
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne pot oxygen mediated syntheses of stable radicals†
Mohit
Kulshrestha
a,
C. N.
Ramachandran
 *a,
Rajesh G.
Gonnade
*b and
Kalyan K.
Sadhu
*a
*a,
Rajesh G.
Gonnade
*b and
Kalyan K.
Sadhu
*a
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee–247667, Uttarakhand, India. E-mail: ramcn@cy.iitr.ac.in; sadhu@cy.iitr.ac.in
bPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Pune–411008, Maharashtra, India. E-mail: rg.gonnade@ncl.res.in
First published on 1st December 2023
Abstract
Air- and thermally-stable emissive carbon-centered radicals (CCRs) were synthesized at room temperature in open air by varying either the base concentration or the dilution factor of the reaction mixture. The carbon radical centers in the synthesized DCP˙, DCPC˙ and CP˙ have two adjacent C(sp2) in the chromenopyridine moiety and are further connected to C(sp3), C(sp2) or H atom, respectively. In situ generated DCPH, which contains contiguous C(sp3)–H bonds in chromenopyridine and dicyanomethyl moieties, is responsible for the base- and oxygen-mediated synthesis of these CCRs. Among these radicals, DCPC˙ having a π-tetramer in its crystal structure shows temperature-dependent electron paramagnetic resonance (EPR) in the solid state. CP˙ behaves differently in the NMR solvents DMSO-d6 and D2O producing nonaromatic and aromatic species, respectively. In water, CP˙ generates kinetically controlled green-red dual emissive cationic species CP(KC)+, which showed pH-dependent reversible absorbance and fluorescence ON–OFF patterns through neutral CP–H.
Introduction
Persistent and isolable organic CCRs, which can tolerate air and water, are always directly bound either by sp or sp2 hybridized carbon atoms or by heteroatoms at adjacent sites.1 Prominent examples of such CCRs have been observed only in the last decade.1,2 Supramolecular host–guest interactions accommodate the air stable radical cationic dimers of tetrathiafulvalene as guests in the catenane host2a and radical anions as guest ligands2b within a metal–organic framework. Zwitterionic tetrathiafulvalene dicarboxylate radical shows metallic conduction behavior even at low temperatures.2c Radical stabilization in an aqueous medium has also been achieved through Zn(II)-metalloprotein containing the semiquinone radical anion.2d Stable organic biradicals show photothermal activity for solar-driven water evaporation.2e The stability of CCRs depends on the temperature and substitution around the radical center. Dicyanomethyl radicals, which are connected to two neighboring sp and one sp2 hybridized carbon atoms, show temperature-dependent radical pairing either through σ or π interactions.2f–i CCRs, which are surrounded with neighboring sp3 hybridized carbon atoms, participate as intermediates in C(sp3)–C(sp3) σ bond synthesis.3In general, most of these stable radicals are not emissive owing to the presence of a single unpaired electron in SOMO. However, there are a few reports on CCRs that show emission in non-aqueous media. Substituted triphenylmethyl radicals or mono-, bis- and tris-pyridyl/phenyl-methyl radicals are emissive in either solid state or solvents with the polarity range between toluene and dichloromethane.3 The radical anion of carbazole together with its isomer as indole-based radical cation result in charge separated phosphorescence.4 Scanning tunneling microscopy has been utilized to measure solid state fluorescence of a single zinc-phthalocyanine radical cation adsorbed on a NaCl-covered Au(111) sample.5 Mechanical stress induced C(sp3)–C(sp3) bond cleavage produces pink colored C(sp2) attached CCR with yellow emission.6 Among heme metabolites, the tripyrridione radical shows red luminescence in THF.7 Recently boron-stabilized CCR results in a sufficiently high quantum yield (ϕ > 0.70) in nonpolar toluene as well as polar acetone solvents.8 In argon-saturated cyclohexane, the azaxanthone ketyl radical having an adjacent O–H bond shows very weak fluorescence (ϕ = 0.05) using flash photolysis.9
