in thin film oxide electrolytes. To investigate the fundamental factors influencing the ionic conductivity at interfaces, we have developed a high-throughput strategy to compute thousands of nearest neighbor activation energies for oxygen vacancy migration in the vicinity of Gd3+ dopants at STO/BZO interfaces. The computation of the rate of these events will facilitate the future development of a kinetic lattice Monte Carlo (KLMC) model78 for vacancy diffusion at oxide heterointerfaces, which will further allow for the determination of oxygen ion conductivity. Although the migration energy barriers are calculated at 0 K, temperature effects will be included in the KLMC simulations to understand the trends in ionic conductivity as a function of temperature. We have used a similar strategy to study and design bulk electrolyte materials as well as compare with experimental observations.78,79As mentioned earlier, we strictly focus on doping the TiO2 or ZrO2 layer for ionic conductivity applications. Moreover, since we are primarily interested in the interplay between dopants and oxygen vacancies precisely at the interface layer, the SrO–BaO interface has not been considered for studying oxygen vacancy migration. For SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2 (both sides) interfaces, we studied 4 different configurations for placing 10 dopants and 5 resulting vacancies. Among these configurations, 3 were chosen with dopants placed as nearest neighbors, whereas in the fourth configuration, dopants were isolated. In a large heterostructure supercell with thousands of cations and anions, (21900 atoms in the current system) 10 dopants indicate a rather dilute doping limit. Since we have deliberately placed all the dopants at the interface layer, the dopant fraction of the interface layer is a bit higher ranging from 1.7–1.9%, which is nevertheless in the dilute limit, but reasonable for comparison with realistic scenarios. Nonetheless, the low dopant and vacancy fraction studied in this work can assist in unravelling the atomic scale mechanisms for vacancy migration at interfaces. Via high-throughput simulations, for each of these interface configurations, ∼1000–1400 nearest neighbor barriers were computed for a single side of the interface. In two different cases, the full structure was studied, which accounted for ∼100000 nearest neighbor barriers. In total, ∼250000 activation energy barriers were computed to obtain a comprehensive understanding of oxygen vacancy migration.
Given in Fig. 2a are 8 possible nearest neighbor jump directions for the oxygen vacancy starting from BO2 (B = Ti, Zr) layer. In Fig. 2b, the normal view of the BO2 layer is offered, which depicts the 4 nearest neighbor pathways for in-layer migration of an oxygen vacancy. Since the focus is on understanding BO2 layer migration pathways specifically at the interface, data showing activation energy barriers for oxygen vacancy migration will be shown for the network consisting of the 4 pathways given in Fig. 2b. It is important to note that though activation energy barriers were computed for vacancy migration throughout the supercell, we limit the discussion to interface layer. This strategy ensures that we capture the effect of misfit dislocations and their interplay with dopants and oxygen vacancies.