Synthetic routes for all these CCRs involve either photochemical, electrochemical or reagent-based oxidation reactions.1–10 Despite being an oxidizing agent, oxygen generates unstable CCRs as reaction intermediates during syntheses.10,11 Recently, Tang and coworkers synthesized oxygen-mediated radical cations from pyrrole-substituted derivatives (Scheme 1a and b).12 However, to the best of our knowledge, there is no report of oxygen-mediated synthesis of air and thermally stable neutral organic radicals. Immediate separation of this reactive oxygen species would be one of the potential ways to overcome the limitation of air-stable CCR synthesis. We have isolated three neutral radicals (Scheme 1c) by tuning the base and solvent in the presence of oxygen. Till date, to the best of our knowledge, there is no report of stable organic radicals in the solid state and these molecules show diamagnetic NMR in the solution state. In this work, we are establishing such observation for the first time along with a detailed analysis of both the phases.
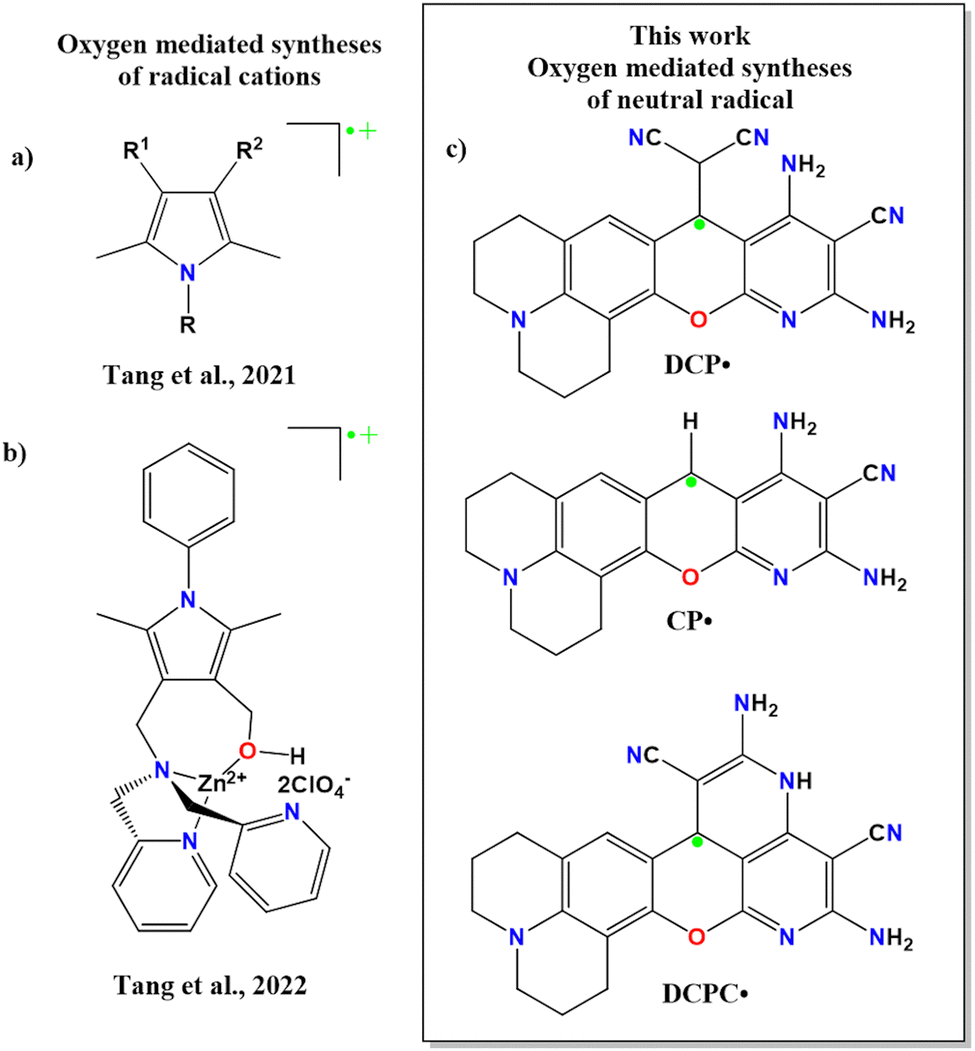 | ||
| Scheme 1 Oxygen-mediated syntheses of (a), (b) pyrrole-based radical cations and (c) neutral radicals. | ||
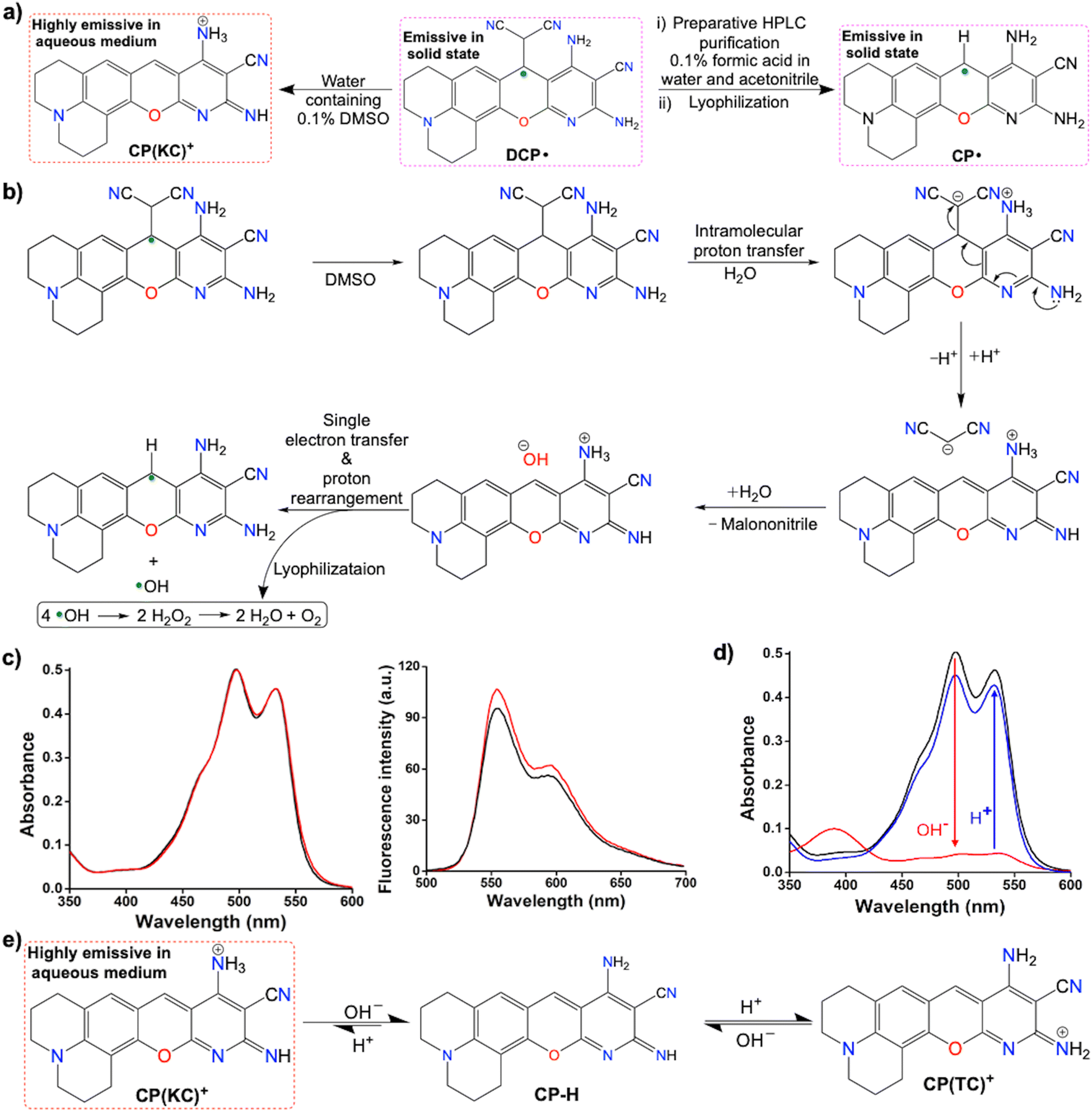
In this study, we have attempted to synthesize a fluorescent dye similar to our previous study (Scheme 2a)13 by changing the active methylene-based ester to malononitrile and a differently substituted salicylaldehyde (Scheme 2b). Fortuitously, we have obtained emissive radicals as a precipitate from the room temperature one-pot reaction between 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 malononitrile and 8-hydroxyjulolidine-9-carboxaldehyde (julolidine hereafter) with piperidine as a base in ethanol. The careful analysis confirmed the oxygen-mediated synthesis of thermally and air-stable emissive DCP˙ (Scheme 2c) in the reaction mixture. By changing the base concentration or the solvent in other batches of the reaction mixture, emissive CP˙ or DCPC˙ were obtained (Schemes 2d and 2e). Interestingly, upon the addition of the CP˙ radical in water, one electron was released to generate green-red dual emissive kinetically controlled cation CP(KC)+ species (Scheme 2f), which showed pH-dependent reversible absorbance and fluorescence ON–OFF patterns through neutral molecules for a few cycles.
:1 malononitrile and 8-hydroxyjulolidine-9-carboxaldehyde (julolidine hereafter) with piperidine as a base in ethanol. The careful analysis confirmed the oxygen-mediated synthesis of thermally and air-stable emissive DCP˙ (Scheme 2c) in the reaction mixture. By changing the base concentration or the solvent in other batches of the reaction mixture, emissive CP˙ or DCPC˙ were obtained (Schemes 2d and 2e). Interestingly, upon the addition of the CP˙ radical in water, one electron was released to generate green-red dual emissive kinetically controlled cation CP(KC)+ species (Scheme 2f), which showed pH-dependent reversible absorbance and fluorescence ON–OFF patterns through neutral molecules for a few cycles.
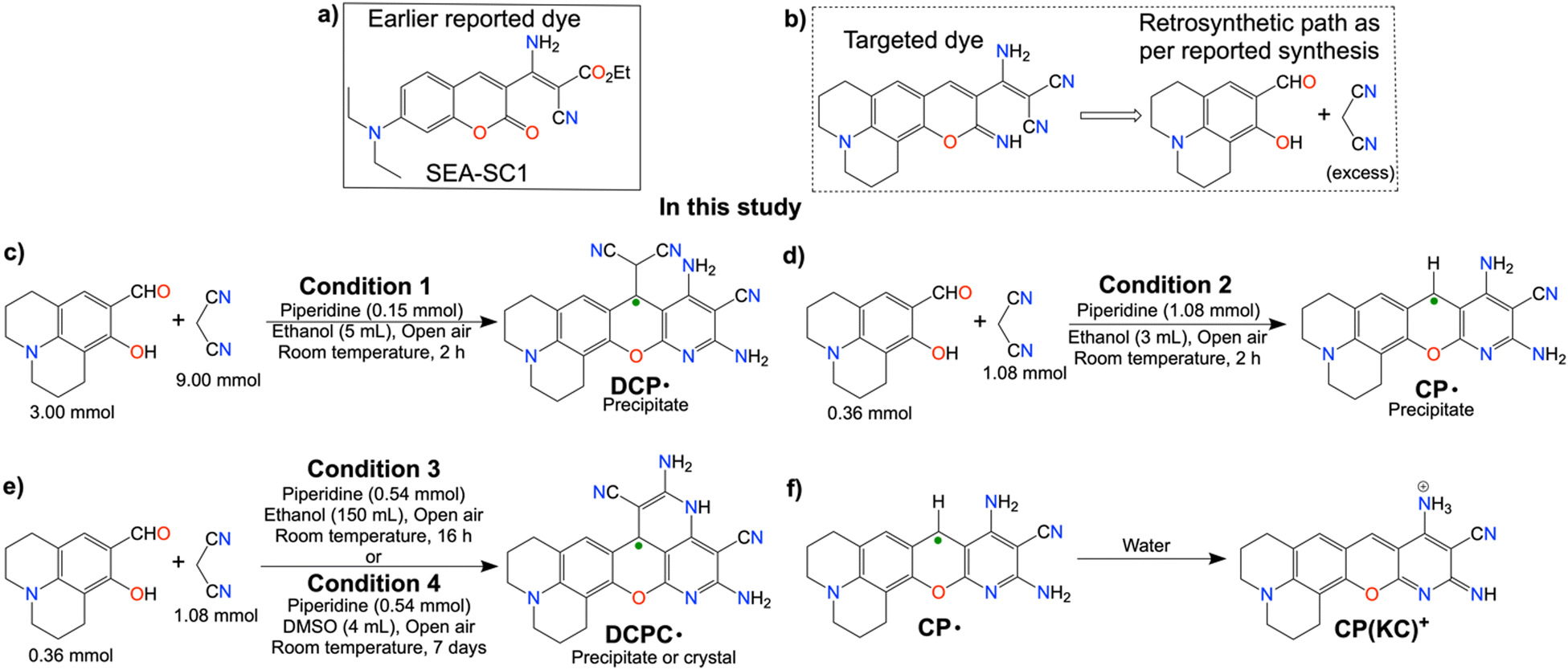 | ||
| Scheme 2 (a) Previously reported fluorescent dyes from our lab; (b) targeted synthesis of fluorescent dye by replacing the active methylene compound; (c)–(e) current study for the emissive radical synthesis: one pot room temperature synthesis of (c) DCP˙, (d) CP˙ and (e) DCPC˙ by varying piperidine equivalent to two different concentrated solutions containing malononitrile and 8-hydroxy julolidine-9-carboxaldehyde either in concentrated ethanolic solution, diluted ethanolic solution or DMSO solution, and (f) conversion of CP˙ to the CP(KC)+ cation by the addition of the radical in water. | ||
Results and discussion
Radical syntheses
In this study, three equivalents of malononitrile and one equivalent of julolidine are converted into an EPR-active red-colored precipitate in the presence of a base. Piperidine as a base was added to the solutions containing malononitrile and julolidine at different strengths in ethanol (Table S1, ESI†). Stirring of 1.80 M malononitrile and 600 mM julolidine in the reaction mixture with 30 mM piperidine in 5 mL ethanol for 2 h in air produced red precipitate of DCP˙ (Scheme 2c). Addition of 3.0 equivalent of TEMPO, the scavenger for the superoxide dismutation reaction,14 partially inhibits the radical synthesis as it interacts mostly with the superoxide after the rapid precipitation of CCR. The role of O2 in the DCP˙ synthesis was verified by performing the reaction in dry ethanol under continuous flow of nitrogen. Although the solution turns red due to the Knoevenagel condensation reaction, no precipitation was observed even after 4 h of the reaction. However, the same reaction precipitates DCP˙ within the next 15 min in the presence of air. The reaction in dry CD3OD in the absence of O2 was performed to find out the condensation product, which was producing the red solution (Fig. S1, ESI†).The reaction mixture of 360 mM malononitrile and 120 mM julolidine with 360 mM piperidine in 3 mL ethanol produces CP˙ as a red precipitate (Scheme 2d and Table S1, ESI†) by breaking the C(sp3)–C(sp3) bond. CP˙, which is sufficiently water soluble, forms CP(KC)+ cationic species (Scheme 2f) by losing one electron in water.15 Multiple fast reactions from the hydrated electron result in hydroxide ions in the solution as the counter anion.15
The synthetic attempt of CP˙ in DMSO by lowering the base concentration results in a single crystal of yellow cyclized DCPC˙ (Scheme 2e) after 6 days. The same quantity of the reactants in 150 mL ethanol also produces DCPC˙ as the precipitate after 16 h. HRMS studies of all these three radicals show the characteristic molecular weight of the radicals (Fig. S2–S4, ESI†). 4-aminopiperidine behaves similarly to piperidine and does not affect the yields much. Radical yields were significantly lowered in the presence of 1.5 or 3.0 eq. of trimethylamine, and trimethylamine, for the reactions between 360 mM malononitrile and 120 mM julolidine in 3 mL EtOH. Aniline does not give a precipitate under both DCP˙ and CP˙ synthetic conditions. Strong bases such as DBU and tetramethyl guanidine also give comparable yields of piperidine (Table S1, ESI†).