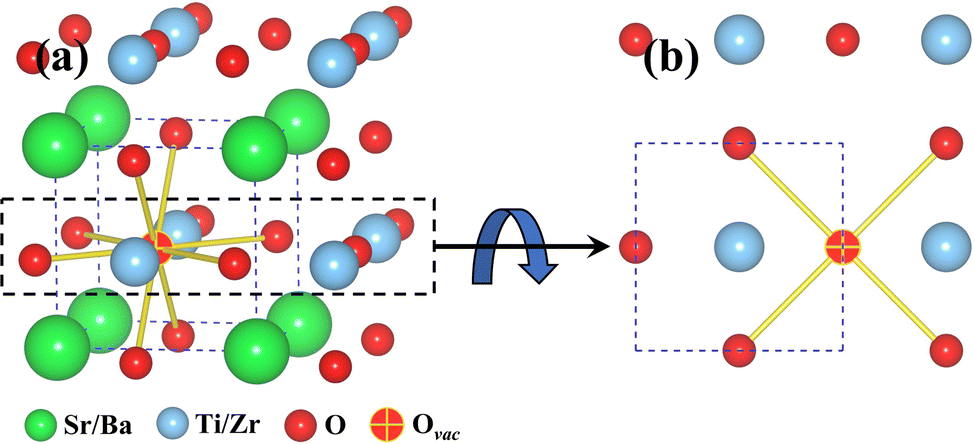 | ||
| Fig. 2 (a) Possible 8 nearest neighbor jump directions for an oxygen vacancy in either TiO2 (STO) layer or ZrO2 (BZO) layer are shown. (b) Normal view for the TiO2 or ZrO2 layer depicting the 4 nearest neighbor in-layer jump directions. These 4 directions will be used in the following figures to show the magnitude of activation energy barriers on each side of the heterointerface. The atomic color scheme is given below the figure. | ||
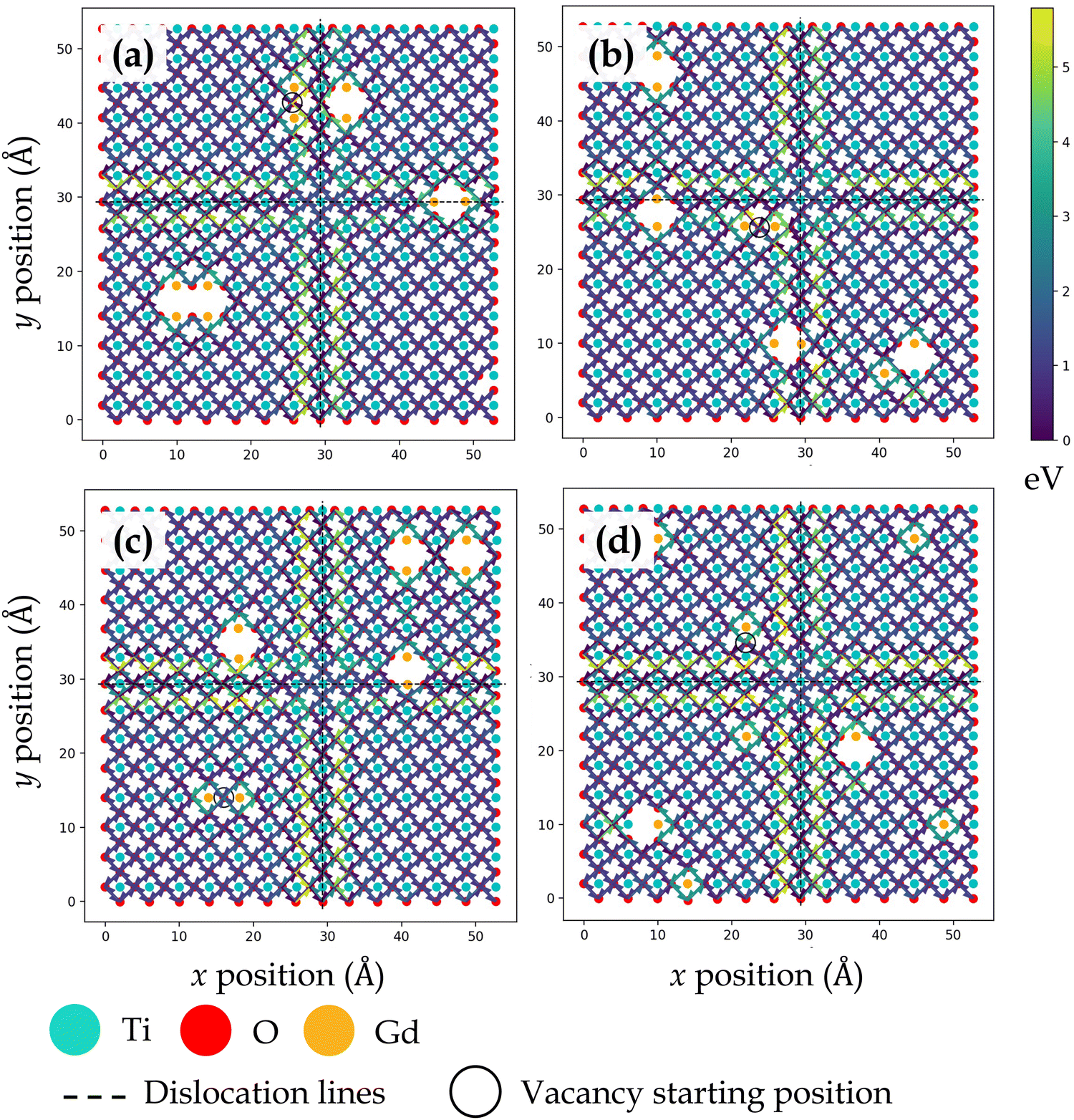
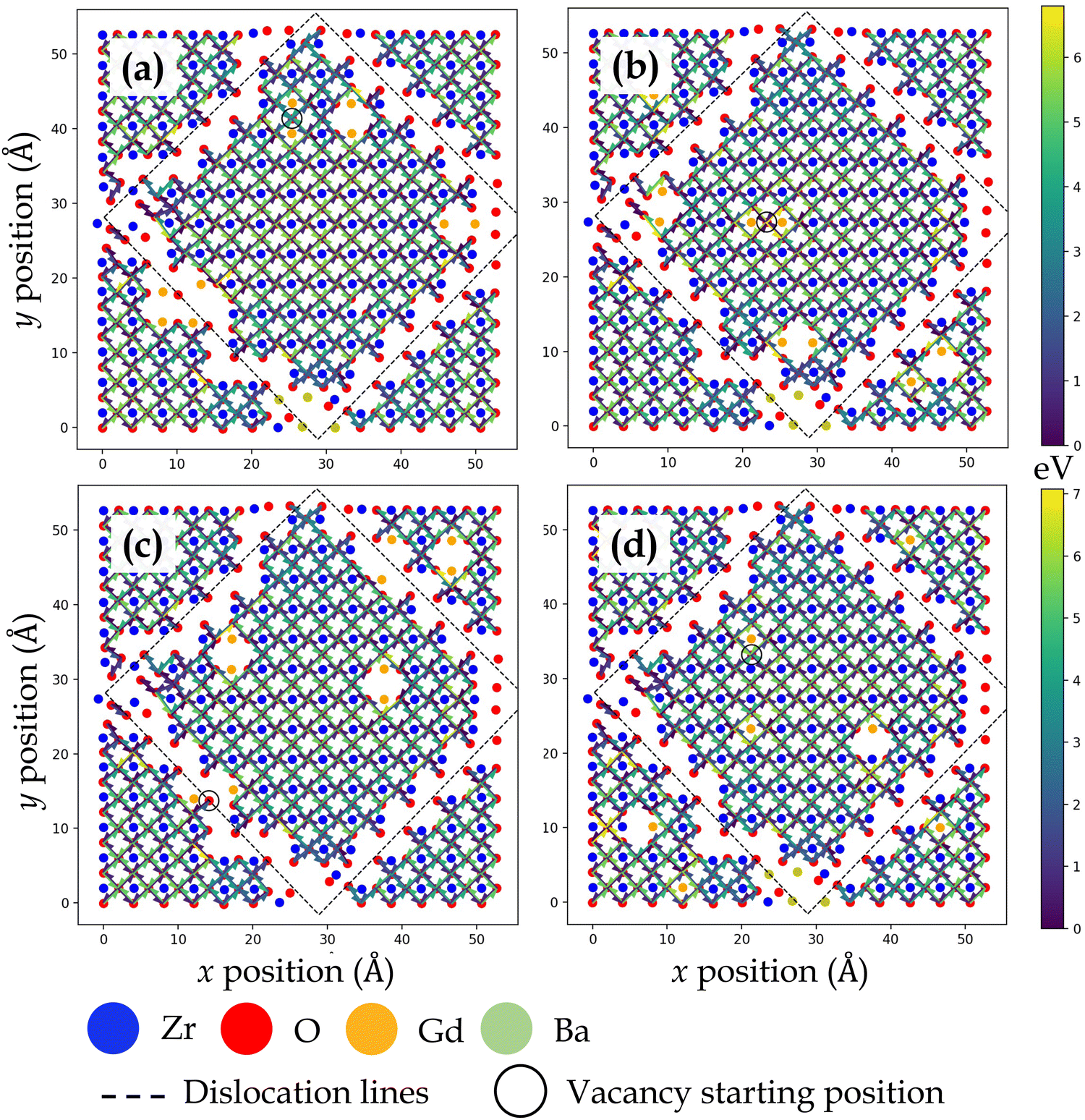
Fig. 3, 4, 5 and 6 display the activation energies for nearest neighbor oxygen vacancy migration jumps in the vicinity of different dopant distributions at ZrO2 side of SrO–ZrO2, TiO2 side of TiO2–BaO, TiO2 side of TiO2–ZrO2, and ZrO2 side of TiO2–ZrO2 interfaces, respectively. Migration barriers connecting longer jumps (longer than the next nearest neighbor vacancy hops) across the misfit dislocations were not computed, which is evident in all figures. In these figures, for respective configurations given in a, b, and c, the gadolinium dopants are placed in nearest neighbor pairs, whereas in d, they are isolated. The primary reason to place dopants in pairs at nearest neighbors to the oxygen vacancy is that this configuration was found to be most favorable energetically at misfit dislocations in STO/BZO heterostructures.70 Activation energies for oxygen vacancy migration are depicted via arrows pointing in a particular jump direction and their colors indicate the absolute magnitude given in the respective energy scale. The activation energies reported in this work are a bit higher in magnitude, especially near misfit dislocations. However, interfaces are likely to induce such high barriers since high activation energy barriers from DFT have also been recently reported for perovskite surfaces.48 Furthermore, activation energies for oxygen vacancy migration in bulk STO and bulk BZO were found to be 1.20 eV and 1.0 eV, respectively. The computed value for STO is in reasonable agreement with reported DFT,80 atomistic simulations,81 and experimental values,82 which offers confidence in the current method. In addition, it has been shown that the methodology used in this work is effective in predicting transport mechanisms in perovskites.83 Vitally, the main aim of this work is to offer physically meaningful qualitative trends to describe the behavior of oxygen vacancies at misfit dislocations, which necessitate large supercells that are not feasible in DFT calculations.57 In this section, we will strictly focus on qualitative trends and discuss their implications.
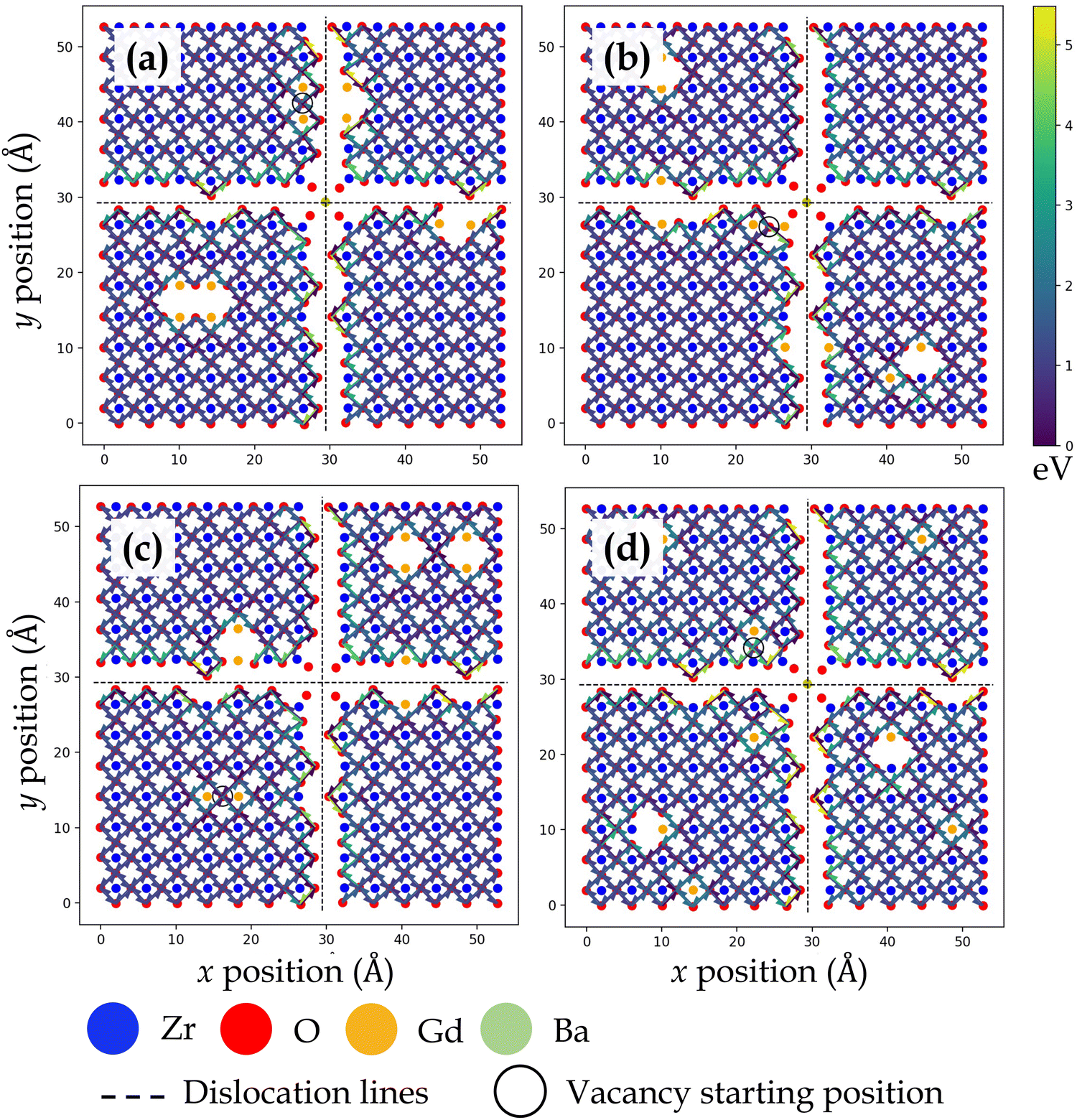 | ||
| Fig. 3 Activation energy barriers for oxygen vacancy defect migration on the ZrO2 side of the SrO–ZrO2 interface. In (a)–(c), two Gd3+ dopants are placed next to the oxygen vacancy in different locations. In (d), each Gd3+ dopant ion is isolated, but the total number of dopants and resultant oxygen vacancy concentration is consistent with previous configurations. The atomic color scheme is given below the figure panel and the energy scale is given on the right in Fig. 3–6. The vacancy starting location is indicated with an open circle and misfit dislocations are indicated by dashed lines. The view is normal to the interface, same as in Fig. 1. | ||
 | ||
| Fig. 4 Activation energy barriers for oxygen vacancy defect migration on the TiO2 side of the BaO–TiO2 interface. In (a)–(c), two Gd3+ dopants are placed next to the oxygen vacancy in different locations. In (d), each Gd3+ dopant ion is placed isolated, but the total number of dopants and resultant oxygen vacancy concentration is consistent with previous configurations. | ||
 | ||
| Fig. 5 Activation energy barriers for oxygen vacancy defect migration on the ZrO2 side of the TiO2–ZrO2 interface. In (a)–(c), two Gd3+ dopants are placed next to the oxygen vacancy in different locations. In (d), each Gd3+ dopant ion is placed isolated, but the total number of dopants and resultant oxygen vacancy concentration is consistent with previous configurations. | ||
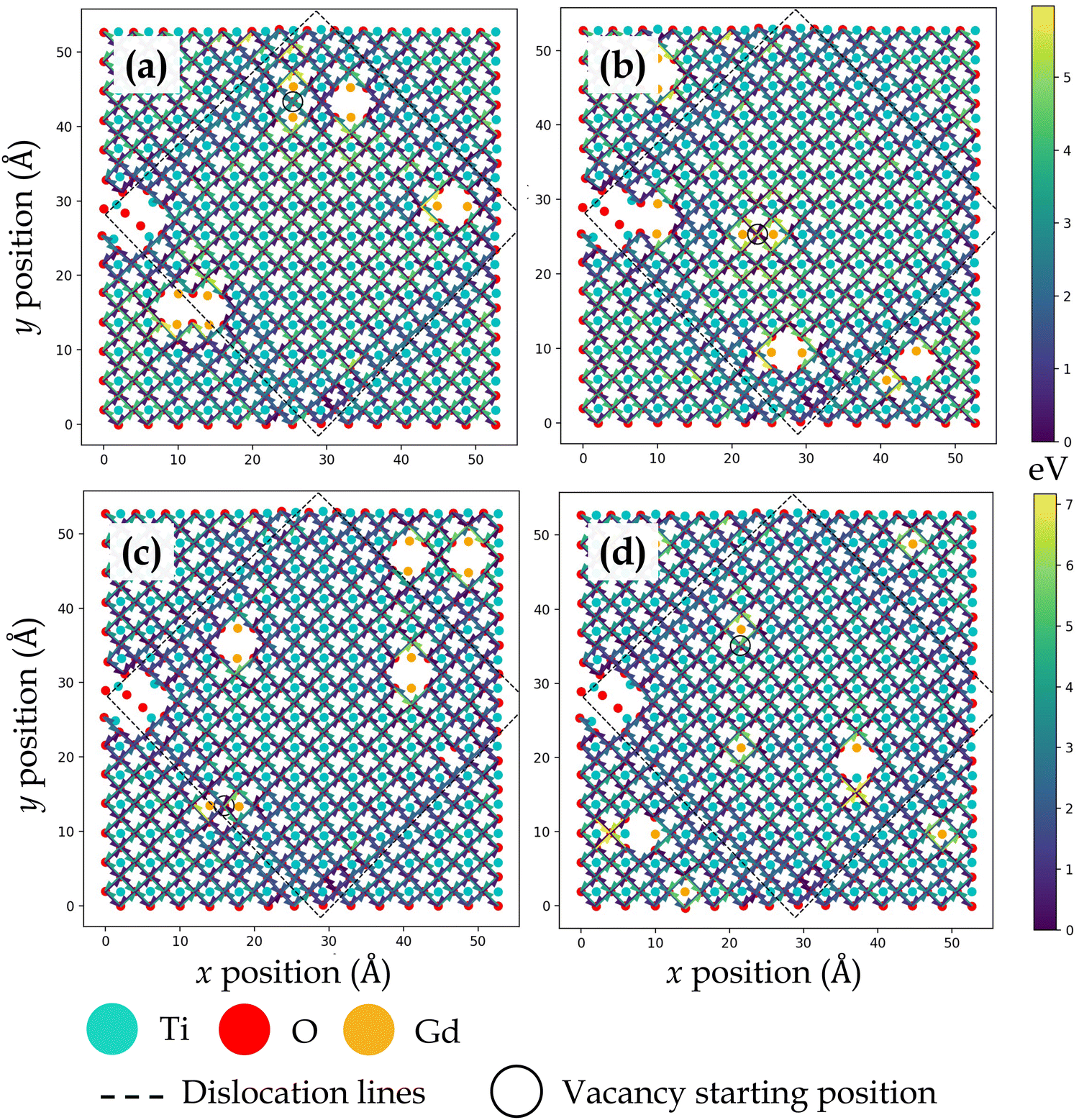 | ||
| Fig. 6 Activation energy barriers for oxygen vacancy defect migration on the TiO2 side of the TiO2–ZrO2 interface. In (a)–(c), two Gd3+ dopants are placed next to the oxygen vacancy in different locations. In (d), each Gd3+ dopant ion is placed isolated, but the total number of dopants and resultant oxygen vacancy concentration is consistent with previous configurations. | ||
TiO2–BaO and SrO–ZrO2 interfaces have comparable chemical environments due to AO–BO2 interface chemistry. At the ZrO2 side of SrO–ZrO2 interface (Fig. 3), throughout the coherent (Fig. 1b) terrace (stacking), activation energies are comparable in magnitude. Analogously, at the TiO2 side of TiO2–BaO interface (Fig. 4), activation energies are more or less similar in magnitude throughout the coherent terrace (Fig. 1c). This can be understood in terms of similar nearest neighbor environments in coherent terraces in SrO–ZrO2 and TiO2–BaO interfaces. For respective coherent regions in SrO–ZrO2 and TiO2–BaO interfaces, OBZO and OSTO do not have a nearest neighbor cation or anion across the interface, which results in similar barrier heights when oxygen vacancies hop around in this landscape. That is, oxygen vacancy migration from {[ ]/OBZO} → {[ ]/OBZO} and {OSTO/[ ]} → {OSTO/[ ]} exhibit similar migration tendencies due to comparable nearest neighbor environments across the interface. The barrier height for a given vacancy to migrate in one direction and hop back to the same site (forward and backward jumps) is nearly equal except for the case where the vacancy is next to the dopants. Though the interface layers in the coherent terrace have somewhat more open space as compared to the grain interior, the migration barriers are comparable to those computed in the bulk.