DCP˙ radical: adjacent C(sp3) substituted CCR
Formation of DCP˙ in the solid state was confirmed from the EPR signal (g = 2.00), which remained unaltered within 300 K to 100 K (Fig. 1a and Fig. S5, ESI†). The solid state 1H and 13C NMR (Fig. S6, ESI†) of DCP˙ show broad peaks. The thermal stability study of DCP˙ in air showed no decomposition up to 140 °C (Fig. S7, ESI†). The visible region absorption of DCP˙ in the solid state showed peaks at 435 nm, 510 nm, and 545 nm (Fig. 1b). The radical shows three emission peaks at 495 nm, 520 nm, and 595 nm (Fig. 1c, ϕabs = 0.71) in the solid state by exciting the sample at 435 nm. Theoretical studies were carried out on both DCP˙ and CPD˙ (Fig. S8, ESI†) with radical centers on couminopyridine (CP) and dicyanomethyl (D) moieties, respectively, to identify the radical center. Comparison of TDDFT (Fig. S9, ESI†) results with the experimental absorption study confirmed the formation of DCP˙, where calculated spin density is mostly localized on the carbon center (Fig. 1d).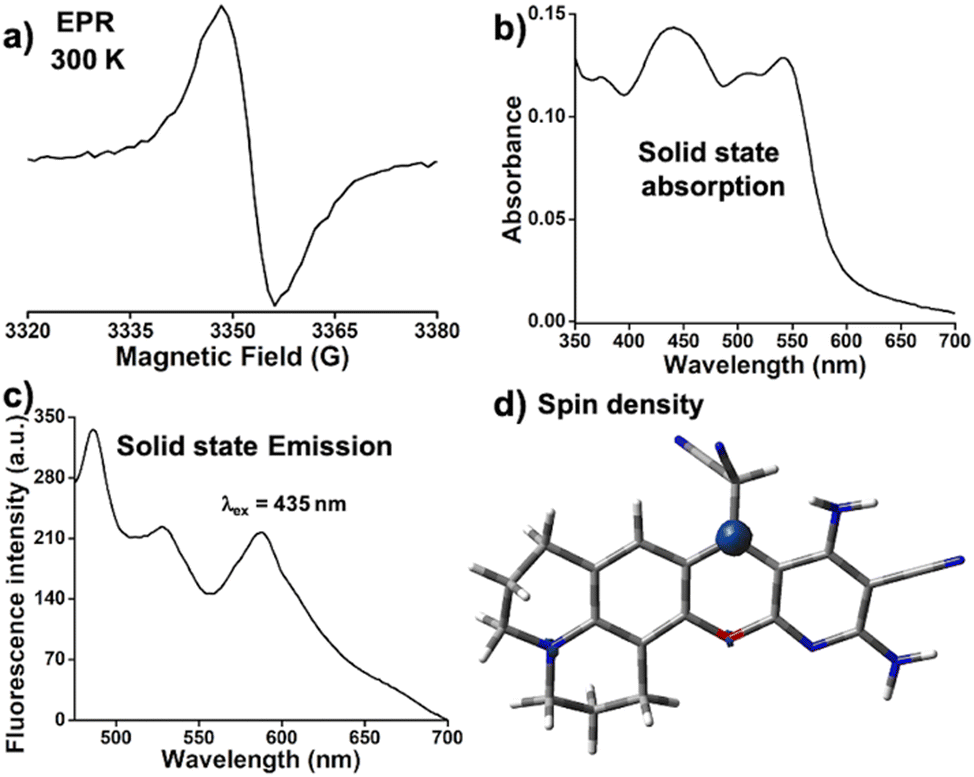 | ||
| Fig. 1 DCP˙ radical: (a) EPR signal (g = 2.00) at 300 K, solid-state (b) absorption and (c) emission (λex = 435 nm) of DCP˙ and the (d) calculated spin density of DCP˙. | ||
Among the common organic solvents, DCP˙ is only soluble in DMSO and DMF, but both solutions showed no EPR signal (Fig. S10, ESI†). The NMR study of DCP˙ in DMSO-d6 showed diamagnetic NMR (Fig. S11, ESI†) due to the formation of DCPH (Fig. 2a) by the overall abstraction of the hydrogen atom from the solvent. Poor EPR signal (Fig. S12, ESI†) was observed immediately after the addition of DCP˙ in DMSO due to the formation of DMSO radicals, which disappeared with time (Fig. S10, ESI†). A few minor peaks in NMR were present due to the degradation of the DMSO-d6 radical (Fig. S13, ESI†). Other substituted DCPH syntheses were reported without any radical formation.16 The DCP˙ radical in DMSO was confirmed by observing EPR in the presence of the spin-trapping agent DMPO at 100 K. (Fig. S14a, ESI†). Furthermore, the NMR of DCP˙ was taken in the presence of DMPO in DMSO-d6 at room temperature, it was observed broad, and one of the characteristic peaks was diminished due to the adduct formation of DMPO with DCP˙. (Fig. S15, ESI†)
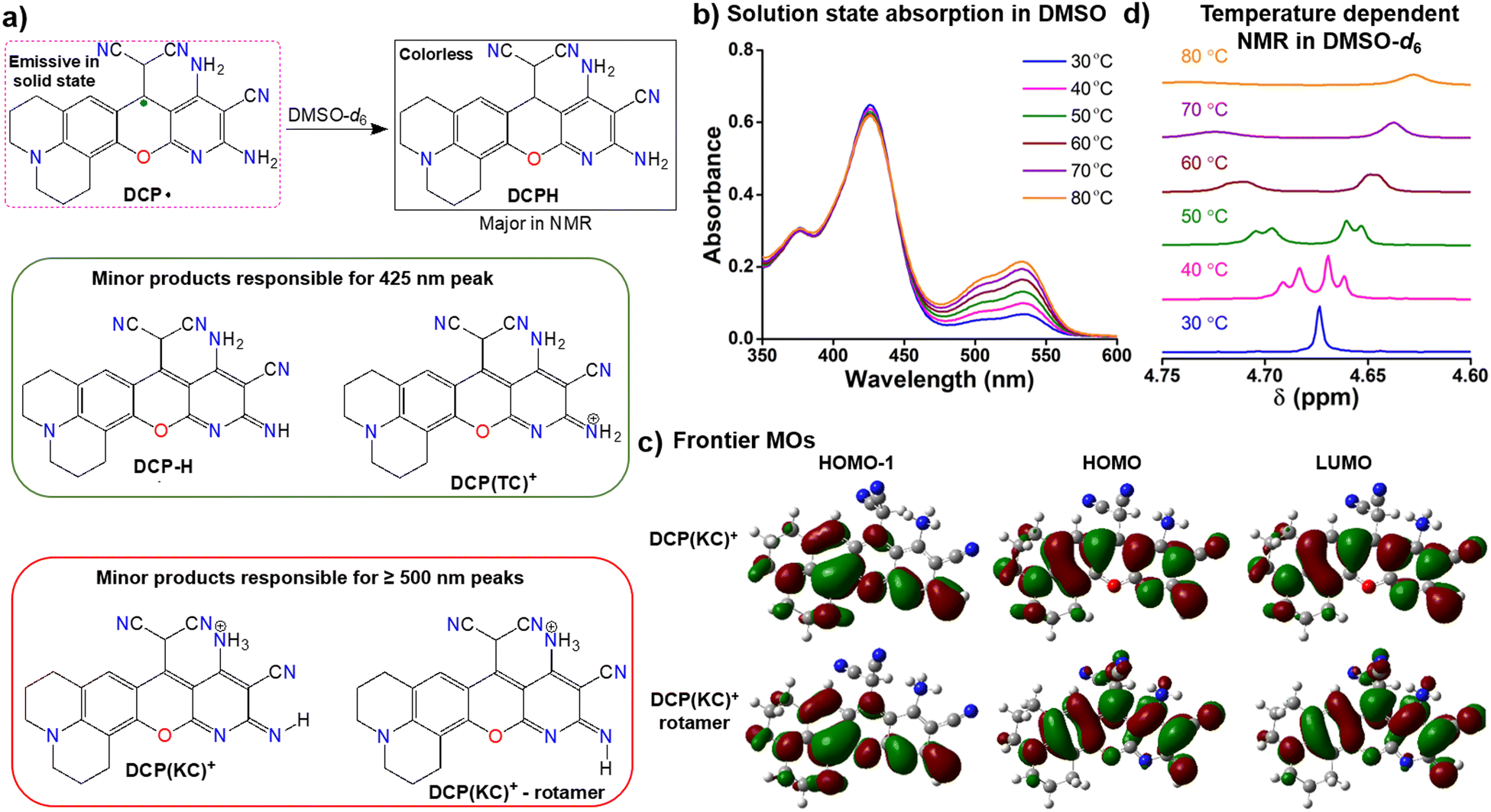 | ||
Fig. 2 DCP˙ radical: solution state studies and theoretical correlation. (a) A schematic representation of the speciation from DCP˙ in DMSO solution; (b) absorption studies of DCP˙ as DCP(KC)+ in DMSO till 80 °C; (c) frontier molecular orbitals involved in the electronic transition for DCP(KC)+ and its imine (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–H) rotamer; (d) temperature-dependent 1H-NMR studies for the conversion of DCPH to DCP(KC)+ till 80 °C in DMSO solution. N–H) rotamer; (d) temperature-dependent 1H-NMR studies for the conversion of DCPH to DCP(KC)+ till 80 °C in DMSO solution. | ||
The reaction was carried out at the NMR scale in DMSO-d6 to understand the reaction pathway. During the reaction, two characteristic peaks corresponding to C(sp3)–H for the in situ generated DCPH (Fig. S16, ESI†) were observed. The presence of organic base and oxygen is responsible for the conversion of DCPH to DCP˙ as per the reported method.17 Theoretical studies predict the synthetic route of DCP˙ from the in situ generated DCPH through the base-mediated synthesis of CPD−, followed by a single electron transfer to oxygen and radical center conversion to energetically favorable DCP˙ (Fig. S17 and S18, ESI†). The high energy barrier for the conversion of CPD− to DCP− by proton transfer (Fig. S19, ESI†) restricts the conversion via an alternate route. However, the energetically unfavorable (ΔG = +69.32 kcal mol−1) process for the radical formation from DCPH is only possible due to the phase separation of DCP˙ in the reaction condition.