Near misfit dislocations in SrO–ZrO2 and TiO2–BaO interfaces, the migration tendencies of oxygen vacancies are different than those observed in the coherent regions. At SrO–ZrO2 (Fig. 3) and TiO2–BaO (Fig. 4) interfaces, oxygen vacancies exhibit lower activation energies migrating toward the misfit dislocations but higher energies migrating away from them. At the TiO2–BaO interface, a characteristic difference emerges at the column of atoms next to misfit dislocations as oxygen vacancies reveal higher barriers migrating toward this column, but lower barriers migrating away from it, creating a distinct pattern in the spread of activation energies near misfit dislocations. In general, this trend reveals that oxygen vacancies might get trapped since high thermal energy would be necessitated to overcome these barriers on either side of the column. The barriers for in-layer jumps across the misfit dislocations on the ZrO2 side of SrO–ZrO2 (Fig. 3) interface were not computed because the jump distances involved are larger than the next neighboring distances.
At TiO2 and ZrO2 side of the TiO2–ZrO2 interface, both layers across the interface have comparable chemical environments due to BO2–BO2 interface chemistry. At ZrO2 side (Fig. 5) and TiO2 side (Fig. 6), oxygen vacancy migration in coherent terraces reveals a peculiar trait owing to oxygen ions having two types of nearest neighbor environment across the interface (Fig. 1d). At the ZrO2 side, the coherent terrace contains {Ti/OBZO} and {[ ]/OBZO} nearest neighbors, whereas at the TiO2 side, it involves {OSTO/Zr} and {OSTO/[ ]}. At the ZrO2 side, when oxygen vacancy migrates from neighborhood {Ti/OBZO} → {[ ]/OBZO}, the migration barriers are low, but when it migrates from {[ ]/OBZO} → {Ti/OBZO} or for the reverse jump, the migration barriers are high. An analogous trend is uncovered at the TiO2 side, wherein the vacancy migration from neighborhood {OSTO/Zr} → {OSTO/[ ]} involves low barriers, but the reverse jump or migration from {OSTO/[ ]} → {OSTO/Zr} exhibit high barriers. Because one potential explanation for the observed trend can be offered on the basis of oxygen vacancy formation, we computed relative oxygen vacancy formation energies for a few cases at the interface layers in TiO2–ZrO2 interface. Oxygen vacancy formation was found to be significantly more favorable at neighborhoods {[ ]/OBZO} and {OSTO/[ ]} as compared to {Ti/OBZO} and {OSTO/Zr}, respectively. The observed outcome is plausible since it is energetically favorable to remove an oxygen ion from a site where it has no nearest neighbor across the interface ({[ ]/OBZO} or {OSTO/[ ]}) as compared to the location where it has a counterion ({Ti/OBZO} or {OSTO/Zr}) in an electrostatically favorable region. This result is in reasonable agreement with reported results for oxygen vacancy formation at oxide heterostructures.54–56,68 The observed trend reveals a fundamental influence of interface layer chemistry on migration barriers since oxygen vacancies exhibit strong preference to migrate at sites that do not have nearest neighbor ions located across the interface.
Oxygen vacancy migration trends in close proximity to misfit dislocations at both sides of the TiO2–ZrO2 interface are dissimilar to their respective coherent regions. At both ZrO2 side (Fig. 5) and TiO2 side (Fig. 6), lower activation energies are uncovered when oxygen vacancies hop toward the misfit dislocations, but higher activation energies are uncovered when they migrate away from them. For a few oxygen ions bordering the misfit dislocations (hanging oxygens in Fig. 5) on the ZrO2 side, a contrasting behavior is witnessed, wherein the activation energies are higher moving toward the misfit dislocations but lower moving away from them. However, once the vacancies move one jump distance from this location, they reach a site where all the barriers diffusing the vacancy away from it are noticeably higher. Essentially, the oxygen vacancies have a high likelihood of getting trapped at those sites. As stated earlier, the barriers for in-layer jumps across the misfit dislocations on the ZrO2 side were not computed since these jump distances were larger than the next nearest neighbor.
As compared to the coherent terraces, the discernable patterns in activation energy barriers emerging at misfit dislocations across the interfaces can be attributed to the atomic scale structure of misfit dislocations, which in turn depends on the interface layer chemistry as well as the concomitant strain at the interface. For instance, the respective interface layer arrangement of atoms in ZrO2 (Fig. S1b, ESI†) and TiO2 (Fig. S1c, ESI†) layers in SrO–ZrO2 and TiO2–BaO interfaces is noticeably different, which ultimately influence the pattern of activation energy barriers displayed in Fig. 3 and 4. Since SrO–ZrO2 and TiO2–BaO interfaces exhibit misfit dislocations in the 〈100〉 crystal direction (Fig. 1b and c), the variations in activation energies create a distinct pattern along the 〈100〉 direction. On the other hand, the respective interface layer arrangement of atoms in ZrO2 (Fig. S2d, ESI†) and TiO2 (Fig. S2c, ESI†) layers in TiO2–ZrO2 interface is dissimilar since the misfit dislocations are formed in the 〈110〉 direction (Fig. 1d), which dictates the trend in activation energies depicted in Fig. 5 and 6. The SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2 interfaces exhibit a common behavior, wherein misfit dislocations push the interface layer atoms away from them, which creates somewhat larger space in their vicinity. This extra room makes it energetically favorable for larger oxygen ions to hop toward the misfit dislocations and less favorable to move away from them.
Since there are fewer atoms on the BZO side (13 unit cells) than the STO side (14 unit cells), there is a bit more room in the vicinity of misfit dislocations on the BZO side (Fig. S1 and S2, ESI†), which further influences the observed trends. Select few calculations of relative oxygen vacancy formation at misfit dislocations reveal that they exhibit lower formation energies at misfit dislocations due to the presence of chemically frustrated regions that are conducive to defect formation and availability of open space. The favorability of oxygen vacancy formation at misfit dislocations can also be inferred from activation energies for vacancy migration since these two quantities are fundamentally correlated.84 Because oxygen vacancy formation is energetically favorable at misfit dislocations in SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2 interfaces, coupled with the effect that migration of these species toward misfit dislocations is favorable, a significant fraction of oxygen vacancies is expected in this locality. However, despite their higher concentration, these mobile species would have a lower probability of diffusing away from misfit dislocations owing to high activation barriers, which is likely to impact ionic transport in the vicinity of misfit dislocations.55
Dopants display a noticeable influence on activation energies. Coherent regions in SrO–ZrO2, TiO2–BaO, and TiO2–ZrO2 interfaces that exhibit a change in activation energies are primarily in the neighborhood of dopants, which in general lead to an increase in activation energies. Placing the dopants in nearest neighbor pairs versus isolated does not have any significant impact on the migration behavior. Owing to electrostatic interactions and vacancy-dopant association, oxygen vacancies have low activation energy barriers to hop toward or between the two Gd3+ dopants in the coherent terrace (Fig. 3c, 4c, 5b and 6c) but would require higher thermal energy to move out of the vacancy–dopant complex. This behavior is uncovered at misfit dislocations as well, where the barriers to hop toward the two Gd3+ dopants are lower but those hopping away are higher (Fig. 3a, b, 4a, b, 5a, b, 6a and c). To offer a glimpse of the distribution of energy barriers that exhibit the highest, lowest, most accessible, and least accessible values at interface locations, 2D histograms for migration energy barriers at each of the four interfaces are given in Fig. 7. Here, data are aggregated from all four scenarios performed for each particular interface layer and the distance is calculated using the migration pathway's closest point to the nearest dislocation line. Evidently, for the SrO–ZrO2 and TiO2–BaO interfaces, energy barriers remain fairly consistent (∼1–1.5 eV), with some spread closer to the dislocation lines. For the TiO2–ZrO2 interface, an alternating pattern explained earlier is visible but further from the dislocation lines, with the barrier value converging towards ∼2–3 eV closer to misfit dislocations. In general, the SrO–ZrO2 and TiO2–BaO interfaces exhibit lower barriers. Largely, the influence of dopants in increasing the activation energies is more pronounced at misfit dislocations as compared to coherent terraces, a behavior in contrast with SOFC electrolytes, wherein trivalent dopants induce low energy pathways for vacancy migration in the grain interior of these materials.85–87
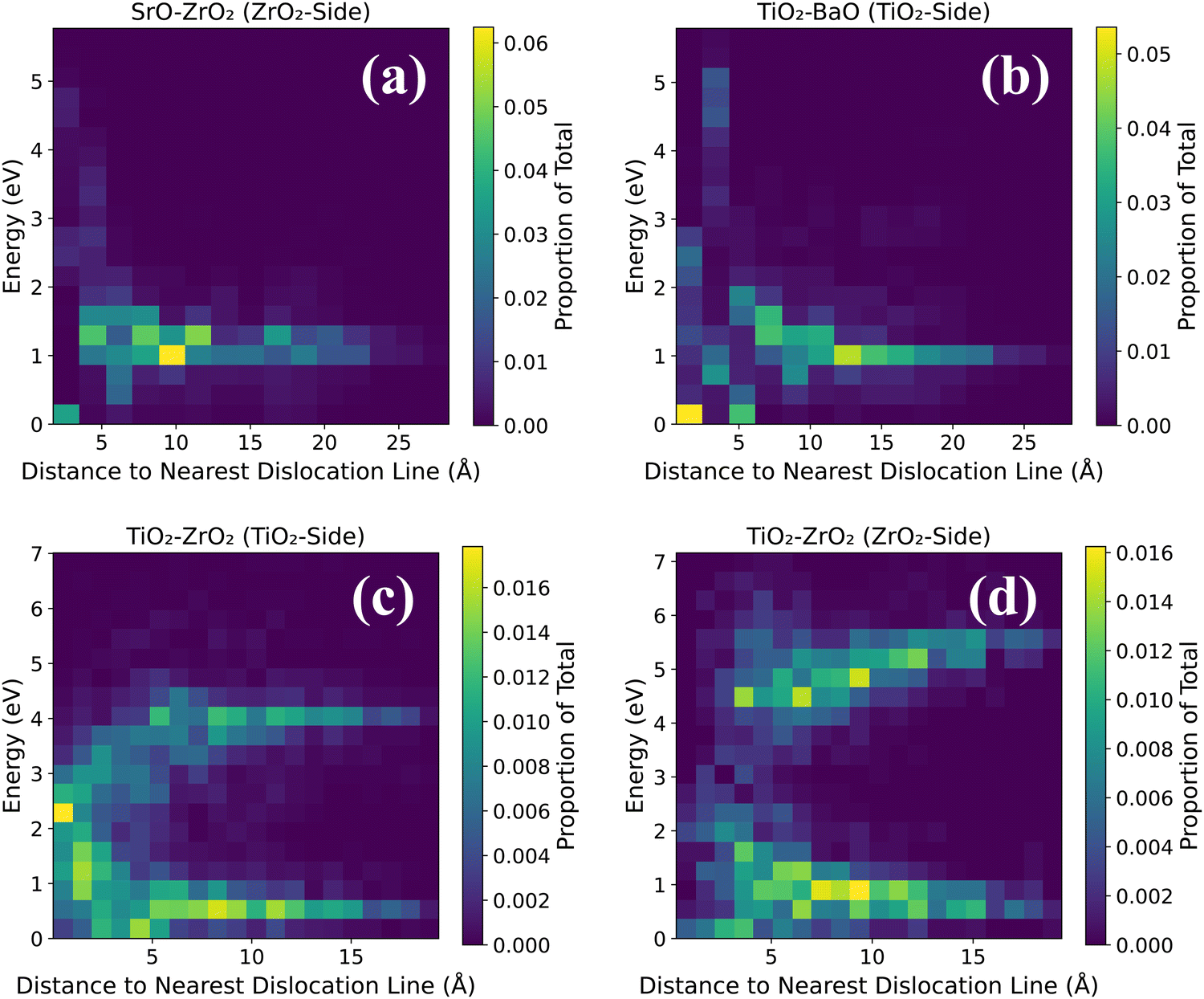 | ||
| Fig. 7 2D histograms for migration energy barriers at the four interfaces (a) SrO–ZrO2 (ZrO side), (b) TiO2–BaO (TiO2 side), (c) TiO2–ZrO2 (TiO2 side), and (d) TiO2–ZrO2 (ZrO2 side). Data are aggregated from all four scenarios performed for each particular interface layer. Distance is calculated using the migration pathway's closest point to the nearest dislocation line. | ||
Literature work focusing on examining the role of oxide interfaces for ionic conductivity applications has reported polarizing results. Several experiments have reported superionic conductivity in oxide heterostructures,8,19,51–53,88,89 where misfit dislocations are hypothesized as potential pathways for fast ionic diffusion. In contrast, few experiments have demonstrated that misfit dislocations slow down ionic transport in oxides.90–92 Adding to the mystery are computational studies that report slower ionic diffusion at misfit dislocations55 and dislocation lines in homophase oxides.93,94 Nonetheless, the atomistic origin for this enhancement or impediment at misfit dislocations is not well understood. Fundamentally, analogous to the atomic arrangement uncovered in perovskite oxide heterostructures, mismatched oxide heterostructures containing misfit dislocations at the interface are likely to have electrostatically balanced coherent terraces separated by misfit dislocations having chemically frustrated neighborhoods and complex atomic stacking.54,57,60,68 As a result, atomic level insights obtained herein for oxygen vacancy migration near misfit dislocations are germane to a broader class of mismatched oxide heterostructures. Dependence of activation energies on the atomic scale structure of misfit dislocations, especially as a function of interface layer chemistry, could be one of the fundamental factors responsible for reported inconsistencies in ionic conductivity measurements. For instance, owing to the challenges involved in experimentally characterizing individual columns of misfit dislocations, it is likely that for the same thin film oxide heterostructure,19,51,52,90–92 measurements performed for differing interface layer chemistry could yield varying ionic conductivity values since the dynamic behavior of oxygen vacancies would be altered as a function of the atomic structure of misfit dislocations.
Present findings demonstrate a broad range of activation energies for oxygen vacancy defect migration at oxide interfaces that are contingent on the nearest neighbor environment and interface layer chemistry. This work sheds light on the intricacy associated with disentangling the fundamental aspects of oxide ion migration at mismatched oxide interfaces, which is noticeably different from that observed in the grain interior. Vitally, it suggests that to develop design principles for thin film oxide electrolytes with enhanced ionic conductivity, it is imperative to understand the atomic scale structure of extended defects at interfaces and study their influence on vacancy defect migration since the basic understanding of bulk behavior is not sufficient. Lastly, it is essential to note that these results do not directly provide insights into diffusion kinetics. However, the rate of these events for nearest neighbor migration, that is the activation energies for oxygen vacancy migration at interfaces, can be incorporated into a KLMC model to study diffusion kinetics of oxygen vacancies as a function of temperature. To this end, a KLMC model is currently being developed by our group, which will utilize these input rates to simulate ionic conductivity at misfit dislocations in oxide heterostructures.
4. Conclusions
Formation and migration of oxygen vacancy defects at oxide interfaces is the heart of the functioning of thin film oxide electrolytes. To this end, we have developed a high-throughput framework to predict the activation energy barriers for oxygen vacancy defect migration at misfit dislocations in perovskite oxide heterostructures. The present work offers a comprehensive account of activation energy barriers for diverse interface layer chemistries amid two lattice mismatched perovskite oxides. We have demonstrated the dependence of the migrating defect on the nearest neighboring atomic environment across the oxide interface, which in turn is contingent on the atomic scale structure of misfit dislocations. Contrasting migration tendencies of oxygen vacancies were uncovered at coherent terraces as compared to misfit dislocations revealing the fundamental effect of extended defects on ionic transport. Activation energy barriers for vacancies hopping toward misfit dislocations exhibit lower magnitude, whereas moving away from them necessitates higher energy, indicating that oxygen vacancies would get trapped at misfit dislocations impacting ionic conductivity. Atomic scale insights offered herein pertaining to the energy landscape for oxygen vacancy migration at oxide interfaces will be instrumental in designing next-generation SOFC electrolytes, wherein the role of misfit dislocations at interfaces is central, but not well understood.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation (NSF) CAREER Award grant number 2042311 in the Division of Materials Research. The authors gratefully acknowledge resources from the Research Computing at Rochester Institute of Technology. This work used Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number ACI-1548562.References
- P. Zubko, S. Gariglio, M. Gabay, P. Ghosez and J.-M. Triscone, Interface Physics in Complex Oxide Heterostructures, Ann. Rev. Condens. Matter Phys., 2011, 2, 141 CrossRef CAS.