At room temperature, the red NMR solution of DCP˙ exists as DCPH form in DMSO-d6. However, the TDDFT study on DCPH suggests no significant absorption in the visible region (Fig. S20, ESI†) due to a lack of π-conjugation. Absorption of the DMSO solution at 30 °C shows one major peak at 425 nm and two minor peaks at 500 and 535 nm (Fig. 2b). TDDFT studies indicate thermodynamically-controlled cation DCP(TC)+ or neutral DCP–H (Fig. 2a) for the 425 nm absorption and kinetically controlled cation DCP(KC)+ and its imine N–H rotamer (Fig. 2a and Fig. S21, ESI†) for the dual absorption peaks ≥500 nm. These cationic species originate from the radical disproportionation reaction18 (Fig. S22, ESI†). The corresponding anion receives a proton from DMSO to form DCPH and the dimsyl anion.19 The cationic species accepts an electron from the dimsyl anion to generate DCP˙ and DMSO radicals (Fig. S22, ESI†).20 This overall conversion produces major DCPH as confirmed in NMR (Fig. S11, ESI†). Minor cationic DCP(KC)+ and DCP(TC)+ or neutral DCP–H (Fig. 2a) products are responsible for coloration. The n → π* and π → π* electronic transitions within these cationic and neutral species involve HOMO−1 to LUMO orbitals (Fig. 2c and Fig. S23, ESI†).
Upon heating the DMSO solution to 80 °C, a significant increase in the absorption intensities of the two peaks ≥ 500 nm was observed (Fig. 2b). This change in the absorbance can be correlated with temperature-dependent 1H-NMR studies, where a 20% decrease in the CP-based C(sp3)–H peak (Fig. 2d) was observed at 80 °C due to the formation of DCP(KC)+ species. By exciting the DMSO solution at 535 nm for DCP(KC)+ species, dual emissions were observed at 560 nm and 600 nm (ϕrel = 0.56, Fig. S24, ESI†). An enhancement of the emission intensities was observed upon heating the solution (Fig. S25, ESI†).
DCPC˙ radical: three C(sp2) substituted CCR
A single electron transfer reaction from CPD− to oxygen produces the CPD˙ and superoxide, which is unstable to show the EPR signal in ethanol. In the case of the reaction in DMSO for DCPC˙, the filtrate shows an EPR signal (Fig. 3a) due to the high stability of superoxide in DMSO.21 Thermal stability of DCPC˙ in the presence of air is significantly enhanced in comparison to the recently reported highest thermally stable organic radicals22 and DCP˙ (Fig. S26, ESI†). Solid state NMR shows broad peaks (Fig. S27, ESI†) and a redical nature was observed in EPR (Fig. 3b).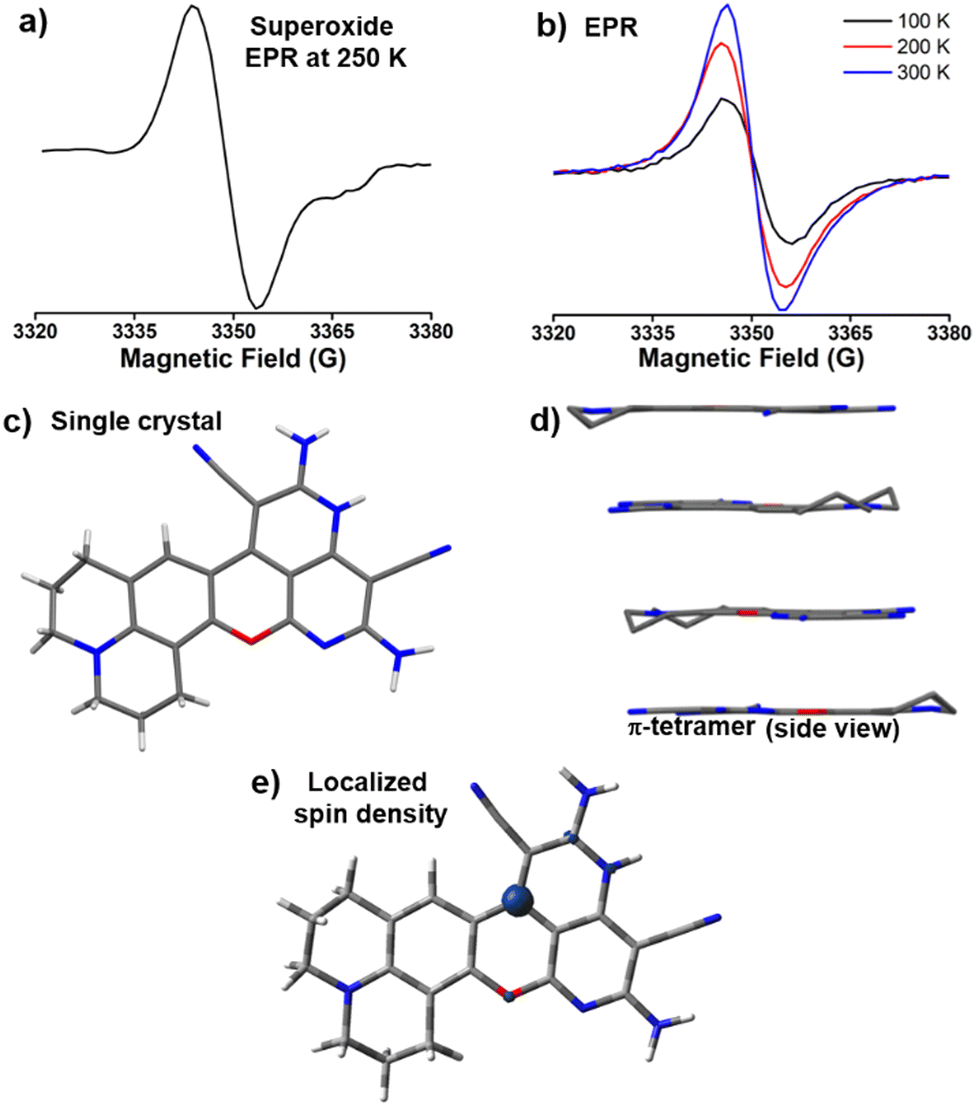 | ||
| Fig. 3 DCPC˙ radical: spectroscopy, crystal structure and spin density. (a) The EPR signal at 250 K for the superoxide (g = 2.00) generated in DMSO solution after 6 days of the reaction with 3:1 malononitrile and 8-hydroxy julolidine-9-carboxaldehyde in the presence of 1.5 equivalent piperidine, (b) temperature dependent EPR signals (g = 2.00) for DCPC˙, (c) the DCPC˙ single crystal asymmetric unit (solvent DMSO molecules are omitted for clarity), (d) side view of the π-tetramer in DCPC˙ and (e) the localized spin density of DCPC˙. | ||
DCPC˙ from the DMSO reaction mixture crystallizes in orthorhombic centrosymmetric Pbcn space group containing one molecule of DCPC˙ (Fig. 3c) and two DMSO solvent molecules in the asymmetric unit. In the unit cell, a total of eight DCPC˙ molecules are present. Crystal packing reveals the presence of the radical π-tetramer, which are related by screw axes and center of symmetry (Fig. 3d). The presence of screw axes between the π-tetramer generates residual non-zero electron spin, which is responsible for the EPR signal of DCPC˙ even at 100 K (Fig. 3b). The repulsion between the unpaired electrons within π-tetramer of DCPC˙ results in slightly higher π–π distances (∼3.55 to 3.77 Å) than the reported π-tetramer23 (3.34 Å) in N-butoxyphenyl(naphthalene)diimide. The temperature-dependent boost in the EPR intensities of DCPC˙ takes place due to the enhancement of π–π distances at 300 K as observed in the reported π-tetramer.23