- J. A. Sulpizio, S. Ilani, P. Irvin and J. Levy, Nanoscale Phenomena in Oxide Heterostructures, Ann. Rev. Mater. Res., 2014, 44, 117 CrossRef CAS.
- N. Pryds and V. Esposito, When Two Become One: An Insight into 2D Conductive Oxide Interfaces, J. Electroceram., 2017, 38, 1 CrossRef CAS.
- P. Yu, Y.-H. Chu and R. Ramesh, Oxide Interfaces: Pathways to Novel Phenomena, Mater. Today, 2012, 15, 320 CrossRef CAS.
- Z. Huang, Ariando, X. R. Wang, A. Rusydi, J. Chen, H. Yang and T. Venkatesan, Interface Engineering and Emergent Phenomena in Oxide Heterostructures, Adv. Mater., 2018, 30, 1802439 CrossRef.
- H. Y. Hwang, Y. Iwasa, M. Kawasaki, B. Keimer, N. Nagaosa and Y. Tokura, Emergent Phenomena at Oxide Interfaces, Nat. Mater., 2012, 11, 103 CrossRef CAS PubMed.
- J. A. Kilner and M. Burriel, Materials for Intermediate-Temperature Solid-Oxide Fuel Cells, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Ionic Conductivity in Oxide Heterostructures: The Role of Interfaces, Sci. Technol. Adv. Mater., 2010, 11, 054503 CrossRef.
- K. E. Sickafus, R. W. Grimes, J. A. Valdez, A. Cleave, M. Tang, M. Ishimaru, S. M. Corish, C. R. Stanek and B. P. Uberuaga, Radiation-induced Amorphization Resistance and Radiation Tolerance in Structurally Related Oxides, Nat. Mater., 2007, 6, 217–223 CrossRef CAS.
- M. Salluzzo, S. Gariglio, D. Stornaiuolo, V. Sessi, S. Rusponi, C. Piamonteze, G. M. De Luca, M. Minola, D. Marré, A. Gadaleta, H. Brune, F. Nolting, N. B. Brookes and G. Ghiringhelli, Origin of Interface Magnetism in BiMnO3/SrTiO3 and LaAlO3/SrTiO3 Heterostructures, Phys. Rev. Lett., 2013, 111, 087204 CrossRef CAS.
- B. Zhu, L. Fan and P. Lund, Breakthrough Fuel Cell Technology Using Ceria-based Multi-functional Nanocomposites, Appl. Energy, 2013, 106, 163–175 CrossRef CAS.
- J. Wang, G. Ji, Y. Liu, M. A. Gondal and X. Chang, Cu2O/TiO2 Heterostructure Nanotube Arrays Prepared by an Electrodeposition Method Exhibiting Enhanced Photocatalytic Activity for CO2 Reduction to Methanol, Catal. Commun., 2014, 46, 17–21 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Materials for Fuel-cell Technologies, Nature, 2001, 414, 345 CrossRef CAS PubMed.
- A. Boudghene Stambouli and E. Traversa, Solid Oxide Fuel Cells (SOFCs): A Review of an Environmentally Clean and Efficient Source of Energy, Renewable Sustainable Energy Rev., 2002, 6, 433 CrossRef.
- S. C. Singhal and K. Kendall, High-temperature Solid Oxide Fuel Cells: Fundamentals, Design and Applications, Elsevier, Oxford, 2003 Search PubMed.
- E. D. Wachsman and K. T. Lee, Lowering the Temperature of Solid Oxide Fuel Cells, Science, 2011, 334, 935 CrossRef CAS.
- A. Jaiswal, A. Pesaran, S. Omar and E. D. Wachsman, Ceria/Bismuth Oxide Bilayer Electrolyte Based Low-Temperature SOFCs with Stable Electrochemical Performance, ECS Trans., 2017, 78, 361 CrossRef CAS.
- J. Santiso, J. Roqueta, N. Bagues, C. Frontera, Z. Konstantinovic, Q. Lu, B. Yildiz, B. Martínez, A. Pomar, L. Balcells and F. Sandiumenge, Self-Arranged Misfit Dislocation Network Formation upon Strain Release in La0.7Sr0.3MnO3/LaAlO3(100) Epitaxial Films under Compressive Strain, ACS Appl. Mater. Interfaces, 2016, 8, 16823–16832 CrossRef CAS.
- C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Ionic conductivity and activation energy for oxygen ion transport in superlattices—the semicoherent multilayer system YSZ-(ZrO2 + 9.5 mol% Y2O3)/Y2O3, Phys. Chem. Chem. Phys., 2008, 10, 4623–4635 RSC.
- G. F. Harrington, A. Cavallaro, D. W. McComb, S. J. Skinner and J. A. Kilner, The Effects of Lattice Strain, Dislocations, and Microstructure on the Transport Properties of YSZ Films, Phys. Chem. Chem. Phys., 2017, 19, 14319–14336 RSC.
- D. Choudhury, B. Pal, A. Sharma, S. V. Bhat and D. D. Sarma, Magnetization in Electron- and Mn-doped SrTiO3, Sci. Rep., 2013, 3, 1433 CrossRef CAS PubMed.
- R. A. Maier and C. A. Randall, Low-Temperature Ionic Conductivity of an Acceptor-Doped Perovskite: I. Impedance of Single-Crystal SrTiO3, J. Am. Ceram. Soc., 2016, 99(10), 3350–3359 CrossRef CAS.
- J. Cheng, Y. Wang, J. Luo and K. Yang, δ-Doping Effects on Electronic and Energetic Properties of LaAlO3/SrTiO3 Heterostructure: First-Principles Analysis of 23 Transition-Metal Dopants, Adv. Mater. Interfaces, 2017, 4, 1700579 CrossRef.
- E. W. McFarland and H. Metiu, Catalysis by Doped Oxides, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- Y. Pai, A. Tylan-Tyler, P. Irvin and J. Levy, Physics of SrTiO3-based Heterostructures and Nanostructures: A Review, Rep. Prog. Phys., 2018, 81, 036503 CrossRef.
- W. Luo, W. Duan, S. G. Louie and M. L. Cohen, Structural and Electronic Properties of n-doped and p-doped SrTiO3, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 214109 CrossRef.
- P. C. Bowes, J. N. Baker and D. L. Irving, Survey of acceptor dopants in SrTiO3: factors limiting room temperature hole concentration, J. Am. Ceram. Soc., 2020, 103, 1156–11733 CrossRef CAS.
- C. Y. R. Vera, H. Ding, D. Peterson, W. T. Gibbons, M. Zhou and D. Ding, A mini-review on proton conduction of BaZrO3-based perovskite electrolytes, J. Phys. Energy, 2021, 3, 032019 CrossRef CAS.
- P. P. Dholabhai, E. Martinez and B. P. Uberuaga, Influence of Chemistry and Misfit Dislocation Structure on Dopant Segregation at Complex Oxide Heterointerfaces, Adv. Theory Simul., 2019, 2, 1800095 CrossRef.
- S. Hui and A. Petric, Electrical Properties of Yttrium-Doped Strontium Titanate under Reducing Conditions, J. Electron. Soc., 2002, 149, J1–J10 CrossRef CAS.
- C. Ohly, S. Hoffmann-Eifert, K. Szot and R. Waser, Electrical Conductivity and Segregation Effects of Doped SrTiO3 Thin Films, J. Eur. Ceram. Soc., 2001, 21, 1673–1676 CrossRef CAS.
- D. V. Azamat, A. G. Badalyan, A. Dejneka, V. A. Trepakov, L. Jastrabik and Z. Frait, High-frequency Electron Paramagnetic Resonance Investigation of Mn3+ Centers in SrTiO3, J. Phys. Chem. Solids, 2012, 73, 822–826 CrossRef CAS.
- H. Yang, H. S. Lee, P. G. Kotula, Y. Sato, Y. Ikuhara and N. D. Browning, Amphoteric Doping of Praseodymium Pr31 in SrTiO3 Grain Boundaries, Appl. Phys. Lett., 2015, 106, 121904 CrossRef.