The absorbance spectrum of DCPC˙ showed a major peak at 425 nm (Fig. S28, ESI†) with a broad emission (Fig. S29, ϕabs = 0.95, ESI†) in the solid state. TDDFT studies confirmed the absorbance of the radical species (Fig. S30, ESI†). Similar to the localized spin density for DCP˙, DCPC˙ has also localized spin density on the same carbon center (Fig. 3e). In the NMR study, unlike hydrogen abstraction for DCP˙ at room temperature, the DCPC˙ is soluble in DMSO-d6 in hot conditions and exists as DCPC+ (Fig. 4a) in diamagnetic NMR at 130 °C (Fig. S31, ESI†) and has no EPR signal (Fig. S32, ESI†). The difference in the solubility between DCP˙ and DCPC˙ might be due to the absence of any hydrogen adjacent to the radical position in DCPC˙. No change in the characteristic NMR peaks in the presence of TEMPO (Fig. S33, ESI†) confirmed the cationic form in the solution instead of the radical form. EPR of DCPC˙ in DMSO was observed in the presence of DMPO (Fig. S14b, ESI†). The temperature-dependent NMR of DCP˙ in DMSO-d6 shows the disappearance of both characteristic C(sp3)–H peaks for DCPH (Fig. S34, ESI†) and the appearance of new peaks at higher temperatures due to cyclization towards DCPC+ (Fig. 4b). Similar studies have been observed for the temperature-dependent absorption of DCP˙ in DMSO at >80 °C (Fig. S35, ESI†). The solution state absorption and emission due to DCPC˙ are obtained at 425 and 485 nm, respectively (ϕrel = 0.39, Fig. 4c) in DMSO. TDDFT studies (Fig. S36, ESI†) confirmed the origin of the absorption peak for DCPC+ species. However, in the aqueous medium, very weak absorption was obtained without any significant emission (Fig. S37, ESI†).
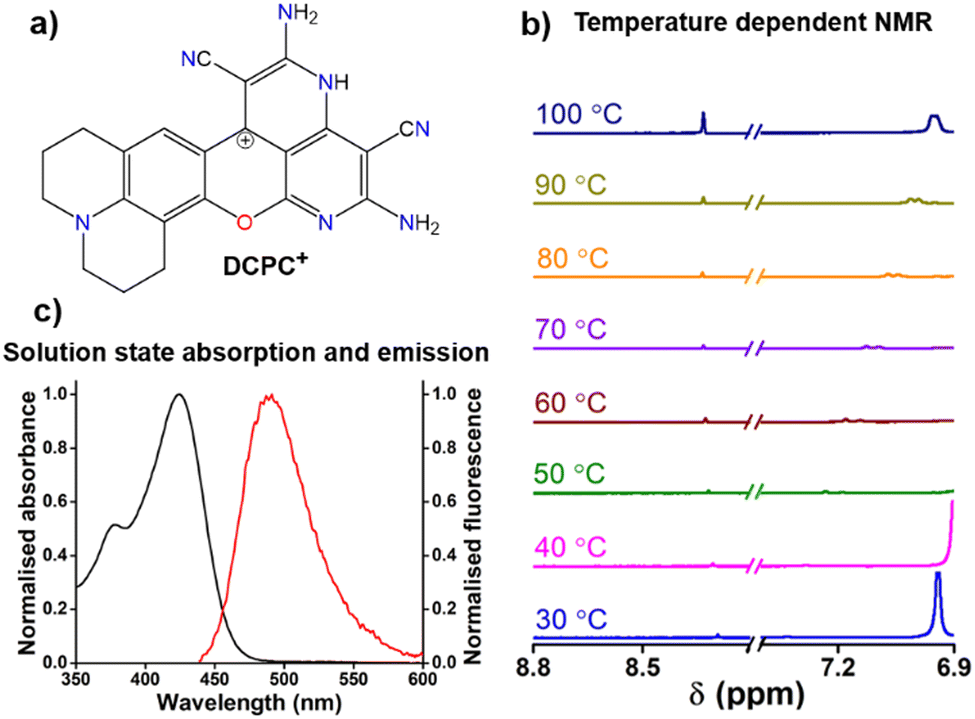 | ||
| Fig. 4 (a) Chemical structure of DCPC+; (b) temperature-dependent NMR data of DCP˙ radical as DCPH in DMSO-d showing generation of new peaks due to conversion to DCPC+ at high temperature; (c) normalized absorbance and emission properties of DCPC˙ as DCPC+ in DMSO. | ||
CP˙ radical: H atom substituted CCR and kinetically-controlled CP(KC)+ cation in water
Similar to DCP˙, CP˙ also provides temperature independent EPR signals (Fig. S38, ESI†), typical solid-state broad peaks NMR (Fig. S39, ESI†), and solid state absorbance and emission (ϕabs = 0.62, Fig. S40, ESI†). However, the thermal stability of CP˙ in the presence of air is greater than DCP˙ and comparable to DCPC˙ (Fig. S41, ESI†) due to the absence of any C(sp3) atom in CP˙ and DCPC˙. CP˙ also has localized spin density at the carbon centre (Fig. S42, ESI†). The solution state NMR of CP˙ also shows nonaromatic CPH by abstracting hydrogen from DMSO-d6 (Fig. 5a and Fig. S43, ESI†). Interestingly in D2O, CP˙ shows distinctly different diamagnetic NMR peaks (Fig. 5b and Fig. S44, ESI†) due to the formation of the CP(KC)+ cationic species. Also, EPR has been observed in DMSO in the presence of DMPO (Fig. S14c, ESI†). Nucleus-independent chemical shift calculations for all the rings containing these three new CCRs have been found to be anti-aromatic (Fig. S45, ESI†).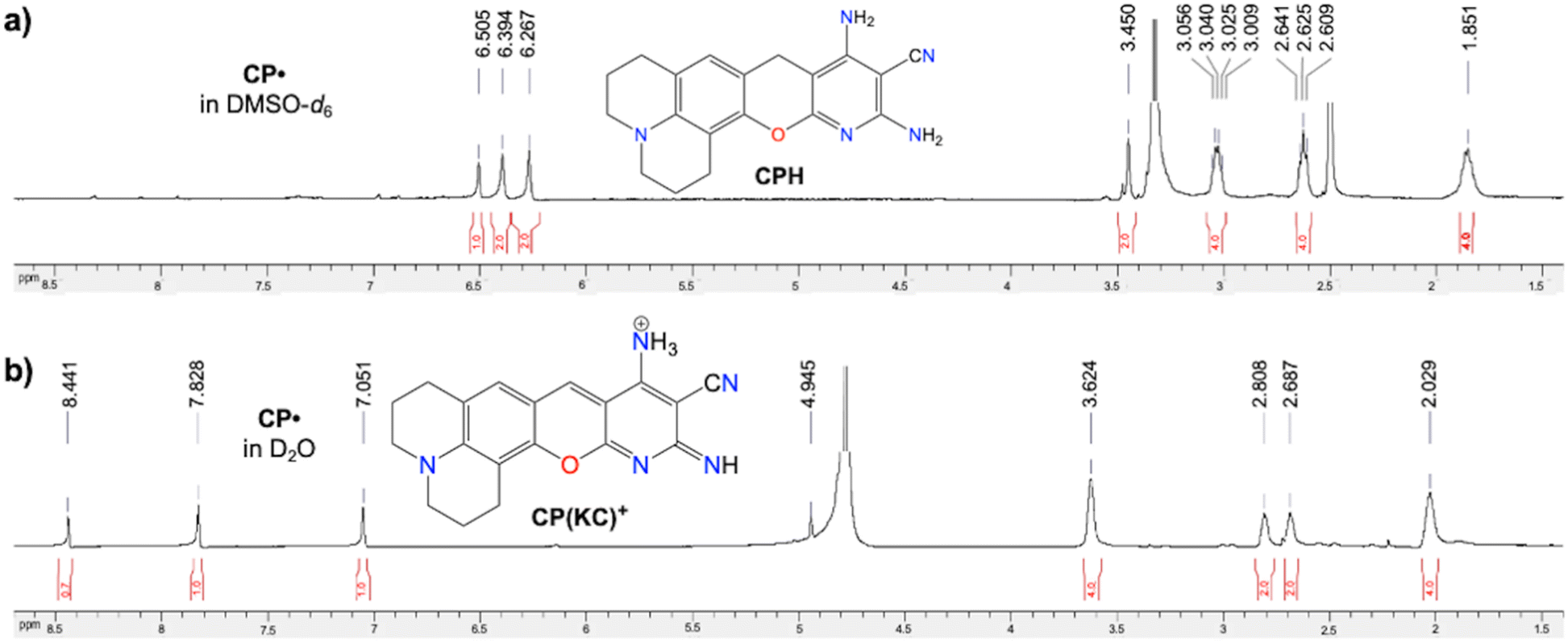 | ||
| Fig. 5 CP˙ radical: 1H NMR spectra of (a) nonaromatic CPH in DMSO-d6 and (b) aromatic CP(KC)+ in D2O. | ||
Color-based purification of DCP˙ was performed to obtain the emissive species selectively from its DMSO solution via the gradient mode using the preparative HPLC with acidic water and acetonitrile combination. Surprisingly, the characterization of the lyophilized solid after purification confirmed the conversion of DCP˙ to CP˙ (Fig. 6a). This is due to the C–C bond cleavage in water-mediated (Fig. 6b) or base-guided heterolytic cleavage24 (Fig. S46, ESI†) in the presence of radicals. The proposed formation of the CP(KC)+ cationic species in the water-mediated reaction was confirmed by the change of absorption with increasing amounts of water (Fig. S47, ESI†). The presence of a clear absorbance peak at 446 nm (Fig. S48 and S49, ESI†) before the generation of the dual peak during the synthesis of CP˙ indicates the formation of a CP–H intermediate due to the base-mediated breaking of the C–C bond. The emission generated from DCP˙ in water containing 0.1% DMSO (ϕrel = 0.77, Fig. 6c) was enhanced by DCP(KC)+ in DMSO (ϕrel = 0.56, Fig. S24, ESI†). The absorption (λabs = 500 nm and 535 nm) and emission (λem = 555 and 595 nm; λex = 535 nm) patterns from DCP˙ in water containing 0.1% DMSO are identical to the patterns of CP˙ in pure water due to the formation of CP(KC)+ in both the cases (Fig. 6d). The TDDFT studies of the optimized CP(KC)+ and its rotamer confirms the transition from HOMO to LUMO (Fig. S50, ESI†). The slight deviations in emission originating from DCP˙ are due to the presence of 0.1% DMSO, which significantly changes the absorption (Fig. S47, ESI†). Remarkably, the absorption and emission intensities of the CP˙ in the water solution containing 0.1% DMSO drastically decreased (Fig. S51, ESI†) due to the formation of non-emissive colorless CPH in DMSO (Fig. S52, ESI†).