- L. Dai, L. Wu, H. Li, H. Hu, Y. Zhuang and K. Liu, Evidence of the Pressure-induced Conductivity Switching of Yttrium-doped SrTiO3, J. Phys.: Condens. Matter, 2016, 28, 475501 CrossRef.
- R. Merkle and J. Maier, Defect Association in Acceptor-doped SrTiO3: Case Study for FeTiVO and MnTiVO, Phys. Chem. Chem. Phys., 2003, 5, 2297–2303 RSC.
- M. Longo, M. G. S. Costa, A. Z. Simoes, I. L. V. Rosa, C. O. P. Santos, J. Andres, E. Longo and J. A. Varela, On the Photoluminescence Behavior of Samarium-doped Strontium Titanate Nanostructures Under UV Light. A Structural and Electronic Understanding, Phys. Chem. Chem. Phys., 2010, 12, 7566–7579 RSC.
- E. Gilardi, E. Fabbri, L. Bi, J. L. M. Rupp, T. Lippert, D. Pergolesi and E. Traversa, Effect of Dopant−Host Ionic Radii Mismatch on Acceptor-Doped Barium Zirconate Microstructure and Proton Conductivity, J. Phys. Chem. C, 2017, 121, 9739–9747 CrossRef CAS.
- D. Han, K. Shinoda, S. Sato, M. Majimad and T. Tetsuya, Uda. Correlation between electroconductive and structural properties of proton conductive acceptor-doped barium zirconate, J. Mater. Chem. A, 2015, 3, 1243 RSC.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, Cation Size Mismatch and Charge Interactions Drive Dopant Segregation at the Surfaces of Manganite Perovskites, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS.
- M. Shirpour, B. Rahmati, W. Sigle, P. A. van Aken, R. Merkle and J. Maier, Dopant Segregation and Space Charge Effects in Proton-Conducting BaZrO3 Perovskites, J. Phys. Chem. C, 2012, 116, 2453–2461 CrossRef CAS.
- S. Chung, S. L. Kang and V. P. Dravid, Effect of Sintering Atmosphere on Grain Boundary Segregation and Grain Growth in Niobium-Doped SrTiO3, J. Am. Ceram. Soc., 2002, 85, 2805–2810 CrossRef CAS.
- R. B. Wexler, G. S. Gautam, E. B. Stechel and E. A. Carter, Factors Governing Oxygen Vacancy Formation in Oxide Perovskites, J. Am. Chem. Soc., 2021, 143, 13212–13227 CrossRef CAS PubMed.
- Q. Yang, J. X. Cao, Y. Ma, Y. C. Zhou, L. M. Jiang and X. L. Zhong, Strain effects on formation and migration energies of oxygen vacancy in perovskite ferroelectrics: A first-principles study, J. Appl. Phys., 2013, 113, 184110 CrossRef.
- R. Merkle, Y. A. Mastrikov, E. A. Kotomin, M. M. Kuklja and J. Maier, First Principles Calculations of Oxygen Vacancy Formation and Migration in Ba1−xSrxCo1−yFeyO3−δ Perovskites, J. Electrochem. Soc., 2012, 159, B219–B226 CrossRef CAS.
- I. Kagomiya, K. Jimbo, K. Kakimoto and M. Nakayama, and Olivier Masson. Oxygen vacancy formation and the ion migration mechanism in layered perovskite (Sr,La)3Fe2O7, Phys. Chem. Chem. Phys., 2014, 16, 10875 RSC.
- J. M. Börgers and R. A. De Souza., The surprisingly high activation barrier for oxygen-vacancy migration in oxygen-excess manganite perovskites, Phys. Chem. Chem. Phys., 2020, 22, 14329 RSC.
- L. Zhang, B. Liu, H. Zhuang, P. R. C. Kent, V. R. Cooper, P. Ganesh and H. Xu, Oxygen vacancy diffusion in bulk SrTiO3 from density functional theory calculations, Comput. Mater. Sci., 2016, 118, 309–315 CrossRef CAS.
- J. J. Brown, Z. Ke, W. Geng and A. J. Page, Oxygen Vacancy Defect Migration in Titanate Perovskite Surfaces: Effect of the A-Site Cations, J. Phys. Chem. C, 2018, 122, 14590–14597 CrossRef CAS.
- M. Cherry, M. S. Islam and C. R. A. Catlow, Oxygen Ion Migration in Perovskite-Type Oxides, J. Sol. State Chem., 1995, 118, 125 CrossRef CAS.
- Y. Pai, A. Tylan-Tyler, P. Irvin and J. Levy, Physics of SrTiO3-based Heterostructures and Nanostructures: A Review, Rep. Prog. Phys., 2018, 81, 036503 CrossRef.
- M. Sillassen, P. Eklund, N. Pryds, E. Johnson, U. Helmersson and J. Bøttiger, Low-temperature Superionic Conductivity in Strained Yttria-stabilized Zirconia, Adv. Funct. Mater., 2010, 20, 2071–2076 CrossRef CAS.
- J. Garcia-Barriocanal, A. Rivera-Calzada, M. Varela, Z. Sefrioui, E. Iborra, C. Leon, S. J. Pennycook and J. Santamaria, Colossal Ionic Conductivity at Interfaces of Epitaxial ZrO2:Y2O3/SrTiO3 Heterostructures, Science, 2008, 321, 676–680 CrossRef CAS.
- S. M. Yang, S. Lee, J. Jian, W. Zhang, P. Lu, Q. Jia, H. Wang, T. W. Noh, S. V. Kalinin and J. L. MacManus-Driscoll, Strongly Enhanced Oxygen Ion Transport Through Samarium-doped CeO2 Nanopillars in Nanocomposite Films, Nat. Commun., 2015, 6, 8588 CrossRef CAS.
- P. P. Dholabhai, G. Pilania, J. A. Aguiar, A. Misra and B. P. Uberuaga, Termination Chemistry-driven Dislocation Structure at SrTiO3/MgO Heterointerfaces, Nat. Commun., 2014, 5, 5043 CrossRef CAS PubMed.
- P. P. Dholabhai, E. Martinez, N. T. Brown and B. P. Uberuaga, On the Mobility of Carriers at Semi-coherent Oxide Heterointerfaces, Phys. Chem. Chem. Phys., 2017, 19, 23122 RSC.
- P. P. Dholabhai, J. A. Aguiar, A. Misra and B. P. Uberuaga, Defect Interactions with Stepped CeO2/SrTiO3 Interfaces: Implications for Radiation Damage Evolution and Fast Ion Conduction, J. Chem. Phys., 2014, 140, 194701 CrossRef PubMed.
- P. P. Dholabhai and B. P. Uberuaga, Beyond Coherent Oxide Heterostructures: Atomic-scale Structure of Misfit Dislocations, Adv. Theory Simul., 2019, 2, 1900078 CrossRef.
- A. Lopez-Bezanilla, P. Ganesh and P. B. Littlewood, Plentiful magnetic moments in oxygen deficient SrTiO3, APL Mater., 2015, 3, 100701 CrossRef.
- A. B. Muñoz-García, A. M. Ritzmann, M. Pavone, J. A. Keith and E. A. Carter, Oxygen Transport in Perovskite-Type Solid Oxide Fuel Cell Materials: Insights from Quantum Mechanics, Acc. Chem. Res., 2014, 47(11), 3340 CrossRef.
- G. Pilania, P. P. Dholabhai and B. P. Uberuaga, Role of Symmetry, Geometry, and Termination Chemistry on Misfit Dislocation Patterns at Semicoherent Heterointerfaces, Matter, 2020, 2, 1324–1337 CrossRef.
- S. Plimpton, Fast Parallel Algorithms for Short-range Molecular Dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- R. A. Buckingham, The Classical Equation of State of Gaseous Helium, Neon and Argon, Proc. R. Soc. London, Ser. A, 1938, 168, 264 CAS.
- P. P. Ewald, Evaluation of Optical and Electrostatic Lattice Potentials, Ann. Phys., 1921, 64, 253–287 CrossRef.
- G. Busker, A. Chroneos, R. W. Grimes and I. Chen, Solution Mechanisms for Dopant Oxides in Yttria, J. Am. Ceram. Soc., 1999, 82, 1553–1559 CrossRef CAS.
- M. O. Zacate, L. Minervini, D. J. Bradfield, R. W. Grimes and K. E. Sickafus, Defect Cluster Formation in M2O3-doped Cubic ZrO2, Solid State Ionics, 2000, 128, 243–254 CrossRef CAS.
- P. R. Choudhury and S. B. Krupanidhi, Dielectric response of BaZrO3/BaTiO3 and SrTiO3/BaZrO3 superlattices, J. Appl. Phys., 2008, 104, 114105 CrossRef.
- N. A. Richter, S. Sicolo, S. V. Levchenko, J. Sauer and M. Scheffler, Concentration of Vacancies at Metal-Oxide Surfaces: Case Study of MgO(100), Phys. Rev. Lett., 2013, 111, 045502 CrossRef PubMed.
- P. P. Dholabhai, Oxygen Vacancy Formation and Interface Charge Transfer at Misfit Dislocations in Gd-Doped CeO2/MgO Heterostructures, J. Phys. Chem. C, 2022, 126, 11735–11750 CrossRef CAS.
- P. P. Dholabhai, Atomic-scale Structure of Misfit Dislocations in CeO2/MgO Heterostructure and Thermodynamic Stability of Dopant-defect Complexes at the Heterointerface, Phys. Chem. Chem. Phys., 2019, 21, 20878 RSC.
- C. Marzano and P. P. Dholabhai, High-throughput prediction of thermodynamic stabilities of dopant-defect clusters at misfit dislocations in perovskite oxide heterostructures, J. Phys. Chem. C, 2023, 127, 15988 CrossRef CAS.
- P. P. Dholabhai, R. Perriot and B. P. Uberuaga, Atomic-Scale Structure and Stability of the Low-Index Surfaces of Pyrochlore Oxides, J. Phys. Chem. C, 2016, 120, 10485 CrossRef CAS.
- M. M. Hasan, P. P. Dholabhai, R. H. R. Castro and B. P. Uberuaga, Stabilization of MgAl2O4 Spinel Surfaces Via Doping, Surf. Sci., 2016, 649, 138–145 CrossRef CAS.
- J. A. Aguiar, P. P. Dholabhai, Z. Bi, Q. Jia, E. G. Fu, Y. Q. Wang, T. Aoki, J. Zhu, A. Misra and B. P. Uberuaga, Linking interfacial step structure and chemistry with locally enhanced radiation-induced amorphization at oxide heterointerfaces, Adv. Mater. Interfaces, 2014, 1(4), 1300142 CrossRef.
- P. P. Dholabhai, J. A. Aguiar, L. Wu, T. G. Holesinger, T. Aoki, R. H. R. Castro and B. P. Uberuaga, Structure and Segregation of Dopant–defect Complexes at Grain Boundaries in Nanocrystalline Doped Ceria, Phys. Chem. Chem. Phys., 2015, 17, 15375 RSC.
- R. Perriot, P. P. Dholabhai and B. P. Uberuaga, Disorder-induced Transition from Grain Boundary to Bulk Dominated Ionic Diffusion in Pyrochlores, Nanoscale, 2017, 9, 6826 RSC.
- M. M. Hasan, P. P. Dholabhai, S. Dey, B. P. Uberuaga and R. H. R. Castro, Reduced Grain Boundary Energies in Rare-earth Doped MgAl2O4 Spinel and Consequent Grain Growth Inhibition, J. Eur. Ceram. Soc., 2017, 37, 40433 CrossRef.
- N. Kuwata, N. Sata, S. Saito, T. Tsurui and H. Yugami, Structural and electrical properties of SrZr0.95Y0.05O3/SrTiO3 superlattices, Solid State Ionics, 2006, 177, 2347–2351 CrossRef CAS.
- P. P. Dholabhai, S. Anwar, J. B. Adams, P. Crozier and R. Sharma, Kinetic lattice Monte Carlo model for oxygen vacancy diffusion in praseodymium doped ceria: Applications to materials design, J. Sol. State Chem., 2011, 184, 811–817 CrossRef CAS.
- P. P. Dholabhai and J. B. Adams, A Blend of First-principles and Kinetic Lattice Monte Carlo Computation to Optimize Samarium-doped Ceria, J. Mater. Sci., 2012, 47, 7530 CrossRef CAS.
- L. Zhang, B. Liu, H. Zhuang, P. R. C. Kent, V. R. Cooper, P. Ganesh and H. Xu, Oxygen vacancy diffusion in bulk SrTiO3 from density functional theory calculations, Comput. Mater. Sci., 2016, 118, 309–315 CrossRef CAS.
- M. Schie, A. Marchewka, T. Müller, R. A. De Souza and R. Waser, Molecular dynamics simulations of oxygen vacancy diffusion in SrTiO3, J. Phys.: Condens. Matter, 2012, 24, 485002 CrossRef.
- J. Claus, M. Leonhardt and J. Maier, Tracer diffusion and chemical diffusion of oxygen in acceptor doped SrTiO3, J. Phys. Chem. Solids, 2000, 61, 1199–1207 CrossRef CAS.
- B. P. Uberuaga and L. J. Vernon, Interstitial and vacancy mediated transport mechanisms in perovskites: a comparison of chemistry and potentials, Solid State Ion., 2013, 253, 18 CrossRef CAS.
- S. L. Shang, B. C. Zhou, W. Y. Wang, A. J. Ross, X. L. Liu, Y. J. Hu, H. Z. Fang, Y. Wang and Z. K. Liu, A comprehensive first-principles study of pure elements: Vacancy formation and migration energies and self-diffusion coefficients, Acta Mater., 2016, 109, 128 CrossRef CAS.
- D. A. Andersson, S. I. Simak, N. V. Skorodumova, I. A. Abrikosov and B. Johansson, Optimization of Ionic Conductivity in Doped Ceria, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3518 CrossRef CAS PubMed.
- P. P. Dholabhai, J. B. Adams, P. Crozier and R. Sharma, Oxygen Vacancy Migration in Ceria and Pr-doped Ceria: A DFT+U Study, J. Chem. Phys., 2010, 132, 094104 CrossRef PubMed.
- M. Nakayama and M. Martin, First-principles Study on Defect Chemistry and Migration of Oxide Ions in Ceria Doped with Rare-earth Cations, Phys. Chem. Chem. Phys., 2009, 11, 3241 RSC.
- Y. Saito, J. Cheng, K. Crabb, H. Huang, R. Pornprasertsuk, P. C. Su and F. Prinz, Ion Conductivity Enhancement Effect by Introduction of Dislocations in Yttria-stabilized Zirconia, ECS Trans., 2008, 11, 3 CrossRef CAS.
- S. Lee, W. Zhang, F. Khatkhatay, H. Wang, Q. Jia and J. L. MacManus-Driscoll, Ionic Conductivity Increased by Two Orders of Magnitude in Micrometer-Thick Vertical Yttria-Stabilized ZrO2 Nanocomposite Films, Nano Lett., 2015, 15, 7362 CrossRef PubMed.
- X. Guo, Comment on Colossal Ionic Conductivity at Interfaces of Epitaxial ZrO2:Y2O3/SrTiO3 Heterostructures, Science, 2009, 324, 465 CrossRef CAS.
- X. Guo, E. Vasco, S. Mi, K. Szot, E. Wachsman and R. Waser, Ionic Conduction in Zirconia Films of Nanometer Thickness, Acta Mater., 2005, 53, 5161 CrossRef CAS.
- D. Pergolesi, E. Fabbri, S. N. Cook, V. Roddatis, E. Traversa and J. A. Kilner, Tensile Lattice Distortion Does Not Affect Oxygen Transport in Yttria-Stabilized Zirconia CeO2 Heterointerfaces, ACS Nano, 2012, 6, 10524 CrossRef CAS.
- D. Marrocchelli, L. Sun and B. Yildiz, Dislocations in SrTiO3: Easy to Reduce but Not so Fast for Oxygen Transport, J. Am. Chem. Soc., 2015, 137, 4735 CrossRef CAS.
- S. P. Waldow and R. A. De, Souza. Computational Study of Oxygen Diffusion along a[100] Dislocations in the Perovskite Oxide SrTiO3, ACS Appl. Mater. Interfaces, 2016, 8, 12246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Interface atomic arrangements for STO and BZO layers in STO/BZO heterostructures for different interface chemistry. See DOI: https://doi.org/10.1039/d3ma00850a |
| This journal is © The Royal Society of Chemistry 2024 |