 | ||
| Fig. 6 CP˙ radical: solution state spectroscopy. (a) A schematic representation of the conversion of DCP˙ to CP˙ and CP(KC)+; (b) the proposed mechanism for the water-mediated conversion of DCP˙ to the CP˙ radical via CP(KC)+; (c) absorbance and emission of CP(KC)+ obtained from 20 μM CP˙ in water (red) and DCP˙ in water containing 0.1% DMSO (black); (d) reversible acid–base cycle for the conversion of CP(KC)+ and CP–H and (e) a schematic representation of equilibrium shifting between CP(KC)+vs. CP(TC)+ and CP–H after a few rounds of deprotonation and protonation studies from CP(KC)+. | ||
The absorbance and emission properties of CP(KC)+ in the aqueous medium decreased (Fig. 6d) upon the addition of the base due to the formation of neutral CP–H (Fig. 6e). The shift in the absorbance of CP–H is due to the use of a strong base. The process is reversible with ∼10% loss of intensities in the presence of HCl (Fig. 6e). Fluorescence lifetime data confirm the enhancement of non-radiative decay rate constant upon the base treatment (Fig. S53 and Table S2, ESI†). Performing these acid–base reversible cycles 6 or more times, there is negligible change in the absorbance at 500 nm (Fig. S54, ESI†) and emission at 555 nm (Fig. S55, ESI†). This behavior is due to the equilibrium with the other thermodynamically-controlled CP(TC)+ cation (Fig. 6f and Fig. S56, ESI†), which has a similar absorption pattern to neutral CP–H. The pH-dependent absorption and emission studies further show that the change of the emission profile depending upon the excitation wavelengths at highly basic pH due to the complete conversion to neutral CP–H (Fig. S57 and S58, ESI†).
Conclusion
We exploited aerial O2 unknowingly as an electron acceptor for synthesizing three distinct air-stable fluorescent CCRs. We show that neighboring C(sp3)–H bonds are not only involved in the radical synthesis on the chromenopyridine unit but also play a key role in the emissive property. The solvent or concentration-dependent cyclization process shows the formation of C(sp2) atoms stabilized second radical, which is also emissive and stabilized by the tetrameric π–π interaction in the crystal structure. The excess amount of base or water content in the reaction mixture is accountable for the production of unique one adjacent H atom stabilized third radical, which in the presence of water produces kinetically controlled green-red dual emissive cationic species. We believe that these oxygen and water-mediated air-stable fluorescent organic radical synthetic routes will open new avenues in the synthesis of fluorescent dyes. We are currently focusing on the bioimaging applications of the green-red dual-emissive cationic and radical species.Author contributions
K.K.S. conceived the idea and wrote the manuscript. M.K. performed the synthesis, characterization, photophysical studies, and EPR experiments. C.N.R. performed the computational studies. R.G.G. solved the crystal structure. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. K. S. acknowledges the DST Nanomission (DST/NM/NB/2018/237) and SERB-DST Grant (CRG/2023/001415) for research funding. M. K. acknowledges the same two funds for his fellowship. The authors thank DST-FIST (SR/FST/CS-II/2018/72(C)) India for 500 MHz NMR support. The authors also thank Prof. Nicolas Winssinger from the University of Geneva, Switzerland for the helpful discussion.Notes and references
- (a) K. Cai, L. Zhang, R. D. Astumian and J. F. Stoddart, Nat. Rev. Chem., 2021, 5, 447 CrossRef CAS PubMed; (b) O. Haze, B. Corzilius, A. A. Smith, R. G. Griffin and T. M. Swager, J. Am. Chem. Soc., 2012, 134, 14287 CrossRef CAS PubMed.
- (a) J. M. Spruell, A. Coskun, D. C. Friedman, R. S. Forgan, A. A. Sarjeant, A. Trabolsi, A. C. Fahrenbach, G. Barin, W. F. Paxton, S. K. Dey, M. A. Olson, D. Benítez, E. Tkatchouk, M. T. Colvin, R. Carmielli, S. T. Caldwell, G. M. Rosair, S. G. Hewage, F. Duclairoir, J. L. Seymour, A. M. Z. Slawin, W. A. Goddard III, M. R. Wasielewski, G. Cooke and J. F. Stoddart, Nat. Chem., 2010, 2, 870 CrossRef CAS PubMed; (b) B. Lü, Y. Chen, P. Li, B. Wang, K. Müllen and M. Y. Lü, Nat. Commun., 2019, 10, 767 CrossRef PubMed; (c) Y. Kobayashi, T. Terauchi, S. Sumi and Y. Matsuhita, Nat. Mater., 2017, 16, 109 CrossRef CAS PubMed; (d) G. Ulas, T. Lemmin, Y. Wu, G. T. Gassner and W. F. DeGrado, Nat. Chem., 2016, 8, 354–359 CrossRef CAS PubMed; (e) G. Chen, J. Sun, Q. Peng, Q. Sun, G. Wang, Y. Cai, X. Gu, Z. Shuai and B. Z. Tang, Adv. Mater., 2020, 32, 1908537 CrossRef CAS PubMed; (f) R. Zhang, A. Ellern and A. H. Winter, Angew. Chem., Int. Ed., 2021, 60, 25082 CrossRef CAS PubMed; (g) B. Adinarayana, K. Kato, D. Shimizu, T. Tanaka, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2020, 59, 4320 CrossRef CAS PubMed; (h) K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. R. Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, J. Am. Chem. Soc., 2020, 142, 4329–4340 CrossRef CAS PubMed; (i) K. Okino, S. Hira, Y. Inoue, D. Sakamaki and S. Seki, Angew. Chem., Int. Ed., 2017, 56, 16597 CrossRef CAS PubMed.
- (a) Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091 CrossRef CAS PubMed; (b) X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536 CrossRef CAS PubMed; (c) H. Guo, Q. Peng, X. K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. L. Brédas and F. Li, Nat. Mater., 2019, 18, 977 CrossRef CAS PubMed; (d) A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224 CrossRef CAS PubMed; (e) S. Kimura, R. Matsuoka, S. Kimura, H. Nishihara and T. Kusamoto, J. Am. Chem. Soc., 2021, 143, 5610 CrossRef CAS PubMed; (f) K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175 CrossRef CAS PubMed.
- B. Doppagne, M. C. Chong, H. Bulou, A. Boeglin, F. Scheurer and G. Schull, Science, 2018, 361, 251 CrossRef CAS PubMed.
- S. Kato, S. Furukawa, D. Aoki, R. Goseki, K. Oikawa, K. Tsuchiya, N. Shimada, A. Maruyama, K. Numata and H. Otsuka, Nat. Commun., 2021, 12, 126 CrossRef CAS PubMed.
- E. Tomat and C. J. Curtis, Acc. Chem. Res., 2021, 54, 4584 CrossRef CAS PubMed.
- M. Ito, S. Shirai, Y. Xie, T. Kushida, N. Ando, H. Soutome, K. J. Fujimoto, T. Yanai, K. Tabata, Y. Miyata, H. Kita and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, 61, e202201965 CrossRef CAS PubMed.
- M. Sakamoto, X. Cai, M. Hara, S. Tojo, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2005, 127, 3702 CrossRef CAS PubMed.
- K. Masuda, M. Nagatomo and M. Inoue, Nat. Chem., 2017, 9, 207 CrossRef CAS PubMed.
- C. Tang, X. Qiu, Z. Cheng and N. Jiao, Chem. Soc. Rev., 2021, 50, 8067 RSC.
- (a) L. Zheng, W. Zhu, Z. Zhou, K. Liu, M. Gao and B. Z. Tang, Mater. Horiz., 2021, 8, 3082 RSC; (b) Z. Zhou, J. Qian, K. Liu, Y. Zhang, M. Gao and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202212671 CrossRef CAS PubMed.
- M. Saini, A. Verma, K. Tomar, P. K. Bharadwaj and K. K. Sadhu, Chem. Commun., 2018, 54, 12836 RSC.
- M. C. Krishna, D. A. Grahame, A. Samuni, J. B. Mitchell and A. Russo, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5537–5541 CrossRef CAS PubMed.
- B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, K. H. Bowen Jr., S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. LaVerne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L. S. Wang and T. S. Zwier, Chem. Rev., 2005, 105, 355–389 CrossRef CAS PubMed.
- (a) J. E. O’Brien, T. B. H. McMurry and C. N. O’Callaghan, J. Chem. Soc., Perkin Trans. 1, 1995, 417–420 Search PubMed; (b) M. N. Elinsona, S. V. Gorbunova, A. N. Vereshchagina, R. F. Nasybullina, A. S. Goloveshkin, I. S. Bushmarinov and M. P. Egorov, Tetrahedron, 2014, 70, 8559–8563 CrossRef; (c) S. Yadav, M. Srivastava, P. Rai, J. Singh, K. P. Tiwari and J. Singh, New J. Chem., 2015, 39, 4556–4561 RSC; (d) S. Nagaraju, K. Sathish and D. Kashinath, J. Heterocycl. Chem., 2021, 58, 1252–1258 CrossRef CAS.
- J. Li, M. J. Lear and Y. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 9060 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 7546 CrossRef CAS PubMed.
- E. Buncel, K.-T. Park, J. M. Dust and R. A. Manderville, J. Am. Chem. Soc., 2003, 125, 5388 CrossRef CAS PubMed.
- M. C. R. Symons, J. Chem. Soc., Perkin Trans. 2, 1976, 908 RSC.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS PubMed.
- X. Ye, L.-H. Chung, K. Li, S. Zheng, Y.-L. Wong, Z. Feng, Y. He, D. Chu, Z. Xu, L. Yu and J. He, Nat. Commun., 2022, 13, 6116 CrossRef CAS PubMed.
- M. Dharmarwardana, B. M. Otten, M. M. Ghimire, B. S. Arimilli, C. M. Williams, S. Boateng, Z. Lu, G. T. McCandless, J. J. Gassensmith and M. A. Omary, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106572118 CrossRef CAS PubMed.
- P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2211819. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ma00868a |
| This journal is © The Royal Society of Chemistry 2024 |